
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceπ-Conjugated nanostructured materials: preparation, properties and photonic applications
Dönüs
Tuncel

Department of Chemistry, UNAM-National Nanotechnology Research Center, Institute of Materials Science and Nanotechnology, Bilkent University, 06800 Ankara, Turkey. E-mail: dtuncel@fen.bilkent.edu.tr
First published on 31st October 2018
Abstract
This article reviews recent advances in π-conjugated nanostructures based on conjugated oligomers and polymers, focusing on their preparation, energy transfer abilities, optoelectronic and laser applications, and photophysical properties including light harvesting. This is a rapidly evolving field as these materials are expected to have many important applications in areas such as light-emitting diodes, solid-state lighting, photovoltaics, solid-state lasers, biophotonics, sensing, imaging, photocatalysis, and photodynamic therapy. Other advantages of these materials are their versatility, and consequently, their adaptability to diverse fields.
 Dönüs Tuncel | Dönüs Tuncel received her PhD degree from Imperial College, London in the area of supramolecular polymer chemistry and worked for 3 years as a postdoctoral research fellow in the Chemistry Department of Oxford University in the area of 2D fluorescent polymer networks and polymeric nanotubes. In 2003, she joined the Department of Chemistry at Bilkent University. She is a fellow of the Royal Society of Chemistry and currently serves as an associate director at the National Nanotechnology Research Center (UNAM) and Institute of Materials Science and Nanotechnology. Her research involves the design and synthesis of functional organic and hybrid organic–inorganic nanomaterials and the use of these materials in optoelectronic, biomedical, and photocatalytic applications. |
1. Introduction
π-Conjugated polymers contain a backbone in which electrons are delocalized and are made electrically conducting by adding or removing an electron through doping.1–3 These electrically and optically active materials possess large extinction coefficients and good light-harvesting activity. Owing to these features, π-conjugated polymers have been used in optoelectronic devices, including field-effect transistors, light-emitting diodes, and photovoltaic cells.4–10π-Conjugated polymers are also appealing because of their low cost, versatile functionalization, thin film flexibility, and ease of processing.10 Another useful and fascinating aspect of these materials is that their optical properties can be tuned by modifying their structures, which can produce materials that absorb and emit across the visible and near-infrared (NIR) region. This feature is particularly important as it is more difficult to tune the optical properties of inorganic materials. Bandgap engineering can be used to synthesize materials with a narrow band gap; for example, materials that absorb in the NIR region with a band gap smaller than 1.6 eV are good candidates for organic photovoltaics, photodetectors, and ambipolar field-effect transistors.11
When π-conjugated polymers are converted into nanostructures, the polymer properties are augmented by the additional features of nanodimensionality.12–14 Nanostructures may offer additional advantages, including functionalization of the interior or exterior of the nanostructures, and solubility in a variety of solvents, including water. Conjugated nanostructured materials exhibit many useful properties, such as high fluorescent yield, molar absorptivity, brightness, photostability, and light harvesting. Because of these features, these materials have been used in a wide range of optoelectronic and biomedical applications.15–22
Accordingly, this review provides an overview of recent works on π-conjugated oligomer- and polymer-based nanostructured materials and focuses on their preparation, properties, light harvesting and energy transfer properties, and their photonic applications. A brief section on their applications in bionanotechnology is also included. The review concludes with a summary and the outlook for these materials.
2. Preparation of π-conjugated nanostructured materials
π-Conjugated nanostructures are prepared either from pre-synthesized conjugated polymers or by miniemulsion or dispersion polymerization.15–19The pre-synthesized conjugated polymers are prepared by miniemulsion, reprecipitation (or nanoprecipitation), or self-assembly. The miniemulsion method requires a biphasic immiscible solvent system, in which one solvent is usually water and the other is chloroform or toluene. The polymer dissolved in the good solvent (e.g., chloroform) is miniemulsified in the surfactant-containing water. During rapid stirring, the hydrophobic polymer chains collapse into micelles, thereby forming nanoparticles. After removal of the good solvent, the nanoparticles are stabilized further by the surfactant. The particle size is in the range of 20 nm to several hundreds of nanometers. The concentration of the conjugated polymer affects the size of the nanoparticles.17,23,24
Nanoparticles smaller than 20 nm containing either a single or a maximum of a few polymer chains are called semiconducting polymer dots.21 Polymer dots are prepared by reprecipitation or nanoprecipitation. New-generation polymer nanoparticles containing various functional groups can also be prepared by these methods. The nanoparticles are prepared by dissolving the conjugated polymer in a good solvent that is miscible with water and the polymer solution is injected into a large excess of water while stirring. This method does not require stabilizing agents, and nanoparticles as small as 5 nm can be obtained due to the collapse of a single polymer chain. The nanoparticles can also be coated with an extra polymer layer to decorate the surface with functional groups and stabilize the nanoparticles.25,26
Nanostructures, such as micelles, vesicles, hollow capsules, and nanoparticles, can be prepared from amphiphilic π-conjugated oligomers or polymers through self-assembly in water.27,28 These oligomers or polymers can contain a hydrophobic conjugated backbone and hydrophilic pendants, or they can be conjugated block copolymers with a hydrophobic and a hydrophilic block that can self-assemble in water. Alternatively, ionic groups containing conjugated polyelectrolytes can form complexes with opposite charge-carrying conjugated polyelectrolytes. The complexes, called coacervates, are charge-neutral nanoparticles.29–31
Nanoparticles can also be prepared starting from monomers that polymerize and collapse into nanoparticles. In this in situ miniemulsion method, appropriate monomers are miniemulsified in a surfactant or emulsifier containing an aqueous phase and are polymerized by the addition of a catalyst in the stabilized droplets to obtain conjugated polymer nanoparticles (CPNs). In the dispersion method, the polymerization is carried out in a solvent that is good for the monomers but poor for the conjugated polymer, allowing the conjugated polymer to collapse into nanoparticles. The nanoparticles are further stabilized by adding a surfactant or other stabilizers.32–37
Conjugated oligomers or polymers with various functional groups are being used to prepare new-generation nanostructures. Furthermore, hybrid nanoparticles of conjugated polymers with metals (e.g., Ag, Au, Pd) can also be prepared for use in plasmonic lasing, photocatalysis, and sensing.38 Due to their multifunctionality and unprecedented properties, the new generation of these materials have more potential applications than existing materials, especially in optoelectronics.
3. Photophysical, light harvesting, and energy transfer properties of nanostructured π-conjugated materials
For optoelectronic, imaging and sensing applications, the photophysical properties of the conjugated materials are important, and these properties depend strongly on the properties and morphologies (chain conformation, aggregation state, shape and size) of the polymers.13,39,40During the nanoparticle preparation, hydrophobic conjugated polymer chains collapse in a poor solvent, such as water, and bringing π-conjugated multichromophoric units into close proximity with each other, affecting the inter-chain and intra-chain interactions. These interactions are mainly responsible for the excited-state dynamics and the intra-chain or inter-chain energy transfer.39–49 Similar to other nanostructured materials, the increased surface area is an important feature that allows for the decoration of the surfaces with various functionalities. The surface charges or other functional groups strongly affect the material's properties.
There are a number of systematic studies on the conjugated polymer structure-dependent spectral properties.50–53 These studies mostly use single-molecule spectroscopy to reveal the conformational changes associated with the photophysical properties at the single polymer chain level.
Depending on the structure and the chemical properties of the conjugated polymer backbone, the conformation of polymers and the extent of the inter- and intra-chain interactions vary.54–63 For example, polymers with a rigid conjugated backbone tend to bend and twist during the nanostructure formation, resulting in strong intra-chain interactions and a decreased conjugation length.55,61–63 These changes cause a blueshift in the absorption band (π–π* transition) spectrum. However, the blueshift is more evident in nanoparticles containing a single chain or a few polymer chains.19 As the number of chains increases, nanoparticles behave more like the bulk and a redshift in the absorption band is observed. In polymers with a less rigid backbone and more polar pendant groups, there is less bending and less decrease in the backbone length, but there are more chain–chain interactions. As a result, redshifted optical absorption bands (π–π* transition) are observed.54,56–60
Usually, the emission wavelengths of nanoparticles are redshifted compared with their spectrum in organic solvents and are more similar to their solid-state emission bands. This occurs because the exciton relaxation of nanoparticles is faster than that of a polymeric solution owing to the efficient energy transfer (inter-chain energy transfer) between adjacent chromophores. Moreover, the solvent polarity also causes spectral changes.40,41
The photophysical properties of poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] (MEH-PPV) nanoparticles with sizes ranging from 10 to 100 nm were studied using single-particle spectroscopy techniques, including static and dynamic measurements.64,65 The effect of particle size on the frequency-resolved photoluminescence (PL) spectra, photobleaching dynamics, and triplet–triplet annihilation kinetics were investigated. Nanoparticles with a diameter greater than 10 nm, which contain more than one polymer chain, exhibited a bulk-like structure and properties. These changes were explained by the distance dependence of the four main processes, which are electronic energy transfer between blue and red sites, triplet–triplet annihilation, singlet exciton quenching by triplets, and singlet exciton quenching by hole polarons.
Molecular dynamics simulations and ultrafast spectroscopy were used to study nanoparticle formation mechanism during the gradual addition of a poor solvent to a tetrahydrofuran (THF) solution of MEH-PPV, in which the polymer is in its extended state (Fig. 1a). Free energy calculations revealed that the thermodynamically stable state of the polymer in water and 75% (v/v) water/THF mixture is a collapsed state. The redshift of the absorption band of the collapsed state was due to a change in the solvent polarity. The change in intensity of the blue and red emission bands with the change in the solvent polarity was caused by a change in polymer conformation from the extended state to the collapsed state. Ultrafast spectroscopy showed a systematic decrease of the faster component at 554 nm (33 to 2 ps), indicating an energy transfer process. The faster component (150 fs) of the time-resolved anisotropy decay due to the fast depolarization process confirmed the interchain energy transfer in the collapsed state.66
 | ||
| Fig. 1 (a) Morphologies and sizes of MEH-PPV polymer-based nanostructures in solutions containing various amounts of poor solvent (water) and exciton dynamics of the extended and the collapsed states of MEH-PPV in a good solvent and poor solvent, respectively. Adapted with permission from ref. 66. Copyright 2017 American Chemical Society. (b) Cartoon of the effect of solvent-induced swelling on the kinetics of multiple energy transfer in PFBT and MEH-PPV nanoparticles and changes in their photophysical properties upon swelling by increasing the good solvent (THF) content. Adapted with permission from ref. 67. Copyright 2017 American Chemical Society. | ||
Conjugated nanostructured materials are promising for light harvesting because their nanostructured multichromophoric nature allows the formation of a number of transient and persistent species that interact with the excited state, including H- or J-aggregates, excimers, and exciplexes.43,58,59
To design appropriate conjugated materials for photocatalysis and photovoltaic applications, it is important to understand the light harvesting process, which involves a series of important optical phenomena, including exciton generation, radiative recombination of excitons, nonradiative relaxation, internal conversion, intersystem crossing, exciton transfer, and charge transfer.40,41 Conjugated nanostructures make a suitable model for studying energy transfer in complex nanoscale systems because of their high absorption cross sections and high sensitivity for the excitation light due to mobile excitons. This model may also clarify the underlying photophysics in such systems, which would help improve the performance of nanoparticle-based sensing schemes and nanostructured electroluminescent device layers.
The effects of solvent-induced swelling on the kinetics of multiple energy transfer in PFBT and MEH-PPV nanoparticles were investigated (Fig. 1b).67 Multiple energy transfer between equivalent chromophores increases the rate of quenching by defects due to a cascading or funneling effect. The effects of swelling on energy transfer between polymer chromophores and the resulting exciton dynamics were modelled using a random walk on a lattice of chromophores. The simulation results agreed well with the experimental fluorescence quantum yield and decay kinetics results at low to moderate THF concentrations. The time scale for the energy transfer between chromophores (∼5 ps for MEH-PPV nanoparticles and ∼100 ps for PFBT nanoparticles) was highly sensitive to swelling, slowing by an order of magnitude or more for swelled particles. The results indicate quenching by defects or polarons, amplified by multiple energy transfer or a cascade effect, as a likely explanation for the typically low fluorescence quantum yield of conjugated polymer particles compared with the free polymer in solution. Similar effects were observed in thin films.67
The molecular properties of the positive charge carriers (holes or oxidized species) in organic semiconductors were studied by single-molecule spectroelectrochemistry (SMS-EC).43,68 The oxidation of nanoparticles of conjugated polymer poly(9,9-dioctylfluorene-co-benzothiadiazole) (PFBT) has been used to study the complicated electrochemical process in heterogeneous media. A reversible hole-injection charging process that occurs primarily by initial injection of shallow (untrapped) holes has been observed; however, soon after injection, a small fraction of holes becomes deeply trapped. Good agreement between the experimental data and simulations strongly supports the presence of deep traps in the nanoparticles and highlights the ability of SMS-EC to study the energetics and dynamics of deep traps in organic materials at the nanoscale.68
Luminescent blinking is a reversible quenching in the fluorescent intensity of a single nanoparticle. To understand the underlying photophysical effect of this quenching better, the motion of individual charge carriers in organic nanostructures was tracked by video rate fluorescence microscopy by McNeill and co-workers.69 Fluctuations in the fluorescence intensity and nanoscale displacements in the fluorescence centroid of single CPNs made out of the green-emitting conjugated polymer, PFBT, were observed. This effect is also called twinkling due to its resemblance to the twinkling of stars and was explained by charge carrier-induced superquenching that forms a dark spot in the nanoparticle and moves with the carrier, causing the displacement in the fluorescence (Fig. 2). Simulations confirmed that the displacement in the fluorescence centroid can provide a measure of the polaron displacement. Analysis of the fluorescence displacement trajectories yielded hole polaron mobility values in a range consistent with highly trapped charge carriers, as expected based on the degree of disorder present in the particles.69
 | ||
| Fig. 2 (a) Scheme showing the setup of the single-particle tracking microscope; (b) schematic image of the quenching of the exciton fluorescence by a single hole polaron on a single molecule chain in a single CPN; (c) a typical fluorescence CCD image of PFBT CPNs; (d) intensity histogram for CPNs at the excitation intensity of ∼70 W cm−2 (in the center of the laser spot). The red curve is obtained by fitting a 1D Gaussian function and gives 1.4 ± 0.6 × 105 detected photons per 20 ms exposure. Adapted with permission from ref. 69. Copyright 2012 American Chemical Society. | ||
In the subsequent study by McNeill and co-workers, the time resolution (1 ms) was improved substantially, and a broad range of polaron dynamics at kilohertz frame rates, ranging from the millisecond to second scale was investigated.70 The position trajectories were analysed using multiscale segmented correlation and mean-square displacement (MSD) analysis to obtain the distribution of the polaron hopping dynamics. In long segments, the polaron was trapped in an area that roughly matched the size of a particle, whereas in short segments, the polaron was trapped in a much smaller region. In some segments, they observed single-exponential dynamics that likely indicated reversible hopping between two local traps. They estimated polaron hopping times and distances from the autocorrelation time constants and MSD confinement lengths of those segments. The hopping time followed a power law distribution and the hopping distance roughly followed a single-exponential distribution, indicating that fast and short-ranged hops occurred more frequently. The energy barrier height for polaron hopping in CPNs was estimated from the hopping time distribution as 430 to 570 meV, suggesting the presence of deep traps with nearest-neighbor distances of 2–5 nm, consistent with a low or moderate density of traps (probably chemical or structural defects) dominating charge transport at low carrier densities (Fig. 3).70
 | ||
| Fig. 3 (a) Typical fluorescence microscopy image of PFBT CPNs. (b) Trajectory of centroid positions of a CPN. The trajectory was originally acquired at a 1 kHz frame rate. For plotting, every 10 frames in the kHz trajectory were binned together to reduce noise. (c) Illustration of a hole polaron quenching the local fluorescence in a CPN. Adapted with permission from ref. 70. Copyright 2018 American Chemical Society. | ||
Energy transfer
Using conjugated nanostructures, a number of different systems have been designed for the energy transfer process. Either polymers are doped with small organic dyes, which behave as the acceptor and the polymer matrix behaves as the donor, or polymers are blended with different conjugated polymers that act as an acceptor.Dye-doped CPNs for energy transfer
Fluorescent dye encapsulated CPNs are appealing for a range of applications, including photonics and biophotonics, due to their high brightness and photostability. The photostability, fluorescent quantum yields, brightness, and extinction coefficients of the conjugated polymers were improved by doping CPNs with fluorescent dyes, which act as an energy acceptor.71–78Dye-doped CPNs were prepared from blue-emitting polyfluorene derivatives by using the anionic surfactant sodium dodecyl sulfate during the miniemulsion process and incorporating two cationic rhodamine-based fluorescent dyes (rhodamine 6G and tetramethylrhodamine ethyl ester perchlorate, rhodamine TM). The energy transfer between the dyes and the polyfluorenepolymer matrix was investigated.72 The blue fluorescence of the dye-coated polyfluorene nanoparticles was slightly quenched after doping with the dyes and a new emission band of the surface-bound dye (rhodamine 6G or rhodamine TM) appeared in the wavelength region of 530–600 nm, indicating an effective excitation energy transfer from the excited polyfluorene chromophores to the fluorescent dye.
Nanoparticles of polyfluorene doped with perylene, tetraphenylporphyrin, coumarin 6, or Nile red (NR) around 30 nm in diameter were prepared and the energy transfer from the donor polyfluorene matrix to the dyes was studied by steady-state and time-resolved spectroscopy.62 The experimental results were compared with a model that was developed considering the combined effect of Förster energy transfer, energy diffusion and particle size. The measurements indicated efficient energy transfer from the host (donor) polyfluorene to the dopant (acceptor) dye molecules. The fluorescence quantum yield and peak extinction coefficients were measured as 40% and 1.5 × 109 M−1 cm−1, respectively, for an aqueous dispersion of polymer nanoparticles doped with perylene or coumarin 6 (2 wt%).62
Although NIR fluorescence sensing is highly sought after for in vivo biological measurements, the number of NIR fluorescent dyes is limited and they show poor performance, such as self-aggregation and low fluorescence, in a physiological environment.73 To tackle this problem, nanoparticles were prepared from PFBT by doping with the NIR dye, silicon 2,3-naphthalocyanine bis(trihexylsilyloxide) (NIR775), that emits at 800 nm with a narrow emission peak. Although free molecules of NIR775 aggregate in aqueous solution, encapsulating them in a hydrophobic PFBT matrix prevented their direct contact with aqueous solution. The brightness of NIR775 was increased substantially because of the efficient energy transfer from PFBT to NIR775.73
Energy transfer and confined motion of dyes trapped in CPNs were investigated by preparing nanoparticles based on poly(N-vinylcarbazole) (PVK) and doped with coumarin 153 (C153) (Fig. 4a).74 The fluorescent quantum yield of the dye increased with the particle size owing to the higher PVK concentration. Steady-state and time-resolved fluorescence spectroscopies were employed to investigate the energy transfer between the host (PVK) and guest (C153 dye molecules), and an efficient energy transfer (around 83%) from PVK to C153 was observed. The radiative decay rate of the dye increased as the nanoparticle size increased, indicating the increase in the refractive index inside the polymer nanoparticle. The increase of the lateral diffusion constant (DL) and the decrease of the diffusion coefficient for wobbling motion (Dw) of the dye molecules with an increase in the polymer nanoparticles size suggested an increase in the microviscosity and rigidity of the system.74
 | ||
| Fig. 4 (a) Cartoon of the energy transfer in CPNs based on the host, energy donor polymer PVK, and doped with an energy acceptor dye, C153. Adapted with permission from ref. 74. Copyright 2011 American Chemical Society. (b) (A) Electron transfer and energy transfer processes involved in [R6G][HFL]–PVK nanoparticles and (B) calculated HOMO–LUMO structures for [R6G][HFL] in the aqueous phase. Adapted with permission from ref. 78. Copyright 2017 American Chemical Society. | ||
To understand the effect of particle size and the concentration of the entrapped dye on the photophysical properties of dye-doped nanoparticles, PVK nanoparticles encapsulating NR dye were prepared and their photophysical properties were studied by steady-state, time-resolved fluorescence spectroscopy and fluorescence correlation spectroscopy (FCS).75 The hydrodynamic diameter, particle brightness, and population of NR molecules inside the particle as functions of particle size and NR concentration were quantitatively analyzed by using FCS. Increasing the concentration of NR from 0.5 to 1.8 wt% at the time of nanoparticle preparation increased the average fluorescence intensity of a single nanoparticle, namely, the per particle brightness value, from 80 to 500 kHz, and increased the number of NR molecules per nanoparticle from 5 to 22. The rotational dynamics of the encapsulated NR molecules for different particle sizes and NR concentrations inside the nanoparticles were investigated in a fluorescence anisotropy study. The particle brightness and quantum yield were increased by an increase in the radiative decay rate.75
The location and freedom of single and double guests in dye-doped (C153 and NR) PVK polymer nanoparticles were investigated by time-resolved fluorescence anisotropy studies using the wobbling-in-a-cone model, which provided details of the restricted movements of the encapsulated molecules.76 When the C153 concentration was increased, the diffusional relaxation (τD) times decreased (τD = 4.6–1.6 ns), suggesting the increased preference of C153 dye for less rigid environments. In contrast, for NR, τD was affected by the dopant content (τD = 12.2–3.09 ns), suggesting it was in a more rigid environment. For the two-dye-doped PVK nanoparticles, where the content of one of the trapped guests was fixed while the concentration of the other was varied, NR slowed down (τR = 0.30–0.37 ns and τD = 3.09–7 ns), whereas C153 accelerated (τR = 0.14–0.03 ns and τD = 1.57–0.69 ns). This suggests that the guest molecules assumed different positions, with C153 preferentially occupying a less rigid environment closer to the nanoparticle surface, whereas NR was in a more rigid environment closer to the core. Thus, the dye distribution in PVK nanoparticles is governed by a combination of the Marangoni effect and consecutive particle swelling. Furthermore, in the two-dye-doped systems, competition for the available less rigid sites was observed when the C153 co-dopant content was increased, whereas this effect was smaller when the NR content was increased.76
To study the cascaded energy transfer ability of the dye-doped CPNs, multi-dye-doped CPNs 50–70 nm in diameter were prepared using PVK as an energy donor host and the dyes, C153 and NR, as the energy acceptor guests. The photophysical properties of the CPNs were studied by steady state and picosecond time-resolved emission spectroscopy.75 The multistep cascaded energy transfer occurred from the excited host PVK to the NR dye molecules through C153. The energy transfer time constants were 180 ps for PVK → C153, 360 ps for PVK → NR, and 140 ps for the overall energy transfer process from PVK to NR through the C153 dye molecules. Through this cascaded energy transfer and by varying the relative concentrations of the encapsulated dye molecules, emission wavelengths covering the region of 350 to 700 nm were tuned and consequently, white light emission was generated in 14% quantum yield.77
Similarly, Das et al. designed a donor–acceptor system through tuning the energy level alignment of interacting counterparts to create an efficient light harvesting system in which PVK nanoparticles act as the donor and the bicolor fluorophore ([R6G][HFL]) acts as the acceptor (Fig. 4b).78 The photophysical interaction between the polymer nanoparticle and bicolor fluorophore was investigated by a spectroscopic study and density functional theory calculations. Dynamic quenching and the decreased decay time of the PVK polymer nanoparticles were caused by the energy and electron transfer process from the PVK nanoparticle to the bicolor fluorophore on the nanoparticle surface. An ultrafast spectroscopic study was performed to investigate the decay dynamics of [R6G][HFL] in the presence and absence of PVK. Fig. 3b shows a schematic of the electron transfer and energy transfer processes involved in [R6G][HFL]–PVK nanoparticles and the calculated HOMO–LUMO structures for [R6G][HFL] in the aqueous phase. White light generation with CIE coordinates (0.31, 0.39) was achieved at a PVK![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[R6G][HFL] donor–acceptor weight ratio of 5.39:1.78
:[R6G][HFL] donor–acceptor weight ratio of 5.39:1.78
Energy transfer through donor–acceptor polymeric systems
Instead of fluorescent dyes, conjugated polymers have been used as dopants. Because the leakage of the small-molecule dyes from the polymer host may be problematic, conjugated polymer blends offer advantages over dye-doped nanoparticles.61,79–85Nanoparticles with diameters of 20–30 nm were prepared by using blue-emitting polyfluorene as a host and doped with varying amounts of green-, yellow-, and red-emitting conjugated polymers (PFPV, PFBT, MEH-PPV, respectively) to study the energy transfer from the host polymer to the dopant polymers.61,79 The particle size and morphology were not affected by the presence of the dopants. Polyfluorene emission decreased with increasing dopant concentration, whereas the emission intensity of the dopants increased and reached a maximum around 6 wt%. However, dopant concentrations higher than 6% caused a decrease in the fluorescent quantum yield. Using the Stern–Volmer relation, the dopant (acceptor) concentration-dependent fluorescence quenching of the host polymer was studied and a considerable deviation from the Stern–Volmer relation in the composition-dependent nanoparticle fluorescence was observed in some cases. To understand the underlying photophysical reasons for the deviation, a nanoparticle energy transfer model was proposed that included the statistical variability in nanoparticle composition. The results from this model were consistent with experimental results, particularly for the dopants with high quenching efficiencies. Both the Stern–Volmer relation and the nanoparticle energy transfer model suggested a highly efficient energy transfer between the donor (host polymer) and the acceptor (dopant polymers) molecules, indicating that 100 or more host molecules were quenched by a single dopant molecule. These findings indicate long-range (>10 nm) energy transport through multiple energy transfer.
Redshifted emission through an efficient energy transfer was achieved by preparing nanoparticles using PFBT as a host (energy donor) and doping with limited amount of MEHPPV (energy acceptor).79 Photobleaching results showed better photostability in the blended sample compared with undoped MEH-PPV nanoparticles, and photoactivation of the donor emission, which could be useful for single-molecule localization super-resolution microscopy (Fig. 5). Single blended nanoparticles exhibited bright fluorescence and saturation behaviour at low excitation intensities.79
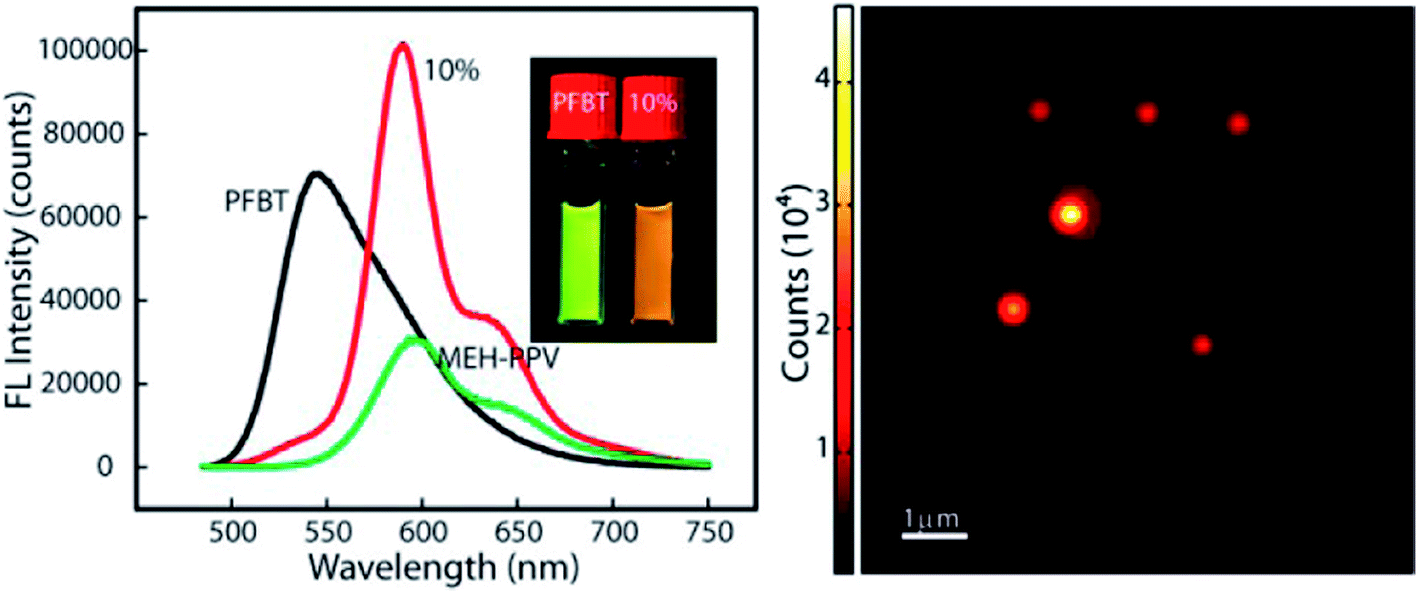 | ||
| Fig. 5 (Right) PL spectra of PFBT, MEH-PPV and doped nanoparticles. Redshifted emission through efficient energy transfer in nanoparticles using PFBT as a host (energy donor) doped with a limited amount of MEHPPV (energy acceptor). (Left) AFM image of doped-nanoparticles. Adapted with permission from ref. 79. Copyright 2013 American Chemical Society. | ||
Four binary polymeric nanoparticle systems were designed using poly[(9,9-dihexylfluorene)] as a donor and MEH-PPV as an acceptor, and non-radiative resonance energy transfer processes were studied.80 The first approach involved the preparation of the polyfluorene and MEH-PPV nanoparticles separately and mixing them at a certain ratio. In the second approach, solutions of the polyfluorene and MEH-PPV were mixed before nanoparticle formation, and then nanoparticles were prepared from the mixture. The third and fourth approaches involved sequential nanoparticle preparation. In the third approach, nanoparticles with a polyfluorene core and a MEH-PPV shell were prepared. The fourth approach was the reverse of the third, in which the core was MEH-PPV and the shell was the polyfluorene. Steady-state fluorescence spectra and time-resolved fluorescence measurements indicated different energy transfer efficiencies from the polyfluorene host polymer to the MEH-PPV acceptor polymer, depending on the donor–acceptor distances and structural properties of the nanoparticles. The highest energy transfer efficiency was 35% and was obtained from the last system, in which a polyfluorene layer was sequentially formed on MEH-PPV nanoparticles. Non-radiative resonance energy transfer occurred in bi-polymer nanoparticles of fluorescent conjugated polymers.80
4. Photonic applications
Conjugated polymer nanostructures exhibit light harvesting properties, which is an important feature for optoelectronic devices. Although the light harvesting abilities of conjugated polymer nanostructures have been investigated, examples of their applications in photonic device are limited.Scherf, Landfester, List, and co-workers used CPNs in optoelectronic device fabrication including monolayer polymer light-emitting diodes (PLEDs), multilayer PLEDs, light-emitting electrochemical cells (LECs), and solar cells.86–91
A light-emitting diode constructed from a homogeneous single layer of nanoparticles based on a methyl-substituted ladder-type poly(p-phenylene) had a lower onset and slightly higher efficiency compared with the one constructed directly from the conjugated polymer films. These differences were explained by enhanced electron injection from the cathode as a consequence of the in situ formation of a stalactite-type nanostructured cathode during aluminium evaporation. Optical spectroscopy showed that the electronic defects were not created by converting conjugated polymers into aqueous nanoparticle dispersions, and the CPNs displayed similar photophysical properties to the bulk state.86
Photovoltaic diodes were prepared from CPNs using two designs. In the first design, the nanoparticles of hole-accepting polymer, PFB [poly(9,9-dioctylfluorene-2,7-diyl-co-bis-N,N0-(4-butylphenyl)-bis-N,N0-phenyl-1,4-phenylenediamine)], and electron accepting polymer PFBT were prepared first, and then the nanoparticles were mixed at a certain ratio. In the second design, polymer solutions of PFB and PFBT were first mixed, and then converted into nanoparticles, forming binary polymer nanoparticles. The relation between the external quantum efficiency and layer composition of these devices was investigated. The properties of devices containing a blend of single-component PFB and PFBT particles differed substantially from those of solar cells based on blend particles, even for the same layer composition. An external quantum efficiency of approximately 4% was measured for a device made from polymer blend nanoparticles composed of PFB:F8BT at a weight ratio of 1:2.85 Multilayer optoelectronic devices were also fabricated using CPNs with varying dimensions.88
PLED and LECs with planar surface cell geometry, with a gold anode and cathode at an interelectrode spacing of 1 mm, were fabricated by inkjet printing. The active material for the PLED was an aqueous MEHPPV dispersion, and for the LECs, MEHPPV blends with poly(ethylene oxide) and lithium triflate formed the solid-state electrolyte.89 In addition, a polymer surface patterned by soft embossing was coated with an aqueous dispersion of CPNs by inkjet printing.90,91
We used a single type of conjugated polymer to generate white light emission. First, polymer nanoparticles based on polyfluorene derivatives containing azide groups were prepared. The surface decoration azide groups were allowed to decompose under UV light to generate reactive species, which allowed the inter- and intra-crosslinking of polymer chains to form shells. The emission kinetics were tuned by controlling the shell formation. The solid-state behaviours of these CPNs were investigated, and photometric calculations were performed for films and dispersions to determine the colour quality. Moreover, to show proof-of-principle of white light generation from these nanoparticles, a light-emitting diode was fabricated.92
Following our previous work on white light-emitting nanoparticles, we achieved better quality white light emission by constructing multi-layered white-emitting organic nanoparticles by the click reactions of azide- and alkyne-containing polymers emitting in the blue, green, and red regions of the spectrum.93 Four designs were selected for preparing the nanoparticles. The first approach involved preparing PFBN3 and PFBT nanoparticles separately and mixing them in a certain ratio. In the second approach, solutions of PFBN3 and PFBT were mixed before nanoparticle formation; the nanoparticles were prepared from a mixture of two polymer solutions. In the third and fourth approaches, sequentially formed nanoparticles were designed. The third approach, in which the core was made of the donor and the shell was composed of acceptor polymers, was the most efficient, allowing us to tune the emission colour and quality of the white light by adjusting the nanoparticle composition and the energy transfer. Using this approach, we prepared bi-layered and tri-layered core–shell nanoparticles by alkyne–azide 1,3-dipolar cycloaddition of polymers with appropriate functional groups to obtain stable, shape-persistent nanoparticles that remained intact in different solvents because of the crosslinking of functional groups. Interestingly, even in the absence of Cu(I)-catalysts, alkyne–azide 1,3-dipolar cycloaddition took place because of the appropriate alignment of the alkyne–azide functional groups in these nanoparticles. Although bi-layered polymer nanoparticles produced white emission, tri-layered polymer nanoparticles or tandem nanoparticles displayed better white light qualities due to their wider coverage of the spectrum. Moreover, light-emitting diodes fabricated using tandem nanoparticles in the absence of Cu(I)-catalysts exhibited superior colour rendering compared with those prepared from Cu(I)-catalyzed nanoparticles. These tandem nanoparticles can be used as colour converters to realize a white light-emitting diode with a colour quality comparable to existing phosphor-based white light-emitting diodes (Fig. 6).93
 | ||
| Fig. 6 (a) PL spectrum, SEM image, cartoon representation and light excited photographs of white light emission obtained from tri-layered CPNs; (b) molecular structures of conjugated polymers and four different designs to generate white light emission; (c) spectra; (d) CRI, CQS, and CCT of the tandem nanoparticle integrated white light-emitting diode at varying current levels; and (e) corresponding CIE coordinates. Adapted with permission from ref. 93. Copyright 2015 Royal Society of Chemistry. | ||
Highly efficient, stable, fully organic colour-converting monoliths suitable for solid-state lighting were prepared by incorporating oligomer nanoparticles into an organic host, sucrose.94
Conjugated polymer-based nano/microparticles have started to attract attention as optical nano/microcavities for lasing applications because of their high refractive indices, high absorptivity, high PL efficiency, and simple preparation methods.95–103 Microcavities confine light with narrow resonance frequencies and have a variety of applications, including laser resonators, photon pair generation, and biosensors. A microcavity consists of a dielectric as a resonator and a fluorescent dye to provide the optical activity. Conjugated polymer-based nano/micro cavities are intrinsically fluorescent and act also as dielectrics; therefore, extra materials are unnecessary.98
To explore the lasing properties of conjugated polymer particles, nano/micro photonic crystals with diameters of 400 nm to 2 μm were synthesized by in situ dispersion polymerization of a mixture of m-/p-divinylbenzene and fluorene derivatives by Heck coupling. These particles were encapsulated with a silica sol–gel precursor, and then processed using inkjet printing to obtain self-assembled photonic crystals (Fig. 7). These conjugated polymer photonic crystals provided the necessary gain medium and exhibited single-line laser emission upon optical pumping.99
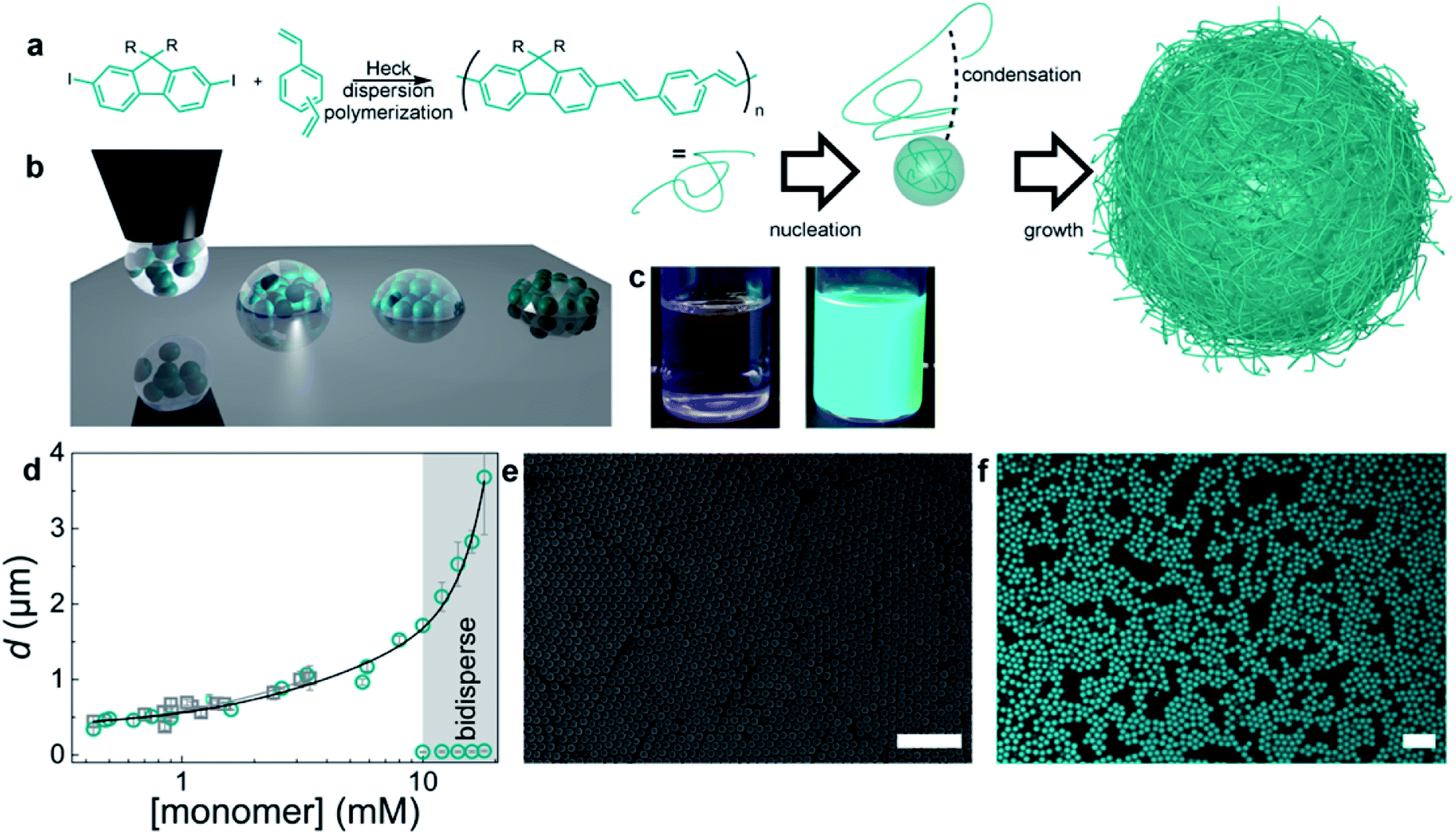 | ||
| Fig. 7 Preparation of monodisperse conjugated polymers. (a) Synthetic scheme and dispersion polymerization mechanism for generating monodisperse conjugated polymer particles. Monomers: (a) mixture of m-/p-divinylbenzene and fluorene with R = octyl or dodecyl. (b) Schematic diagram of the BTES sol–gel coassembly process. Deposition of the liquid (left), self-assembly of the particles, and concurrent evaporation of the solvent and condensation and formation of the sol–gel network (middle) to obtain crystalline, fully encapsulated photonic crystals (right). (c) Photographs of the reaction mixture before (left) and dispersion (right) after polymerization. (d) Particle diameter versus monomer concentration. Gray squares represent particles from the didodecylfluorene monomer, and cyan circles represent particles from the dioctylfluorene monomers. The error bars represent the dispersity of the particles, as measured by DLS or SEM image analysis of more than 100 particles. (e) SEM image of monodisperse particles. (f) Confocal microscopy image of conjugated polymer particles. The scale bars represent 10 μm. Adapted with permission from ref. 99. Copyright 2016 American Chemical Society. | ||
Using a vapor diffusion method, microspheres have been prepared through self-assembly of a series of π-conjugated polymers, and their behaviours as optical resonators have been investigated. Optical resonators play a crucial role in optical devices because they confine, sharpen and amplify light. Whispering gallery modes (WGMs) are excited by laser irradiation of a single isolated nano/microsphere to form sharp, periodic peaks in the PL spectrum that are characterized as transverse electric and magnetic modes.85 WGM-mediated long-range energy propagation has also been observed in microspheres composed of a binary mixture of energy donating polymer F8TPD and energy-accepting polymer PBDTCCz. When the molecular weights and precipitation times (nucleation and growth rates) were similar, these two polymers blended well to form identical microspheres. In contrast, when the molecular weights differed greatly, the polymers only produced irregular aggregates with macroscopic phase separation. PL spectroscopy measurements revealed that well-defined microspheres had efficient intra-sphere donor-to-acceptor energy transfer, indicating that the polymers were miscible with one another in the microspheres. Inter-sphere energy transfer occurred when several microspheres were coupled and one side of a microsphere was photoexcited. WGM-mediated long-range energy propagation was observed (Fig. 8), and energy transfer occurred at the point of contact of the energy-donating and -accepting microspheres. WGM was observed even after energy transfer to the energy-accepting microspheres, indicating that WGM-to-WGM PL conversion took place.85
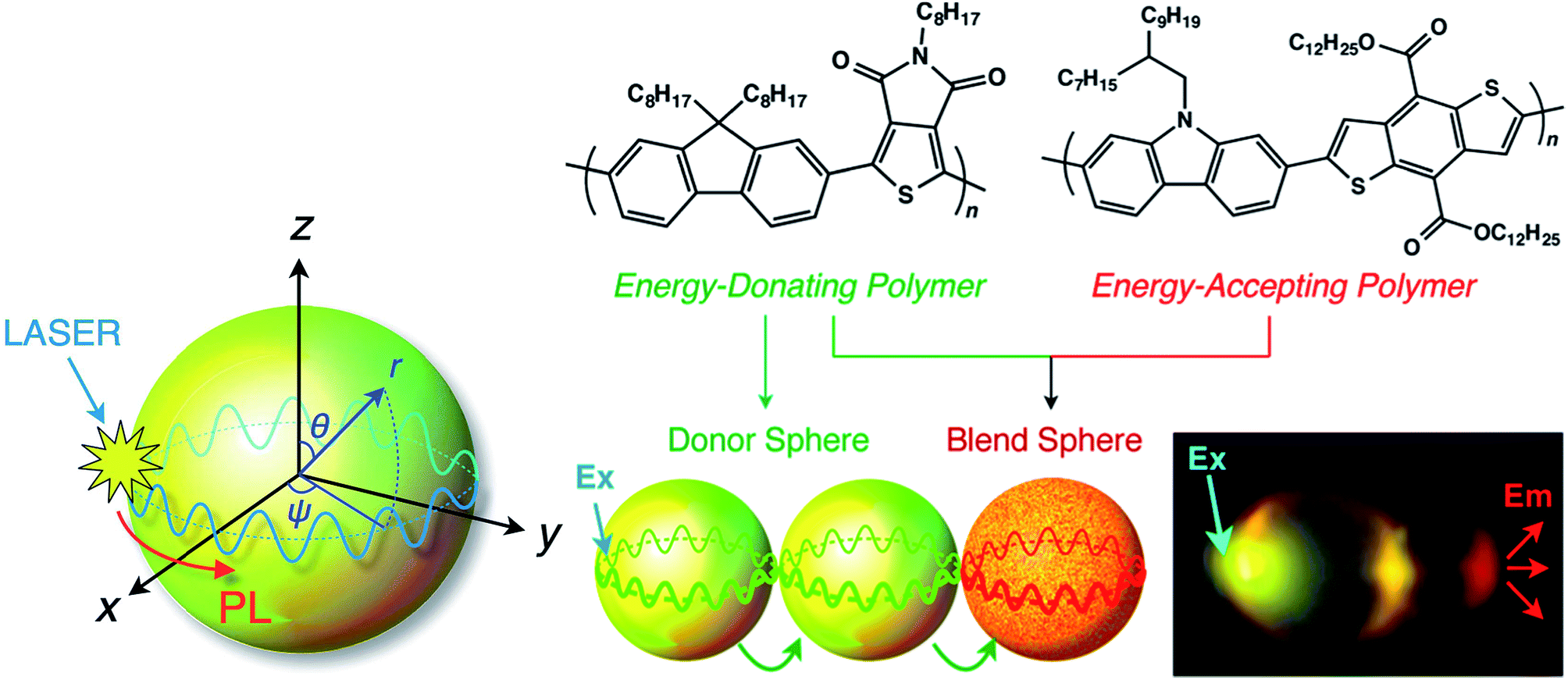 | ||
| Fig. 8 Schematic diagram of a WGM inside a microsphere. PL: photoluminescence; WGM: whispering gallery mode. Adapted with permission from ref. 85. Copyright 2016 American Chemical Society. | ||
Using the same approach, NIR optical microsphere resonators were prepared from a binary mixture of the energy-donating polymer, (poly[(9,9-dioctylfluorene-2,7-diyl)-alt-(5-octylthieno[3,4-c]pyrrole-4,6-dione-1,3-diyl)]), and energy-accepting conjugated polymer, (poly[(5-(2,4,6-triisopropylphenyl)thieno[3,4-c]phosphole-4,6-dione)-alt-(4,4-bis(2-ethylhexyl)-silolo-[3,2-b:4,5-b0]dithiophene)]). In the microspheres, sharp, periodic PL peaks from WGMs in the NIR region with quality factors (Q-factors) as high as 600 were observed, mediated by fluorescence resonance energy transfer from the energy-donating polymer to the energy-accepting polymer. The average Q-factor is defined as the peak wavelength divided by the full-width at the half maximum of the observed WGM PL peaks (Qav).100–103
Conjugated polymer-based microspheres have been used in single-component white-colour resonant micro-photoemitters. White PL was generated from microspheres composed of self-assembled polycarbazole polymer chains (PCz). Microspheres exhibited WGM PL, where resonant PL lines appeared over the whole visible spectral range (400–800 nm) owing to partial oxidation of the polymer main chains upon strong photoexcitation. In contrast, upon weak photoexcitation with stationary light, the PCz microspheres showed only blue PL.103
5. Biomedical applications
CPNs exhibit high photostability, high molar extinction coefficients, high fluorescent quantum yields and negligible toxicity. These features make them good candidates for a wide range of biomedical applications, and thus there are already numerous studies of their applications in nanobiotechnology.104–118A number of review articles20–22 have been published giving an overview of the biomedical applications of these nanostructured conjugated materials; therefore, we present a brief summary of recent work on their biomedical applications to complete this review.
CPNs can be used in long-term cell imaging and as a drug/biomolecules carrier because they are ideal nanocarriers in image-guided drug and biomolecule delivery due to their good photo stability and intrinsic luminescence properties.104–112 The nanoparticle surfaces can be decorated with a variety of functional groups to allow the attachment of biologically relevant species for targeting and recognition. Moreover, carefully selecting the structure of the oligomer or polymer can produce stimuli-responsive nanoparticles105–107 suitable for controlled cargo release or as light-induced sensitizers to generate reactive oxygen species for use in photodynamic therapy and antibacterial applications.113–115
Fig. 9 illustrates some of the biomedical applications of conjugated nanoparticles.
 | ||
| Fig. 9 Biomedical applications of conjugated nanoparticles. (a) Image-guided anticancer drug delivery. Adapted with permission from ref. 106. Copyright 2014, American Chemical Society. (b) NIR-modulated cancer thermo-chemotherapy. Reproduced with permission from ref. 115. Copyright 2016, American Chemical Society. (c) In vivo photoacoustic imaging coupled with photothermal therapy. Reproduced with permission from ref. 117. Copyright 2016, American Chemical Society. (d) Image-guided photodynamic and photothermal therapy. Reproduced with permission from ref. 114. Copyright 2017, John Wiley and Sons. (e) Augmented light reaction of isolated chloroplasts. Reproduced with permission from ref. 118. Copyright 2017, John Wiley and Sons. | ||
Fig. 9a shows an example of the use of conjugated oligomer/polymer nanostructures in image-guided anticancer drug delivery.106 Nanostructures were made out of a red-emitting, fluorene-benzothiodiazole-based amphiphilic conjugated oligomer to carry anticancer drugs and allow pH-triggered drug release. The pH of the nanoparticles and their toxicities were regulated by using the macrocyle, cucurbituril, as a capping group. In the acidic microenvironment of cells, the amine groups were protonated, causing the swelling and disintegration of the particles to release the drug molecules.
Fig. 9b shows NIR-modulated cancer thermo-chemotherapy using nanoparticles prepared from the donor–acceptor conjugated polymer PBIBDF-BT, which contained alternating isoindigo derivative (BIBDF) and bithiophene (BT) units, as a novel photothermal transducer. The nanoparticles exhibited strong NIR absorbance due to their low band gap (1.52 eV). The photothermal effect produced heat in the particles causing a response in the co-assembled polyethylene glycol-block-poly(hexyl ethylene phosphate) (PEG-block-PHEP) polymer and the release of the drug, doxorubicin.115
Fig. 9c shows a theranostic application of CPNs, in which photoacoustic imaging was coupled with photothermal therapy.117 The addition of photothermally active substances, such as fullerenes, can be used to harvest excited states via photoinduced electron transport and thermally relaxing the excited state. This leads to improved heat generation and enhanced photoacoustic contrast.
Fig. 9d shows an elegant use of CPNs as a multiplexed probe for image-guided photodynamic therapy or photothermal therapy.114 The nanoparticles contained a long-lived fluorescent conjugated polymer (poly(fluorenedivinylene-benzothiadiazole) (PFVBT)) and a photothermally active polymer (poly((tetrakis(octyloxyphenyl)-indacenol-dithiophene)-alt-(bis(hexyloxyphenyl)-thiadiazolo-quinoxaline)) (PIDTTTQ)). Depending on the excitation wavelength, either fluorescence imaging and photodynamic therapy or photothermal therapy could be performed. The particles carried surficial affibodies to home in on cancer cells.
Fig. 9e shows the strategy for augmenting the light reaction in isolated chloroplasts using CPNs for artificial photosynthesis.118
6. Outlook and conclusions
Since their discovery and the award of the Nobel Prize in 2000, conjugated polymers have received great attention in academia and industry because of their superior properties and wide range of possible applications. These materials combine the best features of organic and inorganic materials; they are flexible, lightweight, and easily processable with interesting electrical and optical properties. Applications of these materials have been explored in a wide range of areas, spanning optoelectronics, artificial muscles, actuators, and energy. These materials are even more attractive as nanostructures owing to the augmentation of their properties. These new-generation nanostructured materials possess many unusual features and interesting properties. Their optical properties are significantly altered and their uses have been demonstrated in applications including imaging, image-guided delivery of therapeutic agents, photothermal and photodynamic therapeutic agents, photocatalysis, and lasing materials.Accordingly, in this review, recent works on π-conjugated oligomer and polymer-based nanostructured materials were discussed, focusing on their preparation, properties, light harvesting and energy transfer properties, and their photonic applications, as well as examples of their applications in bionanotechnology.
The chemical structures of these materials can be tailored by adding various functionalities to tune their optical, electronic, and mechanical properties. However, examples of their optoelectronic applications are scarce. It is expected that in their use in this area will grow rapidly, as well as in energy conversion, storage and efficiency applications and biophotonics.
Further research on hybrid nanoparticles of conjugated oligomers and polymers with metals (Ag, Au, Pd, etc.) is also anticipated. These new-generation hybrid nanostructures should offer more opportunities than current materials, especially in optoelectronics, plasmonic lasing, photocatalysis, and sensing, due to their multifunctional nature and unprecedented properties.
Conflicts of interest
There are no conflicts to declare.References
- Handbook of Conducting Polymers, ed. T. A. Skotheim and J. Reynolds, 3rd edn, CRC Press, Boca Raton, FL, USA, 2007 Search PubMed.
- T. Swager, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS.
- A. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- J. G. Ibanez, M. E. Rincon, S. Gutierrez-Granados, M. Chahma, O. A. Jaramillo-Quintero and B. A. Frontana-Uribe, Chem. Rev., 2018, 118, 4731–4816 CrossRef CAS PubMed.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS.
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712–757 CrossRef CAS PubMed.
- J. Xie, C. Zhao, Z.-Q. Lin, P.-Y. Gu and Q. Zhang, Chem.–Asian J., 2016, 11, 1489–1511 CrossRef CAS PubMed.
- D. Tuncel and H. V. Demir, Nanoscale, 2010, 2, 484–494 RSC.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- A. J. C. Kuehne, Adv. Biosyst., 2017, 1, 1700100 CrossRef.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- X. Xu, R. Liu and L. Li, Chem. Commun., 2015, 51, 16733–16749 RSC.
- J. Yu, Y. Rong, C.-T. Kuo, X.-H. Zhou and D. T. Chiu, Anal. Chem., 2017, 89, 42–56 CrossRef CAS PubMed.
- C. F. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- K. Landfester, R. Montenegro, U. Scherf, R. Guntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Adv. Mater., 2003, 14(9), 651–655 CrossRef.
- T. Kietzke, D. Nehrer, K. Landfester, R. Montenegro, R. Guntner and U. Scherf, Nat. Mater., 2003, 2, 408 CrossRef CAS PubMed.
- C. Wu, C. Szymanski and J. McNeill, Langmuir, 2006, 22, 2956–2960 CrossRef CAS PubMed.
- W. Changfeng, B. Barbara, S. Craig, C. Kenneth and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef PubMed.
- I. Fischer, K. Petkau-Milroy, Y. L. Dorland, A. P. H. J. Schenning and L. Brunsveld, Chem.–Eur. J., 2013, 19, 16646–16650 CrossRef CAS PubMed.
- I. Fischer, A. Kaeser, M. A. M. Peters-Gumbs and A. P. H. J. Schenning, Chem.–Eur. J., 2013, 19, 10928–10934 CrossRef CAS PubMed.
- J. Liu, G. Feng, R. Liu, N. Tomczak, L. Ma, G. G. Gurzadyan and B. Liu, Small, 2014, 10, 3110 CrossRef CAS PubMed.
- N. Zaquen, H. Lu, T. Chang, R. Mamdooh, L. Lutsen, D. Vanderzande, M. Stenzel and T. Junkers, Biomacromolecules, 2016, 17, 4086 CrossRef CAS PubMed.
- H.-F. Jiao, X. Wang, K. Yao, P. Chen, Z. Jia, Z. Peng and F. Li, J. Mater. Chem. B, 2016, 4, 7882 RSC.
- J. Pecher, J. Huber, M. Winterhalder, A. Zumbusch and S. Mecking, Biomacromolecules, 2010, 11, 2776–2780 CrossRef CAS PubMed.
- J. Huber, C. Jung and S. Mecking, Macromolecules, 2012, 45, 7799–7805 CrossRef CAS.
- J. Deng, B. Chen, X. Luo and W. Yang, Macromolecules, 2009, 42, 933–938 CrossRef CAS.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267–14273 CrossRef CAS PubMed.
- A. J. C. Kuehne, M. C. Gather and J. Sprakel, Nat. Commun., 2012, 3, 1088 CrossRef PubMed.
- S. Ciftci and A. J. C. Kuehne, Macromolecules, 2015, 48, 8389–8393 CrossRef CAS.
- T. Balkan, S. Kizir and D. Tuncel, ACS Omega, 2017, 2, 5470–5477 CrossRef CAS.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, Chem. Rev., 2017, 117, 796–837 CrossRef CAS PubMed.
- Y. Jiang and J. McNeill, Chem. Rev., 2017, 117, 838–859 CrossRef CAS PubMed.
- B. Jana, A. Ghosh and A. Patra, J. Phys. Chem. Lett., 2017, 8, 4608–4620 CrossRef CAS PubMed.
- S. Bhattacharyya and A. Patra, J. Photochem. Photobiol., C, 2014, 20, 51–70 CrossRef CAS.
- P. F. Barbara, A. J. Gesquiere, S.-J. Park and Y. J. Lee, Acc. Chem. Res., 2005, 38, 602–610 CrossRef CAS PubMed.
- I. Hwang and G. D. Scholes, Chem. Mater., 2011, 23, 610–620 CrossRef CAS.
- E. Collini and G. D. Scholes, Science, 2009, 323, 369–373 CrossRef CAS PubMed.
- D. Beljonne, G. Pourtois, C. Silva, E. Hennebicq, L. M. Herz, R. H. Friend, G. D. Scholes, S. Setayesh, K. Müllen and J. L. Brédas, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10982–10987 CrossRef CAS PubMed.
- S. K. Saikin, A. Eisfeld, S. Valleau and A. Aspuru-Guzik, Nanophotonics, 2013, 2, 21–38 CAS.
- E. N. Bodunov, M. N. Berberan-Santos and J. M. G. Martinho, Chem. Phys. Lett., 2001, 340, 137–141 CrossRef CAS.
- S. F. Burlatsky, G. S. Oshanin and A. V. Mogutov, Phys. Rev. Lett., 1990, 65, 3205–3208 CrossRef PubMed.
- J. C. Bolinger, M. C. Traub, J. Brazard, T. Adachi, P. F. Barbara and D. A. V. Bout, Acc. Chem. Res., 2012, 45, 1992–2001 CrossRef CAS PubMed.
- Z. Hu, B. Shao, G. T. Geberth and D. A. V. Bout, Chem. Sci., 2018, 9, 1101–1111 RSC.
- J. M. Lupton, Adv. Mater., 2010, 22, 1689–1721 CrossRef CAS PubMed.
- H. Kobayashi, S. Onda, S. Furumaki, S. Habuchi and M. Vacha, Chem. Phys. Lett., 2012, 528, 1–6 CrossRef CAS.
- N. Kurokawa, H. Yoshikawa, N. Hirota, K. Hyodo and H. Masuhara, ChemPhysChem, 2004, 5, 1609–1615 CrossRef CAS PubMed.
- I. O. Huyal, T. Ozel, D. Tuncel and H. V. Demir, Opt. Express, 2008, 16, 13391–13397 CrossRef CAS PubMed.
- R. Potai and R. Traiphol, J. Colloid Interface Sci., 2013, 403, 58–66 CrossRef CAS PubMed.
- H. Shimizu, M. Yamada, R. Wada and M. Okabe, Polym. J., 2008, 40, 33–36 CrossRef CAS.
- F. Wang, M.-Y. Han, K. Y. Mya, Y. Wang and Y.-H. Lai, J. Am. Chem. Soc., 2005, 127, 10350–10355 CrossRef CAS PubMed.
- S. Siddiqui and F. Spano, Chem. Phys. Lett., 1999, 308, 99–105 CrossRef CAS.
- H. Yamagata and F. C. Spano, J. Chem. Phys., 2012, 136, 184901 CrossRef CAS PubMed.
- C. Wu, H. Peng, Y. Jiang and J. McNeill, J. Phys. Chem. B, 2006, 110, 14148–14154 CrossRef CAS PubMed.
- C. Wu, Y. Zheng, C. Szymanski and J. McNeill, J. Phys. Chem. C, 2008, 112, 1772–1781 CrossRef CAS PubMed.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- J. K. Grey, D. Y. Kim, B. C. Norris, W. L. Miller and P. F. Barbara, J. Phys. Chem. B, 2006, 110, 25568–25572 CrossRef CAS PubMed.
- S. J. Park, A. J. Gesquiere, J. Yu and P. F. Barbara, J. Am. Chem. Soc., 2004, 126, 4116–4117 CrossRef CAS PubMed.
- A. Ghosh, B. Jana, S. Chakraborty, S. Maiti, B. Jana, H. N. Ghosh and A. Patra, J. Phys. Chem. C, 2017, 121, 21062–21072 CrossRef CAS.
- L. C. Groff, Y. Jiang, X. Wang and J. D. McNeill, J. Phys. Chem. C, 2017, 121, 7549–7557 CrossRef CAS.
- R. E. Palacios, F.-R. F. Fan, J. K. Grey, J. Suk, A. J. Bard and P. F. Barbara, Nat. Mater., 2007, 6, 680–685 CrossRef CAS PubMed.
- J. Yu, C. Wu, Z. Tian and J. McNeill, Nano Lett., 2012, 12, 1300–1306 CrossRef CAS PubMed.
- Y. Jiang, T. Nongnual, L. Groff and J. McNeill, J. Phys. Chem. C, 2018, 122, 1376–1383 CrossRef CAS.
- W.-C. Wu, C.-Y. Chen, Y. Tian, S.-H. Jang, Y. Hong, Y. Liu, R. Hu, B. Z. Tang, Y.-T. Lee and C.-T. Chen, Adv. Funct. Mater., 2010, 20, 1413–1423 CrossRef CAS.
- S. Grigalevicius, M. Forster, S. Ellinger, K. Landfester and U. Scherf, Macromol. Rapid Commun., 2006, 27, 200–202 CrossRef CAS.
- Y. Jin, F. Ye, M. Zeigler, C. Wu and D. T. Chiu, ACS Nano, 2011, 5, 1468–1475 CrossRef CAS PubMed.
- S. Bhattacharyya, B. Paramanik and A. Patra, J. Phys. Chem. C, 2011, 115, 20832–20839 CrossRef CAS.
- S. Bhattacharyya, S. Prashanthi, P. R. Bangal and A. Patra, J. Phys. Chem. C, 2013, 117, 26750–26759 CrossRef CAS.
- C. Martín, M. Rosaria di Nunzio, B. Cohen and A. Douhal, Photochem. Photobiol. Sci., 2014, 13, 1580 RSC.
- C. Martin, S. Bhattacharyya, A. Patra and A. Douhal, Photochem. Photobiol. Sci., 2014, 13, 1241 RSC.
- S. Das, B. Jana, T. Debnath, A. Ghoshal, A. K. Das and A. Patra, J. Phys. Chem. C, 2017, 121, 4050–4059 CrossRef CAS.
- X. Wang, L. C. Groff and J. D. McNeill, Langmuir, 2013, 29, 13925–13931 CrossRef CAS PubMed.
- I. O. Ozel, T. Ozel, H. V. Demir and D. Tuncel, Opt. Express, 2010, 18, 670–684 CrossRef CAS PubMed.
- B. Guzelturk, P. L. Hernandez-Martinez, V. K. Sharma, Y. Coskun, V. Ibrahimova, D. Tuncel, A. O. Govorov, X. W. Sun, Q. Xiong and H. V. Demir, Nanoscale, 2014, 6, 11387–11394 RSC.
- T. Erdem, I. Ibrahimova, D. W. Jeon, I. H. Lee, D. Tuncel and H. V. Demir, J. Phys. Chem. C, 2013, 117, 18613–18619 CrossRef CAS.
- H. Keita, B. Guzelturk, J. Pennakalathil, T. Erdem, H. V. Demir and D. Tuncel, J. Mater. Chem., 2015, 3, 10277–10284 CAS.
- T. Erdem, I. Ibrahimova, D. W. Jeon, I. H. Lee, D. Tuncel and H. V. Demir, J. Phys. Chem. C, 2013, 117, 18613–18619 CrossRef CAS.
- S. Kushida, D. Braam, T. D. Dao, H. Saito, K. Shibasaki, S. Ishii, T. Nagao, A. Saeki, J. Kuwabara, T. Kanbara, M. Kijima, A. Lorke and Y. Yamamoto, ACS Nano, 2016, 10, 5543–5549 CrossRef CAS PubMed.
- T. Piok, S. Gamerith, C. Gadermaier, H. Plank, F. P. Wenzl, S. Patil, R. Montenegro, T. Kietzke, D. Nehrer, U. Scherf, K. Landfester and E. J. W. List, Adv. Mater., 2003, 15, 800–804 CrossRef CAS.
- T. Kietzke, D. Nehrer, M. Kumke, R. Montenegro, K. Landfester and U. Scherf, Macromolecules, 2004, 37, 4882 CrossRef CAS.
- T. Piok, H. Plank, G. Mauthner, S. Gamerith, C. Gadermaier, F. P. Wenzl, S. Patil, R. Montenegro, M. Bouguettaya, J. R. Reynolds, U. Scherf, K. Landfester and E. J. W. List, Jpn. J. Appl. Phys., 2005, 44, 479 CrossRef CAS.
- G. Mauthner, K. Landfester, A. Kock, H. Bruckl, M. Kast, C. Stepper and E. J. W. List, Org. Electron., 2008, 9, 164 CrossRef CAS.
- E. Fisslthaler, S. Sax, U. Scherf, G. Mauthner, E. Moderegger, K. Landfester and E. J. W. List, Appl. Phys. Lett., 2008, 92, 183305 CrossRef.
- E. Fisslthaler, A. Blumel, K. Landfester, U. Scherf and E. J. W. List, Soft Matter, 2008, 4, 2448 RSC.
- E. J. Park, T. Erdem, V. Ibrahimova, S. Nizamoglu, H. V. Demir and D. Tuncel, ACS Nano, 2011, 5, 2483–2492 CrossRef CAS PubMed.
- H. Keita, B. Guzelturk, J. Pennakalathil, T. Erdem, H. V. Demir and D. Tuncel, J. Mater. Chem., 2015, 3, 10277–10284 CAS.
- Z. Soran-Erdem, T. Erdem, K. Gungor, J. Pennakalathil, D. Tuncel and H. V. Demir, ACS Nano, 2016, 10(5), 5333–5339 CrossRef CAS PubMed.
- H. Kim, N. Schulte, G. Zhou, K. Müllen and F. Laquai, Adv. Mater., 2011, 23, 894–897 CrossRef CAS PubMed.
- A. Rose, Z. Zhu, C. F. Madigan, T. M. Swager and V. Bulović, Nature, 2005, 434, 876–879 CrossRef CAS PubMed.
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- Y. Yamamoto, Polym. J., 2016, 48, 1045–1050 CrossRef CAS.
- A. Mikosch, S. Ciftci and A. J. C. Kuehne, ACS Nano, 2016, 10, 10195–10201 CrossRef CAS PubMed.
- O. Oki, S. Kushida, A. Mikosch, K. Hatanaka, Y. Takeda, S. Minakata, J. Kuwabara, T. Kanbara, H. D. Dao, S. Ishii, T. Nagao, A. J. C. Kuehne, F. Deschler, R. H. Friend and Y. Yamamoto, Mater. Chem. Front., 2018, 2, 270 RSC.
- D. Okada, T. Nakamura, D. Braam, T. D. Dao, S. Ishii, T. Nagao, A. Lorke, N. Tatsuya and Y. Yamamoto, ACS Nano, 2016, 10, 7058–70639 CrossRef CAS PubMed.
- D. Braam, S. Kushida, R. Niemöller, G. M. Prinz, H. Saito, T. Kanbara, J. Kuwabara, Y. Yamamoto and A. Lorke, Sci. Rep., 2016, 6, 19635, DOI:10.1038/srep19635.
- S. Kushida, S. Okabe, T. D. Dao, S. Ishii, T. Nagao, A. Saeki, M. Kijima and Y. Yamamoto, RSC Adv., 2016, 6, 52854 RSC.
- C. F. Wu, T. Schneider, M. Zeigler, J. B. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- J. Pennakalathil, A. Özgün, I. Durmaz, R. Cetin-Atalay and D. Tuncel, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 114–122 CrossRef CAS.
- J. Pennakalathil, E. Jahja, E. S. Ozdemir, O. Konu and D. Tuncel, Biomacromolecules, 2014, 15, 3366–3374 CrossRef CAS PubMed.
- O. Gezici, I. Durmaz, E. B. Guven, O. Unal, A. Ozgun, R. Cetin-Atalay and D. Tuncel, RSC Adv., 2014, 4, 1302–1309 RSC.
- D. Ding, K. Li, Z. S. Zhu, K. Y. Pu, Y. Hu, X. Q. Jiang and B. Liu, Nanoscale, 2011, 3, 1997–2002 RSC.
- J. H. Moon, E. Mendez, Y. Kim and A. Kaura, Chem. Commun., 2011, 47, 8370–8372 RSC.
- K. Y. Pu, A. J. Shuhendler, M. P. Valta, L. N. Cui, M. Saar, D. M. Peehl and J. H. Rao, Adv. Healthcare Mater., 2014, 3, 1292–1298 CrossRef CAS PubMed.
- T. Repenko, A. Rix, S. Ludwanowski, D. Go, F. Kiessling, W. Lederle and A. J. C. Kuehne, Nat. Commun., 2017, 8, 470 CrossRef PubMed.
- J. Liu, K. Li and B. Liu, Adv. Sci., 2015, 2, 1500008 CrossRef PubMed.
- L. Xu, L. Cheng, C. Wang, R. Peng and Z. Liu, Polym. Chem., 2014, 5, 1573 RSC.
- G. Feng, Y. Fang, J. Liu, J. Geng, D. Ding and B. Liu, Small, 2017, 13, 1602807 CrossRef PubMed.
- D.-D. Li, J.-X. Wang, Y. Ma, H.-S. Qian, D. Wang, L. Wang, G. Zhang, L. Qiu, Y.-C. Wang and X.-Z. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 19312–19320 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- Y. Lyu, Y. Fang, Q. Miao, X. Zhen, D. Ding and K. Pu, ACS Nano, 2016, 10, 4472–4481 CrossRef CAS PubMed.
- Y. Wang, S. Li, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5308–5311 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |