
Delineating the critical role of acid additives in Mn-catalysed C–H bond functionalisation processes†
L. Anders
Hammarback
 a,
Alan
Robinson
b,
Jason M.
Lynam
*a and
Ian J. S.
Fairlamb
*a
a,
Alan
Robinson
b,
Jason M.
Lynam
*a and
Ian J. S.
Fairlamb
*a
aDepartment of Chemistry, University of York, York, YO10 5DD, UK. E-mail: jason.lynam@york.ac.uk; ian.fairlamb@york.ac.uk
bSyngenta Crop Protection AG, Münchwilen, Breitenloh 5, 4333, Switzerland
First published on 26th February 2019
Abstract
Addition of co-catalytic Cy2NH to Mn-catalysed C–H bond activation reactions suggests that the conjugate acid, Cy2NH2X, influences catalysis. Here, acids are shown to positively influence C–H bond alkenylation catalysis involving alkynes. For certain types of alkynes an acid additive is critical to catalysis. In stark contrast, acids retard catalysis involving acrylates. [Cy2NH2]X salts also play a key role in thwarting catalyst degradation to manganese clusters. Our findings enable unreactive substrates to be alkenylated.
The Earth abundant element, manganese, holds much promise in catalysis and applied chemical synthesis, as demonstrated by an eclectic array of recently discovered reactions.1 Underpinning this rise to fame is a rich 40-year history2 of stoichiometric organomanganese chemistry, particularly cyclomanganation and subsequent reactions of manganacycles. Catalytic C–H bond activation and functionalisation has grown from this base over the past 5 years, primarily inspired by Chen and Wang's3 work reported in 2013, from which many important contributions have followed.4 MnI-catalysed ‘redox neutral’ C–H bond functionalisation of, for example 2-phenylpyridine (1), by BrMn(CO)5 (2), invoke Mn(ppy)(CO)4 (3, ppy = 2-phenylpyridyl) formation. Reaction with an unsaturated ‘acceptor’ substrate (4), for example, alkenes, alkynes, carbonyls, isocyanates and cyanides, have been widely reported (Scheme 1).5 Indeed, the synthetic methodologies developed far exceed detailed mechanistic studies, which have arguably lagged behind. However, recent mechanistic work6,7 has shed light on the importance of CO liberation (from 3), acceptor substrate (4) coordination (to give transient intermediate I) and migratory insertion leading to formation of 7-membered manganacycle (II), that can be characterised but only have a short relative lifetime.6 Very little is known about the last step in the general catalytic scheme, namely protonation of II by substrate 1 (the ‘acid’), to bring about liberation of product 5.
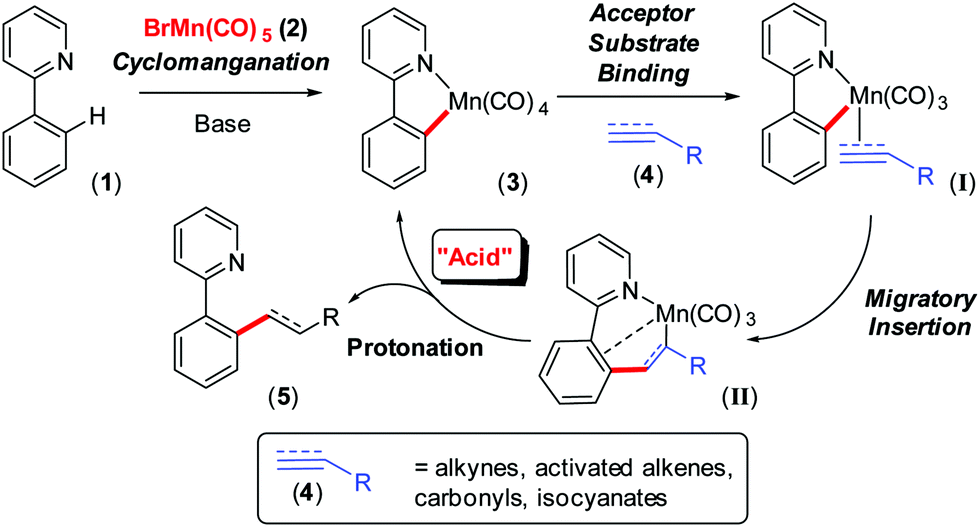 | ||
| Scheme 1 Mechanistic picture for MnI-catalysed C–H bond activation and functionalisation, using a redox-neutral process. | ||
The mechanistic picture detailed in Scheme 1 is not complete, as several reaction components can play the role of the acid,8 from the donor C–H substrate, acceptor substrate, H2O and the conjugate acid of the co-catalytic base, Cy2NH, typically needed for catalysis.
In some reported methodologies, stronger carboxylic acid additives are required for productive catalysis,9 or to increase the rate of protonation to avoid alternative reaction pathways.7,10 It can also be envisaged that the conjugate acids of metal acetate type bases will have a similar effect in most reactions, when employed. The reason behind the effects of conjugate acids and acid additives in MnI-catalysed reaction have not been studied in detail, particularly in improving current promising synthetic methodologies.
To investigate the effect of additives in MnI-catalyzed C–H functionalisation reactions, Mn(ppy)(CO)4 (3) was initially chosen as the entry point to probe subsequent steps, as no base additive nor ‘Mn activation’ is required. We have confirmed that 3 is a validated intermediate in the activation pathway for internal alkynes,8 and a feasible entry point into the catalytic cycle for terminal alkynes. The reactions of 2-phenylpyridine 1 with unsaturated acceptor substrates (4a–c) mediated by 3, in the presence of various additives, are collated in Fig. 1. Reaction of 1 + 4a → 5a under standard conditions (at 100 °C, in n-Bu2O, 3 h) Cy2NH as the base, in the presence of BrMn(CO)5 (10 mol%) as precatalyst, gave 5a in 76% conversion (reported yield = 76%3). Employment of 3 as pre-catalyst gave a lower conversion to 5a (55%). Addition of Cy2NH did not noticeably affect the conversion to 5a (58%), which is in line with previous proposals that the amine base is only required for activation of the MnI in the case of terminal alkynes.
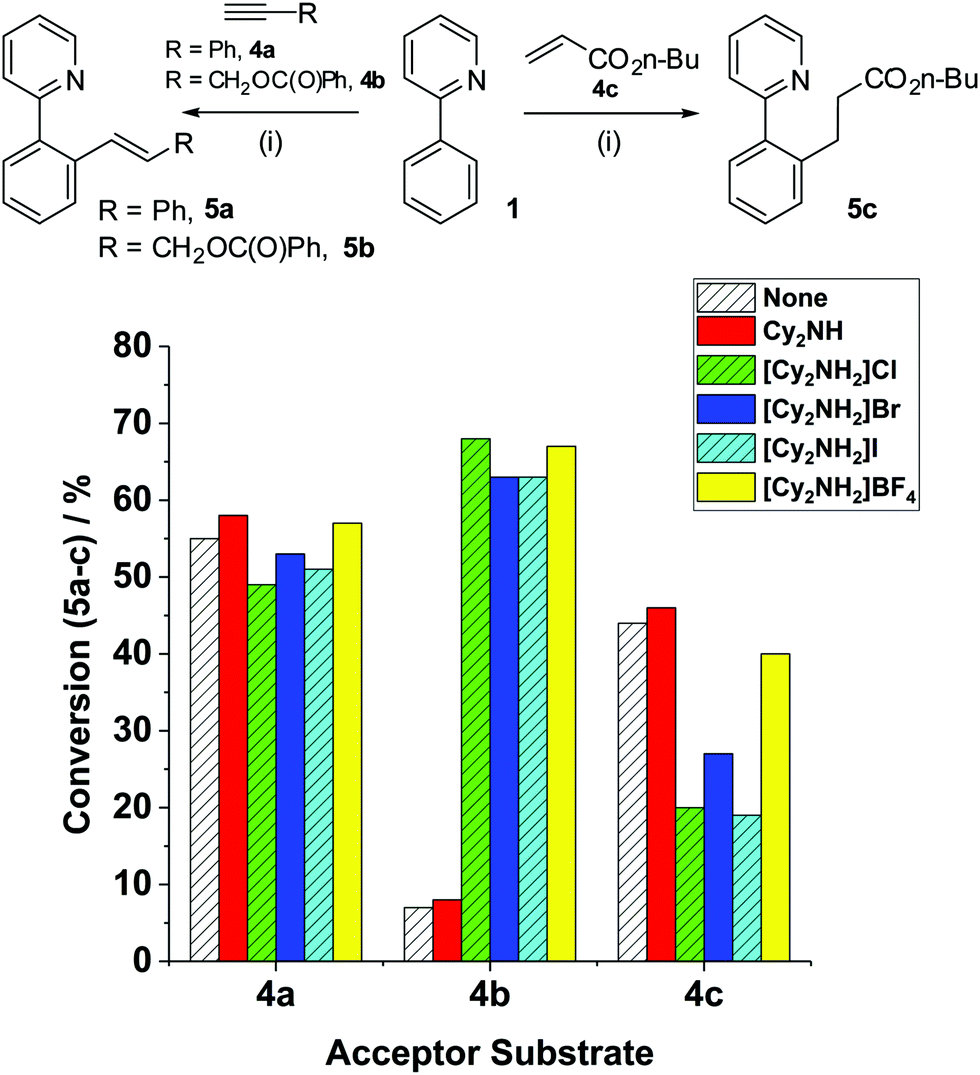 | ||
| Fig. 1 The effect of base/conjugate acid additives in the reactions of 1 + 4a–c → 5a–c. Reaction conditions (i): Mn(ppy)(CO)4 (3) (2, 10 mol%), 1 (2 equiv.), 4a–c (1 equiv.), additive (20 mol%) – see figure legend key for details, n-Bu2O, 100 °C, 3 h. | ||
Adding the conjugate acid of Cy2NH, i.e. [Cy2NH2]Br, a side-product resulting from the initial C–H bond deprotonation in the reaction 1 + 4a → 5a, did not impact upon the conversion to 5a (53%). The reaction is insensitive to changes in the anion, irrespective of the type of halide/pseudohalide (Fig. 1).
The employment of an alternative terminal alkyne, propargyl benzoate 4b with 3 as the precatalyst, formed only a small amount of product 5b, in the presence and absence of Cy2NH {Conv. to 5b 7% and 8% respectively (2.5 × 10−5 mol)}. The extent of product conversion appears to tally with the [MnTOTAL] = 10 mol% (2.5 × 10−5 mol), indicating that there is limited turnover of the Mn catalyst. By contrast, the reaction mediated by BrMn(CO)52 and Cy2NH gave 5b with 54% conversion, in keeping with Chen and Wang's3 observations using Et2O solvent instead of n-Bu2O (under equivalent reaction conditions obtaining 48% yield of 5b). Limited catalyst turnover indicates an issue in either the protonation or recycling steps required for catalysis vide supra, leading to low product conversion and rapid catalyst degradation. The addition of [Cy2NH2]Br had a profound and unexpected effect on product conversion to 5b, increasing the conversion to 63%. The response was equally dramatic using [Cy2NH2]Cl, [Cy2NH2]I or [Cy2NH2]BF4 as acid additives.
In the original reaction of 1 + 4c → 5c, reported by Wang and co-workers,11 it was suggested that [Cy2NH2]Br acted as a proton source for the protonation step (not experimentally proven, but reasonably postulated within a reaction mechanism scheme). We thus assessed the impact of the base/conjugate acid additives in the reaction of 1 + 4c → 5c, employing 3 as the precatalyst (Fig. 1). When no additive was used in this reaction a lower conversion to 5c (44%) was recorded than when BrMn(CO)52 and Cy2NH were employed (79%). The lower product conversion can be rationalised as arising from no [Cy2NH2]Br being present in the reaction mediated by 3. Surprisingly, however, addition of [Cy2NH2]Br was found to negatively affect conversion to product 5c (27%). The negative influence of the conjugate acid is further compounded by altering the anion in [Cy2NH2]X to chloride or iodide. Intriguingly, the less coordinating BF4− counter-ion led to a product conversion similar to the reaction mediated by Cy2NH (40% versus 46%, respectively).
In operando studies using infrared spectroscopic analysis: to gain insight into how [Cy2NH2][X] (X = Br, Cl, I or BF4) affects catalyst efficacy, the reactions of 1 + 4b–c → 5b–c were monitored in operando employing in situ IR spectroscopy, using a Mettler-Toledo ReactIR® instrument with Si-probe. This method allows for changes in metal carbonyl peaks to be monitored (qualitatively and quantitatively), being excellent spectroscopic handles for the observation of the dynamic processes occurring at the manganese centre. The reaction responding positively to acid, of 1 + 4b → 5b, mediated by precatalyst 3 (Fig. 2a) results in the initial appearance of two new species, with overlapping carbonyl bands, depicted by gold stars and closed red circles. After 30 minutes, a minor species with carbonyl bands at ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) 1996 and 1944 cm−1 (black triangles) forms once catalysis is complete. We note the relative slow loss of 3 (trace quantities seen after ca. 5 minutes, depicted by closed blue circles), which is consistent with an independent mechanistic study examining the reaction of 1 + 4a → 5a.3 The species depicted by gold stars is most likely a deactivation product, i.e. manganese hydroxyl-containing clusters previously proposed to be deactivation products for catalysis (see ESI†).12 While the species depicted by closed red circles evolves and then rapidly depletes, suggesting its involvement as a transient intermediate.
1996 and 1944 cm−1 (black triangles) forms once catalysis is complete. We note the relative slow loss of 3 (trace quantities seen after ca. 5 minutes, depicted by closed blue circles), which is consistent with an independent mechanistic study examining the reaction of 1 + 4a → 5a.3 The species depicted by gold stars is most likely a deactivation product, i.e. manganese hydroxyl-containing clusters previously proposed to be deactivation products for catalysis (see ESI†).12 While the species depicted by closed red circles evolves and then rapidly depletes, suggesting its involvement as a transient intermediate.
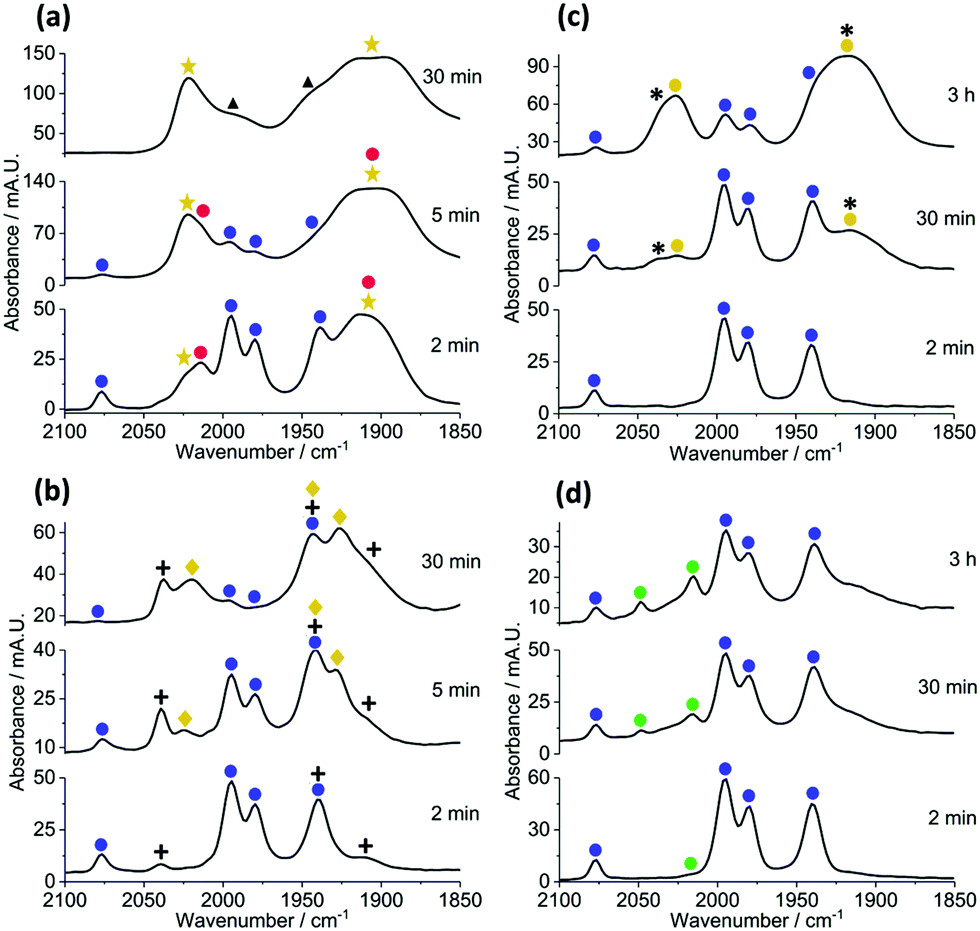 | ||
| Fig. 2 Reaction monitoring using in situ IR monitoring using 3 as the pre-catalyst. (a) Reaction 1 + 4b → 5b, without Cy2NH. (b) Reaction 1 + 4b → 5b, with [Cy2NH2]BF4 (20 mol%). (c) Reaction 1 + 4c → 5c, without Cy2NH. (d) Reaction 1 + 4c → 5c with [Cy2NH2][Br] (20 mol%). | ||
However, when [Cy2NH2][BF4] was added to the reaction (Fig. 2b) the rate of formation of the previously observed deactivation species (gold stars) was greatly diminished. We note that another carbonyl-containing species is formed in this reaction (depicted by + symbols), which remains prominently after 30 minutes reaction time. Manganacycle 3 remains for the duration of the reaction (depleting to 30 minutes). The formation of manganese alkynyl-containing clusters is observed after 5 minutes reaction time (depicted by gold diamonds).
Monitoring the reaction of 1 + 4c → 5c without Cy2NH (Fig. 2c) showed that two new significant overlapping carbonyl bands (depicted by closed gold circles) formed, i.e. similar bands to the manganese hydroxyl-containing clusters seen when employing alkyne 4b but not identical. Furthermore, manganacycle 3 persists throughout the reaction. Addition of [Cy2NH2]Br to the reaction 1 + 4c → 5c (Fig. 2d), once again reduces the amount of manganese hydroxyl-containing clusters formed. Over longer reaction times Mn2(CO)10 is formed (2048 and 2015 cm−1, depicted by closed green circles), which is also known to be a competent precatalyst for this type of transformation. The formation of Mn2(CO)10 was not detected for the reaction conducted in the absence of [Cy2NH2]Br.
The take home messages from the in operando IR studies are:
• Addition of [Cy2NH2]Br hinders the formation of manganese hydroxyl-containing clusters, and related clusters. In the case of the acrylate 4c we note formation of Mn2(CO)10, after just 2 minutes, which becomes inactive as catalyst under these reaction conditions.
• [Cy2NH2]Br increases the lifetime of manganacycle 3 in both reactions 1 + 4b–c → 5b–c, respectively.
Further studies on the acid additives – effect of pK a : we extended our studies on the reaction of 1 + 4b → 5b, mediated by precatalyst 3 along with propionic acid (pKa = 4.9 in H2O13) as a co-catalyst (20 mol%), affording 5b in 82% conversion – an enhancement of 75% when compared against the reaction without any additive (7%, vide supra, see Fig. 3). Switching to HCl (pKa = −8 in H2O) as the acid additive led to no enhancement in catalysis (5% conversion to 5b recorded), while even stronger acids HBr (pKa = −9 in H2O13) and HI (pKa = −10 in H2O13) resulted in negligible product formation (≤1%). Interestingly, HBF4·OEt2 (pKa = −4.9 in H2O13) enhanced product formation, affording 5b in 62% conversion. This latter result possibly indicates that the relatively non-coordinating BF4− can assist catalysis in some way, i.e. providing protons without catalyst degradation or by minimising halide coordination to MnI, consistent with the results presented in Fig. 1.
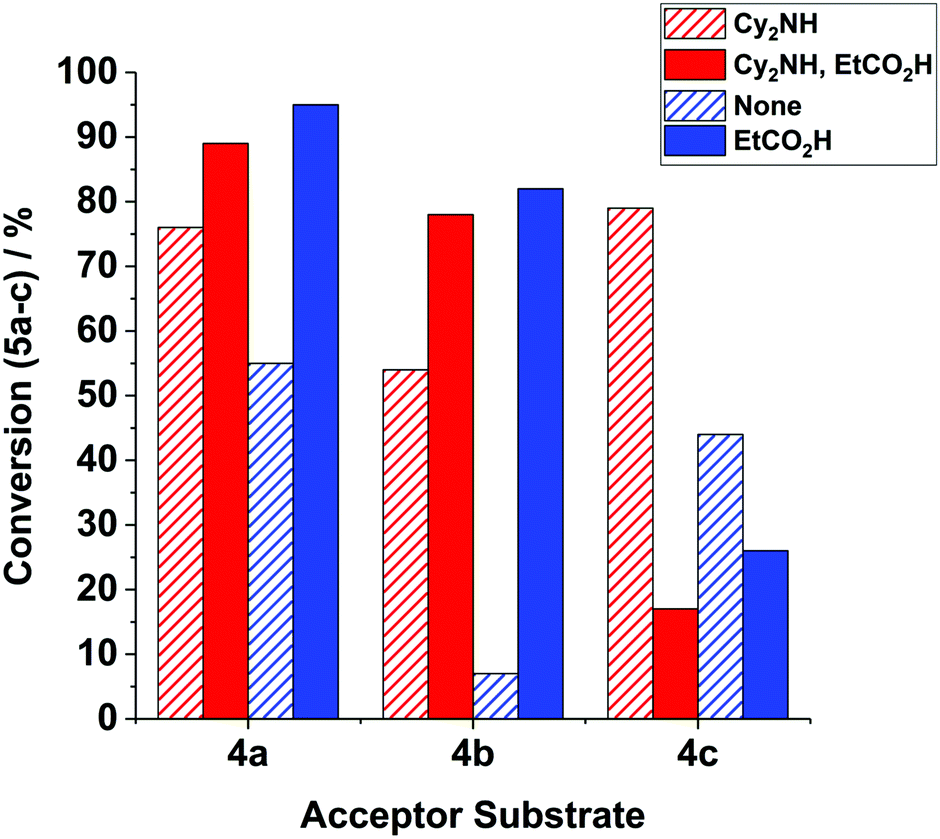 | ||
| Fig. 3 The effect of additives in the reactions of 1 + 4a–c → 5a–c (see Fig. 1). General reaction conditions are detailed in Fig. 1, with the exception of the Mn precatalyst used: reactions mediated by BrMn(CO)5 (10 mol%) are highlighted in red, and those mediated by manganacycle 3 (10 mol%) are highlighted in blue – see figure legend key for details of the additive. | ||
Examination of the reaction of 1 + 4b → 5b mediated by BrMn(CO)52, with Cy2NH (20 mol%) in the presence and absence of propionic acid (20 mol%), provided further evidence for an acid effect in enhancing the conversion of 5b from 54% to 78% (Fig. 3).
The positive promoting effect of propionic acid in the reaction of 1 + 4b → 5b led us to examine the other acceptor substrates 4a and 4c (Fig. 3), for both BrMn(CO)52 and manganacycle 3 precatalysts. For phenyl acetylene 4a we find that propionic acid enhances catalysis for both 2 and 3, in the case of the latter, significantly (i.e. 95% conversion to 5a, as compared to 55% in the absence of propionic acid). It is striking that the conjugate acids [Cy2NH2]X do not positively influence catalysis under the same reaction conditions.
In the reaction of 1 + 4c → 5c, mediated by both BrMn(CO)52 and manganacycle 3 precatalysts, led to a significant reduction in product 5c formation, which is consistent with the net negative effects of the conjugate acids [Cy2NH2]X vide supra.
Promotion of substrates previously determined to be problematic for catalytic C–H bond alkenylation using Mn I catalysis: we have previously determined that internal alkyne 6 strongly prefers reductive elimination pathways over protonation (in a reaction with 2-phenylpyridine 1), thus rendering formation of alkenylated product 7 inaccessible.8 Drawing on what had been learnt from this study we postulated that the protonation step could be assisted by EtCO2H, bringing about the formation of alkenylated product 7. Pleasingly, under the reaction conditions given in Scheme 2, 7 is formed in 20% yield (isolated product after chromatography), showcasing the ability of acid additives to form otherwise unachievable products for the MnI catalysis. The modest amount of 7 formed highlights the difficulty associated with the protonation step within the catalytic cycle, explaining why substrate sensitivity is sometimes observed.
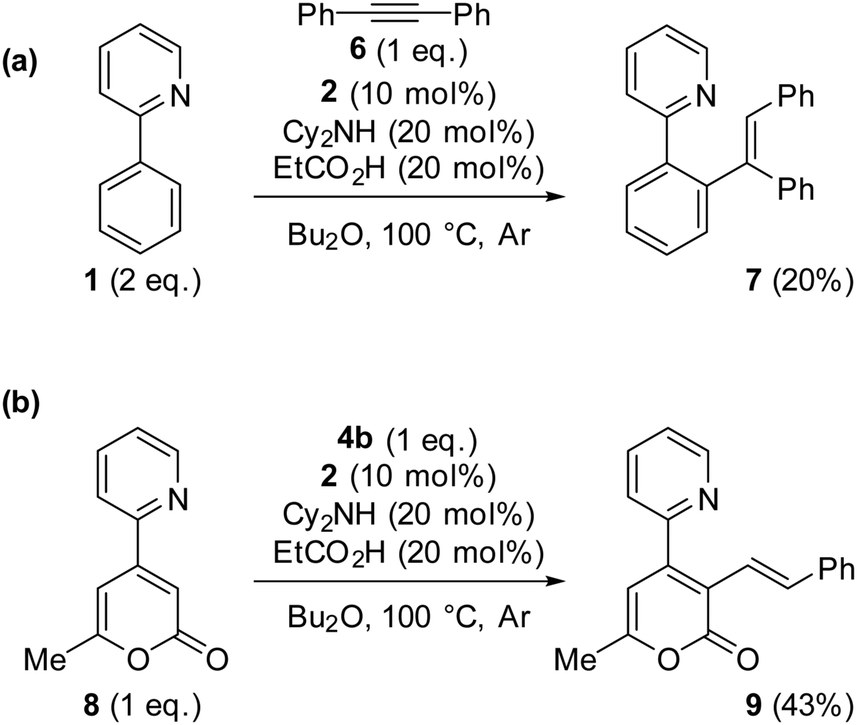 | ||
| Scheme 2 (a) Effect of addition of EtCO2H to a reaction with diphenylacetylene 6. (b) Effect of addition of EtCO2H to a reaction with 8. | ||
We next turned our attention to the reaction with substrate 8, which had previously allowed us to characterise a key 7-membered manganacyclic intermediate, the anvil point to subsequent protonation or reductive elimination,6 with the latter being preferred under typical catalytic reaction conditions (Scheme 2b). Quite remarkably we discovered that the reaction 8 + 4a → 9 was made feasible by the addition of propionic acid affording 9 in 43% yield (isolated product after chromatography) (Scheme 2b). This result highlights the ability of acid additives to promote the protonation pathway, with concomitant formation of alkenylated products.
To summarise, we set out to understand the role of the conjugate acid, [Cy2NH2]X, formed by protonation of co-catalytic Cy2NH base, in influencing C–H bond activation catalysis at MnI. We conclude that conjugate acids positively influence C–H bond alkenylation catalysis involving terminal and internal alkynes, which are found to be critical to productive catalysis. Importantly, the promotional effect of acid did enable unreactive substrates to be alkenylated involving both internal and terminal alkynes (Scheme 2). A dichotomy in behaviour is seen on switching from alkynes to an acrylate substrate, where catalysis is hindered by the presence of the conjugate acid containing a halide anion (cf. not a non-coordinating BF4 anion). A secondary benefit of the [Cy2NH2]X salts is to thwart MnI catalyst degradation to form inactive manganese clusters, which is an issue requiring attention in future catalyst design studies.8
We thank Syngenta and the Engineering and Physical Sciences Research Council (EPSRC) for iCASE funding for LAH, ref. EP/H011455.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews concerning manganese catalysis in general, see: (a) J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Org. Chem., 2016, 3912–3929 CrossRef CAS; (b) R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS PubMed; (c) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed; (d) M. Stanbury, J.-D. Compain and S. Chardon-Noblat, Coord. Chem. Rev., 2018, 361, 120–137 CrossRef CAS.
- M. I. Bruce, B. L. Goodall, M. Z. Iqbal, F. G. A. Stone, R. J. Doedens and R. G. Little, J. Chem. Soc. D, 1971, 1595–1596 RSC.
- B. Zhou, H. Chen and C. Wang, J. Am. Chem. Soc., 2013, 135, 1264–1267 CrossRef CAS PubMed.
- Selected examples: (a) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Angew. Chem., 2014, 126, 5050–5053 CrossRef; (b) W. Liu, D. Zell, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 4092–4096 CrossRef CAS PubMed; (c) B. Zhou, Y. Hu and C. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659–13663 CrossRef CAS PubMed; (d) W. Liu, J. Bang, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 14137–14140 CrossRef CAS PubMed; (e) C. Wang, A. Wang and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 9935–9938 CrossRef CAS PubMed; (f) Q. Lu, S. Cembellin, S. Greßies, S. Singha, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 1399–1403 CrossRef CAS PubMed.
- Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS PubMed.
- N. P. Yahaya, K. M. Appleby, M. Teh, C. Wagner, E. Troschke, J. T. W. Bray, S. B. Duckett, L. A. Hammarback, J. S. Ward, J. Milani, N. E. Pridmore, A. C. Whitwood, J. M. Lynam and I. J. S. Fairlamb, Angew. Chem., Int. Ed., 2016, 55, 12455–12459 CrossRef CAS PubMed.
- L. A. Hammarback, I. P. Clark, I. V. Sazanovich, M. Towrie, A. Robinson, F. Clarke, S. Meyer, I. J. S. Fairlamb and J. M. Lynam, Nat. Catal., 2018, 1, 830–840 CrossRef.
- L. A. Hammarback, A. Robinson, J. M. Lynam and I. J. S. Fairlamb, J. Am. Chem. Soc., 2019, 141, 2316–2322 CrossRef CAS PubMed.
- H. Wang, F. Pesciaioli, J. C. A. Oliveira, S. Warratz and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15063–15067 CrossRef CAS PubMed.
- L. Shi, X. Zhong, H. She, Z. Lei and F. Li, Chem. Commun., 2015, 51, 7136–7139 RSC.
- B. Zhou, P. Ma, H. Chen and C. Wang, Chem. Commun., 2014, 50, 14558–14561 RSC.
- For ‘Mn(OH)(CO)3’ type clusters, see: (a) M. D. Clerk and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1991, 1607–1608 RSC ; For ‘Mn(SR)(CO)3’ type clusters, see: ; (b) S. Gläser, R. Mede, H. Görls, S. Seupel, C. Bohlender, R. Wyrwa, S. Schirmer, S. Dochow, G. U. Reddy, J. Popp, M. Westerhausen and A. Schiller, Dalton Trans., 2016, 45, 13222–13233 RSC ; For ‘Ru(σ,μ2-alkynyl)(CO)3’ type clusters, see: ; (c) S. Bock, C. F. Mackenzie, B. W. Skelton, L. T. Byrne, G. A. Koutsantonis and P. J. Low, J. Organomet. Chem., 2016, 812, 190–196 CrossRef CAS.
- P. Y. Bruice, Organic Chemistry, 5th edn, Pearson, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterisation. See DOI: 10.1039/c9cc00257j |
| This journal is © The Royal Society of Chemistry 2019 |