
Development of a reactor with carbon catalysts for modular-scale, low-cost electrochemical generation of H2O2†
Zhihua
Chen‡
ab,
Shucheng
Chen‡
ab,
Samira
Siahrostami
a,
Pongkarn
Chakthranont
a,
Christopher
Hahn
ab,
Dennis
Nordlund
c,
Sokaras
Dimosthenis
c,
Jens K.
Nørskov
*ab,
Zhenan
Bao
*ab and
Thomas F.
Jaramillo
 *ab
*ab
aSUNCAT Center for Interface Science and Catalysis, Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: norskov@stanford.edu; zbao@stanford.edu; jaramillo@stanford.edu
bSUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, CA 94025, USA
cStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA
First published on 1st March 2017
Abstract
The development of small-scale, decentralized reactors for H2O2 production that can couple to renewable energy sources would be of great benefit, particularly for water purification in the developing world. Herein, we describe our efforts to develop electrochemical reactors for H2O2 generation with high Faradaic efficiencies of >90%, requiring cell voltages of only ∼1.6 V. The reactor employs a carbon-based catalyst that demonstrates excellent performance for H2O2 production under alkaline conditions, as demonstrated by fundamental studies involving rotating-ring disk electrode methods. The low-cost, membrane-free reactor design represents a step towards a continuous, modular-scale, de-centralized production of H2O2.
Introduction
Limited access to clean water is one of the major issues for billions of people in the developing world. According to a recent UN Global issue report, millions of people lack access to drinking water just as in Sub-Saharan Africa.1 The problem is often not access to water itself, but its contamination with urban, industrial, and agricultural waste.2 Conventionally, chlorine has been widely used for disinfection and oxidation in drinking water treatment. It is added to water as chlorine gas, sodium hypochlorite solution, or dry calcium hypochlorite to eliminate pathogenic organisms and remove any color, taste, and odor compounds.3 Unfortunately, excess chlorine residues and by-products of treatment can have negative effects, including eye irritation4 and harm to the growth and reproduction of aquatic life, even at low concentrations.5Hydrogen peroxide (H2O2) is an attractive alternative for water treatment. It is a stronger oxidizing agent capable of removing disease-causing organisms as well as persistent organic pollutants. Importantly, its intrinsic decomposition route results in the production of harmless by-products, water and oxygen. Despite these favorable properties, low-cost, decentralized H2O2 production is a challenge as industrial production is performed through an anthraquinone oxidation process that requires multiple energy-intensive reaction steps and is not conducive to small-scale operation.6 In addition, the instability of H2O2 poses a safety issue for transportation, which further hinders the use of H2O2 for water treatment in developing countries. The cost and safety issues of H2O2 make this potentially important chemical largely inaccessible to a large number of people who need it the most.
One promising method to produce H2O2 on-site is by electrochemical advanced oxidation processes (EAOPs).7 These processes have been recently developed for water purification, where hydrogen peroxide is generated on-site from a two-electron reduction of injected O2 where it can be immediately used as an oxidizing agent for water treatment.7 More importantly, an EAOP device can be coupled with intermittent power sources such as wind and solar and thus can be used even in remote regions that lack access to conventional energy for process heat and electricity. However, EAOP devices often exhibit low efficiency due to the lack of an active and selective catalyst and instability due to decomposition of cell components from the active radicals generated from H2O2. As a result, there is a need to develop improved EAOP devices and catalysts for efficient, stable, scalable, and decentralized H2O2 production; ideally, one that can operate in either a fuel cell mode or an electrolyzer mode (Fig. 1).
 | ||
| Fig. 1 Schematic illustration of an on-site electro-synthesis device for H2O2 production from O2 for water purification. | ||
Several device designs for the production of H2O2 have been proposed; however, the devices were operated at limited efficiency or stability.8,9 A previous report shows a flow-cell design operated under acidic conditions at a pH value of ∼2. Although a high overall current was achieved, the selectivity of ∼60% for H2O2 could be significantly improved with better catalysts.8 In a later report, an improved solid-polymer-electrolyte (SPE) electrolysis method under neutral pH conditions was used to produce a H2O2 solution from O2 and water directly at higher concentrations (8 wt%) by using a small electrolyte volume (0.5 ml). Similar to the aforementioned study, poor catalyst selectivity led to a current efficiency (CE) of only 31%.9 In both setups, product crossover was prevented by using a polymer electrolyte membrane (PEM), which is known to be degraded by radicals generated from the self-decomposition of H2O2, limiting the long term stability of the device.10
One of the major challenges for two-electron O2 reduction to H2O2 is the competing four-electron O2 reduction pathway to water. Recent understanding indicates that catalysts capable of preserving the O–O bond during electrochemical O2 reduction are selective for H2O2 over water.11 Some of the promising examples are Pd and Pt alloyed with mercury11 and Pd–Au alloys.12 While some of these alloys have been shown to be active, selective and stable for two-electron O2 reduction, mercury is toxic and precious metals such as Pd, Pt, and Au limit their implementation into EAPO devices. Recent studies have revealed that porous carbon-based materials are promising candidates for generating H2O2 through the two-electron O2 reduction reaction (ORR).13–16 These materials are inexpensive, nontoxic, stable, and active for H2O2 production, making them suitable for practical applications. In this work, we have identified an exceptionally active and selective catalyst, CMK-3 (ACS Materials), made of highly defective carbon with ordered mesoporous structures.17 We also show that CMK-3 is a very stable catalyst under electrochemical reduction of O2 to H2O2. These properties make CMK-3 an excellent candidate to be incorporated into a device for H2O2 production. To achieve a low-cost device, all of the materials in such a device should be readily available and manufacturable with low-cost methods. We demonstrate a low cost device design using a polycarbonate cell body, a carbon catalyst, and an alkaline electrolyte. The device shows high efficiency and stability under operation due to the exceptional electrocatalytic properties of the carbon-based catalyst and the membrane-less configuration.
Results and discussion
Physical, structural and chemical characterization
Hypothesizing that pore-size and the types and concentrations of defects (e.g. sp2-type defects) affect the overall performance of H2O2 production catalysts, we investigated a commercial mesoporous carbon, CMK-3 (ACS Materials). This material is an ordered mesoporous carbon with hierarchical pores, synthesized using SBA-15 silica as the template, sucrose as the carbon source, and sulfuric acid as the carbonization catalyst.17To characterize the hierarchically porous structure of CMK-3, gas absorption was used to determine the size of the mesopores and micropores. The pore size distribution, calculated by nonlinear density functional theory (NLDFT), revealed that CMK-3 has a micropore volume of 0.163 cm3 g−1 and a mesopore volume of 1.017 cm3 g−1 (Fig. S1a†). Bright-field TEM images confirm the ordered fringes of CMK-3 (Fig. S2†). The material exhibits a high BET surface area of 936 m2 g−1. Raman spectroscopy showed that CMK-3 has both intense G-band and D-band contributions (Fig. S1b†) with an ID/IG ratio of 0.98. The chemical composition of CMK-3 was further analyzed with XPS (Fig. S1c†). The chemical analysis shows a low oxygen content possibly due to its preparation method, during which the sucrose precursor can be easily dehydrated to completely remove the oxygen and hydrogen. The near-edge X-ray absorption fine structure spectroscopy (NEXAFS) spectrum for CMK-3 confirms a low oxygen content (Fig. S1d†), based on the small peak in the region between 286 and 290 eV that can be attributed to electronegative functional groups, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. Such a peak is absent in the spectrum for highly ordered pyrolytic graphite (HOPG), which shows a characteristic π* resonance from sp2-like carbon at 285.3 eV, while the core-exciton peak at 291.65 eV is associated with long-range sp2 order and σ and extended oscillations from a well-ordered sample.
O. Such a peak is absent in the spectrum for highly ordered pyrolytic graphite (HOPG), which shows a characteristic π* resonance from sp2-like carbon at 285.3 eV, while the core-exciton peak at 291.65 eV is associated with long-range sp2 order and σ and extended oscillations from a well-ordered sample.
The spectrum for CMK-3 shows a significant intensity in the π* region, suggesting a primarily graphitic network. The broad π* feature for CMK-3 compared to that of HOPG suggests the presence of a larger variety of sp2 carbon sites in CMK-3. As observed in aromatic compounds, the lower energy states can be associated with more unstable aromatic functionalities, supporting the prevalence within CMK-3 of defect states in an sp2-dominated matrix.18 The absence of core-hole exciton in CMK-3 indicates that the long-range sp2 network has been broken, also confirming a large density of defect states.19 The high surface area, the presence of ordered mesoporous structures, as well as the existence of a large number of sp2 type carbon defects indicate that CMK-3 might be promising as an electrocatalyst for H2O2 generation.
ORR activity and stability
Rotating ring disk electrode (RRDE) measurements with a four-electrode configuration are used to investigate catalyst performance. Fig. 2a shows the voltammograms at 1600 rpm in an O2-saturated electrolyte with the ring current density adjusted for the measured collection efficiency. The disk current shows that CMK-3 reaches a mass transport-limited current at ∼0.65 V vs. RHE, demonstrating high ORR activity for CMK-3. The ring current tracks with the disk current, demonstrating that CMK-3 is also selective for two-electron ORR to H2O2. The earlier onset of the ring current than the standard thermodynamic equilibrium potential of 0.7 V vs. RHE may be attributed to local effects involving pH and the concentration of H2O2. Using the ring and disk currents, a high selectivity of over 90% for H2O2 is confirmed (Fig. 2b). Koutecky–Levich analysis was performed by controlling the electrode rotation rates to further examine the selectivity of CMK-3 for the two-electron ORR (Fig. 2c). The calculated number of electrons transferred per O2 molecule was approximately 2, consistent with the high selectivity for two-electron ORR observed in RRDE measurements. As shown by both techniques, CMK-3 has a high selectivity towards the two-electron ORR pathway across a wide range of applied potentials.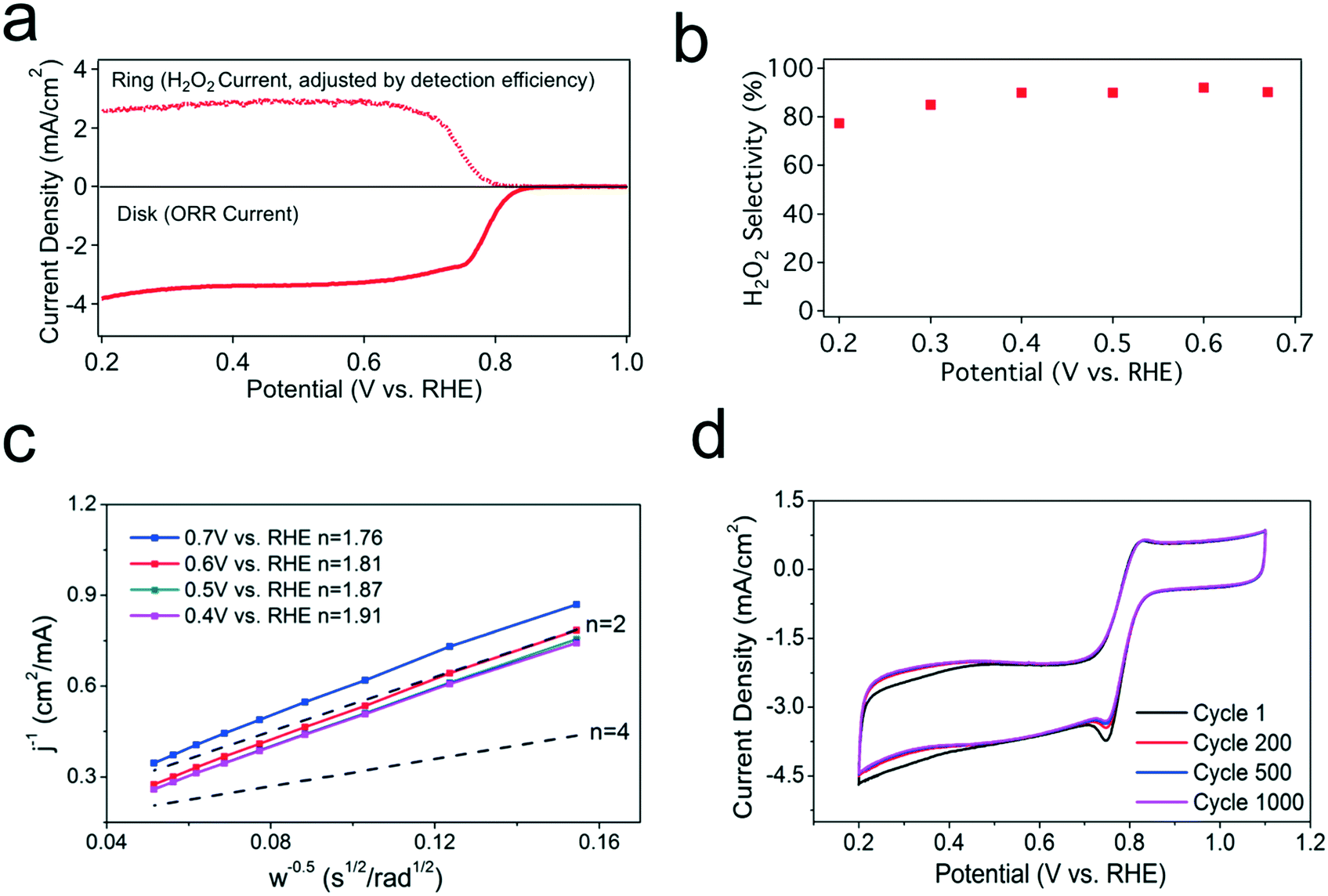 | ||
| Fig. 2 (a) RRDE voltammograms at 1600 rpm in an O2-saturated 0.1 M KOH electrolyte with the disc current density and adjusted ring current density. (b) H2O2 selectivity as a function of the applied potential. (c) Stability performance for CMK-3 in 0.1 M KOH. (d) Koutecky–Levich plots for CMK-3 at different potentials. The theoretical lines for n = 2 and n = 4 are shown for comparison. | ||
Apart from the activity and selectivity, the stability is another key catalyst performance metric, particularly in the presence of H2O2, which is a highly oxidizing agent. An accelerated durability test was conducted by sweeping the potential between 0.2 and 1.1 V vs. RHE at a scan rate of 200 mV s−1 for 1000 cycles. The 1st, 200th, 500th, and 1000th cyclic voltammograms, each measured at a scan rate of 10 mV s−1, are reported in Fig. 2d. CMK-3 showed excellent stability over 1000 cycles with negligible decay in current, indicating the intrinsic stability of defect sites to oxidation from the highly active radicals created by the self-disproportionation of H2O2.20 Overall, these electrochemical measurements demonstrate that CMK-3 is a high-performance two-electron ORR catalyst with fast kinetics, high selectivity for H2O2, and promising long-term stability.
Device design and performance
In developing a device for H2O2 production, we considered four design criteria listed as follows:1. The decomposition of H2O2 should be minimized. As H2O2 can be readily decomposed by transition metals and reductive organic compounds,21 the main body of the device must be composed of an inert plastic of a relatively simple design that will also help reduce manufacturing costs.
2. The device must be operated at sufficiently high current densities and low applied cell potentials to achieve large production rates with a high current efficiency. Thus, in addition to the suitable H2O2 production catalyst at the cathode, the anode must also be active and stable for oxygen evolution reaction (OER).
3. The device must be stable and product crossover should be minimized to prevent oxidation of H2O2 on the anode. In previously reported designs, membranes have been used for product separation but might also be a limiting factor for stability.10 A membrane-free system that can also prevent product crossover could greatly improve cell performance. Moreover, a membrane-free design could potentially reduce required applied voltages by lowering the overall ohmic loss.22
4. Ideally, the device should operate at a temperature and pressure near ambient conditions; doing so reduces energy consumption and obviates the need for strict temperature/pressure controls, facilitating low-cost, decentralized production. Generally, reactor designs operating at high conversion are desired. The cell presented herein was deliberately designed for lower O2 conversion (ca. 5%, see the ESI†), however, in order to maintain more consistent reaction conditions that facilitate analysis.
Taking these criteria into consideration, we have developed a membrane-free electrolyzer consisting of two carbon paper electrodes, the cathode coated with CMK-3 and the anode with an OER catalyst. Catalysts are drop-cast on the Sigracet® Graphite carbon paper from Ion Power Inc., with the backside of the gas diffusion layer (GDL) coated with a hydrophobic polymer which will allow the diffusion of gas but prevent water diffusion to a desired level. The polycarbonate cell is composed of three main chambers: the cathode (compartment 1, working electrode), the product storage (compartment 2), and the anode (compartment 3, counter electrode) compartments as shown in Fig. 3a. The entire cell is filled with 0.1 M KOH electrolyte (Sigma-Aldrich semiconductor grade, pellets).
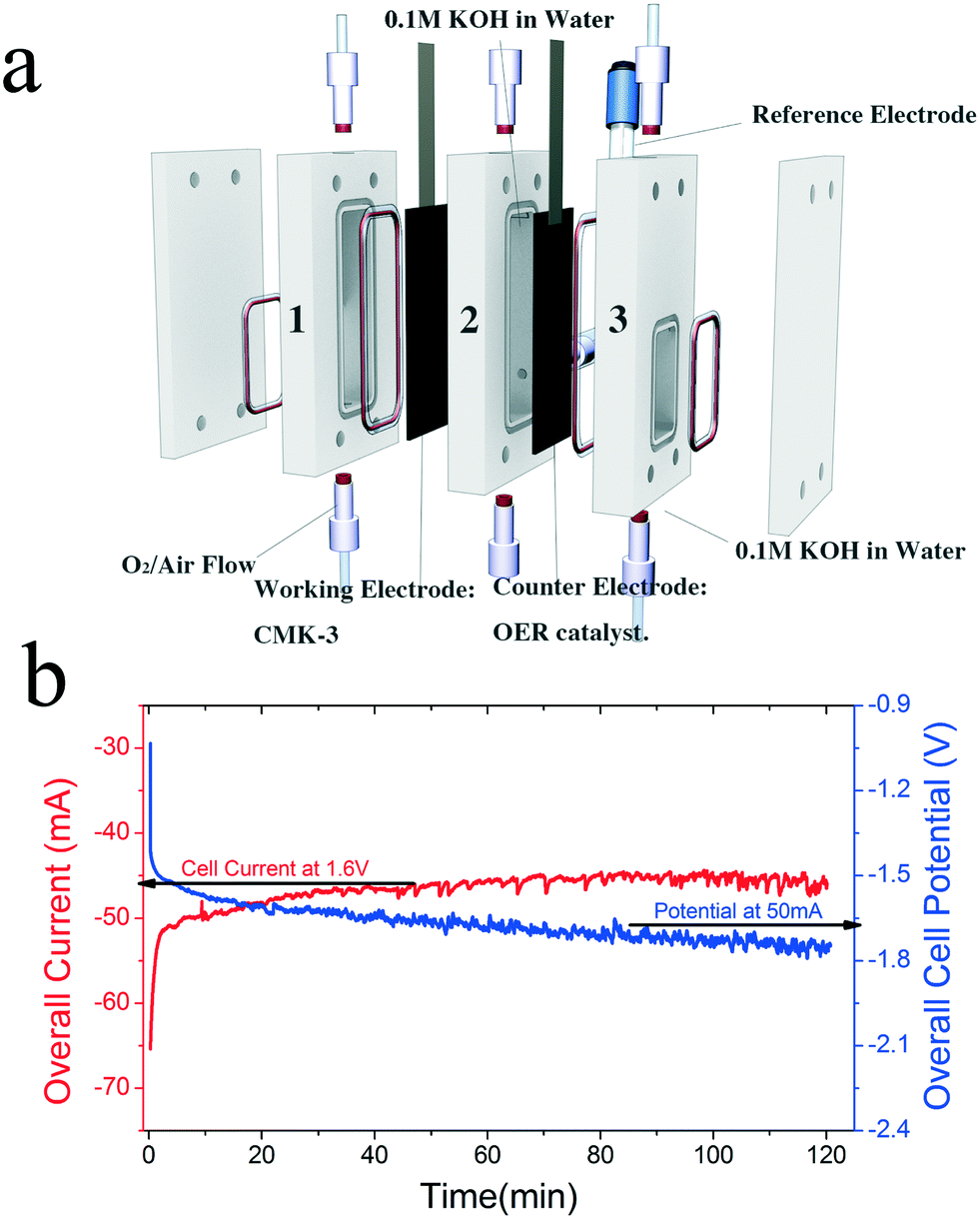 | ||
| Fig. 3 (a) Schematic diagram of the H2O2 generation device in an electrolyzer mode. (b) Electrochemical performance by chronoamperometry and chronopotentiometry. | ||
On the cathode, the oxygen flow (oxygen (O2), 99.993%, ultra high purity grade, T Style Cylinder, CGA 540) comes into contact with the ORR catalyst coated on the side facing compartment 2 and is reduced into H2O2 with an O2 conversion of approximately 5%. The product is dissolved into the electrolyte in compartment 2. On the anode, the catalyst is coated on the side facing compartment 3 and drives the OER. The hydrophobic layer on the back of the carbon paper blocks the diffusion of H2O2, creating a unidirectional flow of H2O2 through the carbon paper. By orienting cathode carbon papers so that the backs are facing compartment 2, product crossover to the anode is mitigated and a high concentration of H2O2 can be accumulated. In this study, three distinct electrolyzer configurations were investigated, labeled electrolyzers 1, 2, and 3, each with CMK-3 as the cathode catalyst for the ORR paired with a different anode catalyst for the OER: Pt, N-doped carbon23 and NiFeOx, respectively (Table 1). The reactions carried out in this work were conducted as batch liquid processes with flowing gaseous reagents, but it is also possible to flow the liquid, operating the device as a continuous electrolyte flow reactor.
| Electrolyzer 1 | Electrolyzer 2 | Electrolyzer 3 (120 min stability) | |
|---|---|---|---|
| Cathode | 0.0848 mg cm−2 CMK-3 + oxygen | 0.0848 mg cm−2 CMK-3 + oxygen | 0.0848 mg cm−2 CMK-3 + oxygen |
| Anode | 0.2544 mg cm−2 Pt + H2O | 0.4240 mg cm−2 N–C + H2O | 0.007 mg cm−2 Ni0.75Fe0.25Ox + H2O |
| Product/efficiency | 518 mg L−1 (∼0.05 wt%)/Faradaic efficiency 100% | 425 mg L−1 (∼0.04 wt%)/Faradaic efficiency 100% | 2795 mg L−1 (∼0.3 wt%) |
Table 1 and Fig. 4 summarize the testing conditions of the three different electrolyzer configurations. All electrolysis experiments are performed at one atmosphere pressure, at room temperature, with continuous O2 flow. The electrolyte is rinsed out at the end of each experiment and refilled with a fresh batch, and the carbon-paper based electrode is replaced if any leakage is detected, typically occurring after three to four rounds of testing mainly due to mechanical damage from the sampling process. Experiments involving electrolyzers 1 and 2 were conducted for 10 minutes each. Recognizing that H2O2 can readily self-decompose in alkaline media without the presence of chemical stabilizers, the 10 minute experimental duration was short enough such that the H2O2 concentration could be measured directly and used to quantify Faradaic efficiency.8 The concentration of H2O2 was determined by test paper (EMD Millipore MQuant™ Peroxide Test Strips) and iodometric titration. During the 10 minute test, the concentration of H2O2 in electrolyzers 1 and 2 were 518 mg L−1 and 425 mg L−1, respectively, indicating that the measured Faradaic efficiencies are 100%, within the error margin of the titration technique.24
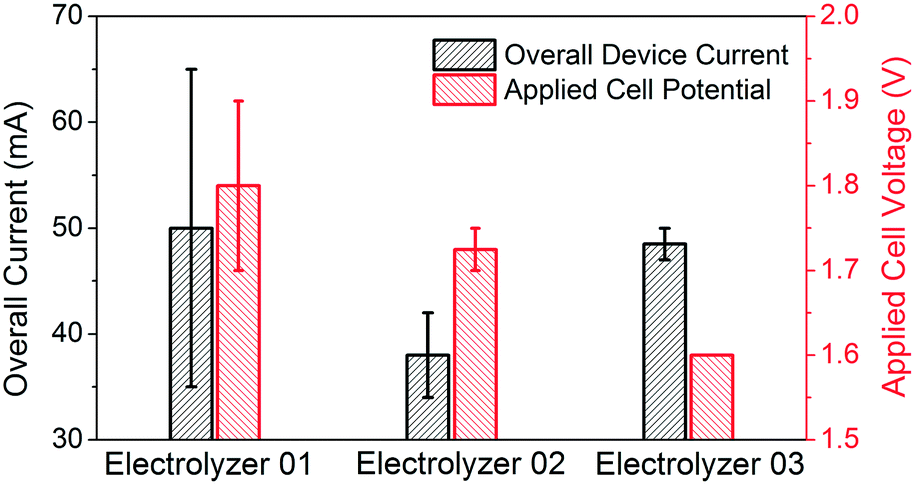 | ||
| Fig. 4 Overall current and applied cell voltages for the electrolyzer mode with different OER catalysts. | ||
For electrolyzer 3, NiFeOx was investigated as the anode catalyst due to its superior activity and stability for OER. Fig. 3b shows a longer-term stability test of electrolyzer 3 over 120 minutes (also without any chemical stabilizers). Chronoamperometric testing at an applied cell potential of 1.6 V yields an average current of approximately 46 mA over 2 hours, beginning with a modest decrease in current of 7% during the first hour of operation and then a stable current flow thereafter, indicating stable electrochemical generation of H2O2. As expected during this longer-term experiment, self-decomposition of H2O2 into O2 and H2O occurred, as evidenced by bubble formation observed within the first 30 minutes of operation. Despite the natural decomposition of the desired product, which takes place at an unknown rate and thus prevents an accurate determination of Faradaic efficiency, at the end of the two hours the measured concentration of H2O2 was 2795 mg L−1, approximately 0.3 wt%, a value commonly used in many commercial applications.
Conclusion
In conclusion, we have developed a small-scale, low-cost electrochemical reactor for the production of hydrogen peroxide (H2O2). In this device, we employed an ordered mesoporous carbon, CMK-3, that exhibits >90% selectivity for 2e− ORR under alkaline conditions, with excellent activity. One manifestation of the reactor employed a carbon catalyst at the cathode and a NiFeOx water oxidation catalyst at the anode, operating in 0.1 M KOH. This electrolyzer demonstrated an overall current of approximately 46 mA at a stable uncompensated applied cell potential of ∼1.6 V for over 2 hours to achieve approximately 0.3 wt% H2O2, without employing any chemical stabilizer additives. Higher product concentrations can be achieved, if desired, by operating beyond two hours and by adding chemical stabilizers, e.g. EDTA, that can mitigate the self-decomposition of H2O2 over long periods of time. Such a product solution in 0.1 M KOH can be used directly for a number of applications, e.g. in the bleaching industry and in the treatment of acid waste streams, or be neutralized first, e.g. with HCl, for broader applications that require near-neutral or acidic conditions. Future work will involve reactor development for direct H2O2 production in a broad range of pH environments and in other configurations designed for continuous electrolyte flow.The high performance of the device demonstrated herein can be attributed to both the intrinsic performance of the CMK-3 catalyst and the durable membrane-free cell design. This cell design allows for flexibility in catalyst material selection, such that a wide range of catalysts for oxygen evolution reaction (OER) or hydrogen oxidation reaction (HOR) can be utilized depending on the mode of operation, electrolyzer or fuel cell, respectively. The simple cell design is also amenable to low-cost manufacturing, and the low total cell potential of 1.6 V is promising for device integration with many electricity sources, including those that are decentralized, e.g. solar panels and portable rechargeable batteries. These advantages allow the device to be practical and readily scalable for use in decentralized applications, e.g. in developing areas for clean drinking water.
Author contributions
Z. C., S. C., C. H., P. C., J. K. N., Z. B., and T. F. J. conceived and designed the experiments. Z. C. carried out electrochemical testing and device design. S. C. performed catalyst selection and chemical characterization. D. N., S. D. and S. N. performed NEXAFS tests. All authors discussed the results and co-wrote the paper.Methods
RRDE measurements were carried out by sweeping the disk potential between 0.2 V and 1.1 V vs. RHE at 10 mV s−1 while holding the Pt ring at 1.2 V vs. RHE to oxidize the hydrogen peroxide species formed on the disc electrode, allowing for ORR product quantification. The system's ring collection efficiency was determined to be 0.2545 using the reversible [Fe(CN)6]4−/3− redox couple (+0.36 vs. SHE). The selectivity of H2O2 can be calculated according to eqn (1):
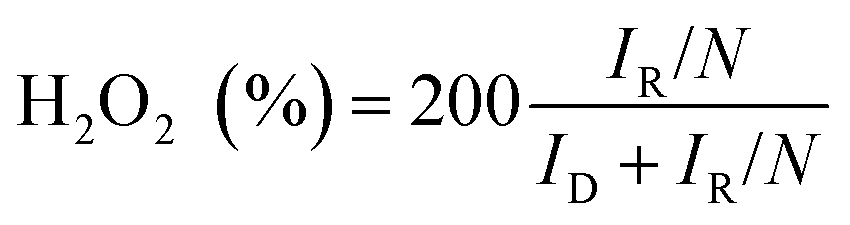 | (1) |
For the RDE measurements, the shaft was rotated at 1600 rpm and the potential was cycled between 0.2 V and 1.1 V vs. RHE at 10 mV s−1. The ORR activity was determined by subtracting the current obtained in an N2-saturated electrolyte from that obtained in an O2-saturated electrolyte. Also, multiple 10 mV s−1 runs were conducted at different rotation rates ranging from 400 to 2500 rpm for Koutecky–Levich analysis. The potential scale was calibrated to the reversible hydrogen electrode (RHE) using a Pt wire (Sigma-Aldrich) as the working electrode in a H2-saturated electrolyte, and a value of 0.959 V was obtained.
Accelerated stability testing was performed by sweeping the potential between 0.2 and 1.1 V vs. RHE at a scan rate of 200 mV s−1 for 1000 cycles. A regular 10 mV s−1 scan between 0.2 and 1.1 V vs. RHE was conducted after 100, 300, 500 and 1000 cycles to ascertain the change in activity over time.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume of 0.1 M HClO4 and measured with the test strip to determine the range of concentrations. At a higher concentration range, iodometric titration was used for more accurate measurements.24 The sample was mixed with 50 mL of demineralized water, 10 mL of 3 M sulfuric acid solution, 10–15 ml of 1% w/v potassium iodide solution, and two drops of ammonium molybdate solution inside an Erlenmeyer flask. The ammonium molybdate solution was prepared by dissolving 9 grams of ammonium molybdate in 10 ml of 6 M NH4OH. Then, 24 grams of NH4NO3 was added and the solution was diluted to 100 ml. The sample was then titrated with 0.1 M sodium thiosulfate to faint yellow or straw color. The sample was swirled or stirred gently during titration to minimize iodine loss. After that, about 2 ml of starch indicator was added and titration was continued until the blue color just disappeared; a similar procedure was repeated for the blank solution. The concentration is calculated by the following equation:
:1 volume of 0.1 M HClO4 and measured with the test strip to determine the range of concentrations. At a higher concentration range, iodometric titration was used for more accurate measurements.24 The sample was mixed with 50 mL of demineralized water, 10 mL of 3 M sulfuric acid solution, 10–15 ml of 1% w/v potassium iodide solution, and two drops of ammonium molybdate solution inside an Erlenmeyer flask. The ammonium molybdate solution was prepared by dissolving 9 grams of ammonium molybdate in 10 ml of 6 M NH4OH. Then, 24 grams of NH4NO3 was added and the solution was diluted to 100 ml. The sample was then titrated with 0.1 M sodium thiosulfate to faint yellow or straw color. The sample was swirled or stirred gently during titration to minimize iodine loss. After that, about 2 ml of starch indicator was added and titration was continued until the blue color just disappeared; a similar procedure was repeated for the blank solution. The concentration is calculated by the following equation:| Weight% H2O2 = (A − B) × (normality of Na2S2O3) × 1.7/sample weight in grams |
Acknowledgements
This work was supported by the US Department of Energy, Office of Science, and Office of Basic Energy Sciences under Award Number DE-SC0008685 and by the SUNCAT Center for Interface Science and Catalysis, a partnership between the SLAC National Accelerator Laboratory and the Department of Chemical Engineering at Stanford University. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Special thanks are given to John W. F. To and Jia Wei Desmond Ng for their valuable suggestions and Dr. Jakob Kibsgaard for his schematic illustrations. Thanks are also given to Taeho Roy Kim for his help in TEM characterization. We also thank Dr. Stanislaw Nowak for his helpful suggestions and assistance in NEXAFS characterization.References
- UN Global issue report, http://www.un.org/en/globalissues/water/index.shtml.
- I. Sires and E. Brillas, Remediation of water pollution caused by pharmaceutical residues based on electrochemical separation and degradation technologies: A review, Environ. Int., 2012, 40, 212–229 CrossRef CAS PubMed.
- Drinking Water Treatability Database, https://iaspub.epa.gov/tdb/pages/treatment/treatmentOverview.do?treatmentProcessId=-1118142891.
- Drinking Water Contaminants – Standards and Regulations, https://www.epa.gov/dwstandardsregulations.
- W. A. Brungs, Effects of Residual Chlorine on Aquatic Life, J. - Water Pollut. Control Fed., 1973, 45(10), 2180–2193 CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45(42), 6962–6984 CrossRef CAS PubMed.
- C. A. Martinez-Huitle and S. Ferro, Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes, Chem. Soc. Rev., 2006, 35(12), 1324–1340 RSC.
- A. Alvarez-Gallegos and D. Pletcher, The removal of low level organics via hydrogen peroxide formed in a reticulated vitreous carbon cathode cell, Part 1, The electrosynthesis of hydrogen peroxide in aqueous acidic solutions, Electrochim. Acta, 1998, 44(5), 853–861 CrossRef CAS.
- T. Murayama and I. Yamanaka, Electrosynthesis of Neutral H2O2 Solution from O-2 and Water at a Mixed Carbon Cathode Using an Exposed Solid-Polymer-Electrolyte Electrolysis Cell, J. Phys. Chem. C, 2011, 115(13), 5792–5799 CAS.
- J. L. Qiao, M. Saito, K. Hayamizu and T. Okada, Degradation of perfluorinated ionomer membranes for PEM fuel cells during processing with H2O2, J. Electrochem. Soc., 2006, 153(6), A967–A974 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Enabling direct H2O2 production through rational electrocatalyst design (vol 12, pg 1137–1143, 2013), Nat. Mater., 2014, 13(1), 97–97 CrossRef CAS.
- J. S. Jirkovsky, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, Single Atom Hot-Spots at Au-Pd Nanoalloys for Electrocatalytic H2O2 Production, J. Am. Chem. Soc., 2011, 133(48), 19432–19441 CrossRef CAS PubMed.
- Y. M. Liu, X. Quan, X. F. Fan, H. Wang and S. Chen, High-Yield Electrosynthesis of Hydrogen Peroxide from Oxygen Reduction by Hierarchically Porous Carbon, Angew. Chem., Int. Ed., 2015, 54(23), 6837–6841 CrossRef CAS PubMed.
- J. Park, Y. Nabae, T. Hayakawa and M. A. Kakimoto, Highly Selective Two-Electron Oxygen Reduction Catalyzed by Mesoporous Nitrogen-Doped Carbon, ACS Catal., 2014, 4(10), 3749–3754 CrossRef CAS.
- Y. H. Lee, F. Li, K. H. Chang, C. C. Hu and T. Ohsaka, Novel synthesis of N-doped porous carbons from collagen for electrocatalytic production of H2O2, Appl. Catal., B, 2012, 126, 208–214 CrossRef CAS.
- T. P. Fellinger, F. Hasche, P. Strasser and M. Antonietti, Mesoporous Nitrogen-Doped Carbon for the Electrocatalytic Synthesis of Hydrogen Peroxide, J. Am. Chem. Soc., 2012, 134(9), 4072–4075 CrossRef CAS PubMed.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, Synthesis of new, nanoporous carbon with hexagonally ordered mesostructure, J. Am. Chem. Soc., 2000, 122(43), 10712–10713 CrossRef CAS.
- M. L. Gordon, D. Tulumello, G. Cooper, A. P. Hitchcock, P. Glatzel, O. C. Mullins, S. P. Cramer and U. Bergmann, Inner-shell excitation spectroscopy of fused-ring aromatic molecules by electron energy loss and X-ray Raman techniques, J. Phys. Chem. A, 2003, 107(41), 8512–8520 CrossRef CAS.
- P. Brühwiler, A. Maxwell, C. Puglia, A. Nilsson, S. Andersson and N. Mårtensson, π* and σ* Excitons in C 1s Absorption of Graphite, Phys. Rev. Lett., 1995, 74(4), 614 CrossRef PubMed.
- S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, Carbon support oxidation in PEM fuel cell cathodes, J. Power Sources, 2008, 176(2), 444–451 CrossRef CAS.
- Z. W. Chen, D. Higgins, A. P. Yu, L. Zhang and J. J. Zhang, A review on non-precious metal electrocatalysts for PEM fuel cells, Energy Environ. Sci., 2011, 4(9), 3167–3192 CAS.
- J. J. Baschuk and X. H. Li, Modelling of polymer electrolyte membrane fuel cells with variable degrees of water flooding, J. Power Sources, 2000, 86(1–2), 181–196 CrossRef CAS.
- J. W. F. To, J. J. He, J. G. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. C. Chen, W. G. Bae, L. J. Pan, J. B. H. Tok, J. Wilcox and Z. N. Bao, Hierarchical N-Doped Carbon as CO2 Adsorbent with High CO2 Selectivity from Rationally Designed Polypyrrole Precursor, J. Am. Chem. Soc., 2016, 138(3), 1001–1009 CrossRef CAS PubMed.
- Technologies, U. Iodometric Titration, http://www.h2o2.com/technical-library/analytical-methods/default.aspx?pid=70&name=Iodometric-Titration.
- J. W. D. Ng, M. Garcia-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Gold-supported cerium-doped NiOx catalysts for water oxidation, Nat. Energy, 2016, 1, 53 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6re00195e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |