
Photomediated oxime ligation as a bioorthogonal tool for spatiotemporally-controlled hydrogel formation and modification†
Payam E.
Farahani
a,
Steven M.
Adelmund
a,
Jared A.
Shadish
a and
Cole A.
DeForest
 *abcd
*abcd
aDepartment of Chemical Engineering, University of Washington, 4000 15th Ave NE, Seattle, WA 98195, USA. E-mail: ProfCole@uw.edu
bDepartment of Bioengineering, University of Washington, 3720 15th Ave NE, Seattle, WA 98195, USA
cInstitute for Stem Cell & Regenerative Medicine, University of Washington, 850 Republican Street, Seattle, WA 98109, USA
dMolecular Engineering & Sciences Institute, University of Washington, 3946 W Stevens Way NE, Seattle, WA 98105, USA
First published on 28th March 2017
Abstract
Click chemistry has proved a valuable tool in biocompatible hydrogel formation for 3D cell culture, owing to its bioorthogonal nature and high efficiency under physiological conditions. While traditional click reactions can be readily employed to create uniform functional materials about living cells, their spontaneity prohibits spatiotemporal control of material properties, thereby limiting their utility in recapitulating the dynamic heterogeneity characteristic of the in vivo microenvironment. Photopolymerization-based techniques gain this desired level of 4D programmability, but often at the expense of introducing propagating free radicals that are prone to non-specific reactions with biological systems. Here we present a strategy for bioorthogonal hydrogel formation and modification that does not rely on propagating free radicals, proceeding through oxime ligation moderated by a photocaged alkoxyamine. Upon mild near UV light exposure, the photocage is cleaved, liberating the alkoxyamine and permitting localized condensation with an aldehyde. Multi-arm crosslinkers, functionalized with either benzaldehydes or photocaged alkoxyamines, formed oxime-based hydrogels within minutes of light exposure in the presence of live cells. Polymerization rates and final mechanical properties of these gels could be systematically tuned by varying crosslinker concentrations, light intensity, aniline catalyst equivalents, and pH. Moreover, hydrogel geometry and final mechanical properties were controlled by the location and extent of UV exposure, respectively. Photomediated oxime ligation was then translated to the biochemical modification of hydrogels, where full-length proteins containing photocaged alkoxyamines were immobilized in user-defined regions exposed to UV light. The programmability afforded by photomediated oxime ligation can recapitulate dynamically anisotropic mechanical and biochemical aspects of the native extracellular matrix. Consequently, photopolymerized oxime-based hydrogels are expected to enable an enhanced understanding of cell-matrix interactions by serving as improved 4D cell culture platforms.
1. Introduction
Hydrogels are water-absorbent polymer networks that have attracted wide use in the areas of 3D cell culture, drug delivery, and regenerative medicine.1–3 Their biocompatibility and mechanical similarity to multiple tissue types make hydrogels suitable tissue engineering scaffolds. These materials have particular importance in probing 3D cell behavior by recapitulating critical aspects of the native extracellular matrix (ECM).4 Cells adapt and respond to mechanical and biochemical cues which spatiotemporally vary throughout their local microenvironment. Consequently, next-generation cell culture platforms are needed to mimic the dynamic heterogeneity of native ECM.The rigidity of the ECM, and its variability over time and 3D space (i.e., 4D), play crucial roles in cellular phenomena such as migration, proliferation, and differentiation.5–7 Spatiotemporal alteration of matrix stiffness within hydrogels can be accomplished by controlling polymer crosslinking densities via photoinitiated free-radical polymerization, a technique for hydrogel fabrication and post-gelation modification.8 While free-radical polymerization allows for the dictation of hydrogel geometry and mechanical properties, propagating radicals employed in this mechanism jeopardize cell viability and function due to non-specific reactions with biological species and generation of reactive oxygen species.9,10 Furthermore, many photoinitiators used to produce free radicals have been shown to be cytotoxic.11
Click reactions have emerged as versatile tools for hydrogel formation and modification, providing a powerful alternative to radical-based chemistries.12,13 Reactions falling under the family of click chemistry are characterized by their rapid procession and high yield under mild conditions, inoffensive byproduct formation, and orthogonality towards chemical species found in biology.13 Numerous click reactions, such as strain-promoted azide–alkyne cycloaddition (SPAAC), tetrazine–alkene addition, and the thiol-based Michael-type reaction have been used for polymerization.14–16 However, the spontaneous nature of these reactions prohibits their use in spatiotemporally presenting biochemical and mechanical cues throughout hydrogel networks. The thiol-ene reaction can also be controlled by light exposure in conjunction with an appropriate photoinitiator, which enables the anisotropic modulation of mechanical and biochemical properties within hydrogels.17,18 However, the required propagating thiyl radical and photoinitiator present similar shortcomings to other photoinitiated free-radical polymerizations. Given the limitations of each of these chemistries, there remains the need for a bioorthogonal, photo-driven approach to hydrogel formation and modification.
Oxime ligation, and similar hydrazone and imine condensations, are spontaneous click reactions of growing interest for hydrogel formation.19,20 In oxime ligation, an alkoxyamine (–ONH2) reacts with an aldehyde (–CHO) or ketone to yield an oxime linkage. The kinetics of oxime ligation can be controlled by adjusting the pH and the amount of an aniline catalyst.20 In addition, alkoxyamines and aldehydes are rare in biology, minimizing undesired biological interactions. The reaction proceeds quickly and effectively irreversibly under physiological conditions, offering a distinct advantage over hydrazone/imine-based reactions whose reversible nature leads to hydrolytic cleavage on the order of days at physiological pHs.21,22 Given its tunable kinetics, bioorthogonality, and stability, oxime ligation has utility in bioconjugation and in vitro labeling.23 These characteristics are equally important in the context of engineering cellular microenvironments.
Efforts to afford spatiotemporal control over oxime ligation and similar chemistries have relied on photocages, photolabile molecules permitting the reaction between complementary species only after photoliberation of one of the reacting groups. The photolabile ortho-nitrobenzyl moiety has been used previously to generate aldehydes in hydrogel photopolymerization and modification strategies that result in hydrazone and imine linkages.24,25 To control oxime formation, 2-(2-nitrophenyl) propyloxycarbonyl (NPPOC) has been utilized as a direct photocage for alkoxyamines. The NPPOC-photocaged alkoxyamine (ONH-NPPOC) can participate in a deprotection-ligation sequence referred to as photomediated oxime ligation, whereby photoliberation of the alkoxyamine allows its reaction with an aldehyde to form a covalent oxime linkage (Fig. 1a).26,27 Importantly, this photochemical reaction sequence proceeds through a mechanism absent of propagating free radicals,28,29 providing high reaction specificity and biocompatibility. We have previously employed photomediated oxime ligation to pattern full-length proteins within synthetic hydrogel biomaterials, which we demonstrated as useful towards directing dynamic stem cell fate in 4D.26
 | ||
| Fig. 1 (a) Alkoxyamines (–ONH2, green) protected by an NPPOC photocage (red) can be utilized for photomediated oxime ligation. Upon near UV irradiation (λ = 365 nm), the alkoxyamine is freed for reaction with a benzaldehyde (–CHO, blue) to form a stable oxime linkage. (b) 8-Arm PEG crosslinkers end-functionalized with either ONH-NPPOC or benzaldehyde moieties provide complementary monomeric units that can be used for hydrogel photopolymerization. (c) Aqueous solutions containing PEG–ONH-NPPOC and PEG–CHO hydrogel precursors undergo step-growth polymerization via photomediated oxime ligation. Cells included in the polymer solution are encapsulated within bulk 3D hydrogels. | ||
Herein, we establish the efficacy of photomediated oxime ligation as an approach to hydrogel photopolymerization and post-gelation modification. Distinct multi-arm poly(ethylene glycol) (PEG) crosslinkers end-functionalized with either ONH-NPPOC or benzaldehyde moieties (denoted respectively as PEG–ONH-NPPOC and PEG–CHO) represent macromolecular precursors capable of polymerizing via oxime ligation, only after ultraviolet (UV) liberation of the alkoxyamine (Fig. 1b and c). Aqueous solutions containing complementary crosslinkers gel rapidly following UV exposure (λ = 365 nm). In addition to crosslinker concentration, we demonstrate that varied UV intensity, aniline concentration, and pH significantly influence polymerization rates and final mechanical properties of formed gels. Moreover, light-based modulation of oxime ligation provides control over the location and extent of polymer crosslinking. After functionalizing full-length proteins with ONH-NPPOC, we demonstrate a new avenue for biochemical modification of hydrogels through protein photopatterning. Cells encapsulated within photopolymerized oxime-based hydrogels exhibited high viability over multiple days, demonstrating this chemistry's potential for future use in cell-based experimentation. Spatiotemporal control over hydrogel mechanics and biochemical cues, afforded by a bioorthogonal click reaction, renders photomediated oxime ligation a promising approach to engineering well-defined cellular microenvironments for tissue engineering applications.
2. Materials and methods
2.1 Small molecule and crosslinker syntheses
Small molecule and multi-arm PEG crosslinker syntheses and characterizations are detailed in the ESI.†2.2 Hydrogel formation and in situ rheological characterization
Polymer solutions were prepared by combining 20 mM solutions of PEG–ONH-NPPOC and PEG–CHO dissolved in deionized water (dH2O) with phosphate buffered saline (PBS) and aniline to yield the desired equimolar crosslinker concentrations [5, 7.5, 10 weight percentage (wt%)], aniline concentration (0, 5, 10 mM), and pH (pH = 6.0, 7.0, or 7.4, measured using an Accumet AB250 pH probe). Except for rheological characterizations, hydrogels were formed by first pipetting 10 μL solutions between Rain-X®-coated glass slides spaced at 500 μm. Solutions were then exposed to light (λ = 365 nm, 10 mW cm−2) for 10 min using an OmniCure® Series 1500 UV lamp, and left to fully cure for 15 min. Hydrogels were equilibrated in PBS (pH = 7.0) before further experimentation.For in situ rheological characterization, 40 μL polymer solutions were placed on a TA Instruments Discovery HR-2 rheometer equipped with a parallel-plate geometry (diameter = 8 mm). Mineral oil was pipetted around the polymer solution to prevent evaporation during measurements. The storage modulus (G′) and loss modulus (G′′) were measured at a gap height of 500 μm and temperature of 25 °C. 365 nm light at varying intensities (2.5, 5, 10 mW cm−2) was irradiated from the bottom plate of a TA instruments UV curing accessory. Except where noted, UV exposure persisted for the entire measurement duration. G′ and G′′ measurements were conducted at a sweep frequency of 1 rad s−1 and strain amplitude of 1%, parameters which were found to lie within the linear viscoelastic region (Fig. S4, ESI†).
2.3 Fluorescent protein production
The sequence for superfolder green fluorescent protein30 (GFP) and mCherry31 were independently placed into PET21-a(+) expression vectors using NdeI and XhoI cut sites added via polymerase chain reaction during C-terminal 6xHis purification tag introduction. The plasmid was then transformed into chemically competent E. coli [BL21(DE3)pLysS] for expression. 20 mL culture was inoculated into 1 L of lysogeny broth supplemented with 100 μg mL−1 ampicillin and grown at 37 °C. When the culture reached an optical density of ∼0.4 at 600 nm, the culture was induced with isopropyl β-D-1-thiogalactopyranoside (0.5 mM). The temperature was reduced to 18 °C and cells were allowed to grow for 24 h. After 24 h, cells were centrifuged at 4000 × g and the supernatant was decanted. The cell pellet was suspended in lysis buffer (20 mM Tris, 50 mM NaCl, 10 mM imidazole, pH 7.4) and sonicated 6 times (3 min on, 3 min off, with a 30% duty cycle). The solution was then centrifuged at 5000 × g for 20 min to pellet the insoluble fraction. The soluble portion was applied to 5 mL of Ni–nitrilotriacetic acid resin, washed 5 times with 20 mL of wash buffer (20 mM Tris, 50 mM NaCl, 25 mM imidazole, pH 7.4) and eluted with 50 mL elution buffer (20 mM Tris, 50 mM NaCl, 250 mM imidazole, pH 7.4). The elution fraction was dialyzed [molecular weight cutoff (MWCO) = 10 kDa] against dH2O (pH 7.4) containing 20 mM Tris and 50 mM NaCl to remove imidazole, yielding a final product of 2.0 mg mL−1 (74 μM) fluorescent protein solution.2.4 Protein modification with ONH-NPPOC
GFP in Tris buffer (0.21 μmol, 2.8 mL) was exchanged into a sodium bicarbonate buffer (2 mL, 0.1 M, pH 8.3) using a centrifugal filter (Amicon Ultra-4, 10 kDa MWCO). NPPOC-ONH–NHS in anhydrous dimethylformamide (DMF) (2.1 μmol, 222 μL) was added to the dissolved protein. The mixture was placed on a shaker wheel for 2 h at 25 °C, protected from light. The mixture was then transferred to a centrifugal filter, where the modified GFP (GFP–ONH-NPPOC) was washed three times with PBS (pH 7.4). GFP–ONH-NPPOC was isolated in PBS at a final concentration of 1.4 mg mL−1 (50 μM). ONH-NPPOC-functionalized mCherry (mCherry–ONH-NPPOC) was synthesized using the same protocol, starting with unmodified mCherry.2.5 Biochemical patterning of hydrogels
Precursor solutions were prepared containing PEG–CHO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEG–ONH-NPPOC at a molar ratio of 1:0.9 (3.33 mM PEG–CHO, 3 mM PEG–ONH-NPPOC, in PBS, 10 mM aniline, pH 7.0). Solutions were irradiated with light (λ = 365 nm, 10 mW cm−2) for 10 min. Hydrogels formed from UV exposure were then incubated overnight in PBS containing GFP–ONH-NPPOC (50 μM) and aniline (10 mM). Protein-swollen gels were irradiated with UV light (365 nm, 10 mW cm−2, 5 min) through a chrome-quartz photomask (Advance Reproduction Corporation). Hydrogels were then washed in PBS for 8 h. Final gels were imaged using a Leica SP8X laser scanning confocal microscope.
:PEG–ONH-NPPOC at a molar ratio of 1:0.9 (3.33 mM PEG–CHO, 3 mM PEG–ONH-NPPOC, in PBS, 10 mM aniline, pH 7.0). Solutions were irradiated with light (λ = 365 nm, 10 mW cm−2) for 10 min. Hydrogels formed from UV exposure were then incubated overnight in PBS containing GFP–ONH-NPPOC (50 μM) and aniline (10 mM). Protein-swollen gels were irradiated with UV light (365 nm, 10 mW cm−2, 5 min) through a chrome-quartz photomask (Advance Reproduction Corporation). Hydrogels were then washed in PBS for 8 h. Final gels were imaged using a Leica SP8X laser scanning confocal microscope.
Using the protocol outlined above, simultaneous and sequential patternings were conducted with modifications to the protein incubation solution. Simultaneous patterning was achieved by incorporating both GFP–ONH-NPPOC and mCherry–ONH-NPPOC (25 μM of each protein, 10 mM aniline) into the incubation solution. Sequential patterning was achieved by first patterning mCherry–ONH-NPPOC (2.5 μM, 10 mM aniline), and washed in PBS for 8 h. mCherry-patterned gels were then patterned with GFP–ONH-NPPOC (2.5 μM, 10 mM aniline).
2.6 Cell encapsulation and viability
NIH/3T3 fibroblasts (3 × 106 cells per mL) were suspended in 7.5 wt% crosslinker solutions dissolved in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) containing fetal bovine serum (FBS, 10%) and penicillin–streptomycin (PS, 1%). 10 μL cell suspensions were pipetted between Rain-X®-treated glass slides separated by a 500 μm spacer. Cell suspensions were then treated with light (λ = 365 nm, 10 mW cm−2) for 5 min to form hydrogels. 30 min after UV treatment, hydrogels were removed from glass slides, individually placed into 6-well plates containing DMEM (5 mL, 10% FBS, 1% PS) and incubated at 37 °C and 5% CO2. The viability of NIH/3T3 fibroblasts within hydrogels was assessed 24, 48, and 72 h after encapsulation. Cells were stained with calcein and Hoechst 33342 (Thermo Fisher Scientific), and imaged using a Leica SP8X confocal laser scanning microscope. Cells stained by both calcein and Hoechst were determined to be alive, while cells only stained by Hoechst were determined to be dead. Cell viability was quantified using ImageJ.3. Results and discussion
3.1 Characterization of hydrogel formation
Photomediated oxime ligation is a step-growth polymerization method that provides four tunable parameters that dictate hydrogel formation rates and final material moduli. These parameters include the concentration of PEG crosslinkers, UV light intensity, aniline concentration, and pH. A baseline set of parameters for equimolar PEG crosslinker solutions was determined based on previous reports of hydrogel formation via oxime ligation or photopolymerizations (7.5 wt% PEG, 365 nm light at 10 mW cm−2, 10 mM aniline, pH 7.0).20,24 Oxime bonds are essentially irreversible under physiological conditions,20 and, as expected, hydrogels formed under these conditions remained intact when stored in PBS at room temperature for at least two weeks. The storage modulus (G′) and loss modulus (G′′) for all oxime-based hydrogels were measured by in situ parallel-plate rheometry under continuous light exposure (λ = 365 nm), except where noted otherwise. Irradiation of polymer solutions elicited a rapid increase in G′ (Fig. S5, ESI†). For the standard set of conditions, the crossover point between G′ and G′′, indicative of the gel point, was found to occur after 56 ± 4 s (Fig. S5, ESI†). The gel reached 90% of its final storage modulus within 515 ± 35 s of UV light exposure, with an average storage modulus of 9.5 ± 1.3 kPa. To assess the tunable nature of this hydrogel platform, a series of experiments was designed to vary PEG wt%, UV intensity, aniline concentration, or pH, while the other variables were maintained at the baseline condition.The storage modulus of hydrogels is proportional to the crosslinking density of the comprising polymer network.32 Thus, increasing the total crosslinker wt% in polymer solutions can lead to increasingly stiffer gels due to a higher crosslinking density, in addition to a lower degree of swelling. Faster polymerization rates are often observed with increasing crosslinker wt%, resulting from an increased concentration of crosslinking end-groups. As expected, increasing rates of G′ evolution were observed with higher PEG wt% amounts (Fig. 2a). Varying PEG wt% between 5–10% allowed for the formation of hydrogels with storage moduli between ∼1.8–17 kPa (Fig. 2a). The final moduli of hydrogels for each PEG wt% were comparable to values reported for similar systems.22,33
 | ||
| Fig. 2 In situ rheological measurements of hydrogel storage moduli (G′). Four variables were identified with the potential to impact the polymerization rates and mechanical properties of hydrogels formed by photomediated oxime ligation: (a) PEG weight percentage, (b) UV light intensity, (c) aniline concentration, and (d) pH. Individual parameters were varied from a baseline set of conditions (7.5 wt% PEG, 10 mW cm−2, 10 mM aniline, pH 7.0). Light irradiation begins at t = 0 s and persists for the duration of the experiment. The mechanical properties (G′ evolution profiles and final G′ values) of each hydrogel were obtained via parallel-plate rheometry. Error bars represent one standard deviation. *: p < 0.05 determined by a one-way ANOVA. | ||
Since the formation of oxime-based linkages is contingent upon NPPOC cleavage, it was expected that UV intensity would influence polymerization kinetics through the rate of alkoxyamine liberation. Our previous studies of NPPOC cleavage kinetics26 and extensive prior work in the field15,18,34,35 led to the selection of 10 mW cm−2 as the standard intensity for rheological studies. Decreasing UV intensity from this standard value led to moderately slower responses in the evolution of G′ (Fig. 2b). However, intensities as low as 2.5 mW cm−2 displayed polymerization rates comparable to those at 10 mW cm−2. These comparable polymerization rates despite a 75% decrease in intensity are rationalized by the rapid cleavage kinetics of NPPOC,26 relative to the rate-limiting oxime ligation responsible for increases in G′. Minimizing intensity and exposure time may serve to ameliorate potential deleterious effects of UV light on biological systems, without significant influence on the time required for oxime-based hydrogel formation.36
In the absence of a catalyst, the time required for complete oxime formation is generally on the order of hours.37 This reaction timescale may be prohibitively slow for 3D cell encapsulation and other contexts that require rapid and complete formation of covalent linkages. Aromatic amines, including aniline, have garnered usage as biocompatible catalysts for oxime ligation, providing the reaction utility in time-sensitive bioconjugations and labeling applications.38,39 In the case of photomediated oxime ligation, the presence of aniline led to complete hydrogel formation in a matter of minutes, while lowering the concentration of aniline in solution led to a dramatic decrease in the rates of polymerization (Fig. 2c). In addition to slower polymerization rates, lowering aniline concentration led to a decrease in the final G′ (Fig. 2c). These trends are consistent with previous work probing the influence of aniline on oxime-based hydrogel formation.20 These findings suggest the potential for aniline to be used as a handle for tuning hydrogel mechanical properties in addition to polymerization kinetics.
Oxime ligation is extremely sensitive to pH; under acidic conditions (pH < 6.5), the rate of reaction is several orders of magnitude faster than that under more basic conditions (pH > 7.0).20 Rapid hydrogel formation at a physiological pH (7.4) is desired for this approach to have utility in cell-based applications. The pH-dependence of photomediated oxime ligation was explored by assessing the rate of hydrogel formation at pH values of 6.0, 7.0, and 7.4. Hydrogels formed at pH 7.0 and 7.4 were comparable in both polymerization kinetics and final G′, suggesting photomediated oxime ligation can be used between pH 7.0–7.4 without significantly influencing polymerization rates and mechanical properties (Fig. 2d). Hydrogels formed at pH 6.0 reached their final G′ significantly faster than hydrogel samples of pH 7.0 and 7.4 (Fig. 2d). However, the final G′ of hydrogels at pH 6.0 was statistically lower than those of hydrogels formed at pH 7.0 and 7.4 (Fig. 2d). Since oxime ligation can be reversed under highly acidic conditions, we hypothesize the reaction equilibrium at pH 6.0 shifts away from the oxime.23 Consequently, an increase in unreacted end-groups would lead to a lower crosslinking density and G′. A similar decrease in G′ was observed for hydrogels formed at pH 6.0 via hydrazone condensation.24
3.2 Spatiotemporal control over hydrogel polymerization
The crosslinking mechanisms of free-radical and thiol–ene chemistries rely on propagating radicals whose activities are difficult to moderate once polymerization is initiated. These propagating mechanisms necessitate the introduction of additional crosslinkers after complete hydrogel formation in order to spatially stiffen regions via site-selective light exposure.6 Conversely, the use of an NPPOC photocage to mask a non-propagating reactive group provides control of the number of liberated alkoxyamines via the duration of UV exposure. Thus, the non-propagating mechanism of photomediated oxime ligation makes it possible to temporally control the extent of polymer crosslinking. Limiting the irradiation dosage dictates the degree of polymerization, providing control of mechanical properties without modifying the hydrogel precursor composition (Fig. 3a). Moderating the duration of UV exposure affords control of G′ over at least two orders of magnitude (Fig. 3b). An initial 10 s dose of UV light followed by sufficient time for equilibration led to a hydrogel with G′ of ∼75 Pa. This process was repeated by pulsing 5 s of UV light and allowing the gel to re-equilibrate to a G′ of ∼850 Pa. Each successive pulse produced additional stiffening until the storage modulus approached that of the fully crosslinked networks exhibited in Fig. 2. Seconds of light exposure lead to dramatic increases in polymer crosslinking, occurring on the order of minutes. These results suggest that oxime ligation, as opposed to alkoxyamine uncaging, is the rate-limiting step in this reaction sequence. Correlation between irradiation time and G′ could be used to tune the final mechanical properties of these materials. | ||
| Fig. 3 (a) Photopolymerized oxime-based hydrogels can be partially crosslinked by limiting UV exposure. Subsequent photomasked irradiation dictates the location and extent of additional crosslinking. (b) Storage modulus evolution of hydrogel solutions increases in response to an initial 10 s of UV exposure (10 mW cm−2), followed by 3 successive pulsed light exposures (5 s each). Purple lines denote times of UV exposure. (c) Hydrogel after exposure to UV light through a parallel-line photomask. Opaque regions correspond to areas of NPPOC cleavage and oxime ligation, while dark regions correspond to the unreacted polymer solution (scale bar = 1 mm). (d) Brightfield microscope image of a hydrogel patterned to display light 25 μm-wide sol regions dispersed throughout the dark gel (scale bar = 100 μm). | ||
In addition to temporal control over crosslinking, the light-driven nature of photomediated oxime ligation allows polymerization to be confined to user-defined regions within samples. Photomasked light exposure to polymer solutions induced gelation exclusively in UV-irradiated regions (Fig. 3c and d). The NPPOC photocleavage led to an increase in opacity, making it convenient to visualize patterned hydrogels without the aid of fluorescent markers. Hydrogel polymerization was controlled with geometries whose features spanned tens to several hundreds of microns (Fig. 3c and d). Directing micron-scale polymerization events holds promise in engineering mechanically heterogeneous microenvironments, where spatial variations in matrix rigidity over sizes relevant to single cells can be used to probe mechanobiological responses.
3.3 Protein patterning within 3D hydrogels
Similar to mechanical stimuli, biochemical cues throughout the ECM orchestrate cell behavior in spatiotemporal manners. Therefore, we translated photomediated oxime ligation into a biomolecular patterning strategy within hydrogel networks. Providing excess benzaldehyde end-groups in the gel formulation, GFP–ONH-NPPOC was immobilized within the polymer network following UV light exposure (Fig. 4a and b). A photomask was used to demonstrate the spatial patterning capabilities of this technique, providing protein patterns with sharp, micron-scale features that extended throughout the 3D gel, as visualized by confocal microscopy (Fig. 4c and d). This patterning approach was extended to the sequential and simultaneous immobilization of multiple fluorescent proteins (GFP–ONH-NPPOC and mCherry–ONH-NPPOC) (Fig. 4e and f). From these results, we expect any ONH-NPPOC-functionalized biomolecules smaller than the mesh size of the hydrogel network could be introduced at user-defined times and locations via photomediated oxime ligation. Control of biomolecule immobilization on this length-scale allows this patterning strategy to be used in experiments that biochemically probe cell behavior within a well-defined microenvironment. In its current state, the use of NPPOC-functionalized biomolecules would prevent simultaneous modification of biochemical and mechanical properties in the same area by inadvertently stiffening a partially-crosslinked hydrogel. However, it is expected that wavelength-orthogonal photocages could be used to independently tune biochemical and mechanical stimuli.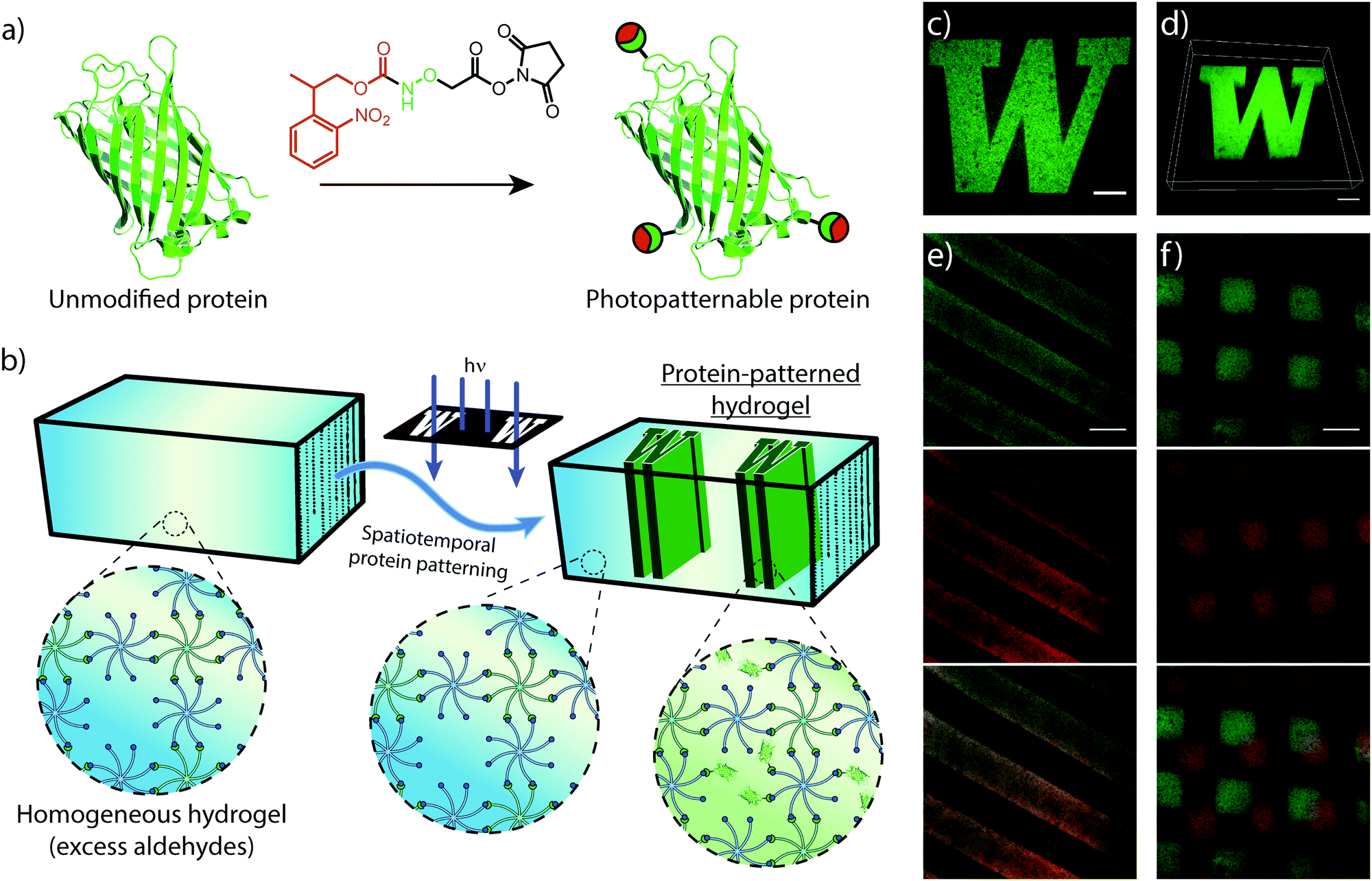 | ||
| Fig. 4 (a) Fluorescent proteins are functionalized with ONH-NPPOC through NHS chemistry. (b) Off-stoichiometric hydrogels are prepared (1:0.9, CHO:ONH-NPPOC), leaving unreacted benzaldehydes uniformly distributed throughout the polymer network. Subsequent treatment of the gel with ONH-NPPOC-functionalized fluorescent protein and photomasked UV irradiation immobilizes the protein in irradiated regions. (c) Micron-resolution patterns obtained by spatially immobilized GFP (scale bar = 100 μm). (d) 3D z-stack confocal image displaying uniform protein concentrations over 100 μm (scale bar = 100 μm). This patterning approach can be applied to (e) simultaneously, and (f) sequentially, pattern multiple ONH-NPPOC-functionalized biomolecules. (e) Simultaneous patterning of GFP and mCherry (top to bottom: GFP, mCherry, and overlay, scale bar = 200 μm). (f) Sequential patterning of GFP and mCherry (top to bottom: GFP, mCherry, and overlay, scale bar = 200 μm). | ||
3.4 Cell encapsulation and viability within hydrogels formed by photomediated oxime ligation
Cells have demonstrated long-term viability when encapsulated in 3D PEG networks containing proper environmental cues.4 The capability to tune mechanical and biochemical properties within hydrogels is particularly advantageous for engineering cellular microenvironments in vitro. Ideally, the chemistries for such processes would react orthogonally to chemical moieties found in biology, allowing their use in cell-laden materials. NIH/3T3 fibroblasts were encapsulated in hydrogels formed by photomediated oxime ligation, and assessed for viability over a 72 h period (Fig. 5). Cells displayed high viability (90 ± 8%) 24 h post-encapsulation, with no significant decrease in viability seen at 48 h (90 ± 5%) and 72 h (82 ± 8%) time points. Cells within hydrogels adopted rounded morphologies similar to those observed in other hydrogel cell culture platforms, attributed to the physical confinement of cells by the surrounding non-degradable polymer network and the lack of substrates promoting cell adhesion.14,24 Future cell-based applications incorporating enzyme-degradable crosslinkers and adhesive motifs (e.g., RGD peptides40) are expected to allow cells to proliferate and remodel the substrate, leading to a well-spread, physiologically-relevant morphology.16 These results demonstrate a biocompatible photochemistry for hydrogel formation, from which mechanical and biochemical cues can be modulated post-gelation. | ||
| Fig. 5 (a–c) NIH/3T3 fibroblasts were encapsulated in hydrogels (3 × 106 cells per mL) formed by photomediated oxime ligation and their viability assayed at 24, 48, and 72 h. Live cells were visualized with a calcein stain (green) and all cells were stained with Hoechst 33342 (blue). High viability was observed 24 h after encapsulation: representative calcein, Hoechst, and merged images are shown in panels a, b, and c, respectively (scale bar = 200 μm). (d) Cell viability remained high throughout the 72 h culture, over which no statistically significant decrease in viability was observed (determined by a one-way ANOVA). Error bars represent one standard deviation. | ||
4. Conclusion
We have presented a click-type photochemistry with utility in hydrogel photopolymerization and 4D post-gelation modification that proceeds in the absence of propagating free radicals. Light-based moderation of oxime ligation provides control over the location and extent of polymer crosslinking. This system responds rapidly to light, resulting in gelation minutes after UV exposure. Several parameters (e.g., polymer wt%, UV exposure, aniline concentration, pH) can be used to control the mechanical properties of hydrogels formed via photomediated oxime ligation. By dictating the duration of UV exposure, we have demonstrated that the systematic tuning of mechanical properties can be achieved without altering hydrogel precursor composition. The same photo-driven oxime chemistry can be used to immobilize multiple biomolecules with spatiotemporal control and micron-scale resolution. The bioorthogonal nature of this chemistry enables its use in fabricating cell-laden hydrogels, as seen by the high viability of NIH/3T3 fibroblasts following encapsulation. While radical-based polymerization is a mature technology with its own strengths, photomediated oxime ligation represents a promising alternative with distinct advantages. Harnessing the mechanical and biochemical versatility of these hydrogel platforms will enable the design of well-defined environments, opening doors to improved studies interrogating cellular biology.Acknowledgements
The authors thank Alshakim Nelson's laboratory at the University of Washington (UW) for the generous use of their rheometer. We acknowledge the support from the NIH to the UW W. M. Keck Microscopy Center (S10 OD016240). P. E. F. was supported by the Mary Gates Research Scholarship, Washington NASA Space Grant Consortium (NASA Grant #NNX15AJ98H), and Washington Research Foundation Fellowship. This work was supported by a UW Faculty Startup Grant (C.A.D.).References
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 421–444 CrossRef CAS PubMed.
- B. C. Isenberg, P. A. DiMilla, M. Walker, S. Kim and J. Y. Wong, Biophys. J., 2009, 97, 1313–1322 CrossRef CAS PubMed.
- A. Banerjee, M. Arha, S. Choudhary, R. S. Ashton, S. R. Bhatia, D. V. Schaffer and R. S. Kane, Biomaterials, 2009, 30, 4695–4699 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- M. S. Hahn, J. S. Miller and J. L. West, Adv. Mater., 2006, 18, 2679–2684 CrossRef CAS.
- V. A. Bhanu and K. Kishore, Chem. Rev., 1991, 91, 99–117 CrossRef CAS.
- O. I. Aruoma, J. Am. Oil Chem. Soc., 1998, 75, 199–212 CrossRef CAS.
- C. G. Williams, A. N. Malik, T. K. Kim, P. N. Manson and J. H. Elisseeff, Biomaterials, 2005, 26, 1211–1218 CrossRef CAS PubMed.
- C. M. Nimmo and M. S. Shoichet, Bioconjugate Chem., 2011, 22, 2199–2209 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS PubMed.
- S. P. Singh, M. P. Schwartz, J. Y. Lee, B. D. Fairbanks and K. S. Anseth, Biomater. Sci., 2014, 2, 1024–1034 RSC.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- F. Lin, J. Yu, W. Tang, J. Zheng, A. Defante, K. Guo, C. Wesdemiotis and M. L. Becker, Biomacromolecules, 2013, 14, 3749–3758 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- N. Boehnke, C. Cam, E. Bat, T. Segura and H. D. Maynard, Biomacromolecules, 2015, 16, 2101–2108 CrossRef CAS PubMed.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2014, 20, 34–41 CrossRef CAS PubMed.
- M. A. Azagarsamy, I. A. Marozas, S. Spaans and K. S. Anseth, ACS Macro Lett., 2016, 5, 19–23 CrossRef CAS.
- Y. Yang, J. Zhang, Z. Liu, Q. Lin, X. Liu, C. Bao, Y. Wang and L. Zhu, Adv. Mater., 2016, 28, 2724–2730 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
- N. Dendane, A. Hoang, L. Guillard, E. Defrancq, F. Vinet and P. Dumy, Bioconjugate Chem., 2007, 18, 671–676 CrossRef CAS PubMed.
- W. P. H. Giegrich, S. Eisele-Bühler, C. Hermann, E. Kvasyuk and R. Charubala, Nucleosides Nucleotides, 1998, 17, 1987–1996 Search PubMed.
- W. Xi, M. Krieger, C. J. Kloxin and C. N. Bowman, Chem. Commun., 2013, 49, 4504–4506 RSC.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef PubMed.
- N. C. Shaner, R. E. Campbell, P. A. Steinbach, B. N. G. Giepmans, A. E. Palmer and R. Y. Tsien, Nat. Biotechnol., 2004, 22, 1567–1572 CrossRef CAS PubMed.
- S. K. Patel, S. Malone, C. Cohen, J. R. Gillmor and R. H. Colby, Macromolecules, 1992, 25, 5241–5251 CrossRef CAS.
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef CAS PubMed.
- L. A. Sawicki and A. M. Kloxin, Biomater. Sci., 2014, 2, 1612–1626 RSC.
- A. M. Rosales, K. M. Mabry, E. M. Nehls and K. S. Anseth, Biomacromolecules, 2015, 16, 798–806 CrossRef CAS PubMed.
- M. P. Carty, M. Zernik-Kobak, S. McGrath and K. Dixon, EMBO J., 1994, 13, 2114–2123 CAS.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- A. R. Blanden, K. Mukherjee, O. Dilek, M. Loew and S. L. Bane, Bioconjugate Chem., 2011, 22, 1954–1961 CrossRef CAS PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb03400d |
| This journal is © The Royal Society of Chemistry 2017 |