
Early diagnosis of Alzheimer's disease using infrared spectroscopy of isolated blood samples followed by multivariate analyses†
S.
Mordechai
*a,
E.
Shufan
b,
B. S.
Porat Katz
c and
A.
Salman
*b
aDepartment of Physics, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel. E-mail: shaulm@bgu.ac.il; Fax: +972-8-6472903; Tel: +972-8-6461749
bDepartment of Physics, SCE – Shamoon College of Engineering, Beer-Sheva 84100, Israel. E-mail: ahmad@sce.ac.il; Fax: +972-8-8519161; Tel: +972-8-6475794
cThe Hebrew University of Jerusalem, the Robert H. Smith Faculty of Agriculture, Food and Environment School of Nutritional Sciences, Rehovot, Israel
First published on 9th November 2016
Abstract
Alzheimer's disease (AD) is the most common cause of dementia, particularly in the elderly. The disease is characterized by cognitive decline that typically starts with insidious memory loss and progresses relentlessly to produce global impairment of all higher cortical functions. Due to better living conditions and health facilities in developed countries, which result in higher overall life spans, these countries report upward trends of AD among their populations. There are, however, no specific diagnostic tests for AD and clinical diagnosis is especially difficult in the earliest stages of the disease. Early diagnosis of AD is frequently subjective and is determined by physicians (generally neurologists, geriatricians, and psychiatrists) depending on their experience. Diagnosing AD requires both medical history and mental status testing. Having trouble with memory does not mean you have AD. AD has no current cure, but treatments for symptoms are available and research continues. In this study, we investigated the potential of infrared microscopy to differentiate between AD patients and controls, using Fourier transform infrared (FTIR) spectroscopy of isolated blood components. FTIR is known as a quick, safe, and minimally invasive method to investigate biological samples. For this goal, we measured infrared spectra from white blood cells (WBCs) and plasma taken from AD patients and controls, with the consent of the patients or their guardians. Applying multivariate analysis, principal component analysis (PCA) followed by linear discriminant analysis (LDA), it was possible to differentiate among the different types of mild, moderate, and severe AD, and the controls, with 85% accuracy when using the WBC spectra and about 77% when using the plasma spectra. When only the moderate and severe stages were included, an 83% accuracy was obtained using the WBC spectra and about 89% when using the plasma spectra.
1. Introduction
“Dementia” is commonly used to refer to significant cognitive disorders, with Alzheimer's disease (AD) being the most common cause.1 Approximately 10% of the population over the age of 65 is affected by this form of progressive dementia. AD is one of the 70 known types of dementia commonly found in the elderly population.2 In the US, AD is considered as the sixth leading cause of death;3 there are 5.3 million people suffering from AD,4 and by 2025 this figure is expected to increase by more than 40%.1,5 Although other major causes of death have been on the decrease, deaths due to AD have been rising dramatically. Between 2000 and 2008 deaths because of AD increased by 66%.6As such, AD represents a major medical challenge. The annual cost of this disease in 2015 was $226 billion,1 which means that nearly one in five Medicare dollars is spent on dementia. By 2050, this cost will increase to one in three.3 AD incidence is forecast to grow by 62% between 2015 and 2030 to around 1 billion worldwide.7
AD results in severe loss of memory and progressive deterioration of intellectual and emotional functions.8 The causes of AD are multifactorial in nature. The probable causes of AD are due to the formation of senile plaques, hyperphosphorylated tau proteins in neurofibrillary tangles (NFTs), mitochondrial dysfunction, and environmental toxins.9
There is no single clinical method available to diagnose AD unambiguously. There are no motor, sensory, or coordination deficits early in the disease. The diagnosis cannot be determined by validated clinical tests. Only 45% of people with AD, a figure that includes their caregivers, report being told of their diagnosis. AD can be diagnosed with complete accuracy only after death.10–13 Presently, cognitive tests, such as the mini-mental state examination (MMSE) and medical imaging, such as positron emission tomography (PET) and magnetic resonance imaging (MRI), computerized tomography (CT),10 electroencephalogram (EEG)12 and cerebrospinal fluid (CSF)10 techniques are combined to give a probable diagnosis of AD.14
New techniques were developed like single photon-emission computed tomography (SPECT),12 and FDG PET.15 These tests may be repeated to give doctors information about how the person's memory and other cognitive functions are changing over time.
Several cerebrospinal fluid (CSF) biomarkers like tau, phosphorylated tau and the 24 amino acid forms of amyloid-β have been developed for the diagnosis of AD.16–18 Different spectral methods were used to analyze CSF biomarkers like ELISA,19 mass spectrometry, Raman,20–23 and the two-photon Rayleigh scattering properties of gold nanoparticle24 polymerase-chain-reaction.25
Brain tissue samples were used in many studies to diagnose AD like Raman spectroscopy22,23, proton magnetic resonance (MR) spectroscopy,26 and FTIR microscopy.27 A new method using near-infrared fluorescent probes for the imaging purpose of amyloid plaques in AD patients’ brains28 was also studied.
Recently a very interesting technique was developed.29,30 This technique relies on extracting a small amount of the amyloid beta peptides and analyzing their secondary structure. Due to AD, the secondary structure of the abeta peptide starts to change to a beta-sheet reflecting the spectral changes in the secondary structure of proteins, which could be used for diagnosis at early stages.
Even though the underlying AD process cannot be stopped or reversed,11 early and accurate diagnosis is beneficial for several reasons. A multidisciplinary research yielded detailed knowledge of the molecular pathogenesis of AD16 resulting in new therapeutic strategies with putative disease-modifying effects.16 These treatments might be most effective when starting at the beginning of AD. Moreover, early treatments may help preserve daily functioning for some time. The available treatments can improve the quality of a patient's life by slowing the worsening of dementia symptoms. Our previous studies have shown that IR spectroscopy can detect the early development of diseases or cell transformations at a stage when standard microscopic evaluation still produces normal results.31,32
FTIR based studies for medical purposes have been reported for different diseases, including cancer and certain infectious diseases.31,33–44 By employing FTIR spectroscopy, it is possible to acquire mid-infrared absorption spectra, which are considered as fingerprints of the samples, enabling both qualitative and quantitative analysis of the measured biological samples. Infrared spectroscopy is rapid, cheap, and label free and yields objective analysis as a clinical tool. The development of advanced instrumentation in the field of infrared spectroscopy besides the development of new bioinformatics methods for spectral analysis in the past twenty years allows the examination of a large number of samples with different diseases using biological fluids.45 Analysis of biofluids like urine, serum,46 plasma etc. has become a diagnostic tool for future healthcare47via spectral marker signatures. Biofluid analyses are considered to be cheap and minimally invasive and can be used routinely in clinics as it is familiar to patients, and also because it is a low risk method.47
The combination of FTIR microscopy with bioinformatics analysis has become a powerful new technique with the ability to detect protein secondary structural changes associated with AD.45
Using FTIR spectroscopy to gain an understanding of cell and plasma molecular changes due to AD may lead to early diagnosis and effective treatment. AD may cause the secretion of amyloid beta20,48 peptides into the blood stream, as well as changes in the structure of white blood cells (WBCs). According to blood tests, lymphocytes were found to be consistently low for AD patients.49 Large numbers of senile plaques and neurofibrillary tangles are characteristic features of AD. Abnormal neurites in senile plaques are composed primarily of paired helical filaments, a component of neurofibrillary tangles. In AD they are primarily composed of amyloid beta peptides.50
The initiation of AD starts with different changes. Part of these changes are known (amyloid beta peptides) but the majority are not known yet.51 Many studies reported changes that occur in the lymphocytes in AD patients51–53 and in another study they proceeded and suggested it to be a biomarker for AD diagnosis.54
While the use of vibrational spectroscopy with plasma to diagnose AD was reported in several studies,55,56 to our knowledge this is the first time that the potential of infrared spectroscopy for AD diagnosis based on peripheral blood WBCs has been studied.
It is important to mention that although the changes due to AD are minute our experience showed that using multivariate analysis it is possible to reveal these changes for classification purposes.
We used PCA followed by LDA, on the mid-infrared absorption spectra measured from WBCs, and plasma classification and differentiation procedures.
The main goal of this preliminary study is to examine the potential of FTIR microscopy as a spectroscopic technique for early diagnoses of AD patients regardless of the stage, by analyzing blood components isolated from patients suffering due to neurodegenerative disorders.
2. Materials and method
2.1 Preparation of samples
Blood samples from AD patients were retrieved with the patients’ (or patients’ guardian) consent and the approval of the Institutional Review Helsinki Board. The blood samples were processed within 2 h of collection, following the method of Hudson and Hay, to isolate the various components of the blood such as serum and WBCs.57 Briefly, 3 ml of blood was collected from each patient to be separated into WBCs and serum. Three ml of Histopaque solution (Sigma Chemical Co., St Louis, MO) was added to the blood tube and centrifuged at 300g for 30 min at 23 °C. The lymphocyte layer, located at the middle of the tube, was isolated and the lymphocytes were washed again with 10 ml of PBS by centrifugation at 300g for 10 min at 23 °C. Thereafter, 1 μl of plasma was mounted on a ZnSe crystal and air-dried for 0.25 h under laminar flow. One μl of the cleaned lymphocytes was also mounted on a zinc selenide crystal to create a uniform layer, and then air-dried. The samples mounted on the zinc selenide crystals were used for FTIR measurements.2.2 FTIR measurements and spectral manipulation
All the measurements were performed using an Equinox 55 infrared spectrometer (BRUKER Germany) in the transmission mode, with 4 cm−1 spectral resolution. An MCT liquid nitrogen detector was used. The spectrometer was attached to a microscope that enabled inspection of the sample and had the ability to choose carefully the measured sites on the sample. All the spectra were measured in the 600–4000 cm−1 wavenumber range. Each spectrum is an average of 128 scans and the measurement time was about one minute for each spectrum. We set the circular diameter of the measured spot at 100 μm.We manipulated the spectra using OPUS 7 software (BRUKER Germany). All the spectra were smoothed using the Savitzky–Golay algorithm with 13 points; then the spectra were bisected into two regions, one with a wavenumber range of 600–1800 cm−1, the other with a wavenumber range of 2800–3010 cm−1. We made baseline corrections for each region separately, using the “concave rubberband” method, with 64 sections and 5 iterations in the first, 600–1800 cm−1 region, and 10 sections with two iterations in the second, 2800–3010 cm−1 region.
Using the concave rubberband method, a polynomial fit is generated based on the minima derived from each section. This polynomial fit is then subtracted from the spectra to obtain the baseline corrected spectra. This procedure is repeated as many times as the number of iterations.
Each region was normalized using the vector normalization method and then underwent offset correction by shifting the minimal intensities to zero.
Using vector normalization, the average intensity of the spectrum is calculated and subtracted from the spectrum, and then the acquired spectrum is normalized to 1.
2.3 Statistical analysis
The main goal was to analyze the infrared spectra of the measured cells to determine the cell type. The sample type belongs to one of the two categories—the control, and AD. The AD category was designed in three different manners; AD012 includes mild, moderate, and severe stages; AD12 includes moderate and severe stages; and AD2 includes the severe stage only. Each sample was characterized by two blood components: WBCs and serum. The details of patients and measurements included in this study are summarized in Table 1.| Lymphocytes | Plasma | |||
|---|---|---|---|---|
| Patients number | No. of measurements | Patients number | No. of measurements | |
| Controls | 26 | 145 | 23 | 111 |
| AD0/mild stage | 4 | 23 | 4 | 20 |
| AD1/moderate stage | 4 | 23 | 3 | 14 |
| AD2/severe stage | 12 | 75 | 11 | 53 |
| AD012/all stages | 20 | 121 | 18 | 87 |
| AD12/mild & moderate stages | 16 | 98 | 14 | 67 |
3. Results
Fig. 1 shows the average infrared absorption spectra for AD patients (severe) and the controls, in the 900–1800 cm−1 range, for both (a) WBCs and (b) plasma. The centroids of the major bands are displayed in the figure. The bands centered at 1741 cm−1 are contributed due to phospholipids.62 Amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and amide II (C–N stretch and H–N–C bend) bands are contributed mainly by protein vibrations.63–66 Lipids are the main contributors to the band centered at 1456 cm−1, due to antisymmetric vibrations of CH3. The band centered at 1395 cm−1 is related mainly due to the COO− symmetric stretching of amino acids and the symmetric bending mode of the methyl group (CH3) in proteins. Proteins contribute to the amide III band at 1252 cm−1.63–66
O) and amide II (C–N stretch and H–N–C bend) bands are contributed mainly by protein vibrations.63–66 Lipids are the main contributors to the band centered at 1456 cm−1, due to antisymmetric vibrations of CH3. The band centered at 1395 cm−1 is related mainly due to the COO− symmetric stretching of amino acids and the symmetric bending mode of the methyl group (CH3) in proteins. Proteins contribute to the amide III band at 1252 cm−1.63–66
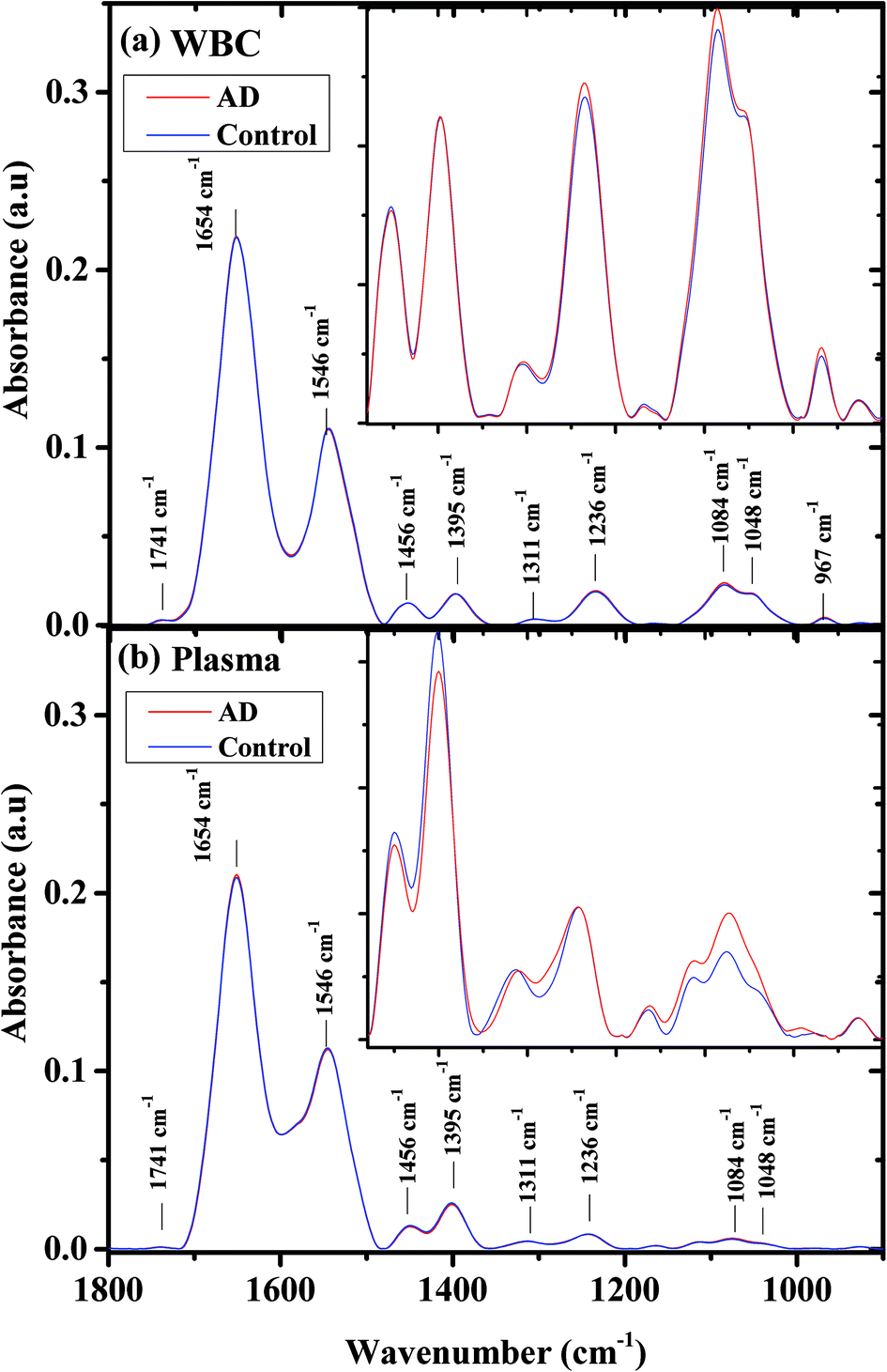 | ||
| Fig. 1 AD and controls’ average infrared absorption spectra in the 900–1800 cm−1 region; WBCs (a) and plasma (b). Each spectrum is an average of measurements of the same type for all the patients. The expanded region where the differences between the categories are large is clearly displayed in the insets. | ||
The biological molecules, proteins, lipids, and phosphate compounds absorb in the 1185–1485 cm−1 range of the infrared absorption spectrum, due to their CH2, CH3, and PO functional groups; while in the 900–1185 cm−1 range, the carbohydrates are the main contributors.
In the higher wavenumber region (2800–3010 cm−1) of the infrared absorption spectrum (not shown), the main contributors are lipids, due to their CH2 and CH3 symmetric and anti-symmetric vibrations.67 As can be seen from the figure, the spectra averages are very similar and the major differences are in the 900–1550 cm−1 wavenumber region. This region is displayed as an inset in Fig. 1. In the WBC spectra, the main spectral differences between controls and AD samples are in the PO2− symmetric (1084 cm−1) and antisymmetric vibrations (1236 cm−1). In the plasma spectra, the spectral differences between the controls and the AD patients are larger and exist in the PO2− symmetric (1084 cm−1) vibrations, COO− symmetric stretching of amino acids, and CH3 symmetric bending of the protein methyl group (1395 cm−1).
Second derivative spectra have some advantages because they can reveal hidden bands under a broadband peak in the original spectrum, and because they reduce the Mie scattering effect and many other effects.68
Fig. 2 shows the second derivative spectra for controls and AD samples (a) WBCs and (b) plasma in the 1480–1000 cm−1 wavenumber range. Also here the spectral differences between the control and AD patients are more significant in the plasma spectra.
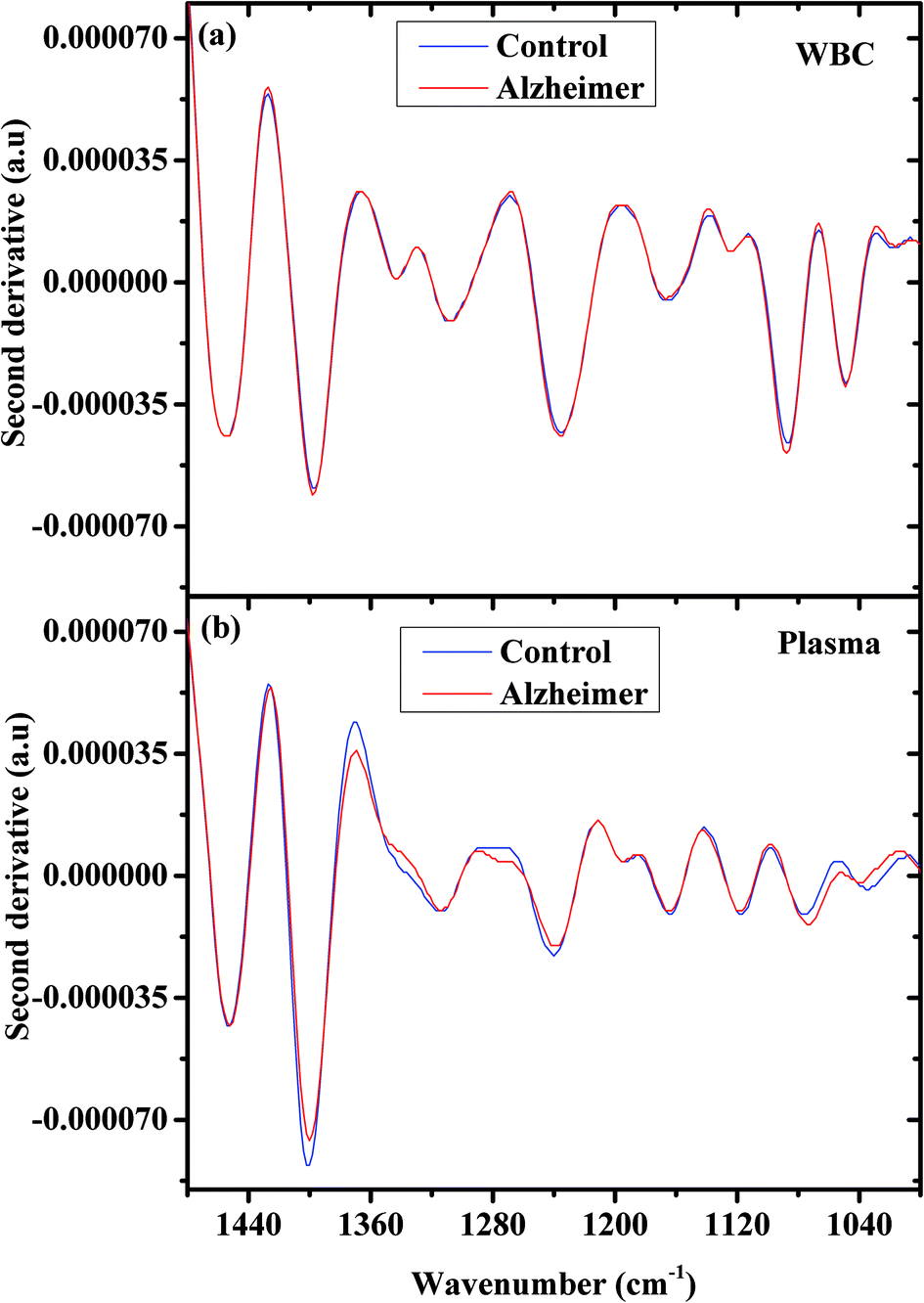 | ||
| Fig. 2 AD and controls’ second derivative of the average infrared absorption spectra in the 900–1480 cm−1 region; WBCs (a) and plasma (b). | ||
For cases where the changes between the biological samples are minute as in our study, special attention should be given to the quality and reproducibility of the measured spectra. ESI Fig. 1a† shows five spectra obtained from the same WBC sample at different sites. As can be seen from the figure the spectra almost lie on each other and the reproducibility of the spectra is good. A similar test of reproducibility was also performed for the plasma sample and the results are presented in ESI Fig. 1b.†
As can be seen from Fig. 2, the spectral differences between the different categories are minute, thus we applied multivariate analysis using PCA followed by LDA. We used the manipulated spectra and not the second derivative spectra for the PCA and LDA analysis.
We calculated the percentages of the classification success of the results as controls and AD, and as a function of the PC number, using three different experiments. In performing the calculations, we used the Fischer model (LDA) and LOO was used for validation. In experiment I the AD category was designed to include only the AD severe stage (AD2). In experiment II, the AD category was designed to include the AD severe and moderate stages (AD21). In experiment III the AD category was designed to include all three measured stages of AD—mild, moderate, and severe (AD210).
The percentage success rates were deduced for the three different experiments and plotted in Fig. 3(a) for plasma measurements and Fig. 3(b) for lymphocyte measurements.
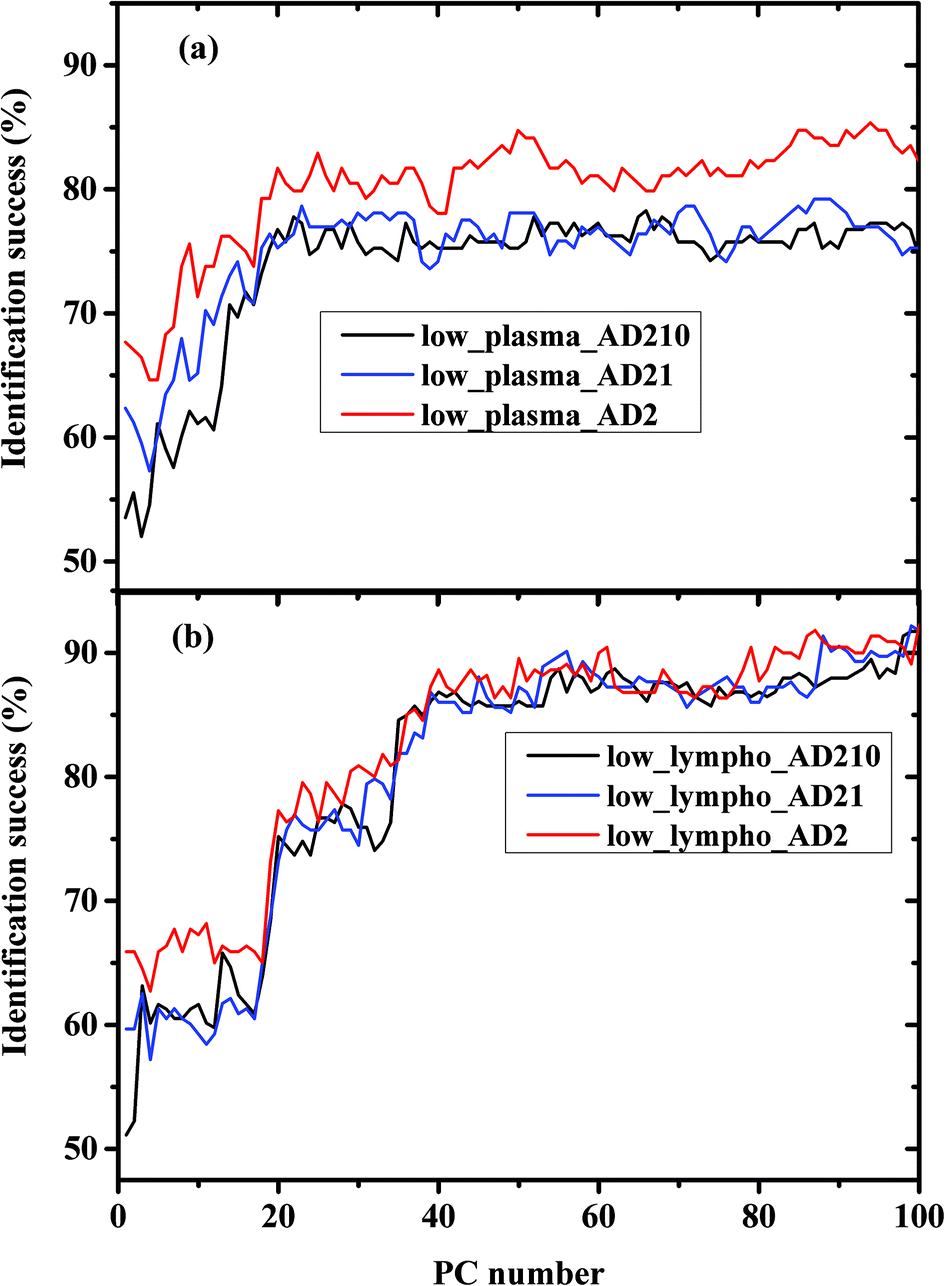 | ||
| Fig. 3 Classification results of control and AD categories based on LDA calculations using the LOO method in the lower 900–1800 cm−1 wavenumber region; (a) plasma and (b) lymphocytes. LDA classification rates versus PC numbers are displayed in the figure. The AD categories were designed to include different combinations of AD stages as shown in the figure. | ||
In order to compare between the lymphocytes and plasma measurements regarding the classification procedure, the percentages of classification successes using the same experiment were plotted in Fig. 4(a–c) for AD210, AD21, and AD2 categories, respectively.
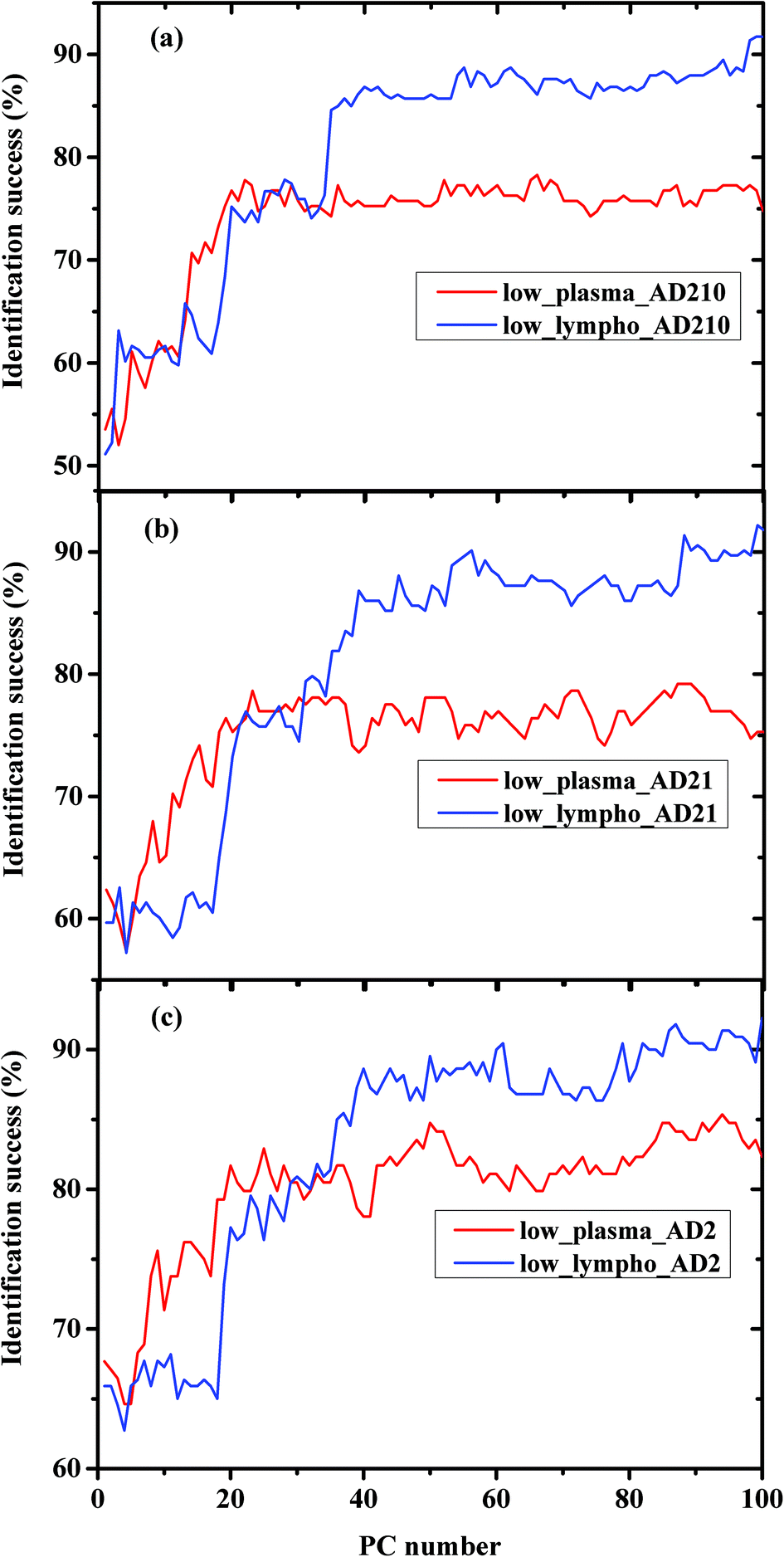 | ||
| Fig. 4 Comparison between the performance of LDA using plasma and WBC measurements with different combinations of AD patients. (a) The AD012 category included all stages of AD—mild, moderate, and severe. (b) The AD12 category included both AD moderate and severe stages. (c) The AD2 category included the AD severe stage only. | ||
Table 2 preliminarily summarizes the specificity (SP), the sensitivity (SE), and the accuracy of our method for classification of the measurements into two categories—controls and AD—using two blood components—plasma and lymphocytes. The results were deduced based on LDA calculations using 25 PCs for plasma and 36 PCs for lymphocytes.
| Used measurements | SE | SP | Acc |
|---|---|---|---|
| Lymphocyte (controls, AD = AD012) | 0.82 | 0.88 | 0.85 |
| Lymphocyte (controls, AD = AD12) | 0.76 | 0.86 | 0.81 |
| Lymphocyte (controls, AD = AD2) | 0.75 | 0.90 | 0.85 |
| Plasma (controls, AD = AD012) | 0.70 | 0.79 | 0.75 |
| Plasma (controls, AD = AD12) | 0.67 | 0.83 | 0.77 |
| Plasma (controls, AD = AD2) | 0.74 | 0.87 | 0.83 |
The accuracy (Acc) column in the table measures the probability with which our method successfully identifies and locates the sample in its correct group. The ability of the method to identify the AD measurements as being in the AD category is defined as sensitivity (SE), while the specificity (SP) corresponds to correctly classifying control measurements as the control.
A similar analysis was performed from the higher 2800–3100 cm−1 wavenumber region; however, the results were not as good as for the lower region. When the two regions were combined, the performance of the method was also less successful than when using only the lower region. Thus, the high wavenumber region was excluded from the analysis.
We tried to classify between the controls and AD categories' samples in the three different AD210, AD21, and AD2 experiments, using the 900–1480 cm−1 range, where major spectral differences occur, as shown in Fig. 1.
Using this range, the performance of LDA was slightly lower using the lymphocyte measurements, while using the plasma measurements, the performance was improved using higher PCs (Fig. 5).
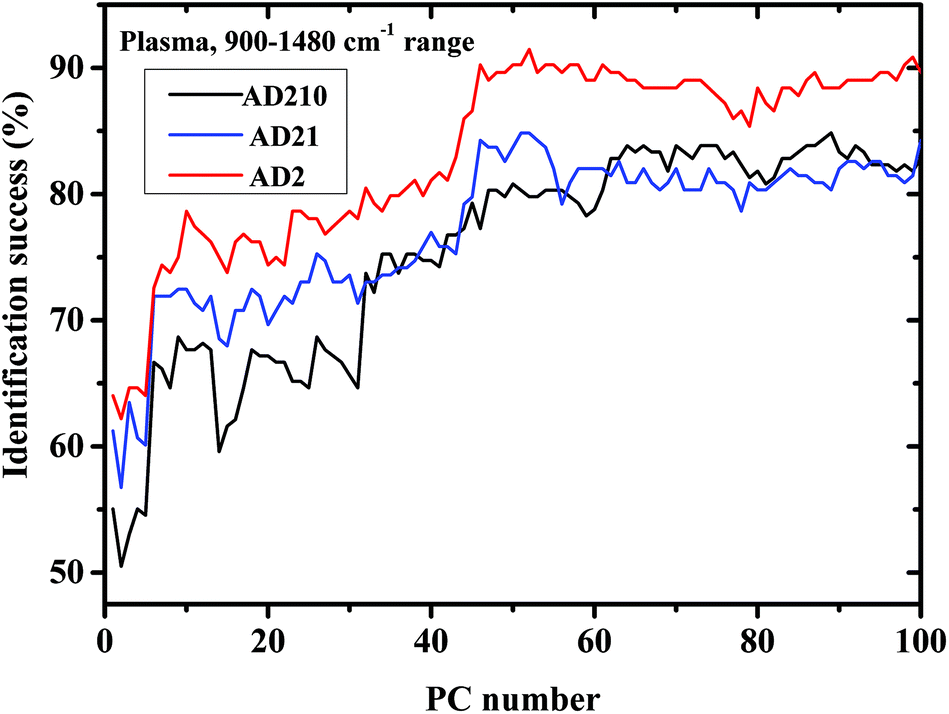 | ||
| Fig. 5 Classification results of controls and AD categories based on LDA calculations using the LOO method in the 900–1480 cm−1 region, using plasma measurements. | ||
Table 3 summarizes the Acc, SE, and SP of the classification results derived by LDA using the plasma measurements in the 900–1480 cm−1 range.
| Used measurements | SE | SP | Acc |
|---|---|---|---|
| Plasma (controls, AD = AD012) | 0.72 | 0.81 | 0.77 |
| Plasma (controls, AD = AD12) | 0.78 | 0.88 | 0.89 |
| Plasma (controls, AD = AD2) | 0.83 | 0.94 | 0.90 |
4. Discussion
Currently there is no validated clinical test that can diagnose AD. Thus there is a clear need to develop a simple, inexpensive, and accurate method that can be objectively used to determine the presence of AD. Such a test may inform patients, their caregivers, and their physicians about their AD status.Infrared microscopy is known for its simplicity and sensitivity to molecular and chemical changes in the blood components due to the secretion of amyloid beta peptides into the blood stream, as well as changes in the WBC structure (such as protein conformation29,30), due to AD. Thus, infrared microscopy may suggest a new method for diagnosing AD, thereby providing a second opinion to physicians seeking to support their current cognitive MMSE tests in order to enhance the probable diagnosis of AD. Today, PET and MRI techniques are used for this purpose,14 but these methods are expensive and not always available. Although the CSF biomarkers are used increasingly, they have drawbacks since they are not standardized at the level of sample handling, concentration and well defined criteria.69 Different neuroimaging markers, genetic and biochemical markers have been developed which may highlight the changes that happen while developing AD. However, their pathognomonic and diagnostic values have not yet been proven significantly. Moreover, it is difficult to perform many of these techniques that are used and the equipment/reagents are expensive or potentially hazardous (CSF extraction).70 Thus there is a clear need to develop a new objective, reliable and valid method for the clinical screening and follow-up of a large number of patients that can be repeated routinely.70 The most promising biomarker is the peripheral blood.71
Our method is very simple and relies on a simple blood test (2–4cc), which is a commonly used routine in each clinic. There is no need either for complex and expensive instrumentation or for a trained technician. Moreover, it has minimal risk compared with CSF and brain tissues and it is fast and objective. All these advantages make it suitable for routine clinic tests.
As can be seen from Fig. 1, the infrared absorption spectra for AD and controls are similar in their WBCs and plasma. However, small changes can be observed if we expand the 900–1500 cm−1 region (inset).
The classification results obtained in Fig. 4 and 5 are very interesting. In Fig. 4 we compared between the same kind of measurements (a) plasma and (b) lymphocytes, while the AD category was designed to include different AD stages. As shown in the figures, the classification success reaches a plateau after a certain PC number—36 PCs for lymphocytes and 25 PCs for plasma. The LDA is a supervised method, thus it should be trained before validation. The training results are based on the prognosis of the physician (which serves as the gold standard), while in general the confidence of the physician's prognosis does not exceed 80%. This can help explain the classification errors without regard to other error factors such as statistics and the process of preparing the samples and sample purification.
Using PCA, the calculated loadings are arranged in descending order based on their variance, which is calculated as the ratio between certain PC eigenvalues and the sum of the eigenvalues of all the loadings. Thus, PC1 has the largest variance, followed by PC2, and so on. Thus, it may be suggested that the spectral differences based on PC1 are the largest, followed by differences based on the first and second PCs, and so on. As can be seen from Fig. 5, the classification results of plasma are better than those of lymphocytes almost up to PC25, beyond this PC number there is a crossover in the results, with the lymphocytes yielding better success rates than plasma. Thus, it may be suggested that the differences in the plasma samples are larger than those in the lymphocytes72 for the three experiments of AD categories using different combinations of AD stages (mild, moderate, and severe). Using a greater number of PCs (>25), the classification results based on the lymphocyte measurements are superior. Using the first 36 PCs and the lymphocyte measurements, the classification rate was about 85% when the AD category included the severe stage, 81.9% when the AD category included both severe and moderate stages, and 85% when the AD category included all three AD stages—mild, moderate, and severe. The heterogeneity among the AD categories when they included all three stages of AD was larger compared to the same categories when they included severe stages only, even though the classification rate is almost the same. This may be explained due to the statistics of the LDA method. When the category of AD included all three stages, 20 patients were included in the category (132 measurements), while only 12 patients were diagnosed as being at the severe stage.
Using plasma measurements, the classification results of the severe stage patients were found to be the highest in the plateau region. When the lymphocyte measurements, rather than plasma measurements, were taken from patients with different stages of AD categories, the classification rates were almost the same in the plateau region. This may be explained due to the dependence of the classification process on the spectral changes and statistics. In the plasma measurements, the spectral changes were larger and dominant relative to the influence of statistics; thus the classification rate was higher when severe stages were used in the AD category (12 patients), although the statistics are higher in the AD category when using all three stages.
In Fig. 3a and Fig. 5 it can be seen that the identification success was indeed higher when only the severe cases were included in the AD category (labeled AD2). When the AD category included mild, moderate and severe cases (labeled AD21 & AD210) the classification success was lower as expected. This is true although AD21 and AD210 are larger groups than AD2. This emphasizes the fact that the largest differences exist between control and severe AD.
It is interesting to speculate why the crossover of the classification success rates occurred when the lymphocyte measurements were used. We believe that this may be explained due to the nature of the samples. The lymphocytes are separated and washed, thus they are homogeneous, while the plasma is a complex fluid that contains different types and sizes of proteins and other biomolecules. Because the spectral changes are very small, the use of a large number of PCs will enhance the contribution of different molecules (even the molecules with a low concentration).
As can be seen from Table 2, the performance of the method using lymphocytes is better than the plasma. Employing controls and AD012 or AD2, the accuracy is 85% for both cases. The specificity using AD012 is higher, while the sensitivity is higher for the AD2 group.
Narrowing the measurements into the 900–1480 cm−1 range improves the performance of the method, as can be seen from Fig. 5 and Table 3. It is interesting to note that when the amide I and amide II regions were removed, it resulted in an overall improved performance. This may be explained due to the heterogeneity (complexity) of the plasma. Proteins that are the main contributors to the amide I and amide II bands may be the major causes for lymphocytes heterogeneity and by removing the amide region, we eliminate most of their contribution to the spectra, resulting in an improvement in the plasma results. The fact that the classification results using lymphocytes were not improved using the same procedure strengthens this hypothesis.
The small number of patients in our data set is a restriction in this study and in order to make our statistics more solid, a larger number of patients should be included,73,74 thus our findings will be more solid in this difficult and controversial field. Such an extensive study with a sufficient number of patients72 will strengthen our findings and make the statistical results unimpeachable.
In case the results for a patient using our method are positive, the patient should be referred to more sophisticated tests like the method developed by Gerwert's group29,30 in order to determine the AD stage. This method is more complicated and may be used as a second phase for patients that were diagnosed as having AD using our method.
In this study, we used the two components of blood, plasma and WBCs to diagnose AD. It is difficult, using this method, to point out which exact biological changes occur during the disease, but this does not deduce from the importance of the ability of classification of the samples into AD and not AD which is the main goal of this study.
5. Conclusions
FTIR microcopy combined with multivariate analysis of simple blood tests makes it possible to differentiate between AD and controls with reasonable success rates, in the time span of a few minutes.The spectral changes between controls and AD patients in the plasma measurements are larger than those observed for lymphocytes. It is therefore surprising that for classification purposes it is better to use the lymphocyte measurements.
Increasing the number of patients (which is time consuming and not an easy task) could improve the results and increase the identification success rates.
Acknowledgements
This research work was supported by the Israel Science Foundation (ISF) and the Israel Cancer Association (ICA). Many thanks are due to Keren Kanterowitch for her help in the data measurements.References
- M. D. Llorente, J. Nerv. Ment. Dis., 2015, 203, 978 CrossRef.
- M. A. Boss, Biochim. Biophys. Acta, 2000, 1502, 188–200 CrossRef CAS.
- http://www.alz.org/facts/overview.asp .
- A. Alzheimer's, Alzheimer's Dementia, 2015, 11, 332–384 CrossRef.
- D. P. Rice, H. M. Fillit, W. Max, D. S. Knopman, J. R. Lloyd and S. Duttagupta, Am. J. Manag. Care, 2001, 7, 809–818 CAS.
- 2013 Alzheimer's disease facts and figures, Alzheimers Dement., 2013, 9(2), 208–245 CrossRef PubMed.
- US Department of Commerce, Mid-year population by older age groups and sex – world, US Department of Commerce, 2015.
- P. H. Robert and I. Medecin, Ann. Med. Interne, 1998, 149, 216–220 CAS.
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741–766 CAS.
- http://www.mayoclinic.org/diseases-conditions/alzheimers-disease/diagnosis-treatment/diagnosis/dxc-20167109 .
- https://www.nia.nih.gov/alzheimers/topics/diagnosis .
- http://www.nhs.uk/Conditions/Alzheimers-disease/Pages/Diagnosis.aspx .
- https://www.ucsfhealth.org/conditions/alzheimers_disease/diagnosis.html .
- P. H. Scheltens, Aging, 2001, 13, 203–209 CAS.
- K. Herholz, Biomarkers Med., 2012, 6, 431–439 CrossRef CAS PubMed.
- K. Blennow, H. Hampel, M. Weiner and H. Zetterberg, Nat. Rev. Neurol., 2010, 6, 131–144 CrossRef CAS PubMed.
- M. Pitschke, R. Prior, M. Haupt and D. Riesner, Nat. Med., 1998, 4, 832–834 CrossRef CAS PubMed.
- U. Andreasson, E. Vanmechelen, L. M. Shaw, H. Zetterberg and H. Vanderstichele, Biomarkers Med., 2012, 6, 377–389 CrossRef CAS PubMed.
- H. Mori, K. Hosoda, E. Matsubara, T. Nakamoto, Y. Furiya, R. Endoh, M. Usami, M. Shoji, S. Maruyama and S. Hirai, Neurosci. Lett., 1995, 186, 181–183 CrossRef CAS PubMed.
- C. Humpel, Trends Biotechnol., 2011, 29, 26–32 CrossRef CAS PubMed.
- C. Mulder, N. A. Verwey, W. M. van der Flier, F. H. Bouwman, A. Kok, E. J. van Elk, P. Scheltens and M. A. Blankenstein, Clin. Chem., 2010, 56, 248–253 CAS.
- C. D. Sudworth and N. Krasner, Raman spectroscopy of Alzheimer's diseased tissue, 2004 Search PubMed.
- R. Michael, C. Otto, A. Lenferink, E. Gelpi, G. A. Montenegro, J. Rosandić, F. Tresserra, R. I. Barraquer and G. F. J. M. Vrensen, Exp. Eye Res., 2014, 119, 44–53 CrossRef CAS PubMed.
- A. Neely, C. Perry, B. Varisli, A. K. Singh, T. Arbneshi, D. Senapati, J. R. Kalluri and P. C. Ray, ACS Nano, 2009, 3, 2834–2840 CrossRef CAS PubMed.
- E. Majounie, Y. Abramzon, A. E. Renton, R. Perry, S. S. Bassett, O. Pletnikova, J. C. Troncoso, J. Hardy, A. B. Singleton and B. J. Traynor, N. Engl. J. Med., 2012, 366, 283–284 CrossRef CAS PubMed.
- T. K. Shonk, R. A. Moats, P. Gifford, T. Michaelis, J. C. Mandigo, J. Izumi and B. D. Ross, Radiology, 1995, 195, 65–72 CrossRef CAS PubMed.
- N. Benseny-Cases, O. Klementieva, M. Cotte, I. Ferrer and J. Cladera, Anal. Chem., 2014, 86, 12047–12054 CrossRef CAS PubMed.
- H. Tong, K. Lou and W. Wang, Acta Pharm. Sin. B, 2015, 5, 25–33 CrossRef PubMed.
- A. Nabers, J. Ollesch, J. Schartner, C. Kötting, J. Genius, U. Haußmann, H. Klafki, J. Wiltfang and K. Gerwert, J. Biophotonics, 2016, 9, 224–234 CrossRef CAS PubMed.
- A. Nabers, J. Ollesch, J. Schartner, C. Kötting, J. Genius, H. Hafermann, H. Klafki, K. Gerwert and J. Wiltfang, Anal. Chem., 2016, 88, 2755–2762 CrossRef CAS PubMed.
- A. Salman, V. Erukhimovitch, M. Talyshinsky, M. Huleihil and M. Huleihel, Biopolymers, 2002, 67, 406–412 CrossRef CAS PubMed.
- E. Bogomolny, S. Mordechai, A. Zwielly and M. Huleihel, Eur. Biophys. J., 2009, 38, 971–980 CrossRef CAS PubMed.
- H. Mantsch and D. Chapman, Infrared spectroscopy of biomolecules, Wiley-Liss, New York, 1996 Search PubMed.
- A. Zwielly, S. Mordechai, I. Sinielnikov, A. Salman, E. Bogomolny and S. Argov, Med. Phys., 2010, 37, 1047–1055 CrossRef CAS PubMed.
- M. M. Mariani, L. J. Maccoux, C. Matthaus, M. Diem, J. G. Hengstler and V. Deckert, Anal. Chem., 2010, 82, 4259–4263 CrossRef CAS PubMed.
- J. Sulé-Suso, D. Skingsley, G. D. Sockalingum, A. Kohler, G. Kegelaer, M. Manfait and A. J. El Haj, Vib. Spectrosc., 2005, 38, 179–184 CrossRef.
- S. Argov, J. Ramesh, A. Salman, I. Sinelnikov, J. Goldstein, H. Guterman and S. Mordechai, J. Biomed. Opt., 2002, 7, 248–254 CrossRef PubMed.
- V. R. Kondepati, H. M. Heise and J. Backhaus, Anal. Bioanal. Chem., 2007, 390, 125–139 CrossRef PubMed.
- M. J. Baker, E. Gazi, M. D. Brown, J. H. Shanks, P. Gardner and N. W. Clarke, Br. J. Cancer, 2008, 99, 1859–1866 CrossRef CAS PubMed.
- G. Bellisola and C. Sorio, Am. J. Cancer Res., 2012, 2, 1–21 CAS.
- P. D. Lewis, K. E. Lewis, R. Ghosal, S. Bayliss, A. J. Lloyd, J. Wills, R. Godfrey, P. Kloer and L. A. Mur, BMC Cancer, 2010, 10, 1–10 CrossRef PubMed.
- F. Severcan, N. Simsek Ozek and S. Gok, Biophys. J., 2015, 108, 479a–480a CrossRef.
- V. Erukhimovitch, M. Talyshinsky, Y. Souprun and M. Huleihel, in DNA Viruses: Methods and Protocols, ed. P. M. Lieberman, Humana Press, Totowa, NJ, 2005, pp. 161–172 Search PubMed.
- F. T. Lee-Montiel, K. A. Reynolds and M. R. Riley, J. Biol. Eng., 2011, 5, 16 CrossRef CAS PubMed.
- S. Caine, P. Heraud, M. J. Tobin, D. McNaughton and C. C. A. Bernard, NeuroImage, 2012, 59, 3624–3640 CrossRef PubMed.
- R. González-Domínguez, T. García-Barrera and J. L. Gómez-Ariza, J. Pharm. Biomed. Anal., 2015, 107, 75–81 CrossRef PubMed.
- M. J. Baker, S. R. Hussain, L. Lovergne, V. Untereiner, C. Hughes, R. A. Lukaszewski, G. Thiefin and G. D. Sockalingum, Chem. Soc. Rev., 2016, 45, 1803–1818 RSC.
- D. H. J. Lopes, A. Meister, A. Gohlke, A. Hauser, A. Blume and R. Winter, Biophys. J., 2007, 93, 3132–3141 CrossRef CAS PubMed.
- K. F. Shad, Y. Aghazadeh, S. Ahmad and B. Kress, Synapse, 2013, 67, 541–543 CrossRef CAS PubMed.
- A. Cedazo-Minguez and B. Winblad, Exp. Gerontol., 2010, 45, 5–14 CrossRef CAS PubMed.
- G. S. Leandro, R. R. Lobo, D. V. Oliveira, J. C. Moriguti and E. T. Sakamoto-Hojo, Int. J. Mol. Sci., 2013, 14, 12380–12400 CrossRef PubMed.
- E. Richartz-Salzburger, A. Batra, E. Stransky, C. Laske, N. Köhler, M. Bartels, G. Buchkremer and K. Schott, J. Psychiatr. Res., 2007, 41, 174–178 CrossRef PubMed.
- S. E. Marsh, E. M. Abud, A. Lakatos, A. Karimzadeh, S. T. Yeung, H. Davtyan, G. M. Fote, L. Lau, J. G. Weinger, T. E. Lane, M. A. Inlay, W. W. Poon and M. Blurton-Jones, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1316–E1325 CrossRef CAS PubMed.
- U. Wojda, Biomarkers Med., 2015, 10, 1–4 CrossRef PubMed.
- P. Carmona, M. Molina, E. Lopez-Tobar and A. Toledano, Anal. Bioanal. Chem., 2015, 407, 7747–7756 CrossRef CAS PubMed.
- P. Carmona, M. Molina, M. Calero, F. Bermejo-Pareja, P. Martínez-Martín and A. Toledano, J. Alzheimer's Dis., 2013, 34, 911–920 CAS.
- L. Hudson and F. C. Hay, Practical immunology, Blackwell Scientific, Oxford, 1976 Search PubMed.
- C. M. Bishop, Pattern recognition and machine learning, Springer, New York, 2006 Search PubMed.
- R. O. Duda, P. E. Hart and D. G. Stork, Pattern classification, Wiley, 2001 Search PubMed.
- A. Salman, I. Lapidot, A. Pomerantz, L. Tsror, E. Shufan, R. Moreh, S. Mordechai and M. Huleihel, J. Biomed. Opt., 2012, 17, 017002 CrossRef PubMed.
- A. Salman, E. Shufan, L. Zeiri and M. Huleihel, Biochim. Biophys. Acta, 2013, 1830, 2720–2727 CrossRef CAS.
- R. K. Dukor, in Handbook of Vibrational Spectroscopy, John Wiley & Sons, Ltd, 2006 Search PubMed.
- G. Kos, R. Krska, H. Lohninger and P. R. Griffiths, Anal. Bioanal. Chem., 2004, 378, 159–166 CrossRef CAS PubMed.
- N. Stone, C. Kendall, J. Smith, P. Crow and H. Barr, Faraday Discuss., 2004, 126, 141–157 RSC ; discussion 169–183.
- E. O. Faolain, M. B. Hunter, J. M. Byrne, P. Kelehan, H. A. Lambkin, H. J. Byrne and F. M. Lyng, J. Histochem. Cytochem., 2005, 53, 121–129 CrossRef CAS PubMed.
- D. Naumann, Infrared and NIR Raman spectroscopy in medical microbiology, Bellingham, Washington, 1998 Search PubMed.
- Z. Movasaghi, S. Rehman and D. I. ur Rehman, Appl. Spectrosc. Rev., 2008, 43, 134–179 CrossRef CAS.
- P. Lasch, Chemom. Intell. Lab. Syst., 2012, 117, 100–114 CrossRef CAS.
- N. Mattsson, I. Zegers, U. Andreasson, M. Bjerke, M. A. Blankenstein, R. Bowser, M. C. Carrillo, J. Gobom, T. Heath, R. Jenkins, A. Jeromin, J. Kaplow, D. Kidd, O. F. Laterza, A. Lockhart, M. P. Lunn, R. L. Martone, K. Mills, J. Pannee, M. Ratcliffe, L. M. Shaw, A. J. Simon, H. Soares, C. E. Teunissen, M. M. Verbeek, R. M. Umek, H. Vanderstichele, H. Zetterberg, K. Blennow and E. Portelius, Biomarkers Med., 2012, 6, 409–417 CrossRef CAS PubMed.
- P. Carmona, M. Molina and A. Toledano, Curr. Alzheimer Res., 2016, 13, 450–464 CrossRef CAS PubMed.
- S. E. O'Bryant, G. Xiao and R. Barber, et al. , Arch. Neurol., 2010, 67, 1077–1081 CrossRef PubMed.
- E. Barlev, U. Zelig, O. Bar, C. Segev, S. Mordechai, J. Kapelushnik, I. Nathan, F. Flomen, H. Kashtan, R. Dickman, O. Madhala-Givon and N. Wasserberg, J. Gastroenterol., 2016, 51, 214–221 CrossRef CAS PubMed.
- C. Beleites, U. Neugebauer, T. Bocklitz, C. Krafft and J. Popp, Anal. Chim. Acta, 2013, 760, 25–33 CrossRef CAS PubMed.
- X. Mu, M. Kon, A. Ergin, S. Remiszewski, A. Akalin, C. M. Thompson and M. Diem, Analyst, 2015, 140, 2449–2464 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an01580h |
| This journal is © The Royal Society of Chemistry 2017 |