
Trifluoromethylthiolative 1,2-difunctionalization of alkenes with diselenides and AgSCF3†
Perumal
Saravanan
and
Pazhamalai
Anbarasan
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai-600036, India. E-mail: anbarasansp@iitm.ac.in; Web: https://home.iitm.ac.in/anbarasansp/
First published on 23rd March 2019
Abstract
An efficient regioselective difunctionalization of alkenes via trifluoromethylthiolation has been accomplished employing diaryl diselenide and AgSCF3 in the presence of BF3·OEt2. Various substituted 1,2-dichalcogenated products having the SCF3 moiety were synthesized in good to excellent yields under mild conditions. The preliminary mechanistic investigation revealed the possible reaction pathway and unique combination of diselenide and AgSCF3 for successful transformation.
Fluorinated molecules have found widespread application in various fields. Notably, a significant number of drugs in pharmaceuticals and agrochemicals contain at least one fluorine atom/group in the form of –F, –CF3, –SCF3, or –SOCF3.1 Recently, trifluoromethylthio (–SCF3) containing organic molecules gained significant attention, due to their unique properties such as high lipophilicity, bioavailability, and metabolic stability. The representative examples of –SCF3 containing drug molecules include toltrazuril, cefazaflur, and fipronil. Thus, the development of an elegant strategy for the construction of trifluoromethylthiolated molecules has been of continuing interest in organic synthesis and other fields.2 Consequently, enormous efforts have been dedicated towards the development of various strategies for the construction of trifluoromethylthiolated compounds such as F-exchange3 and direct introduction of –CF34 and –SCF35 groups. Among them, direct trifluoromethylthiolation, using electrophilic or nucleophilic ‘SCF3’ reagents, is the most efficient and viable strategy in the synthesis of trifluoromethyl sulfides via the direct construction of C–SCF3 bonds.
On the other hand, difunctionalization of readily accessible substituted alkenes with electrophiles and nucleophiles is an efficient multi-component approach for the synthesis of structurally complex frameworks.6 In this context, the incorporation of the trifluoromethylthio (–SCF3) group along with other functional groups, such as amines, amides, and acids,7 is a useful concept for the synthesis of trifluoromethylthiolated molecules, which could provide the excellent opportunity for medicinal chemists in the drug evolution. Billard and co-workers reported for the first time trifluoromethylthiolative functionalization of alkenes using electrophilic trifluoromethylthiolating reagents.8 Subsequently, numerous methods9 employing electrophilic trifluoromethylthiolating reagents have been documented (Scheme 1a). However, use of nucleophilic trifluoromethylthiolating reagents is rather limited.10 For instance, the groups of Wang10a and Qing10d utilized AgSCF3 in combination with superstiochiometric amounts of persulfate for the difunctionalization of alkenes. Wang and co-workers10b utilized a substoichiometric amount of copper acetate and AgSCF3. On the other hand, the combination of AgSCF3 and trichloroisocyanuric acid was exploited by Yang and co-workers.10c However, to the best of our knowledge, difunctionalization of alkenes with nucleophilic AgSCF3 in the absence of a metal catalyst or oxidant was not documented in the literature. Thus, we envisioned an arylselenative trifluoromethylthiolation of alkenes with diaryl diselenide and nucleophilic AgSCF3 for the synthesis of 1,2-dichalcogenated compounds, where the potential intermediate ArSeSCF3 might afford the expected product in the presence of Lewis acids (Scheme 1b). We herein disclose an elegant arylselenative trifluoromethylthiolation of substituted alkenes.
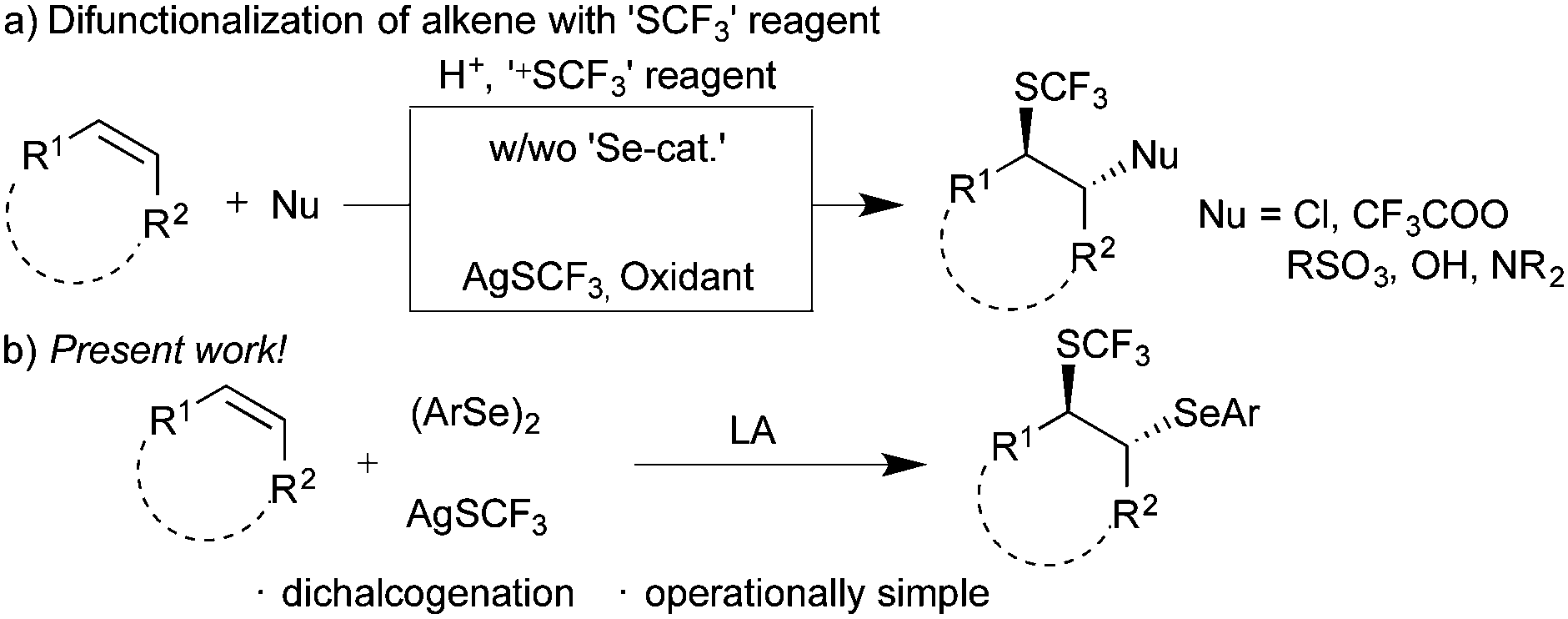 | ||
| Scheme 1 Trifluoromethylthiolative difunctionalization of alkenes. | ||
Initially, the difunctionalization of styrene 1a with diphenyl diselenide 2a and AgSCF3 was chosen as a model reaction. Initial screening of Lewis acids suggested that BF3·Et2O was the most suitable promoter of the expected difunctionalization (see the ESI†). Thus, the reaction of 1 equiv. of 1a with 1 equiv. of both 2a and AgSCF3 in the presence of 1 equiv. of BF3·Et2O in acetonitrile at room temperature afforded the expected product 3a in 44% yield after 12 h. Having identified the formation of the expected product 3a, various conditions were examined to improve the yield of 3a. Thus, different solvents such as DCM, DCE, and CH3CN were screened at room temperature to enhance the transformation (Table 1, entries 1–5). Among them, only acetonitrile showed a better yield.
|
|
|||
|---|---|---|---|
| Entry | BF3·OEt2 (equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: styrene 1a (0.24 mmol, 1.0 equiv.), 2a (0.24 mmol, 1.0 equiv.), AgSCF3 (0.24 mmol, 1.0 equiv.), BF3·OEt2, solvent (0.24 M), rt, 1 h. b Isolated yield. c At 50 °C. d 0.5 equiv. of styrene. e 2.0 equiv. of styrene. | |||
| 1 | 1.0 | CH3CN | 44 |
| 2 | 1.0 | DCE | <5 |
| 3 | 1.0 | DCM | 20 |
| 4 | 1.0 | THF | 20 |
| 5 | 1.0 | DMF | <5 |
| 6 | 2.0 | CH3CN | 40 |
| 7 | 0.5 | CH3CN | 26 |
| 8 | 0.2 | CH3CN | 24 |
| 9c | 1.0 | CH3CN | 48 |
| 10d | 1.0 | CH3CN | 20 |
| 11e | 1.0 | CH3CN | 96 |
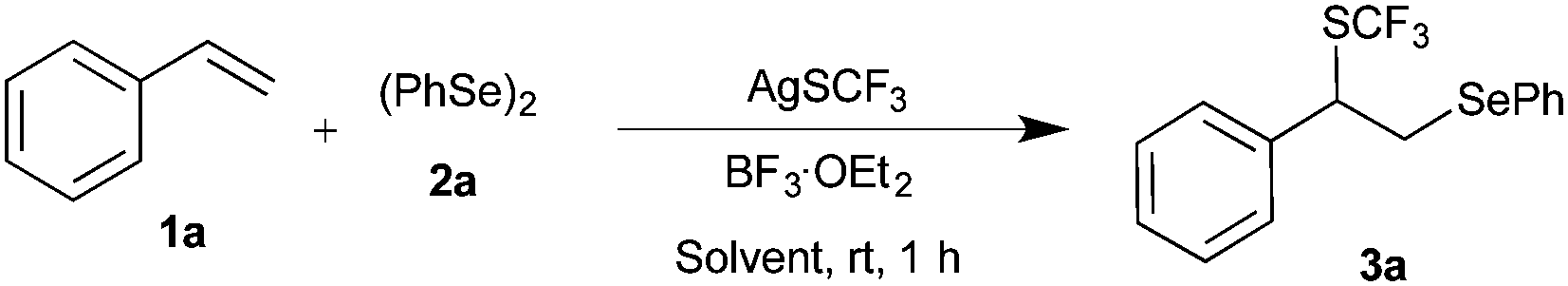
Next, increasing the BF3·OEt2 to 2.0 equivalents doesn’t show much improvement in the yield, but decreasing the equivalents of BF3·OEt2 shows drastic reduction in the yield (Table 1, entries 6 and 7). Subsequently, the focus was directed to study the effect of temperature. Increasing the reaction temperature to 50 °C with one equivalent of BF3·OEt2 furnished product 3a in only comparable yield (Table 1, entry 9). On the other hand, altering the equivalents of styrene showed a drastic change in the outcome (Table 1, entries 10 and 11). The best yield of 96% for the formation of 3a was observed with 2.0 equivalents of styrene in the presence of one equivalent of 2a, AgSCF3 and BF3·OEt2 at room temperature after 1 h; the same conditions were used for further studies.
Having achieved the suitable conditions for the tri-component difunctionalization of 1a for the synthesis of the highly functionalized molecule, the scope and generality of the transformation were investigated. For example, 4-alkyl (methyl/tert-butyl) substituted styrenes gave products 3b and 3c in 92% and 75% yield, respectively (Scheme 2). Similarly, electron donating group (3,4-dimethoxy) and halogen (bromo/chloro) substituted styrenes were also well tolerated under the optimized conditions to afford the corresponding difunctionalized products 3d, 3e, 3f, and 3g in excellent yields. The reaction of 2-vinylnaphthalene also furnished a similar product, 3h, in 73% yield. Also, sterically hindered 2-methylstyrene underwent smooth reaction to give 3i in 88% yield. However, actively coordinating cyano and NMe2 group substituted styrenes do not afford difunctionalized products.
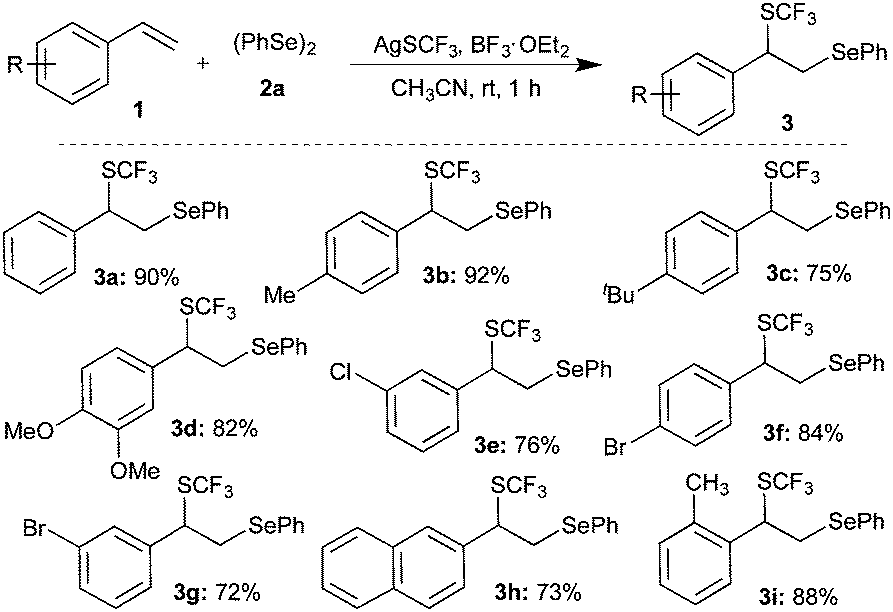 | ||
| Scheme 2 Difunctionalization of substituted styrenes. | ||
Further, to confirm the regioselectivity of the difunctionalized product, oxidative elimination of –SePh was envisioned. Thus, the isolated difunctionalized compound 3h was treated with m-CPBA in DCM at room temperature. Interestingly, the formation of α-(trifluoromethylthio)vinylnaphthalene 4 was observed in 95% yield (eqn (1)). The formation of 4 further confirms that SCF3 and SePh were attached at α-carbon and β-carbon, respectively.
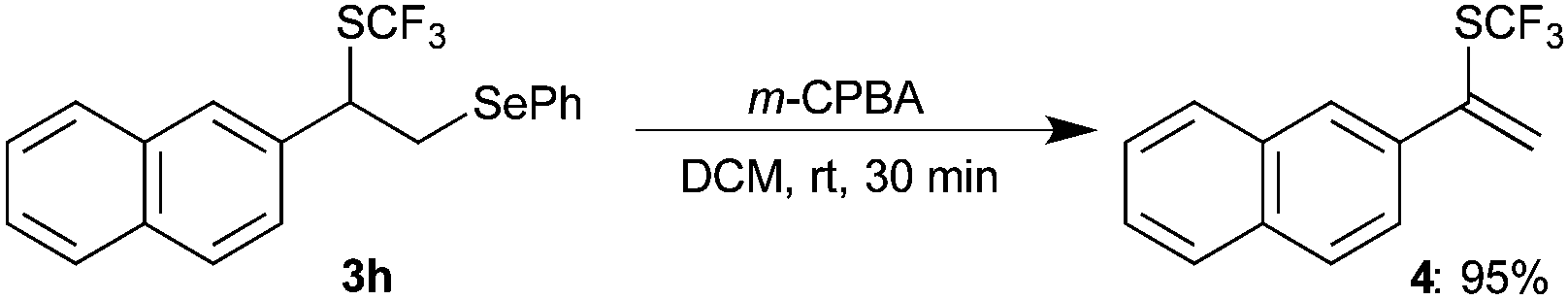 | (1) |
 | ||
| Scheme 3 Difunctionalization of substituted alkenes. | ||
Subsequently, various substituted diaryl diselenides11 were subjected to the difunctionalization of styrene under the optimized conditions. Methyl, methoxy, and halogen substituted diaryl diselenides gave the corresponding difunctionalized products 3j, 3k, 3l, 3m and 3p in 70, 81, 51, 55 and 79% yields, respectively (Scheme 4).
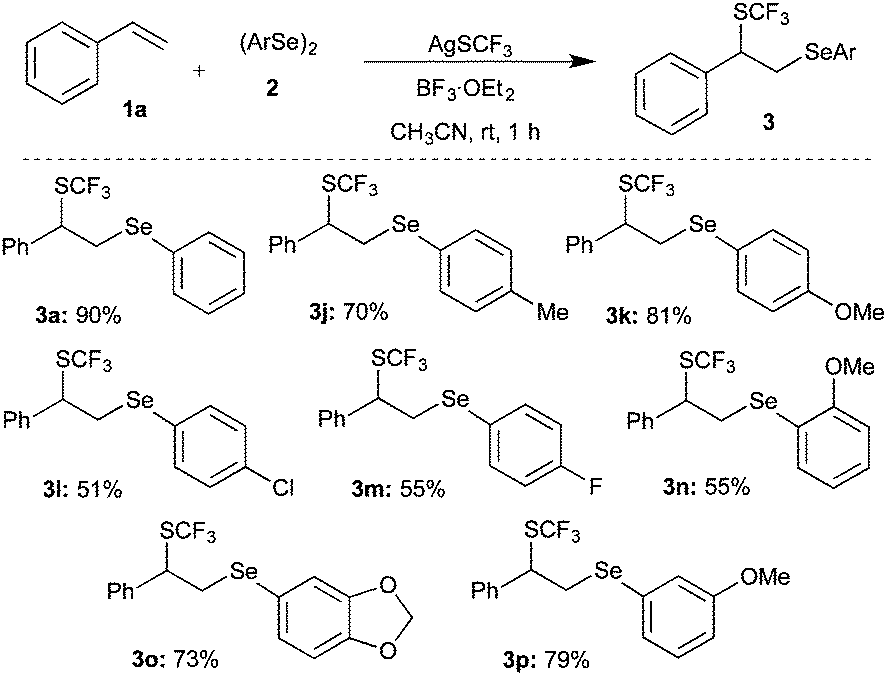 | ||
| Scheme 4 Difunctionalization of styrene with diaryl diselenide 2 and AgSCF3. | ||
Interestingly, sterically hindered o-methoxy substituted diaryl diselenide furnished the corresponding product 3n in good yield. Acid-sensitive acetal containing difunctionalized product 3o was synthesized in 73% yield from the corresponding diaryl diselenide.
After the successful demonstration of BF3·OEt2 induced difunctionalization of styrenes and alkenes with diaryl diselenides and AgSCF3, the formation of potential intermediates and the plausible reaction mechanism of the developed transformation were investigated. 19F NMR analysis of the optimized reactions showed an additional singlet at δ −45.3 along with AgSCF3 and difunctionalized product 3a. Subsequently, GCMS analysis revealed the formation of possible intermediate (phenylselanyl)(trifluoromethyl)sulfane (PhSeSCF3) from the corresponding diaryl diselenide and AgSCF3. Consequently, ((4-methoxyphenyl)selanyl)-(trifluoromethyl)sulfane 7 was synthesized from bis(4-methoxyphenyl)diselenide 2k and AgSCF3 in acetonitrile in 84% yield (Scheme 5a).12 After the successful synthesis of 7, the reactivity of 7 with styrene was investigated under the optimized conditions. The initial reaction of styrene 1a with 7 in the presence of BF3·OEt2 in CH3CN didn’t afford the expected product. A detectable amount of difunctionalized product 3k was observed in 19F NMR upon addition of an additive such as silver salt or NaOTs (Scheme 5b).
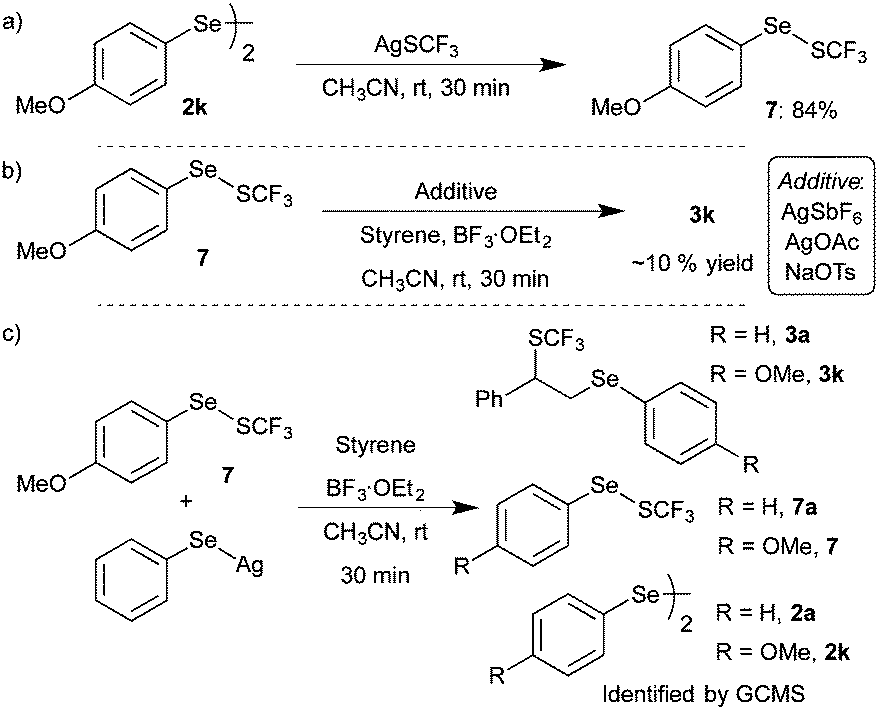 | ||
| Scheme 5 Control experiments. | ||
However, the formation of 7, via treatment of diselenide and AgSCF3 in CH3CN in the presence of BF3·OEt2, followed by the addition of styrene afforded the expected difunctionalized product 3k in good yield. These studies suggested that ArSeSCF3 might not be the intermediate of the developed transformation and the formed ArSeSCF3 might exist in equilibrium with AgSCF3 and diaryl diselenide. Further, to confirm the reversible formation of ArSeSCF3 from AgSCF3 and diselenides, compound 7 was treated with silver phenylselenate and styrene in the presence of BF3·OEt2. Interestingly, a mixture of difunctionalized products 3a and 3k was observed along with –SCF3 exchanged products 7 and 7a and possible diselenides 2a and 2k in GCMS and 19F NMR analysis (Scheme 5c). This –SCF3 exchange could be explained via the initial formation of diselenides followed by reaction with AgSCF3. All the above observations confirm the possible reversible reaction between diselenides and AgSCF3.
Furthermore, difunctionalization was performed with other dichalcogenides in place of diselenides. Unfortunately, both diphenyl disulfide and diphenyl ditelluride did not afford the expected product (Scheme 6). Further analysis of the reaction mixture suggested the possible reasons that the disulfides failed to get activated due to the high bond energy of the S–S bond and no availability of ditelluride due to the rapid, irreversible formation of PhTeSCF3. Thus, based on the observed regioselectivity and mechanistic investigation, in the present strategy diselenide and AgSCF3 acted as an electrophilic –SeAr source and a nucleophilic –SCF3 source, respectively.
 | ||
| Scheme 6 Reaction of styrene with different dichalcogenides and AgSCF3. | ||
Based on the mechanistic study, the possible pathway for the difunctionalization of alkenes was proposed as shown in Scheme 7. Initially, diphenyl diselenide 2a and AgSCF3 convert into 7a, which exists in equilibrium with 2a and AgSCF3. The remaining diselenide on reaction with styrene in the presence of BF3·OEt2 would afford the episelenonium intermediate A. Subsequently, regioselective ring opening of episelenonium ion A with AgSCF3 would furnish the difunctionalized product 3. Even though the concentration of AgSCF3 is lower at equilibrium, the overall reaction yield was satisfactory, because of the reversible reaction between AgSCF3 and diphenyl diselenide.
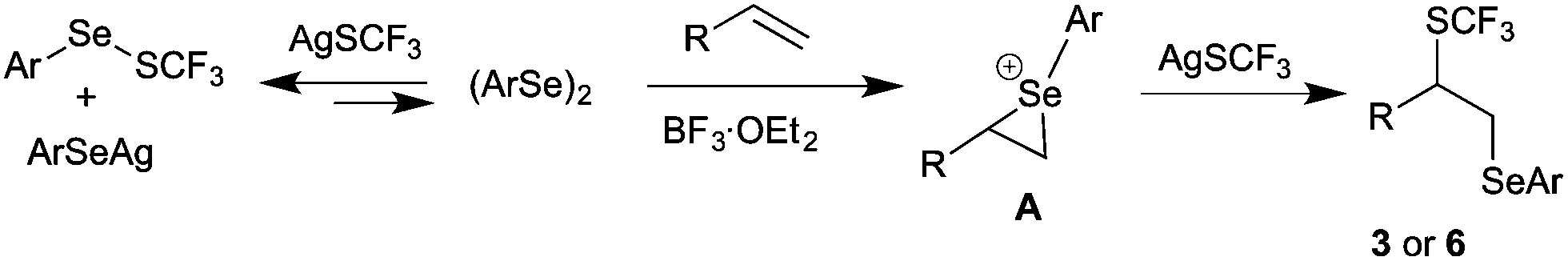 | ||
| Scheme 7 Plausible mechanism. | ||
In conclusion, we have developed an efficient regioselective difunctionalization of alkenes with diaryl diselenide and AgSCF3 in the presence of BF3·OEt2 as an activator. The developed reaction tolerates various functional groups and allows the synthesis of diverse 1,2-dichalcogenated products having a trifluoromethylthio moiety in good to excellent yield. The preliminary mechanistic investigation revealed the possible reaction pathway and unique combination of diselenide and AgSCF3 for successful transformation.
We thank the Indian Institute of Technology Madras for financial support. P. S. thanks CSIR, New Delhi, for a fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) F. Toulgoat, S. b. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415 CrossRef CAS; (b) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS; (c) X. Shao, C. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227 CrossRef CAS PubMed; (d) H. Zheng, Y. Huang and Z. Weng, Tetrahedron Lett., 2016, 57, 1397 CrossRef CAS; (e) S. Barata-Vallejo, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 7150 RSC.
- (a) T. Umemoto and S. Ishihara, J. Fluorine Chem., 1998, 92, 181 CrossRef CAS; (b) B. Gutmann, D. Obermayer, B. Reichart, B. Prekodravac, M. Irfan, J. M. Kremsner and C. O. Kappe, Chem. – Eur. J., 2010, 16, 12182 CrossRef CAS PubMed; (c) E. A. Nodiff, S. Lipschutz, P. N. Craig and M. Gordon, J. Org. Chem., 1960, 25, 60 CrossRef CAS; (d) B. R. Langlois, T. Billard, J.-C. Mulatier and C. Yezeguelian, J. Fluorine Chem., 2007, 128, 851 CrossRef CAS.
- (a) V. G. Koshechko, L. A. Kiprianova and L. I. Fileleeva, Tetrahedron Lett., 1992, 33, 6677 CrossRef CAS; (b) A. Harsányi, É. Dorkó, Á. Csapó, T. Bakó, C. Peltz and J. Rábai, J. Fluorine Chem., 2011, 132, 1241 CrossRef; (c) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS PubMed; (d) N. J. W. Straathof, B. J. P. Tegelbeckers, V. Hessel, X. Wang and T. Noel, Chem. Sci., 2014, 5, 4768 RSC; (e) G. Danoun, B. Bayarmagnai, M. F. Gruenberg and L. J. Goossen, Chem. Sci., 2014, 5, 1312 RSC; (f) M. V. Riofski, A. D. Hart and D. A. Colby, Org. Lett., 2013, 15, 208 CrossRef CAS PubMed; (g) J.-J. Ma, W.-B. Yi, G.-P. Lu and C. Cai, Catal. Sci. Technol., 2016, 6, 417 RSC.
- (a) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164 CrossRef CAS; (b) C. Matheis, V. Wagner and L. J. Goossen, Chem. – Eur. J., 2015, 22, 79 CrossRef PubMed; (c) A. B. Durr, G. Yin, I. Kalvet, F. Napoly and F. Schoenebeck, Chem. Sci., 2016, 7, 1076 RSC; (d) J.-B. Liu, X.-H. Xu, Z.-H. Chen and F.-L. Qing, Angew. Chem., Int. Ed., 2014, 54, 897 CrossRef; (e) P. Zhu, X. He, X. Chen, Y. You, Y. Yuan and Z. Weng, Tetrahedron, 2014, 70, 672 CrossRef CAS; (f) Y. Huang, X. He, H. Li and Z. Weng, Eur. J. Org. Chem., 2014, 7324 CrossRef CAS; (g) Y.-D. Yang, A. Azuma, E. Tokunaga, M. Yamasaki, M. Shiro and N. Shibata, J. Am. Chem. Soc., 2013, 135, 8782 CrossRef CAS PubMed; (h) A. Ferry, T. Billard, E. Bacque and B. R. Langlois, J. Fluorine Chem., 2012, 134, 160 CrossRef CAS; (i) S. Alazet, L. Zimmer and T. Billard, Angew. Chem., Int. Ed., 2013, 52, 10814 CrossRef CAS PubMed; (j) T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856 CrossRef CAS; (k) X. Wang, T. Yang, X. Cheng and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 12860 CrossRef CAS PubMed; (l) S. Alazet, L. Zimmer and T. Billard, Chem. – Eur. J., 2014, 20, 8589 CrossRef CAS PubMed.
- (a) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (b) M. R. Heinrich, Chem. – Eur. J., 2009, 15, 820 CrossRef CAS PubMed; (c) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (d) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333 RSC; (e) X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821 CrossRef CAS; (f) Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727 CrossRef CAS; (g) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC; (h) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem. – Asian J., 2018, 13, 2277 CrossRef CAS PubMed; (i) S. R. Chemler, Org. Biomol. Chem., 2009, 7, 3009 RSC; (j) T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed; (k) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC.
- (a) J. Sheng, C. Fan and J. Wu, Chem. Commun., 2014, 50, 5494 RSC; (b) S. Pan, H. Li, Y. Huang, X.-H. Xu and F.-L. Qing, Org. Lett., 2017, 19, 3247 CrossRef CAS PubMed.
- A. Ferry, T. Billard, B. R. Langlois and E. Bacqué, Angew. Chem., Int. Ed., 2009, 48, 8551 CrossRef CAS PubMed.
- (a) J. Luo, Z. Zhu, Y. Liu and X. Zhao, Org. Lett., 2015, 17, 3620 CrossRef CAS PubMed; (b) Y. Yang, X. Jiang and F.-L. Qing, J. Org. Chem., 2012, 77, 7538 CrossRef CAS PubMed; (c) C.-C. Xi, Z.-M. Chen, S.-Y. Zhang and Y.-Q. Tu, Org. Lett., 2018, 20, 4227 CrossRef CAS PubMed; (d) L. Jiang, T. Ding, W.-b. Yi, X. Zeng and W. Zhang, Org. Lett., 2018, 20, 2236 CrossRef CAS PubMed; (e) X. Liu, Y. Liang, J. Ji, J. Luo and X. Zhao, J. Am. Chem. Soc., 2018, 140, 4782 CrossRef CAS PubMed; (f) Y. Jia, H. Qin, N. Wang, Z.-X. Jiang and Z. Yang, J. Org. Chem., 2018, 83, 2808 CrossRef CAS PubMed; (g) Z. Zhu, J. Luo and X. Zhao, Org. Lett., 2017, 19, 4940 CrossRef CAS PubMed; (h) J. Luo, Y. Liu and X. Zhao, Org. Lett., 2017, 19, 3434 CrossRef CAS PubMed; (i) P. Zhang, M. Li, X.-S. Xue, C. Xu, Q. Zhao, Y. Liu, H. Wang, Y. Guo, L. Lu and Q. Shen, J. Org. Chem., 2016, 81, 7486 CrossRef CAS PubMed; (j) C. Xu and Q. Shen, Org. Lett., 2015, 17, 4561 CrossRef CAS PubMed; (k) J. Luo, Q. Cao, X. Cao and X. Zhao, Nat. Commun., 2018, 9, 527 CrossRef PubMed; (l) Y. Li, T. Koike and M. Akita, Asian J. Org. Chem., 2016, 6, 445 CrossRef; (m) G. Dagousset, C. Simon, E. Anselmi, B. Tuccio, T. Billard and E. Magnier, Chem. – Eur. J., 2017, 23, 4282 CrossRef CAS PubMed; (n) H. Li, C. Shan, C.-H. Tung and Z. Xu, Chem. Sci., 2017, 8, 2610 RSC; (o) X. Liu, R. An, X. Zhang, J. Luo and X. Zhao, Angew. Chem., Int. Ed., 2016, 55, 5846 CrossRef CAS PubMed.
- (a) F. Yin and X.-S. Wang, Org. Lett., 2014, 16, 1128 CrossRef CAS PubMed; (b) L. Zhu, G. Wang, Q. Guo, Z. Xu, D. Zhang and R. Wang, Org. Lett., 2014, 16, 5390 CrossRef CAS PubMed; (c) H. Xiang and C. Yang, Org. Lett., 2014, 16, 5686 CrossRef CAS PubMed; (d) S. Pan, Y. Huang, X.-H. Xu and F.-L. Qing, Org. Lett., 2017, 19, 4624 CrossRef CAS PubMed.
- Z. Li, F. Ke, H. Deng, H. Xu, H. Xiang and X. Zhou, Org. Biomol. Chem., 2013, 11, 2943 RSC.
- M. Jereb and D. Dolenc, RSC Adv., 2015, 5, 58292 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00815b |
| This journal is © The Royal Society of Chemistry 2019 |