DOI:
10.1039/C7QM00574A
(Research Article)
Mater. Chem. Front., 2018,
2, 566-572
A phenol-fused tetrathiafulvalene: modulation of hydrogen-bond patterns and electrical conductivity in the charge-transfer salt†
Received
12th December 2017
, Accepted 19th January 2018
First published on 22nd January 2018
Abstract
A tetrathiafulvalene (TTF) derivative fused with a phenol moiety, PhOH-EDT-TTF, was designed and synthesized as a new electron-donor molecule with hydrogen-bonding (H-bonding) abilities. Electrochemical oxidation of this donor molecule in the presence of Bu4NClO4 gave a charge-transfer salt, β-(PhOH-EDT-TTF)2ClO4, that contains PhOH-EDT-TTF+0.5 and ClO4− in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. In this salt, the donor and anion molecules are connected with three-centered H-bonds through the phenol hydroxy group, which is in contrast to the linear H-bonds observed in an analogue salt based on the catechol-fused TTF. This variation in the donor-anion H-bonds modulated the arrangement of the donor molecules in the conducting layer, leading to better conducting properties than those of the analogue salt.
:1 ratio. In this salt, the donor and anion molecules are connected with three-centered H-bonds through the phenol hydroxy group, which is in contrast to the linear H-bonds observed in an analogue salt based on the catechol-fused TTF. This variation in the donor-anion H-bonds modulated the arrangement of the donor molecules in the conducting layer, leading to better conducting properties than those of the analogue salt.
Introduction
Hydrogen-bonded (H-bonded) molecular systems have continuously attracted much attention, because of their unique structural and functional features attributable to the electrostatic and dynamic properties of the H-bond interactions.1–3 For example, in the field of molecular conductors,3 Murata et al. have realized a purely organic molecular metal based on a H-bonded charge-transfer (CT) complex, where the H-bonds controlled the redox activity and ratio of the component electron donor/acceptor molecules.3a,c Our group has also found several kinds of interesting phenomena and properties based on H-bonds coupled to the π-electrons in molecular conductors.4–8 In particular, H-bond dynamics-based unique electronic properties were observed in the catechol-fused ethylenedithiotetrathiafulvalene (H2Cat-EDT-TTF; Fig. 1 left)-based conductors.5–7 What is important here is that these features are fundamentally based on the interplay of the electron donating and H-bonding abilities of the component molecule, H2Cat-EDT-TTF.
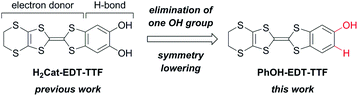 |
| | Fig. 1 Molecular design of PhOH-EDT-TTF (right) on the basis of the catechol analogue, H2Cat-EDT-TTF4a (left). | |
These results have encouraged us to further explore such TTF-based electron donor molecules with H-bonding abilities. Thus, in this study, we have designed phenol-fused ethylenedithiotetrathiafulvalene, PhOH-EDT-TTF (Fig. 1 right), by eliminating one OH group from H2Cat-EDT-TTF. This type of symmetry-lowered, mono-substituted TTF derivatives has also extensively been investigated as an interesting component of molecular conductors.3a,c–i,8a,b,d,9,10 Here, we report the synthesis, structures, and properties of PhOH-EDT-TTF and its CT salt, β-(PhOH-EDT-TTF)2ClO4. Interestingly, this chemical modification modulated not only the donor-anion H-bond pattern but also the donor arrangement in the conducting layer, which resulted in significant improvement of the conducting properties.
Results and discussion
Synthesis, structure, and properties of phenol-fused EDT-TTF electron donor
The synthetic scheme for PhOH-EDT-TTF is depicted in Scheme 1 (for details, see the Experimental section). The Stille coupling reaction of 1,2-dibromo-4-methoxybenzene 111 with nBu3SnS(CH2)2CN12 provided the bis(cyanoethylthio) derivative 2, which was then transformed into the 1,3-dithiole-2-thione derivative 3 through decyanoethylation and subsequent cyclization. Demethylation of 3 followed by tert-butyldimethylsilylation gave compound 4, which was then subjected to the Wittig-type reaction with 4,5-ethylenedithio-1,3-dithiole-2-one, to afford the compound 5. Finally, removal of the silyl group from 5 provided the desired compound PhOH-EDT-TTF as an orange powder.
 |
| | Scheme 1 Synthesis of PhOH-EDT-TTF. Reagents and conditions: (i) nBu3SnS(CH2)2CN, Pd(PPh3)4, toluene, 120 °C; (ii) NaOMe, ZnCl2, nBu4Br, CH2Cl2/MeOH, room temperature; (iii) 1,1′-thiocarbonyldiimidazole, AcOH, THF, −78 °C–room temperature; (iv) BBr3, CH2Cl2, −5 °C to 30 °C; (v) tert-butyldimethylsilyl chloride, Et3N, N,N′-dimethylamino-4-pyridine, CH2Cl2, room temperature; (vi) P(OEt)3, 115 °C; (vii) nBu4NF, AcOH, THF, room temperature. | |
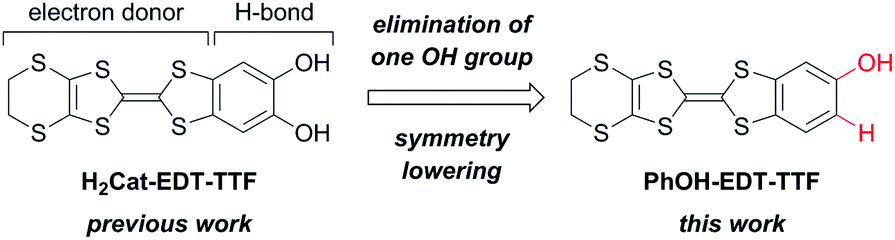
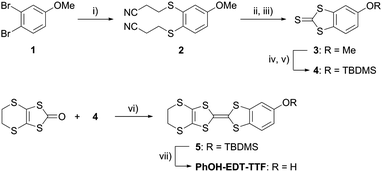
Single crystals of PhOH-EDT-TTF suitable for X-ray structure analysis (Table 1) were obtained by recrystallization from a methanol solution. In the crystal, the TTF skeleton of PhOH-EDT-TTF is slightly distorted in a boat-like fashion (Fig. 2a), which is typical for neutral TTF derivatives. The phenol hydroxy group is disordered in two positions (O1, H1A, and H4 vs. O2, H2A, and H3) with site occupancy factors of 0.74(1) and 0.26(1), respectively. The dominant hydroxy group (O1–H1A) participates in a C–H⋯O-type hydrogen-bond with a methylene group in the neighboring molecule (dashed blue lines in Fig. 2b; dH⋯O ∼ 2.5 Å), as the hydrogen-bond acceptor, resulting in the formation of one-dimensional chain structures along the a + c direction. Also, the OH proton (H1A) is directed to sulfur atoms on the EDT-TTF skeleton in the neighboring chain with dH⋯S ∼ 3.0 Å (dashed purple lines in Fig. 2b). Furthermore, S⋯S short contacts are found between the chains (dashed brown lines in Fig. 2b; dS⋯S ∼ 3.5 Å). As shown in Fig. 2c, the donor molecules form dimeric pairs in a head-to-tail manner (∼3.4 Å), constructing a two-dimensional (2D) sheet-like arrangement.
Table 1 Crystallographic data for PhOH-EDT-TTF and β-(PhOH-EDT-TTF)2ClO4
| Compound |
PhOH-EDT-TTF
|
β-(PhOH-EDT-TTF)2ClO4
|
| Formula |
C12H8O1S6 |
C24H16Cl1O6S12 |
| FW |
360.55 |
820.56 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c (#14) |
C2/c (#15) |
|
a (Å) |
12.369(5) |
33.29(4) |
|
b (Å) |
11.196(4) |
7.227(7) |
|
c (Å) |
10.011(4) |
13.232(14) |
|
α (°) |
90 |
90 |
|
β (°) |
92.537(5) |
105.227(18) |
|
γ (°) |
90 |
90 |
|
V (Å3) |
1385.1(9) |
3072(6) |
|
Z value |
4 |
4 |
|
T (K) |
273 |
293 |
|
d
calc (g cm−3) |
1.729 |
1.774 |
|
λ (Å) |
0.71073 |
0.71073 |
| Goodness of fit (GOF) |
1.010 |
1.134 |
| # of observations |
3146 |
3476 |
| # of variables |
184 |
196 |
|
R
1 (I > 2.0σ(I)) |
0.0819 |
0.0754 |
| wR2 (all data) |
0.2098 |
0.2367 |
| CCDC number |
1590325
|
1590324
|
 |
| | Fig. 2 Crystal structure of the neutral donor PhOH-EDT-TTF at 273 K. (a) ORTEP drawings of the molecular structure (top: top view, bottom: side view; minor disordered components (O2, H2A, and H3) are shown in green), (b) intermolecular short contacts, and (c) two-dimensional sheet arrangement. In (a), the thermal ellipsoids are scaled to the 50% probability level. | |
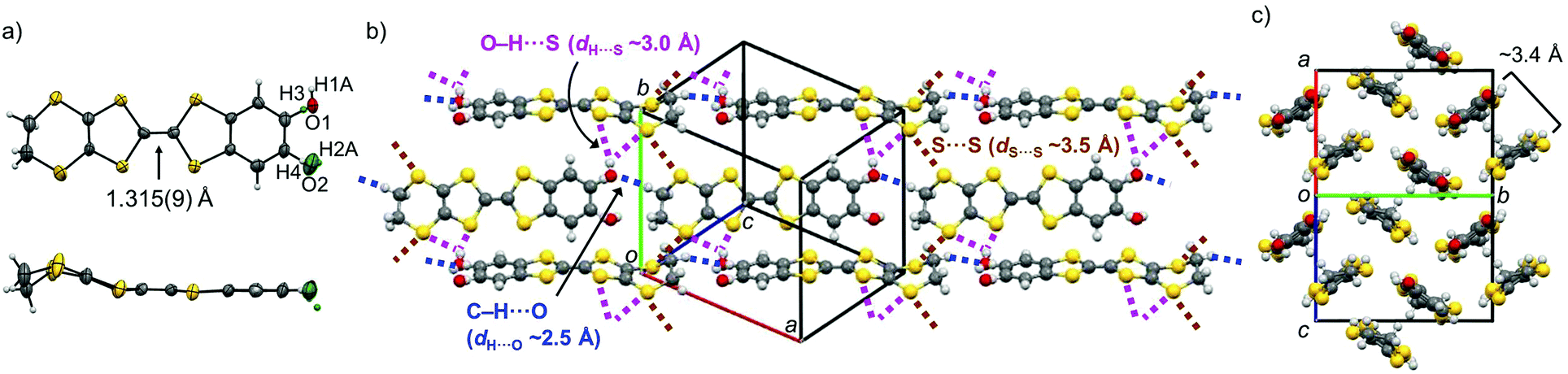
The cyclic voltammetry of PhOH-EDT-TTF in CH3CN showed two reversible oxidation waves (E11/2 = +0.48 V, E21/2 = +0.77 V; vs. SCE), whose values are higher than those of the catechol analogue H2Cat-EDT-TTF (E11/2 = +0.40 V, E21/2 = +0.65 V; vs. SCE)4a (Fig. 3). This result indicates that the electron-donating ability is significantly decreased by the elimination of one hydroxy group. In fact, a similar trend is predicted by DFT calculations (Fig. 3, right column):13 the calculated HOMO energy level of PhOH-EDT-TTF (−4.77 eV) is lower than that of H2Cat-EDT-TTF (−4.72 eV).4a Also, the ΔE value (= E21/2 − E11/2) is increased from 0.25 V in H2Cat-EDT-TTF4a to 0.29 V in PhOH-EDT-TTF, which corresponds to the fact that the degree of HOMO delocalization, especially onto the benzene ring, is decreased with this chemical modification (Fig. 3). Nevertheless, PhOH-EDT-TTF has a sufficiently high electron-donating ability comparable to BEDT-TTF (bis(ethylenedithio)tetrathiafulvalene) (E11/2 = +0.49 V, E21/2 = +0.72 V; vs. SCE), probably thanks to the residual hydroxy group.
 |
| | Fig. 3 Comparison of electronic properties of the donor molecules, PhOH-EDT-TTF and H2Cat-EDT-TTF.4a The oxidation potentials, E11/2 and E21/2, were estimated from CV experiments (in CH3CN with 0.1 M Bu4NClO4, vs. SCE). The energy levels and distributions of HOMO were obtained from DFT calculations (B3LYP/6-31G(d)).13 | |
Synthesis, structure, and properties of the CT salt, β-(PhOH-EDT-TTF)2ClO4
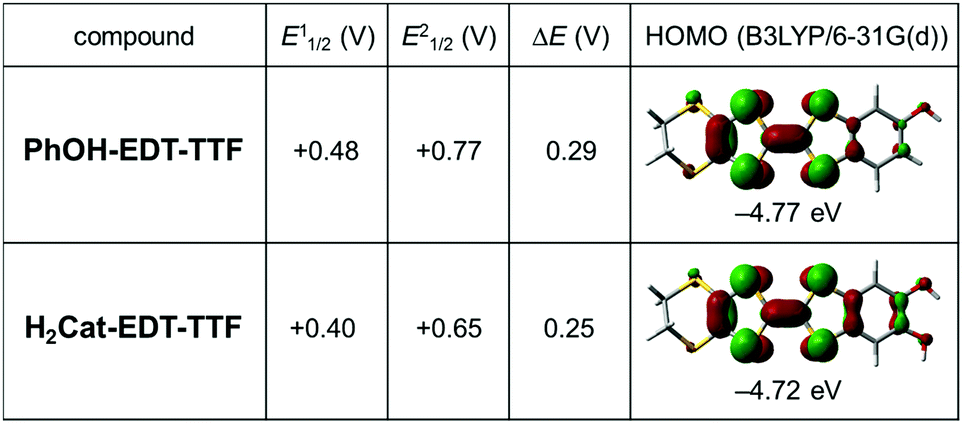
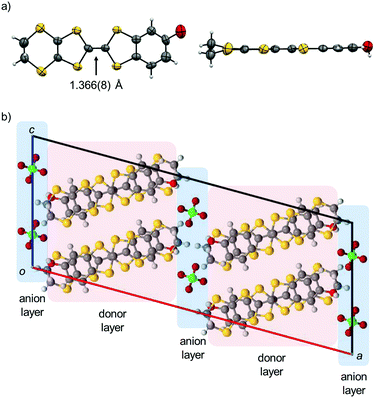
Electrochemical oxidation of PhOH-EDT-TTF in the presence of nBu4N·ClO4 in PhCl–CH3CN gave brown plate crystals of the titled CT salt. The X-ray crystal structure analysis at room temperature (Table 1) revealed that the space group is C2/c and the asymmetric unit consists of one donor molecule and a half of the ClO4 anion. Therefore, the chemical formula of this salt is given as (PhOH-EDT-TTF)2ClO4, where the donor molecule should be in a +0.5 state, PhOH-EDT-TTF+0.5. In fact, the TTF skeleton of the donor molecule in this salt (Fig. 4a) is highly planar and has a longer central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (1.366(8) Å) compared to that in the neutral crystal (1.315(9) Å, Fig. 2a), which are characteristic of oxidized TTF derivatives. The unit cell contains eight donor molecules and four anions (Fig. 4b). These two species separately form a layer structure, as represented by pink and blue colors in Fig. 4b, and these layers are alternately arranged along the a axis. Such a layered crystal structure is frequently seen in TTF-based CT salts and actually the previous H2Cat-EDT-TTF salts also have a similar structure.4a
C bond (1.366(8) Å) compared to that in the neutral crystal (1.315(9) Å, Fig. 2a), which are characteristic of oxidized TTF derivatives. The unit cell contains eight donor molecules and four anions (Fig. 4b). These two species separately form a layer structure, as represented by pink and blue colors in Fig. 4b, and these layers are alternately arranged along the a axis. Such a layered crystal structure is frequently seen in TTF-based CT salts and actually the previous H2Cat-EDT-TTF salts also have a similar structure.4a
 |
| | Fig. 4 Crystal structure of the CT salt, β-(PhOH-EDT-TTF)2ClO4. (a) Molecular structure of the donor molecule PhOH-EDT-TTF+0.5 (left: top view, right: side view) and (b) unit cell. In (a), the thermal ellipsoids are scaled to the 50% probability level. | |
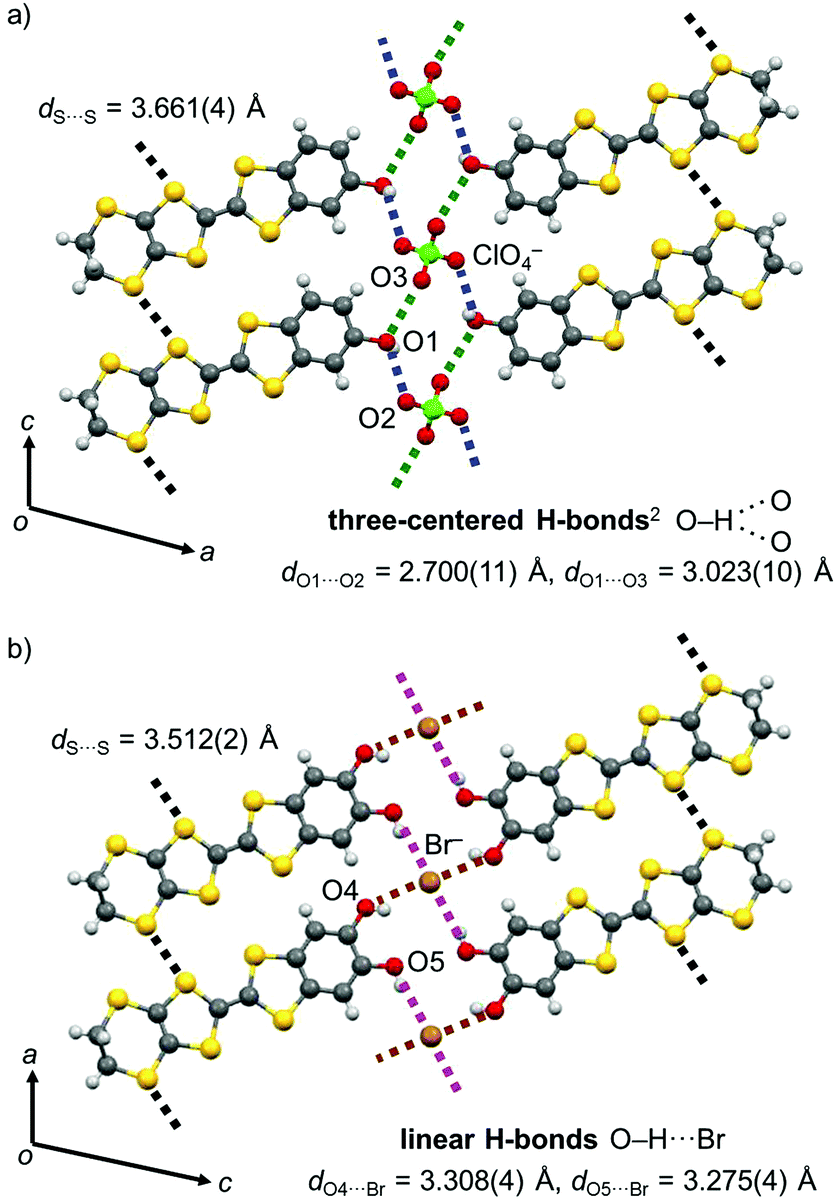
Also, the present PhOH-EDT-TTF+0.5 molecules form two O–H⋯O-type H-bonds with ClO4 anions through the phenol hydroxy group (Fig. 5a; dO1⋯O2 = 2.700(11) Å (blue dashed lines) and dO1⋯O3 = 3.023(10) Å (green dashed lines)). Although the position of the OH proton is not completely determined (owing to the quality of the diffraction data), this type of H-bonds can be categorized as a three-centered (or bifurcated) H-bond, where one proton donor is connected to two proton acceptors.2 Thanks to these donor-anion H-bonds, the donor molecules are arranged along the c axis, accompanied by lateral S⋯S interactions between the EDT-TTF moieties (Fig. 5a). The shortest S⋯S distance in this H-bonded array is 3.661(4) Å (black dashed lines), close to the sum of the van der Waals radii (3.60 Å).14 Such a H-bonded array is also seen in CT salts of the catechol analogue, β-(H2Cat-EDT-TTF)2Br and β′′-(H2Cat-EDT-TTF)2Cl; however, the H-bonds are not three-centered but linear (Fig. 5b, O–H⋯Br).4a Therefore, the difference in the number of hydroxy groups in the donor molecules might make the H-bond pattern different. In addition, the difference in the counter anion in these salts (i.e., ClO4vs. Br) might also contribute to the difference in the H-bond patterns.15 The shortest S⋯S distance in the catechol system, β-(H2Cat-EDT-TTF)2Br (3.512(2) Å, Fig. 5b), is shorter than that in the present salt (3.661(4) Å, Fig. 5a), which is probably related to the H-bond structure differences mentioned above.
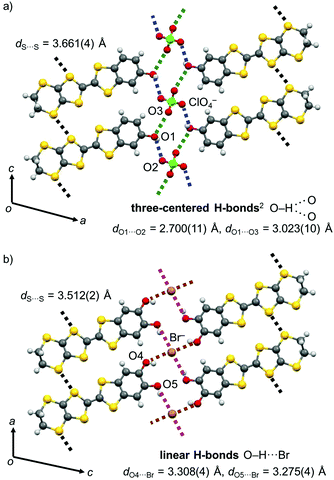 |
| | Fig. 5 Donor-anion H-bonded arrays in the crystal of (a) β-(PhOH-EDT-TTF)2ClO4 and (b) β-(H2Cat-EDT-TTF)2Br.4a | |
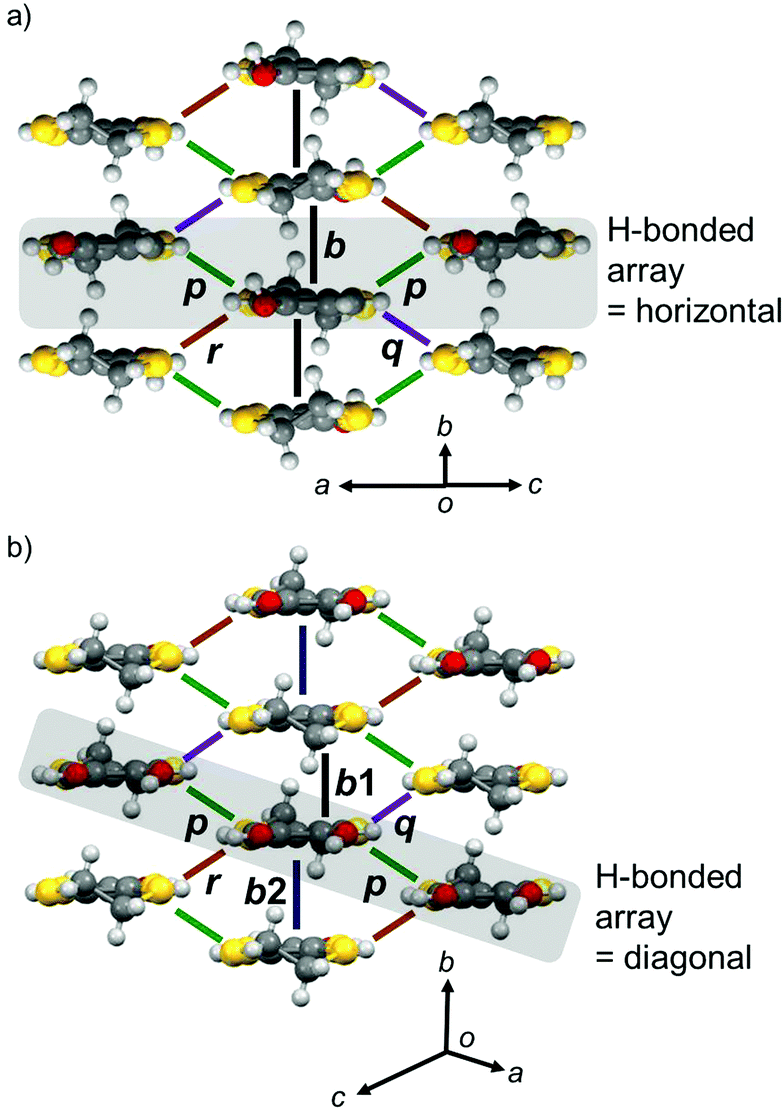
In the donor layer (Fig. 4b, pink areas), PhOH-EDT-TTF+0.5 molecules construct a two-dimensional (2D) sheet structure (Fig. 6a), categorized as the β-type arrangement in the BEDT-TTF-based CT salts.16 Thus, this salt is named β-(PhOH-EDT-TTF)2ClO4. The donor molecules are uniformly stacked along the b axis in a head-to-tail manner, where the interplanar separation is about 3.6 Å. These donor stacks are arranged along the c axis through the above-mentioned H-bonds (Fig. 5a). Similar to this compound, the catechol analogue, β-(H2Cat-EDT-TTF)2Br,4a also form a β-type donor arrangement (Fig. 6b) by utilizing the H-bonds shown in Fig. 5b. However, as shown in gray in Fig. 6a and b, the arrangement manners of the H-bonded molecules in these two systems are significantly different from each other: the H-bonded molecules in β-(PhOH-EDT-TTF)2ClO4 are arranged horizontally in the 2D layer (Fig. 6a), whereas those in β-(H2Cat-EDT-TTF)2Br are arranged diagonally (Fig. 6b). This difference is clearly due to the difference in their donor-anion H-bond patterns (i.e., three-centered or linear; Fig. 5a and b).
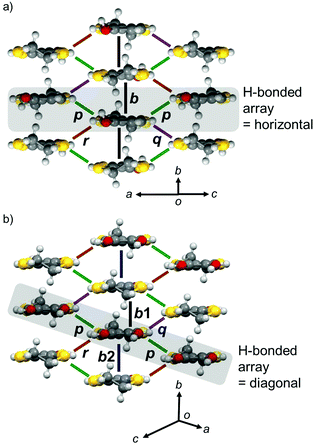 |
| | Fig. 6 Donor arrangement in (a) β-(PhOH-EDT-TTF)2ClO4 and (b) β-(H2Cat-EDT-TTF)2Br.4a The symbols, b, b1, b2, p, q, and r, represent the intermolecular transfer integrals: b = 117, p = −30, q = −31, and r = −19 meV for (a) β-(PhOH-EDT-TTF)2ClO4 and b1 = −229, b2 = −60, p = −23, q = −11, and r = 78 meV for (b) β-(H2Cat-EDT-TTF)2Br.4a Gray-colored areas represent the H-bonded molecular arrays in Fig. 5a and b. | |
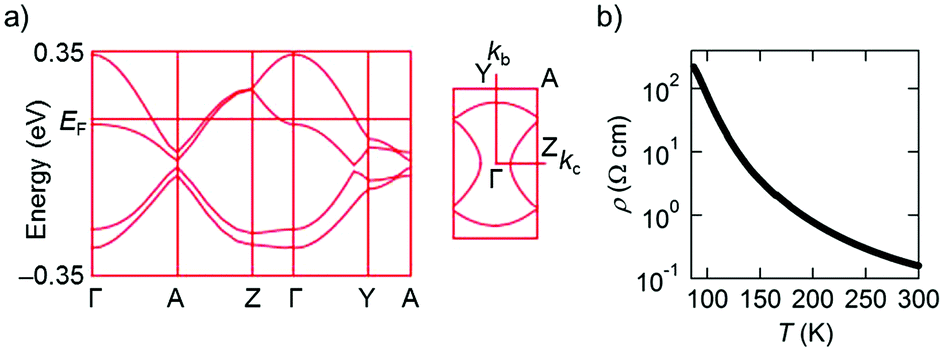
Intermolecular transfer integrals in the donor layer of β-(PhOH-EDT-TTF)2ClO4 are calculated17 to be b = 117, p = −30, q = −31, and r = −19 meV (Fig. 6a), indicating the occurrence of a strong interaction in the stack direction (b). In addition, the interstack directions (p, q, and r) also have substantial interactions (up to a quarter of the stack direction), which should be due to the lateral S⋯S interactions resulted from the H-bond formation (Fig. 5a). Reflecting these intermolecular interactions, a quasi-two-dimensional (Q2D) Fermi surface was obtained (Fig. 7a, right) by tight-binding band calculations.17 Despite having such a 3/4-filled band structure, the single crystal of this salt showed not metallic but semiconducting behavior (Fig. 7b).18 The room-temperature electrical conductivity and activation energy are 6 S cm−1 and 0.08 eV, respectively. These values indicate that the conducting properties are significantly enhanced compared to those of the catechol analogue, β-(H2Cat-EDT-TTF)2Br (0.02 S cm−1 and 0.10 eV, semiconducting).4a This is because the uniform stacks of donor molecules are realized in the present system β-(PhOH-EDT-TTF)2ClO4, in contrast to the strongly dimerized stacks in β-(H2Cat-EDT-TTF)2Br (i.e., b1/b2 = 3.8 (≫1), Fig. 6 caption). Thus, one can imagine that the variation in the arrangement manners of the H-bonded donor molecules (Fig. 6a and b) allows this salt to have the uniform stacks, which results in the improvement of the conducting properties. Namely, the H-bond interactions have played an important role in controlling not only the donor-anion contacts, but also the donor arrangement and electronic structure in the conducting layer.
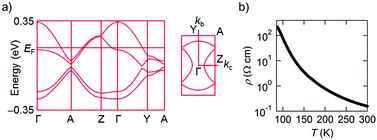 |
| | Fig. 7 (a) Electronic band structure and (b) temperature dependence of electrical resistivity (single crystal, four-probe) of β-(PhOH-EDT-TTF)2ClO4. | |
Conclusions
Phenol-fused ethylenedithiotetrathiafulvalene, PhOH-EDT-TTF, was designed and synthesized as a new TTF derivative with H-bonding abilities. Cyclic voltammetry measurements and DFT calculations indicate that this molecule has a sufficiently high electron-donating ability comparable to BEDT-TTF, enabling us to obtain the charge-transfer salt, β-(PhOH-EDT-TTF)2ClO4, composed of PhOH-EDT-TTF+0.5 and ClO4− in a 2:1 ratio. In this salt, the donor molecule forms three-centered H-bonds with two neighboring ClO4− anions through the phenol hydroxy group, which is in contrast to the catechol-fused analogue that constructs the linear H-bonds. Interestingly, this difference in the H-bond patterns modulated the arrangement of the donor molecules in the conducting layer, leading to uniform stacks of the donors and better conducting properties in the present system, β-(PhOH-EDT-TTF)2ClO4. These results indicate that PhOH-EDT-TTF is a promising component molecule in electronic functional materials with H-bonds. Further exploration of PhOH-EDT-TTF-based H-bonded molecular conductors and also the investigation of dynamic properties based on the H-bonds5–7 are currently underway.
Experimental
Materials and methods
Triethyl phosphite was distilled over sodium before use. Other commercially available materials were used without further purification. All the reactions were carried out under argon atmosphere. 1H NMR (300 MHz) spectra were measured with a JEOL JNM-AL300 spectrometer with CDCl3 or acetone-d6 as a solvent. The residual solvent was used as the internal standard. APCI-MS spectra were recorded on a Bruker micrOTOF II spectrometer. Melting points were measured with a hot-stage apparatus and are uncorrected. Elemental analyses were performed at the Department of Chemistry, Graduate School of Science, The University of Tokyo. Cyclic voltammetry (CV) measurements were performed with an ALS Electrochemical Analyzer Model 610DB in a CH3CN solution containing 0.1 M nBu4NClO4 as a supporting electrolyte at room temperature (working electrode: glassy carbon, counter electrode: Pt wire, reference electrode: saturated calomel electrode (SCE), scan rate: 0.1 V s−1).
Synthesis of 1,2-bis(2′-cyanoethylthio)-4-methoxybenzene (2).
1,2-Dibromo-4-methoxybenzene 111 (6.4 g, 24 mmol) and 3-[(tributylstannyl)thio]propanenitrile12 (33 g, 89 mmol) were placed in a 300 mL two-necked round-bottomed flask and dissolved in toluene (200 mL, dehydrated). To the solution was added Pd(PPh3)4 (4.2 g, 3.6 mmol) at room temperature. The resulting mixture was stirred at 120 °C for 18 h and then cooled down to 50–60 °C. The insoluble material was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 and filtered through silica gel. The filtrate was concentrated under reduced pressure, and the residue was washed with hexane and methanol, to give 2 (4.7 g, 70%) as a light brown powder. This material was used for the next reaction without further purification. [Analytically pure sample was obtained as a beige powder by silica gel column chromatography using hexane:ethyl acetate = 1:1 as eluent.] Rf 0.61 (hexane:ethyl acetate = 1:1); mp 66–68 °C; 1H NMR (300 MHz, CDCl3) δ 2.56 (t, J = 7.2 Hz, 2H), 2.71 (t, J = 7.2 Hz, 2H), 3.06 (t, J = 7.2 Hz, 2H), 3.20 (t, J = 7.2 Hz, 2H), 3.83 (s, 3H), 6.75 (dd, J = 8.4, 2.4 Hz, 1H), 6.83 (d, J = 2.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 17.70, 18.14, 28.38, 30.53, 55.55, 111.88, 115.05, 117.73, 118.04, 122.98, 137.17, 140.65, 160.72; anal. calcd for C13H14N2O1S2(H2O)0.3: C, 55.02; H, 5.19; N, 9.87. Found C, 54.96; H, 5.03; N, 9.77.
Synthesis of 5-methoxybenzo[d][1,3]dithiole-2-thione (3).
The bis(cyanoethylthio) derivative 2 (4.0 g, 14 mmol) was placed in a 500 mL round-bottomed flask and dissolved in CH2Cl2 (80 mL, dehydrated). To this solution was added sodium methoxide (10 mL, 50 mmol, 5 M methanol solution) at room temperature, and the resulting mixture was stirred for several minutes at the same temperature. To the reaction mixture was then added a mixture of ZnCl2 (2.7 g, 20 mmol) and Bu4NBr (5.5 g, 17 mmol) in methanol (20 mL, dehydrated), and the resulting mixture was stirred for 1 h at room temperature. The reaction mixture was concentrated under reduced pressure and dried in vacuo at room temperature for 1 h. The residue was then suspended in THF (150 mL, stabilizer free) and acetic acid (50 mL). To this suspension, 1,1′-thiocarbonyldiimidazole (3.9 g, 22 mmol) in THF (50 mL, stabilizer free) was added dropwise at −78 °C for 30 min. The resulting mixture was stirred overnight (16 h) with gradual warming to room temperature. To the reaction mixture were added 28% NH3 aqueous solution and satd NaCl aqueous solution. The organic layer was separated, dried over Na2SO4, filtered through silica gel, and concentrated under reduced pressure. The residue was washed with methanol, collected by filtration and then dried in vacuo at room temperature, to give 3 (1.4 g, 47%) as a deep yellow powder. This material was used for the next reaction without further purification. [Analytically pure sample was obtained as a light yellow powder by silica gel column chromatography using hexane:ethyl acetate = 1:1 as eluent.] Rf 0.83 (hexane:ethyl acetate = 1:1); mp 142.0–142.5 °C; 1H NMR (300 MHz, CDCl3) δ 3.85 (s, 3H), 6.96–7.01 (m, 2H), 7.36 (d, J = 8.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 55.77, 106.14, 115.28, 122.35, 132.14, 142.40, 159.48, 192.27; anal. calcd for C8H6O1S3: C, 44.83; H, 2.82; N, 0.00. Found C, 44.87; H, 2.94; N, 0.00.
Synthesis of 5-((tert-butyldimethylsilyl)oxy)benzo[d][1,3]dithiole-2-thione (4).
The methoxy derivative 3 (1.4 g, 6.5 mmol) was placed in a 500 mL two-necked round-bottomed flask and dissolved in CH2Cl2 (200 mL, dehydrated). To the solution was added BBr3 (12 mL, 12 mmol, 1 M CH2Cl2 solution) at −5 °C. The resulting mixture was stirred at the same temperature for 15 min and then at 30 °C for 13 h. To the mixture were further added BBr3 several times (74 mL (in total), 74 mmol, 1 M CH2Cl2 solution) with stirring at 30 °C. After the methoxy derivative 3 completely disappeared (on TLC), methanol and satd NaCl aqueous solution were added to the reaction mixture. The organic layer was separated, dried over Na2SO4, filtered through silica gel, and concentrated under reduced pressure. After drying in vacuo at room temperature for 15 min, the residue was suspended in CH2Cl2 (200 mL, dehydrated). To this suspension were successively added Et3N (1.7 mL, 12 mmol), N,N′-dimethylamino-4-pyridine (0.32 g, 2.6 mmol), and tert-butyldimethylsilyl chloride (1.8 g, 12 mmol), and the resulting mixture was stirred at room temperature for 1.5 h. To the reaction mixture was added water, and the resulting organic layer was separated, dried over Na2SO4, filtered through silica gel, concentrated under reduced pressure, and dried in vacuo at room temperature for 1 h, to give 4 (1.85 g, 90%) as a deep yellow solid. Rf 0.80 (hexane:ethyl acetate = 1:1); mp 32–34 °C; 1H NMR (300 MHz, acetone-d6) δ 0.27 (s, 6H), 1.01 (s, 9H), 7.08 (dd, J = 8.7, 2.4 Hz, 1H), 7.36 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 8.7 Hz, 1H); 13C NMR (75 MHz, acetone-d6) δ −4.51, 18.74, 25.88, 114.35, 121.46, 123.91, 133.71, 143.02, 156.69, 191.39.
Synthesis of TBDMS-protected PhOH-EDT-TTF5.
A mixture of thione 4 (629 mg, 2.0 mmol), 4,5-(ethylenedithio)-1,3-dithiole-2-one (625 mg, 3.0 mmol), and triethyl phosphite (8 mL) was stirred at 115 °C for 3 h. The reaction mixture was concentrated in vacuo, and to the residue was then added hexane. The resulting insoluble solid was removed by filtration and the filtrate was concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography with a 3:1 mixture of hexane and ethyl acetate as eluent, to give 5 (396 mg, 42%) as an orange powder. Rf 0.64 (hexane:ethyl acetate = 3:1); mp 114–115 °C; 1H NMR (300 MHz, CDCl3) δ 0.18 (s, 6H), 0.97 (s, 9H), 3.30 (s, 4H), 6.61 (dd, J = 8.4, 2.4 Hz, 1H), 6.76 (d, J = 2.4 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H); HRMS (APCI), calcd for C18H22O1S6Si1+: 473.9759 [M]+; found: 473.9750; anal. calcd for C18H22O1S6Si1: C, 45.53; H, 4.67; N, 0.00. Found C, 45.82; H, 4.97; N, 0.00.
Synthesis of PhOH-EDT-TTF.
TBDMS-protected PhOH-EDT-TTF2 (150 mg, 0.32 mmol) was placed in a 50 mL round-bottomed flask and dissolved in THF (6 mL, stabilizer free). To this solution were added acetic acid (2 drops) and nBu4NF (0.48 mL, 0.48 mmol, 1 M THF solution) at room temperature. After stirring for 20 minutes at the same temperature, the reaction mixture was directly subjected to silica gel column chromatography with ethyl acetate as eluent. The obtained crude material was washed with hexane and dried in vacuo at room temperature, to give PhOH-EDT-TTF (74 mg, 64%) as a reddish orange powder. A single crystal suitable for X-ray analysis was obtained as an orange plate by slow evaporation of a methanol solution at 4 °C. Rf 0.70 (ethyl acetate); mp 187 °C (dec.); 1H NMR (300 MHz, acetone-d6) δ 3.44 (s, 4H), 6.75 (dd, J = 8.4, 2.4 Hz, 1H), 6.96 (d, J = 2.4 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H) [the OH proton was not detectable by 1H NMR. A clear 13C NMR spectrum was not obtained because of low solubility of PhOH-EDT-TTF in common organic solvents.]; HRMS (APCI), calcd for C12H8O1S6+: 359.8894 [M]+; found: 359.8886; anal. calcd for C12H8O1S6(C6H14)0.05: C, 40.49; H, 2.40; N, 0.00. Found C, 40.51; H, 2.66; N, 0.00.
Electrochemical crystallization of β-(PhOH-EDT-TTF)2ClO4
PhOH-EDT-TTF (8.0 mg) was placed in one side of an H-shaped cell equipped with a glass filter. Then, nBu4NClO4 (20 mg) was placed in each side of the cell, and the cell was purged with argon gas. These materials were dissolved in a mixed solvent of CH3CN (2 mL) and degassed PhCl (17 mL). The resulting solution was kept at 20 °C for 2 h, and then an annealed Pt electrode was inserted into each side of the cell. A constant current of 1.0 μA was applied to the solution at 20 °C for 1 week, to afford brown plate crystals of β-(PhOH-EDT-TTF)2ClO4 (typical size: 0.30 × 0.05 × 0.01 mm3) suitable for the X-ray crystal analysis and physical property measurements.
Single crystal X-ray structure analysis
X-ray crystallographic measurements were performed on a Rigaku MercuryII CCD X-ray diffractometer (MoKα, λ = 0.71073 Å). The structures were solved by direct methods and refined with a full-matrix least squares technique using CrystalStructure 4.2.4 (Rigaku Corporation (2000–2016)). Anisotropic thermal parameters were applied to all non-hydrogen atoms. The hydrogen atoms were generated geometrically.
CCDC 1590325 (PhOH-EDT-TTF) and 1590324 (β-(PhOH-EDT-TTF)2ClO4).†
Electrical resistivity
Electrical resistivity measurements of a single crystal of β-(PhOH-EDT-TTF)2ClO4 were performed on a Physical Property Measurement System (Quantum Design) by the conventional four-probe method using carbon paste and gold wires.
Theoretical calculations
Density functional theory (DFT) calculations were performed with Gaussian03 program (revision E.01) at the B3LYP/6-31G(d) level of theory.13 Intermolecular transfer integrals were calculated by the extended Hückel method.17 Electronic band structures were calculated by the tight-binding approximation method.17
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (No. 15H00988, 16K05744, 17H05143, 24340074, 16H04010) from Japan Society for the Promotion of Science (JSPS), Japan, and the Canon Foundation. The authors also thank Prof. S. Suzuki (Osaka University) for measurements of the APCI-MS spectra and Prof. H. Yoshizawa (ISSP, The Univ. of Tokyo) for assistance in using his Physical Property Measurement System (Quantum Design).
Notes and references
-
Hydrogen-Transfer Reactions, ed. J. T. Hynes, J. P. Klinman, H.-H. Limach and R. L. Schowen, Wiley-VCH, Weinheim, 2007 Search PubMed;
A. Nangia, in Organic Crystal Engineering: Frontiers in Crystal Engineering, ed. E. R. T. Tiekink, J. Vittal and M. Zaworotko, John Wiley & Sons, Chichester, 2010, ch. 5, pp. 151–189 Search PubMed;
Hydrogen Bonded Supramolecular Structures, ed. Z.-T. Li and L.-Z. Wu, Springer, Berlin-Heidelberg, 2015 Search PubMed;
Hydrogen Bonded Supramolecular Structures Materials, ed. Z.-T. Li and L.-Z. Wu, Springer, Berlin-Heidelberg, 2015 Search PubMed.
-
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed;
G. Gilli and P. Gilli, The Nature of the Hydrogen Bond, Oxford University Press, New York, 2009 Search PubMed.
- Recent examples, see:
(a) T. Murata, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui, M. Maesato, H. Yamochi, G. Saito and K. Nakasuji, Angew. Chem., Int. Ed., 2004, 43, 6343 CrossRef CAS PubMed;
(b) T. Akutagawa, S. Takeda, T. Hasegawa and T. Nakamura, J. Am. Chem. Soc., 2004, 126, 291 CrossRef CAS PubMed;
(c) T. Murata, Y. Morita, Y. Yakiyama, K. Fukui, H. Yamochi, G. Saito and K. Nakasuji, J. Am. Chem. Soc., 2007, 129, 10837 CrossRef CAS PubMed;
(d) Y. Kobayashi, M. Yoshioka, K. Saigo, D. Hashizume and T. Ogura, J. Am. Chem. Soc., 2009, 131, 9995 CrossRef CAS PubMed;
(e) A. El-Ghayoury, C. Mézière, S. Simonov, L. Zorina, M. Cobián, E. Canadell, C. Rovira, B. Náfrádi, B. Sipos, L. Forró and P. Batail, Chem. – Eur. J., 2010, 16, 14051 CrossRef CAS PubMed;
(f) S. Oliveira, J. Ministro, I. C. Santos, D. Belo, E. B. Lopes, S. Rabaça, E. Canadell and M. Almeida, Inorg. Chem., 2015, 54, 6677 CrossRef CAS PubMed;
(g) S. Rabaça, S. Oliveira, A. C. Gonçalves, V. Gama, I. C. Santos, D. Belo and M. Almeida, Cryst. Growth Des., 2017, 17, 2801 CrossRef . See also review articles;
(h) M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379 CrossRef PubMed;
(i) Y. Morita, T. Murata and K. Nakasuji, Bull. Chem. Soc. Jpn., 2013, 86, 183 CrossRef CAS.
-
(a) H. Kamo, A. Ueda, T. Isono, K. Takahashi and H. Mori, Tetrahedron Lett., 2012, 53, 4385 CrossRef CAS;
(b) A. Ueda, H. Kamo and H. Mori, Chem. Lett., 2015, 44, 1538 CrossRef CAS.
- T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakao, K. Kobayashi, Y. Murakami and H. Mori, Nat. Commun., 2013, 4, 1344 CrossRef CAS PubMed; A. Ueda, S. Yamada, T. Isono, H. Kamo, A. Nakao, R. Kumai, H. Nakao, Y. Murakami, K. Yamamoto, Y. Nishio and H. Mori, J. Am. Chem. Soc., 2014, 136, 12184 CrossRef PubMed; T. Isono, H. Kamo, A. Ueda, K. Takahashi, M. Kimata, H. Tajima, S. Tsuchiya, T. Terashima, S. Uji and H. Mori, Phys. Rev. Lett., 2014, 112, 177201 CrossRef PubMed; A. Ueda, A. Hatakeyama, M. Enomoto, R. Kumai, Y. Murakami and H. Mori, Chem. – Eur. J., 2015, 21, 15020 CrossRef PubMed; S. Yamashita, Y. Nakazawa, A. Ueda and H. Mori, Phys. Rev. B: Condens. Matter Mater. Phys., 2017, 95, 184425 CrossRef; M. Shimozawa, K. Hashimoto, A. Ueda, Y. Suzuki, K. Sugii, S. Yamada, Y. Imai, R. Kobayashi, K. Itoh, S. Iguchi, M. Naka, S. Ishihara, H. Mori, T. Sasaki and M. Yamashita, Nat. Commun., 2017, 8, 1821 CrossRef PubMed.
- K. Yamamoto, Y. Kanematsu, U. Nagashima, A. Ueda, H. Mori and M. Tachikawa, Phys. Chem. Chem. Phys., 2016, 18, 29673 RSC; K. Yamamoto, Y. Kanematsu, U. Nagashima, A. Ueda, H. Mori and M. Tachikawa, Chem. Phys. Lett., 2017, 674, 168 CrossRef CAS.
- A. Ueda, Bull. Chem. Soc. Jpn., 2017, 90, 1181 CrossRef.
-
(a) S. C. Lee, A. Ueda, H. Kamo, K. Takahashi, M. Uruichi, K. Yamamoto, K. Yakushi, A. Nakao, R. Kumai, K. Kobayashi, H. Nakao, Y. Murakami and H. Mori, Chem. Commun., 2012, 48, 8673 RSC;
(b) S. C. Lee, A. Ueda, A. Nakao, R. Kumai, H. Nakao, Y. Murakami and H. Mori, Chem. – Eur. J., 2014, 20, 1909 CrossRef CAS PubMed;
(c) J. Yoshida, A. Ueda, A. Nakao, R. Kumai, H. Nakao, Y. Murakami and H. Mori, Chem. Commun., 2014, 50, 15557 RSC;
(d) S. J. Krivickas, C. Hashimoto, J. Yoshida, A. Ueda, K. Takahashi, J. D. Wallis and H. Mori, Beilstein J. Org. Chem., 2015, 11, 1561 CrossRef PubMed;
(e) J. Yoshida, A. Ueda, R. Kumai, Y. Murakami and H. Mori, CrystEngComm, 2017, 19, 367 RSC;
(f) T. Higashino, A. Ueda, J. Yoshida and H. Mori, Chem. Commun., 2017, 53, 3426 RSC.
- Recent examples of molecular conductors based on asymmetrically mono-substituted TTF derivatives, see: M. M. Matsushita, H. Kawakami, Y. Kawada and T. Sugawara, Chem. Lett., 2007, 36, 110 CrossRef CAS; X. Shao, Y. Nakano, H. Yamochi, A. D. Dubrovskiy, A. Otsuka, T. Murata, Y. Yoshida, G. Saito and S. Koshihara, J. Mater. Chem., 2008, 18, 2131 RSC; J. Lieffrig, O. Jeannin, T. Guizouarn, P. Auban-Senzier and M. Fourmigué, Cryst. Growth Des., 2012, 12, 4248 Search PubMed; M. Ishikawa, Y. Nakano, M. Uruichi, A. Otsuka, K. Yakushi and H. Yamochi, Eur. J. Inorg. Chem., 2014, 3941 CrossRef . See also ref. 3a, c–i, 8a, b, d and 10.
- Representative books and review articles, see: TTF Chemistry: Fundamentals and Applications of Tetrathiafulvalene, ed. J. Yamada and T. Sugimoto, Kodansha-Springer, Tokyo, 2004 CrossRef CAS PubMed;
M. Fourmigué, in Halogen Bonding: Fundamentals and Applications, ed. P. Metrangolo and G. Resnati, Springer-Verlag, Berlin-Heidelberg, 2008, pp. 181–207 CrossRef CAS PubMed; J. M. Fabre, Chem. Rev., 2004, 104, 5133 CrossRef CAS PubMed; N. Avarvari and J. D. Wallis, J. Mater. Chem., 2009, 19, 4061 RSC.
- B. Andersh, D. L. Murphy and R. J. Olson, Synth. Commun., 2000, 30, 2091 CrossRef CAS.
- D. Demeter, P. Blanchard, I. Grosu and J. Roncali, Electrochem. Commun., 2007, 9, 1587 CrossRef CAS.
-
M. J. Frisch, et al., Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- To reveal this, the synthesis of the Br salt of PhOH-EDT-TTF and/or the ClO4 salt of H2Cat-EDT-TTF is effective and thus is currently underway in our group.
- T. Mori, Bull. Chem. Soc. Jpn., 1998, 71, 2509 CrossRef CAS.
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627 CrossRef CAS.
- This might be due to strong electron correlations resulting from a relatively narrow bandwidth (∼0.6 eV; Fig. 7a, left). Further details of the electronic structure are currently being investigated by magnetic measurements.
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Hatsumi
Mori
*
* and
Hatsumi
Mori
*