
Chemoprotective activity of mixed valence polyoxovanadates against diethylsulphate in E. coli cultures: insights from solution speciation studies†
K. Postal a,
D. F. Malufa,
G. Valdamerib,
A. L. Rüdigera,
D. L. Hughesc,
E. L. de Sáa,
R. R. Ribeiroa,
E. M. de Souzab,
J. F. Soaresa and
G. G. Nunes*a
a,
D. F. Malufa,
G. Valdamerib,
A. L. Rüdigera,
D. L. Hughesc,
E. L. de Sáa,
R. R. Ribeiroa,
E. M. de Souzab,
J. F. Soaresa and
G. G. Nunes*a
aDepartamento de Química, Universidade Federal do Paraná, 81530-900 – Curitiba-PR, Brazil. E-mail: nunesgg@ufpr.br
bDepartamento de Bioquímica e Biologia Molecular, Universidade Federal do Paraná, 81530-900 – Curitiba-PR, Brazil
cSchool of Chemistry, University of East Anglia, Norwich NR4 7TJ, UK
First published on 5th December 2016
Abstract
The mixed valence polyoxovanadates [(CH3)4N]6[V15O36(Cl)] (A) and K(NH4)4[H6V14O38(PO4)]·11H2O (B) were evaluated for their chemoprotective activity against the alkylating agent diethylsulphate (DES) in Escherichia coli DH5α cultures. Compound A was synthesized previously by our and other research groups and product B was prepared in this work by reaction of NH4VO3, mannitol and KH2PO4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5:0.1) in water under reflux at atmospheric pressure. Polyoxovanadate A showed to be non-toxic to E. coli up to the concentration of 10.0 mmol L−1, and its presence in cultures treated with a fixed concentration of DES (6.0 mmol L−1) led to a chemoprotective effect of up to 40% compared to the control cells without A. In contrast, product B exhibited growth inhibitory activity against E. coli with a GI50 value of 8.2 mmol L−1 and was unable to protect the bacteria from the alkylating agent in conditions similar to those employed for A. Speciation studies were carried out for A and B by 51V NMR and EPR spectroscopies in aqueous solution and Luria–Bertani (LB) broth. The results indicate that the polynuclear structure of A is more stable in LB than in pure aqueous solution, and that A is able to react with increasing amounts of DES. Polyoxovanadate B, in turn, suffers rapid breakage and concentration-/pH-dependent rearrangements in both water and LB, forming from simple mononuclear complexes to larger aggregates such as [H4VV14O38(PO4)]5−, [HVV10O28]5− and one or more mixed valence polyoxovanadates. The dominant vanadium(V) species in LB solutions of B, [HV10O28]5−, is poorly reactive towards DES, being also known to inhibit biological processes in living cells; this may relate to the lack of chemoprotection by B and to the actual increase in toxicity observed when E. coli cultures were exposed to both DES and B. Taken together, these results indicate that the observed chemoprotective effect is not only highly dependent on the solution stability of the polyoxometalates, but is also limited by the formation of decomposition products, such as decavanadate and mononuclear VO2+ complexes, which are not able to react with and therefore do not deactivate the alkylating agent in the culture media.
:0.5:0.1) in water under reflux at atmospheric pressure. Polyoxovanadate A showed to be non-toxic to E. coli up to the concentration of 10.0 mmol L−1, and its presence in cultures treated with a fixed concentration of DES (6.0 mmol L−1) led to a chemoprotective effect of up to 40% compared to the control cells without A. In contrast, product B exhibited growth inhibitory activity against E. coli with a GI50 value of 8.2 mmol L−1 and was unable to protect the bacteria from the alkylating agent in conditions similar to those employed for A. Speciation studies were carried out for A and B by 51V NMR and EPR spectroscopies in aqueous solution and Luria–Bertani (LB) broth. The results indicate that the polynuclear structure of A is more stable in LB than in pure aqueous solution, and that A is able to react with increasing amounts of DES. Polyoxovanadate B, in turn, suffers rapid breakage and concentration-/pH-dependent rearrangements in both water and LB, forming from simple mononuclear complexes to larger aggregates such as [H4VV14O38(PO4)]5−, [HVV10O28]5− and one or more mixed valence polyoxovanadates. The dominant vanadium(V) species in LB solutions of B, [HV10O28]5−, is poorly reactive towards DES, being also known to inhibit biological processes in living cells; this may relate to the lack of chemoprotection by B and to the actual increase in toxicity observed when E. coli cultures were exposed to both DES and B. Taken together, these results indicate that the observed chemoprotective effect is not only highly dependent on the solution stability of the polyoxometalates, but is also limited by the formation of decomposition products, such as decavanadate and mononuclear VO2+ complexes, which are not able to react with and therefore do not deactivate the alkylating agent in the culture media.
Introduction
Contemporary interest in polyoxovanadates reflects not only their attractive catalytic,1,2 magnetic3,4 and electronic5 properties, but also their interactions with a wide variety of biological targets6 and their potential biomedical applications.7,8 As examples, among polyoxometalates (POM) of different transition metals, vanadium-containing complexes such as [NiV13O38]7− and [V18O42(PO4)]9− present good efficacy as antitumor agents on human melanoma, lung and cervical cancer cells.9–11 Such a positive effect against chemical carcinogenesis in human and animal models seems to be due to modification of xenobiotic enzymes and inhibition of the formation of carcinogenic metabolites.12 In addition, vanadium-based oxometalates such as [V10O28]6− and its protonated derivatives have been extensively studied in their antidiabetic, antibacterial and antitumor activities.13 A remarkable property of polyoxovanadates, as far as speciation and biological studies are concerned, is the establishment of multiple equilibria among different chemical species14 – from mononuclear vanadates(V) to polyoxovanadates – in extra and intracellular environments.13–15An important study by Hamilton and Wilker has demonstrated that, among inorganic salts of metals of the main and d-transition blocks, oxocompounds such as vanadate, molybdate and selenite were capable of inhibiting supercoiled DNA alkylation by diethylsulphate (DES).16,17 The proposed mechanism suggested that chemoprotection occurs when the affinity of the alkylating agent for the oxometalate is higher than for the DNA molecule.16,17 Later on, studies with oxidovanadium compounds of different nuclearities such as [V3O9]3−, [V4O12]4−, [V5O14]3−, [V10O26]6− and [H3V10O28]3− showed that the reactivity of these complexes is dictated by charge density, in which a higher negative charge provides greater nucleophilicity and facilitates the reaction of the vanadium compound with the alkylating agent.16
In our research group, the alkylation of pUC19 DNA has been employed as a model reaction for studies on the chemoprotective activity of the highly charged, mixed valence polyoxovanadate [(CH3)4N]6[V15O36(Cl)]18 (A, Fig. S1†) against DNA alkylation. Complex A was capable of preventing alkylation of pUC19 by both dimethylsulphate (DMS) and diethylsulphate (DES), showing chemoprotective activities of 48% and 70%, respectively.18 This effect proved to be dependent on specific reaction conditions, vanadium concentrations and chemical nature of the buffering agent. The complexity of these results suggested the need of more extensive studies on similar systems.
Exogenous alkylating agents such as diethylnitrosamine (DEN),19 1,2-dimethylhydrazine (1,2-DMH),20 dimethylsulphate (DMS) and diethylsulphate (DES)21 represent one of the major classes of toxic compounds generated in various industrial and environmental processes, being widespread in natural surroundings.22,23 It is known that dimethylsulphate (DMS) is able to enter living cells and react with biological macromolecules such as DNA and RNA.24–26 Other alkylating agents, namely S-adenosylmethionine (SAM), acetaldehyde, malondialdehyde (MDA), N-methyl-N-nitrosourea (MNU) and by-products of glycine nitrosation,27 are present in intracellular media of prokaryotic and eukaryotic organisms as endogenous products of primary and secondary metabolism.27 The most common targets in DNA and RNA alkylation reactions are nucleophilic oxygen and nitrogen sites in nucleosides, as well as oxygen atoms in phosphodiester moieties.28 Alkylation can cause DNA mispairing or even block DNA replication or transcription, impacting cell division and leading to mutations or misreading that can be deleterious and frequently lethal to cells.29 A number of chemicals have been tested to oppose or minimize this effect,7 including studies of simple vanadates such as NH4VO3 against chemically induced genotoxicity in rats. Results have shown that the presence of vanadium limits early molecular damage and preneoplastic lesions by inhibiting the action of DEN19 and 1,2-DMH.20
After the early recognition of the potentially serious danger represented by alkylation reactions, a number of experiments were performed to access the outcomes of DNA adduct formation27 and the functioning of natural cell defence mechanisms.23 In this context, the classical SOS system of DNA repair30 and the inducible adaptive (Ada) response to alkylating agents have been largely investigated in Escherichia coli and other bacterial species.27 Further studies revealed that bacterial and eukaryotic responses to treatments are significantly different.31 Recently, E. coli strains have also been used as models for the investigation of RNA alkylation lesions and restoration;32 for the study of chemically-stable, sugar-phosphate backbone damage that resists DNA repair;33,34 and the development of mass spectrometry-based strategies to identify the blocking and mutagenic sites of DNA lesions.35
In this work, the ability of mixed-valence (+IV/+V) polyoxovanadates A and [H6V14O38(PO4)]5− (B) to prevent Escherichia coli death after exposition to the lethal effects of diethylsulphate was studied for the first time. Whole bacterial cells constitute a more complex model system than the direct DNA alkylation reactions employed earlier by our research group,18 and this in vivo approach demanded the establishment of a number of new experimental conditions, as well as the consideration of a possibly large number of chemical interactions of both DES and the polyoxovanadates with the culture medium and E. coli cells that could interfere with the chemoprotective activity.36–39
In this context, the aims of this work were to (i) evaluate the general response of whole bacterial cells to the damaging effects of DES in the presence and absence of the two polyoxovanadates, and (ii) get insights, from solution speciation studies of A and B in Luria–Bertani (LB) medium, on the nature of the vanadium-containing species possibly related with the presence or absence of chemoprotection.
Our studies revealed very distinct solution behaviours for the two polyoxovanadates in pure LB, which appear to correlate well with their effect on the whole E. coli cells subjected to DES. Spectroscopic analyses indicated that the biological action on the bacteria depends on the stability of the two distinct POV aggregates in the culture medium, and on the decomposition species formed from each of them in solution. An attempt to correlate the speciation results with the known biological activities of the V-containing products, their ability to penetrate or form inside living cells, and the occurrence (or absence) of chemoprotection has also been made. Additional studies are surely needed to further explore this speciation–activity relationship, in the light of (necessary) new information on solution stabilities of other polyoxovanadates in both extra and intracellular media. In such a context, our present contribution may be seen as a first link between the chemical speciation of mixed valence (VIV/VV) POV molecules and their possible deleterious or protecting effects upon prokaryotic cells.
Materials and methods
Solutions and chemicals
All solutions, flasks and materials employed in the biological analysis, including ultrapure water (MilliQ, Millipore type 1, resistivity of 18.2 MΩ cm at 25 °C), were autoclave-sterilized. Potassium dihydrogen phosphate (KH2PO4) and Cu(SO4)·5H2O were purchased from Merck, mannitol from USB, ammonium vanadate (NH4VO3) from Vetec and diethylsulphate (DES) and Hg[Co(NCS)4] from Aldrich. All chemicals and solvents were analytical reagent grade and were used without further purification. The polyoxovanadate (Me4N)6[V15O36(Cl)] (A) was prepared according to the method reported by our research group.18Analytical methods
FTIR spectra were recorded from KBr pellets in the range of 400–4000 cm−1 in a BIORAD FTS 3500GX instrument. Raman spectra were obtained from a Renishaw Image spectrophotometer coupled to a Leica optical microscope, which focuses the incident radiation on a 1 μm2 area. Spectra were recorded using Ar+ (514 nm) and He–Ne (632.8 nm) laser excitation over the range of 200 to 4000 cm−1 and with an incident power of 0.2 mW. Elemental analyses were run by MEDAC Laboratories Ltd. (Chobham, Surrey, UK). C, H and N contents were determined by combustion analysis on a Thermal Scientific Flash EA 1112 Series Elemental Analyser, while K, P and V contents were obtained by Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-OES) with a Varian Vista MPX ICP-OES system. Vanadium(IV) contents were also determined by titration with a 0.002 mol L−1 KMnO4 solution, while aqueous (NH4)2[Fe(SO4)2]·6H2O (0.01 mol L−1) was used for the quantification of VV.40,41 EPR data were recorded from solid samples and solutions (in water and Luria–Bertani medium) at room temperature and 77 K on an X-band Bruker ELEXSYS E-500 (9.5 GHz) spectrometer. Spectral intensities were normalized with the use of a chromium(III) standard and simulations were carried out with the EasySpin software package.42 1H, 31P and 51V NMR spectra were acquired at 295 K in D2O from a Bruker 400 MHz Avance III spectrometer operating at 9.4 T and equipped with a 5 mm multinuclear direct detection probe. 51V was measured at 105.2 MHz, 31P at 161.9 MHz and 1H at 400.1 MHz. 51V NMR analyses were performed approximately 10 to 30 min after sample preparation (0.5 mL), and selected samples were analysed again after 3 h. Spectra were acquired using calibrated 90° pulses and 2048 to 10240 scans with a recycling delay of 0.1 s and acquisition times of 0.157 s, on a spectral width of 990 ppm (+44 to −946 ppm). 31P NMR spectra were registered with 2048 scans with a recycling delay of 0.2 s and acquisition times of 0.34 s, on a spectral width of 590 ppm (+408 to −182). Tetramethylsilane (TMS, internal reference), 85% H3PO4 (neat, capillary) and VOCl3 (neat, capillary) were employed as references for 1H, 31P and 51V respectively. Spectral intensities were normalized on each experiment by comparison with the reference signal. Magnetic susceptibility measurements in the solid state were run at 296 K on a Johnson-Matthey MKII magnetic susceptibility balance, using a modified Gouy method43,44 and Hg[Co(NCS)4] and Cu(SO4)·5H2O as calibration standards. Pascal constants45 were employed to correct the data for the diamagnetism of the ligands (diamagnetic correction, χdia for H44KN4O53PV14 = −767.4 × 10−6 cm3 mol−1). UV/Vis/NIR spectra were acquired at room temperature in the range of 250–2500 nm from 0.01 to 2.0 mmol L−1 solutions of A and B in ultrapure water. This employed a PerkinElmer LAMBDA 1050 UV/Vis/NIR spectrophotometer equipped with a three-detector PMT/InGaAs/PbS setup. Thermogravimetric (TGA) data were collected on a Netzsch STA449 F3 Jupiter thermal analyser equipped with a silicon carbide furnace and using dinitrogen as carrier gas. A weighed portion (ca. 4 mg) of each sample was heated in an aluminium pan at 10 °C min−1 from 20 to 800 °C.
Preparation of K(NH4)4[H6V14O38(PO4)]·11H2O (product B)
The reaction was carried out in a round bottom flask containing 1.16 g (9.92 mmol) of NH4VO3, 0.920 g (5.05 mmol) of mannitol and 0.150 g (1.10 mmol) of KH2PO4 in 50 mL of water. The reaction mixture was stirred under reflux for 24 h, producing a deep bluish-green solution that was cooled down and kept at room temperature for two weeks to give deep green crystals suitable for single-crystal X-ray diffraction analysis. Yield: 0.920 g (75% based on NH4VO3). Product B is soluble in water and insoluble in polar and nonpolar organic solvents. Analysis calcd for H44KN4O53PV14: H 2.56; K 2.26; N 3.24; P 1.79; V 41.19%. Found: H 2.59; K 2.01; N 3.15; P 1.78; V 38.83%. The found hydrogen, potassium and nitrogen contents are average values from two independent determinations, with standard deviations (σn−1) of 0.12, 0.40 and 0.08 respectively. Although the vanadium content determined by ICP-OES (reported above) is slightly lower than the expected figures, the average result obtained by titrimetry41 was equal to 41.27 ± 0.40% (triplicate analysis), in very good agreement with the calculated value. Results from the combustion (CHN) analysis carried out in duplicate also revealed a small amount of carbon (0.97 ± 0.33%), probably coming from an oxidized mannitol residue deposited on the crystals. This carbon content is equivalent to ca. 1.3 carbon atoms per [H6V14O38(PO4)]5− aggregate. IR spectrum (cm−1): 3357(s), 3135(s), 1628(m), 1399(s), 1053(w), 936(s), 859(w), 726(w), 588(m), 479(w). Raman spectrum (cm−1): 142(s), 194(w), 280(m), 405(w), 691(w), 990(s).Single-crystal X-ray diffraction analysis
Data for complex B were collected on a Bruker D8 Venture diffractometer equipped with a Photon 100 CMOS detector, Mo-Kα radiation and graphite monochromator. From a sample under oil, one deep green block, ca. 0.024 × 0.162 × 0.228 mm, was mounted on a MicroMount™ 100 μM (MiTeGen), fixed in the cold nitrogen stream and cooled to 100 K. Data were processed using the APEX2 program. The structure was determined by the direct methods routines in the SHELXS program46 and refined by full-matrix least-squares methods, on F2's, in SHELXL.46 The non-hydrogen atoms of the cluster were refined with anisotropic thermal parameters. Three (independent) hydrogen atoms were located on the cluster and these were refined freely. Scattering factors for neutral atoms were taken from the literature.47 Computer programs used have been noted above, and were run through WinGX.48 At the conclusion of the refinement, wR2 = 0.069 and R1 = 0.036 for all 4720 reflections weighted w = [σ2(Fo2) + (0.0387P)2 + 9.61P]−1 with P = (Fo2 + 2Fc2)/3; for the ‘observed’ data only, R1 = 0.025. In the final difference map, the highest peak (ca. 0.7 e Å−3) was on the V(7)–O(7) bond.Bacterial growth conditions and chemoprotection bioassays
The chemoprotective effect of polyoxovanadates A and B against alkylation induced by diethylsulphate (DES) was evaluated in Escherichia coli strain DH5α by adaptation of a standard protocol for bacterial growth as described below.49 The optical densities50 of all batch cultures were read at 540 nm (18518 cm−1, OD540) to avoid the absorption bands typical of the culture media and those observed in the electronic spectra of A or B (see ESI, Fig. S3 and S4†). The polyoxovanadates stock solutions (20.0 mmol L−1) were prepared in ultrapure water without pH adjustment, resulting in a pH of 6.3 for A and 3.2 for B. Solutions of the two polyoxovanadates at suitable concentrations were employed as blanks in each experiment. Cells were grown49 in Luria–Bertani (LB) broth at 37 °C from stock cultures kept in 50 (v/v) % glycerol at −20 °C. Assays were performed three times employing three independent batch cultures for each concentration of DES or polyoxovanadate.
In a typical procedure (Scheme S1†), bacteria were grown at 37 °C at 120 rpm in 10 mL of fresh LB medium containing 10 μg mL−1 of nalidixic acid (Nal) until the OD540 of the cultures reached 1.0. Cells were subsequently collected by centrifugation (5000 rpm for 10 min), resuspended and incubated for 15 min at 37 °C in 1 mL of saline solution (0.9% NaCl) containing different amounts of DES, A or B depending on the specific experiment as mentioned below. These samples were then transferred to 10 mL of LB medium, incubated at 37 °C and 120 rpm for 3 h (OD540 of 1.0). The concentration of DES able to inhibit the growth of bacteria by 50% (GI50) was determined after incubation of the cells with different amounts of DES and used in the chemoprotection assays. Similar growth inhibition assays were conducted with the polyoxovanadates. GI50 values for DES and for complexes A and B were calculated using the GraphPad Prism® 5.1 software51 and refer to the whole cell cultures for which they were determined.
Chemoprotection assays were carried out by resuspending bacterial pellets in saline containing a fixed concentration of DES (6.0 mmol L−1) and increasing amounts of polyoxovanadates (0.01, 0.05, 0.1, 1.0, 5.0, and 10.0 mmol L−1). All samples were incubated for 15 min at 37 °C, transferred to 10 mL of LB and then incubated again at 37 °C and 120 rpm. Growth was assessed by OD540 measurements (Scheme S1†).
Results and discussion
In this work, chemical and solid state spectroscopic characterisation is described only for B, because the synthesis and characterisation of A have already been reported by our research group.18 Results of the biological assays in E. coli cultures and of speciation studies in LB medium will be presented and discussed for both polyoxovanadates, A and B.Characterisation of product B
The pseudo-spherical polyoxovanadate B was prepared in this work by partial reduction of vanadium(V) by mannitol in hot, weakly acid medium, employing PO43− as a structural reference to direct the aggregation of the oxidovanadium units. This synthetic route, described earlier by our group,18 is very simple, accessible and does not depend on inert atmosphere, UV irradiation or hydrothermal conditions.The X-ray structure of the polyoxoanion in B (Fig. 1) is well-resolved and analogous to that reported by Kanoo and coworkers for [VV12VIV2O38(PO4)]11−.52 The surrounding counter ions (potassium and ammonium cations) and water molecules have proven harder to identify and locate precisely, because of structural disorder and lower resolution. In some cases, it has been difficult to distinguish between ammonium ions and water molecules from diffraction data alone, and with a lack of resolution of some groups, it is not possible to use the available single-crystal X-ray data to count up the charges accurately. A number of attempts to get higher quality diffraction data were not successful. Considering all this, crystallographic information and selected structural parameters for the best possible structural model provided by the X-ray diffraction analysis are listed in Tables S1 and S2;† they were used together with other analytical, spectroscopic, magnetic and theoretical data to build the proposed formulation for B in this work.
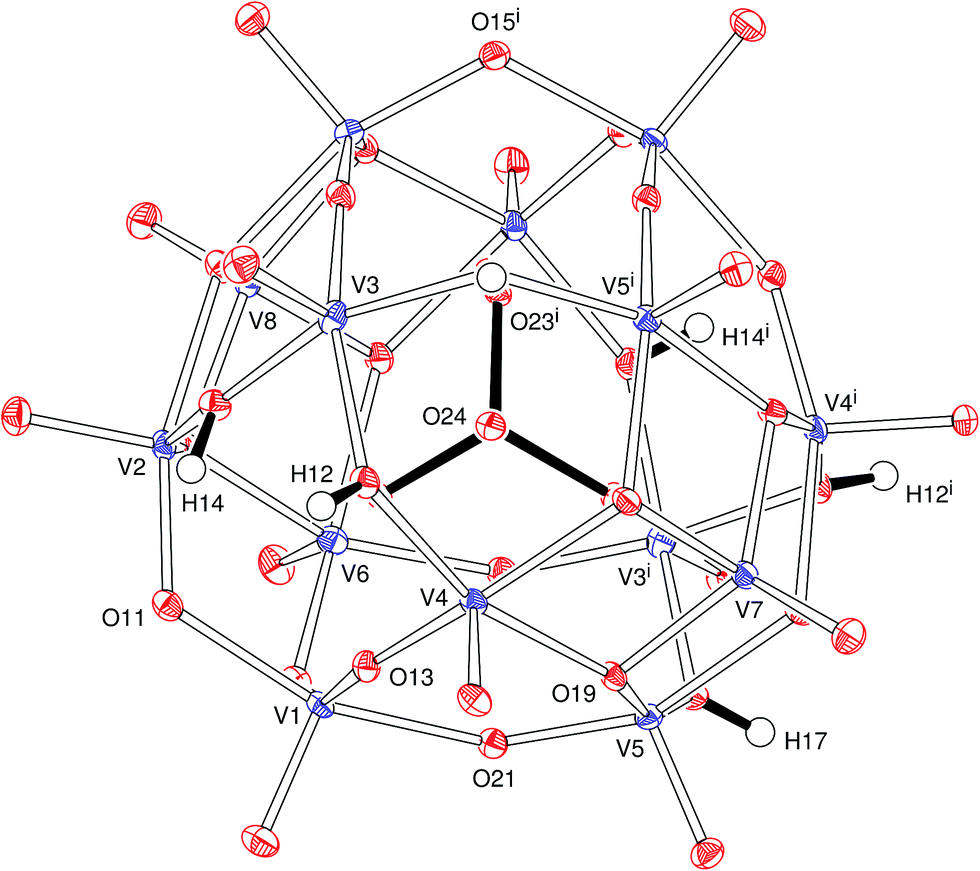 | ||
| Fig. 1 ORTEP-3 plot of the polynuclear, mixed-valence oxidovanadium anion in B, with the atom numbering scheme (50% ellipsoids). Cations, water molecules and hydrogen bonds are omitted. Bonds in the phosphate anion are depicted by the solid lines at the centre of the ORTEP plot. | ||
In the anion, the 14 oxidovanadium moieties are linked by 24 bridging groups of which 10 are μ-O, 8 are μ3-O and 6 are μ-OH (Fig. 1). The metal aggregate lies around a twofold symmetry axis that passes through O(7), V(7), P(1), V(8) and O(8). The tetrahedral PO43− group lies at the centre of the cluster with P–O bonds of 1.536(2) and 1.549(2) Å. The oxygens of this group are directed towards the centres of the V3O3 faces of the cluster and present O–V distances in the range 2.302(2) to 2.427(2) Å, significantly longer than all the other V–O bonds. Vanadium atoms are five-coordinate with a distorted square pyramidal pattern and apical oxygens (directed out of the cluster) 1.601(2) to 1.616(2) Å away from the bonded vanadium centres. The three crystallographically independent hydrogen atoms (H12, H14 and H17, Fig. 1) in the anion were clearly identified and were refined satisfactorily.
The water molecules and counter ions that fill the space around and between the cluster anions are linked by an extensive three-dimensional network of hydrogen and K–O coordination bonds that also involve the oxide and hydroxide groups of the polyoxovanadate (Fig. S2 and Table S3†). The potassium ion is coordinated by eight oxygen atoms (from the cluster and water molecules) with K–O distances in the range of 2.814(13) to 3.144(15) Å.
The polyoxoanion in B is analogous to that reported for [H5(H2pip)3][VV12VIV2O38(PO4)]·8H2O, (pip = piperazine)52 and similar to the fully oxidized53–57 [HxVV14O38(PO4)]9−x; in the three cases, the pseudo-spherical framework is based on the Keggin structure.58 Our proposition of the mixed valence [H6VV12VIV2O38(PO4)]5− formulation for the anion B is based on a combination of results provided by elemental analysis (including metal contents, see Experimental), single-crystal X-ray diffraction, EPR and electronic spectroscopies (UV-vis-NIR), magnetic susceptibility measurements and bond valence sum calculations, as discussed below, and finds support in literature data.52 Because of the precision and accuracy limits imposed by experimental deviations and data quality, taken together with the intrinsic limitations that characterize each experimental technique, no available method was able to provide ‘final evidence’ or establish alone an indisputable structural model for the product. Despite this, all results, considered together and within experimental error, indicate K(NH4)4[H6VV12VIV2O38(PO4)]·11H2O as the best total formulation for product B.
The [H6V14O38(PO4)]5− formula implies an average charge of +4.86 for the vanadium centres in B, which is the same described by Kanoo and co-workers52 and indicates +IV and +V as the formal oxidation states (namely 12 VV and 2 VIV) for the vanadium centres. This in turn agrees with the results of bond valence sum (BVS) calculations based on the X-ray data,59,60 which gave an average BVS value of 4.81 for vanadium in B, starting from the individual values of 4.49, 4.79, 4.88, 4.77, 4.83, 4.80, 5.07 and 5.10 for V(1), V(2), V(3), V(4), V(5), V(6), V(7) and V(8) respectively. This calculation considered that each of the V(1)–V(6) centres has two symmetry-equivalent sites in the anion (Fig. 1 and Table S2†), differently from V(7) and V(8) that sit on the twofold symmetry axis. The presence of the five counter ions (K+, 4NH4+) is in turn supported by potassium, nitrogen and hydrogen contents determined by ICP-OES (Inductively Coupled Plasma-Optical Emission Spectroscopy) and combustion analyses, which also gave very compatible results for phosphorus (see Experimental). Triplicate vanadium determinations by titration analysis, in turn, gave an excellent agreement with the transition metal content calculated for B.
Results of magnetic susceptibility measurements carried out for B in the solid state at 296 K (χMT = 0.789 cm3 K mol−1) indicate two unpaired electrons per polyoxoanion, which is fully compatible with the presence of two vanadium(IV) (d1) ions amongst the total 14 vanadium centres in the cluster. This experimental value is very close to the expected from the Curie law for two S = 1/2 uncorrelated spins at room temperature, 0.75 cm3 K mol−1 (assuming g = 2),45,61 and therefore corroborates the [H6VV12VIV2O38(PO4)]5− formulation.
The mixed-valence composition proposed for the polyoxovanadate B also finds support in the UV-vis-NIR spectrum registered for a 1.5 mmol L−1 aqueous solution of B. It shows a broad band with maximum at 11500 cm−1 (ε = 340 L mol−1 cm−1) assigned to an intervalence charge transfer transition (IVCT, VIV → VV) in the [H6VV12VIV2O38(PO4)]5− anion (Fig. S4†). The theoretical and experimental values of the half-height bandwidth, Δ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/char_e0ce.gif)
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) 1/2, of 5154 and 6861 cm−1 respectively, indicate that product B belongs to Class II of the Robin–Day classification for mixed valence compounds.62,63 The same classification was reported for other polyoxovanadates such as K9[V16O38(CN)]·13H2O,64 [V6O8(OCH3)11]65 and [V6O8(OCH3)11][SbCl6].65
1/2, of 5154 and 6861 cm−1 respectively, indicate that product B belongs to Class II of the Robin–Day classification for mixed valence compounds.62,63 The same classification was reported for other polyoxovanadates such as K9[V16O38(CN)]·13H2O,64 [V6O8(OCH3)11]65 and [V6O8(OCH3)11][SbCl6].65
Additional supporting evidence comes from the EPR spectrum recorded for B in the solid state at 77 K (Fig. S5†). It presents a clearly isotropic Lorentzian line shape (Δpp = 10.06 mT and g = 1.968), typical of a polynuclear exchange-narrowed species,66 that is, a system containing exchange-coupled paramagnetic ions with extensive spin density delocalization. This result not only confirms the partial reduction of the vanadium(V) ions provided by the NH4VO3 starting material, but is also in accordance with the Class II Robin–Day assignment discussed above and with the presence of magnetically-interacting vanadium(IV) ions in the anionic V14 aggregate.62,63
The complete K(NH4)4[H6VV12VIV2O38(PO4)]·11H2O formulation is also supported by results of thermogravimetric (TG) analysis, which show two weight losses bellow 250 °C corresponding to the release of the 11 water molecules and four ammonium counter ions (Fig. S6†). In this temperature range, the total weight loss of 15.8% shows excellent agreement with the calculated value (15.6%). The thermal decomposition of the aggregate became evident above 350 °C, producing an oxide residue at 550 °C. From an independent thermal treatment that employed a higher amount of B, the nature of this oxide was confirmed as V2O5 by powder X-ray diffractometry (JCPDS card number 41-1426) (Fig. S7†).
In summary, the contributions coming from all solid state analyses discussed above, taken together, indicate the K(NH4)4[H6VV12VIV2O38(PO4)]·11H2O as the best balanced formulation for B. This also agrees with the solution EPR and 51V NMR characterisation described in the next sections, which complement each other for the identification of the VIV and VV species formed from B in aqueous and LB solutions. The behaviour of B in the biological assays with E. coli and DES, in turn, reflects its relatively low stability towards structural breakage in LB medium, and this conclusion derives directly from the spectroscopic studies. Overall, all results described in this work are consistent with one another as far as the relationship between the solid state structure and the solution reactivity of polyoxovanadate B is concerned.
Finally, the FTIR spectrum of B presents bands at 588, 726, 936 and 1053 cm−1 assigned to ν(V–O), νas(V–O–V), ν(V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and ν(P–O), respectively (Fig. S8 and Table S4†).67,68 Bands at 1399, 3135 and 3357 cm−1, in turn, were assigned to the deformation and stretching vibrations of the NH4+ cation,69 and their broadening agrees with the occurrence of hydrogen bonding in the crystal structure of B. The band at 990 cm−1 in the Raman spectrum of the product (Fig. S9†) was assigned to the VO stretching of terminal oxidovanadium(IV) units, and vibrations at 405, 523 and 691 cm−1 were attributed to δ, νs and νas of (V–O–V),67,70 respectively.
O) and ν(P–O), respectively (Fig. S8 and Table S4†).67,68 Bands at 1399, 3135 and 3357 cm−1, in turn, were assigned to the deformation and stretching vibrations of the NH4+ cation,69 and their broadening agrees with the occurrence of hydrogen bonding in the crystal structure of B. The band at 990 cm−1 in the Raman spectrum of the product (Fig. S9†) was assigned to the VO stretching of terminal oxidovanadium(IV) units, and vibrations at 405, 523 and 691 cm−1 were attributed to δ, νs and νas of (V–O–V),67,70 respectively.
Spectroscopic characterisation of the equilibrium species obtained from B in aqueous solutions
The complexity of aqueous solutions containing vanadium(V), which are characterized by the co-existence of multiple species in equilibrium depending on pH, ionic strength and vanadium concentration, is well understood and involves the fully oxidized [V10O28]6− anion and other vanadates of lower nuclearity.71 On the other hand, spectroscopic studies of aqueous solutions of mixed-valence (IV/V) polyoxovanadates are much less common, and the solution speciation of these complexes still lacks complete characterisation. The few studies available point to the partial breakage of the polynuclear aggregate as reported by our research group for [V15O36Cl]6−,18 giving a mixture of simple vanadates(V) and the hydrated oxidovanadium(IV) complex [VO(H2O)5]2+. Very few works report the complete preservation of the polynuclear structure in aqueous solution as described for [V6O8(OCH3)11]65 and [HV12O32(Cl)]4−.72Spectroscopic studies performed with [H6V14O38(PO4)]5−, B, in aqueous solution evidenced the breakage of the polynuclear aggregate and the formation of a variable mixture of vanadium species as the solution concentration was increased from 0.1 to 10.0 mmol L−1 (Scheme 1 and Fig. 2). The 51V NMR spectrum of diluted aqueous solutions of B (0.1 mmol L−1) showed resonances at δ −560, −574, −577 and −593 ppm assigned respectively to the oxometalates “V1” (H2VO4−), “V2” (H2V2O72−), “V4” (V4O124−) and “V5” (V5O155−).14
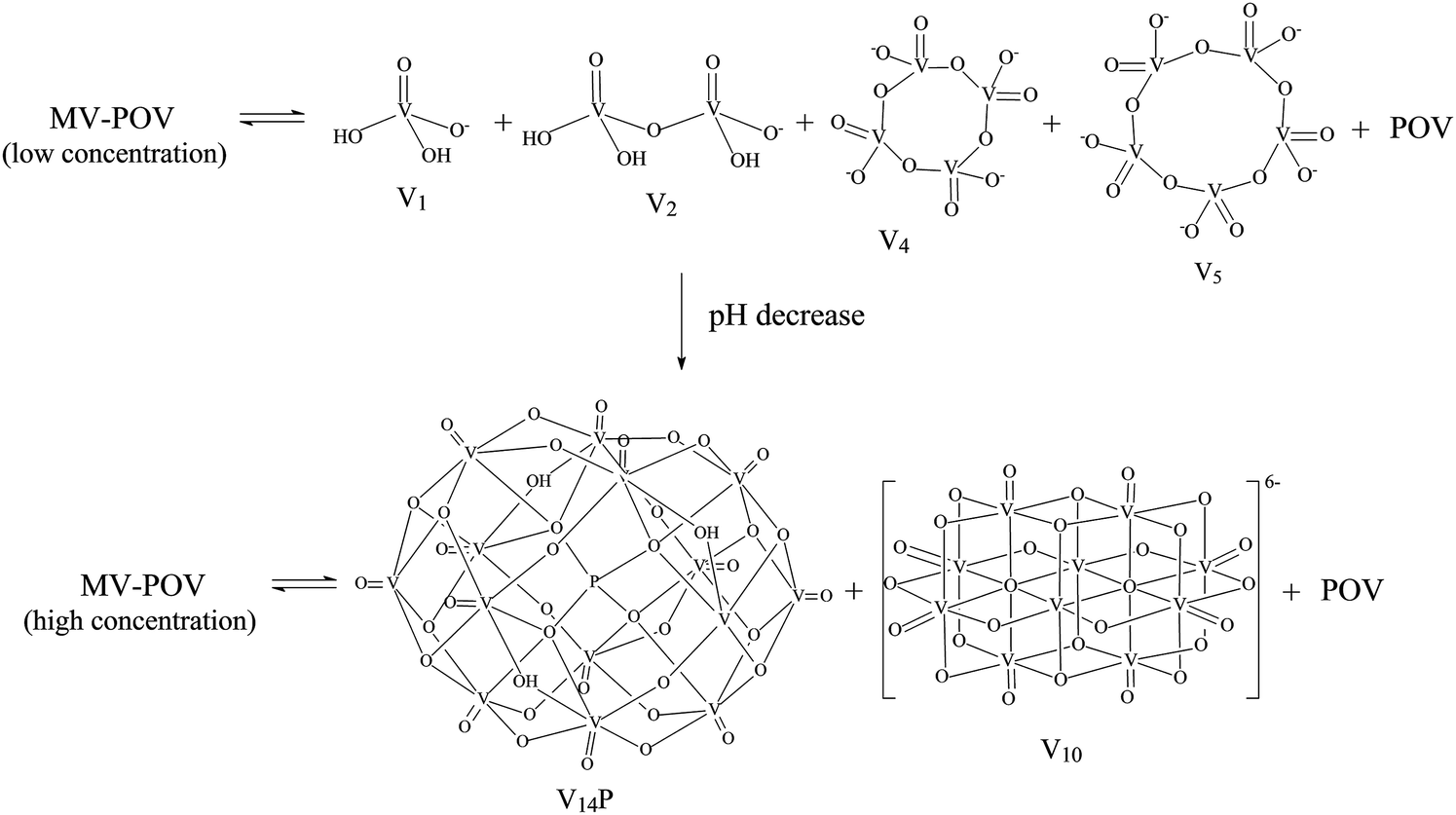 | ||
| Scheme 1 Representation of the proposed equilibria in aqueous solutions from the lowest to the highest concentration of B employed in this work (0.1 to 10.0 mmol L−1). “V1” (H2VO4−), “V2” (H2V2O72−), “V4” (V4O124−), “V5” (V5O155−), “V10” ([HV10O28]5−) and “V14P” ([H4V14O38(PO4)]5−) correspond to chemical species detected by 51V NMR analysis, while VO2+ = [VO(H2O)5]2+ and POV (unknown polyoxovanadate) are species detected by EPR, as discussed below. MV-POV stands for more than one mixed-valence polyoxovanadate; in this case, derived from B. | ||
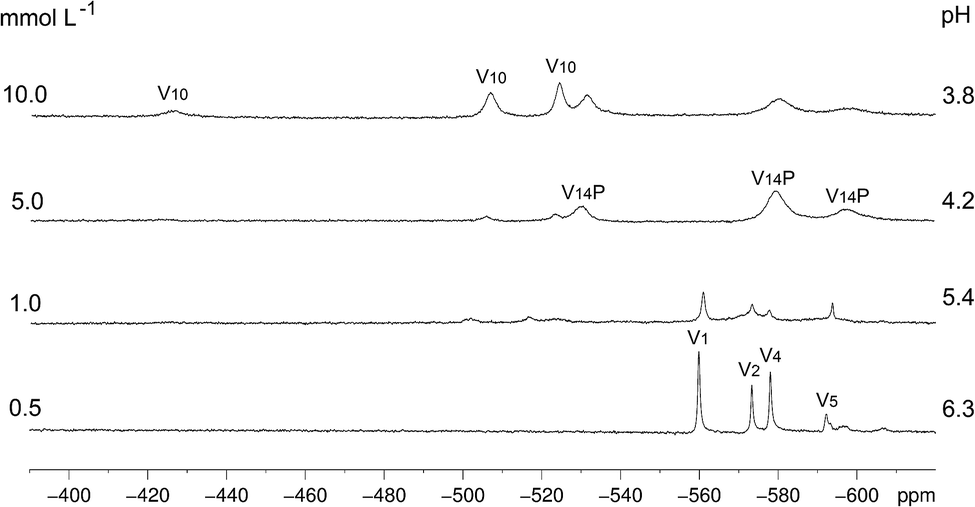 | ||
| Fig. 2 51V NMR spectra (105.2 MHz) recorded in D2O for variable concentrations of B (0.5 to 10.0 mmol L−1), following pH decrease (without adjustment) of 6.3 to 3.8 as a function of the concentration of B. The identified low nuclearity vanadium(V) species were “V1” = H2VO4−, “V2” = H2V2O72−, “V4” = V4O124− and “V5” = V5O155−. The three signals assigned to “V10”, HV10O285−,14 and the ones assigned to [H4V14O38(PO4)]5−, “V14P”,73 are generated by non-equivalent vanadium(V) centres in the structures. | ||
The increase in concentration of B from 0.5 to 10 mmol L−1 led to a decrease in the solution pH from 6.3 to 3.8, which was observed due to the acidic nature of this polyoxovanadate. When the concentration reached 5.0 mmol L−1, the spectral profile changed to give signals attributed to the fully oxidized [H4VV14O38(PO4)]5− (“V14P”) species, which has been reported in solution at pH values ranging from 1.5 to 5.5.73 This tetradecavanadophosphate has a trans-bicapped α-Keggin structure (Scheme 1) and presents 51V NMR signals at δ (ppm) −531 (capping), −580 (connected to capping vanadium by oxo bridges) and −598 (central) with an integral ratio of 2:8:4. Solutions of B whose concentrations ranged between 5 and 10 mmol L−1, in turn, were marked by the appearance of a set of signals characteristic of [HV10O28]5− (“V10”) at δ (ppm) −425 (two central VV ions), −506 (four peripheral ions located at the corners of the molecule) and −524 (four capping VO units), Fig. 2 and Scheme 1. This decavanadate anion is the predominant oligonuclear species in vanadate(V) solutions whose pH values range from 3 to 5,14 although a recent Raman study has revealed that V10 can also occur in alkaline medium after being formed upon acidification.74 This reinforces the complexity of vanadium chemistry in aqueous solution.
EPR analyses in aqueous solutions at 77 K were also performed with increasing concentrations of B (0.5, 1.0, 5.0 and 10.0 mmol L−1), in order to allow inferences on the chemical nature of the different species in equilibrium. All spectra (Fig. S10†) showed a broad band – similar to the solid-state spectrum presented in Fig. S5† – for which spectral simulation revealed the co-existence of at least two different types of polynuclear aggregates in all concentrations. It is relevant to emphasize that NMR and EPR results are complementary in this work, as the first technique reveals mainly diamagnetic species while the latter only detects paramagnetic ones, being both types of molecules present in the solutions of B.
In summary, the chemical equilibrium established in diluted aqueous solutions of B involves mono- and oligonuclear vanadates(V) expected to exist in neutral to slightly acidic pH, together with mixed-valence polynuclear aggregates. The acidic nature of B apparently dictates the composition of the equilibrium mixtures generated by the breakage of the original polynuclear structure, because the gradual decrease of pH with increasing concentration of B leads to the formation of the most stable vanadium(V) species at each specific condition.
Chemoprotective activity of mixed-valence polyoxovanadates against the alkylating agent DES
Studies of the chemoprotective activity of the polyoxovanadates A and B against the mutagenic action of diethylsulphate (DES) were started by evaluating the toxic effect of each of the three compounds (A, B and DES) on the growth of E. coli DH5α cultures. Results of the dose–response curves after 3 h of incubation with DES indicated a GI50 value of 5.8 mmol L−1 (Fig. S11†). Attempts to work with 7.0 mmol L−1 of DES in the cultures killed about 85% of the bacteria, while lower concentrations gave variable survival rates, even in carefully controlled culture conditions. The concentration of 6.0 mmol L−1 of DES was then chosen for the subsequent chemoprotection experiments, as described below.Growth inhibition assays in the presence of polyoxovanadates (Scheme S1†) evidenced that A was nontoxic in all concentrations employed in this work (0.05 to 10.0 mmol L−1, Fig. 3). This is in accordance with our earlier reports on the absence of a deleterious effect of A after the direct reaction with pUC19 plasmid DNA.18 Product B, in turn, was nontoxic only in concentrations up to 1.0 mmol L−1 compared to the control sample, showing an increasing bacterial growth inhibitory effect at higher concentrations and an estimated GI50 value of 8.2 mmol L−1. These results are probably associated with the lower stability of B in aqueous solutions as compared to A, a feature that could generate a higher number or a higher concentration of vanadium species potentially toxic to E. coli.
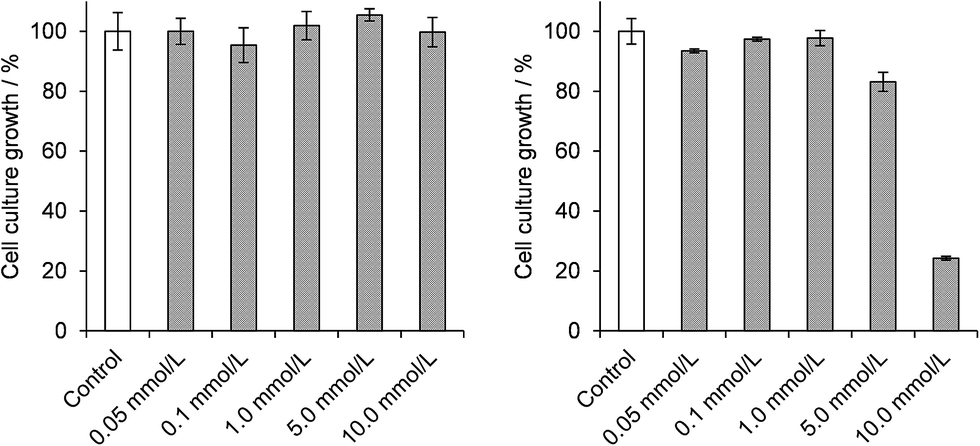 | ||
| Fig. 3 Cell culture growth assays in E. coli suspensions treated with [(CH3)4N]6[V15O36(Cl)] (A, left) and K(NH4)4[H6V14O38(PO4)]·11H2O (B, right), respectively, with concentrations varying from 0.05 to 10.0 mmol L−1 in both cases. Data were obtained after the third hour of incubation at 37 °C and 120 rpm, and are given as average values and standard deviations of triplicates. OD540 values of 0.926 ± 0.062 and 0.980 ± 0.042 in the control cultures were taken as 100% growth for assays with A and B respectively. | ||
For the chemoprotection assays, bacterial cultures were incubated with DES at the final concentration of 6.0 mmol L−1 in the presence of varying concentrations (0.01 to 10.0 mmol L−1) of the polyoxovanadates A and B for 3 h in LB (Fig. 4 left and right respectively). The chemoprotective activity shown by A does not appear to be dose-dependent, as an increase in bacterial growth of 30–40% (as compared with the DES control, second column, Fig. 4) was observed in the range of 0.1 to 5.0 mmol L−1 of A, which corresponds to DES:V molar proportions of 4:1 to 0.08:1.
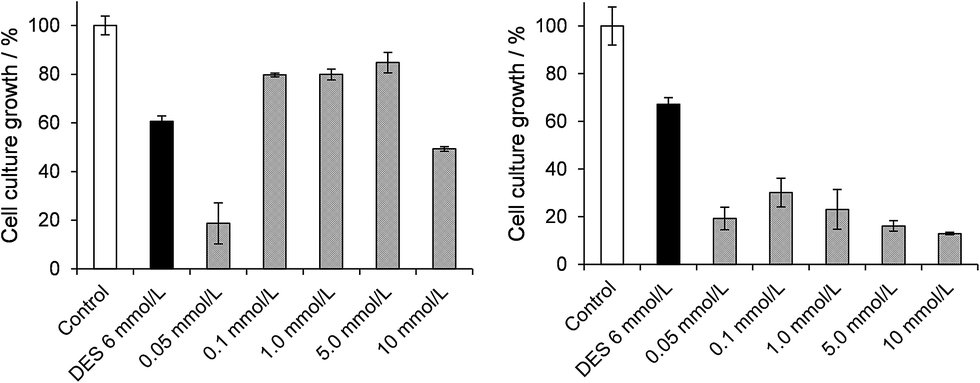 | ||
| Fig. 4 Cell culture growth assays in bacterial suspensions treated with increasing concentrations (0.05 to 10.0 mmol L−1) of [(CH3)4N]6[V15O36(Cl)] (A, left hand side) and K(NH4)4[H6V14O38(PO4)]·11H2O (B, right), along with 6.0 mmol L−1 of the alkylating agent DES. Data were obtained after the third hour of incubation at 37 °C and 120 rpm, and are given as average values and standard deviations of triplicates. OD540 values of 0.805 ± 0.038 and 0.862 ± 0.080 in the control cultures were taken as 100% growth for assays with A and B respectively. In this experiment, the control culture (first column in each graph) did not receive any addition of POV or DES. The second column in each graph corresponds to cultures treated with 6.0 mmol L−1 of DES in the absence of A or B. | ||
For the highest concentration of A (10.0 mmol L−1, DES:V = 0.04:1) protection was not observed, possibly because of the production of toxic vanadium-containing species from A itself in this specific culture condition. The lowest concentrations of A (0.05 mmol L−1, or DES:V molar proportions of 8:1) also appear to reinforce the toxic effect of DES. In these cases, large fluctuations in culture growth were observed, suggesting exposure of the cells to varying adverse conditions. This result gives support to the hypothesis that a chemical reaction between A and DES could be responsible for chemoprotection, as differences in molar proportions appear to determine positive or negative results.
In sharp contrast, results evidenced the absence of chemoprotective activity and even an increase in toxicity when the polyoxovanadate B was added to the DES-treated cultures. In this case, the decrease in bacterial growth was higher than 55% (as compared to the DES control) for all concentrations of B. This means that the presence of both DES and B in LB medium is more deleterious to the cells than each of them alone, either because their intact molecules potentiate the toxic action of one another, or because their interaction produces new compounds that are themselves more toxic to the bacteria.
Spectroscopic study of direct reactions between the alkylating agent and the polyoxovanadates in LB medium
Spectroscopic investigations performed by 51V NMR and EPR, in the presence and absence of DES, assessed the reasons for the distinct effects of the two polyoxovanadates in E. coli cultures. Based on the assumption that the nature of the potentially toxic or chemoprotective oxidovanadium species produced from the polyoxovanadates in solution is highly dependent on the reaction medium and on the stability of the parent compound, speciation studies were performed for both A and B in LB, in order to differentiate the results from those previously obtained in aqueous solutions. The spectroscopic analyses were carried out with DES:V molar proportions varying from 0:1 to 5:1 at room temperature; this contains the proportion range in which A has shown chemoprotective activity. 51V NMR and EPR spectroscopic studies carried out for A (4.6 mmol L−1) and B (5.0 mmol L−1) showed that the stability of the polynuclear aggregates in LB is indeed different from that reported earlier in water. For clarity, data will be presented (and discussed) first for A and then for B.
Speciation studies of A in LB in the absence and presence of DES, performed by 51V NMR and EPR spectroscopies
While the 51V NMR spectrum of A in D2O evidenced structural breakage (signals at δ −559.9, −573.3 and −578.5 ppm for “V1”, “V2” and “V4” respectively),18 the spectrum recorded in LB shows only low intensity signals in the region between −450 and −600 ppm (Fig. 5, first and second spectra from the bottom for D2O and LB respectively).14 The absence of small nuclearity vanadium species generated from the breakage of A in this pH range suggests the possible preservation of the polynuclear structure in LB. Caution should be exercised in this case, as 51V NMR analyses usually give good evidence only for diamagnetic vanadium(V) species, while a possible breakage can also generate paramagnetic VIV-containing compounds that would not be easily identified by the technique. EPR results obtained for solutions of A in LB were therefore relevant to complement this analysis; in fact, they give support to the formation of a mononuclear, paramagnetic species from A that coexists with the remaining polynuclear molecules, as described below.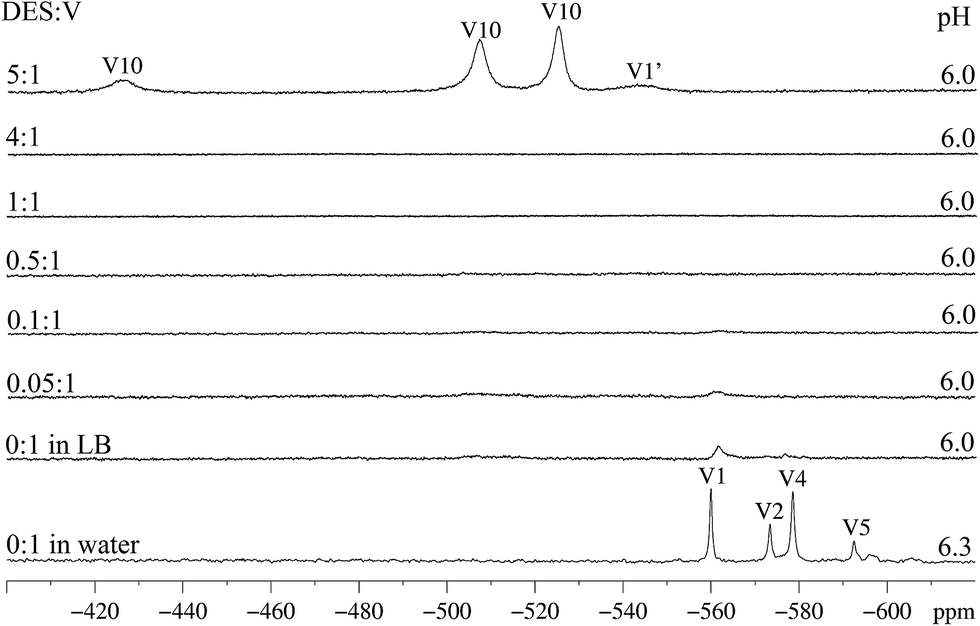 | ||
| Fig. 5 51V NMR spectra of product A (4.6 mmol L−1, pH 6.3) in water (first spectrum from the bottom), in LB (0:1 DES/V, pH 6.0); and with addition of increasing amounts of DES in LB medium (0.05:1 to 5:1 DES/V). Vanadium(V) species were identified as follows: “V1” = H2VO4−, “V1′” = HVO42− and “V10” = HV10O285−. pH Values measured in the reaction mixtures and DES:V proportions are presented on the right and left hand sides respectively. | ||
The addition of small amounts of DES to the bluish-green solutions of A in LB (up to the DES:V molar ratio of 4:1), did not cause any significant change in colour or in the 51V NMR spectra. In these solutions, only very weak signals given by small nuclearity vanadium(V) species are seen (Fig. 5). Only the highest proportion of DES to vanadium (5:1) leads to the (quick) formation of the decavanadate anion, HV10O285−, for which high intensity signals at −525, −507 and −425 ppm are observed (Fig. 5, first spectrum from the top). The formation of a hydrolysis product of decavanadate, hereafter called “V1′” (H2VO4−), was also observed (δ −544 ppm). For the highest proportion of DES:V (8:1), a white solid precipitated from a yellowish-green solution; this suspension was not analysed by 51V NMR because of shimming problems.
It is interesting to notice that, for A, the chemoprotective activity shown in Fig. 4 was observed when the DES:V proportion ranged from 0.08 to 4 (the concentration of 0.1 mmol L−1 of A corresponds to the DES:V molar proportion of 4:1). In addition, for the condition in which a high proportion of DES to vanadium was employed (8:1), chemoprotection was not observed. These results, together with the 51V NMR data, may indicate that the expressive breakage of the polyoxovanadate structure to form the decavanadate anion (top spectrum in Fig. 5) could be related to the negative chemoprotection by A in high DES:V molar ratios.
In our view, the polynuclear A, or other MV-POV species formed from it, appear to be the active species in chemoprotection and their conversion into the decavanadate anion is one of possible pathways to the deactivation of A occurring in solution. In this context, chemoprotection in the observed concentration range (Fig. 4) could occur in the extracellular medium by the interception mechanism suggested by Hamilton and Wilker,16 with A quickly reacting with DES and avoiding its penetration in the cells. More extensive considerations on this subject will be made below.
In an attempt to understand the result shown in Fig. 4 for the highest concentration of A (10.0 mmol L−1; DES:V = 0.04:1), the same proportion and conditions employed in the biological assay were used in the direct reaction of A with DES in LB medium. A blank experiment was run with the same concentration of A in pure LB, that is, without the addition of DES. The reaction mixtures were analysed by 51V NMR and, for both spectra, broad low intensity signals centred at −466, −510 and −562 ppm were observed (Fig. S12†). These yet unidentified species, produced in high enough amounts to be detected by NMR, were probably formed by reaction of A with components of the LB medium (mainly tryptone and yeast extract, that is, peptides, free aminoacids and sugars). It is possible that these species are also formed from the lower concentrations of A in LB, but in this case their amounts are too low for detection, particularly considering the usual quadrupolar broadening and moderate signal/noise ratios in 51V NMR spectra. Although the chemical nature of these species is very difficult to determine – especially considering the chemical complexity of the LB medium – the observation of chemoprotective activity in the DES:V window in which they are not formed, or, in other words, in which the polynuclear structure of A appears to be at least partially preserved, is a significant finding.
In the EPR spectra recorded at 77 K for solutions of A both in water (Fig. S13†) and in LB (Fig. 6), it was possible to identify the presence of a broad band, usually given by oligo/polynuclear complexes with magnetic interaction between vanadium(IV) centres. This system could include the intact molecule of A, which contains both vanadium(V) and vanadium(IV), and derived MV-POV produced in solution. Superimposed to this broad line, the parallel and perpendicular spectra given by a magnetically isolated, mononuclear oxidovanadium(IV) complex (I = 7/2) (Fig. 6) are seen. For the mononuclear species detected from A in aqueous solution, anisotropic parameters obtained by spectral simulation (gx = 1.9777, gy = 1.9746, gz = 1.9351, Ax = 69.58 × 10−4 cm−1, Ay = 59.60 × 10−4 cm−1 and Az = 177.01 × 10−4 cm−1) are compatible with those reported in the literature for [VO(H2O)5]2+.75 In LB medium, in turn, the mononuclear component shows slightly different EPR parameters (gx = 1.9755, gy = 1.9746, gz = 1.9410, Ax = 62.96 × 10−4 cm−1, Ay = 55.76 × 10−4 cm−1 and Az = 169.60 × 10−4 cm−1, Table S5†) that can be generated by replacement of water in the metal coordination sphere by a component of the LB medium. Despite this difference, the EPR pattern and the numerical parameters are all compatible with that expected for a mononuclear oxidovanadium(IV) complex.
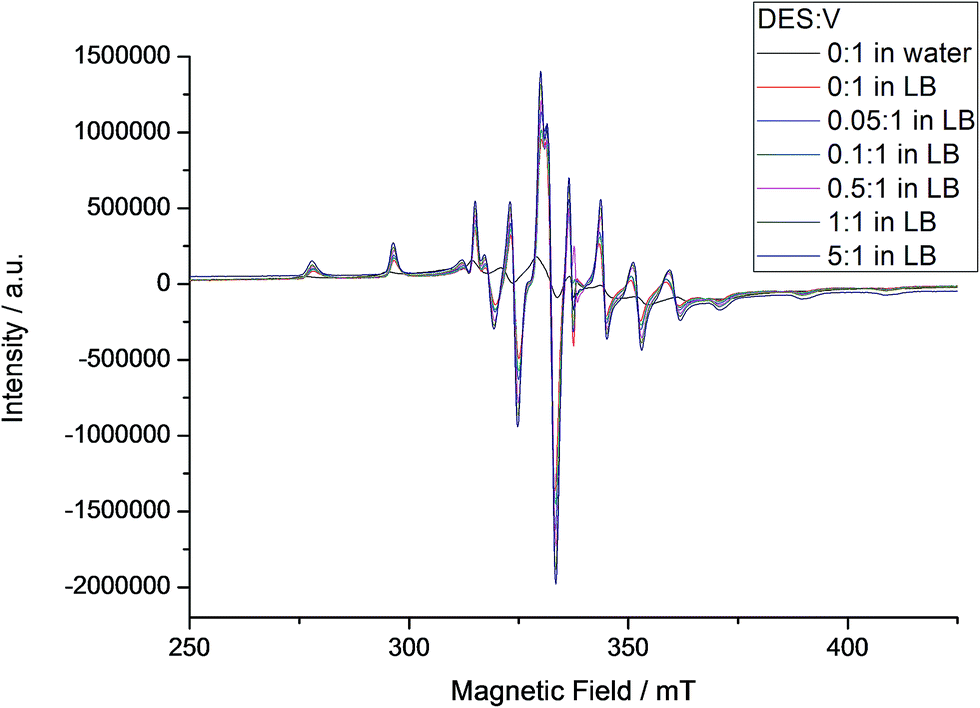 | ||
| Fig. 6 X-band EPR spectra recorded for A in aqueous and LB solutions at 77 K, with addition of increasing amounts of DES (see internal legend). The intensities of the different spectra were normalized by comparison with the signal given by the MgO/Cr3+ marker. EPR simulation showed the presence of mononuclear and polynuclear species in the samples; parameters for the mononuclear species are gathered in Table S5.† | ||
Differently from the result obtained in water, the EPR spectra registered for A in LB, also shown in Fig. 6, are characterized by the predominance of a mononuclear oxidovanadium(IV) complex over the polynuclear species, although their ratio is difficult to quantify because of spectral broadness. This observation is corroborated by the results expressed in Fig. S13† and discussed below. Despite this difficulty, data obtained by both EPR and 51V NMR analyses (Fig. 5 and 6) strongly indicate the establishment, in LB solution without the addition of DES, of an equilibrium between polynuclear species (possibly intact A and derived mixed-valence polyoxovanadates) and two mononuclear complexes, “V1” (diamagnetic, evidenced by NMR, Fig. 5) and an oxidovanadium(IV) complex (paramagnetic, revealed by EPR, Fig. 6). The extension of this equilibrium is still to be quantified.
The detection of the aforementioned vanadium-containing species in solution – either mononuclear or in higher nuclearities – made by the combination of 51V NMR and X-band EPR spectroscopies agrees well with other studies performed by our and other research groups.16,18 It is clear that A gives distinct spectroscopic results when dissolved in water or in LB, and the detection of new species in LB was indeed expected because of the composition of the culture medium.
Interestingly, sequential additions of DES do not change the EPR parameters of the mononuclear species produced from A, which seems to be non-reactive towards the alkylating agent. This finding is relevant in the context of the present work. The polynuclear spectral component, in turn, changes non-linearly and produces distinct g tensor values and spectral linewidths (from 56.8 to 72.4 mT, Fig. 6 and S14†) by simulation, suggesting the possibility of interconversion of different chemical species in solution. The system may therefore be composed by a mixture of different nuclearity species, some of them polynuclear, produced by addition of increasing proportions of DES. Wilker and co-workers16 have reported such type of rearrangement after the direct reaction of several simple vanadates(V) with DES in acetonitrile, yielding ethanol and a new metal aggregate that is one oxygen-atom-deficient in relation to the parent polyoxovanadate and is stable in specific conditions (eqn (1)). As an example, (V3O9)3− was converted into (V5O14)3− upon alkylation.16
| C2H5+ + [VxOy]n− + H+ → [VxOy−1](n−2)− + C2H5OH | (1) |
Taking into consideration the whole set of results obtained for A, both in this and in our previous work, the main vanadium(IV) and vanadium(V) products obtained after the addition of the alkylating agents DMS18 and DES to solutions of A (in water and LB) are mononuclear oxidovanadium(IV) complexes and the decavanadate(V) anion, HV10O285−, although the latter is only formed after addition of an excess of alkylating agent. The effect of chemoprotection promoted by intact A (or other MV-POV species derived from it) may be severely limited by the formation of these two compounds ([VO(H2O)5]2+ and V10) because, once formed, they appear to be unreactive towards the alkylating agents. As soon as these evidences became clear, we focused our efforts on the study of other polyoxovanadate systems, in an attempt to investigate the generality of these observations and to help understand the role of the different products in promoting or quenching the chemoprotective effect.
Speciation studies of B in LB in the absence and presence of DES, carried out by 51V NMR and EPR spectroscopies
Spectroscopic studies were also performed for compound B, which clearly differs from A because of not preventing cell death in the presence of DES. Indeed, a contrasting 51V NMR data set was obtained for B in pure LB (Fig. 7). While few signals, and of low intensity, were obtained for A (Fig. 5, second to seventh line from the bottom), in the case of B three strong signals at δ = −524, −506 and −425 ppm, typical of the three different coordination environments of vanadium(V) in the decavanadate anion [HV10O28]5− were clearly observed (Fig. 7). These signals appear immediately after the dissolution of B in LB. The main change observed in the 51V NMR spectra with increasing concentration of DES was the shift of these three signals to δ = −517, −501 and −424 ppm, which can be assigned to the second protonation of the anion in pH near 5, generating [H2V10O28]4−.14 Residual signals for the all-vanadium(V) species “V14P”, [H4V14O38(PO4)]5−, detected in the spectrum of B in aqueous solution (Fig. 7, first line from the bottom), can also be observed in LB after addition of DES. They appear as three low intensity signals with chemical shifts at δ = −596, −579 and −532 ppm.73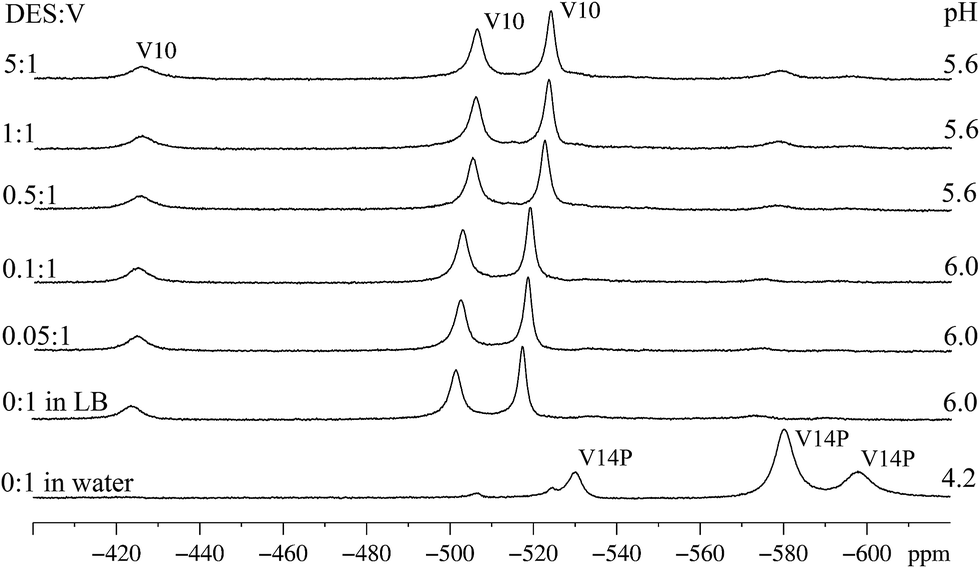 | ||
| Fig. 7 51V NMR spectra of a 5 mmol L−1 solution of product B in water (first line from the bottom, pH 4.2), in LB medium (pH 6.0), and in LB with increasing amounts of DES (from 0.05:1 to 5:1 DES/V). The identified vanadium(V) species were: “V14P” = [H4V14O38(PO4)]5− and “V10” = HV10O285−, both with three signals generated by non-equivalent vanadium(V) centres. The pH values of the reaction mixtures and the DES:V proportions are presented on the right and left hand sides of the spectra respectively. | ||
Reasons for this significant formation of decavanadate(V) from B in LB, even at the relatively high pH 6 (compare Fig. 2 and 7 for aqueous and LB solutions), are still to be established. A recent study in aqueous medium showed that, once decavanadate is formed in acidic conditions, it remains the dominant species up to pH 6.45 for 5 h, and continues to be detectable up to pH 8.59.74,76 In this context, one possible explanation for decavanadate detection in LB could be its formation in the acidic stock solution of B (20.0 mmol L−1, pH = 3.2) later added to the culture media. However, this anion was also quickly formed as the dominant species in freshly prepared solutions of B in LB, which is itself a buffered medium (see Fig. 7, second spectrum from the bottom). A full explanation for this occurrence will require further formation and stability studies from other mixed valence POV systems.
It is also possible that the hard-donor, chelating components of the LB medium (peptides, free aminoacids and sugars) facilitate the disaggregation of the vanadium ions from the polynuclear structure. This could also be related to the higher proportion of the harder vanadium(V) to vanadium(IV) ions in B, [H6VV12VIV2O38(PO4)]5−, as compared to A, [VV8VIV7O36(Cl)]6−, taking into consideration that decavanadate ions are not so easily formed from the latter. After an eventual breakage of the molecular aggregate in LB, this free VV could reaggregate to form decavanadate. Expanding this view to the biological context, this reaggregation could occur either outside or inside E. coli cells, as it is known that low nuclearity vanadium(V) species travel through cell membranes in various pathways depending on their ionic charge and inorganic or organic ligands.77 It is also accepted that decavanadate itself and other polyoxometalates are able to cross cell membrane bilayers, when for example they target (and inhibit) prokaryotic and eukaryotic ion pumps at the cytoplasmic side.78
As far as reactivity with alkylating agents is concerned, the “V10” anion has been reported to be a relatively poor reactant in contact with DES in acetonitrile.16 Its apparently favoured formation from B in LB medium, both in the absence and presence of DES, is therefore assumed to be one of the reasons why B does not show activity in the protection of E. coli against the alkylating agent in LB medium. Indeed, this anion is reported to be the most stable vanadate(V) oligomer formed in acidic pH range, and also to be kinetically inert for hours in neutral pH.14
Additionally, decavanadate has demonstrated a moderate to strong antimicrobial activity, which is modulated by the counterion, against Gram-positive and Gram-negative bacteria.79–82 The toxicity is improved by synergistic interaction with toxic molecules or by substances that facilitate vanadate transportation to the inside of cells.83,84 In superior eukaryotic organisms, the interaction of decavanadate(V) with proteins such as actin,76 myosin and Ca2+-ATPase85 has been reported to lead to protein oxidation with consequent formation of oxidovanadium(IV) complexes. In fact, the induction of redox reactions with biological macromolecules seems to be an important mode of action of vanadates.38,86,87
In the specific case of E. coli, the antibacterial activity of decavanadate species has been reported to relate to the inhibition of ion transportation across cell membranes, which drastically affects cell metabolism.83,88 These features could at least partially explain not only the poor chemoprotective performance of B in E. coli cultures, but also its intrinsic toxicity towards the bacteria in the experiments summarized in Fig. 3 and 4.
EPR spectra registered for B in LB at 77 K (Fig. 8 and Table S5†) also showed the breakage of the polynuclear structure. Spectra simulation again revealed the presence of a mononuclear oxidovanadium(IV) complex with the same anisotropic parameters found from A (gx = 1.9752, gy = 1.9739, gz = 1.9379, Ax = 63.91 × 10−4 cm−1, Ay = 57.69 × 10−4 cm−1 and Az = 171.93 × 10−4 cm−1) and, differently from the aqueous solutions, only one polynuclear component with a g value of 1.9723 and Δpp = 34.0 mT. Reactions of B in LB with different proportions of DES gave no changes in the spectral profile or in the individual VO2+ complex and MV-POV contributions to the total EPR spectrum of the frozen solution (Fig. 8 and S15†). This again agrees with the low chemoprotective activity of B in E. coli cultures, which is probably due to the high stability of the decavanadate(V) and oxidovanadium(IV) species formed from B after dissolution in LB.
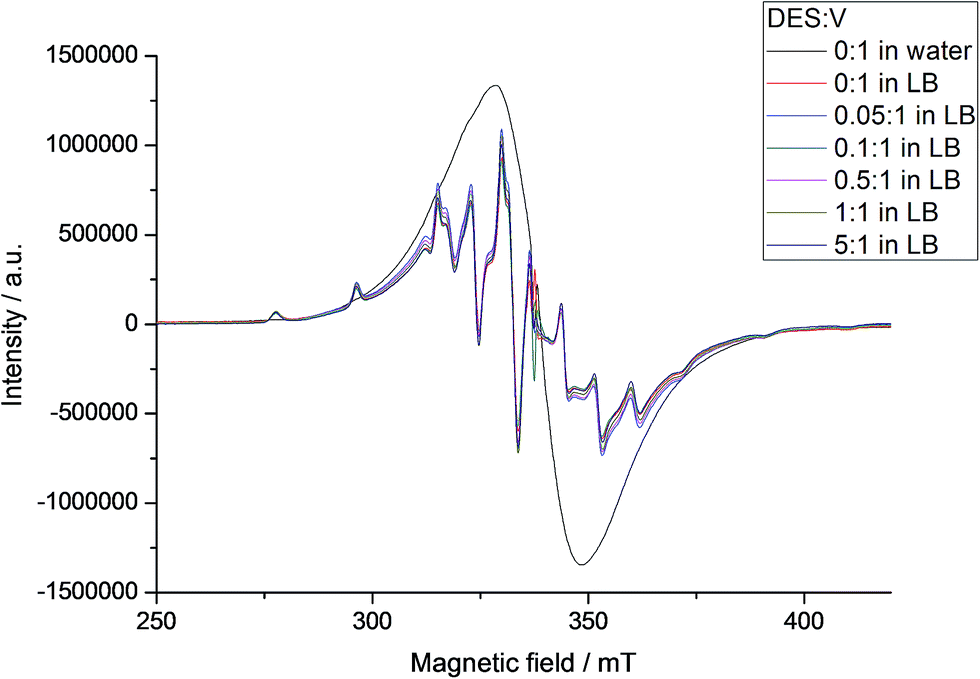 | ||
| Fig. 8 X-band EPR spectra of B at 77 K in aqueous solution and in LB with addition of increasing amount of DES (see internal legend). Spectral intensities were normalized by comparison with the MgO/Cr3+ marker. EPR simulation showed the presence of mononuclear and polynuclear species in LB; parameters for the mononuclear species are gathered in Table S5.† | ||
The reactivity of B with DES in LB was also investigated by 31P NMR analysis. After addition of DES, the single resonance signal registered for the sample at 0.11 ppm disappeared and gave place to a new signal at 0.13 ppm (Fig. S16†). Both chemical shifts are in the region reported for “V14P” (from 1.2 to −0.58 ppm depending on the degree of protonation, pH and ionic strength of medium).56 It is not known if “V14P” alone is able to protect the bacteria from the alkylating agent, but in the case of B (Fig. 7, first spectrum from the top) this species was formed in a small amount and its effect, if existing, is certainly minimal in comparison with the damage apparently caused by the formation of decavanadate.
Individual contributions of the MV-POV and mononuclear species for the total EPR spectrum of A and B in aqueous and LB solutions
The relative contributions of the distinct chemical species formed from A and B to the total, frozen solution EPR spectrum of these polyoxovanadates in solution (aqueous and LB) were quantified in EPR measurements. This was carried out by integration of the broad MV-POV and the sharper mononuclear lines separately (Fig. S14 and S15†) and expressed as the contribution of each species to the total magnetization of the system. For A, the proportion of the polynuclear to the mononuclear component of the mixture is much higher in water than in LB, decreasing and then remaining approximately constant in the latter when zero or lower concentrations of DES are employed, and decreasing further when larger amounts of DES are added to the medium (Fig. S14†). In other words, solutions of A in LB respond differently to increasing amounts of the alkylating agent and this agrees with our earlier considerations on the capacity of A to effectively react with DES and therefore protect the bacterial cells from its action.For B, on the other hand, no changes were observed in the proportion of the two contributions (mononuclear and polynuclear in Fig. S15†), independently of the concentration of DES in the LB medium. This again corroborates the results of the 51V NMR and previous EPR studies, which pointed out to quick breakage of the original polynuclear structure of B in LB and the poor reactivity of the resulting species in solution (MV-POV, “V10” and oxidovanadium(IV) complex) with the alkylating agent.
In Fig. S14 and S15,† differences in the intensities of the mononuclear (EPR) spectra generated from A and B at the same concentrations of vanadium could not be quantitatively evaluated due to the difference in the proportions of vanadium(IV) and vanadium(V) produced by the two polyoxovanadates in each reaction condition.
Conclusions
The methodology of synthesis that applies mannitol as reducing agent for vanadium(V), employed earlier by our research group to produce the polyoxovanadate A under mild conditions and good yield, has been successfully applied again in this work to give B, [H6VV12VIV2O38(PO4)]5−. This reinforces the versatility of this simple synthetic route.Studies of the chemoprotective activity of polyoxovanadates A and B against the toxic effect of diethylsulphate (DES) as an alkylating agent in E. coli cultures revealed disparate results; while A plays a chemoprotective role (30–40%) in the biological assays, B enhanced the deleterious effect of DES on bacterial growth. In both cases, however, E. coli cultures appeared to function as a suitable biological model system. 51V NMR and EPR data obtained from reaction mixtures that simulated the biological assay conditions allowed significant advances in the rationalization of these results.
The observation or absence of chemoprotective activity by polyoxovanadates A and B against DES was clearly dependent on the chemical nature and stability of the soluble species formed from the polynuclear aggregates in the culture medium after addition of the alkylating agent. The buffered culture (LB) medium was effective in inhibiting the formation of multiple, low nuclearity vanadium(V)-containing species usually detected in aqueous solution (“V1”, “V2”, “V4” and “V5”). This speciation equilibrium is directly controlled by pH changes and the ionic strength of the medium, which are drastically different in LB as compared to water. This gives rise to a very different composition of the reaction medium in LB, as far as the vanadium-containing components are concerned.
In the case of B, the complete absence of chemoprotective activity against DES might be correlated with the formation of the very stable decavanadate(V) anion (“V10” = HV10O285−) and the mononuclear oxidovanadium(IV) species in LB medium, both in the presence and the absence of DES. Once formed, these compounds react poorly or even do not react with the alkylating agent, and therefore do not contribute to its elimination from the culture medium. Also, decavanadate itself is believed to penetrate living cells or even be assembled inside them,78 and has been shown to inhibit a number of metabolic pathways.13 In eukaryotic cells, for example, decavanadate affects oxidative stress processes related to membrane depolarization and oxygen consumption, and inhibits myosin ATPase activity and actin polymerization.39,88 In prokaryotic cells, in turn, the antibacterial activity has been associated with the inhibition of ion pumps, constraining transport across the membrane and deeply compromising bacterial metabolism.88 In the light of these findings, the facile formation of decavanadate from B revealed in this work in the presence of DES could be associated with the enhanced toxicity shown by this POV towards E. coli cells.
Polyoxovanadate A, on the other hand, only forms “V10” in LB medium at the highest DES:V proportions employed in this work. Also, in LB, the cage-like structure of A appears to be much more resistant to breakage than in aqueous solution, as it does not form significant amounts of the vanadate(V) species “V1” to “V5” when dissolved in LB. Although A has also been shown to form mononuclear oxidovanadium(IV) complexes in LB, which are again unreactive towards DES, this formation does not appear to inhibit E. coli culture growth – LB solutions of A were not toxic towards the bacteria in most concentrations employed in this work (Fig. 3).
In summary, it is possible that a significant amount of A, or of a structurally-related polyoxovanadate, is kept unbroken in LB and then reacts with the alkylating agent to prevent cell death. Evidence for this type of reaction comes from previous works reported in the literature16,17 and from the fact that the chemoprotective activity revealed for A in this work is dependent on the reactants (DES:V) proportion. Such an “interception” reaction with the alkylating agent could occur directly in the extracellular medium and therefore prevent the access of the toxin to the cytoplasm. However, because of the lack of information on the ability of large POM aggregates, particularly mixed valence ones, to penetrate cell membranes, it is not possible to exclude the occurrence of intracellular interactions between A and DES. If, on one hand, this positive activity of A on prokaryotic cells appears to be consistent and deserves further investigation, the interaction of this polyoxovanadate with eukaryotic cell components and the existence of a possible chemoprotective effect of A on this type of cell remains an open issue.
Finally, the chemistry of mixed valence polyoxovanadates in solution remains largely unknown and needs to be further clarified. As an example, the influence of the size of the polyoxovanadate aggregate on its stability, and the role played by the central anion (Cl− or PO4−) in the molecular self-assembly process is not yet fully understood. In face of the above, a particular challenge of using polyoxovanadates in biological studies, especially mixed-valence compounds, lies in the rationalization of their solution chemistry to permit the selection of the most promising aggregates for chemoprotection studies. Other fundamental pieces of work are studies on the interaction of such molecules with membrane proteins, ion pumps and channels and specific intracellular targets. These studies are on their beginning and their findings will surely clarify the mechanism of action of these POM molecules in biological systems.
Acknowledgements
This work was financially supported by Fundação Araucária (project number 20171010), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant 307592/2012-0), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, PVE A099/2013), the National Institute of Science and Technology-Biological Nitrogen Fixation (INCT-FBN), Fundação Araucária and Universidade Federal do Paraná (UFPR). Authors thank Mr Angelo Roberto dos Santos Oliveira (UFPR) for the TGA analyses. K. P, A. L. R, G. V, G. G. N, D. L. H, J. F. S and E. M. S thank CNPq, CAPES and Fundação Araucária for research grants and scholarships.References
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- K. Y. Monakhov, W. Bensch and P. Kogerler, Chem. Soc. Rev., 2015, 44, 8443–8483 RSC.
- A. Müller, R. Rohlfing, A.-L. Barra and D. Gatteschi, Adv. Mater., 1993, 5, 915–917 CrossRef.
- K. Y. Monakhov, O. Linnenberg, P. Kozłowski, J. van Leusen, C. Besson, T. Secker, A. Ellern, X. López, J. M. Poblet and P. Kögerler, Chem.–Eur. J., 2015, 21, 2387–2397 CrossRef CAS PubMed.
- J. J. Chen, M. D. Symes, S.-C. Fan, M. S. Zheng, H. N. Miras, Q. F. Dong and L. Cronin, Adv. Mater., 2015, 27, 4649–4654 CrossRef CAS PubMed.
- C. C. McLauchlan and D. C. Crans, Dalton Trans., 2013, 42, 11744–11748 RSC.
- E. Kioseoglou, S. Petanidis, C. Gabriel and A. Salifoglou, Coord. Chem. Rev., 2015, 301–302, 87–105 CrossRef CAS.
- J. C. Pessoa, S. Etcheverry and D. Gambino, Coord. Chem. Rev., 2015, 301–302, 24–48 CrossRef CAS.
- P. P. Fu, X. L. Wang, E. B. Wang, C. Qin and L. Xu, Chem. Res. Chin. Univ., 2005, 21, 381–385 CAS.
- Y. Y. Liu, S. Y. Tian, S. X. Liu and E. B. Wang, Transition Met. Chem., 2005, 30, 113–117 CrossRef CAS.
- L. Roubatis, N. C. Anastasiadis, C. Paratriantafyllopoulou, E. Moushi, A. J. Tasiopoulos, S. C. Karkabounas, P. G. Veltsistas, S. P. Perlepes and A. M. Evangelou, Inorg. Chem. Commun., 2016, 69, 85–88 CrossRef CAS.
- A. M. Evangelou, Crit. Rev. Oncol. Hematol., 2002, 42, 249–265 CrossRef PubMed.
- M. Aureliano, Oxid. Med. Cell. Longevity, 2016, 2016, 6103457 CAS.
- M. Aureliano and D. C. Crans, J. Inorg. Biochem., 2009, 103, 536–546 CrossRef CAS PubMed.
- J. Costa Pessoa, J. Inorg. Biochem., 2015, 147, 4–24 CrossRef CAS PubMed.
- E. E. Hamilton, P. E. Fanwick and J. J. Wilker, J. Am. Chem. Soc., 2006, 128, 3388–3395 CrossRef CAS PubMed.
- E. E. Hamilton and J. J. Wilker, J. Biol. Inorg. Chem., 2004, 9, 894–902 CrossRef CAS PubMed.
- G. G. Nunes, A. C. Bonatto, C. G. de Albuquerque, A. Barison, R. R. Ribeiro, D. F. Back, A. V. C. Andrade, E. L. de Sá, F. d. O. Pedrosa, J. F. Soares and E. M. de Souza, J. Inorg. Biochem., 2012, 108, 36–46 CrossRef CAS PubMed.
- T. Chakraborty, A. Chatterjee, A. Rana, D. Dhachinamoorthi, P. A. Kumar and M. Chatterjee, Biochim. Biophys. Acta, 2007, 1772, 48–59 CrossRef CAS PubMed.
- S. Samanta, V. Swamy, D. Suresh, M. Rajkumar, B. Rana, A. Rana and M. Chatterjee, Mutat. Res., 2008, 650, 123–131 CAS.
- P. Villani, M. Spanò, F. Pacchierotti, M. Weimer and E. Cordelli, Reprod. Toxicol., 2010, 30, 44–49 CrossRef CAS PubMed.
- J. Nieminuszczy and E. Grzesiuk, Acta Biochim. Pol., 2007, 54, 459–468 CAS.
- D. T. Beranek, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1990, 231, 11–30 CrossRef CAS PubMed.
- O. Fernández-Miragall and E. Martínez-Salas, J. Gen. Virol., 2007, 88, 3053–3062 CrossRef PubMed.
- A. Méreau, R. Fournier, A. Grégoire, A. Mougin, P. Fabrizio, R. Lührmann and C. Branlant, J. Mol. Biol., 1997, 273, 552–571 CrossRef PubMed.
- A. J. Zaug and T. R. Cech, RNA, 1995, 1, 363–374 CAS.
- D. Mielecki, M. Wrzesiński and E. Grzesiuk, Mutat. Res., Rev. Mutat. Res., 2015, 763, 294–305 CrossRef CAS PubMed.
- B. Singer and D. Grunberger, in Molecular Biology of Mutagens and Carcinogens, Springer US, Boston, MA, 1983, pp. 45–96 Search PubMed.
- K. Kleibl, Mutat. Res., Rev. Mutat. Res., 2002, 512, 67–84 CrossRef CAS.
- C. Janion, Int. J. Biol. Sci., 2008, 4, 338–344 CrossRef CAS PubMed.
- B. Sedgwick and T. Lindahl, Oncogene, 2002, 21, 8886–8894 CrossRef CAS PubMed.
- P. Ø. Falnes, A. Klungland and I. Alseth, Neuroscience, 2007, 145, 1222–1232 CrossRef CAS PubMed.
- A. N. Suhasini, J. A. Sommers, S. Yu, Y. Wu, T. Xu, Z. Kelman, D. L. Kaplan and R. M. Brosh Jr, J. Biol. Chem., 2012, 287, 19188–19198 CrossRef CAS PubMed.
- J. Musarrat, J. Arezina-Wilson and A. A. Wani, Biochim. Biophys. Acta, 1995, 1263, 201–211 CrossRef.
- C. You and Y. Wang, Acc. Chem. Res., 2016, 49, 205–213 CrossRef CAS PubMed.
- J. M. Fautch, P. E. Fanwick and J. J. Wilker, Eur. J. Inorg. Chem., 2009, 2009, 33–37 CrossRef.
- N. Samart, J. Saeger, K. J. Haller, M. Aureliano and D. C. Crans, J. Mol. Eng. Mater., 2014, 02, 1440007 CrossRef.
- S. Ramos, J. J. G. Moura and M. Aureliano, Spectrosc. Int. J., 2012, 27, 5 Search PubMed.
- M. Aureliano and C. A. Ohlin, J. Inorg. Biochem., 2014, 137, 123–130 CrossRef CAS PubMed.
- L. S. A. Dikshitulu and G. G. Rao, Fresenius' Z. Anal. Chem., 1962, 189, 421–426 CrossRef CAS.
- D. X. Wang, H. H. Kung and M. A. Barteau, Appl. Catal., A, 2000, 201, 203–213 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- J. Woolcock and A. Zafar, J. Chem. Educ., 1992, 69, A176 CrossRef CAS.
- Z. S. Teweldemedhin, R. L. Fuller and M. Greenblatt, J. Chem. Educ., 1996, 73, 906 CrossRef CAS.
- O. Kahn, Molecular Magnetism, Wiley, 1993, p. 375 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 (Acta Crystallogr., Sect. C: Struct. Chem., 2014, 71, 3–8) CrossRef CAS PubMed.
- C. Colliex, J. M. Cowley, S. L. Dudarev, M. Fink, J. Gjønnes, R. Hilderbrandt, A. Howie, D. F. Lynch, L. M. Peng, G. Ren, A. W. Ross, V. H. Smith, J. C. H. Spence, J. W. Steeds, J. Wang, M. J. Whelan and B. B. Zvyagin, in International Tables for Crystallography Volume C: Mathematical, Physical and Chemical Tables, ed. E. Prince, Springer, Netherlands, Dordrecht, 2004, pp. 259–429 Search PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- J. Sambrook and D. W. Russell, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, vol. 1, 2001 Search PubMed.
- V. S. Fluxa, N. Maillard, M. G. P. Page and J.-L. Reymond, Chem. Commun., 2011, 47, 1434–1436 RSC.
- GraphPad Prism Version 5.1 for Windows, GraphPad Software, La Jolla California USA, https://www.graphpad.com Search PubMed.
- P. Kanoo, A. C. Ghosh and T. K. Maji, Inorg. Chem., 2011, 50, 5145–5152 CrossRef CAS PubMed.
- M. I. Khan, J. Zubieta and P. Toscano, Inorg. Chim. Acta, 1992, 193, 17–20 CrossRef CAS.
- R. Kato, A. Kobayashi and Y. Sasaki, Inorg. Chem., 1982, 21, 240–246 CrossRef CAS.
- A. T. Harrison and O. W. Howarth, J. Chem. Soc., Dalton Trans., 1985, 1953–1957 RSC.
- A. Selling, I. Andersson, L. Pettersson, C. M. Schramm, S. L. Downey and J. H. Grate, Inorg. Chem., 1994, 33, 3141–3150 CrossRef CAS.
- D. Wang, W. Zhang, K. Grüning and D. Rehder, J. Mol. Struct., 2003, 656, 79–91 CrossRef CAS.
- J. F. Keggin, Nature, 1933, 131, 908–909 CrossRef CAS.
- H. H. Thorp, Inorg. Chem., 1992, 31, 1585–1588 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- R. L. Carlin, Magnetochemistry, Springer-Verlag, 1986 Search PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef CAS.
- P. Day, N. S. Hush and R. J. H. Clark, Philos. Trans. R. Soc., A, 2008, 366, 5–14 CrossRef CAS PubMed.
- T. D. Keene, D. M. D'Alessandro, K. W. Krämer, J. R. Price, D. J. Price, S. Decurtins and C. J. Kepert, Inorg. Chem., 2012, 51, 9192–9199 CrossRef CAS PubMed.
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2009, 131, 5101–5114 CrossRef CAS PubMed.
- A. Bencini and D. Gatteschi, EPR of Exchange Coupled Systems, Dover Publications, Incorporated, 2012 Search PubMed.
- R. L. Frost, K. L. Erickson, M. L. Weier and O. Carmody, Spectrochim. Acta, Part A, 2005, 61, 829–834 CrossRef PubMed.
- M. Klähn, G. Mathias, C. Kötting, M. Nonella, J. Schlitter, K. Gerwert and P. Tavan, J. Phys. Chem. A, 2004, 108, 6186–6194 CrossRef.
- E. I. Voit, V. A. Davydov, A. A. Mashkovskii and A. V. Voit, J. Struct. Chem., 2008, 49, 13–20 CrossRef CAS.
- B. Dong, J. Peng, Y. Chen, Y. Kong, A. Tian, H. Liu and J. Sha, J. Mol. Struct., 2006, 788, 200–205 CrossRef CAS.
- A. Gorzsas, I. Anderson and L. Pettersson, J. Inorg. Biochem., 2009, 103, 517–526 CrossRef CAS PubMed.
- K. Okaya, T. Kobayashi, Y. Koyama, Y. Hayashi and K. Isobe, Eur. J. Inorg. Chem., 2009, 2009, 5156–5163 CrossRef.
- I. Andersson, A. Gorzsas, C. Kerezsi, I. Toth and L. Pettersson, Dalton Trans., 2005, 3658–3666 RSC.
- M. Aureliano, C. A. Ohlin, M. O. Vieira, M. P. M. Marques, W. H. Casey and L. A. E. Batista de Carvalho, Dalton Trans., 2016, 45, 7391–7399 RSC.
- M. M. Iannuzzi and P. H. Rieger, Inorg. Chem., 1975, 14, 2895–2899 CrossRef CAS.
- S. Ramos, M. Manuel, T. Tiago, R. Duarte, J. Martins, C. Gutiérrez-Merino, J. J. G. Moura and M. Aureliano, J. Inorg. Biochem., 2006, 100, 1734–1743 CrossRef CAS PubMed.
- E. Garribba and D. Sanna, in Binding, Transport and Storage of Metal Ions in Biological Cells, RSC, 2014, pp. 153–187 Search PubMed.
- M. Aureliano, G. Fraqueza and C. A. Ohlin, Dalton Trans., 2013, 42, 11770–11777 RSC.
- F. Yraola, S. García-Vicente, L. Marti, F. Albericio, A. Zorzano and M. Royo, Chem. Biol. Drug Des., 2007, 69, 423–428 CAS.
- M. Shahid, P. K. Sharma, A. S. Chibber and Z. A. Siddiqi, J. Cluster Sci., 2014, 25, 1435–1447 CrossRef CAS.
- N. Fukuda and T. Yamase, Biol. Pharm. Bull., 1997, 20, 927–930 CAS.
- S. Toumi, N. Ratel-Ramond and S. Akriche, J. Cluster Sci., 2015, 26, 1821–1831 CrossRef CAS.
- S. P. Chen, G. Z. Wu, D. W. Long and Y. D. Liu, Carbohydr. Polym., 2006, 64, 92–97 CrossRef CAS.
- M. Aureliano, World J. Biol. Chem., 2011, 2, 215–225 CrossRef PubMed.
- G. Fraqueza, C. A. Ohlin, W. H. Casey and M. Aureliano, J. Inorg. Biochem., 2012, 107, 82–89 CrossRef CAS PubMed.
- S. Ramos, R. M. Almeida, J. J. G. Moura and M. Aureliano, J. Inorg. Biochem., 2011, 105, 777–783 CrossRef CAS PubMed.
- S. Ramos, R. O. Duarte, J. J. G. Moura and M. Aureliano, Dalton Trans., 2009, 7985–7994 RSC.
- M. Aureliano, Oxid. Med. Cell. Longevity., 2016, 2016, 6103457 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complementary spectroscopic, crystallographic and thermogravimetric data for compounds A and B. Crystallographic data for product B. CCDC 1468753. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra15826a |
| This journal is © The Royal Society of Chemistry 2016 |