
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic layer deposition of Co3O4 nanocrystals on N-doped electrospun carbon nanofibers for oxygen reduction and oxygen evolution reactions†
Mohammad Aref
Khalily
 *ab,
Bhushan
Patil
a,
Eda
Yilmaz
*a and
Tamer
Uyar
*a
*ab,
Bhushan
Patil
a,
Eda
Yilmaz
*a and
Tamer
Uyar
*a
aInstitute of Materials Science and Nanotechnology, National Nanotechnology Research Center (UNAM), Bilkent University, Ankara, 06800, Turkey. E-mail: uyar@unam.bilkent.edu.tr; eda.yilmaz@gmail.com
bLaboratory of Biomolecular Nanotechnology, MESA+ Institute for Nanotechnology, University of Twente, Enschede 7500 AE, The Netherlands. E-mail: m.a.khalily@utwente.nl
First published on 26th December 2018
Abstract
The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are considered as the two crucial reactions in key renewable-energy technologies including fuel cells and water splitting. Despite promising research progress in the preparation of various non-noble metal based electrocatalysts, it is still highly challenging but desirable to develop novel fabrication strategies to synthesize highly active and cost-effective ORR/OER bifunctional electrocatalysts in a precisely controlled manner. Herein, we report atomic layer deposition (ALD) of highly monodisperse Co3O4 nanocrystals of different sizes on N-doped electrospun carbon nanofibers (nCNFs) as high performance bifunctional catalysts (Co@nCNFs) for the ORR and OER. Co@nCNFs (with an average Co3O4 particle size of ∼3 nm) show high ORR performance exhibiting an onset potential of 0.87 V with a low Tafel slope of 119 mV dec−1 approaching that of commercial Pt/C. Similarly, the Co@nCNF electrocatalyst showed remarkable catalytic activity in the OER. The turnover frequency (TOF) value determined at an overpotential of 550 mV for the Co@nCNFs is ∼0.14 s−1 which is ca. 3 and ca. 15-fold higher than those of bulk Co (∼0.05 s−1) and the standard state-of-the-art IrOx (0.0089 s−1) catalyst, respectively. This work will open new possibilities for fabrication of inexpensive non-noble metal materials in highly controlled manner for applications as bifunctional ORR/OER electrocatalysis.
Introduction
Increasing energy demands and environmental concerns have stimulated researchers to seek alternative energy conversion and storage systems which could be cleaner, cost-effective and sustainable. The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are considered as the two crucial reactions in key renewable-energy technologies including fuel cells and water splitting.1–4 However, both the ORR and OER suffer from sluggish reaction kinetics resulting in huge overpotentials which limits the broad utilization of electrochemical devices.4 State-of-the-art catalysts developed for the ORR are based on expensive and rare metals such as platinum (Pt) and its alloys.5 Likewise, precious and rare ruthenium (Ru) and iridium (Ir)-based electrocatalysts are best performing for the OER or water oxidation.6 Therefore, the development of cost-effective and stable electrocatalytic systems which can catalyze both the ORR and OER at appreciable rates is highly desirable.Within this context, a wide range of non-noble metal and metal-free electrocatalysts such as carbon materials,7 transition metal oxides,8 sulfides,9,10 and nitrides11,12 have been reported to show promising electrocatalytic activities towards the OER/ORR. Among the aforementioned electrocatalysts, cobalt-based materials have attracted huge research interest owing to their higher stability in electrocatalytic reactions and unusual 3d electronic configurations.13–16 Cobalt oxide (CoOx) suffers from low intrinsic conductivities; thus Co-based electrocatalysts are commonly coupled with conductive supports such as graphene and carbon nanotubes.17–19 Despite promising research progress in the preparation of various CoOx-decorated graphene and carbon nanotubes as ORR/OER electrocatalysts, it is still highly challenging but desirable to develop novel fabrication strategies to synthesize highly active and cost-effective ORR/OER electrocatalysts in a precisely controlled manner.
Atomic layer deposition (ALD) is a thin film growth technique which applies self-limiting chemical reactions between gaseous metal/metal oxide precursors and support surfaces allowing precise control over the film thickness and composition.20 One of the remarkable characteristics of ALD is depositing thin films in a highly uniform, conformal and reproducible manner on various supports including flat surfaces, and porous, high surface area and three-dimensional materials.21 Meanwhile, ALD has shown to be successful in producing discrete metallic and metal oxide nanoparticles having a tunable size, shape and composition. A wide range of monometallic,22,23 bimetallic24 and core–shell25 nanosized particles produced by ALD have been employed as catalysts in a variety of chemical reactions. Recently, the ALD technique has been increasingly utilized in design and synthesis of novel catalytic systems because it offers high reproducibility and precise control over their size, shape and composition.26,27 Electrospinning is another versatile nanofabrication technique which employs electrostatic forces to produce a wide range of one-dimensional (1D) nanostructures from polymer solutions.28 This cost-effective technique offers a range of marvelous advantages such as the controlled size, morphology, chemical composition, porosity and surface area of electrospun 1D nanostructures.29 A number of promising electrocatalysts produced by electrospinning have already been reported.30–32
Herein, we utilize electrospinning and ALD nanofabrication techniques to synthesize a novel bifunctional electrocatalyst nanosystem in a highly precise and reproducible manner which is highly active towards the ORR and OER. Electrospinning was used to prepare well-defined 1D N-doped electrospun carbon nanofibers (nCNFs). The as-synthesized conductive nCNF support was further decorated with discrete Co3O4 nanoparticles using the ozone-assisted ALD technique, and the resulting product is hereafter referred to as Co@nCNFs. We studied systematically the effect of the number of Co ALD cycles on the catalytic activity of Co@nCNFs. The electrocatalyst obtained with 100 cycles of Co deposition having ∼3 nm Co3O4 nanoparticles (Co100@nCNFs) shows high ORR performance with a positive half wave potential of 700 mV approaching that of commercial Pt/C. Co100@nCNFs also exhibit superior OER catalytic activity with a low overpotential of 550 mV at a current density of 10 mA cm−2. The TOF value determined at an overpotential of 550 mV for the Co100@nCNFs is ∼0.14 s−1 which is ca. 3 and ca. 15-fold higher than those of bulk Co (∼0.05 s−1) and the standard state-of-the-art IrOx (0.0089 s−1) catalyst, respectively.
Experimental section
Materials
Polyacrylonitrile (PAN, Mw ≈ 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Scientific Polymer Products, Inc. Dimethylformamide (DMF) and 20% platinum on graphitized carbon (<5 nm (Pt); Pt/C) were purchased from Sigma-Aldrich, and KOH was purchased from Alfa Aesar. All chemicals were used as received without further purification.
000) was purchased from Scientific Polymer Products, Inc. Dimethylformamide (DMF) and 20% platinum on graphitized carbon (<5 nm (Pt); Pt/C) were purchased from Sigma-Aldrich, and KOH was purchased from Alfa Aesar. All chemicals were used as received without further purification.
Electrospinning
A 13% (w/v, with respect to the solvent) polyacrylonitrile (PAN, Mw ∼ 150000, Scientific Polymer Products, Inc.) polymer solution was prepared in DMF at 50 °C. The clear PAN solution was loaded into a 3 mL syringe having a needle of 0.4 mm inner diameter. 0.5 mL h−1 flow rate was maintained by a pump (KD Scientific, KDS 101) and a voltage of 15 kV was applied by a high voltage power supply (Matsusada, AU Series) to initiate the electrospinning. The PAN nanofibrous web was collected on aluminium foil which was positioned at 10 cm from the end of the tip. The electrospun PAN nanofibers were left in the hood for 72 h to get rid of residual DMF.
Synthesis of electrospun carbon nanofibers
The as-prepared electrospun PAN was loaded into a furnace and heated up to 280 °C at a heating rate of 1 °C min−1 and held for 1 h under air flow. The sample was allowed to cool down to room temperature followed by passing Ar gas (100 sccm) for 30 minutes before the carbonization step. The sample was carbonized at 800 °C in an Ar environment with a heating rate of 5 °C min−1 and held for 1.5 h at 800 °C.Environmental scanning electron microscopy
The morphology of electrospun PAN nanofibers and nCNFs was imaged with an FEI Quanta 200 FEG environmental scanning electron microscope with an ETD detector. Electrospun PAN nanofibers were sputter coated with 5 nm gold/palladium prior to imaging.X-ray photoelectron spectroscopy
The samples were stabilized on copper tape and then were analyzed with a Thermo K-alpha monochromatic high performance X-ray photoelectron spectrometer. Survey analyses were performed at 2 scans while high resolution XPS was performed at 50 scans. The pass energy, step size and spot size were adjusted to 30 eV, 0.1 eV and 400 μm, respectively.X-ray diffraction
The samples were analyzed with a PANalytical X'Pert powder diffractometer. All data were recorded by using CuKα radiation in the 2θ range of 10°–80°.Elemental (CHNS–O) analysis
2 mg of electrospun carbon nanofibers plus 8 mg of vanadium(V) oxide were loaded into a tin container. BBOT (2,5-bis(5-tert-butyl-2-benzo-oxazol-2-yl)) was used as the standard for calibrations. The measurements were performed with a Thermo Scientific FLASH 2000 Series CHNS–O analyzer.Brunauer–Emmett–Teller analysis
A small amount of nCNFs (∼50 mg) was weighed into an analysis tube and degassed under high vacuum at 80 °C for 720 min. The analysis was conducted after reweighing the degassed sample. The Brunauer–Emmett–Teller (BET) surface areas were determined from N2 adsorption isotherms by multipoint analysis.Transmission electron microscopy
An FEI Tecnai G2 F30 transmission electron microscope (TEM) was used to image the samples. Minute amounts of samples were first dispersed in ethanol followed by adding dropwise 10 μL of the mixture on a carbon-covered copper grid and letting it dry at room temperature.Atomic layer deposition
Co3O4 nanoparticles were grown on nCNFs using a Savannah S100 ALD reactor (Ultratech Inc.). Approximately 10 mg of nCNFs was dispersed in ethanol and deposited on a clean silicon wafer substrate and was left to dry at room temperature. Cobaltocene was used as the cobalt precursor while ozone was utilized as the reactant gas to grow Co3O4 nanoparticles. The cobalt precursor was preheated to 70 °C to produce and maintain the vapor pressure of the organometallic precursor. The deposition of cobalt oxide was carried out at 230 °C. The sample was loaded into the ALD reaction chamber having a temperature of 230 °C. N2 was used as the carrier gas with a flow rate of 20 sccm. O3 was produced from a pure O2 flow with a Cambridge NanoTech Savannah Ozone Generator.Inductively coupled plasma-mass spectroscopy
2 mg of Co@nCNFs was kept in 2 mL of aqua regia for 4 days. Standards of Co having 500 ppb, 250 ppb, 125 ppb and 62.5 ppb concentrations were prepared in a 2% solution of HNO3:HCl (1:1) for obtaining the calibration curve. The 2% solution of HNO3:HCl (1:1) was used as the blank. Co@nCNFs in aqua regia were passed through a cellulose filter to get rid of undissolved nCNFs and then were diluted ∼100 times with the 2% solution of HNO3:HCl (1:1) for ICP-MS analysis. A Thermo X series II inductively coupled plasma-mass spectrometer was used to perform the measurements. The ICP-MS operating parameters were as follows: dwell time – 10000 ms, channel per mass – 1, acquisition duration – 7380, channel spacing – 0.02, and carrier gas – argon.
Electrochemical measurements
All experiments were performed at room temperature using a Biologic SP-150 Potentiostat with a standard three-electrode electrochemical cell. The catalyst-modified glassy carbon electrode (GC; 3 mm diameter and 0.07068 cm2 geometric surface area), a Pt spiral wire and Ag|AgCl|KCl(sat.) were used as working, counter and reference electrodes, respectively. The ORR and OER were performed in 20 mL 0.1 M KOH solution where prior to each measurement, the electrolyte solution was saturated with either N2 or O2 gas (99.999% purity) for 45 min. The rotating disk electrode (RDE) linear sweep voltammetry (LSV) technique was employed to determine the ORR mechanism and kinetics. The Nernst equation used to convert all the potentials measured vs. Ag|AgCl|KCl(sat.) to the reversible hydrogen electrode (RHE) scale.Results and discussion
The conductivity and extent of nitrogen (N)-doping of electrospun carbon fibers play an essential role in electrocatalysis particularly in the ORR.33 N-doped carbon materials also exhibit enhanced stability against corrosion during electrocatalysis. The relationship between conductivity and N-doping is inversely proportional for the synthesis of electrospun carbon fibers. In other words, higher carbonization temperatures produce more graphitic phase resulting in more conductive carbon fibers while the extent of N-doping decreases.33To this end, nCNFs were synthesized by first electrospinning a solution of polyacrylonitrile (PAN, Mw ≈ 150000) into well-defined 1D PAN nanofibers having diameters in the range of 300–500 nm (Fig. S1†) as imaged by scanning electron microscopy (SEM). Subsequently, PAN nanofibers were converted into nCNFs by a two-step carbonization process. Electrospun PAN nanofibers were first stabilized by heating to 280 °C under an air atmosphere. Then, the stabilized nanofibers were converted into nCNFs by performing carbonization at 800 °C in an argon environment. The SEM image of nCNFs (Fig. 1a) clearly shows the formation of well-defined 1D nanostructures having relatively thinner diameters in the range of 200–350 nm. The X-ray photoelectron spectroscopy (XPS) spectrum of nCNFs displays that they consist of carbon (C), nitrogen (N) and oxygen (O) species (Fig. 1b). The chemical composition of nCNFs was quantified by elemental (CHNS–O) analysis showing the presence of 71.3% C, 14.4% N, 13.2% O and 1.1% H. The powder X-ray diffraction (XRD) pattern of nCNFs exhibits a wide interlayer distance between the graphene sheets (002 planes) which can be attributed to the typical feature of turbostratic carbon (Fig. 1c).33 Not only the extent of N-doping but also the chemical nature of the nitrogen present in the structure of nCNFs plays a vital role in the electrocatalysis. Thus, we conducted high resolution XPS of nCNFs to determine different N species both qualitatively and quantitatively. The deconvoluted N 1s spectrum of nCNFs (Fig. 1d) clearly reveals the presence of four different N species including pyridinic (397.9 eV), nitrile (399.6 eV), quaternary (400.9 eV) and oxidized (403.0 eV) nitrogen.34 Finally, we estimated the surface area of nCNFs to be 63.4 m2 g−1 using BET analysis (Fig. S2†).
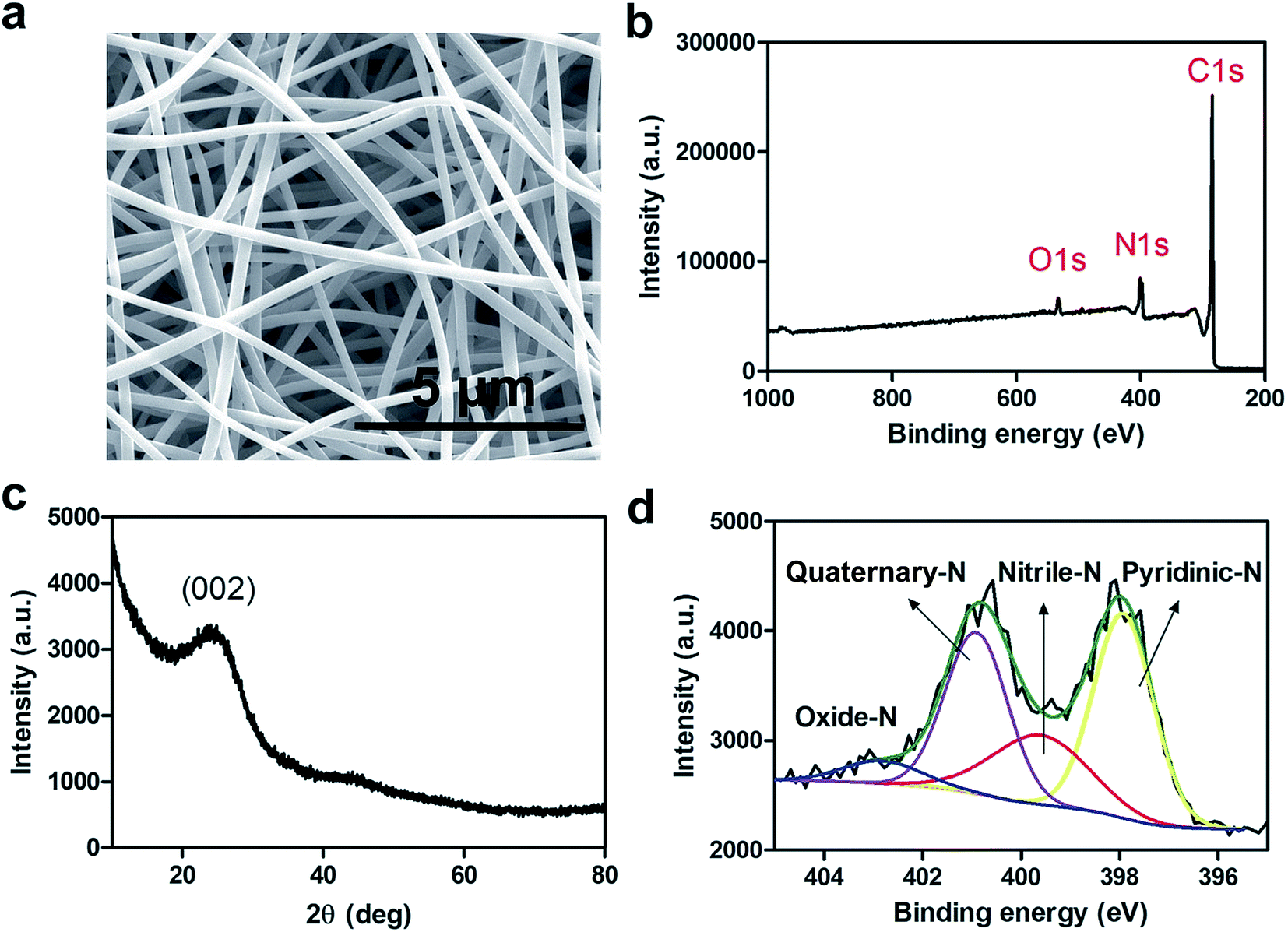 | ||
| Fig. 1 SEM image of nCNFs produced at 800 °C (a). Survey XPS spectrum (b), XRD spectrum (c) and deconvoluted XPS spectrum (d) of N1s of nCNFs. | ||
ALD was utilized to grow well-defined and monodisperse Co3O4 nanoparticles on nCNFs. Cobaltocene was used as the cobalt precursor while ozone was utilized as the reactant gas to grow Co3O4 nanoparticles. The cobalt precursor was preheated to 70 °C to produce and maintain the vapor pressure of the organometallic precursor. The deposition of Co was carried out at 230 °C. In order to investigate systematically the impact of the number of Co ALD cycles on electrocatalytic activity, we prepared three different electrocatalysts by performing 50, 100 and 150 cycles of Co deposition hereafter referred to as Co50@nCNFs, Co100@nCNFs and Co150@nCNFs, respectively.
The growth of discrete Co3O4 nanoparticles on nCNFs was confirmed by transmission electron microscopy (TEM) for Co100@nCNFs (Fig. 2a), Co50@nCNFs (Fig. S3a†) and Co150@nCNFs (Fig. S3b†). Energy dispersive X-ray spectroscopy-scanning TEM (EDS-STEM) revealed clearly the presence of Co species on nCNFs (Fig. S4a and b†). Moreover, the existence of Co species on nCNFs was verified by XPS (Fig. S5†). High resolution TEM (HRTEM) exhibited the crystalline spinel structure of Co3O4 nanoparticles for all catalysts (Fig. 2b, S3c and S3d†).18 Co100@nCNF and Co150@nCNF samples analyzed by XRD showed a weak signal in the range of 35–40° (Fig. S6†) which can be attributed to the (311) lattice phase of Co3O4 (ref. 35) while Co50@nCNFs did not show any recognizable peaks (Fig. S6†). Owing to the low loading and small size of the nanoparticles, we were not able to observe other distinct signals associated with the crystalline phases of Co3O4. ALD for 50 cycles produced Co3O4 nanoparticles with an average size of ∼3 nm. Increasing the number of deposition cycles to 100 did not increase the Co3O4 nanoparticle size significantly but the increase of the population of ∼3 nm nanoparticles was noticeably observed. On the other hand, deposition for 150 cycles produced Co3O4 nanoparticles with an average size of ∼5 nm. These observations were further supported by inductively coupled plasma-mass spectroscopy (ICP-MS) measurements. Co loadings of 0.1%, 0.2% and 0.35% were determined for Co50@nCNFs, Co100@nCNFs and Co150@nCNFs, respectively.
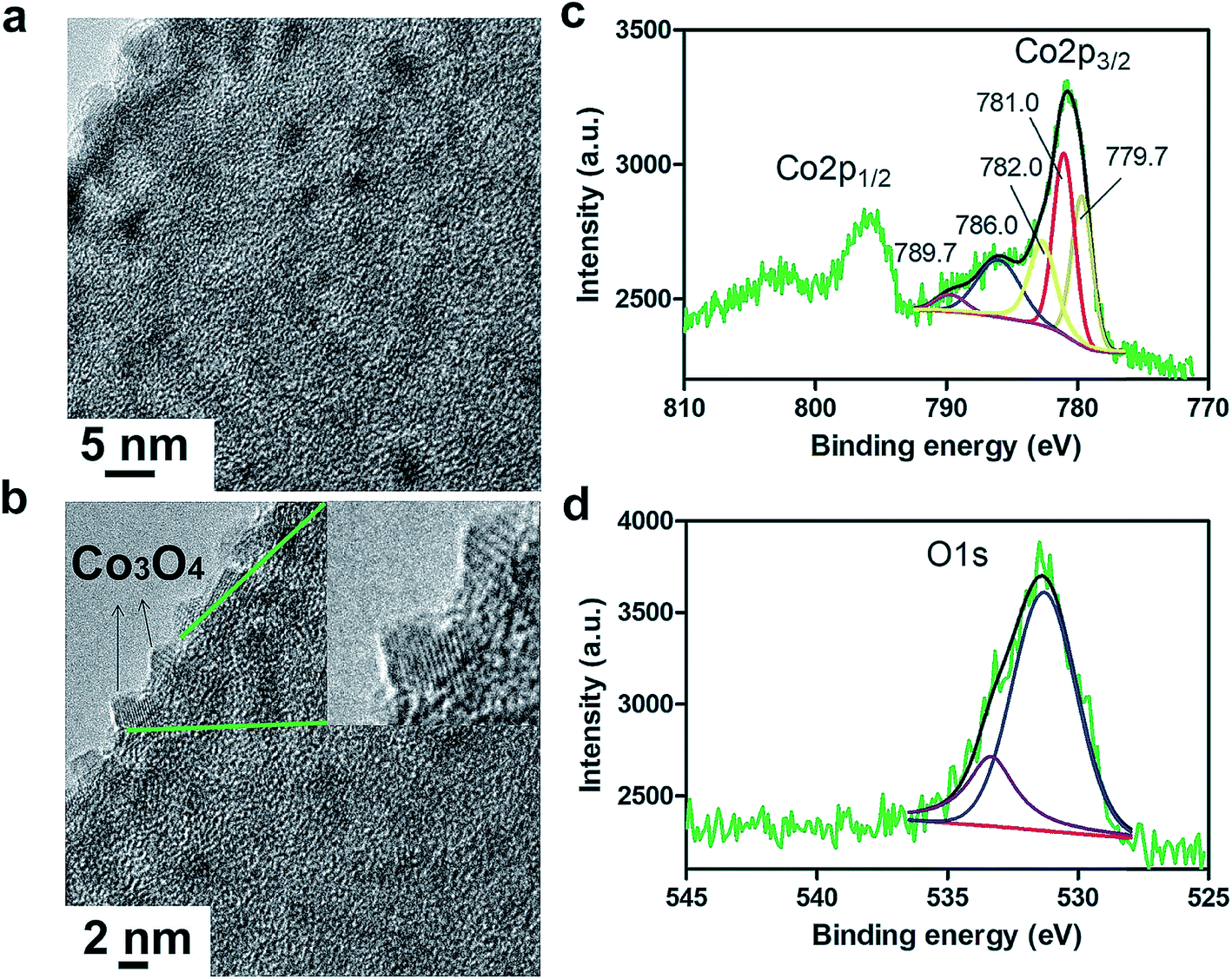 | ||
| Fig. 2 TEM (a) and HRTEM (b) images of Co100@nCNFs. The inset shows the crystalline spinel structure of Co3O4 nanoparticles. Deconvoluted XPS spectra of Co2p (c) and O1s (d) for Co100@nCNFs. | ||
To gain more insights into the chemical composition of the as-deposited Co3O4 nanoparticles, we conducted high resolution XPS for Co (Fig. 2c, S7a and S7b†) and O (Fig. 2d, S7c and S7d†). The deconvolution of the Co2p XPS spectrum in Fig. 2c shows spin–orbit splitting into 2p1/2 and 2p3/2 components with shakeup peaks displaying mixed oxidation states of Co2+/Co3+.35 Since the 2p3/2 signal has higher intensity, it was chosen for curve fitting and qualitative analysis. The satellite lines can be used to distinguish between Co2+ and Co3+ chemical states. Pure Co2+ typically shows peaks at 786 and 790 eV whereas Co3+ generally shows a peak at 790 eV. Binding energies at 779.7 and 781.0 eV correspond to Co3+ and Co2+ species, respectively, and their shakeup signals emerge at 786.0 and 789.7 eV.35 Likewise, the O 1s peak at 531.3 eV (Fig. 2d) corresponds to the lattice oxygen in the Co3O4 spinel structure.35 ALD studies on growth of CoOx films using cobaltocene and ozone have shown formation of mainly polycrystalline Co3O4 structures up to a deposition temperature of about 285 °C.36 In these ALD studies, very low XRD peak intensities even after 1000 cycles of Co deposition have also been observed.36 Overall, our results confirm the formation of cobalt oxide with a dominantly Co3O4 chemical structure which is consistent with the literature.
To assess and compare the ORR catalytic activities of our three electrocatalysts, we first loaded nCNFs, Co50@nCNFs, Co100@nCNFs and Co150@nCNFs (with the same mass loading) on glassy carbon electrodes.
Fig. 3a shows CVs measured at the nCNFs and Co100@nCNFs in N2 and O2 saturated KOH solution. An increase in the current density at the nCNFs shows catalytic activity towards the ORR; however, after the Co deposition onset potential of the ORR shows an anodic shift of ca. 120 mV. Thus, this clearly proves the catalytic effect of Co towards the ORR. The CV obtained at the Co50@nCNFs was cathodic to Co100@nCNFs whereas it is almost similar to that of the Co150@nCNFs in O2 saturated KOH (Fig. 3a and S8†). Results of the ORR are summarized in Table 1 and compared with those of a standard 20 wt% Pt/C catalyst. The nCNF catalyst without Co3O4 showed an ORR onset potential of 0.75 V vs. RHE (reversible hydrogen electrode) with the number of electrons of ∼2.0, whereas after deposition of Co3O4 nanocrystals, the onset potential shifted anodically to 0.87 V vs. RHE and the number of electrons turned out to be 4.0. Production of H2O2 can drastically decrease the efficiency of energy devices due to its strong oxidizing nature which harms the electrolyte and the electrode surface.37 Thus, the path of the ORR (i.e. the ORR mechanism) and its kinetics is the prime key to selecting the catalyst. Furthermore, to analyze the ORR mechanism, RDE experiments were used, and the results are shown in Fig. 3b and S9.† In all these catalysts, loading densities (i.e. ≥350 μg cm−1) were well above the limit required to promote the four-electron ORR (i.e. 200 μg cm−1).38
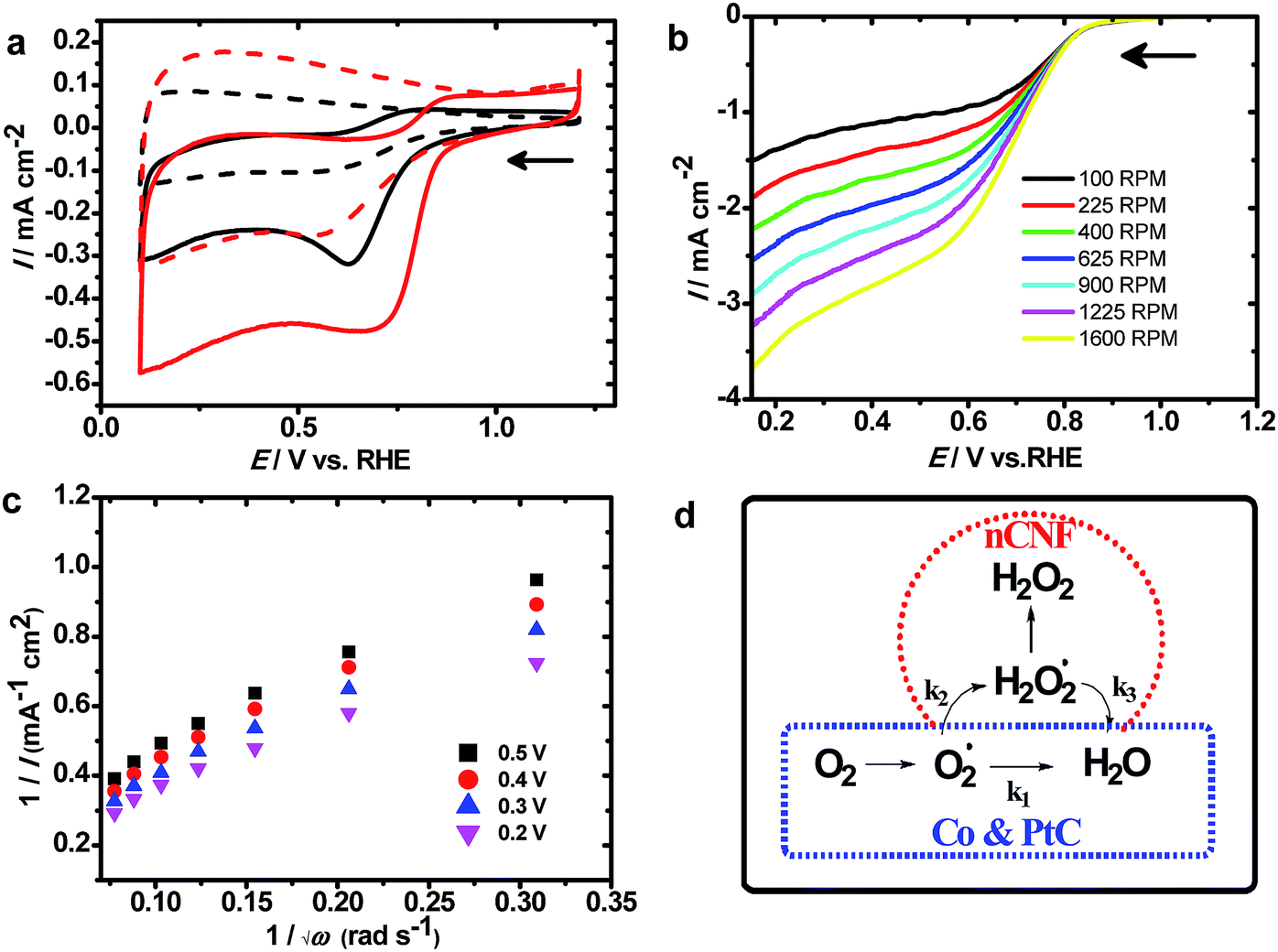 | ||
| Fig. 3 CVs of oxygen reduction obtained at the Co100@nCNFs (red) and nCNFs (black) in N2 (dotted lines) and O2-saturated (solid lines) 0.1 M KOH solution at a scan rate of 10 mV s−1 (a), RDE measurements at the Co100@nCNFs from 100 to 1600 rpm in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 (b), the Koutecky–Levich plot at 0.5, 0.4, 0.3, and 0.2 V vs. RHE (using data from (b)) (c), and schematic of the ORR mechanism (d). | ||
| Sample | Onset potential V vs. RHE | No. of electrons (n) | Tafel slope, mV dec−1 | E 1/2 at 400 rpm mV vs. RHE |
|---|---|---|---|---|
| nCNFs | 0.75 | 2.2 | 125 | 646 |
| Co50@nCNFs | 0.87 | 3.9 | 174 | 705 |
| Co100@nCNFs | 0.87 | 3.95 | 119 | 700 |
| Co150@nCNFs | 0.87 | 4.0 | 158 | 680 |
| Pt/C | 0.98 | 3.99 | 121 | 900 |
The calculation details are given in the ESI.† Based on the RDE plots, the K–L plot (Fig. 3c) is derived to estimate the number of electrons involved in the ORR. This can reveal the ORR kinetics; furthermore, the production of H2O2 can be determined from the K–L plot. The ORR kinetics can follow serial or parallel pathways as shown in the mechanism (Fig. 3d). In all these catalysts, ORR kinetics proceeds through a serial pathway (i.e. k1 = k2 and k3 = 0; Fig. 3d, blue dashed line) resulting in ∼4 electron oxygen reduction like the Pt/C (Fig. S9 and S10†) while in the case of nCNFs, it follows a parallel pathway (i.e. k2 = 2 × k1 and k3 = 0; Fig. 3d, red dashed line).38 These results clearly show that cobalt oxide-modified nCNFs follow a similar ORR mechanism to the Pt/C. Although the onset potential of cobalt-modified nCNFs is slightly cathodic to Pt/C, their low cost and abundant availability can replace such a noble metal like Pt for the ORR. In addition to the ORR mechanism, ORR kinetics is equally important to evaluate the efficiency of the catalyst towards the ORR. Among the Co50@nCNFs, Co100@nCNFs, and Co150@nCNFs, Co100@nCNFs are kinetically enhanced with a Tafel slop of 119 mV dec−1 which indicates that step one is the rate determining step in the ORR which is comparable with the standard Pt/C. Thus, overall ORR catalysis is efficient at the Co100@nCNFs. A dual-site mechanism has been proposed for cobalt–polypyrrole/C39 and Co3O4/N-rmGO18 where peroxide forms by O2 reduction at Co–N–C sites which were further reduced by cobalt oxides to OH−. We expect a similar mechanism at the ALD deposited-Co oxides on the nCNFs.
Bifunctional catalysts that are light weight and can promote efficient catalysis are always in demand for energy devices to make them light weight. Thus, optimization of the nanoparticle size and catalyst amount or weight is one of the key aspects for such bifunctional catalysts. Conventionally, it has been accepted that 10% efficient solar water-splitting devices should operate at 10 mA cm−2 and below ∼0.45 V overpotential for the overall OER and hydrogen evolution reaction (HER).40 In order to meet the requirement of a higher overpotential for the OER than the HER, highly active catalysts need to be developed for the OER. To determine the catalytic activities of these different ALD-deposited cobalt oxides on the nCNFs towards the OER, LSV was performed in the N2 saturated KOH solution (Fig. 4a). A small value of the Tafel slope of 35 mV dec−1 (Fig. 4b) was obtained for the Co100@nCNFs, proving their efficient catalytic activity among these three Co-modified catalysts. The overpotential towards the OER was calculated as η = E vs. RHE – 1.23 V.41 The important analysis results of the OER such as the onset overpotential, potential to reach this 10 mA cm−2 (for the OER) based on the geometric area (jg), turnover frequency (TOF) and mass activity were compared and are summarized in Table 2 (calculation details are elaborated in the ESI†). Fig. 4a shows a cathodic shift of 250 mV in the OER potential for Co100@nCNFs compared to the Co50@nCNFs and a slight anodic shift (∼30 mV) compared to the Co150@nCNFs at the 10 mA cm−2. However, comparison of mass activity at 0.55 V vs. RHE obtained at the Co100@nCNFs shows the highest current per gram (the cobalt catalyst weight measured from the ICP-MS is used for the mass activity calculations) against the Co50@nCNFs and the Co150@nCNFs (Fig. 4c). The OER mechanism at the cobalt oxide-modified nCNFs has been schematically presented in Fig. 4d.42
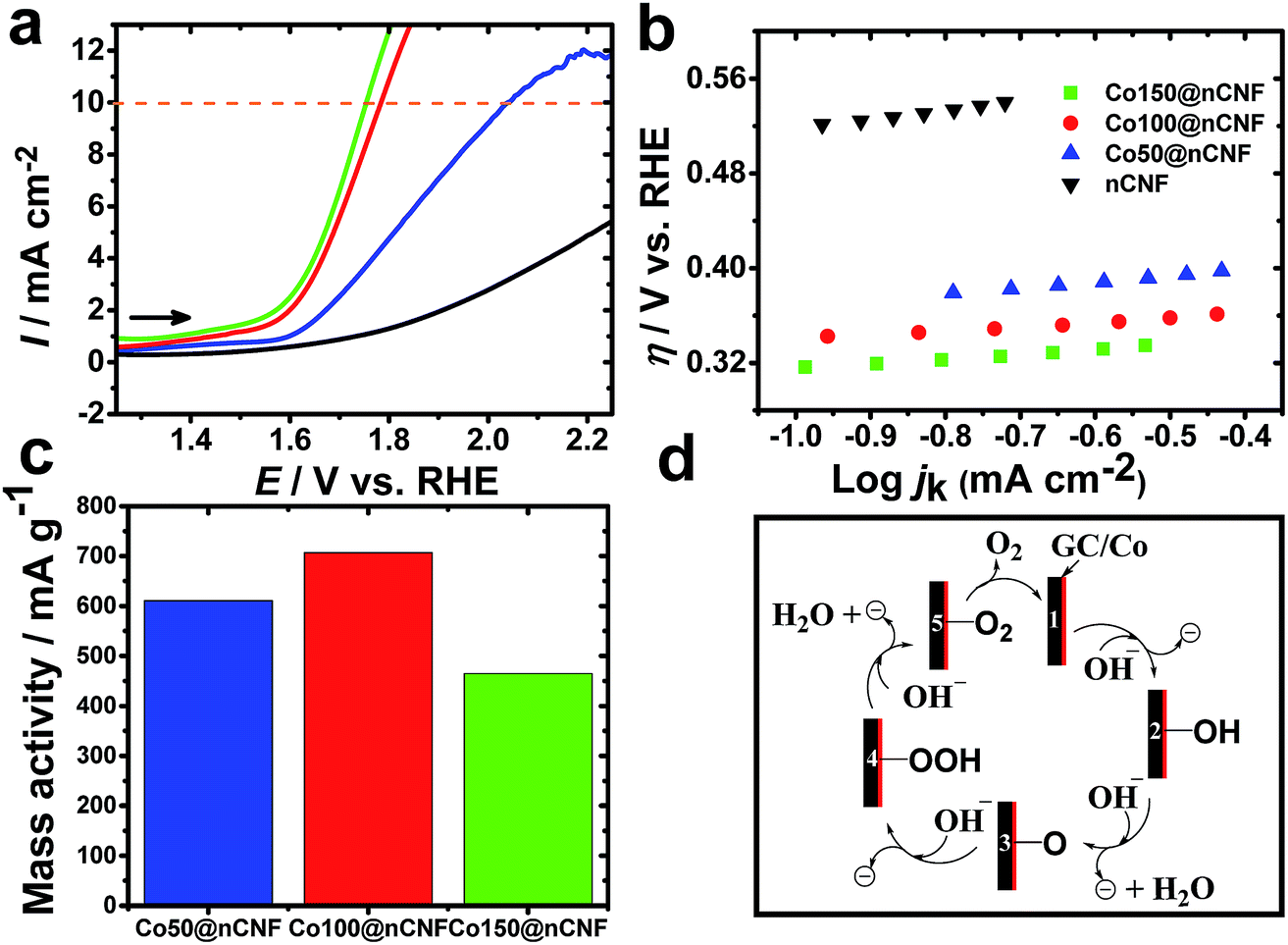 | ||
| Fig. 4 LSVs of the OER obtained at the nCNFs (black), Co50@nCNFs (blue), Co100@nCNFs (red), and Co150@nCNFs (green) in N2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 (a), Tafel plots of the nCNFs (black), Co50@nCNFs (blue), Co100@nCNFs (red), and Co150@nCNFs (green) (using data from (a)) (b), the mass activity of Co50@nCNFs (blue), Co100@nCNFs (red), and Co150@nCNFs (green) normalized with the amount of the Co loading (c), and schematic of the OER mechanism at the cobalt oxide-modified electrodes (d). | ||
| Sample | Onset overpotential η/V | η@10 mA cm−2/V | Tafel slope/mV dec−1 | j g@η = 0.55 V/mA cm−2 | TOF per active site/s−1 | Mass activity/mA g−1 |
|---|---|---|---|---|---|---|
| nCNFs | ∼0.57 | — | 76 | 1.17 | — | — |
| Co50@nCNFs | 0.38 | 0.8 | 51 | 4.32 | 0.118 | 610.67 |
| Co100@nCNFs | 0.34 | 0.55 | 35 | 10 | 0.137 | 706.80 |
| Co150@nCNFs | 0.32 | 0.52 | 40 | 11.5 | 0.090 | 464.46 |
As postulated by Yeo et al., Co2+/Co3+ and Co4+ states of cobalt influence the OER mechanism due to differences in the electrophilicity of adsorbed O (Fig. 4d, step 3). Furthermore, the more cationic state of Co can likely promote the deprotonation of the OOH species resulting in O2 formation via the electron withdrawing inductive effect (Fig. 4d, steps 4 and 5).42 The XPS results clearly show a decrease in the Co3+/Co2+ ratio i.e. an increase in the Co2+ with an increase in the number of cycles of the Co ALD deposition, which might be one of the reasons for the enhanced OER catalysis of Co100@nCNFs compared to the Co50@nCNFs.17 Moreover, the less loading of cobalt active sites in the Co50@nCNFs might also influence the poor catalytic performance in terms of the OER activity. Notably, the higher OER activity of Co100@nCNFs than Co150@nCNFs can be explained by the presence of smaller particle size Co3O4 nanocrystals (∼3 nm) with a higher surface on the Co100@nCNF electrocatalyst.43 The TOF value determined at an overpotential of 550 mV for the Co100@nCNFs was ∼0.14 s−1 (Table 2) which is ca. 3 and ca. 15-fold higher than those of bulk Co (∼0.05 s−1)42 and the standard state-of-the-art IrOx (0.0089 s−1) catalyst.44 The TOF value shows the trend of Co100@nCNFs > Co50@nCNFs > Co150@nCNFs thus further proving the importance of ALD optimization of Co catalysts for the OER. In comparison with Co3O4/N-rGO, the overpotential at 10 mA cm−2 is cathodic (i.e. ∼70 mV) at the Co100@nCNFs and close to that of RuO2 (ref. 44) proving its better catalytic activity towards the OER. The overall bifunctional catalytic activity of Co100@nCNFs for the ORR and OER is compared and summarized in Table S1.† We can clearly observe that the overall electrocatalytic activity of Co100@nCNFs is comparable with that of other Co-based catalysts. The performance of Co100@nCNFs can be further enhanced by producing nCNFs with higher conductivities.
Conclusions
In summary, we utilized two versatile nanofabrication techniques namely electrospinning and ALD to synthesize a series of novel bifunctional electrocatalysts. Well-defined 1D electrospun carbon nanofibers were produced using electrospinning. The as-prepared conductive 1D carbon support was decorated with highly monodisperse Co3O4 nanocrystals. The ORR and OER activities of these catalysts were measured and compared under basic conditions. 100 cycles of Co deposition resulted in the formation of ∼3 nm Co3O4 nanocrystals which showed the highest catalytic activity towards both the ORR and OER. Co100@nCNFs exhibited high ORR performance with a positive half-wave potential of 700 mV approaching that of commercial Pt/C. Co@nCNFs also exhibited an onset potential of 0.87 V with a low Tafel slope of 119 mV dec−1 for the ORR. Likewise, Co100@nCNFs showed remarkable catalytic activity in the OER. The TOF value determined at an overpotential of 550 mV for the Co100@nCNFs is ∼0.14 s−1 which is ca. 3 and ca. 15-fold higher than those of bulk Co (∼0.05 s−1) and the standard state-of-the-art IrOx (0.0089 s−1) catalyst, respectively. This work will open new possibilities for fabrication of inexpensive non-noble metal materials in highly controlled manner for applications as bifunctional ORR/OER electrocatalysis.Conflicts of interest
There are no conflicts to declare.References
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. K. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS.
- A. A. Gewirth, J. A. Varnell and A. M. DiAscro, Chem. Rev., 2018, 118, 2313–2339 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W. G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225 CrossRef CAS.
- S. Sui, X. Y. Wang, X. T. Zhou, Y. H. Su, S. Riffatc and C. J. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- N. Y. Cheng, Q. Liu, A. M. Asiri, W. Xing and X. P. Sun, J. Mater. Chem. A, 2015, 3, 23207–23212 RSC.
- L. L. Feng, G. T. Yu, Y. Y. Wu, G. D. Li, H. Li, Y. H. Sun, T. Asefa, W. Chen and X. X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- P. Z. Chen, K. Xu, Z. W. Fang, Y. Tong, J. C. Wu, X. L. Lu, X. Peng, H. Ding, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed.
- K. Xu, P. Z. Chen, X. L. Li, Y. Tong, H. Ding, X. J. Wu, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- X. H. Deng and H. Tuysuz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- Y. Y. Liang, H. L. Wang, P. Diao, W. Chang, G. S. Hong, Y. G. Li, M. Gong, L. M. Xie, J. G. Zhou, J. Wang, T. Z. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 15849–15857 CrossRef CAS PubMed.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- Y. C. Wang, T. Zhou, K. Jiang, P. M. Da, Z. Peng, J. Tang, B. A. Kong, W. B. Cai, Z. Q. Yang and G. F. Zheng, Adv. Energy Mater., 2014, 4, 1400696 CrossRef.
- H. Y. Jin, J. Wang, D. F. Su, Z. Z. Wei, Z. F. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Y. Tong, P. Chen, T. Zhou, K. Xu, W. Chu, C. Wu and Y. Xie, Angew. Chem., 2017, 56, 7121–7125 CrossRef CAS PubMed.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- M. A. Khalily, H. Eren, S. Akbayrak, H. H. Susapto, N. Biyikli, S. Ozkar and M. O. Guler, Angew. Chem., 2016, 55, 12257–12261 CrossRef CAS PubMed.
- M. A. Khalily, M. Yurderi, A. Haider, A. Bulut, B. Patil, M. Zahmakiran and T. Uyar, ACS Appl. Mater. Interfaces, 2018, 10, 26162–26169 CrossRef CAS PubMed.
- S. T. Christensen, H. Feng, J. L. Libera, N. Guo, J. T. Miller, P. C. Stair and J. W. Elam, Nano Lett., 2010, 10, 3047–3051 CrossRef CAS PubMed.
- S. F. Xie, S. I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. G. Wang, M. J. Kim, Z. X. Xie, M. Mavrikakis and Y. N. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- J. L. Lu, J. W. Elam and P. C. Stair, Acc. Chem. Res., 2013, 46, 1806–1815 CrossRef CAS PubMed.
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef.
- J. J. Xue, J. W. Xie, W. Y. Liu and Y. N. Xia, Acc. Chem. Res., 2017, 50, 1976–1987 CrossRef CAS PubMed.
- S. J. Peng, G. R. Jin, L. L. Li, K. Li, M. Srinivasan, S. Ramakrishna and J. Chen, Chem. Soc. Rev., 2016, 45, 1225–1241 RSC.
- S. Abouali, M. A. Garakani, B. Zhang, Z. L. Xu, E. K. Heidari, J. Q. Huang, J. Q. Huang and J. K. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 13503–13511 CrossRef CAS PubMed.
- S. Surendran, S. Shanmugapriya, A. Sivanantham, S. Shanmugam and R. K. Selvan, Adv. Energy Mater., 2018, 8, 1800555 CrossRef.
- N. Wu, Y. D. Wang, Y. P. Lei, B. Wang, C. Han, Y. Z. Gou, Q. Shi and D. Fang, Sci. Rep., 2015, 5, 17396 CrossRef CAS PubMed.
- D. S. Yang, S. Chaudhari, K. P. Rajesh and J. S. Yu, Chemcatchem, 2014, 6, 1236–1244 CrossRef CAS PubMed.
- M. Einert, C. Wessel, F. Badaczewski, T. Leichtweiss, C. Eufinger, J. Janek, J. Y. Yuan, M. Antonietti and B. M. Smarsly, Macromol. Chem. Phys., 2015, 216, 1930–1944 CrossRef CAS.
- J. Kim, T. Iivonen, J. Hamalainen, M. Kemell, K. Meinander, K. Mizohata, L. D. Wang, J. Raisanen, R. Beranek, M. Leskela and A. Devi, Chem. Mater., 2017, 29, 5796–5805 CrossRef CAS.
- M. Diskus, O. Nilsen and H. Fjellvag, Chem. Vap. Deposition, 2011, 17, 135–140 CrossRef CAS.
- D. Harman, J. Gerontol., 1956, 11, 298–300 CrossRef CAS PubMed.
- R. Mishra, B. Patil, F. Karadas and E. Yilmaz, ChemistrySelect, 2017, 2, 8296–8300 CrossRef CAS.
- T. S. Olson, S. Pylypenko, P. Atanassov, K. Asazawa, K. Yamada and H. Tanaka, J. Phys. Chem. C, 2010, 114, 5049–5059 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- T. G. U. Ghobadi, B. Patil, F. Karadas, A. K. Okyay and E. Yilmaz, ACS Omega, 2017, 2, 8319–8329 CrossRef.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CrossRef CAS.
- T. T. Zhang, C. S. He, F. Z. Sun, Y. Q. Ding, M. C. Wang, L. Peng, J. H. Wang and Y. Q. Lin, Sci. Rep., 2017, 7, 43638 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00330k |
| This journal is © The Royal Society of Chemistry 2019 |