
Palladium-catalyzed Si–C bond-forming silylation of aryl iodides with hydrosilanes: an enhanced enantioselective synthesis of silicon-stereogenic silanes by desymmetrization†
Li
Chen
a,
Jiang-Bo
Huang
a,
Zheng
Xu
a,
Zhan-Jiang
Zheng
a,
Ke-Fang
Yang
*a,
Yu-Ming
Cui
a,
Jian
Cao
a and
Li-Wen
Xu
*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 311121, P. R. China. E-mail: liwenxu@hznu.edu.cn; licpxulw@yahoo.com; Fax: +86 2886 5135; Tel: +86 2886 5135
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, P. R. China
First published on 11th July 2016
Abstract
An enantioselective Pd-catalyzed silicon–carbon bond-forming silylation reaction of aryl iodides with hydrosilanes for the synthesis of silicon-stereogenic silanes has been developed, in which a systematic optimization of a TADDOL-derived monodentate phosphoramidite ligand set resulted in the identification of a new TADDOL-derived phosphoramidite ligand that accesses chiral silanes with moderate to good yield and enantioselectivity under mild conditions.
The selective construction of aryl carbon–silicon bonds is one of the most important and challenging reactions in element-organic chemistry and especially in the field of organosilicon chemistry,1 which enables the transformation of earth-abundant silicon resources to synthetically useful organosilicon compounds. Accordingly, extensive investigation of silicon–carbon bond-forming transformations has been carried out for the synthesis and application of valuable organosilicon compounds in versatile research fields.2 In this regard, the classic method for the preparation of aryl silanes is the traditional alkali metals-involved Wurtz-type coupling of chlorosilanes with organic halides. An alternative and improved method is the aryl magnesium reagent-based Grignard addition to chlorosilanes.3 Despite the direct nucleophilic substitution of chlorosilanes using an organometallic reagent is a useful approach to the synthesis of aryl silanes, the nucleophilic substitution reaction suffers from serious drawbacks such as the requirement of strictly anhydrous conditions, possibly toxic, difficult to handle, or less practical reaction temperatures. And especially, it is impossible to be used for the construction of chiral silanes by Wurtz- or Grignard-type addition. Gratifyingly, recent efforts have focused on the development of catalytic silylation of aryl halides with hydrosilanes by transition-metal complexes, which can widen the applicability of silicon–carbon bond-forming silylation reactions to catalytic asymmetric transformation with various aryl halides and functional silanes. Meanwhile, optically active organosilicon compounds bearing a silicon-stereogenic center have attracted much attention in the fields of organic synthesis, functional material, and bioorganic chemistry.4
Although the potential of palladium-catalyzed Si–C cross coupling approach with hydrosilanes has been revealed since 1994,5 limited progress has been reported in catalytic silicon–carbon-bond-forming silylation of aryl halides with hydrosilanes.6 In this context, Masuda et al. described the first example of palladium(0)-catalyzed Si–C bond-forming silylation of aryl halides with hydrosilane.7 And then in the subsequent decade, other groups have also reported a series of catalytic strategies for the palladium-promoted silylation of aryl halides with hydrosilanes in the presence of various phosphine ligands.8 However, the synthesis of silicon-stereogenic organosilicon compounds through palladium-catalyzed silicon–carbon bond-forming silylation with aryl halides and hydrosilanes is not an easy task because there is no successful example in the past decade.4c Notably, as the only example in this context, Yamanoi and Nishihara9 have ever reported an enantioselective Pd2(dba)3-catalyzed silicon–carbon bond-forming silylation of aryl halides with dihydrosilanes in the presence of chiral TADDOL-derived phosphoramidite ligand (Scheme 1), which afforded the optically active tertiary silanes with low to moderate enantioselectivities (11–77% ee for 13 examples) as well as low to moderate yields. This pioneering work also revealed the difficulty and challenge in the stereoselective construction of silicon-stereogenic silanes through palladium-catalyzed silicon–carbon cross-coupling of aryl halides with dihydrosilanes. Therefore, the development of an efficient or improved asymmetric method for the enantioselective silicon–carbon bond-forming silylation of aryl halides with dihydrosilanes is eagerly awaited. Herein we report an improved synthetic strategy with the development of highly efficient TADDOL-based chiral P-ligands – enhanced palladium-catalyzed silicon–carbon bond-forming silylation of aryl iodides with dihydrosilanes on the basis of previous findings in chiral phosphoramidite ligand chemistry.
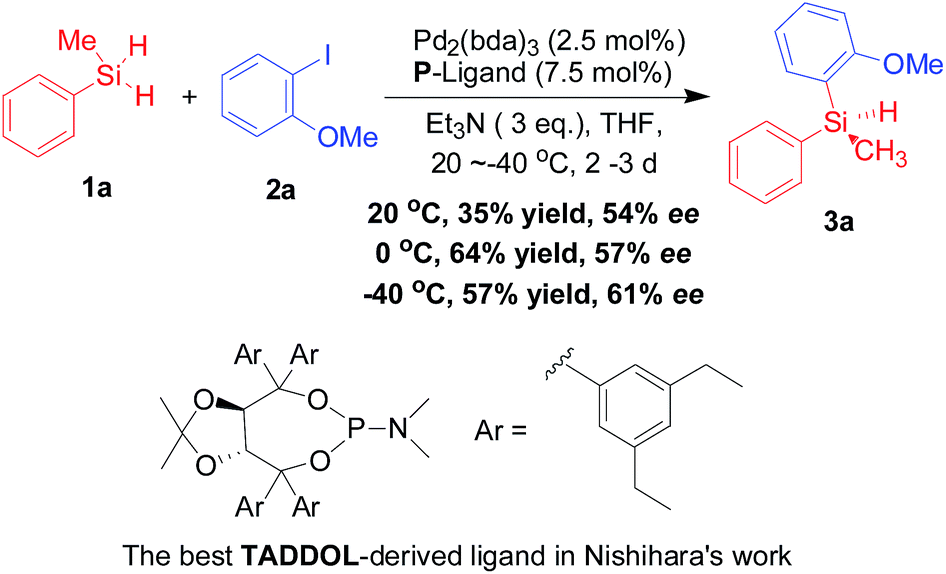 | ||
| Scheme 1 Previous and best result in the asymmetric palladium-catalyzed Si–C bond-forming silylation of 2a with 1a that reported by Nishihara and Yamanoi.9 | ||
Drawing inspiration from the powerful silicon–carbon cross-coupling strategies, in which the hydrosilanes are merged with aryl halides to generate aryl silanes, we envisioned that general phosphine ligands could be used in this reaction. However, in the preliminary investigation on the palladium-catalyzed silicon–carbon bond-forming silylation of aryl iodides with dihydrosilanes in the presence of commercially available P-ligands or our ligands10 that could be applied in various catalytic asymmetric transformations, almost no product was detected in these cases (Scheme 2).
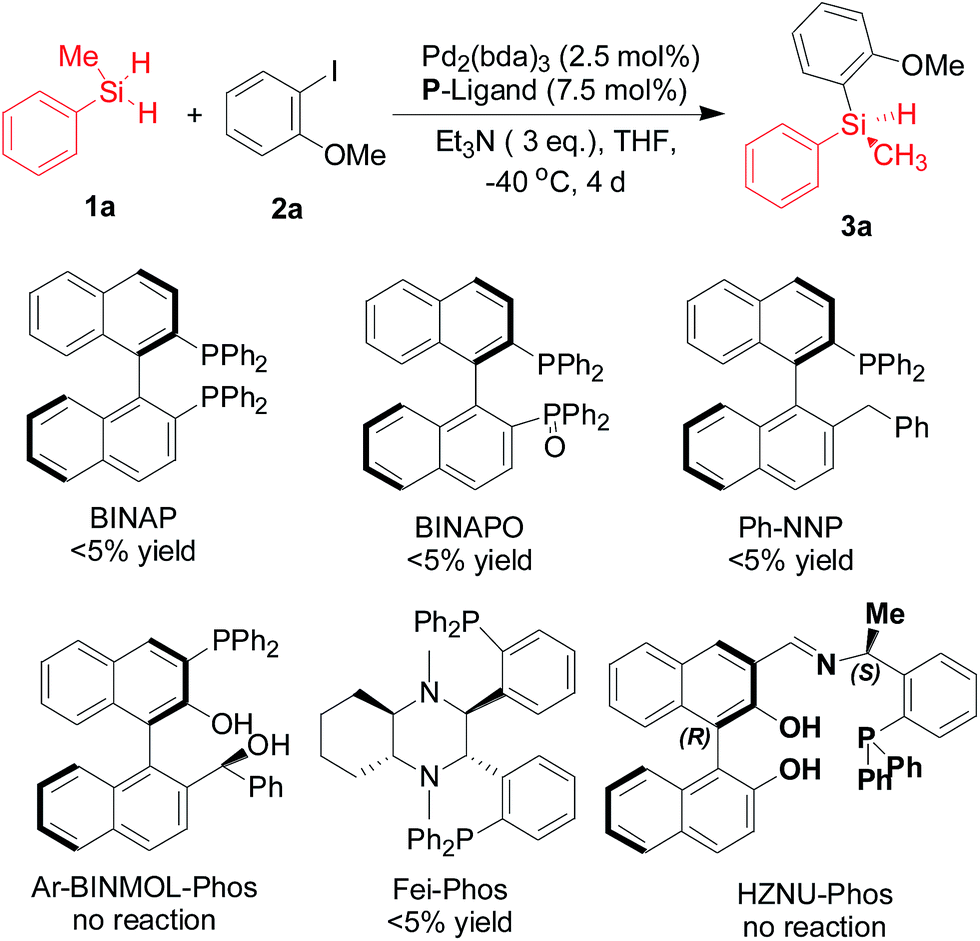 | ||
| Scheme 2 The evaluation of commercially available BINAP, BINAPO, and our ligands10 in the palladium-catalyzed silicon–carbon bond-forming silylation of aryl iodide 2a with dihydrosilane 1a. | ||
In light of previous progress in the palladium-catalyzed silicon–carbon bond-forming silylation of aryl iodides,5–9 we speculated that common TADDOL-derived phosphoramidite ligands with alkyl substituents in the 3,5-position of phenyl rings are not enough large with the aid of bulky group to control the enantioselectivity of such reactions. For high levels of stereocontrol, the TADDOL-derived phosphoramidite ligand would be needed to extend substituents by introduction of bulky group on ketal part and modification of nitrogen-center steric repulsion and electronic effect that could engage in a potential space interaction between ligand and substrate. In this way, the TADDOL-derived phosphoramidite could induce the molecular reaction tube with specific responses to certain type of substrates as a single-molecular reactor. Based on this hypothesis, we synthesized various TADDOL-derived phosphoramidite ligands bearing different substituents on ketal (modification 1) or aryl ring (modification 2) or amine (modification 3) showed in Scheme 3.11 These phosphoramidite ligands inherited the feature of TADDOL-based backbone and contain the features which were important for this reaction in the stereoselective induction of silicon–carbon bond-forming silylation.
 | ||
| Scheme 3 The optimization of TADDOL-based phosphoramidite by exploiting steric repulsion and electronic effect. | ||
Despite the TADDOL-derived chiral phosphoramidites have been widely used as effective P-ligands in various transition-metal-catalyzed asymmetric transformations,11,12 the structural modification of TADDOL-derived phosphoramidites with high level of enantioselectivity for silicon–carbon bond-forming silylation reaction is not an easy task. Then we began our investigations using methylphenylsilane 1a and 2-iodoanisole 2a as model substrates in this reaction (Table 1 and Scheme 1). With eight representative TADDOL-based phosphoramidites bearing different substituents in hand (Scheme 3), we investigated the enantioselective induction of these chiral phosphoramidites L1–L8 in silicon–carbon bond-forming silylation reaction of methylphenylsilane 1a and 2-iodoanisole 2a in the absence or presence of Pd2(dba)3. Interestingly, it was not perfect in both catalytic activity and enantioselectivity for all the TADDOL-derived phosphoramidites. As shown in Table 1, the silicon–carbon bond-forming silylation reaction carried out with TADDOL-derived phosphoramidites L1–L8 bearing different substituents on three positions at −40 °C proceeded to give the desired product in varied yields (25–90% yields) and low to good enantioselectivities (10–85% ee), which revealed the great contribution of these substituents on chiral TADDOL-based phosphoramidites to the enantioselective construction of silicon-stereogenic center in this reaction. For example, the TADDOL-derived phosphoramidite L1 (Ar = Ph, X = NMe2, Y1 = Y2 = Me) gave the desired product in good yield (90%) but with only low enantioselectivity (40% ee). Gratifyingly, the use of chiral TADDOL-derived phosphoramidites with N-methylaniline as a substituent, L2, L6, and L8, led to the enantioselectivity enhancement from 40% ee to 85% ee (Table 1, entries 2, 6, and 8). Especially, when the TADDOL-based phosphoramidite L8 (Scheme 3, Ar = 3,5-(t-Bu)2, 4-OMePh, X = NMePh, Y1 = Y2 = cyclohexyl) was used as a chiral ligand, the best enantioselectivity was achieved in this case (85% ee).
|
|
|||
|---|---|---|---|
| Entry | P-Ligand | Yielda (%) | eeb (%) |
| a The reaction was carried out with methylphenylsilane 1a (1.5 mmol). 2-Iodoanisole 2a (1.0 mmol), trimethylamine (3.0 equiv.), Pd2(dba)3 (0.025 mmol), ligand (0.075 mmol), in THF (2.0 mL), at −40 °C, for 4 days. And the isolated yield is purified by flash column chromatography. b The enantioselectivity was determined by chiral HPLC analysis employing a chiral stationary phase. And absolute configuration was determined by comparing with the literature value. | |||
| 1 | L1 | 90 | 40 |
| 2 | L2 | 59 | 75 |
| 3 | L3 | 42 | 28 |
| 4 | L4 | 38 | 10 |
| 5 | L5 | 51 | 42 |
| 6 | L6 | 56 | 79 |
| 7 | L7 | 25 | 36 |
| 8 | L8 | 50 | 85 |
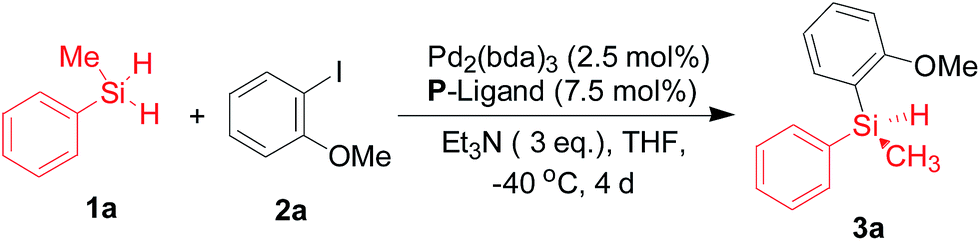
Encouraged by the ligand-controlled enantioselective enhancement outlined above, we turn attention to investigate the effect of different solvents, palladium salts, and bases, on the enantioselective silicon–carbon bond-forming silylation reaction of 2a with 1a, since they had been demonstrated to be important factors for catalytic asymmetric transformations. The screening results from these investigations are presented in Table S1 (see ESI†). With TADDOL-based phosphoramidite L8 as chiral ligand, the solvent effect on the catalytic activity of Pd2(dba)3 was really important and except THF, these solvents were found to the inferior media to promote the enantioselective silicon–carbon bond-forming silylation reaction with varied yields and enantioselectivities (entries 1–8, Table S1 of ESI†). For example, most of solvents led to the deceased enantioselectivity (17–79% ee), whereas the protic solvent, such as methanol, resulted in no reaction (entry 7, Table S1 of ESI†). Notably, although Pd(PPh3)2Cl2, PdCl2, and Pd(OAc)2 were also effective in this reaction (45–60% yields, 9–51% ee), Pd2(dba)3 turned out to be the best catalyst in term of enantioselectivity. In addition, we continued to investigate various bases, such as DBU, DIPEA, K2CO3, TMEDA, in the model silicon-bond forming silylation reaction (entries 14–17, Table S1 of ESI†). Surprisingly, TMEDA or K2CO3 was found to be the suitable base in this reaction (76% ee or 80% ee respectively, entries 16 and 17), which further supported the choice of solvent, base and ligand in Table 1. Notably, strong base, such as DBU, was proved to be non-effective for this reaction (entry 14, Table S1 of ESI†). Comparably, the use of DIPEA as base gave almost no conversion under the present reaction conditions (entries 17 and 18, Table S1 of ESI†). Although the optimized reaction conditions were not perfect in this reaction (up to 85% ee in this model reaction), the importance of chiral TADDOL-based phosphoramidite L8 as well as other factors was confirmed on the basis of these results. To the best of our knowledge, the enantioselectivity achieved in this work reached to the highest level for such silicon–carbon bond-forming silylation reaction.
With the optimized conditions, the catalytic activity of palladium/L8 complex generated in situ from TADDOL-based phosphoramidite (L8) and Pd2(dba)3 was then investigated in the silicon–carbon bond-forming silylation reaction of various aryl iodides. As shown in Scheme 4, the Pd/L8 catalyst system was also applicable to the silicon–carbon bond-forming silylation reaction of various aryl iodides that were converted into corresponding silicon-stereogenic silanes in moderate yields and promising enantioselectivities (up to 86% ee). Similarly to previous report, the silicon–carbon bond-forming silylation reaction of dihydrosilane with aryl iodides bearing methyl or methoxyl groups at p-, m-, and o-position afforded varied isolated yields (28–62% yields). Sterically hindered aryl iodides, such as 2-iodoanisole (2a), 1-iodo-2-methylbenzene (2d), 1-iodo-2,4-dimethylbenzene (2f), 1-iodonaphthalene (2g), 1-iodo-4,5-dimethoxy-2-methylbenzene (2j), and 1-iodo-2-methoxynaphthalene (2k), were also resulted in good ee value of the desired products (3d, 3f, 3j, and 3k), which supported the crucial role of steric repulsion between substrate and catalyst in this reaction. More importantly, the substituted methyl or methoxyl group at different positions on the phenyl ring of aryl iodides gave varied enantioselectivity for this silicon–carbon bond-forming silylation reaction. For example, the Pd/L8 catalyst exhibited different activity in enantioselective induction for the silicon–carbon bond-forming silylation reaction of methoxyl-substituted aryl iodides (2a–c) with methylphenylsilane, in which the order of enantioselectivity for methoxyl-substituted aryl iodides is 2-position (3a, 85% ee) > 3-position (3b, 35% ee) > 4-position (3c, 14% ee). Therefore, these experimental results provided useful and comprehensive information on the steric repulsion and electronic effect of aryl iodides in this reaction. Supplementary findings on the importance of electronic effect were also achieved from the experimental data of 3d–f (the order of ee value: 3f (86% ee) > 3d (78% ee) > 3e (69% ee)). Thus in this reaction, both the electronic effect and steric repulsion of substituted aryl iodides could not be ignored. Furthermore, 3j with 2-methyl substituent on the aryl ring gave better enantioselectivity (80% ee) than that without 2-methyl substituent on the aryl ring (3h, 21% ee), which indicated the crucial role of steric effect in the stereoselective construction of silicon-stereogenic silane. However, it should be noted that the crowded 2-iodo-1,3,5-trimethoxylbenzene (2i) or tert-butyl(phenyl)silane gave the corresponding silicon-stereogenic silane 3i or 3m respectively in low yield and poor enantioselectivity (28% yield and 36% ee for 3i, and 30% yield and 25% ee for 3m).
 | ||
| Scheme 4 Catalytic asymmetric silicon–carbon bond-forming silylation of aryl iodides with methylphenylsilane promoted by Pd/L8. | ||
Thus on the basis of these experimental results outed in Scheme 4, the chiral palladium catalyst system combined with TADDOL-based phosphoramidite ligand L8 still exhibited substrate-sensitive feature in the silicon–carbon bond-forming silylation reaction, which is similarly to previous reports on catalytic synthesis of silicon-stereogenic silanes by arylation of hydrosilanes with aryl iodides. In any event, the enantioselective silylation method described in this reaction is still an effective and improved procedure for the catalytic synthesis of various silicon-stereogenic silanes. Though only the good but not excellent enantioselectivity of this silicon–carbon bond-forming silylation reaction was observed, it is surprising that the catalytic role of palladium/L8 complex was realized on the silicon–carbon cross coupling reaction for the highest level of enantioselectivity with palladium catalysis at present. The probing exploration on the development of new TADDOL-derived phosphoramidite ligands seems to be a good attempt to improve the enantioselectivity in palladium-catalyzed silicon–carbon bond-forming silylation reaction.
In summary, we have developed an arduous investigation for the enantioselective synthesis of silicon-stereogenic silanes via silicon–carbon bond-forming silylation reaction with aryl iodides and dihydrosilane. The screening and optimization of reaction conditions, especially with the development of new P-ligand by modification of TADDOL-derived phosphoramidite ligands, resulted in the determination of an efficient procedure, which provided the corresponding silicon-stereogenic silanes in moderate yields and low to good enantioselectivities (up to 86% ee) under mild reaction conditions. In addition, to the best of our knowledge, it is one of the best examples of asymmetric palladium-catalyzed silicon–carbon cross coupling reaction for a stereoselective synthesis of silicon-stereogenic silanes with good enantioselectivity, in which the adaption of TADDOL-based phosphoramidite L8 with the intermolecular interaction between catalyst and substrate indicated that the control of steric and electronic effect of chiral ligand is very important in asymmetric catalysis. Further efforts will be devoted to develop a chiral P-ligand to asymmetric palladium-catalyzed silicon–carbon bond-forming silylation reaction with high level of enantioselectivity.
Acknowledgements
The authors gratefully thank the financial support of the National Natural Science Foundation of China (51303043, 21472031, and 21503060), Zhejiang Provincial Natural Science Foundation of China (LR14B030001 and LY14B020013), Science and Technology Department of Zhejiang Province (2015C31138), and Hangzhou Science and Technology Bureau of China (20140432B04).Notes and references
- R. West, in The Chemistry of Organic Silicon Compounds, ed. S. Patai and Z. Rappoport, Wiley, New York, vol. II, ch. 19, 1989 Search PubMed.
- For representative reviews, see: (a) T. H. Chan and I. Fleming, Synthesis, 1979, 761 CrossRef CAS; (b) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley-Interscience, New York, 1999 Search PubMed; For selected examples, see: (c) W. Gu, S. Liu and R. B. Silverman, Org. Lett., 2002, 4, 4171 CrossRef CAS; (d) S. E. Demark and J. M. Kallemeyn, Org. Lett., 2003, 5, 3483 CrossRef; (e) J. Beckman, A. Duthie, G. Reeske and M. Schürmann, Organometallics, 2005, 24, 3629 CrossRef; (f) Y. Nakao, J. Chen, M. Tanaka and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 11694 CrossRef CAS; (g) N. Chernyak, A. S. Dudnik, C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 8270 CrossRef CAS; (h) Y. Liang, S. Zhang and Z. Xi, J. Am. Chem. Soc., 2011, 133, 9204 CrossRef CAS; (i) A. Kuznetsov, Y. Onishi, Y. Inamoto and V. Gevorgyan, Org. Lett., 2013, 15, 2498 CrossRef CAS; (j) T. Ishiyama, T. Saiki, E. Kishida, I. Sasaki, H. Ito and N. Miyaura, Org. Biomol. Chem., 2013, 11, 8162 RSC; (k) A. Kajetanowicz, J. Czaban, R. Krishnan, M. Malińska, K. Woźniak, H. Siddique, L. G. Peeva, A. G. Livingston and K. Grela, ChemSusChem, 2013, 6, 182 CrossRef CAS; (l) C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236 CrossRef CAS; (m) H. Hettegger, I. Sumerskii, S. Sortino, A. Potthast and T. Rosenau, ChemSusChem, 2015, 8, 680 CrossRef CAS.
- For selected examples, see: (a) A. S. Manoso, C. Ahn, A. Soheili, C. J. Handy, R. Correia, W. M. Seganish and P. DeShong, J. Org. Chem., 2005, 69, 8305 CrossRef; (b) M. Wander, P. J. C. Hausoul, L. A. J. M. Slidregt, B. J. van Steen, C. van Koten and R. J. M. K. Gebbink, Organometallics, 2009, 28, 4406 CrossRef CAS; (c) A. Lorbach, C. Reus, M. Bolte, H. W. Lerner and M. Wagner, Adv. Synth. Catal., 2010, 352, 3443 CrossRef CAS; (d) N. Hirone, H. Sanjiki, R. Tanaka, T. Hata and H. Urabe, Angew. Chem., Int. Ed., 2010, 49, 7762 CrossRef CAS.
- For recent reviews, see: (a) M. Oestreich, Synlett, 2007, 1629 CrossRef CAS; (b) A. Weickgenannt, M. Wewald and M. Oestreich, Org. Biomol. Chem., 2010, 8, 1497 RSC; (c) L. W. Xu, L. Li, G. Q. Lai and J. X. Jiang, Chem. Soc. Rev., 2011, 40, 1777 RSC; (d) L. W. Xu, Angew. Chem., 2012, 124, 13106 ( Angew. Chem., Int. Ed. , 2012 , 51 , 12932 ) CrossRef; (e) R. Shintani, Asian J. Org. Chem., 2015, 4, 510 CrossRef CAS; For recent examples, see: (f) J. O. Bauer and C. Strohmann, J. Am. Chem. Soc., 2015, 137, 4304 CrossRef CAS; (g) R. Shintani, C. Takagi, T. Ito, M. Naito and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 1616 CrossRef CAS; (h) J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 720 CrossRef CAS; (i) J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 8167 CrossRef CAS; (j) Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520 CrossRef CAS; (k) K. Igawa, D. Yoshihiro, N. Ichikawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2012, 51, 12745 CrossRef CAS; (l) R. Shintani, E. E. Maciver, F. Tamakuni and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 16955 CrossRef CAS; (m) C. D. F. Konigs and M. Oestreich, Synthesis, 2011, 13, 2062 Search PubMed; (n) Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510 CrossRef CAS PubMed; (o) K. Igawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2010, 49, 728 CrossRef CAS.
- A. Kunai, T. Sakurai, E. Toyoda, M. Ishikawa and Y. Yamamoto, Organometallics, 1994, 13, 3233 CrossRef CAS.
- For recent reviews, see: (a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946 CrossRef CAS; (b) Y. Yang and C. Wang, Sci. China: Chem., 2015, 58, 1266 CrossRef CAS; (c) R. Sharma, R. Kumar, I. Kumar, B. Singh and U. Sharma, Synthesis, 2015, 47, 2347 CrossRef CAS; (d) Z. Xu, W.-S. Huang, J. Zhang and L.-W. Xu, Synthesis, 2015, 47, 3645 CrossRef CAS; (e) Z. Xu and L.-W. Xu, ChemSusChem, 2015, 8, 2176 CrossRef CAS.
- M. Murata, K. Suzuki, S. Watanabe and Y. Masuda, J. Org. Chem., 1997, 62, 8569 CrossRef CAS.
- (a) A. S. Manoso and P. DeShong, J. Org. Chem., 2001, 66, 7449 CrossRef CAS; (b) Y. Yamamoi, J. Org. Chem., 2005, 70, 9607 CrossRef PubMed; (c) M. Murata, H. Ohara, R. Oiwa, S. Watanabe and Y. Masuda, Synthesis, 2006, 1771 CrossRef CAS; (d) Y. Yamanoi, T. Taira, J.-I. Sato, I. Nakamula and H. Nishihara, Org. Lett., 2007, 9, 4543 CrossRef CAS; (e) A. Lesbani, H. Kondo, Y. Yabusaki, M. Nakai, Y. Yamanoi and H. Nishihara, Chem.–Eur. J., 2010, 16, 13519 CrossRef CAS; (f) N. Iranpoor, H. Firouzabadi and R. Azadi, J. Organomet. Chem., 2010, 695, 887 CrossRef CAS; (g) Y. Kurihara, Y. Yamanoi and H. Nishihara, Chem. Commun., 2013, 49, 11275 RSC.
- Y. Kurihara, M. Nishikawa, Y. Yamanoi and H. Nishihara, Chem. Commun., 2012, 48, 11564 RSC.
- For the Ph-NNP, see: (a) L. S. Zheng, L. Li, K. F. Yang, Z. J. Zheng, X. Q. Xiao and L. W. Xu, Tetrahedron, 2013, 69, 8777 CrossRef CAS; For Fei-Phos, see: (b) F. Ye, Z. J. Zheng, L. Li, K. F. Yang, C. G. Xia and L. W. Xu, Chem.–Eur. J., 2013, 19, 15452 CrossRef CAS; (c) J. X. Xu, F. Ye, X. F. Bai, J. Zhang, Z. Xu, Z. J. Zheng and L. W. Xu, RSC Adv., 2016, 6, 45495 RSC; For the Ar-BINMOL-Phos, see: (d) T. Song, L. S. Zheng, F. Ye, W. H. Deng, Y. L. Wei, K. Z. Jiang and L. W. Xu, Adv. Synth. Catal., 2014, 356, 1708 CrossRef CAS; (e) T. Song, L. Li, W. Zhou, Z. J. Zheng, Y. Deng, Z. Xu and L. W. Xu, Chem.–Eur. J., 2015, 21, 554 CrossRef CAS; (f) Z. Xu and L. W. Xu, Chem. Rec., 2015, 15, 925 CrossRef CAS; For HZNU-Phos, see: (g) F. Ye, Z. J. Zheng, W. H. Deng, L. S. Zheng, Y. Deng, C. G. Xia and L. W. Xu, Chem.–Asian J., 2013, 8, 2242 CrossRef CAS; (h) W. H. Deng, F. Ye, X. F. Bai, Z. J. Zheng, Y. M. Cui and L. W. Xu, ChemCatChem, 2015, 7, 75 CrossRef CAS; and our Xing-Phos was also not effective in this palladium-catalyzed Si–C bond-forming silylation reaction of aryl halides with hydrosilane, for Xing-Phos, see: (i) X. F. Bai, T. Song, Z. Xu, C. G. Xia, W. S. Huang and L. W. Xu, Angew. Chem., Int. Ed., 2015, 54, 5255 CrossRef CAS; (j) X. F. Bai, Z. Xu, C. G. Xia, Z. J. Zheng and L. W. Xu, ACS Catal., 2015, 5, 6016 CrossRef CAS.
- (a) H. Y. Sun, K. Kubota and D. G. Hall, Chem.–Eur. J., 2015, 21, 19186 CrossRef CAS; (b) S. Klimczyk, A. Misale, X. L. Huang and N. Maulide, Angew. Chem., Int. Ed., 2015, 54, 10365 CrossRef CAS; (c) J. Pedroni, M. Boghi, T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 9064 CrossRef CAS; (d) B. S. Zeng, X. Y. Yu, P. W. Siu and K. A. Scheidt, Chem. Sci., 2014, 5, 2277 RSC.
- For recent examples, see: (a) X. Zhou and G. B. Dong, J. Am. Chem. Soc., 2015, 137, 13715 CrossRef CAS; (b) K. Kitamura, N. Shimada, C. Stewart, A. C. Atesin, T. A. Atesin and M. A. Tius, Angew. Chem., Int. Ed., 2015, 54, 6288 CrossRef CAS; (c) J. Pedroni, T. Saget, P. A. Donets and N. Cramer, Chem. Sci., 2015, 6, 5164 RSC; (d) C. R. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534 CrossRef CAS; (e) A. Ros, B. Estepa, P. Ramirez-Lopez, E. Alvarez, R. Fernandez and J. M. Lassaletta, J. Am. Chem. Soc., 2013, 135, 15730 CrossRef CAS; (f) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 7865 CrossRef CAS PubMed; (g) D. M. Dalton, A. K. Rappe and T. Rovis, Chem. Sci., 2013, 4, 2062 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12873d |
| This journal is © The Royal Society of Chemistry 2016 |