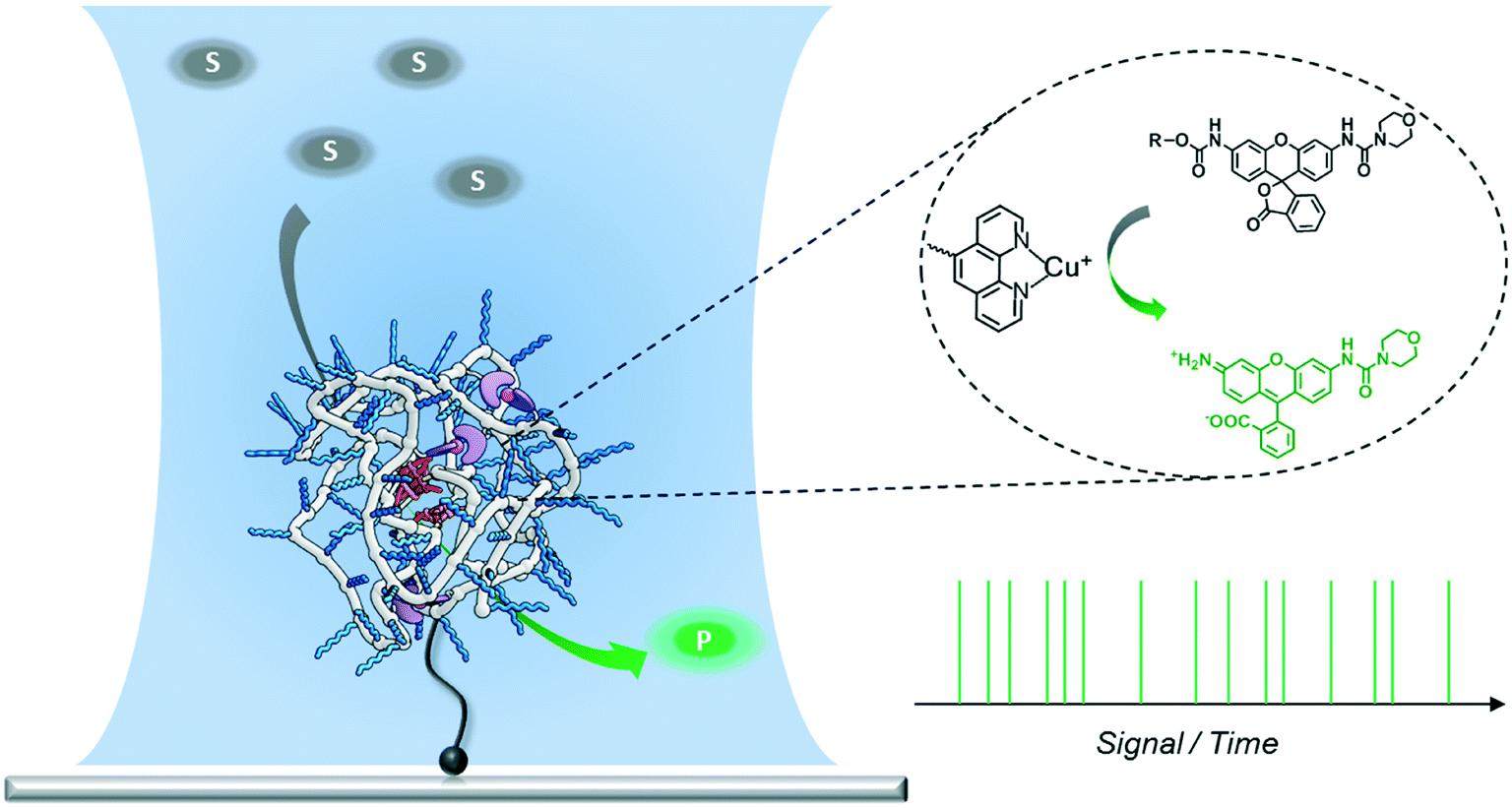
Catalytic single-chain polymeric nanoparticles at work: from ensemble towards single-particle kinetics†
Received
7th April 2018
, Accepted 24th May 2018
First published on 19th June 2018
Abstract
Folding a single polymer chain around catalytically active sites to construct catalytic single chain polymeric nanoparticles (SCPNs) is a novel approach to mimic the activity and selectivity of enzymes. In order to relate the efficiency of SCPNs to their three-dimensional structure, a better understanding of their catalytic activity at an individual level, rather than at an ensemble level, is highly desirable. In this work, we present the design and preparation of catalytic SCPNs and a family of fluorogenic substrates, their characterization at the ensemble level as well as our progress towards analyzing individual SCPNs with single-molecule fluorescence microscopy (SMFM). Firstly, organocopper-based SCPNs together with rhodamine-based fluorogenic substrates were designed and synthesized. The SCPNs catalyze the carbamate cleavage reaction of mono-protected rhodamines, with the dimethylpropargyloxycarbonyl protecting group being cleaved most efficiently. A systematic study focusing on the conditions during catalysis revealed that the ligand acceleration effect as well as the accumulation of substrates and catalytically active sites in SCPNs significantly promote their catalytic performance. Secondly, a streptavidin–biotin based strategy was developed to immobilize the catalytic SCPNs on the surface of glass coverslips. Fluorescence correlation spectroscopy experiments confirmed that the SCPNs remained catalytically active after surface immobilization. Finally, single-SCPN activity measurements were performed. The results qualitatively indicated that fluorescent product molecules were formed as a result of the catalytic reaction and that individual fluorescent product molecules could be detected. So far, no evidence for strongly different behaviors has been observed when comparing individual SCPNs.
Design, System, Application
A new approach to polymer-based catalytic systems has been recently disclosed, which is based on the folding of a polymer chain around catalytically active sites covalently bound to the polymer. In contrast to enzymes, these polymer-based catalysts are structurally heterogeneous, so that the catalytically active sites may reside in different micro-environments. As a result, differences in reactivity may arise due to this diversity of catalytic sites. It is therefore of great interest to investigate this possible diversity and we here design a system to study the catalytic activity at the single-polymer level. In the design, the single-chain polymer nanoparticles are connected to the surface, and substrates are transformed from a luminescent, silent state to a strongly fluorescent state. With these polymers and substrates synthesized it has become possible to study the catalytic activity of individual chains with single-molecule fluorescence microscopy. The new insights obtained in this work can be used to progress the search to an enzyme-mimic for bio-orthogonal chemistry in complex cellular media.
|
Introduction
Folding individual synthetic polymer chains into nanoparticles, which is reminiscent of nature's way of folding polypeptides into proteins, has emerged as a novel approach toward attaining defined polymer architectures.1–6 In general, these polymers are decorated with pendant groups that are capable of forming covalent or supramolecular bonds. After intra-chain (supramolecular) crosslinking between the pendant groups, the individual polymer chains are folded into single-chain polymeric nanoparticles (SCPNs). The incorporation of additional pendant groups on the polymers allows the synthesis of SCPNs with a range of different functionalities. Of particular interest are catalytically active SCPNs, which can be considered as a first step towards catalysts that show the activity and selectivity of natural enzymes.7–14 Organometallic SCPNs, which are reminiscent of metallo-enzymes, have attracted significant attention in recent studies. SCPNs loaded with metal ions, such as those of palladium,7,14 platinum,8 ruthenium,9 rhodium,15 copper,11–13 show efficient and selective catalysis in a variety of reactions. For example, organocopper-based SCPNs were applied in catalyzing the oxidative coupling of terminal acetylenes,11 hydroxylation of phenols,12 and azide–alkyne cycloaddition,13 in which the reaction media ranged from organic solvent to aqueous solution or even an intracellular environment. In this way catalytic SCPNs can be seen as the renaissance of polymer- and dendrimer-based catalysts that have been studied in great detail a few decades ago.16–21
Although significant progress has been achieved in expanding the diversity and versatility of catalytic SCPNs, their catalytic activity lags far behind the performance of enzymes. In order to further improve their catalytic function, it is crucial to gain a deeper insight into the systems currently developed. Our group has developed water-soluble catalytic SCPNs by designing polymers comprising water-soluble oligo(ethylene glycol) side-chains, benzene-1,3,5-tricarboxamides (BTA) supramolecular motifs and catalytic active sites.22–24 A number of techniques have been employed to investigate the BTA-based SCPNs and to improve our understanding of how their 3D structures relate to their functions.25–28 Circular dichroism (CD) spectroscopy and scattering techniques have been utilized to study their inner structure and shape;25,26 single-molecule force spectroscopy has been introduced to provide information on the folding kinetics and pathways.27 Recently, Overhauser dynamic nuclear polarization NMR has been employed to investigate the local water translational diffusion dynamics at the site where catalysis would occur.28 All these experiments have provided detailed information on the properties of SCPNs and the factors that may affect their catalytic performance. While these approaches were mostly focused on structural aspects, methods that are able to directly monitor the catalytic reaction are still lacking. Additionally, ensembles of catalytic SCPNs are intrinsically heterogeneous as the result of the molar mass dispersity of the copolymers in combination with the random incorporation of catalyst sites. As the exact location of the catalysts cannot be controlled, it may be envisaged that every SCPN shows a different activity and selectivity. Therefore, a technique able to monitor the catalytic activity of individual SCPNs is expected to directly reveal if structural heterogeneities are translated into a different catalytic performance.
Single-molecule fluorescence microscopy (SMFM) combined with fluorogenic substrates provides information on the activity of individual catalysts, which is inaccessible in ensemble measurements.29–41 Although mostly applied for investigating enzymatic reactions, the fluorogenic substrate reporter systems also allow for studying non-enzymatic processes in real time.34–38,40,41 Upon cleavage, the initially non-fluorescent substrate is converted into a highly fluorescent product molecule. As every turnover yields one fluorescent product molecule, a detailed and straightforward kinetic analysis is facilitated. In particular, when following the turnover sequence, information about dynamic reaction processes is obtained.42–45 We herein report our first steps toward utilizing SMFM to investigate the catalytic activity of individual SCPNs (Scheme 1). We have developed an experimental strategy to addresses the following challenges: 1) obtaining a catalytically active SCPN platform with modular functionality; 2) synthesizing mono-substituted fluorogenic substrates with different functional groups suitable for ensemble and SMFM experiments; 3) immobilization of active SCPNs on a surface, required for the SMFM experiments; 4) establishing a single-molecule experimental protocol and data analysis.
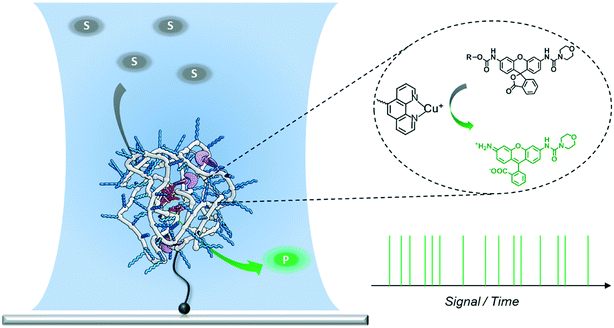 |
| | Scheme 1 Experimental design for measuring the activity of a single SCPN using a confocal fluorescence microscope. The laser is focused onto the position of the SCPN immobilized on a glass coverslip. Every fluorophore produced by the SCPN is recorded in real time. | |
Results and discussion
Design and preparation of catalytic SCPNs
Organocopper-based SCPNs were chosen owing to their efficient catalytic performance in a broad range of reactions. Three ligand-containing polymers (P1–P3) and a catalytically inactive control polymer (P4) were designed (Fig. 1, Table 1). These polymers contain water-soluble polyether side-chains and BTA pendants, which enable them to form SCPNs in aqueous solution. P1–P3 were modified with phenanthroline ligands, which can bind Cu ions to form catalytically active sites, while the control polymer P4 has dodecyl chains instead. To allow surface immobilization, P2 and P3 were decorated with 1% of biotin-containing side chains. Moreover, P3 was labeled with the fluorescent dye Alexa Fluor® 488 to enable its visualization with fluorescence microscopy. The polymers were modularly synthesized using a strategy of post-polymerization modification.46,47 The precursor polymer poly(pentafluorophenyl acrylate) (pPFPA, Mn, SEC = 18.0 kD, Đ = 1.28, DP = 120) was synthesized by reversible addition–fragmentation chain-transfer (RAFT) polymerization and the thiocarbonyl end group was removed upon heating with excess azobisisobutyronitrile (AIBN) and lauroyl peroxide (LPO) (Fig. S1†). P1–P4 were prepared via polymer analogous reactions of pPFPA by sequential addition of amines (Fig. S2–S4†). 5-Amino-N-(1,10-phenanthrolin-5-yl)pentanamide (phen-C4-amine) and the enantiomerically pure, amino-functionalized BTA unit (BTA-amine) were synthesized, the other amines were commercially obtained. After full modification, the final polymers were obtained via dialysis to remove excess amines and byproducts.
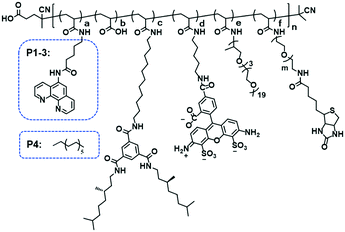 |
| | Fig. 1 Chemical structure of polymer P1–4. P1–P3 were modified with phenanthroline ligands, while the control polymer P4 has dodecyl chains instead. To allow surface immobilization, P2 and P3 were decorated with 1% of biotin-containing side chains. Moreover, P3 was labeled with the fluorescent dye Alexa Fluor® 488 to enable its visualization with fluorescence microscopy. | |
Table 1 Composition and SEC characterization of polymers P1–P4a
| Polymer |
a
|
b
|
c
|
d
|
e
|
f
|
n
|
m
|
M
n (kD) |
Đ
|
|
The values for a–f are determined via19F-NMR. Mn is measured by SEC in DMF with 10 mM LiBr, relative to poly(ethylene oxide) standards.
|
|
P1
|
0.08 |
0.04 |
0.10 |
— |
0.78 |
— |
120 |
— |
31.4 |
1.18 |
|
P2
|
0.08 |
0.08 |
0.10 |
— |
0.73 |
0.01 |
120 |
45 |
28.0 |
1.30 |
|
P3
|
0.08 |
0.10 |
0.10 |
0.01 |
0.70 |
0.01 |
120 |
45 |
27.8 |
1.20 |
|
P4
|
0.08 |
— |
0.10 |
— |
0.82 |
— |
120 |
— |
31.7 |
1.20 |
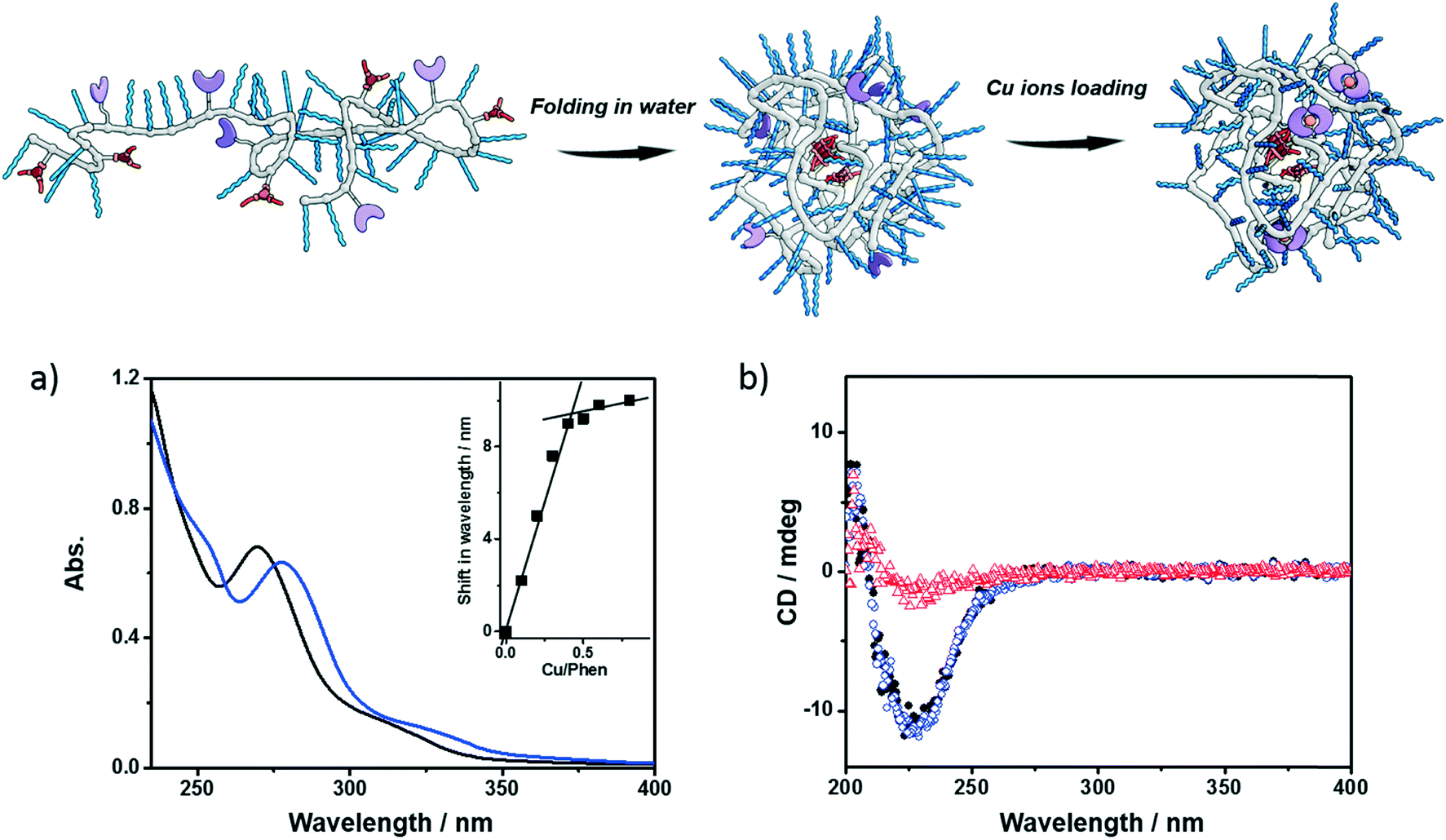
Next, the polymers were complexed with Cu2+ ions. For this purpose, the polymers were dissolved in water, sonicated and subjected to a heating–cooling procedure. Then CuSO4 was added to the solution. Taking P1 as an example, its folding behavior, complexation with Cu2+ ions (P1@Cu(II)) and SCPN formation were characterized using a combination of techniques. The binding of Cu2+ ions to the phenanthroline ligand was studied with UV-vis spectroscopy. As shown in Fig. 2a, the phenanthroline ligand shows a characteristic absorption band around 270 nm. Upon addition of CuSO4 the absorption undergoes a red-shift to around 280 nm. A detailed plot of Cu2+ ratio versus shift in absorption reveals a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry between the phenanthroline ligand and Cu2+ ions. In the CD spectra of P1, a negative Cotton effect was present at 223 nm before and after Cu2+ complexation without a change in value. Moreover, the CD temperature scans of P1 with or without CuSO4 overlap (Fig. S5†). These observations indicate that (i) the BTA pendants form helically stacked dynamic aggregates driving the folding of polymer chains and (ii) Cu2+ complexation is not interfering with BTA aggregation.
:1 binding stoichiometry between the phenanthroline ligand and Cu2+ ions. In the CD spectra of P1, a negative Cotton effect was present at 223 nm before and after Cu2+ complexation without a change in value. Moreover, the CD temperature scans of P1 with or without CuSO4 overlap (Fig. S5†). These observations indicate that (i) the BTA pendants form helically stacked dynamic aggregates driving the folding of polymer chains and (ii) Cu2+ complexation is not interfering with BTA aggregation.
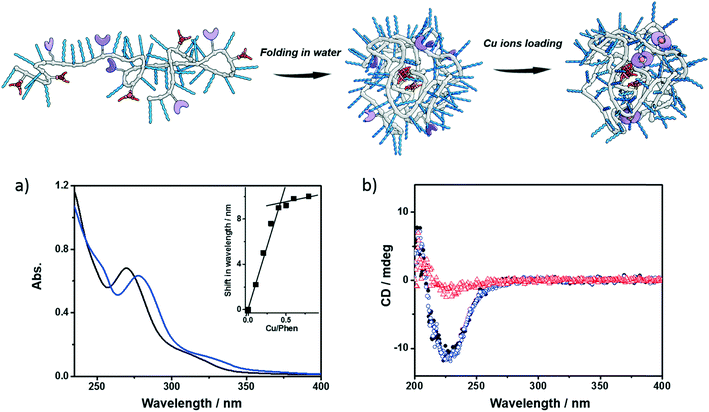 |
| | Fig. 2 Folding and Cu-loading of the polymers; a) UV-vis spectra of P1 (black, 0.6 mg mL−1) and P1@Cu(II) (blue, 0.6 mg mL−1, [phen]:[Cu] = 2:1); b) CD spectra of P1 (0.5 mg mL−1, black dot: 20 °C, red triangle: 90 °C), P1@Cu(II) (blue square, 20 °C, 0.5 mg mL−1, [phen]:[Cu] = 2:1). | |
Small-angle X-ray scattering (SAXS) and dynamic light scattering (DLS) were employed to evaluate the size of the polymeric nanoparticles formed. SAXS measurements indicate that P1 and P1@Cu(II) form nanoparticles with a radius of gyration (Rg) of 5.6 and 5.4 nm, respectively (Fig. S6†). The DLS results of P1 and P1@Cu(II) show a monodisperse distribution of particles with a hydrodynamic radius (Rh) of 7.4 and 8.2 nm, respectively (Fig. S7†). The scattering results are comparable to similar systems reported previously, suggesting that single-polymer chains fold into SCPNs indeed. P2–P4 were characterized in the same way as P1 and similar results were obtained (Fig. S7, Table S1†). P3, which is modified with Alexa Fluor® 488, was further studied with fluorescence correlation spectroscopy (FCS). The FCS experiments were performed at a concentration of 100 nM of P3 in aqueous solution. The diffusion of the P3-based fluorescent nanoparticles was found to be approximately one order of magnitude slower than a reference fluorophore (ATTO488, Fig. S8†). This indicates that the Alexa Fluor® 488 dye was successfully coupled to the polymers. Using the Stokes–Einstein equation, the Rh of P3 was determined to be 8.3 ± 0.8 nm, which is consistent with the light scattering results.
Design and synthesis of fluorogenic substrates
A series of fluorogenic substrates were designed that are predicted to be good substrates for the Cu-phenanthroline functionalized SCPNs. Considering that these substrates should be suitable for both ensemble and single-SCPN kinetic experiments, a number of criteria need to be considered: i) the substrates are stable in aqueous solution, but can undergo fast cleavage in the presence of Cu(I)-based organometallic catalysts, catalysts made out of the Cu(II)-based precursor; ii) the substrates are based on a fluorophore with high brightness, so that a high signal-to-noise ratio can be achieved; iii) the substrates contain only one cleavable group, in order to avoid the formation of reaction intermediates with different fluorescent properties; and iv) the substrates are hydrophobic and are converted into hydrophilic products, which could facilitate the SCPN-based catalysis. With these guidelines in mind, substrates S1–S4 were designed and synthesized (Fig. 3). Rhodamine 110 (Rh110) was chosen as the fluorophore owing to its high brightness. However, it carries two amino groups for further modification. To prepare a substrate with a single reactive site, one amino group of Rh110 was first modified with a morpholine-carbonyl group to obtain MC-Rh110.48–50 The urea bond formed is not reactive and hydrolytically stable. At the same time, MC-Rh110 remains sufficiently bright for SMFM experiments.48 Further modification of the remaining amino group to generate carbamate bonds yields substrates S1–S4, which contain different alkyne or alkene groups. The carbamate bond of S1 was previously shown to be hydrolyzed in the presence of Cu(I),51 while the carbamate bond in S2 is labile in the presence of Pd(II) catalysts.52,53 The hydrolytic stability of the carbamate bonds in S3 and S4 in the presence of Cu(I) is not known.
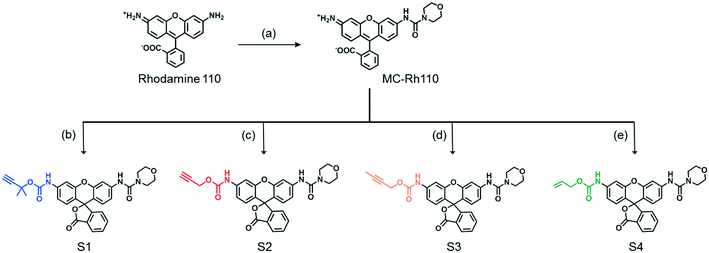 |
| | Fig. 3 Chemical structures and synthesis of the substrates S1–S4. (a) 4-Morpholinecarbonyl chloride; (b) triphosgene, 2-methylbut-3-yn-2-ol; (c) propargyl chloroformate; (d) triphosgene, but-2-yn-1-ol; (e) triphosgene, prop-2-en-1-ol. | |
SCPN-based catalysis in aqueous solution
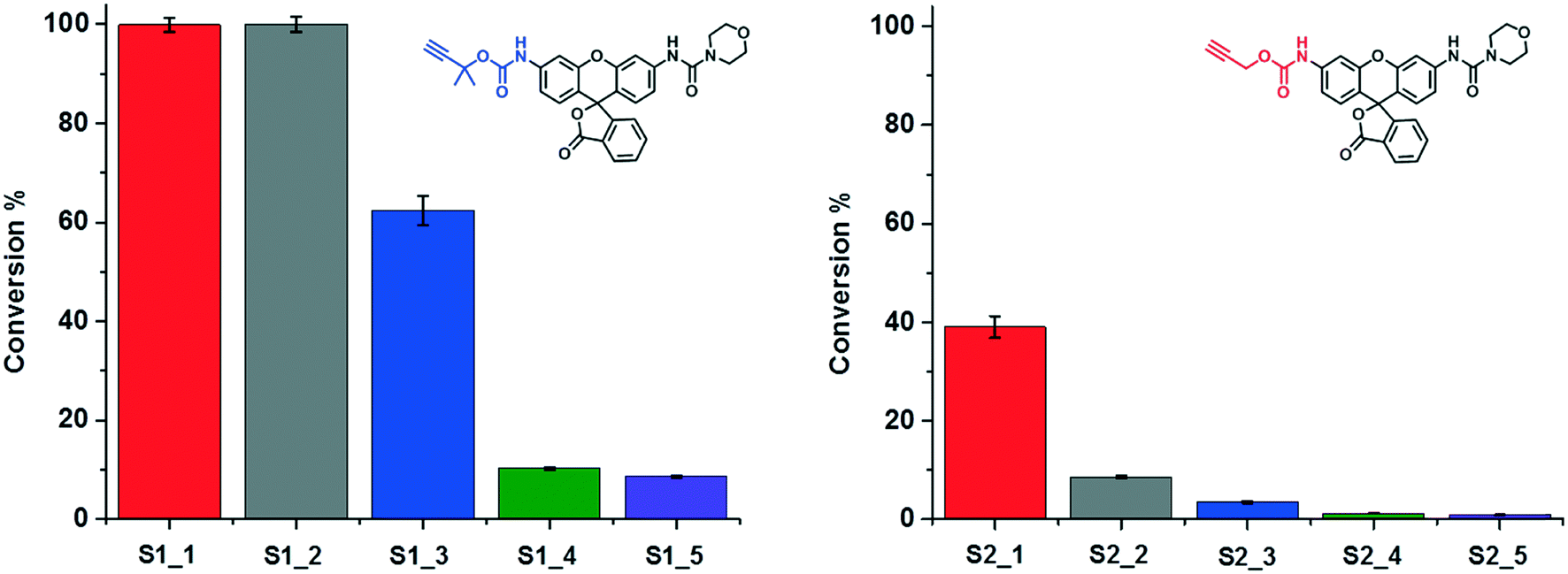
The catalytic performance of SCPNs in the carbamate-cleavage reaction was first studied at the ensemble level in aqueous solution. The progress of the reaction was monitored with fluorescence spectroscopy as well as liquid chromatography-mass spectroscopy (LC-MS) (Fig. S9 and S10†). Whereas the former provides information about the catalytic reaction in real time, LC-MS allows exact quantification of the substrate conversion at set time intervals. As a first step, a substrate activity screening was performed. P1@Cu(I) ([phen]:[Cu] = 2:1) was chosen as a representative catalyst. Generally, a stock solution of the substrate (in DMSO) was added into a P1@Cu(II) solution (2 μM) to reach a final concentration of 30 μM. The reactions were initialized by adding sodium ascorbate (NaAsc, 1 mM) to generate P1@Cu(I)in situ. The fluorescence intensity was recorded at 520 nm for 30 min and substrate conversion was directly analyzed with LC-MS (Fig. S9b†). The fluorescence–time curves were normalized relative to the maximum conversion and are shown in Fig. 4. Substrates S1 and S2, which contain end-functionalized alkyne moieties, are hydrolyzed, with the tertiary alkyne bearing S1 showing the highest TOF (∼200 h−1). In contrast, S3 and S4, which contain internal alkyne and alkene moieties, respectively, show almost no reactivity. It has been proposed that the formation of a Cu-acetylide intermediate is crucial in the catalytic mechanism. The terminal alkyne moieties of S1 and S2 bind Cu(I) to generate the Cu-acetylide. This leads to the formation of a Cu-stabilized propargyl cation thus cleaving the carbamate bond. The methyl groups of the tertiary propargyl moiety in S1 further stabilize the cation, thereby promoting the cleavage reaction.51
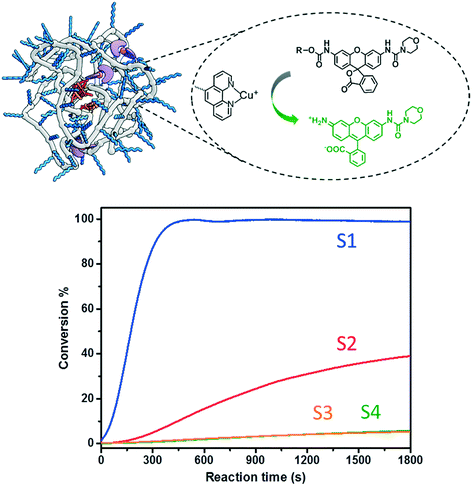 |
| | Fig. 4 Carbamate cleavage reaction of S1–S4 catalyzed by P1@Cu(I) ([P1] = 2 μM, [CuSO4] = 10 μM, NaAsc = 1 mM, substrate = 30 μM). The kinetic curves were recorded via fluorescence spectroscopy (ex. 495 nm; recorded at 520 nm) followed with normalization according to the conversions obtained from LC-MS. | |
The ligand-containing SCPNs have several merits toward aqueous catalysis: α) ligand accelerated catalysis; β) hydrophobic interior; and γ) accumulation of catalytically active sites. Whereas the first feature is determined directly by the mechanism of the Cu-catalyzed reaction, the other two features lead to an enhancement of the local concentration of active sites and/or substrate molecules. To verify in how far these three features are responsible for enhancing the kinetics of the hydrolysis reaction, we performed systematic kinetic experiments on S1 and S2. As catalysts, we selected P1@Cu(I), which possesses contributions from α, β and γ, whereas P4 mixed with Phen@Cu(I) only possesses the α and β contribution. As reference Phen@Cu(I), P4 mixed with Cu(I) and only Cu(I) were used (Table 2). The outcome of these catalysis experiments is shown in Fig. 5 (and Fig. S9c†). The phenanthroline ligand indeed accelerates the Cu(I)-catalyzed carbamate-cleavage reaction. Moreover, the accumulation of catalytic active sites and substrates enhances the reaction to a large extent. In short, the more characteristics the system contains, the more effective the catalytic reaction becomes.
Table 2 Catalysts with different characteristics. α) ligand accelerated catalysis; β) hydrophobic interior; γ) accumulation of catalytically active sites
| Entry (S1/S2) |
Catalyst |
Characteristics |
| 1 |
P1@Cu(I)
|
α, β, γ |
| 2 |
P4 & Phen@Cu(I) |
α, β |
| 3 |
Phen@Cu(I)
|
α |
| 4 |
P4 & Cu(I) |
β |
| 5 |
Cu(I)
|
— |
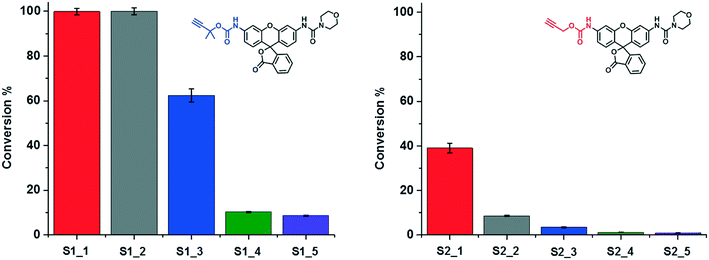 |
| | Fig. 5 A study on the characteristics of SCPN-based catalysis. The conversions of the reactions under different catalytic conditions (Table 2) were obtained though LC-MS. | |
Surface immobilization of catalytic SCPNs
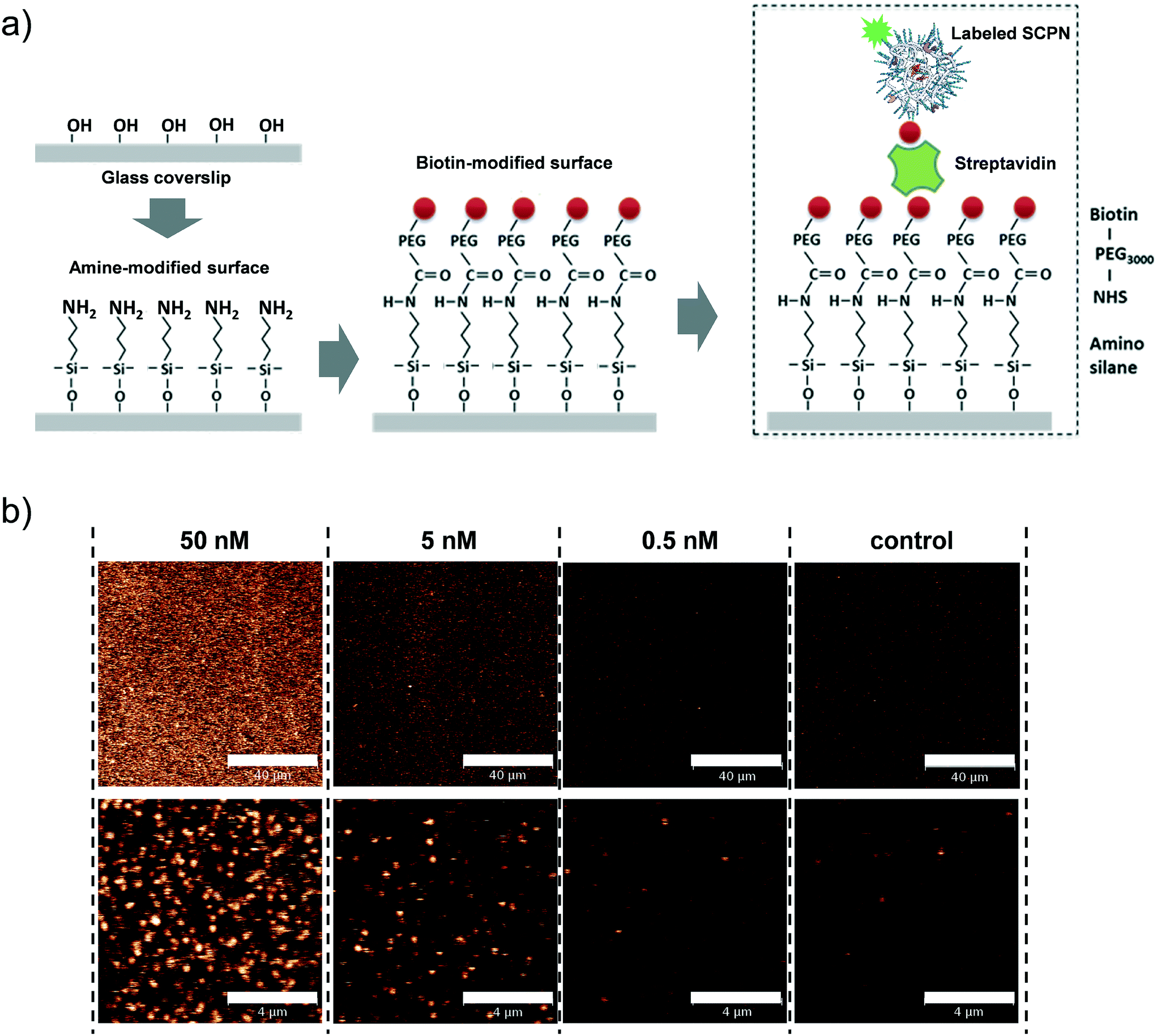
Having obtained insights into the substrate specificity and the kinetics of the SCPN catalyzed reactions, a first step was taken toward single-SCPN experiments. For observing individual SCPNs with a confocal microscope, the SCPNs need to be immobilized on the surface of glass coverslips. We selected an immobilization strategy based on the well-established streptavidin–biotin interaction. In detail, the glass surface was first modified with an amino silane (3-aminopropyl dimethylethoxy silane). In the next step, a hetero-bifunctional poly(ethylene glycol) containing an amino-reactive NHS ester and a biotin group (NHS-PEG3000Da-biotin) was used to obtain a biotin covered surface. The biotin-containing SCPNs were then immobilized on the surface using streptavidin as a bridge (Fig. 6a). P3 was employed to test the SCPN immobilization protocol. P3 contains biotin motifs on its side-chains and is labeled with Alexa Fluor® 488, which allows its visualization with confocal microscopy. Fig. 6b shows confocal images obtained for different surfaces incubated with P3 at concentrations of 50 nM, 5 nM, and 0.5 nM, respectively. Inspection of Fig. 6b shows that the immobilization is successful, as indicated by the concentration-dependent density of SCPNs.
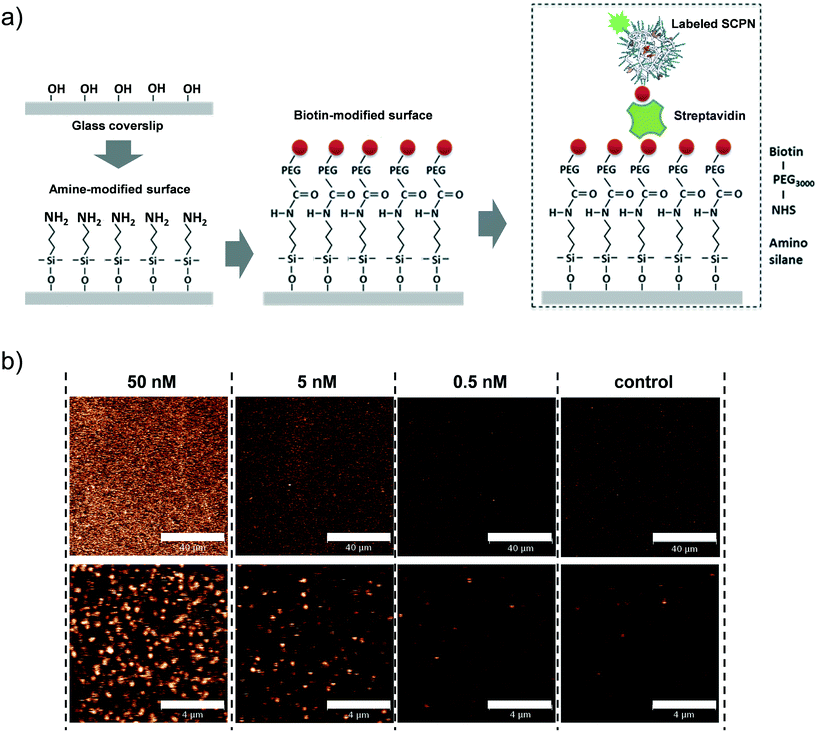 |
| | Fig. 6 Surface immobilization of biotinylated SCPNs. a) Experimental design. b) Confocal images showing biotinylated and Alexa Fluor® 488-labeled SCPN on a functionalized coverslip. The images show SCPNs immobilized in various concentrations and a corresponding control measurement where no SCPNs were added. The bar represents 40 and 4 μm in the top and bottom panel, respectively. | |
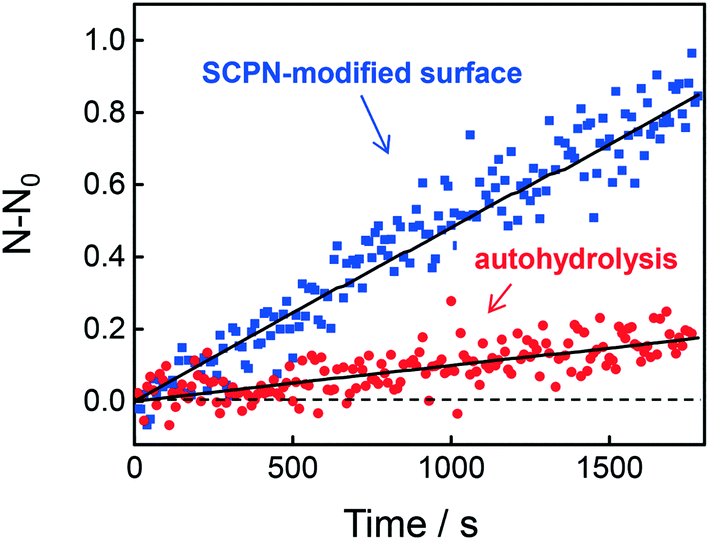
The question then arises whether the surface-immobilized SCPNs are still catalytically active. Using fluorescence correlation spectroscopy (FCS), the carbamate-cleavage reaction was monitored by following the increase in fluorescent products that are released from the immobilized SCPNs and diffuse into the bulk solution (see ESI† for details, Fig. S12). P2@Cu(II) was chosen for these experiments instead of P3@Cu(II) to avoid any influence from the Alexa Fluor® 488 dye used for labeling the SCPNs. The immobilization of P2@Cu(II) followed exactly the same protocol as shown for P3. A solution containing S1 (10 μM) and NaAsc (1 mM) was added onto a surface with or without P2@Cu(II), respectively. As shown in Fig. 7, hydrolysis of S1 is clearly visible on the P2@Cu(I) covered surface. This provides first evidence that at least a fraction of SCPNs remains catalytically active after surface immobilization.
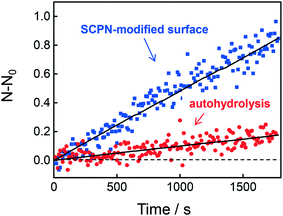 |
| | Fig. 7 Fluorescence correlation spectroscopy (FCS) to determine product accumulation in solution. The graph illustrates the rate of product accumulation for the sample containing immobilized SCPNs (blue) and for a control sample without SCPNs (red, auto-hydrolysis). | |
Single-molecule activity measurements
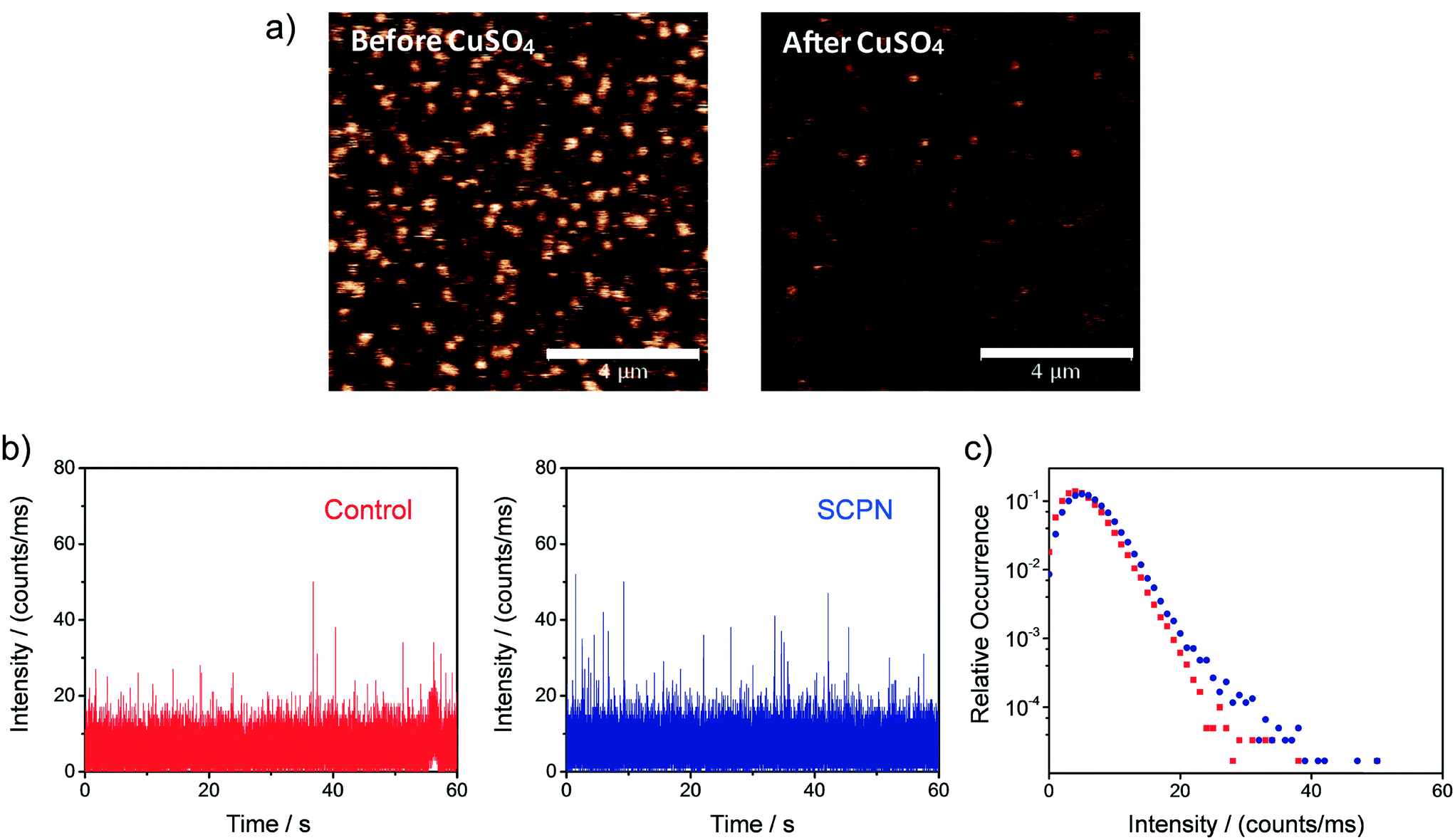
Single-SCPN activity measurements were performed to find out if individual turnovers of a single SCPN can be followed over time. In these experiments, P3@Cu(II) was used in combination with S1 as the substrate. As the complex of the phenanthroline ligand with Cu ions quenches the fluorescence of Alexa Fluor® 488 (Fig. S11†), this fluorescent label is used indirectly to visualize Cu-containing SCPNs on the surface. Therefore, we developed the following protocol to localize the SCPNs on the coverslip surface. P3-based SCPNs (20 nM) were first immobilized on the glass coverslip. A surface area of 10 × 10 μm was scanned before and after incubation with CuSO4 solution (concentration 1 mM). Fluorescent spots that disappeared after the incubation with CuSO4 were considered as locations of Cu-loaded SCPNs (Fig. 8a). It is important to note that the disappearance of the fluorescent signal can also originate from bleaching of Alexa Fluor® 488 during consecutive scans. The degree of bleaching was tested and it was found to be negligible under the experimental conditions used (Fig. S13†).
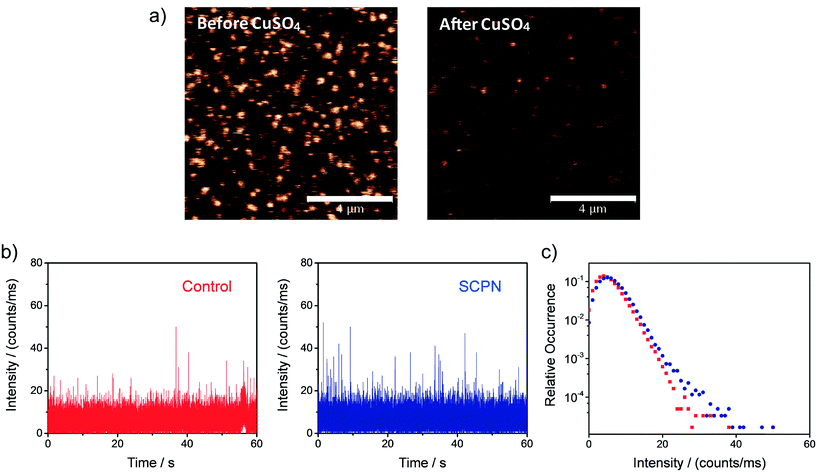 |
| | Fig. 8 Representative single-SCPN activity measurement. a) Confocal images (10 × 10 μm) showing the Alexa Fluor 488-labeled and biotin-functionalized SCPNs immobilized on a streptavidin-functionalized glass coverslip before (left) and after (right) the addition of CuSO4. The bar in the panel represents 4 μm. b) Fluorescent intensity time traces and c) corresponding intensity distributions obtained when binning the data with 1 ms bin size. The data was recorded on the location of one individual SCPN (blue) and on an empty area on the surface (red; control). | |
Fluorescence time traces were recorded at the positions of different SCPNs for 1 minute each after delivering a solution containing 10 μM S1 and NaAsc. One representative time trace is shown in Fig. 8b. Despite the relatively low signal-to-noise ratio, which is expected for MC-Rh110, the results suggest that fluorescent product molecules are produced on the surface and can be detected. When comparing the time traces recorded at the position of one SCPN with the control (empty area which does not contain SCPNs), a small increase in the frequency of fluorescence bursts (with high intensity) was observed, suggesting that the SCPNs are turning over the fluorogenic substrate. However, the number of clearly resolved turnovers detected appears to be low (Fig. 8b), which can be rationalized by the relatively low average TOF of the catalytic SCPN system. Nevertheless, the fact that no individual SCPNs with a significantly higher activity was observed (Fig. S14 and S15†), may imply that structural heterogeneities of the SCPNs do not cause a dramatic diversity in their catalytic performance.
Although a detailed quantitative kinetic analysis proved difficult, the results above provide a practical platform that holds potential for further optimization. Catalytically-active SCPNs with organometallic sites other than copper complexes, e.g. palladium or ruthenium complexes, are possible candidates, which may have higher TOFs and overcome the fluorescence quenching problem. Besides developing new catalysts, we are continuing our efforts on optimizing the experimental setup to obtain higher SNR and throughput. For example, the possibility of using a wide-field fluorescence setup combined with nano-photonic metal structures, such as zero-mode waveguides (ZMWs),54–57 is under exploration. Wide-field fluorescence microscopy, which allows for monitoring many individual SCPNs on the surface simultaneously, may provide insights into possible heterogeneities between individual SCPNs in a more straightforward manner. Meanwhile, ZMWs are able to reduce the detection volume drastically, thus allowing the use of higher substrate concentrations while reducing the background signal. In addition, their ability to enhance the fluorescence signal could increase the signal-to-noise ratio even further.
Conclusion
We have shown the preparation of SCPNs that are capable of catalyzing fluorogenic reactions, as well as provide protocols for their surface immobilization and for monitoring their catalytic activity. The combined design of catalysts and substrates enabled us to obtain novel information about the substrate specificity of SCPNs and allowed to guide the activity of the catalytic reaction at the ensemble level. Similar to changes in reactivity as the result of mutations in enzymes, we expect that not every SCPN will be equally reactive.58 Therefore, we are highly intrigued by the question on how to discriminate the diversity in reactivity at the individual level. The protocol for surface immobilization, which retains the catalytic activity of SCPNs, allows further SMFM measurements on individual SCPNs. In the course of our investigation, we identified the experimental challenges, mainly a low TOF of the catalysts combined with a low signal-to-noise ratio in SMFM measurements, which will allow us to improve the system in future experiments. So far, we have not found any evidence that some SCPNs are more reactive than the others. However, this work has laid a solid foundation for applying SMFM to further understanding the structure–function relation of catalytic SCPNs. We anticipate that these efforts will assist in directing the future of catalytic SCPNs, e.g. if sequence control in the polymer synthesis and regulation of polymer chain's precision folding are the gateway toward accessing enzyme-like activity and selectivity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Y. L., A. R. A. P. and E. W. M. acknowledge financial support from the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035) and the European Research Council (FP7/2007-2013, ERC Grant Agreement 246829). P. T., K. G. B. and A. E. R. acknowledge financial support from the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035), the Dutch National Research School Combination Catalysis Controlled by Chemical Design (NRSCC grant 2009-10016B, A. E. R.), as well as The Netherlands Organization for Scientific Research (NWO; VIDI grant 700.58.430, K. G. B.). Dr. L. Albertazzi is acknowledged for fruitful discussions. Gijs ter Huurne is acknowledged for assistance with the SAXS measurements. The ICMS animation studio is acknowledged for providing the artwork.
References
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef PubMed.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef PubMed.
- M. Gonzalez-Burgos, A. Latorre-Sanchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef.
- J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- N. D. Knöfel, H. Rothfuss, J. Willenbacher, C. Barner-Kowollik and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 4950–4954 CrossRef PubMed.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef PubMed.
- I. Berkovich, S. Mavila, O. Iliashevsky, S. Kozuch and N. G. Lemcoff, Chem. Sci., 2016, 7, 1773–1778 RSC.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef.
- S. Thanneeru, S. S. Duay, L. Jin, Y. Fu, A. M. Angeles-Boza and J. He, ACS Macro Lett., 2017, 6, 652–656 CrossRef.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef PubMed.
- K. Freytag, S. Säfken, K. Wolter, J. C. Namyslo and E. G. Hübner, Polym. Chem., 2017, 8, 7546–7558 RSC.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196–4203 RSC.
- D. E. Bergbreiter, ACS Macro Lett., 2014, 3, 260–265 CrossRef.
- G. Wulff, Chem. Rev., 2002, 102, 1–28 CrossRef PubMed.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef PubMed.
- L. J. Twyman, A. S. H. King and I. K. Martin, Chem. Soc. Rev., 2002, 31, 69–82 RSC.
- D. Méry and D. Astruc, Coord. Chem. Rev., 2006, 250, 1965–1979 CrossRef.
- J. Kofoed and J.-L. Reymond, Curr. Opin. Chem. Biol., 2005, 9, 656–664 CrossRef PubMed.
- E. Huerta, B. van Genabeek, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Macromol. Rapid Commun., 2014, 35, 1320–1325 CrossRef PubMed.
- M. Artar, E. R. J. Souren, T. Terashima, E. W. Meijer and A. R. A. Palmans, ACS Macro Lett., 2015, 4, 1099–1103 CrossRef.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef PubMed.
- M. A. J. Gillissen, T. Terashima, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2013, 46, 4120–4125 CrossRef.
- G. M. ter Huurne, L. N. J. de Windt, Y. Liu, E. W. Meijer, I. K. Voets and A. R. A. Palmans, Macromolecules, 2017, 50, 8562–8569 CrossRef PubMed.
- N. Hosono, A. M. Kushner, J. Chung, A. R. A. Palmans, Z. Guan and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 6880–6888 CrossRef PubMed.
- P. J. M. Stals, C.-Y. Cheng, L. van Beek, A. C. Wauters, A. R. A. Palmans, S. Han and E. W. Meijer, Chem. Sci., 2016, 7, 2011–2015 RSC.
- W. E. Moerner and D. P. Fromm, Rev. Sci. Instrum., 2003, 74, 3597–3619 CrossRef.
- P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2005, 44, 2642–2671 CrossRef PubMed.
- T. G. Terentyeva, J. Hofkens, T. Komatsuzaki, K. Blank and C.-B. Li, J. Phys. Chem. B, 2013, 117, 1252–1260 CrossRef PubMed.
- V. I. Claessen, H. Engelkamp, P. C. M. Christianen, J. C. Maan, R. J. M. Nolte, K. Blank and A. E. Rowan, Annu. Rev. Anal. Chem., 2010, 3, 319–340 CrossRef PubMed.
- P. Chen, X. Zhou, M. Andoy, K.-S. Han, E. Choudhary, N. Zou, G. Chen and H. Shen, Chem. Soc. Rev., 2014, 43, 1107–1117 RSC.
- D. Wöll, H. Uji-i, T. Schnitzler, J. Hotta, P. Dedecker, A. Herrmann, F. C. De Schryver, K. Müllen and J. Hofkens, Angew. Chem., Int. Ed., 2008, 47, 783–787 CrossRef PubMed.
- Q. T. Easter and S. A. Blum, Angew. Chem., Int. Ed., 2017, 56, 13772–13775 CrossRef PubMed.
- Q. T. Easter and S. A. Blum, Angew. Chem., Int. Ed., 2018, 57, 1572–1575 CrossRef PubMed.
- G. K. Hodgson, S. Impellizzeri and J. C. Scaiano, Chem. Sci., 2016, 7, 1314–1321 RSC.
- Z. Ristanović, J. P. Hofmann, G. De Cremer, A. V. Kubarev, M. Rohnke, F. Meirer, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 6559–6568 CrossRef PubMed.
- P. Turunen, A. E. Rowan and K. Blank, FEBS Lett., 2014, 588, 3553–3563 CrossRef PubMed.
- J. D. Ng, S. P. Upadhyay, A. N. Marquard, K. M. Lupo, D. A. Hinton, N. A. Padilla, D. M. Bates and R. H. Goldsmith, J. Am. Chem. Soc., 2016, 138, 3876–3883 CrossRef PubMed.
- B. Dong, Y. Pei, F. Zhao, T. W. Goh, Z. Qi, C. Xiao, K. Chen, W. Huang and N. Fang, Nat. Catal., 2018, 1, 135–140 CrossRef.
- K. Velonia, O. Flomenbom, D. Loos, S. Masuo, M. Cotlet, Y. Engelborghs, J. Hofkens, A. E. Rowan, J. Klafter, R. J. M. Nolte and F. C. de Schryver, Angew. Chem., Int. Ed., 2005, 44, 560–564 CrossRef PubMed.
- N. S. Hatzakis, H. Engelkamp, K. Velonia, J. Hofkens, P. C. M. Christianen, A. Svendsen, S. A. Patkar, J. Vind, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2006, 2012–2014 RSC.
- N. S. Hatzakis, L. Wei, S. K. Jorgensen, A. H. Kunding, P.-Y. Bolinger, N. Ehrlich, I. Makarov, M. Skjot, A. Svendsen, P. Hedegård and D. Stamou, J. Am. Chem. Soc., 2012, 134, 9296–9302 CrossRef PubMed.
- T. G. Terentyeva, H. Engelkamp, A. E. Rowan, T. Komatsuzaki, J. Hofkens, C.-B. Li and K. Blank, ACS Nano, 2012, 6, 346–354 CrossRef PubMed.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef PubMed.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef PubMed.
- L. D. Lavis, T.-Y. Chao and R. T. Raines, ACS Chem. Biol., 2006, 1, 252–260 CrossRef PubMed.
- T. G. Terentyeva, W. Van Rossom, M. Van Der Auweraer, K. Blank and J. Hofkens, Bioconjugate Chem., 2011, 22, 1932–1938 CrossRef PubMed.
- Z.-Q. Wang, J. Liao and Z. Diwu, Bioorg. Med. Chem. Lett., 2005, 15, 2335–2338 CrossRef PubMed.
- A. A. Kislukhin, V. P. Hong, K. E. Breitenkamp and M. G. Finn, Bioconjugate Chem., 2013, 24, 684–689 CrossRef PubMed.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef PubMed.
- P. Zhu and H. G. Craighead, Annu. Rev. Biophys., 2012, 41, 269–293 CrossRef PubMed.
- M. J. Levene, J. Korlach, S. W. Turner, M. Foquet, H. G. Craighead and W. W. Webb, Science, 2003, 299, 682–686 CrossRef PubMed.
- H. Rigneault, J. Capoulade, J. Dintinger, J. Wenger, N. Bonod, E. Popov, T. W. Ebbesen and P.-F. Lenne, Phys. Rev. Lett., 2005, 95, 117401 CrossRef PubMed.
- D. Gérard, J. Wenger, N. Bonod, E. Popov, H. Rigneault, F. Mahdavi, S. Blair, J. Dintinger and T. W. Ebbesen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 45413 CrossRef.
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8me00017d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bd,
Alan E.
Rowan
*bc,
Anja R. A.
Palmans
bd,
Alan E.
Rowan
*bc,
Anja R. A.
Palmans