
Multifunctional mesoporous silica nanocarriers for stimuli-responsive target delivery of anticancer drugs
Yujuan Chena,
Hui Zhanga,
Xiaoqing Caia,
Jianbo Jia,
Shuwang He*b and
Guangxi Zhai
*a
aDepartment of Pharmaceutics, College of Pharmacy, Shandong University, 44 Wenhua Xilu, Jinan 250012, China. E-mail: professorgxzhai@126.com; Tel: +86-531-88382015
bDepartment of Pharmaceutical Development, Shandong Dyne Marine Biopharmaceutical Limited Corporation, 19 Lingming Beilu, Rongcheng 264300, China. E-mail: heshuwang@dynemed.com; Tel: +86-10-85869817
First published on 20th September 2016
Abstract
With the rapid development of nanotechnology, mesoporous silica nanoparticles (MSNs), as a new type of inorganic nanomaterial, have been widely used in biomedical applications especially in drug delivery systems owing to their unique physical–chemical properties such as tunable particle/pore size, high surface area and pore volume, easy surface modification, remarkable stability and biocompatibility, and high drug loading efficiency. By modifying the outer surface of MSNs with various functional groups such as polymers, co-polymers, nanoparticles, quantum dots, supermolecules, ligands, or/and using a combination with other nanomaterials, stimuli-responsive and active targeting nanosystems can be designed for targeted delivery of anticancer drugs. In this review, the recent advances in stimuli-responsive strategies involving pH-sensitive, redox-sensitive, thermo-sensitive, enzyme-sensitive, light-sensitive, magnetic-sensitive, ultrasound-sensitive, and active targeting approaches involving vascular targeting, tumor cell targeting, nuclear targeting and multistage targeting are discussed in detail. The remaining challenges and the possible future directions are also suggested.
 Guangxi Zhai |
Introduction
Cancer is a serious threat to human health and economic constraints in human life, its morbidity is increasing at an alarming rate and it is a major cause of death worldwide.1 Conventional chemotherapy is the most common cancer treatment method based on highly potent anticancer drugs such as vinblastine, camptothecin, doxorubicin (DOX), taxol and curcuma (Cur) which can inhibit cell growth and cause subsequent apoptosis. However, the harmful side-effects due to the nonspecific uptake by normal tissues/organs and high-dose administration of therapeutic agents, non-tumor selective biodistribution, hypersensitivity, acquisition of multidrug resistance (MDR) and toxic metabolites which can lead to cardiotoxicity and nephrotoxicity are all challenges. In addition, the poor solubility of several anticancer agents also limits the ability for intravenous delivery which is the preferred administration route for many anticancer drugs.1 To conquer these limitations, an effective way is to encapsulate the therapeutic agents in nanocarriers with high loading efficiency, which can spatiotemporally deliver the cargos and control the release of them in a smart manner. These nanocarriers can be divided into two groups: organic (such as liposomes, dendrimers, polymeric micelle) and inorganic (such as mesoporous silica, carbon nanomaterials, graphene oxide, quantum dot, gold/silver nanoparticles, magnetic nanoparticles). Organic nanoparticles, such as liposomes, polymeric micelles are able to encapsulate/load both hydrophilic therapeutic agents and hydrophobic agents due to hydrophobic/hydrophilic interactions, chemical reactions and electrostatic interactions and so on, to protect therapeutic agents from destroying. A few conventional organic nanocarriers have been approved by FDA and applied in clinical, by contrast, most inorganic nanocarriers still remain in the preclinical development stage.2 Inorganic nanomaterials, compared to conventional organic nanocarriers, exhibit outstanding advantages such as good biocompatibility and high thermal, mechanical and chemical stabilities under physiological conditions.3 Moreover, relatively simple chemical modifications can provide versatility of these systems and extend their application, which make them suitable for drug delivery.2Among the different inorganic materials, MSNs are considered as one of the most promising nanomaterials for drug delivery due to its unique inherent characteristic: (I) the large surface area (>700 m2 g−1) due to MSNs unique porous, honeycomb-like frame4 and high pore volume (>1 cm3 g−1),5 which makes it possible for drug to be adsorbed and encapsulated within the pore channels at high level. (II) The smart mesoporous structure and an adjustable pore diameter (20–50 nm) which enables better control of drug loading and release kinetics.6 (III) At physiological pH, there is abundant negatively charged silanol groups (SiO−) on the surface of MSNs,4 through interaction with SiO−, the functionalized MSNs can be achieved by decorating with various functional groups, to control and target delivery of anticancer drug with enhanced therapeutic efficacy and reduced toxicity.7,8 (IV) The good biocompatibility, the research on toxicology of MSNs in vitro indicated that these materials are well-tolerated at dosages <100 μg mL−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9,10 and toxicity could be only observed at dosage above hundreds of mg kg−1 in vivo.11,12 (V) When combine with magnetic and/or luminescent compounds, MSNs allow drug delivery and bioimaging simultaneously.
9,10 and toxicity could be only observed at dosage above hundreds of mg kg−1 in vivo.11,12 (V) When combine with magnetic and/or luminescent compounds, MSNs allow drug delivery and bioimaging simultaneously.
Due to these unique intrinsic characteristic, MSNs have been widely used to design multifunctional nanocarrier systems. Such as immediate drug delivery systems (DDSs), sustained DDSs, stimuli-responsive controlled drug delivery systems (CDDSs), and active targeted DDSs.13 In this review, we mainly focus on the most rapid and significant progresses of MSNs as stimuli-responsive CDDSs and active targeted DDSs in the past three years.
Development of MSNs
Until the early 1990s, the MCM-41-type mesoporous silica was first reported and perked up the applications of MSNs in catalysis due to its large surface area and the adjustable pore size.14 In 2001, isoprofen was first successfully loaded into MCM-41 with sustained drug-releasing performance as well as the drug-loading rate up to 30%.15Since then, the studies on MSNs increased rapidly, especially in biomedical applications such as drug delivery. The development of MSNs for drug delivery has experienced three generations.16 The properties of each generation are listed in Table 1. The rapid development of synthetic chemistry allows constructing MSNs with various structures and morphologies such as hollow nanostructures, janus MSNs, yolk shell nanorattles, organic–inorganic hybrid mesoporous silica. In addition, MSNs can conjugate with many types of functional groups due to rich reactive silanol groups on the surface of MSNs, such as targeting ligands, functional polymers/materials, and fluorescent agents. These functional groups can provide MSNs with diverse interesting functionalities including target delivery, controlled release (e.g., stimuli responsive release, on-demand release), co-delivery, bioimaging, synergistic therapy, etc.
| Generation | Features properties |
|---|---|
| Generation I: concept demonstration | 1. Evaluations of the sustained releasing performances of various loaded cargos in vitro |
| 2. Large particle sizes, irregular morphologies and severe aggregations | |
| 3. Limited cell-level evaluations and in vivo applications | |
| Generation II: standard evaluation | 1. Standard MSNs-based nanosystems and systematic in vitro and in vivo evaluations |
| 2. Uniform spherical morphology, tunable pore/particle sizes and compositions | |
| 3. MSNs with hollow nanostructures (HMSNs) enhance the drug-loading capacity of the carriers | |
| Generation III: multifunctionalization | 1. Diverse functionalities, such as targeted delivery, co-delivery, on-demand releasing, synergistic therapy, overcoming the multidrug resistance (MDR) of cancer cells, theranostic, bioimaging |
| 2. The surface of MSNs can be modified with a variety of biocompatible materials which possess unique physical–chemical properties or/and biological recognition ligands to control drug release under stimulation and promote the targeted transport, even co-delivery different therapeutic agent | |
| 3. The combine of mesoporous silica and magnetic nanoparticles, quantum dots, gold/silver nanoparticle, upconverting nanoparticles, photothermal conversion material, fluorescence material broaden the versatility of silica based nanocarriers | |
| 4. Janus MSNs, yolk shell nanorattles and organic–inorganic hybrid mesoporous silica further enhance the drug-loading capacity of the carriers and broaden the types of loading cargo |
Stimuli-responsive MSNs
Stimuli-responsive MSNs only release the cargos in the targeted cancer sites upon triggering by intratumoral stimuli (pH, redox, enzyme, temperature) or exogenous stimuli (magnetic field, ultrasound, light) with nearly no premature drug release (Fig. 1). The stimuli-responsive nanosystems based on MSNs are shown in Tables 2 (pH) and 3. The schematic diagram of stimuli-responsive mechanism is shown in Fig. 2. Stimuli-responsive MSNs have been regarded as promising approach to improve the therapeutic effect of anticancer agent and simultaneously reduce the undesirable side effects to normal cells.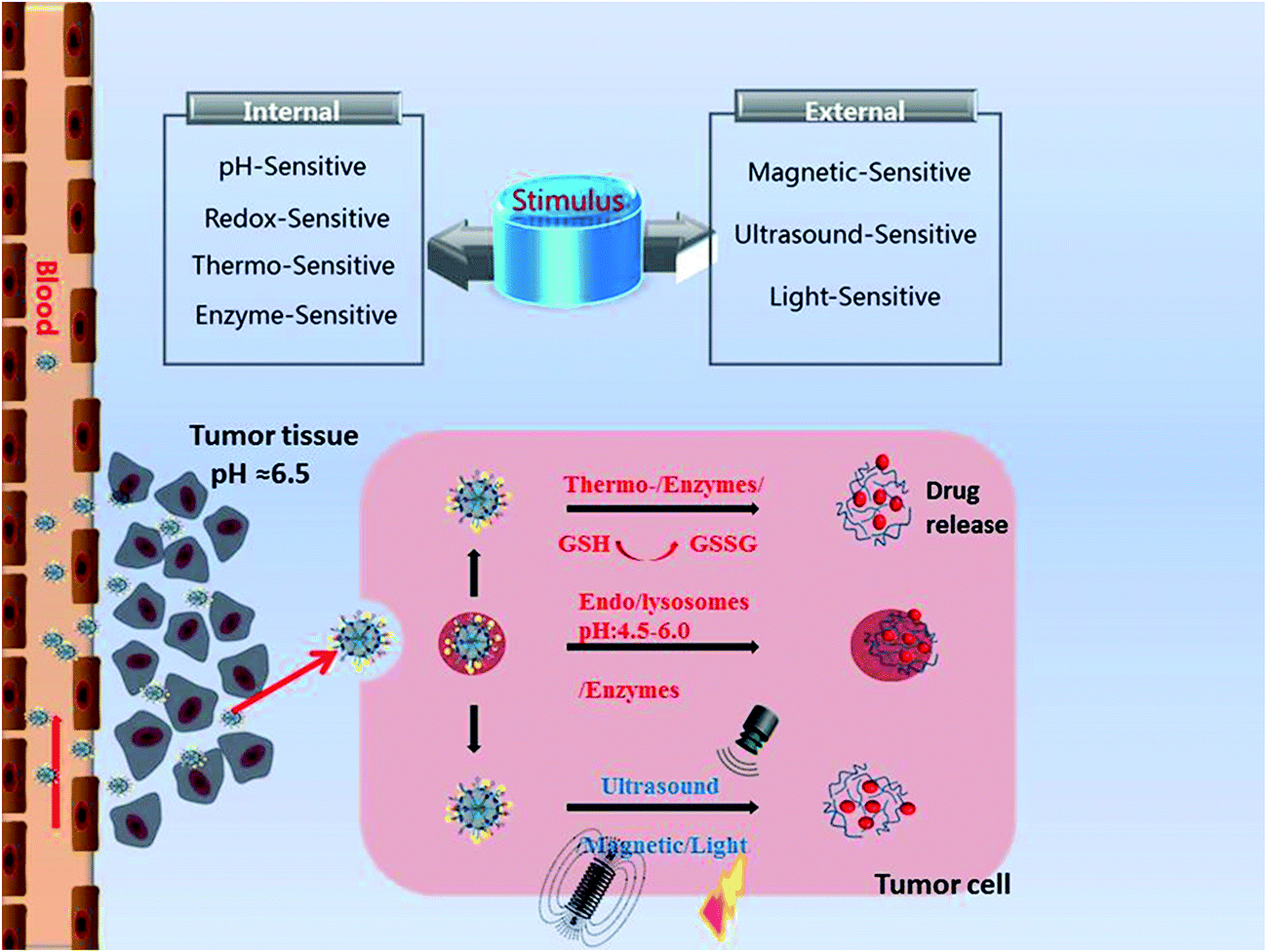 | ||
| Fig. 1 Stimuli-responsive drug delivery based on MSNs in cancer therapy. | ||
| Type | Material | pH-Responsive mechanism | Ref. |
|---|---|---|---|
| Acid cleavable linker/bond | Gold NP–acetal linker–MS | Acetal linker | 18 |
| Lanthanide doped NP–acetal bond–MS | Acetal bond | 19 | |
| Fe3O4 NP–boronate ester bond–MSNs | Boronate ester bond | 20 | |
| Poly(N-succinimidyl acrylate)–acetal linker–MS | Acetal linker | 21 | |
| MSN–hydrazone–Dox | Hydrazine | 22 | |
| DOX@PAA–ACL–MSN | ACL | 23 | |
| Au NPs–DNA–MSNs | DNA | 24 | |
| Polymer gatekeepers | DOX@MSNs–gelatin | Gelatin | 25 |
| DOX@MSNs–chitosan | Chitosan | 26 | |
| DOX@MSNs–PVP–PEG | PVP | 27 | |
| DOX@MSNs–PLGA | PLGA | 28 | |
| PAH/PSS MSNs | PAH/PSS polyelectrolyte multilayers | 29 | |
| Alginate/chitosan–NH2–MSNs | Alginate/chitosan multilayers | 30 | |
| Chitosan/dialdehyde starch–MSNs | Chitosan/dialdehyde starch polyelectrolyte multilayers | 31 | |
| PAH–cit/APTES–MSNs | Charge-reversal polymer PAH-cit | 32 | |
| Cur@PAMAM–MSNs | PAMAM- | 33 | |
| PDEAEMA–MSNs | PDEAEMA | 34 | |
| DOX@PAA–MSNs | PAA | 35 | |
| DOX@PPEMA/PEG–MSNs | PPEMA | 36 | |
| i-motif DNA–MSNs | i-motif DNA | 37 | |
| Supramolecular-nanovalves | β-Cyclodextrin caps–aromatic amines stalks–MSNS | Aromatic amines stalks | 38 and 39 |
| α-Cyclodextrin–p-anisidino stalks–MCM-41 | p-Anisidino stalks | 40 | |
| cucurbit(6)uril–trisammonium stalks–MSNS | Trisammonium stalks | 41 | |
| Acid decomposable gatekeepers | ZnO@MSN | ZnO | 42 |
| DOX–Si–MP-cap | pH-Tunable calcium phosphate | 43 | |
| LDHs–MSNs | LDHs | 44 |
| Type | Material | Mechanism |
|---|---|---|
| Redox responsive | CdS50 Au51 Fe3O452 |
Inorganic NPs–disulfide bonds–MSNs |
| Poly(propylenimine) dendrimers8 PEG13,53,54 heparin55 peptides57 polyethylenimine (PEI)56 cyclodextrin58,59 PDS62 cytochrome c60 PEG–PCL61 | Organic molecules–disulfide bonds–MSNs | |
| mPEG@6-MP@CMS53 | Thiolated drug–disulfide bonds–MSNs | |
| HA@6-MP@CMS63 | ||
| MSN@MnO264 |
Glutathione degradable gatekeepers | |
| Enzymes responsive | MSN–GFLGR7–RGDS/α-CD65 | Protease-sensitive crosslinker |
| DNA68 HA69 gelation70,71 cellulose72 galacto-oligosaccharides73 | Enzyme degradable polymer | |
| Temperature responsive | Poly(ethyleneoxideb-N-vinylcaprolactam),79 zwitterionic sulfobetaine copolymers,80 paraffins,81 supramolecules rotaxane82 copolymer–lipid bilayers83 | Thermo sensitive polymers |
| DNA84,85 peptide sequences86,87 | Thermo sensitive bio-molecules | |
| Magnetic responsive | Poly(N-isopropylacrylamide/N-hydroxymethylacrylamide),98 poly(ethyleneimine)-b-poly(N-isopropylacrylamide),89 lipid bilayer,104 pseudorotaxanes105 | Heat produced by AFM + thermo-sensitive gatekeeper |
| mNPs + DNA + MSNs103 | Heat produced by AFM + thermally unstable chemical linkers | |
| Azo-PEG@Fe3O4@SiO296 |
||
| SPION@MSN-DA95 | Heat produced by AFM + thermally reversible cycloreversion reaction | |
| Ultrasound responsive | MSNC@Au–PFH–PEG111 | Ultrasound sensitive material + cavitation |
| p(MEO2MA)-co-THPMA116 | Ultrasound-cleavable moieties | |
| Fc–CONH–MS117 | Ultrasound sensitive of ferrocene derivative | |
| Light responsive UV-Vis | β-CD and/or Py-β-CD–azobenzene stalks–MSNs118 | The isomerized of azobenzene group from cis to trans |
| α-CD–azobenzene stalks–MSNs119 | ||
| Thymine derivatives–MSNs120 o-nitrobenzyl ester moiety–MSNs121 poly(N-isopropylacrylamide-co-2-nitrobenzyl acrylate)–MSNs122 | Photoresponsive polymers gatekeeper | |
| 7-Amino-coumarin derivative (CD)–MSNs,123 S-coordinated Ru(bpy)2(PPh3)-moieties–MSNs,124 TUNA–MSNs125 | Photoresponsive linkers | |
| NIR | DNA–Au@MSNs91 | NIR-absorbing materials + thermal-responsive materials |
| DNA–Cu1.8S@mSiO292 |
||
| 1-Tetradecanol-GNR@MSNs127 | ||
| sulfonatocalix[4]arene–AuNR@MSN128 | ||
| Au–nanocage@mSiO2@PNIPAM129 | ||
| CuS@mSiO2–PEG130 | ||
| SWNT@MS–PEG131 | ||
| UCNP@mSiO2–Ru132 |
 | ||
| Fig. 2 The schematic diagram of stimuli-responsive mechanism. | ||
Intratumoral stimuli-sensitive MSNs
Various kinds of tumor tissues share similar microenvironment distinct from normal ones, such as leaky vasculatures, acidic and hypoxic environments, high redox potential, increased level of cancer-associated enzymes and higher local temperature. Based on these, stimuli-sensitive delivery system can be designed to response to the tumor microenvironment.pH-Responsive MSNs
Among the various stimuli-sensitive DDSs, pH-responsive CDDSs have been widely researched since the human body exhibits variations in pH. The pH value approximately 7.4 in extracellular of normal tissues and blood, while between 6.0 and 7.0 in tumor microenvironment, which is mainly caused by high level of CO2 and high glycolysis rate.17 The pH value will decrease further inside cancer cell such as endosomes (pH = 5.5–6.0) and lysosomes (pH = 4.5–5.0). Therefore, we can combine the unusual pH gradients with the advantages of MSNs to design pH-responsive nanosystems which can control the release of drug molecules under certain pH conditions. The pH-responsive nanosystems based on MSNs are shown in Table 2.pH-Sensitive linkers such as hydrazine bond, acetal bond, boronate ester bond and ester bond that can be cleaved under acidic pH value, thus providing the possibilities for designing pH-responsive MSNs.
A multifunctional envelope-type DDS based on upconverting nanoparticle-capped mesoporous silica (MS) via pH-sensitive linkers was reported by Chen et al.19 In this study, the gatekeeper (S-NPs), upconverting nanoparticle doped with ultra-small lanthanide (NaGdF4:Yb/Tm@NaGdF4), was grafted onto the orifices of MS with the acid-labile acetals. DOX as model drug was trapped in the pores. At acidic pH, the S-NPs caps were removed due to hydrolysis of acetal group. DOX loaded nanocomposite could accumulate in tumor through enhanced permeation and retention (EPR) effect after intravenous injection into the murine model. DOX was rapidly released in the acidic environment of lysosomes and endosomes, which enhanced the therapeutic efficacy by remarkable inhibition of tumor growth, and the treated mice survived over 30 days without any obviously visible tumor growth. In addition, the encapsulation of gadolinium makes the nanocomposite a promising T1-MRI contrast agent with contrast enhanced MRI performance. Importantly, the nanocomposites are biocompatible and could be metabolized and degraded into nearly nontoxic products within a few days.
The supramolecular nanovalve for controlling drug release, is composed of an immobilized stalk molecule connected to the silica matrix via covalent interaction and a movable cyclic molecule as the gatekeeper encircling the stalk non-covalently. More importantly, the binding process of cyclic gatekeeper/stalk is reversible and the supramolecular gatekeeper can be switched under certain stimuli (pH, light, redox potential, temperature), causing large amplitude movement of the gatekeeper, which in turn leads to the blocked pore open.45
Based on the noncovalent interactions between cyclic molecule caps and immobilized amine stalks, Ling et al.46 successfully constructed a pH-responsive nanosystem in view of CD capped MSN that could control the release of cargo. With DOX as model drug and β-CD as gatekeeper, this constructed nanosystem showed a good pH-sensitive release property. The cumulative release within 4 h was 0.7% at pH 7.4 and 3.6% at pH 6.5, because the tight bind between hydrophobic p-anisidine stalks and CD caps blocked the nanopores limited the release of DOX. When at pH 5.0, the cumulative release increased significantly to 84.2% because the binding between p-anisidine stalks and β-CD decreased and forced the removal of the CD caps, then the cargo released by diffusion. In addition, they investigated the p-anisidine stalks density and type of CD which were critical factors impacting the pH inducing drug release. Result showed that the too high or too low density of the grafted p-anisidine stalk could lead to poor drug release, and the optimal stalk density was ∼8.7 stalks per nm2. Different types of CD capes (α-CD and β-CD) were investigated and results showed that the complex of p-anisidine stalk with β-CD had an excellent pH-sensitive release capability for its largest changed formation constant (ΔKf). Furthermore, the mechanism of pH-sensitivity between CD and p-anisidine stalk was investigated, under neutral or acidic media, the binding process of p-anisidine/α-CD was significantly enthalpy-driven with the main driving force of van der Waals forces and hydrogen bonding. While the complexion process between p-anisidine and β-CD was entropy-driven with strong hydrophobic interaction under neutral environment, but weak hydrogen bonding existed under acidic pH. In short, both the stalk density and type of CD could significantly affect the pH-sensitive release capability.
pH-Sensitive polymer shell contains functional groups with acid/base properties. Thus, the polymer shell, such as poly(2-vinylpyridine) PVP,27 self-fluorescent agents PAMAM (polyamidoamine) dendrimers,33 poly(2-(diethylamino)ethyl-methacrylate) PDEAEMA,34 poly(2-(pentamethyleneimino)ethyl-methacrylate) (PPEMA)36 and i-motif DNA,37 can undergo strong conformational structure transformation with the variation of external pH because of strong hydrophilic change, chargeability change, solubility change.35 The polymer shell also can be easily modified with various targeting ligands to acquire active target delivery, simultaneously.
Containing repeating charged groups, polyelectrolytes that can be either covalently bonded or electrostatically adsorbed to the surface of MSNs and undergo form transition along with the changes of pH values, which have been also utilized to design pH-responsive release system.
Poly(allylamine)–citraconic anhydride (PAH–cit) is a pH responsive charge-reversal polymer, whereby the charge can be readily converted from negative to positive through side-chain hydrolysis upon exposure to acid solutions. Using this property, Zhang et al.,32 firstly developed a PAH–cit coated MSNs (PAH–cit/APTES MSNs) and the MSNs was applied to devise an in situ monitoring system of intracellular pH-responsive DDSs for the treatment of malignant cells. APTES ((3-aminopropyl)triethoxysilane) was modified onto MSNs to obtain amino-functionalized MSNs, then, through electrostatic interactions, PAH–cit was bonded onto the inner and outer surfaces of amino-functionalized MSNs (APTES MSNs). In acidic environments, the loaded DOX was steadily released from the MSNs as a result of strong electrostatic repulsion forces among the positively charged MSNs (APTES–MSNs), PAH and DOX. The result showed that the constructed nanocomposite (PAH–cit/APTES–MSNs) could effectively deliver and release DOX to the nucleus of HeLa (human cervical carcinoma) cells.
Polyelectrolyte multilayers (PEM) are sensitive to various specific physical and chemical conditions of the surrounding medium based on its special properties and structure. In particular, the pH can largely influence the state of the inter-polyelectrolyte complex. Based on this, pH-sensitive drug release systems via electrostatic interaction have been developed in contribution to layer-by-layer assembly of PEM onto MSNs surface, such as poly(allylamine hydrochloride) (PAH)/poly(styrene sulfonate) (PSS),29 alginate/chitosan30 and chitosan (CHI)/dialdehyde starch (DAS).31 The biocompatible polyelectrolyte multilayers functionalized MSNs have stupendous potential to developed efficient and safe drug delivery system.
Some acidic-decomposable materials such as ZnO quantum dots (QDs),42 calcium phosphate (CaP),43 layered double hydroxides (LDHs) which include the positively charged brucite-type layers and exchangeable interlayer anions,44 have recently been reported as gatekeepers to design pH-sensitive DDSs.
ZnO QDs are stable at pH 7.4, but when the pH is less than 5.5, ZnO QDs immediately dissociate into Zn2+ ions which induce the generation of ROS and the ROS can involve lipid peroxidation and damage of DNA. Based on this, Zhang and coworkers42 developed a dual pH-sensitive DDS ZnO@MSN. In this system, cell-penetrating peptide decalysine sequence (K10) covalently covered MSNs via acid-labile β-carboxylic amides and the peptide can also enhance endosomes/lysosome escape capability. After this, ZnO QDs capped MSNs via electrostatic interaction to achieve a synergistic antitumor effect. At pH 7.4, almost no DOX released from the ZnO@MSN DDS after 48 h, however, DOX was released rapidly at pH 5.0 from the system, about 34%, 86% of DOX was released within 12 h, 48 h, respectively, because the dissociation of ZnO QDs and the hydrolysis of acid-labile amides leaded to the uncap of MSNs, which confirmed that ZnO@MSN had great sensitivity to the pH signal of endosomal environment and obtained intracellular drug release. In addition, compared to free DOX, the DOX-loaded ZnO@MSN showed higher cytotoxicity with IC50 of 50 μg mL−1.
An innovative strategy to obtain pH-responsive DDSs is using janus nanoparticles. For example, Shao et al.47 reported a novel janus nanoparticles (Ag–MSNs) with a silver nanosphere head and a mesoporous silica ordered mesostructure body. Within 24 h, more than 40% of DOX released in pH 5.5, while less 5% was released at pH 7.4 due to the protonation and solubility of DOX in acidic environments.
Redox-responsive MSNs
Glutathione (GSH) is an important antioxidant in the human body, protecting cellular components from damage caused by reactive oxygen species (ROS). In particular, the GSH concentration in the intracellular environment is about 103 folds higher than that in the extracellular matrix. Furthermore, the intracellular GSH concentrations in most tumor cells are at least 4-times higher than those in normal cells.48 Therefore, GSH can be a potential trigger for drug release within cancer cells. Given the fact that disulfide bonds are sensitive to GSH reductant, smart designs incorporating disulfide S–S linkages into nanoparticles can lead to redox-triggered release systems.49 Inorganic nanoparticles such as CdS,50 Au,51 Fe3O452 and organic molecules such as poly(propylenimine) dendrimers,8 PEG,13,53,54 heparin,55 polyethylenimine (PEI),56 peptides,57 cyclodextrin,58,59 cytochrome c,60 poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG–PCL),61 can be chemically attached on the MSN surface as biocompatible gatekeepers to control drug release by using GSH-sensitive linkers. When the nanocarrier reaches the intracellular sites, the existence of increased amounts of reductive species can produce the rupture of the disulfide bonds and open the pores.
Palanikumar et al.62 reported an active target and redox-responsive MSNs through one-pot synthesis, which could load DOX or cisplatin or DOX and cisplatin with high efficiency at 44, 33 wt% and 25% for DOX and 14% for cisplatin, respectively. Self-crosslinkable random copolymer containing biocompatible pyridine disulfide hydrochloride (PDS) and PEG side chains noncovalently end-capped MSN without multiple chemical modifications. PDS has multiple functions including wrapping the negatively charged MSNs via temporarily positive charge, stabilizing polymer–MSNs shell as a crosslinker and connecting the targeting ligand (celcyclic (Arg–Gly–Asp–D-Phe–Cys) (cRGDfC), an integrin-targeting ligand) through its functional group. Intracellular reducing agents such as glutathione (GSH) can lead to the cleavage of the wrapped polymer and induce the release of cargo in a concentration-dependent manner.
A new strategy is connecting the thiol-containing/thiolated therapeutic agent to the inside and outside of MSNs via cleavable disulfide bonds to design redox-responsive DDS. Zhao et al.53,63 designed a redox-responsive DDS based on 6-mercaptopurine (6-MP)-conjugated colloidal mesoporous silica (CMS) via disulfide bonds. Hydrophilic polymers mPEG modified the outside of MSNs to improve the biocompatibility and dispersibility of CMS by reducing hemolysis and protein adsorption,53 and hyaluronic acid (HA) as a targeting ligand was grafted on CMS surface through disulfide bonds which also increased the biocompatibility and stability of CMS under physiological conditions.63 CMS was prepared through a co-condensation method, involving the simultaneous condensation of TEOS and MPTMS, to obtain a homogeneous distribution of mercapto groups on the internal and external surface of the CMS carrier. In vitro release studies demonstrated that the CMS possessed the great redox-responsive drug release capability. For the CMS–SSMP@mPEG,53 the cumulative release of 6-MP was less than 3% without GSH, while more than 70% of loaded 6-MP was released within 2 h in the existence of 3 mM GSH. For the CMS–SS–MP/oHA,63 the cumulative release of 6-MP within 2 h was less than 3% without GSH, while more than 80% for that in the existence of 3 mM GSH.
Another new strategy to design redox-responsive DDS is capping outside of MSNs with glutathione degradable gatekeepers. Yang et al.64 have demonstrated a new type of redox-responsive DDS by employing glutathione degradable MnO2 as capping to block the pore of MSNs. In the presence of GSH molecules, the capped MnO2 nanostructure dissociated into Mn2+ via redox reaction, which opened the blocked pores and resulted in the release of the entrapped drugs.
Enzymes-responsive MSNs
Recently, due to mild reaction conditions, high specificity, and less damage to body tissues, enzyme reactions have received widespread attention. More importantly, many specific enzymes show a high level of expression in tumor tissues and cancer cells but no or relatively low level of expression in healthy tissues or normal cells.65 Endogenous enzymes in tumor tissues and cancer cell have used as stimuli to trigger drug release.To obtain enzymes-responsive release, one strategy is coating MSNs with gatekeepers containing protease-sensitive sequences or enzyme-sensitive linkers. Cathepsin B, which overexpressed in late endosomes and lysosomes of cancer cells, could selectively hydrolyze specific peptide sequences.65–67 Cheng et al.65 described an enzyme-induced and tumor-targeted mesoporous silica nanocarrier which was capable of releasing therapeutic drugs in response to increased levels of cathepsin B. In the nanocarrier, the alkoxysilane chain and α-cyclodextrin (α-CD) formed rotaxane structure and anchored onto the pore of MSNs as gatekeeper, subsequently modified by azido-GFLGR7–RGDS which included a cell-penetrating peptide with seven arginine (R7) sequence, a cathepsin B-cleavable peptide of Gly–Phe–Leu–Gly (GFLG) as crosslinker and a tumor-targeting peptide of Arg–Gly–Asp–Ser (RGDS) to stabilize the gatekeeper. The in vitro release results indicated that in the presence of cathepsin B (20 U), 60% and 80% of loaded DOX could be released from the DOX@MSN–GFLGR7–RGDS/α-CD nanoparticles in pH 7.4 and pH 5.0 PBS buffer within 24 h, respectively. By contrast, in the absence of cathepsin B, due to the protection of the gatekeeper on the surface of the MSNs, less than 10% of loaded DOX was released during the same period. Furthermore, in vitro cellular experiments indicated that this nanocarrier had high growth inhibition rate toward αvβ3-positive HeLa cancerous cells.
Biopolymers such as single-stranded DNA,68 HA,69 gelatin70,71 and other polymers such as cellulose,72 galacto-oligosaccharides73 can be grafted on MSN surface, acting as enzyme-responsive nanovalves and hampering the drug diffusion until certain hydrolytic enzymes which are capable of decomposing these polymers are present. Matrix metalloproteinases (MMPs) are up-regulated in most human cancer and they are involved in tumor invasiveness, metastasis, and angiogenesis.74 Based on this, Zou and coworkers71 conjugated the FA–MSN with MMP2-degradable gelatin to develop a smart mesoporous silica nanocarrier (PGFMSN) which showed MMP2-triggered release and FA modified tumor targeting. After functionalizing MSN with target ligand FA, gelatin layer was decorated onto FA–MSN as gatekeeper to control the release of drug and protect the target ligand. In order to prolong blood circulation lifetime, PEG was further introduced to obtain PGFMSN. In these nanosystems, DOX was effectively loaded in PGFMSN with the loading amount as high as 74.3 mmol g−1 SiO2. The in vitro release results indicated that PGFMSN exhibited MMP-2 concentration-dependent release profiles via MMP2-triggered hydrolyzation of the gelatin layer with approximately 7.2%, 19.1%, 43.7% and 75.3% of loaded DOX released at MMP2 concentration of 0, 1.25, 2.5, 5.0 mg mL−1 within 600 min. Owing to the introduction of gelatin, FA and PEG, PGFMSN can specifically target cancer cells by up-regulated extracellular MMP2 and FA receptor, exhibiting enhanced cancer cell internalization. Most importantly, the in vivo therapeutic study indicated that compared with free DOX, non-targeted nanoparticles and non-PEG nanoparticles, DOX@PGFMSN exhibited the better therapeutic efficacy.
Temperature-responsive MSNs
It has been revealed that the local temperature in many tumors, inflamed or infected tissue is higher than normal body temperature by 4–5 °C. Motivated by this, researchers graft temperature-sensitive gatekeepers on MSN surface to retain their cargo at normal body temperature (∼37 °C), and rapidly release at higher temperature (∼40–42 °C) of heated tumor site, which can prolong blood-circulation lifetime of the cargo and avoid washout from the tumor. A common temperature-responsive gatekeeper is thermo-sensitive polymer based on poly-N-isopropylacrylamide (PNIPAM) and its derivatives.11 PNIPAM can undergo a transition from hydrophilic to hydrophobic at “lower critical solution temperature” (LCST) of about 32 °C.75 Below the LCST, PNIPAM chains are hydrophilic and extended, preventing the release of the loaded drugs, when temperature is above the LCST, PNIPAM chains are dehydrated and collapsed, leading to the pore open and the release of the cargo.11The LCST of pure PNIPAM is around 32 °C that is not suitable for biomedical applications. Increasing the LCST to physiological temperature can be achieved by introducing hydrophilic monomers into the polymer composition, such as acrylamide,76 N-isopropylmethacrylamide (the LCST increased to ∼37 °C).77,78 Other thermo-sensitive polymers such as poly(ethyleneoxide-b-N-vinylcaprolactam),79 biocompatible zwitterionic sulfobetaine copolymers,80 paraffins,81 supramolecules rotaxane82 or copolymer–lipid bilayers83 have been also reported. Besides these abiotic thermo-sensitive polymers, some bio-molecules have also been utilized, such as double-stranded DNA (temperature above its melting point could induce the melting of ds-DNA),84 reversible single-stranded DNA85 (as the DNA gatekeeper which could be adsorbed outside of the silica shell via electrostatic interaction and destroyed at high temperature, leading to the release of loaded cargo molecules from the nanocarrier), peptide sequences86,87 (present the disassemble processes or a-helix-to-disordered transformation at specific temperature).
Another strategy was based on the combination of magnetic88–90 or photothermal91,92 materials with thermo-sensitive polymers to reach temperature-triggered drug release.
Exogenous stimuli-sensitive MSNs
In this section, stimuli-responsive DDSs that take advantage of externally applied stimuli, including magnetic fields, ultrasounds and light, were discussed.Magnetic-responsive MSNs
Magnetic particles could be heated up under alternating magnetic field (MF), or be guided to the tumor tissue under a locally applied external MF. Therefore, researchers take advantage of magnetic field and designed magnetic-responsive DDSs through combining MSNs with magnetic particles. Biocompatible superparamagnetic iron oxide nanoparticles (SPION) are the most widely employed magnetic particles. Under an extracorporeal permanent MF focusing on the biological target, injected magnetic responsive nanocarriers can be guided to the target site. The temperature will rise when an alternating magnetic field (AMF) is applied or permanent and alternating MF is alternately used. These magnetic particles transform magnetic energy into heat following two mechanisms, Brownian fluctuations caused by the rapid rotation of the nuclei and Nell fluctuations incited by the spontaneous reorientation of the magnetic moment of particle, which are consequence of the rotation of the magnetic moments.93,94 The heat produced by an AMF can be used to achieve on demand pulsatile drug release. By adjusting the intensity and location of the MF, the accumulation of nanocarriers and the release of loaded drug can be controlled, and harmful side effects on normal tissues can be avoided. Therefore, the combination of magnetic nanoparticles (mNPs) and MSNs with high target specificity, loading capacity and magnetic properties constitute promising DDSs to target and control the drug release.The different possible structures of the combination between MSNs and magnetic nanoparticles (mNPs) can be summarized into four categories (Fig. 3): (I) core–shell structure covering mNPs core with mesoporous silica shell.95–98 The outer mesoporous silica shell provides pore volume and enough surface area for drug store and release, stimuti-responsive gatekeepers placed on the pore outlets controlled the release under an external magnetic field. (II) Rattle-type hollow structure composing of mNPs core and mesoporous silica shell.99,100 Research showed that the saturation magnetization value of rattle-type hollow structure (35.7 emu g−1) is significantly higher than that of the corresponding core–shell structure with an intact middle silica layer (28.8 emu g−1) due to the removal of the in-between silica layer. Furthermore, pore volume and surface area of the rattle-type structure are calculated to be 0.58 cm3 g−1 and 435 m2 g−1, respectively, which are obviously higher than the sample with an intact middle silica layer (274 m2 g−1 and 0.38 cm3 g−1).100 Therefore, the rattle-type structure is more suitable for targeting drug release. (III) Embedded structure encapsulating mNPs in mesoporous silica nanospheres;89,101 (IV) mesoporous silica nanoparticles being capped with mNPs.102,103 mNPs can be immobilized onto the surface of MSNs surface via chemical linkers or polymer.
 | ||
| Fig. 3 The four possibilities structures of the combination between mesoporous silica nanoparticles and magnetic nanoparticles (mNPs). | ||
As mentioned above, there will be a temperature increase when magnetic-responsive MSNs were under an AMF. Therefore, temperature-responsive gatekeepers (such as DNA, thermosensitive polymer) can be used to induce pore opening accompanied by temperature increase. Thermosensitive polymers like poly(N-isopropylacrylamide/N-hydroxymethylacrylamide),98 poly(ethyleneimine)-b-poly(N-isopropylacrylamide),89 lipid bilayer104 and temperature sensitive pseudorotaxanes105 act as thermosensitive nanovalves have been reported. At high temperature achieved by applying an external magnetic field, these temperature-responsive gatekeepers present structural changes, permeability increasing, molecular nanocap disassemble. mNPs or polymers can be placed onto the surface of MSN via thermally unstable chemical linkers like single-stranded DNA with a melting temperature of 47 °C103 or unstable covalent bond azo bond.96 Zink and co-worker96 firstly reported a core–shell Fe3O4@SiO2 nanocarrier coated with thermodegradable azo-functionalised PEG. The thermally decompose capability of the azo compounds makes them widely used as thermal initiators via initiate radical polymerizations. Azo-PEG can be broken at high temperature caused by a high frequency oscillating MF, leading to the release of loaded drug, while restricting cargo in mesopores at body temperature. This strategy exhibits no fibroblast cytotoxicity, indicating its safety and no damage to the surrounding tissues, because the external MF produces locally heat within a nanoscopic volume.
[2 + 4] cycloaddition reaction (Diels Alder reaction) and cycloreversion reaction (retro-Diels Alder reaction) of furan derivatives with maleimide derivatives can be carried out at mild temperature, especially, cycloreversion reaction can proceed at conventional heating or heating triggered by superparamagnetic. Based on this, Ruhle and associate95 constructed a novel magnetic-responsive DDS (SPION@MSN-DA) with thermoresponsive snaptop as gatekeeper by fixing the adamantane group onto maleimide functionalized SPION@MSN (SPION core–mesoporous silica shell) via a thermally reversible [2 + 4] cycloaddition of a furan-modified linker. When under external AMF, cycloreversion reaction could be carried out, leading to cap detach from MSN, pore open and loaded cargo release.
Besides superparamagnetic iron oxide NPs, superparamagnetic manganese ferrite nanoparticles106 and zinc-doped iron oxide nanocrystals105,107 combined with MSNs have been reported to produce novel magnetic responsive drug delivery systems.
Ultrasound-responsive MSNs
Among possible external stimuli, ultrasounds (US) as an effective external trigger can be used to control drug release spatiotemporally. US is appealing due to its non-invasiveness or minimally invasiveness, without ionizing radiations, cost effectiveness and easy control of tissue penetration depth by tuning the frequency, cycles and exposure time.93 High-frequency US can penetrate deeply into the body with focused beams, which allows local therapy avoiding adverse side effects to healthy tissues. Moreover, the US can not only trigger drug release from nanocarriers but also enhance drug-loaded nanoparticle extravasation through blood capillaries, which increase cell membrane permeation and even induce an immune response against tumors.108,109Several physical effects will occur when an US wave propagates through body tissue, so it can be utilized as trigger to design ultrasound-responsive DDSs. These physical effects include cavitation, local higher temperature, simple pressure variation and acoustic fluid streaming. An ideal ultrasound-responsive DDSs typically involves ultrasound sensitive material to respond to one or more of these physical effects.110
Wang and coworkers111 synthesized an Au NPs-capped, perfluorohexane (PFH)-encapsulated and PEGylated mesoporous silica nanocapsules-based enhancement agents (MAPP). Hydrophobic pyrene as model drug was loaded into MAPP (pyr–MAPP) to verify the effect of US on inducing drug release behavior of MAPP. Before US irradiation, nearly no pyrene was released, by contrast, under/after ultrasound irradiation, a plenty of smaller-sized phase-changed PFH micro bubbles in MAPP solution were generated, subsequently further swelled and merged into larger microbubbles which could enhance the release of pyrene. This indicated that the nanosized inorganic MAPP possessed excellent US sensitivity, and the loaded drug release could be trigger-controlled and enhanced via external US.
US waves can induce thermal and/or mechanical effects that trigger the loaded drug release from nanocarriers. Moreover, the advances in sonochemistry have shown that ultrasound-induced chemical reactions can differ from those carried out by bulk heating, implying mechanical or thermal effects at the nanoscale.112 US irradiation can also cleave certain chemical bonds, so-called mechanophores.113,114 This strategy can be exploited in the design of responsive nanoparticles with ultrasound-labile moieties. 2-Tetrahydropyranyl methacrylate (THPMA) can undergo a phase transformation from hydrophobic to hydrophilic by US irradiation. THPMA is a hydrophobic monomer bearing a labile acetal group that can be cleaved by US to yield hydrophilic methacrylic acid (MAA).113,115 Based on this, Paris et al.116 developed a new ultrasound-responsive system with MSNs as carriers and dual-responsive copolymers composed of a thermoresponsive poly(2-(2-methoxyethoxy)ethyl-methacrylate) (p(MEO2MA)) and THPMA (MEO2MA:THPMA = 90:10) as nanogates sensitive to ultrasounds. The thermal response can be used to load and retain the cargo, and the US sensitivity can induce the release of cargo at physiological temperature. Copolymer p(MEO2MA)-co-THPMA bearing US-cleavable hydrophobic tetrahydropyranyl moieties, presents a LCST below physiological temperature. At 4 °C, the polymer is in its coil-like conformation, allowing the cargo to be loaded in the mesopores. When the temperature is increased to physiological temperature, the copolymer changes to a collapsed state and the nanogates are closed retaining the cargo into the pores. Upon US irradiation, the hydrophobic tetrahydropyranyl groups in the polymer backbone were cleaved, leading to hydrophilic methacrylate and increasing the LCST over 37 °C. This induced a change in conformation of the polymer to coil-like, opening the gates of the mesopores of the MSNs and allowing the release of entrapped cargo. These hybrid nanoparticles were no cytotoxic and could also be endocyted by LNCaP cells, retaining their ultrasound-responsive capability because they could release the cargo inside the cells upon US irradiation. When loaded with DOX, the hybrid MSNs only induced cell death when exposed to ultrasound. Dating back to 2007, Kwon and coworkers117 first designed a newly ultrasound-responsive system Fc–CONH–MS by grafting a ferrocene derivative including carboxylic acid group at both ends onto the outside of amino-functionalized MSN. Upon ultrasound irradiation, the ferrocene derivatives could be easily broken up by local hyperthermia or the reaction of oxidized species, allowing the entrapped cargo to be released.
Ultrasound-targeted DDS based on MSNs has achieved some progress, but still in its infancy, need further study.
Light-responsive MSNs
Among various external stimuli, light is chosen as a trigger on account of its rapid activation, low invasiveness and the possibility of remote spatiotemporal control, which has been widely applied, especially in drug delivery systems. In the past few years, a large variety of light-responsive systems have been designed to obtain on-demand drug release in response to illumination of a specific wavelength (in the ultraviolet, visible or near-infrared (NIR) regions).Under UV irradiation, the azobenzene group and/or its derivatives can undergo isomerization from a planar trans form to a non-planar cis form in surrounding aqueous medium. The isomerization from cis to trans via illumination in the visible region enables photo-regulated control of drug release.118,119 Zink and coworkers designed a light-operated nanosystem with an immobilized azobenzene-containing stalk molecule connected to MSNs, light-operated dissociation movable β-CD and/or Py–β-CD cyclic molecule threaded onto the azobenzene-containing stalks, acting as gatekeeper. Without the irradiation of 351 nm light, movable cyclic molecule could bind to trans-azobenzene units to form the pseudorotaxanes, limiting release of the cargo through closing the nanopores. The irradiating with 351 nm light could cause the isomerization of azobenzene to the cis conformation, and the pores were uncapped, the cargo was released.118 MSNs assembled with reversible and reusable nanovalves were designed by the same research group based on the azobenzene/α-CD recognition motif, which is capable of controlling the release of both small cargo (alizarin red S) and larger dye molecules (propidium iodide) because of azobenzene-containing stalks with different lengths.119 The results showed that cargo size-selectivity could be achieved by tuning the length of the azobenzene stalk.
Molecules suffering physicochemical changes under UV irradiation such as thymine possessing photodimerization–cleavage cycle120 and photocleavable o-nitrobenzyl ester moiety and its derivatives121 have also been used as gatekeepers to control drug release. Besides, the structure of the PNIPAM polymers incorporating hydrophilic or hydrophobic light-sensitive monomers could transform from collapsed (insoluble) to coil (soluble) state in the presence of UV irradiation, the transformation can lead to the pores open and subsequently the loaded molecules escape from the pores.122
Another strategy is grafting photoresponsive linkers such as 7-amino-coumarin derivative (CD) (at visible or NIR),123 S-coordinated Ru(bpy)2(PPh3)-moieties (λ = 455 nm),124 and thioundecyl-tetraethylene-glycol-ester-o-nitrobenzylethyl dimethyl ammonium bromide (TUNA) (at UV region)125 to the surface of MSNs. The photoresponsive linkers can suffer physicochemical changes or induced rupture under light irradiation, and then light-induced drug release can be obtained.
Lin and colleague125 reported a gold nanoparticle (AuNP)-capped MSN as light sensitive DDS via photoresponsive linker TUNA which would give rise to the negatively charged thioundecyltetraethyleneglycolcarboxylate (TUEC) and then the capped Au NPs dissociated from the MSNs surface via charge repulsion upon UV irradiation. Thus, the dissociation of the Au NPs leads to the mesopores open and release of cargo molecules.
Although UV/visible light have been widely applied in light-responsive DDSs, its drawbacks limit its clinical application. The UV/visible region of the spectrum (less than 700 nm) usually has low penetration depth (∼10 mm) due to the strong scattering properties of soft tissues, which often limited its application only on skin or external layers of organs. In addition, UV light may cause unwanted reactions including high risks for DNA damage leading to cellular apoptosis. Thus, developing longer wavelength NIR laser (in the range of 700–1000 nm) systems with lower scattering properties, deeper tissue penetration and minimal harm to tissues is required and also desirable for real clinical application.93,126
The property of NIR-absorbing materials such as Au,127–129 CuS92,130 single-walled carbon nanotubes (SWNTs),131 upconverting nanoparticles (UCNPs)132 to convert the adsorbed photon energy through irradiation into hyperthermia, has been used to trigger the release of therapeutic molecules from NIR responsive DDSs. When combined with a thermal-responsive polymeric shell, temperature-sensitive bonds or linkers, drugs could be released under high temperature induced by NIR light irradiation. For example, DNA duplexes have been used as gatekeeper capping mesoporous silica shell and the dehybridization of the DNA duplexes leaded to the release of the loaded molecules.91
UCNPs have the ability to absorb lower energy NIR light photons and emit a single high energy photon of shorter wavelength in the UV or visible region owing to its expanded optical properties. He et al.132 synthesized b-phase NaYF4:TmYb@NaYF4 UCNPs (core: NaYF4: 0.5 mol% Tm3+: 30 mol% Yb3+; shell = NaYF4) coated by MSNs loaded with the anticancer drug DOX and grafted with ruthenium complexes as photoactive molecular valves. Result showed that loaded drug release could be triggered by 974 nm light with 0.35 W cm−2 based on UCNP-assisted photochemistry. The literatures showed that the lowest intensity for UCNP-assisted photochemistry was 0.35 W cm−2. This intensity of light is also lower than the maximum permissible exposure of skin (0.726 W cm−2). Such low intensity of light minimized superheating problems and avoided photo damage to biological samples. After irradiating with 0.35 W cm−2 light (974 nm) for 5 hours, about 42% loaded DOX released, while an increased release of 78% was obtained after treating with 0.64 W cm−2 light (974 nm) for 5 hours. The results showed that drug release kinetics were intensity related to the power of the light irradiation.
Multiple stimuli-responsive MSNs
Compared to single stimuli-responsive drug delivery systems, dual or multi-responsive DDSs are sensitive to two or more stimuli, either in a synergistic fashion or in an independent style (Table 4), which has gained considerable attention in the current decades for its capability of further improving drug delivery.| Mechanism | Material | Release condition | Biological model | Model drug | Ref. |
|---|---|---|---|---|---|
| pH/cellulase | Cellulose | pH 4.0/cellulose | HepG2 | DOX | 72 |
| Redox/enzyme | HA | GSH/hyaluronidases | HCT-116 | DOX | 138 |
| pH/redox/UV | PDEAEMA/disulfide bond/o-nitrobenzyl ester | pH 5.0/DTT/UV | HeLa | DOX | 134 |
| NIR/PH/thermo | Au25(SR)18/P(NIPAM–MAA) | 980 nm (NIR)/tumor sites | A549, HeLa, SKOV3 | DOX | 139 |
| pH/redox | Poly(allylamine hydrochloride)–citraconic anhydride/galactose-modified trimethyl chitosan–cysteine | pH 5.0/Cytoplasmic glutathione | QGY-7703 | DOX/siRNA | 140 |
| Esterase/pH | Poly(β-amino ester) | Liver esterase/pH < 5.0 | MDA-MB-231 | DOX | 141 |
| Enzyme/redox or thermo/redox | AND logic gates (DNA) | DNase I/DTT or 50 °C/DTT | A549 | Calcein | 142 |
| pH/enzyme | AND logic gate (PAA/PCL) | pH 5.5@esterase | SK-N-BE(2), HeLa, MRC-5 | DOX | 137 |
| pH/redox | Disulfide bonds/benzoic–imine bond | Glutathione/pH 5.0 | U87MG | DOX | 143 |
| Magnetic/NIR | Fe3O4@poly-L-lysine@Au@dsDNA | Magnetic target/808 nm NIR | HeLa, nude mice | DOX | 144 |
| Ultrasound/pH/magnetic | Crown-ether/SPION | Ultrasound/pH/magnetic | L929 | DOX | 135 |
Zhao and colleagues133 capped mesoporous silica with hyaluronic acid (HA) through cleavable disulfide (SS) bonds, and the loaded DOX release occurred either in a redox responsive way by addition of glutathione (GSH) or in an enzyme responsive way by introducing of hyaluronidases (HAase). In this system, HA acted as both gatekeeper and targeting ligand owing to the specific affinity with CD44 receptors with a high level of expression on various tumor cells such as human HCT-116 cells. The MSN–SS–HA/DOX had the high drug loading efficiency up to 12.5% and in vitro drug release study showed that the release of DOX was triggered by GSH and HAase. Without GSH and HAase, DOX released from MSN–SS–HA/DOX was obviously inhibited with less than 20% for a period of 48 h. Nevertheless, in the presence of HAase, GSH, GSH and HAase, the cumulative amount of released DOX was significantly increased to 30%, 50% and 60% within 48 h, respectively. In addition, fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM) showed a higher cellular uptake via CD44 receptor-mediated endocytosis with increasing 3.0-times and 2.7-times for 100 and 200 μg mL−1 MSN–SS–HA in HCT-116 cells compared with that in CD44 receptor-negative NIH-3T3 cells. In vitro cytotoxicity studies showed that the IC50 value of MSN–SS–HA/DOX was significantly reduced to 0.6 μg mL−1 in HCT-116 cells from 4.5 μg mL−1 in NIH-3T3 cells. Therefore, MSN–SS–HA/DOX nanosystem could enhance the anticancer efficiency of DOX to CD44 receptor-positive cancer cells and reduce the undesirable side effects to healthy cells/CD44 receptor-negative cells. The results suggested a potential approach via using a single substance to obtain dual-stimuli responsive targeted cancer therapy.
Internal-sensitivity can also be associated with external responsiveness. Very recently, Zhang et al.134 reported a reduction, pH and light triple responsive nanocarriers (HMSNs–PDEAEMA) based on hollow mesoporous silica nanoparticles (HMSNs) coated by poly(2-(diethylamino)-ethyl methacrylate) (PDEAEMA). pH-Sensitive PDEAEMA polymer capped on the surface of HMSNs through linkages including reduction cleavable disulfide bond and light-cleavable o-nitrobenzyl ester. DOX was easily loaded into the nanocarriers with high drug loading efficiency, and the rapidly released of DOX was triggered by the stimuli of acid environment, reducing agent or UV light irradiation. In addition, the results of flow cytometry analysis, CLSM and cytotoxicity indicated that the DOX loaded HSNs–PDEAEMA was efficiently uptaken by HeLa cells, showing (i) smart control on drug delivery and release, (ii) the enhanced DOX release into the cytoplasm under external UV light irradiation, and (iii) higher cytotoxicity against HeLa cells.
Another nanosystem based on supramolecular switches showed the response to ultrasound, pH and magnetic fields. Leung and coworkers135 reported that a nanoparticle with SPION as core, mesoporous silica as shell was functionalized with a series of metal cations complexed dibenzo-crown ether macrocycles which was utilized as gatekeeper controlling the release of cargo through ultrasound waves. In this system, pH-sensitive electrostatic interactions (such as metal chelating) or intramolecular hydrogen bonds were used to control the “ON/OFF” switching of the gatekeeper supramolecules. The dibenzo-crown ether macrocycles nanovalves could be blocked via chelating with Cs+ and Na+ ions and have the capability of loading and controlling the release of different modes of drug. In this work, the release profiles suggested that DOX released from metal cation blocked crown ether-based nanovalves capped NPs could be triggered by (i) the change in electrostatic interaction and hydrogen bonds induced by lower pH, (ii) the dissociation of blocked agents from dibenzo-crown ethers induced by ultrasound. Furthermore, under external magnetic field, particles could be attracted to specific lesion site via MF guiding and the nanoparticles showed great MRI capability, which indicated that this constructed nanosystem is potentially useful as multiple stimuli-responsive DDSs and theranostic agents.
AND logic gates could also be used to design MSNs with dual-stimuli controlled release, a MSN system with “AND” logic gate in respond to pH and light was first reported by Angelos.136 The dual-controlled nanoparticle systems were functioned with a true “AND” logic gate in which drug release only happened in the presence of two stimuli. So the drug was released only at the required site and required time. Recently, Chen et al.137 reported a MSN system with “AND” logic gate in respond to enzyme and pH based on polycaprolactone (PCL) and polyacrylic acid (PAA) functionalized MSNs (PAA–PCL–MSNs). PAA–PCL–MSNs showed the capacity of selectively controlling delivery and release of cargo in cancer cells. Esterase degradable PCL was immobilized into the pores of MSNs while pH responsive PAA was capped the outside of the MSNs to constitute a PAA–PCL–MSNs construct. With DOX as model drug, the PAA–PCL–MSNs@DOX possessed high drug loading efficiency up to 0.3 g g−1, the in vitro drug release study showed that only in the coexistence of low pH and esterase, DOX release could occur. The IC50 of the PAA–PCL–MSNs@DOX in normal fibroblasts MRC-5 was 8-times than that in neuroblastoma SK-N-BE(2) cell. This 8-fold divergence demonstrated that PAA–PCL–MSNs@DOX possessed the higher cytotoxicity to cancer cells than normal cells. The release based on “AND” logic gate could reach smart control in complicated physiological environment, then decreasing undesirable toxic side effects on normal cells and enhancing therapeutic efficacy of target cells.
Active targeting MSNs
Active targeting as a complementary strategy to EPR effect has opened up a new area for traditional chemotherapy to enhance the efficiency of anti-cancer, which known as ligand-mediated targeting involving functionalization of the MSNs surface with active targeting ligands. The active targeting ligands, such as small molecules, peptides, antibodies, proteins, saccharides and aptamers, have specific affinity to the over-expressed receptors on the tumor cells. Typically, there are three paths (targeting to tumor vessels, tumor cells, nuclear) to achieve effective enrichment of drugs in tumor tissues (Fig. 4). The most relevant results derived from the conjugation of active targeting ligands to MSNs-based nanosystems to promote specific recognition and cellular uptake by cancer cells are summarized in Table 5.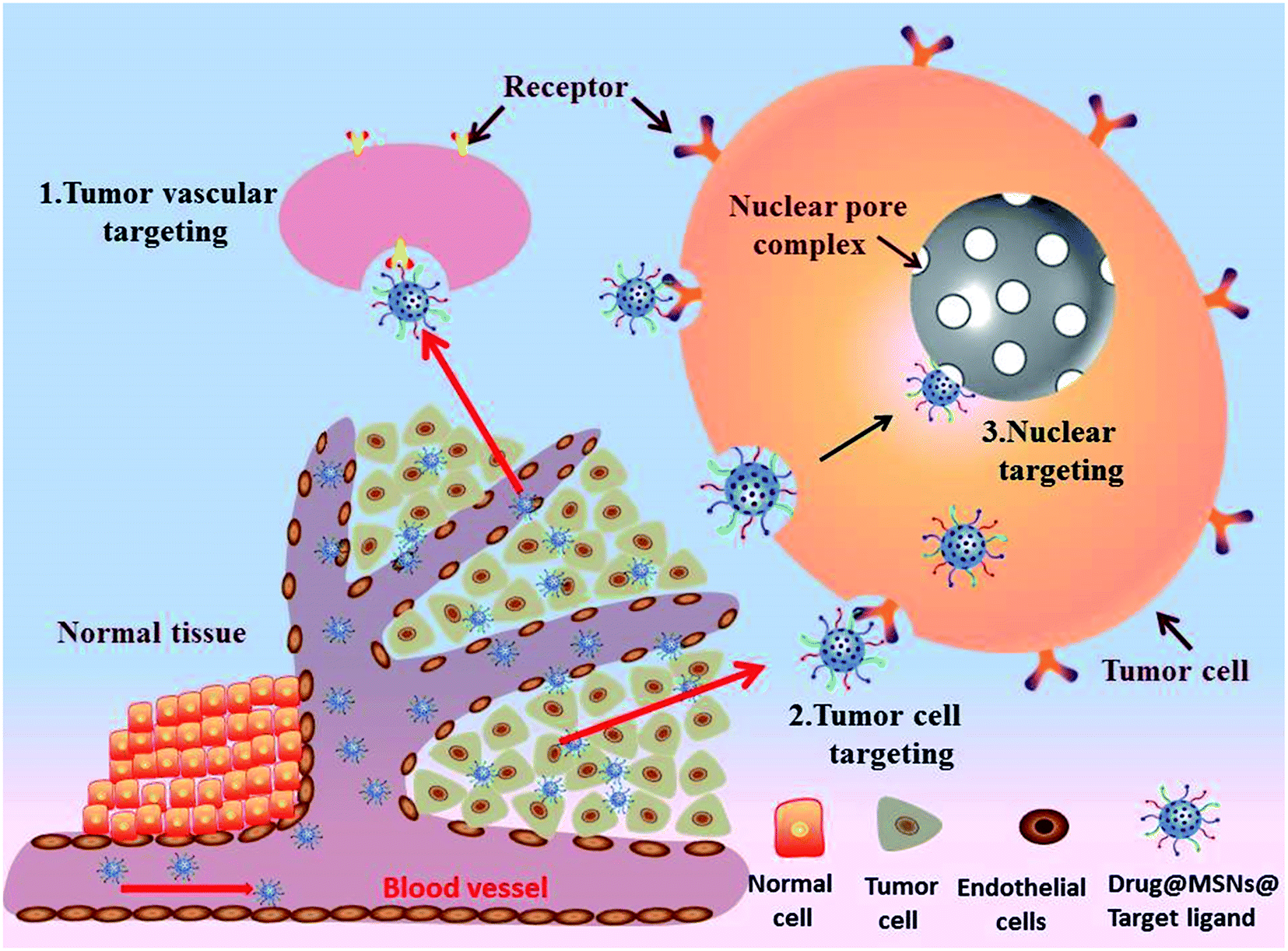 | ||
| Fig. 4 Schematic illustration of active targeted drug delivery based on MSNs for effective cancer therapy: (1) tumor vascular targeting. (2) Tumor cell targeting (3) nuclear targeting. | ||
| Site | Targeting ligand | Targets/mechanism | Model | Model drug | Stimuli-responsive | Ref. |
|---|---|---|---|---|---|---|
| a anti-EpCAMDNA Ap: (5′-amino-CAC TAC AGA GGT TGC GTC TGT CCCACG TTG TCA TGG GGG GTTGGC CTG-3′, MW = 748.70), HB5Ap: (5′-AACCGCCCAAATCCCTAA-GAGTCTGCACTTGTCATTTTGTATATGTATTTGGTTTTTGGCTCTCACA-GACACACTACACACGCACA-3′, 86 bp), tLyp-1: (sequence CGNKRTR) PEGA–pVEC peptides (cCPGPEGAGC-LLIILRRRIRKQAHAHSK-NH2), LA: lactobionic acid, ASGPR: asialoglycoprotein receptor, EpCAM: epithelial cell adhesion molecule, HER2: human epithelial growth factor receptor 2, 6-MP: 6-mercaptopurine, HA: hyaluronic acid, SERS: surface enhanced Raman scattering. | ||||||
| Tumor cell membrane | HA | CD44 receptor | MDA-MB-231 | Rhodamine B | 69 | |
| HA | CD44 receptor | HCT-116 | DOX | 145 | ||
| MSN–SS–HA | CD44 receptor | HCT-116 | DOX | Redox and enzyme redox and enzyme control release, improved cytotoxicity and cellular uptake | 133 | |
| CMS–SS–oHA | CD44 receptor | HCT-116 | 6-MP | Redox improved cytotoxicity and cellular uptake | 63 | |
| FA | Folate receptor | A549, HeLa | DOX | 146 | ||
| DOX@HPSN–Salphdc–FA | Folate receptor | HepG2 | DOX | PH, minimal toxic side effect, induce cell apoptosis, inhibition of tumor growth | 147 | |
| MSN–FA@gelatin–PEG | Folate receptor | HT-29 | DOX | Enzyme, MMP-2 induced release, target endocytosis | 71 | |
| SBA–PEG–FA | Folate receptor | HepG2 | DOX | PH, enhanced cancer cell killing efficacy | 21 | |
| FA–PEI–HMSN@Dox@siRNA | Folate receptor | HeLa | DOX and siRNA | PH, enhanced therapeutic efficacy co-delivery of DOX and siRNA | 148 | |
| MSNs–S-S–HP–LA | Galactose receptor | HepG2 | DOX | Redox induce cell apoptosis, inhibition of tumor growth | 55 | |
| HMSNs–S-S-Ada/β-CD–LA | Asialoglycoprotein receptor | HepG2 | DOX | Redox, inhibited tumor growth with the minimized side effect | 58 | |
| anti-EpCAMDNA aptamer (Ap) | EpCAM | SW620 | DOX | 149 | ||
| YY146 (an anti-CD146 antibody) | CD146 | U87MG | DOX | 150 | ||
| Antibody/single-chain variable fragment (Ab-/scFv) | Specific affinity receptor | OVCAR-5 | Bevacizumab | 151 | ||
| K4YRGD peptide | αvβ3 receptor | HepG2 | DOX | 152 | ||
| Lactose | ASGPR | HepG2 SMMC7721 | Docetaxel | 153 | ||
| AS1411 aptamer | Nucleolin | HeLa | DTX | 154 | ||
| AS1411 aptamer | Nucleolin | HeLa | DOX | ATP | 155 | |
| ATP aptamer | ATP-triggered release, high therapeutic efficacy | |||||
| PTX–MSNs@AgNPs–PEG/sgc8 (aptamer) | — | CEM | Paclitaxel (PTX) | Redox, inhibition of tumor growth | 156 | |
| Tf-Ag@SiO2@mSiO2@CS–PMAA | Transferrin receptor (TfR) | HeLa | DOX | pH-responsive, SERS-traceable, cancer cells target | 157 | |
| N3GPLGRGRGDK-Ad | αvβ3 integrins | SCC-7 | DOX | 158 | ||
| PEI-cRGD | αvβ3 integrins | Blood–brain barrier model | DOX | 159 | ||
| cRGDfK | αvβ3 integrins | MDA-MB-231 triple-negative breast cancer | Arsenic trioxide (ATO) | 160 | ||
| CRGDKGPDC | α2β3 receptor | HeLa | Combretastatin A4, DOX | 161 | ||
| CRGDyK | αvβ3 integrins | U87MG | Sunitinib (SUN) | 162 | ||
| K8(RGD)2 | αvβ3 integrins | U87MG | DOX | 163 | ||
| MSN–GFLGR7RGDS/α-CD | αvβ3 integrins | HeLa | DOX | Enzyme superior tumor targeting, drug internalization, cytotoxicity, and in vivo antitumor efficacy | 65 | |
| cRGD | αvβ3 integrins | SCC-7 | CPT | Tumor tissues (vasculature) | 164 | |
| MSN–SS–RGDFFFFC-MPEG | αvβ3 integrins | U-87 MG | DOX | Redox, PH selectively tumor cell killing | 143 | |
| HB5 aptamer | HER2 | SK-BR-3 | DOX | 165 | ||
| PEGA–pVEC peptide | — | MCF-7 | (−)-Epigallocatechin-3-gallate | 166 | ||
| Vascular endothelial growth factor (VEGF) | VEGF receptor | SKOV3 | siRNA | 167 | ||
| VEGF121 | VEGF receptor | U87MG | Sunitinib (SUN) | 168 | ||
| Anti-TRC105 | CD105 | HUVE-Cs | DOX | 169 | ||
| anti-VCAM-1 | VCAM-1 receptors | HUVEC-CS | Fluorescein isothiocyanate (FITC) | 170 | ||
| Nuclei | TAT peptide | Nuclear membrane receptors | MCF-7/ADR | DOX | 171 | |
| MONs–PTAT | Nuclear membrane receptors | HeLa | DNA | 172 | ||
| MSNSATAT&DMAK11 | Stepwise-acid-active | HeLa | DOX | 173 | ||
| QDs@mSiO2-CPP (TAT, PGFK, oligoanionic-inhibitory domain) | Cathepsin B protease | A549 | DOX | Enzyme selectively release drug into the nucleus of targeted tumor cells with high tumor cytotoxicity and minimum side effects | 67 | |
| Dexamethasone (DEX) | Glucocorticoid receptor (GR) | HeLa | DOX | 174 | ||
|
||||||
| Multistage target | ||||||
| Tumor and nuclei | FA and dexamethasone (DEX) | Folate receptor and glucocorticoid receptor (GR) | HeLa | DOX | 174 | |
| Tumor cells and vessels | tLyp-1 peptide | Neuropilin (NRP) | MDA-MB-231, HUVECs | DOX | 175 | |
| Vascular-cell nuclear | RGD peptides, TAT peptide | αvβ3 integrins | HeLa | DOX | 176 | |
| Nuclear membrane receptors | ||||||
| Tissue-cell-nuclear | Magnetic, FA, TAT peptide | Magnetic target, folate receptor, nuclear-target | HeLa | Camptothecin (CPT) | 177 | |
Vessel targeting MSNs
Different from normal tissues, many proteins show specific over-expression on the surface of tumor-associated endothelial cells due to the abnormal overgrowth of intratumoral vasculature, which can be utilized as targets for targeted drug delivery and cancer treatment. According to the literatures, targeting to the endothelial cells of the tumor vessels and subsequently killing them can lead to the necrosis of tumors because tumor vasculatures are the transport channel of nutrition which guarantees the fast proliferation of tumor cells,178 which has become a promising alternative in treating solid tumors. In 2013, Chen et al.169 reported the first example of in vivo tumor vascular targeted drug delivery system based on MSNs. In this nanosystem, TRC105 antibody targeting to CD105 receptor was conjugated onto MSNs surface. In the 4T1 tumor tissue, the receptors (CD105) only overexpressed in the tumor vasculature but did not express on 4T1 tumor cell. Active vessel targeting giving rise to ∼2 times enhancement of tumor uptake comparing to passive targeting only based on the EPR effect.Since then, various tumor vascular targeting ligands, such as vascular endothelial growth factor (VEGF) specific for VEGF receptors (VEGFRs), arginine–glycine–aspartic acid (RGD) peptides targeting to αvβ3 integrin receptor, HB5 aptamer which was specific for human epithelial growth factor receptor 2 (HER2), anti-VCAM-1 monoclonal antibody specific for vascular cell adhesion molecule-1 (VCAM-1) receptors, have been connected to the surface of MSNs to develop vascular targeted drug delivery systems with enhanced therapeutic efficiency.
Very recently, Li et al.161 reported a novel vascular-targeting co-delivery DDS based on targeting molecules (iRGD peptide) modified MSNs. In this system, antiangiogenic agent (combretastatin A4) and chemotherapeutic drug (DOX) were payloaded, leading to significantly improved anti-cancer efficacy even at a very low DOX dose (1.5 mg kg−1). Furthermore, the disruption of vascular structure caused by combretastatin A4 which was released quickly at tumor vasculatures had a synergetic effect with DOX which released slowly in the subsequent delivery of DOX into tumors.
Tumor cell targeting MSNs
After reaching tumor tissues, drug loaded nanocarriers are often expected to target to tumor cells through specific interaction between ligand and receptor, leading to the enhanced cellular uptake and drug delivery efficiency and the enhanced therapeutic efficacy. Ligand-mediated targeting to tumor cells need meet the following requirements: (I) a threefold overexpression of the targeted receptor on the cancer cell compared with normal cells is generally considered to be sufficient to warrant further investigation, although greater upregulation is preferred.179 (II) The density of active targeting ligands on the surface of nanoparticles should be carefully manipulated and optimized in order to obtain maximized targeting and therapy efficiency. Because the ligand/nanoparticle ratio strongly affects the cell recognition specificity, so greater selectivity and targeting efficiency can be obtained with higher density of active targeting ligands.180 But excessively high density of active targeting ligands may increase the steric hindrance effect and lead to poor cellular uptake efficiency.181 (III) Preventing targeting ligands coated by the plasma protein is also a noteworthy issue because the targeting ligands can be shielded by opsonization, which can result in loss of their targeting ability in a complex in vivo environment.182 (IV) The targeted receptor on the cancer cell can induce endocytosis.Various tumor cell targeting ligands such as antibodies (anti-CD146 antibody, antibody fragment (Ab-/scFv)), proteins (transferrin (Tf)), peptides (K4YRGD peptide), saccharides (hyaluronic acid (HA)), lactobionic acid (LA), lactose, small molecules (folic acid (FA)), aptamers (anti-EpCAMDNA aptamer, AS1411 aptamer), have been conjugated onto MSNs surface to receive tumor cell targeted property.
Quan et al.153 designed a hepatoma targeting DDS based on lactose conjugated MSNs (Lac–MSNs) with anticancer drug DTX loaded. The DTX–Lac–MSNs showed specific targeting to ASGPR-positive SMMC7721 and HepG2 cells, and the cellular uptake of Lac–MSNs was an energy-consuming process and predominated by clathrin-mediated endocytosis. Thanks to active targeting, significantly enhanced inhibition of the growth of HepG2 and SMMC7721 cells in vitro was obtained.
Nuclear-targeting MSNs
The nucleus is important space for the storage, replication and transcription of genetic material and plays an important role in cell metabolism, growth and differentiation. Therapeutic genes must enter the cell nucleus and correct dysfunctional and/or missing genes. Some anti-cancer drugs, such as cisplatin and DOX, must enter into the cell nucleus to induce apoptosis. Therefore, the nuclei of cancer cells has become an ultimate target point and nuclear-targeted treatment systems are hoped to be more efficiently and directly deliver therapeutic drugs to kill tumor cells. There are a lot of nuclear pore complexes distributing on the nuclear membrane, the pore with a diameter value of 20–70 nm. The diameter of the pore is related to the cell type and cell cycle, especially, the nuclear pore diameter of tumor cells is bigger than normal cells.183 The classical cytoplasm-to-nuclear transport of nanocarriers must pass through the nuclear pore, nanocarriers with the aid of proteins which contain nuclear localization sequences (NLS) form complexes with importin α/β, then interact with nucleoporins within the NPCs, and finally transversely enter the nuclei from cytoplasm.183 Therefore, enough small nanocarriers or dilated nuclear pore and conjugated appropriate nuclear targeting ligands (such as cell-penetrating peptides (CPPs) which contain nuclear localization sequences (NLS)), are two critical presuppositions for the nuclear-targeted nanocarriers. Besides these, the nanocarriers should be tumor specific without any harmful side effects and have the ability to escape from endo/lysosomal to avoid the internalized nanocarriers and cargos being degraded.Lin et al.174 reported a cancer-cell-specific nuclear targeted delivery system based on both FA and dexamethasone (DEX) targeting ligand modified MSNs. In which, the nuclear targeting ligand dexamethasone is a potent glucocorticoid with the capability of enlarging nuclear pore up to 60 nm during the translocation process, it can promote transport from cytoplasm to nucleus via specifically binding to the nuclear receptor, glucocorticoid receptor (GR) expressed in almost style cell. FA acting as tumor cell targeting ligand can enhance cancer cellular uptake. The results demonstrated that the constructed FA–MSN–DEX showed higher anticancer efficacy of DOX on HeLa cells via enhanced cellular uptake and active nucleus accumulation with the calculated IC50 of 0.78 μg mL−1 at 48 h.
Conclusions
In the war against cancer, conventional chemotherapy is still the main approach among various types of cancer treatments. While conventional chemotherapy will cause severe harmful side-effects due to nonspecific uptake by healthy cells and high-doses administration of therapeutic agents. The nonspecific cellular uptake of the drug by healthy cells is an important obstacle during the treatment of cancer. Therefore, it is important to target delivery of anticancer drugs with suitable nanocarriers. MSNs are promising candidates in target delivery of anticancer drugs for its unique physical–chemical properties. Through conjugating of various functional groups on MSNs, stimuli-responsive and active targeted anticancer drug delivery systems have been successfully prepared. The wide range of endogenous stimuli (e.g. pH, redox, enzymes, temperature) or exogenous stimuli (e.g. light, magnetic, ultrasound) is able to trigger the release of anticancer drug at the right place and time. And various functional groups or molecules can be combined with MSNs, which allows more flexibility in design of stimuli-responsive DDSs. Yet, the design of sensitive nanocarriers to endogenous or exogenous stimuli may exist severe limitation in practice, because the difference of internal stimuli is not enough big and external stimuli is difficult to accurately locate. To improve practicability, multiple stimuli-responsive drug delivery systems or/and combination with active targeting have been designed to cover the insufficiency of single stimuli-sensitive nanosystems, thereby achieve more accurately controlled release of drug. As for active target, nanocarriers based on MSNs possessing higher targeting efficiency and treatment efficiency are needed, the role and contribution of tumor cell targeting ligands to tumor site specific accumulation at in vivo conditions need more detailed studies.Although a number of stimuli-responsive and active targeting nanosystems have been reported in vitro proofs of concept, only a few have been performed in vivo preclinical research, and none has reached the clinical stage. In addition, researches about the toxicity and benefit-to-risk ratio after long-term use, the feature of pharmacokinetic in vivo are severe insufficient. Therefore, to develop a biodegradable, non-toxic, safety, high targeting efficiency drug delivery system based on MSNs is an important direction and main objective in the future.
Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province, China (No. ZR2015HM032).References
- S. Nazir, T. Hussain, A. Ayub, U. Rashid and A. J. MacRobert, Nanomedicine, 2014, 10, 19–34 CAS.
- W. X. Mai and H. Meng, Integr. Biol., 2013, 5, 19–28 RSC.
- Y. Yang and C. Yu, Nanomedicine: Nanotechnology, Biology and Medicine, 2015, 12, 317–332 CrossRef PubMed.
- D. Douroumis, I. Onyesom, M. Maniruzzaman and J. Mitchell, Crit. Rev. Biotechnol., 2013, 33, 229–245 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS PubMed.
- J. Lu, M. Liong, J. I. Zink and F. Tamanoi, Small, 2007, 3, 1341–1346 CrossRef CAS PubMed.
- L. Sun, Y. Wang, T. Jiang, X. Zheng, J. Zhang, J. Sun, C. Sun and S. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 103–113 CAS.
- P. Nadrah, F. Porta, O. Planinsek, A. Kros and M. Gaberscek, PCCP Phys. Chem. Chem. Phys., 2013, 15, 10740–10748 RSC.
- Y. Zhao, J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Lin, Expert Opin. Drug Delivery, 2010, 7, 1013–1029 CrossRef CAS PubMed.
- S. P. Hudson, R. F. Padera, R. Langer and D. S. Kohane, Biomaterials, 2008, 29, 4045–4055 CrossRef CAS PubMed.
- M. Colilla, B. González and M. Vallet-Regí, Biomater. Sci., 2013, 1, 114–134 RSC.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS PubMed.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang, T. Jiang and S. Wang, Nanomedicine, 2015, 11, 313–327 CAS.
- C. Kresge, M. Leonowicz, W. Roth, J. Vartuli and J. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- M. Vallet-Regi, A. Ramila, R. Del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef CAS.
- Y. Chen, H. Chen and J. Shi, Expert Opin. Drug Delivery, 2014, 11, 917–930 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Preat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- R. Liu, Y. Zhang, X. Zhao, A. Agarwal, L. J. Mueller and P. Feng, J. Am. Chem. Soc., 2010, 132, 1500–1501 CrossRef CAS PubMed.
- Y. Chen, K. Ai, J. Liu, G. Sun, Q. Yin and L. Lu, Biomaterials, 2015, 60, 111–120 CrossRef CAS PubMed.
- Q. Gan, X. Lu, Y. Yuan, J. Qian, H. Zhou, X. Lu, J. Shi and C. Liu, Biomaterials, 2011, 32, 1932–1942 CrossRef CAS PubMed.
- K. Yang, H. Luo, M. Zeng, Y. Jiang, J. Li and X. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 17399–17407 CAS.
- C. H. Lee, S. H. Cheng, I. Huang, J. S. Souris, C. S. Yang, C. Y. Mou and L. W. Lo, Angew. Chem., 2010, 122, 8390–8395 CrossRef.
- M. Chen, X. He, K. Wang, D. He, S. Yang, P. Qiu and S. Chen, J. Mater. Chem. B, 2014, 2, 428–436 RSC.
- L. Chen, J. Di, C. Cao, Y. Zhao, Y. Ma, J. Luo, Y. Wen, W. Song, Y. Song and L. Jiang, Chem. Commun., 2011, 47, 2850–2852 RSC.
- Z. Zou, D. He, X. He, K. Wang, X. Yang, Z. Qing and Q. Zhou, Langmuir, 2013, 29, 12804–12810 CrossRef CAS PubMed.
- X. Hu, Y. Wang and B. Peng, Chem.–Asian J., 2014, 9, 319–327 CrossRef CAS PubMed.
- S. Niedermayer, V. Weiss, A. Herrmann, A. Schmidt, S. Datz, K. Muller, E. Wagner, T. Bein and C. Brauchle, Nanoscale, 2015, 7, 7953–7964 RSC.
- J. Zheng, X. Tian, Y. Sun, D. Lu and W. Yang, Int. J. Pharm., 2013, 450, 296–303 CrossRef CAS PubMed.
- W. Feng, X. Zhou, C. He, K. Qiu, W. Nie, L. Chen, H. Wang, X. Mo and Y. Zhang, J. Mater. Chem. B, 2013, 1, 5886–5898 RSC.
- W. Feng, W. Nie, C. He, X. Zhou, L. Chen, K. Qiu, W. Wang and Z. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 8447–8460 CAS.
- J. Wang, H. Liu, F. Leng, L. Zheng, J. Yang, W. Wang and C. Z. Huang, Microporous Mesoporous Mater., 2014, 186, 187–193 CrossRef CAS.
- P. Zhang, T. Wu and J. L. Kong, ACS Appl. Mater. Interfaces, 2014, 6, 17446–17453 CAS.
- X. Xu, S. Lü, C. Gao, X. Wang, X. Bai, N. Gao and M. Liu, Chem. Eng. J., 2015, 266, 171–178 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, J. Phys. Chem. C, 2010, 114, 12481–12486 CAS.
- L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CAS.
- T. Chen, W. Wu, H. Xiao, Y. Chen, M. Chen and J. Li, ACS Macro Lett., 2016, 5, 55–58 CrossRef CAS.
- C. Chen, F. Pu, Z. Huang, Z. Liu, J. Ren and X. Qu, Nucleic Acids Res., 2011, 39, 1638–1644 CrossRef CAS PubMed.
- H. Meng, M. Xue, T. Xia, Y.-L. Zhao, F. Tamanoi, J. F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690–12697 CrossRef CAS PubMed.
- Y.-L. Zhao, Z. Li, S. Kabehie, Y. Y. Botros, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 13016–13025 CrossRef CAS PubMed.
- L. Du, H. Song and S. Liao, Microporous Mesoporous Mater., 2012, 147, 200–204 CrossRef.
- S. Angelos, N. M. Khashab, Y.-W. Yang, A. Trabolsi, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 12912–12914 CrossRef CAS PubMed.
- J. Zhang, D. Wu, M.-F. Li and J. Feng, ACS Appl. Mater. Interfaces, 2015, 7, 26666–26673 CAS.
- H. P. Rim, K. H. Min, H. J. Lee, S. Y. Jeong and S. C. Lee, Angew. Chem., Int. Ed., 2011, 50, 8853–8857 CrossRef CAS PubMed.
- Q. Zheng, Y. Hao, P. Ye, L. Guo, H. Wu, Q. Guo, J. Jiang, F. Fu and G. Chen, J. Mater. Chem. B, 2013, 1, 1644–1648 RSC.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- L. Bai, Q. Zhao, J. Wang, Y. Gao, Z. Sha, D. Di, N. Han, Y. Wang, J. Zhang and S. Wang, Nanotechnology, 2015, 26, 165704 CrossRef PubMed.
- D. Shao, X. Zhang, W. Liu, F. Zhang, X. Zheng, P. Qiao, J. Li, W. F. Dong and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 4303–4308 CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- K. T. Nguyen and Y. Zhao, Acc. Chem. Res., 2015, 48, 3016–3025 CrossRef CAS PubMed.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- F. Torney, B. G. Trewyn, V. S. Lin and K. Wang, Nat. Nanotechnol., 2007, 2, 295–300 CrossRef CAS PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., 2005, 117, 5166–5172 CrossRef.
- Q. Zhao, C. Wang, Y. Liu, J. Wang, Y. Gao, X. Zhang, T. Jiang and S. Wang, Int. J. Pharm., 2014, 477, 613–622 CrossRef CAS PubMed.
- L. Chen, Z. Zheng, J. Wang and X. Wang, Microporous Mesoporous Mater., 2014, 185, 7–15 CrossRef CAS.
- L. Dai, J. Li, B. Zhang, J. Liu, Z. Luo and K. Cai, Langmuir, 2014, 30, 7867–7877 CrossRef CAS PubMed.
- L. Sun, Y.-J. Liu, Z.-Z. Yang and X.-R. Qi, RSC Adv., 2015, 5, 55566–55578 RSC.
- J. Lee, H. Kim, S. Han, E. Hong, K.-H. Lee and C. Kim, J. Am. Chem. Soc., 2014, 136, 12880–12883 CrossRef CAS PubMed.
- Z. Luo, Y. Hu, K. Cai, X. Ding, Q. Zhang, M. Li, X. Ma, B. Zhang, Y. Zeng and P. Li, Biomaterials, 2014, 35, 7951–7962 CrossRef CAS PubMed.
- Q. Zhang, X. Wang, P. Z. Li, N. K. Truc, X. J. Wang, Z. Luo, H. Zhang, N. S. Tan and Y. Zhao, Adv. Funct. Mater., 2014, 24, 2413 CrossRef CAS.
- B. Zhang, Z. Luo, J. Liu, X. Ding, J. Li and K. Cai, J. Controlled Release, 2014, 192, 192–201 CrossRef CAS PubMed.
- H. He, H. Kuang, L. Yan, F. Meng, Z. Xie, X. Jing and Y. Huang, PCCP Phys. Chem. Chem. Phys., 2013, 15, 14210–14218 RSC.
- L. Palanikumar, E. S. Choi, J. Y. Cheon, S. H. Joo and J. H. Ryu, Adv. Funct. Mater., 2014, 25, 957–965 CrossRef.
- Q. Zhao, H. Geng, Y. Wang, Y. Gao, J. Huang, Y. Wang, J. Zhang and S. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 20290–20299 CAS.
- X. Yang, D. He, X. He, K. Wang, Z. Zou, X. Li, H. Shi, J. Luo and X. Yang, Part. Part. Syst. Charact., 2015, 32, 205–212 CrossRef CAS.
- Y. J. Cheng, G. F. Luo, J. Y. Zhu, X. D. Xu, X. Zeng, D. B. Cheng, Y. M. Li, Y. Wu, X. Z. Zhang, R. X. Zhuo and F. He, ACS Appl. Mater. Interfaces, 2015, 7, 9078–9087 CAS.
- C. de la Torre, L. Mondragon, C. Coll, F. Sancenon, M. D. Marcos, R. Martinez-Manez, P. Amoros, E. Perez-Paya and M. Orzaez, Chemistry, 2014, 20, 15309–15314 CrossRef CAS PubMed.
- J. Li, F. Liu, Q. Shao, Y. Min, M. Costa, E. K. Yeow and B. Xing, Adv. Healthcare Mater., 2014, 3, 1230–1239 CrossRef CAS PubMed.
- G. Zhang, M. Yang, D. Cai, K. Zheng, X. Zhang, L. Wu and Z. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 8042–8047 CAS.
- Z. Chen, Z. Li, Y. Lin, M. Yin, J. Ren and X. Qu, Chem.–Eur. J., 2013, 19, 1778–1783 CrossRef CAS PubMed.
- J.-H. Xu, F.-P. Gao, L.-L. Li, H. L. Ma, Y.-S. Fan, W. Liu, S.-S. Guo, X.-Z. Zhao and H. Wang, Microporous Mesoporous Mater., 2013, 182, 165–172 CrossRef CAS.
- Z. Zou, X. He, D. He, K. Wang, Z. Qing, X. Yang, L. Wen, J. Xiong, L. Li and L. Cai, Biomaterials, 2015, 58, 35–45 CrossRef CAS PubMed.
- A. Hakeem, F. Zahid, R. Duan, M. Asif, T. Zhang, Z. Zhang, Y. Cheng, X. Lou and F. Xia, Nanoscale, 2016, 8, 5089–5097 RSC.
- A. Agostini, L. Mondragon, A. Bernardos, R. Martinez-Manez, M. D. Marcos, F. Sancenon, J. Soto, A. Costero, C. Manguan-Garcia, R. Perona, M. Moreno-Torres, R. Aparicio-Sanchis and J. R. Murguia, Angew. Chem., Int. Ed. Engl., 2012, 51, 10556–10560 CrossRef CAS PubMed.
- L. A. Shuman Moss, S. Jensen-Taubman and W. G. Stetler-Stevenson, Am. J. Pathol., 2012, 181, 1895–1899 CrossRef PubMed.
- Z. Zhou, S. Zhu and D. Zhang, J. Mater. Chem., 2007, 17, 2428 RSC.
- A. Zintchenko, M. Ogris and E. Wagner, Bioconjugate Chem., 2006, 17, 766–772 CrossRef CAS PubMed.
- M. Keerl, V. Smirnovas, R. Winter and W. Richtering, Macromolecules, 2008, 41, 6830–6836 CrossRef CAS.
- T. Hoare, J. Santamaria, G. F. Goya, S. Irusta, D. Lin, S. Lau, R. Padera, R. Langer and D. S. Kohane, Nano Lett., 2009, 9, 3651–3657 CrossRef CAS PubMed.
- M. Karesoja, J. Mckee, E. Karjalainen, S. Hietala, L. Bergman, M. Linden and H. Tenhu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5012–5020 CrossRef CAS.
- J. T. Sun, Z. Q. Yu, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2012, 33, 811–818 CrossRef CAS PubMed.
- E. Aznar, L. Mondragon, J. V. Ros-Lis, F. Sancenon, M. D. Marcos, R. Martinez-Manez, J. Soto, E. Perez-Paya and P. Amoros, Angew. Chem., Int. Ed. Engl., 2011, 50, 11172–11175 CrossRef CAS PubMed.
- H. Yan, C. Teh, S. Sreejith, L. Zhu, A. Kwok, W. Fang, X. Ma, K. T. Nguyen, V. Korzh and Y. Zhao, Angew. Chem., Int. Ed. Engl., 2012, 51, 8373–8377 CrossRef CAS PubMed.
- X. Wu, Z. Wang, D. Zhu, S. Zong, L. Yang, Y. Zhong and Y. Cui, ACS Appl. Mater. Interfaces, 2013, 5, 10895–10903 CAS.
- A. Schlossbauer, S. Warncke, P. M. Gramlich, J. Kecht, A. Manetto, T. Carell and T. Bein, Angew. Chem., Int. Ed. Engl., 2010, 49, 4734–4737 CrossRef CAS PubMed.
- Z. Yu, N. Li, P. Zheng, W. Pan and B. Tang, Chem. Commun., 2014, 50, 3494–3497 RSC.
- G. Martelli, H. R. Zope, M. B. Capell and A. Kros, Chem. Commun., 2013, 49, 9932–9934 RSC.
- C. de la Torre, A. Agostini, L. Mondragon, M. Orzaez, F. Sancenon, R. Martinez-Manez, M. D. Marcos, P. Amoros and E. Perez-Paya, Chem. Commun., 2014, 50, 3184–3186 RSC.
- C. Liu, J. Guo, W. Yang, J. Hu, C. Wang and S. Fu, J. Mater. Chem., 2009, 19, 4764–4770 RSC.
- A. Baeza, E. Guisasola, E. Ruiz-Hernández and M. Vallet-Regí, Chem. Mater., 2012, 24, 517–524 CrossRef CAS.
- C. R. Thomas, D. P. Ferris, J.-H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J.-S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed.
- Y. T. Chang, P. Y. Liao, H. S. Sheu, Y. J. Tseng, F. Y. Cheng and C. S. Yeh, Adv. Mater., 2012, 24, 3309–3314 CrossRef CAS PubMed.
- Y. Zhang, Z. Hou, Y. Ge, K. Deng, B. Liu, X. Li, Q. Li, Z. Cheng, P. Ma, C. Li and J. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 20696–20706 CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- S. Laurent, S. Dutz, U. O. Hafeli and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8–23 CrossRef CAS PubMed.
- B. Ruhle, S. Datz, C. Argyo, T. Bein and J. I. Zink, Chem. Commun., 2016, 52, 1843–1846 RSC.
- P. Saint-Cricq, S. Deshayes, J. I. Zink and A. M. Kasko, Nanoscale, 2015, 7, 13168–13172 RSC.
- F. Liu, J. Wang, Q. Cao, H. Deng, G. Shao, D. Y. Deng and W. Zhou, Chem. Commun., 2015, 51, 2357–2360 RSC.
- E. Guisasola, A. Baeza, M. Talelli, D. Arcos, M. Moros, J. M. de la Fuente and M. Vallet-Regi, Langmuir, 2015, 31, 12777–12782 CrossRef CAS PubMed.
- Y. Wang and H. Gu, Adv. Mater., 2015, 27, 576–585 CrossRef CAS PubMed.
- H. Wu, G. Liu, S. Zhang, J. Shi, L. Zhang, Y. Chen, F. Chen and H. Chen, J. Mater. Chem., 2011, 21, 3037 RSC.
- C. Tao and Y. Zhu, Dalton Trans., 2014, 43, 15482–15490 RSC.
- P.-J. Chen, S.-H. Hu, C.-S. Hsiao, Y.-Y. Chen, D.-M. Liu and S.-Y. Chen, J. Mater. Chem., 2011, 21, 2535 RSC.
- E. Ruiz-Hernandez, A. Baeza and M. Vallet-Regi, ACS Nano, 2011, 5, 1259–1266 CrossRef CAS PubMed.
- E. Bringas, O. Koysuren, D. V. Quach, M. Mahmoudi, E. Aznar, J. D. Roehling, M. D. Marcos, R. Martinez-Manez and P. Stroeve, Chem. Commun., 2012, 48, 5647–5649 RSC.
- C. R. Thomas, D. P. Ferris, J. H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J. S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed.
- B. Sahoo, K. S. Devi, S. Dutta, T. K. Maiti, P. Pramanik and D. Dhara, J. Colloid Interface Sci., 2014, 431, 31–41 CrossRef CAS PubMed.
- D. Yang, K. Wei, Q. Liu, Y. Yang, X. Guo, H. Rong, M. L. Cheng and G. Wang, Mater. Sci. Eng., C, 2013, 33, 2879–2884 CrossRef CAS PubMed.
- N. Rapoport, K. H. Nam, R. Gupta, Z. Gao, P. Mohan, A. Payne, N. Todd, X. Liu, T. Kim, J. Shea, C. Scaife, D. L. Parker, E. K. Jeong and A. M. Kennedy, J. Controlled Release, 2011, 153, 4–15 CrossRef CAS PubMed.
- A. K. Wood and C. M. Sehgal, Ultrasound Med. Biol., 2015, 41, 905–928 CrossRef PubMed.
- S. R. Sirsi and M. A. Borden, Adv. Drug Delivery Rev., 2014, 72, 3–14 CrossRef CAS PubMed.
- X. Wang, H. Chen, Y. Zheng, M. Ma, Y. Chen, K. Zhang, D. Zeng and J. Shi, Biomaterials, 2013, 34, 2057–2068 CrossRef CAS PubMed.
- P. Cintas, S. Tagliapietra, M. Caporaso, S. Tabasso and G. Cravotto, Ultrason. Sonochem., 2015, 25, 8–16 CrossRef CAS PubMed.
- J. Wang, M. Pelletier, H. Zhang, H. Xia and Y. Zhao, Langmuir, 2009, 25, 13201–13205 CrossRef CAS PubMed.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Science, 2011, 333, 1606–1609 CrossRef CAS PubMed.
- J. Xuan, O. Boissiére, Y. Zhao, B. Yan, L. Tremblay, S. Lacelle, H. Xia and Y. Zhao, Langmuir, 2012, 28, 16463–16468 CrossRef CAS PubMed.
- J. L. Paris, M. V. Cabanas, M. Manzano and M. Vallet-Regi, ACS Nano, 2015, 9, 11023–11033 CrossRef CAS PubMed.
- E. J. Kwon and T. G. Lee, Appl. Surf. Sci., 2008, 254, 4732–4737 CrossRef CAS.
- D. P. Ferris, Y. L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686–1688 CrossRef CAS PubMed.
- D. Tarn, D. P. Ferris, J. C. Barnes, M. W. Ambrogio, J. F. Stoddart and J. I. Zink, Nanoscale, 2014, 6, 3335–3343 RSC.
- D. He, X. He, K. Wang, J. Cao and Y. Zhao, Langmuir, 2012, 28, 4003–4008 CrossRef CAS PubMed.
- M. A. Azagarsamy, D. L. Alge, S. J. Radhakrishnan, M. W. Tibbitt and K. S. Anseth, Biomacromolecules, 2012, 13, 2219–2224 CrossRef CAS PubMed.
- J. Lai, X. Mu, Y. Xu, X. Wu, C. Wu, C. Li, J. Chen and Y. Zhao, Chem. Commun., 2010, 46, 7370–7372 RSC.
- Q. Lin, Q. Huang, C. Li, C. Bao, Z. Liu, F. Li and L. Zhu, J. Am. Chem. Soc., 2010, 132, 10645–10647 CrossRef CAS PubMed.
- N. Z. Knezevic, B. G. Trewyn and V. S. Lin, Chem. Commun., 2011, 47, 2817–2819 RSC.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- S. Baek, R. K. Singh, D. Khanal, K. D. Patel, E. J. Lee, K. W. Leong, W. Chrzanowski and H. W. Kim, Nanoscale, 2015, 7, 14191–14216 RSC.
- J. Liu, C. Detrembleur, D. Pauw-Gillet, S. Mornet, C. Jérôme and E. Duguet, Small, 2015, 11, 2323–2332 CrossRef CAS PubMed.
- H. Li, L.-L. Tan, P. Jia, Q.-L. Li, Y.-L. Sun, J. Zhang, Y.-Q. Ning, J. Yu and Y.-W. Yang, Chem. Sci., 2014, 5, 2804 RSC.
- J. Yang, D. Shen, L. Zhou, W. Li, X. Li, C. Yao, R. Wang, A. M. El-Toni, F. Zhang and D. Zhao, Chem. Mater., 2013, 25, 3030–3037 CrossRef CAS.
- X. Liu, Q. Ren, F. Fu, R. Zou, Q. Wang, G. Xin, Z. Xiao, X. Huang, Q. Liu and J. Hu, Dalton Trans., 2015, 44, 10343–10351 RSC.
- J. Liu, C. Wang, X. Wang, X. Wang, L. Cheng, Y. Li and Z. Liu, Adv. Funct. Mater., 2015, 25, 384–392 CrossRef CAS.
- S. He, K. Krippes, S. Ritz, Z. Chen, A. Best, H.-J. Butt, V. Mailänder and S. Wu, Chem. Commun., 2015, 51, 431–434 RSC.
- Q. Zhao, J. Liu, W. Zhu, C. Sun, D. Di, Y. Zhang, P. Wang, Z. Wang and S. Wang, Acta Biomater., 2015, 23, 147–156 CrossRef CAS PubMed.
- Y. Zhang, C. Y. Ang, M. Li, S. Y. Tan, Q. Qu, Z. Luo and Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 18179–18187 CAS.
- S. F. Lee, X. M. Zhu, Y. X. Wang, S. H. Xuan, Q. You, W. H. Chan, C. H. Wong, F. Wang, J. C. Yu, C. H. Cheng and K. C. Leung, ACS Appl. Mater. Interfaces, 2013, 5, 1566–1574 CAS.
- S. Angelos, Y. W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344–11346 CrossRef CAS PubMed.
- X. Chen, A. H. Soeriyadi, X. Lu, S. M. Sagnella, M. Kavallaris and J. J. Gooding, Adv. Funct. Mater., 2014, 24, 6999–7006 CrossRef CAS.
- Q. Zhao, J. Liu, W. Zhu, C. Sun, D. Di, Y. Zhang, P. Wang, Z. Wang and S. Wang, Acta Biomater., 2015, 23, 147–156 CrossRef CAS PubMed.
- R. Lv, P. Yang, F. He, S. Gai, G. Yang, Y. Dai, Z. Hou and J. Lin, Biomaterials, 2015, 63, 115–127 CrossRef CAS PubMed.
- L. Han, C. Tang and C. Yin, Biomaterials, 2015, 60, 42–52 CrossRef CAS PubMed.
- I. R. Fernando, D. P. Ferris, M. Frasconi, D. Malin, E. Strekalova, M. D. Yilmaz, M. W. Ambrogio, M. M. Algaradah, M. P. Hong, X. Chen, M. S. Nassar, Y. Y. Botros, V. L. Cryns and J. F. Stoddart, Nanoscale, 2015, 7, 7178–7183 RSC.
- S. Zhou, X. Du, F. Cui and X. Zhang, Small, 2014, 10, 980–988 CrossRef CAS PubMed.
- D. Xiao, H. Z. Jia, J. Zhang, C. W. Liu, R. X. Zhuo and X. Z. Zhang, Small, 2014, 10, 591–598 CrossRef CAS PubMed.
- W. P. Li, P. Y. Liao, C. H. Su and C. S. Yeh, J. Am. Chem. Soc., 2014, 136, 10062–10075 CrossRef CAS PubMed.
- M. Yu, S. Jambhrunkar, P. Thorn, J. Chen, W. Gu and C. Yu, Nanoscale, 2013, 5, 178–183 RSC.
- J. Pang, L. Zhao, L. Zhang, Z. Li and Y. Luan, J. Colloid Interface Sci., 2013, 395, 31–39 CrossRef CAS PubMed.
- L. Dai, Q. Zhang, J. Li, X. Shen, C. Mu and K. Cai, ACS Appl. Mater. Interfaces, 2015, 7, 7357–7372 CAS.
- X. Ma, Y. Zhao, K. W. Ng and Y. Zhao, Chemistry, 2013, 19, 15593–15603 CrossRef CAS PubMed.
- X. Xie, F. Li, H. Zhang, Y. Lu, S. Lian, H. Lin, Y. Gao and L. Jia, Eur. J. Pharm. Sci., 2016, 83, 28–35 CrossRef CAS PubMed.
- Y. Yang, F. Chen, S. Shi, S. Graves, R. Nickles and W. Cai, J. Nucl. Med., 2015, 56, 117 Search PubMed.
- Y. Zhang, J. Guo, X. L. Zhang, D. P. Li, T. T. Zhang, F. F. Gao, N. F. Liu and X. G. Sheng, Int. J. Pharm., 2015, 496, 1026–1033 CrossRef CAS PubMed.
- Y. T. Liao, C. H. Liu, J. Yu and K. C. Wu, Int. J. Nanomed., 2014, 9, 2767–2778 Search PubMed.
- G. Quan, X. Pan, Z. Wang, Q. Wu, G. Li, L. Dian, B. Chen and C. Wu, J. Nanobiotechnol., 2015, 13, 7 CrossRef PubMed.
- P. Zhang, F. Cheng, R. Zhou, J. Cao, J. Li, C. Burda, Q. Min and J. J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 2371–2375 CrossRef CAS PubMed.
- F.-F. Zheng, P.-H. Zhang, Y. Xi, J.-J. Chen, L.-L. Li and J.-J. Zhu, Anal. Chem., 2015, 87, 11739–11745 CrossRef CAS PubMed.
- C. Liu, J. Zheng, L. Deng, C. Ma, J. Li, Y. Li, S. Yang, J. Yang, J. Wang and R. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 11930–11938 CAS.
- W. Fang, Z. Wang, S. Zong, H. Chen, D. Zhu, Y. Zhong and Y. Cui, Biosens. Bioelectron., 2014, 57, 10–15 CrossRef CAS PubMed.
- J. Zhang, Z. F. Yuan, Y. Wang, W. H. Chen, G. F. Luo, S. X. Cheng, R. X. Zhuo and X. Z. Zhang, J. Am. Chem. Soc., 2013, 135, 5068–5073 CrossRef CAS PubMed.
- J. Mo, L. He, B. Ma and T. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 6811–6825 CAS.
- X. Wu, Z. Han, R. Schur and Z. R. Lu, Biogeochemistry, 2016, 16, 1–42 Search PubMed.
- X. Li, M. Wu, L. Pan and J. Shi, Int. J. Nanomed., 2016, 11, 93 CAS.
- R. Chakravarty, S. Goel, H. Hong, F. Chen, H. F. Valdovinos, R. Hernandez, T. E. Barnhart and W. Cai, Nanomedicine, 2015, 10, 1233–1246 CrossRef CAS PubMed.
- G.-F. Luo, W.-H. Chen, Y. Liu, J. Zhang, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, J. Mater. Chem. B, 2013, 1, 5723–5732 RSC.
- J. J. Hu, L. H. Liu, Z. Y. Li, R. X. Zhuo and X. Z. Zhang, J. Mater. Chem. B, 2016, 4, 1932–1940 RSC.
- K. Wang, H. Yao, Y. Meng, Y. Wang, X. Yan and R. Huang, Acta Biomater., 2015, 16, 196–205 CrossRef CAS PubMed.
- J. Ding, J. Yao, J. Xue, R. Li, B. Bao, L. Jiang, J. J. Zhu and Z. He, ACS Appl. Mater. Interfaces, 2015, 7, 18145–18155 CAS.
- Y. Chen, X. Wang, T. Liu, D. S. Zhang, Y. Wang, H. Gu and W. Di, Int. J. Nanomed., 2015, 10, 2579–2594 CAS.
- S. Goel, F. Chen, H. Hong, H. F. Valdovinos, R. Hernandez, S. Shi, T. E. Barnhart and W. Cai, ACS Appl. Mater. Interfaces, 2014, 6, 21677–21685 CAS.
- F. Chen, H. Hong, Y. Zhang, H. F. Valdovinos, S. Shi, G. S. Kwon, C. P. Theuer, T. E. Barnhart and W. Cai, ACS Nano, 2013, 7, 9027–9039 CrossRef CAS PubMed.
- H. Yang, F. Zhao, Y. Li, M. Xu, L. Li, C. Wu, H. Miyoshi and Y. Liu, Int. J. Nanomed., 2013, 8, 1897–1906 Search PubMed.
- L. Pan, J. Liu, Q. He, L. Wang and J. Shi, Biomaterials, 2013, 34, 2719–2730 CrossRef CAS PubMed.
- M. Wu, Q. Meng, Y. Chen, Y. Du, L. Zhang, Y. Li, L. Zhang and J. Shi, Adv. Mater., 2015, 27, 215–222 CrossRef CAS PubMed.
- Z. Y. Li, Y. Liu, J. J. Hu, Q. Xu, L. H. Liu, H. Z. Jia, W. H. Chen, Q. Lei, L. Rong and X. Z. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 14568–14575 CAS.
- L. Xiong, X. Du, F. Kleitz and S. Z. Qiao, Small, 2015, 11, 5919–5926 CrossRef CAS PubMed.
- Y. Liu, Q. Chen, M. Xu, G. Guan, W. Hu, Y. Liang, X. Zhao, M. Qiao, D. Chen and H. Liu, Int. J. Nanomed., 2015, 10, 1855 CrossRef CAS PubMed.
- L. Pan, J. Liu, Q. He and J. Shi, Adv. Mater., 2014, 26, 6742–6748 CrossRef CAS PubMed.
- Z. Li, K. Dong, S. Huang, E. Ju, Z. Liu, M. Yin, J. Ren and X. Qu, Adv. Funct. Mater., 2014, 24, 3612–3620 CrossRef CAS.
- D. Neri and R. Bicknell, Nat. Rev. Cancer, 2005, 5, 436–446 CrossRef CAS PubMed.
- M. Srinivasarao, C. V. Galliford and P. S. Low, Nat. Rev. Drug Discovery, 2015, 14, 203–219 CrossRef CAS PubMed.
- C. P. Tsai, C. Y. Chen, Y. Hung, F. H. Chang and C. Y. Mou, J. Mater. Chem., 2009, 19, 5737–5743 RSC.
- K. Laginha, D. Mumbengegwi and T. Allen, Biochim. Biophys. Acta, Biomembr., 2005, 1711, 25–32 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- Q. He and J. Shi, Adv. Mater., 2014, 26, 391–411 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |