
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A continuous luminescence assay for monitoring kinase activity: signalling the ADP/ATP ratio using a discrete europium complex†
Sarah H.
Hewitt
,
Jonathan
Parris
,
Romain
Mailhot
and
Stephen J.
Butler
 *
*
Department of Chemistry, Loughborough University, Epinal Way, Loughborough, LE11 3TU, UK. E-mail: s.j.butler@lboro.ac.uk; Tel: +44 (0)1509 222577
First published on 13th November 2017
Abstract
We report the application of a stable cationic europium complex [Eu.1]+ in a continuous-read luminescence assay for kinase activity. [Eu.1]+ binds reversibly to ATP and ADP in water, at neutral pH, in the presence of Mg2+ ions, providing distinctive luminescence responses that permits the kinase-catalysed conversion of ATP to ADP to be monitored in real-time.
Monitoring enzymatic activity in real-time is of fundamental importance to biological and biomedical researchers. It enables enzyme kinetics and thus enzyme mechanism to be determined, a crucial first step in the discovery of new inhibitors and activators of enzymes.1 Nucleoside polyphosphate (NPP) anions such as ATP, ADP, GTP and GDP are important substrates for a number of pharmaceutically relevant enzymes, most prominently in drug discovery programmes involving kinases and GTPases.2,3 Kinases catalyse the phosphorylation of proteins, converting ATP to ADP in the process. Dysregulation of kinase activity, resulting in changes in selectivity and frequency of substrate phosphorylation, is the primary cause of a number of cancers. Therefore, kinases represent one of the most promising targets in oncological drug discovery.4
The search for new kinase inhibitors currently begins with high-throughput screening of potential leads followed by measurements of selectivity and potency. However, the majority of commercial kinase activity assays are restricted to single end-point measurements (e.g. ADPGlo,5 HTRF KinEASE,6 DELFIA7), which give no kinetic and thus no mechanistic information. Current assays typically require the use of either expensive antibodies to detect the phosphorylated peptide substrate, or fluorescent/radiolabelled substrates, which precludes the use of natural peptides or proteins. Crucially, there is no low-cost method available for continuous reporting of kinase activity, which significantly limits our understanding of kinase kinetics and inhibition mechanisms.
In this work we have developed an alternative approach to monitor kinase activity, which involves the use of a stable luminescent europium(III) complex, [Eu.1]+, that binds reversibly to ATP and ADP, providing distinctive emission spectral responses, enabling the change in ADP/ATP ratio to be dynamically followed during a kinase reaction (Fig. 1a). Measuring the ADP/ATP ratio instead of the production of specific phosphorylated substrates, could permit a range of kinases to be screened readily. Crucially, our assay does not require the use of expensive antibodies to recognise a specific phosphorylated substrate. Complex [Eu.1]+ is stable under the conditions used routinely in kinase assays (neutral pH, high concentrations of Mg2+ ions) and does not require modification of the enzyme or substrate with a fluorescent or radioactive label,8,9 avoiding intensive and time consuming synthetic procedures.
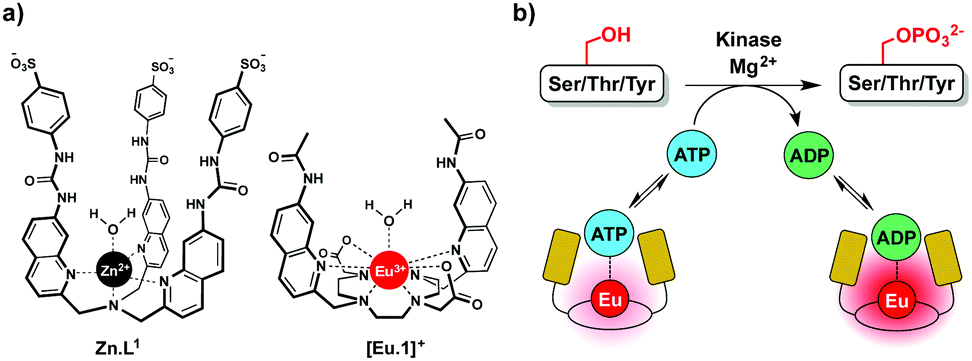 | ||
| Fig. 1 (a) Structures of complexes Zn.L1 and [Eu.1]+; (b) cartoon illustrating continuous monitoring of kinase activity using [Eu.1]+. The Eu(III) complex binds reversibly to ATP and ADP, providing a ratiometric luminescent signal that precisely indicates the ADP/ATP ratio. | ||
A number of synthetic receptors have been developed for binding and sensing ATP (and ADP) in water.10 However, very few exhibit the required selectivity to enable direct monitoring of enzyme activity.11–13 Lanthanide-based probes capable of binding to NPP anions are rare.14 Most bind weakly to NPP anions and behave as ‘on–off’ sensors, whereby anion binding causes quenching of luminescence due to displacement of a weakly bound sensitizing ligand from the complex or via energy transfer to the nucleotide base.12a,15,16 Use of ‘on–off’ type probes in enzyme monitoring have potential problems with calibration, as competitive quenching processes can also give rise to the observed emission response.17
We recently reported a fluorescent tripodal zinc(II) complex, Zn.L1, capable of the selective recognition of ATP at physiological pH (Fig. 1a).13 Strong, selective binding to ATP was achieved by arranging multiple hydrogen-bond donor groups around a coordinatively unsaturated central zinc(II) ion. We used the probe to monitor the apyrase-catalysed hydrolysis of ATP to ADP and AMP in real-time. However, this complex was unsuitable for monitoring kinase activity, due to its susceptibility to quenching by peptide/protein substrates and short-lived fluorescence lifetime. To overcome these issues, we synthesised the cationic europium(III) complex, [Eu.1]+, which offers improvements in performance for enzyme assays. The complex has a typical line-like Eu(III) emission spectrum that enables ratiometric analysis and a long luminescence lifetime (0.5 ms in water), permitting the use of time-resolved detection to eliminate autofluorescence arising from protein substrates.18[Eu.1]+ is based on an octadentate ligand bearing two coordinated quinoline groups that facilitate sensitisation of Eu(III) emission, and a coordinated water molecule. It was envisaged that NPP anions would bind strongly to [Eu.1]+, displacing the bound water and inducing an increase in luminescence. The quinoline units possess amide groups at the 7-position, to strengthen binding to polyphosphate anions via hydrogen bonding interactions.13
The synthesis and selected photophysical properties of [Eu.1]+ were reported previously.19 Addition of 1 mM ADP to [Eu.1]+ in aqueous buffer at pH 7.0 (10 mM HEPES) resulted in direct coordination of the anion to the Eu(III) ion and displacement of the bound water molecule,‡ inducing a dramatic 43-fold enhancement in emission intensity at 616.5 nm within the ΔJ = 2 band (λexc 330 nm) and pronounced changes in spectral form (Fig. 2a). Adding 1 mM ATP caused a comparatively smaller (24-fold) increase in luminescence, whereas pyrophosphate (PPi), HPO42− (Pi), AMP, phosphorylated amino acids (pS, pT, pY), and selected mono-phosphorylated peptides (e.g. AYpYAA, Fig. S6, ESI†) caused much smaller changes in emission intensity. Evidence for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding between [Eu.1]+ and ATP or ADP was provided by high resolution (ESI†) mass spectrometric data, which revealed major signals for the singly charged species [Eu.1 + ATP]− and [Eu.1 + ADP]−, respectively (Fig. S8, ESI†). The effect of pH on the emission response of [Eu.1]+ in the presence of ADP was also shown to be small over the range pH 6.4 to 8.5 (Fig. S9, ESI†). Anion association constants were determined by plotting the change in the intensity ratio of the ΔJ = 2/ΔJ = 1 emission bands (605–630/580–600 nm) as a function of anion concentration, followed by curve fitting based on a 1:1 binding model (Table 1). [Eu.1]+ showed strong binding to ATP and ADP in water (log Ka = 4.4 and 4.6 respectively), approximately 10 times stronger than to PPi and AMP, and at least 100 times stronger than mono-phosphorylated peptides. The nucleoside moiety clearly influences anion binding, as [Eu.1]+ binds to AMP but not to HPO42−, and binds to ADP with 10-fold selectivity over pyrophosphate.
:1 binding between [Eu.1]+ and ATP or ADP was provided by high resolution (ESI†) mass spectrometric data, which revealed major signals for the singly charged species [Eu.1 + ATP]− and [Eu.1 + ADP]−, respectively (Fig. S8, ESI†). The effect of pH on the emission response of [Eu.1]+ in the presence of ADP was also shown to be small over the range pH 6.4 to 8.5 (Fig. S9, ESI†). Anion association constants were determined by plotting the change in the intensity ratio of the ΔJ = 2/ΔJ = 1 emission bands (605–630/580–600 nm) as a function of anion concentration, followed by curve fitting based on a 1:1 binding model (Table 1). [Eu.1]+ showed strong binding to ATP and ADP in water (log Ka = 4.4 and 4.6 respectively), approximately 10 times stronger than to PPi and AMP, and at least 100 times stronger than mono-phosphorylated peptides. The nucleoside moiety clearly influences anion binding, as [Eu.1]+ binds to AMP but not to HPO42−, and binds to ADP with 10-fold selectivity over pyrophosphate.
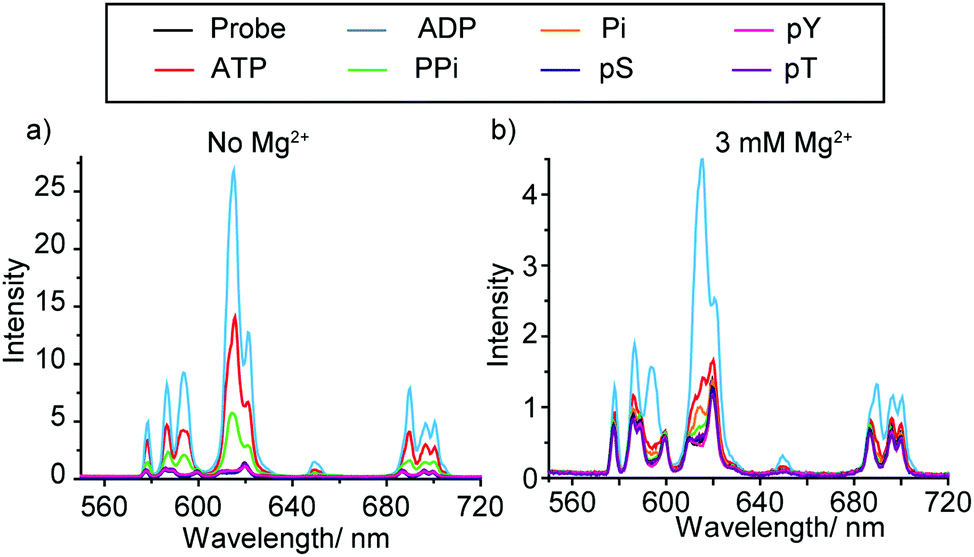 | ||
| Fig. 2 Change in emission spectra of [Eu.1]+ (8 μM) in the presence of different phosphoanions (1 mM, sodium salts) in buffered aqueous solution (10 mM HEPES, pH 7.0), containing: (a) no Mg2+ ions; (b) 3 mM Mg2+ ions. λexc = 330 nm, 298 K. | ||
| Anion | ATP | ADP | AMP | PPi | AYpYAAb |
|---|---|---|---|---|---|
| Each value represents the average of two duplicate titration experiments.a Values in italics were determined in an aqueous solution (10 mM HEPES, pH 7.0) containing 3 mM MgCl2.b Representative mono-phosphorylated peptide containing one phosphotyrosine (pY) residue. | |||||
| Log Ka | 4.4 ± 0.1 | 4.6 ± 0.1 | 3.4 ± 0.1 | 3.5 ± 0.1 | 2.0 ± 0.1 |
| 3.7 ± 0.1 | 4.1 ± 0.1 | ||||
Although [Eu.1]+ showed similar affinities for ATP and ADP in water, the key objective in this work is to discriminate between these anions in the aqueous conditions used commonly in kinase assays (neutral pH, millimolar levels of Mg2+ ions), rather than to achieve thermodynamically selective anion binding. Enzymes that utilise ATP as a substrate (e.g. kinases, ATPases) require high concentrations of Mg2+ ions as a cofactor, to stabilise the enzyme–ATP interaction. Mg2+ ions are known to bind more strongly to ATP compared to ADP in water at neutral pH, with log Ka between Mg2+ and ATP and ADP being 4.2 and 3.6 respectively.21 These affinity constants are similar in magnitude to those determined for [Eu.1]+ and ATP and ADP. Therefore, we reasoned that in an aqueous medium containing high concentrations of Mg2+ ions, competitive binding would perturb the equilibrium speciation such that ATP and ADP could be distinguished even more effectively.
Fig. 2b shows the emission spectral response of [Eu.1]+ in the presence of different phosphate species in a background of 3 mM MgCl2. Under these conditions, excellent discrimination between ATP and ADP was achieved. Adding 1 mM ADP generated a substantial 800% increase in overall emission intensity, whereas adding 1 mM ATP caused a much smaller 250% increase in luminescence. Pyrophosphate, HPO42− and phosphorylated amino acids induced negligible spectral responses. A NaCl titration showed these changes in emission response on addition of Mg2+ cannot solely be accounted for by the additional ionic strength of the solution (Fig. S10 and S11, ESI†), therefore some of the effects seen must come from specific interactions between Mg2+ and ATP/ADP. Previously reported synthetic probes have shown limited discrimination between ATP and ADP at enzyme relevant concentrations of Mg2+ ions.12b Such high discrimination in our system can be attributed to the high affinity of [Eu.1]+ for ADP as well as the higher competition between Mg2+ and ATP compared to ADP. Under these conditions, the majority of ATP is in the form Mg-ATP2−, meaning less ATP is available for interaction with [Eu.1]+. A high level of sensing selectivity between GTP and GDP was also achieved (Fig. S14, ESI†). Apparent association constants were determined for ATP and ADP to be log Ka = 3.7 and 4.1 respectively, slightly lower than those determined in the absence of Mg2+ ions (Table 1). Affinity constants for monophosphate species could not be determined under these conditions as only minor changes in Eu(III) emission took place.
The striking differences between the emission spectral responses to ATP and ADP in a background of MgCl2 allowed us to monitor the kinase-catalysed conversion of ATP to ADP in real-time. Initially, we simulated a kinase reaction by increasing the molar ratio of ADP/ATP systematically, whilst keeping the total concentration of ATP and ADP constant (1 mM). The stepwise increase in emission intensity of [Eu.1]+ with decreasing ATP concentration and increasing ADP concentration is shown in Fig. 3a. The large increase in intensity at 616.5 nm within the hypersensitive ΔJ = 2 manifold precisely indicates the ratio of ADP/ATP, while emission intensity at 599.5 nm within the ΔJ = 1 manifold remains constant, indicating total nucleoside (and [Eu.1]+) present. This is exactly the type of ratiometric signalling that we hoped to achieve in our enzyme assay, as it increases precision, improves signal-to-noise and minimises artefacts arising from variations in concentration of [Eu.1]+ or liquid handling. A plot of the change in the intensity ratio at 616.5/599.5 nm versus mole fraction of ADP is approximately linear (Fig. 3b). Significant modulation of Eu(III) emission is evident at 20% conversion of ATP to ADP, indicating that [Eu.1]+ may be used to determine initial enzyme reaction rates and thus, the effect of kinase inhibitors. Simulation of a GTPase reaction was also possible, whereby the change in ratio of GDP/GTP is signalled by a ratiometric change in luminescence (Fig. S14, ESI†).
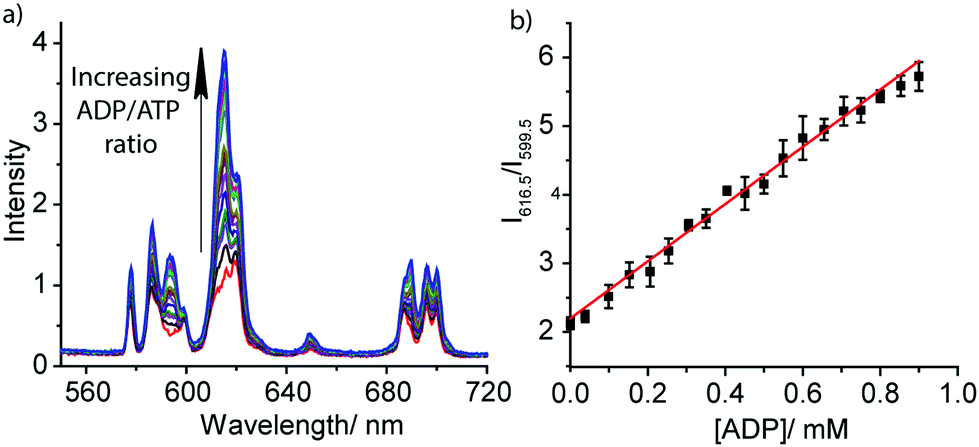 | ||
| Fig. 3 (a) Ratiometric change in emission spectra of [Eu.1]+ (8 μM) in the presence of increasing ratio of ADP/ATP; (b) plot of the emission intensity ratio, 616.5/599.5 nm, versus mole fraction of ADP. Conditions: buffered aqueous solution (10 mM HEPES, pH 7.0) containing MgCl2 (3 mM). Concentration of ADP: 0–0.9 mM, total nucleoside concentration = 1 mM. λexc = 330 nm, 298 K. | ||
Next, we demonstrated the ability of [Eu.1]+ to directly and continuously monitor a kinase reaction using the catalytic subunit of protein kinase A (PKAc). PKAc is a serine-threonine kinase that phosphorylates proteins with the minimal substrate sequence Arg–Arg–Xaa–Ser/Thr (Xaa = any amino acid). Control experiments revealed that the addition of the peptide substrate, kemptide (H2N–LRRASLG–CO2H, 1 mM), DTT (1 mM) or PKAc (0.8 μM) to [Eu.1]+ did not modulate the Eu(III) emission intensity significantly or interfere with the measurement of the ADP/ATP ratio (Fig. S15–S18, ESI†). The enzyme assay was then performed in following order: PKAc was added last to a solution containing all the other assay components and measurements were taken every 96 seconds.§
As the phosphorylation of the substrate takes place, the ratio of ADP/ATP increases. As a result, the intensity of the ΔJ = 2 band centred at 616.5 nm increases, whereas the emission intensity at 599.5 nm within the ΔJ = 1 manifold remains constant (Fig. S19, ESI†), consistent with the ratiometric calibration plot in Fig. 3b. Time-trace plots of the kinase reaction using different concentrations of PKAc (0.05–0.4 μM) are shown in Fig. 4. The plots clearly reveal that using higher concentrations of PKAc results in the overall emission intensity of [Eu.1]+ increasing at a faster rate. Comparable time traces were obtained by following the emission intensity ratio at 616.5/599.5 nm (Fig. S20, ESI†). Importantly, the background reaction in the absence of the enzyme shows essentially no change in emission intensity over time. For the first 10 minutes of each reaction, the increase in emission intensity at 616.5/599.5 nm can be fitted linearly to a monoexponential function (Fig. 4b). The slope of the fit indicates the initial rate of reaction, which depends linearly on the concentration of enzyme used.
 | ||
| Fig. 4 Continuous monitoring of a kinase activity by measuring the conversion of ATP to ADP. (a) Time-trace plots showing the increase in emission intensity of [Eu.1]+ during the PKAc catalysed phosphorylation of the peptide substrate H2N–LRRASLG–CO2H (λem 616.5 nm); (b) linear change in responses over the first 10 minutes. Conditions: buffered aqueous solution (10 mM HEPES, pH 7.0), [Eu.1]+ (8 μM), PKAc (0–0.4 μM), kemptide (1 mM), ATP (1 mM), MgCl2 (3 mM), DTT (2 mM). λexc = 330 nm, 298 K. | ||
To demonstrate the utility of [Eu.1]+ in inhibition experiments, the broad spectrum kinase inhibitor staurosporine was added to the reaction mixture containing 0.4 μM PKAc.¶ The time-trace plots in Fig. 5 and Fig. S22, S23 (ESI†) reveal that PKAc activity is inhibited significantly in the presence of 0.2 μM staurosporine, whereas 0.4 μM of the inhibitor almost completely deactivates the enzyme and very little phosphorylation occurs. This data is consistent with the strong inhibitory effect of staurosporine on PKAc activity (IC50 = 15 nM)22 and demonstrates the utility of [Eu.1]+ for screening of potent inhibitors of protein kinases in real-time.
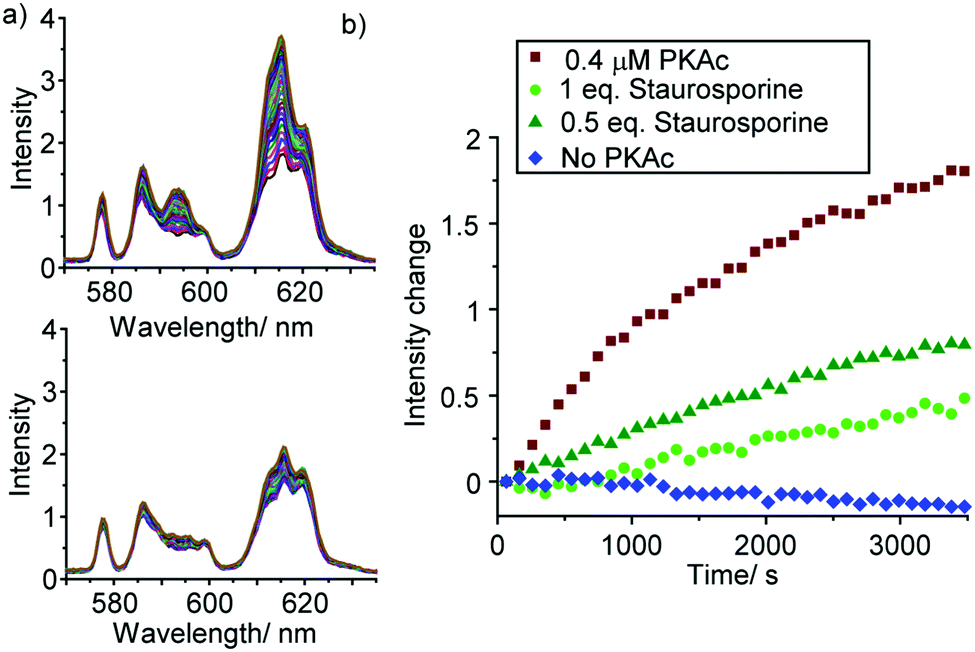 | ||
| Fig. 5 Kinetic analysis of the inhibition of PKAc by staurosporine. (a) Change in emission spectra of [Eu.1]+ (570–635 nm) in the absence of staurosporine (upper) and with 1 eq. of staurosporine (lower). (b) Time traces showing the emission intensity increase during the PKAc catalysed phosphorylation of kemptide, in the presence of staurosporine. Conditions: 10 mM HEPES, pH 7.0, [Eu.1]+ (8 μM), PKAc (0 or 0.4 μM), kemptide (1 mM), ATP (1 mM), MgCl2 (3 mM), DTT (2 mM). λexc = 330 nm, 298 K. | ||
In conclusion, we have developed a versatile luminescence assay for monitoring the activity of kinase enzymes, involving a stable emissive Eu(III) complex [Eu.1]+ that directly signals the ratio of ADP/ATP by a ratiometric change in Eu(III) emission. [Eu.1]+ binds reversibly to ATP and ADP with high affinity in water, and can discriminate between these anions effectively in a competitive aqueous medium containing high concentrations of Mg2+ ions. The Eu(III) complex is very well suited for screening of inhibitors (or activators) of kinase activity, as demonstrated by monitoring the effect of staurosporine on the activity of protein kinase A. Our assay offers a number of advances in performance over commercial kinase assays: it is label-free and does not require chemical modification of the enzyme or substrate with a fluorescent or radioactive label, nor does it require the use of expensive antibodies to recognise a specific phosphorylated substrate.
The use of a discrete Eu(III) complex to discriminate between structurally similar polyphosphate anions during an enzyme reaction is a potentially powerful and general strategy. We are not limited to a specific enzyme; instead we may be able to monitor several classes of enzymes involving NPP anions, including kinases, ATPases, GTPases and DNA polymerases. Furthermore, the Eu(III) complex can be tailored to studying enzymes with a range of activities, by tuning the charge of the Eu(III) complex and the structure and geometry of the macrocyclic ligand, in order to optimise the anion affinity and the desired spectral response. Work is ongoing in this regard.
This work was supported by a Wellcome Trust Seed Award (204500/Z/16/Z) and a Royal Society Research Grant. SJB would like to thank Dr David Worrall for use of fluorescence spectrometry facilities and Dr Charlotte Dodson for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. G. Acker and D. S. Auld, Perspect. Sci., 2014, 1, 56 CrossRef.
- E.-F. Hagit and E. Miriam, Curr. Pharm. Des., 2009, 15, 2463 CrossRef.
- Y. Lin and Y. Zheng, Expert Opin. Drug Discovery, 2015, 10, 991 CrossRef PubMed.
- S. Muller, A. Chaikuad, N. S. Gray and S. Knapp, Nat. Chem. Biol., 2015, 11, 818 CrossRef CAS PubMed.
- H. Zegzouti, M. Zdanovskaia, K. Hsiao and S. A. Goueli, Assay Drug Dev. Technol., 2009, 7, 560 CrossRef CAS PubMed.
- F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, Curr. Chem. Genomics, 2009, 3, 22 CrossRef CAS PubMed.
- Y. Wang and H. Ma, Drug Discovery Today: Technol., 2015, 18, 1 CrossRef CAS PubMed.
- (a) N. P. Oien, L. T. Nguyen, F. E. Jernigan, M. A. Priestman and D. S. Lawrence, Angew. Chem., Int. Ed., 2014, 53, 3975 CrossRef CAS PubMed; (b) E. Lukovic, E. V. Taylor and B. Imperiali, Angew. Chem., Int. Ed., 2009, 48, 6828 CrossRef CAS PubMed.
- (a) J. Mok, H. Im and M. Snyder, Nat. Protoc., 2009, 4, 1820 CrossRef CAS PubMed; (b) H. Zhu and M. Snyder, Curr. Opin. Chem. Biol., 2003, 7, 55 CrossRef CAS PubMed.
- (a) S. E. Schneider, S. N. O'Nei and E. V. Anslyn, J. Am. Chem. Soc., 2000, 122, 542 CrossRef CAS; (b) F. Sancenón, A. B. Descalzo, R. Martínez-Máñez, M. A. Miranda and J. Soto, Angew. Chem., Int. Ed., 2001, 40, 2640 CrossRef; (c) C. Li, M. Numata, M. Takeuchi and S. Shinkai, Angew. Chem., Int. Ed., 2005, 44, 6371 CrossRef CAS PubMed; (d) A. Ojida, H. Nonaka, Y. Miyahara, S. Tamaru, K. Sada and I. Hamachi, Angew. Chem., Int. Ed., 2006, 45, 5518 CrossRef CAS PubMed; (e) A. J. Moro, P. J. Cywinski, S. Korsten and G. J. Mohr, Chem. Commun., 2010, 46, 1085 RSC; (f) Y. Kurishita, T. Kohira, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 18779 CrossRef CAS PubMed; (g) E. A. Weitz, J. Y. Chang, A. H. Rosenfield and V. C. Pierre, J. Am. Chem. Soc., 2012, 134, 16099 CrossRef CAS PubMed; (h) J.-L. Tang, C.-Y. Li, Y.-F. Li and C.-X. Zou, Chem. Commun., 2014, 50, 15411 RSC; (i) L. Wang, L. Yuan, X. Zeng, J. Peng, Y. Ni, J. C. Er, W. Xu, B. K. Agrawalla, D. Su, B. Kim and Y.-T. Chang, Angew. Chem., Int. Ed., 2016, 55, 1773 CrossRef CAS PubMed; (j) D. Maity, M. Li, M. Ehlers and C. Schmuck, Chem. Commun., 2017, 53, 208 RSC; (k) H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928 RSC.
- (a) Z. Xu, N. J. Singh, J. Lim, J. Pan, H. N. Kim, S. Park, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2009, 131, 15528 CrossRef CAS PubMed; (b) T. Sakamoto, A. Ojida and I. Hamachi, Chem. Commun., 2009, 141 RSC.
- (a) E. A. Weitz, J. Y. Chang, A. H. Rosenfield, E. A. Morrow and V. C. Pierre, Chem. Sci., 2013, 4, 4052 RSC; (b) M. Schäfeling, T. Aaritalo and T. Soukka, Chem. – Eur. J., 2014, 20, 5298 CrossRef PubMed.
- S. J. Butler, Chem. – Eur. J., 2014, 20, 15768 CrossRef CAS PubMed.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652 RSC.
- M. Schäferling and O. S. Wolfbeis, Chem. – Eur. J., 2007, 13, 4342 CrossRef PubMed.
- S. Nadella, J. Sahoo, P. S. Subramanian, A. Sahu, S. Mishra and M. Albrecht, Chem. – Eur. J., 2014, 20, 6047 CrossRef CAS PubMed.
- F. Kielar, C. P. Montgomery, E. J. New, D. Parker, R. A. Poole, S. L. Richardson and P. A. Stenson, Org. Biomol. Chem., 2007, 5, 2975 CAS.
- (a) E. M. Surender, S. J. Bradberry, S. A. Bright, C. P. McCoy, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2017, 139, 381 CrossRef CAS PubMed; (b) R. Pal, D. Parker and L. C. Costello, Org. Biomol. Chem., 2009, 7, 1525 RSC; (c) S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750 RSC; (d) H. M. Burke, T. Gunnlaugsson and E. M. Scanlan, Org. Biomol. Chem., 2016, 14, 9133 RSC; (e) B. K. McMahon and T. Gunnlaugsson, J. Am. Chem. Soc., 2012, 134, 10725 CrossRef CAS PubMed.
- S. J. Butler, Chem. Commun., 2015, 51, 10879 RSC.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- (a) R. K. Airas, Biophys. Chem., 2007, 131, 29 CrossRef CAS PubMed; (b) G. D. Williams, T. J. Mosher and M. B. Smith, Anal. Biochem., 1993, 214, 458 CrossRef CAS PubMed; (c) C. T. Burt, H. M. Cheng, S. Gabel and R. E. London, J. Biochem., 1990, 108, 441 CrossRef CAS PubMed.
- F. Meggio, A. D. Deana, M. Ruzzene, A. M. Brunati, L. Cesaro, B. Guerra, T. Meyer, H. Mett, D. Fabbro, P. Furet and G. Dobrowolska, FEBS J., 1995, 234, 317 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of complex characterisation and spectroscopic analysis are available. See DOI: 10.1039/c7cc05887j |
| ‡ Emission lifetimes of [Eu.1]+ measured in H2O and D2O in the absence and presence of ADP were consistent with a hydration state, q, of 1.0 (±20%) and zero respectively, confirming that ADP displaces the coordinated water from the complex.20 |
| § Given that [Eu.1]+ binds strongly to ATP, it is important to consider that this may lower the effective concentration of ATP and thus influence the rate of reaction. However, anion binding is fast and reversible, and due to the highly sensitive nature of the emission response, the concentration of [Eu.1]+ required in the kinase assay is much lower (8 μM) compared to the concentration of ATP (1 mM). This ensures that the rate of the enzyme reaction is not significantly perturbed by [Eu.1]+. |
| ¶ Staurosporine was dissolved in DMSO and added to the enzyme reaction mixture (to give a final DMSO concentration of 1%). The presence of up to 10% DMSO was found to have minimal impact on the ability of [Eu.1]+ to report the conversion of ATP to ADP (Fig. S21, ESI†). |
| This journal is © The Royal Society of Chemistry 2017 |