
Pyrene-based aggregation-induced emission luminogens and their applications†
Md. Monarul
Islam
 ab,
Zhen
Hu
a,
Qingsong
Wang
a,
Carl
Redshaw
c and
Xing
Feng
*a
ab,
Zhen
Hu
a,
Qingsong
Wang
a,
Carl
Redshaw
c and
Xing
Feng
*a
aGuangdong Provincial Key Laboratory of Functional Soft Condensed Matter, School of Material and Energy, Guangdong University of Technology, Guangzhou 510006, P. R. China. E-mail: hyxhn@sina.com
bChemical Research Division, Bangladesh Council of Scientific and Industrial Research (BCSIR), Dhanmondi, Dhaka-1205, Bangladesh
cDepartment of Chemistry & Biochemistry, University of Hull, Cottingham Road, Hull, Yorkshire HU6 7RX, UK
First published on 20th March 2019
Abstract
“Unity is force” – Aesop. It is a common phenomenon that traditional chromophores exhibit high fluorescence in dilute solutions, yet luminescence is quenched at high concentrations or in the aggregate state, i.e. “aggregation-caused quenching” (ACQ). Tang reported the unusual photophysical observation that luminogens can exhibit weak or no fluorescence in solution, yet they are highly emissive in the aggregate or solid state; this is defined as aggregation-induced emission (AIE). The discovery of AIE helped solve the ACQ effect in traditional luminophores. Pyrene is an important polycyclic aromatic hydrocarbon (PAH), which exhibits very different photophysical behavior in solution versus the aggregate state, and the ACQ effect has played a dominant role in pyrene chemistry. The ACQ effect is harmful for some practical applications and is a challenge in organic light-emitting diodes (OLEDs) and light-emitting electrochemical cells, for which the effect is more severe in the solid state. Thus, how to overcome the ACQ effect observed in pyrene chemistry still remains a challenge. In this review, we discuss how following basic AIE mechanisms such as the restriction of intramolecular motion (RIM), excited-state intramolecular proton transfer (ESIPT), and twisted intramolecular charge transfer (TICT), can transform pyrene-based ACQ luminogens to AIE luminogens with excellent optical properties. Furthermore, prospective applications of pyrene-based AIEgens are discussed, as is the potential for designing new organic functional materials.
 Md. Monarul Islam | Md. Monarul Islam is affiliated with the Chemical Research Division (CRD), BCSIR Laboratories, Dhaka. Dr Islam graduated (MS) with an undergraduate degree (BS) in Chemistry from the University of Dhaka and received his PhD (2015) from Saga University (Japan) under the supervision of Prof. Takehiko Yamato. He is interested in initiating science leadership programs for young scientists in Bangladesh to improve research environments and motivation. He is a founding convener of the Young Scientists Committee, BCSIR formed by the BCSIR office and a founding member of the National Young Academy of Bangladesh (NYAB). His research is mainly focused on the development of new functional organic molecules and macrocycles for organic optoelectronics and biological applications. At present, he has been working in Dr Xing Feng's research group (Guangdong University of Technology, GDUT) as a Visiting Scholar since 2018. |
 Zhen Hu | Zhen Hu received his BS degree from Nanchang Hangkong University in 2017. He is currently studying for his master's degree at Guangdong University of Technology under Prof. Wu and Dr Xing Feng's supervision. His research is mainly on the design and synthesis of new organic fluorescent molecules. |
 Qingsong Wang | Qingsong Wang received his BS degree from Gannan Normal University in 2018. He is currently studying for his master's degree at Guangdong University of Technology under Dr Xing Feng's supervision. His research is mainly on the development of new pyrene-based fluorescent molecules. |
 Carl Redshaw | Carl Redshaw is Chair of Inorganic Materials at the University of Hull. He received his PhD at Newcastle University, and was a Welch Fellow at the University of Texas (Austin) and a postdoc with the late Sir G. Wilkinson at Imperial College (IC). Following 2 years at Durham University and a further stint at IC, he was awarded a Leverhulme Special Research Fellowship and moved to UEA as Lecturer, Senior Lecturer and Reader. He was appointed Professor of Inorganic Materials at Hull in 2012. He has been Visiting Professor at the Shanghai Institute of Organic Chemistry (SIOC) and the Institute of Chemistry (ICCAS), Beijing and Guest Professor at Sichuan Normal University in Chengdu. He is currently 100 Talents Professor at Northwest University in Xi’an (China) and Guest Professor at the National Institute of Technology, Akashi (Japan). His research interests include calixarene chemistry, polymerisation catalysis and the use of metal-based anti-cancer/imaging agents. |
 Xing Feng | Xing Feng received his PhD (2013) from Saga University (Japan) under the supervision of Prof. Takehiko Yamato. Then, he joined the School of Beijing Institute of Graphic Communication (BIGC) as a lecturer. He was an established academic researcher in BIGC and held a position at the Ministry of Science and Technology of China, being in charge of the administration of the National Program for International Science and Technology Cooperation in 2015. In 2016 he joined Professor Ben Zhong Tang's group in the Department of Chemistry at the Hong Kong University of Science and Technology (HKUST) as postdoctoral fellow. In 2017, he moved to the School of Materials and Energy, Guangdong University of Technology (GDUT) in Guangzhou as an associate professor. His research interests include the development of novel fluorescence materials and fluorescent bioprobes for application in organic electronics. He was awarded with the Chinese Government Award for Outstanding Self-Financed Students Abroad by the China Scholarship Council in 2013. |
1. Introduction
1.1. Chemistry of pyrene and its derivatives
Pyrene is a representative member of the polycyclic aromatic hydrocarbons (PAHs), and consists of four fused benzene rings with extensive π-electron delocalization. Since Weitzenböck first synthesized pyrene 1 in the laboratory in 1913, pyrene 1 (Fig. 1) and its derivatives have attracted much attention from scientists, primarily due to their blue emissive property, good hole-transporting ability, and their application in organic electronics, such as organic light-emitting diodes (OLEDs), light-emitting electrochemical materials, solar cells etc.1–3 Salient features of pyrene are that its π-system does not require protection during functionalization,4 its moderately long fluorescence lifetime, and excimer formation.5 However, there are two major drawbacks of pyrenes when used in organic fluorescent materials, (i) the absorption and emission of the pyrene fluorophore lies in the near-ultraviolet (UV) region, whereas a good optoelectronic device emits in the visible region and (ii) excimer formation occurs in concentrated solutions or in the solid state, which decreases or quenches the fluorescence intensity, with a large red-shifted emission.6–8 This common phenomenon is defined as aggregation-induced quenching (ACQ). For example, PAH-based luminogens like pyrene and its derivatives emit deep blue emission with high quantum yields at low concentrations, which has limited its application as an efficient solid emitting material.2,9 | ||
| Fig. 1 The molecular structure of pyrene 1. | ||
The intrinsic properties of pyrene alone do not make it a good candidate for light-emitting devices; however, the ACQ effect of pyrene has been overcome by introducing bulky groups with long alkyl chains which enhances the fluorescence quantum efficiency in OLEDs and also reduces the aggregation of the final material.2 For example, a 1,3,6,8-substituted pyrene-based conjugated molecule such as 1,3,6,8-tetrakis(4-butoxyphenyl)pyrene exhibits an active emitting layer in simple solution-processed OLEDs producing deep blue emission (CIE = 0.15, 0.18) and maximum efficiencies and brightness levels of 2.56 cd A−1 and 5000 cd m−2.11
Experimentally, the planar structure of pyrene prefers to adopt a close-packing form in the aggregation state. The π–π stacking interaction prompts the formation of detrimental species such as excimers, resulting in the observed ACQ effect. Pyrene-based highly efficient emitting materials can be obtained by introducing various substituents at the 1-, 3-, 6-, and 8-positions of the pyrene core.2 Generally, these four positions are significantly more active than the other positions (the 4-, 5-, 9-, and 10-positions or the 2- and 7-positions), and it is not easy to directly substitute at these latter positions versus the 1-, 3-, 6-, and 8-positions.9 Theoretically, the energy of the 1-, 3-, 6-, and 8-positions are lower by 8.8 kcal mol−1 than the 2- and 7-positions, and the substitution order follows the trend 1- > 8- > 6- > 3-.12 Pyrene can be easily modified at the 1-, 3-, 6- and 8-positions to form mono-, di-, tri-, and tetra-substituted derivatives13 in a manner that can hinder π–π interactions to effectively suppress the excimer formation.14 The introduction of bulky groups at the 1-, 3-, 6- and 8-positions of pyrene leads to a shift in the main absorption and emission bands into the visible region due to the extension of the π-conjugation system. On the other hand, as the nodal plane of pyrene passes through its carbon atoms at the 2,7-positions in the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), substituents at these positions will interact weakly with the central core.15 For example, the uridine ring exhibited a weak electronic interaction with the pyrene core when it was substituted at the 2-position instead of the 1-position.16 Thus, our group has previously focused on synthetic methodologies for the functionalization of the pyrene core to suppress the ACQ effect based on its special electronic structure, and numerous novel synthetic routes for the functionalization of the pyrene core have been explored. Examples include tert-butylation, electrophilic substitution, formylation, halogenation, oxidation, and borylation. Moreover, other research groups have also attempted to explore a wide variety of methods including physical and engineering approaches and processes, but the results to date are far from satisfactory.17
1.2. Pyrene-based aggregation-induced quenching luminogens
The design and synthesis of efficient, stable pyrene-based fluorescent materials has been an active topic in the area of OLEDs.10b,18 However, the π-aggregates/excimers in concentrated solution and the solid state,19 lead to the formation of π-aggregates/excimers and subsequent quenching of fluorescence with low photoluminescence (PL) quantum yields (Fig. 2).20,21 As a result, much effort has been devoted to improving the photophysical properties of pyrene.1a,22 Thus far, the pyrene derivatives that have been reported still present some degree of fluorescence quenching in the solid state.6,11 Therefore, the preparation of compounds based on pyrene that emit efficiently in the solid state remains an attractive objective. | ||
| Fig. 2 Chart of the molecular structures of pyrene-based blue emitters with the ACQ feature.19,20 | ||
Research to modulate the photophysical properties of pyrenes to enhance fluorescence both in the solid and (or) in thin film is currently being conducted.23 Using synthetic methods for the functionalization of the pyrene core, 1-substituted, 1,3-di-substituted-, 2,7-di-substituted-, 1,3,6,8-tetrakis-substituted-, 1,3,5,9-substituted-, and 4,5,9,10-substituted-pyrenes have been explored. However, all of above-mentioned compounds are ACQ compounds, because of the low quantum yields in the solid (film) state compared with those in solution. For example, 1,3,6,8-tetraphenylpyrene (TPPY; Fig. 2) is highly fluorescent (Φ ca. 0.9) in solution, and the organic light-emitting field-effect transistor devices (OLEFET) based on TPPY have been shown to exhibit electroluminescence (EL) with an external quantum efficiency of only 0.5% due to aggregation.6,24 Thus, conventional dyes suffering from the ACQ effect may not be suitable for constructing highly efficient non-doped OLEDs. However, materials with non-planar propeller-shaped molecular structures exhibiting aggregation-induced emission (AIE) characteristics may be an ideal choice for non-doped OLEDs.
1.3. Aggregation-induced emission
It is essential for organic luminescent materials to exhibit excellent optoelectronic properties (such as high fluorescence, good electronic/hole mobility etc.) for potential application in organic electronics, especially in the solid state or aggregated state. Tang et al. observed an abnormal photophysical phenomenon in hexaphenylsilole (HPS) in 2001 (Fig. 3), whereby weak emission was present in solution but enhanced emission in the solid state.25 This phenomenon was termed “aggregation-induced emission” (AIE), and the discovery of AIE solved the ACQ effect of traditional luminophores.25 Recently, the AIE concept has been widely accepted in the scientific community, and more than 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 papers on AIE research have now been published, including on siloles,26 tetraphenylethylene (TPE),27 9,10-bis(p-dimethylaminostyryl)-anthracene (9,10-MADSA),28 1-cyano-trans-1,2-bis-(40-methyl biphenyl)ethylene (CN-MBE),29 2,5-diphenyl-1,4-distyryl-benzene (DPDSB) derivatives,30 diphenyldibenzofulvene (DPDBF) derivatives,31 conjugated polymers,32 borondipyrromethene (BODIPY) derivatives,33 triazoles,34 and others.35,36
000 papers on AIE research have now been published, including on siloles,26 tetraphenylethylene (TPE),27 9,10-bis(p-dimethylaminostyryl)-anthracene (9,10-MADSA),28 1-cyano-trans-1,2-bis-(40-methyl biphenyl)ethylene (CN-MBE),29 2,5-diphenyl-1,4-distyryl-benzene (DPDSB) derivatives,30 diphenyldibenzofulvene (DPDBF) derivatives,31 conjugated polymers,32 borondipyrromethene (BODIPY) derivatives,33 triazoles,34 and others.35,36
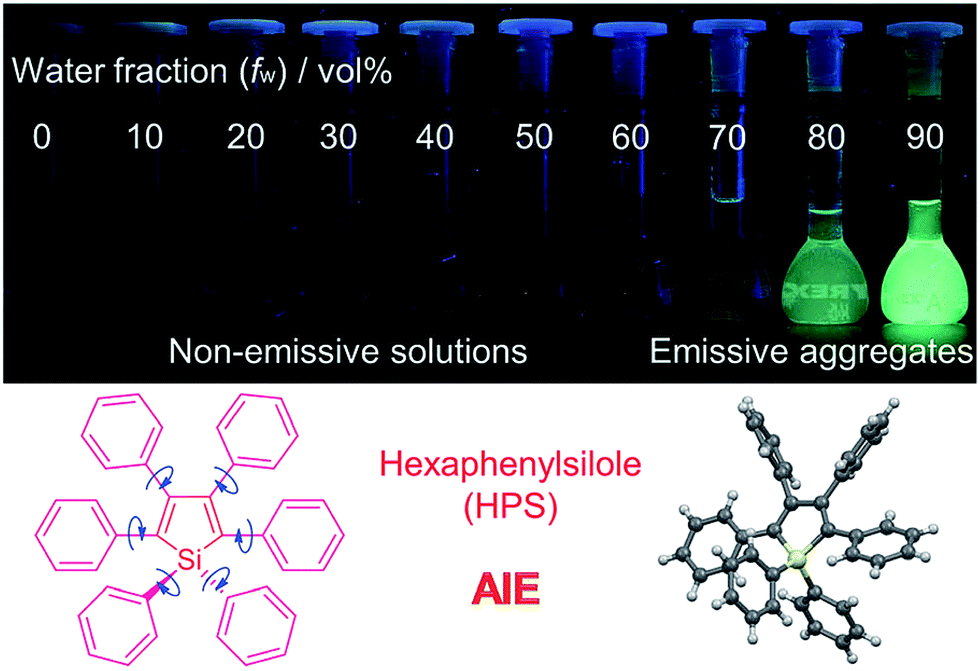 | ||
| Fig. 3 Fluorescence photographs of solutions and suspensions of hexaphenylsilole (HPS; 20 mM) in tetrahydrofuran (THF)/water mixtures with different fractions of water. Reproduced from ref. 25d. Copyright (2014) Wiley-VCH. | ||
Different hypotheses have been put forward for AIE mechanisms, such as J-aggregates, excited-state intramolecular proton transfer (ESIPT), and twisted intramolecular charge transfer (TICT) etc.37 The restriction of intramolecular motion (RIM) was widely accepted to explain the AIE mechanism. Take hexaphenylsilole (HPS) as an example – the six peripheral phenyl rotors were dynamically rotated relative to the central silole, around the C–C or C–Si bond. The rotation of the aromatic rotors generally destroys the excitons in a non-radiative mode in the solution state. Upon aggregate formation, the emission intensity of the AIEgens would be enhanced by the restricted intramolecular rotation (RIR), which blocks the non-radiative pathway for the excitons to decay with the activated radiative transition, thus the radiative decay of the excited state becomes dominant, rendering the luminogens emissive. As a consequence, the fluorescence quantum yields of siloles in aggregates is two orders of magnitude greater than in solution due to the restriction of RIR of the chromophoric compound.25
1.4. Jablonski diagram
A molecule in the excited state can decay in several ways, as shown in the Jablonski diagram (Fig. 4).38 Relaxation to the ground state from these excited states occurs by a number of mechanisms that are either radiation-less, in that no photons are emitted, or involve the emission of a photon. The most likely pathway by which a molecule relaxes back to its ground state is that which gives the shortest lifetime for the excited state, of the order of 10−15 s.38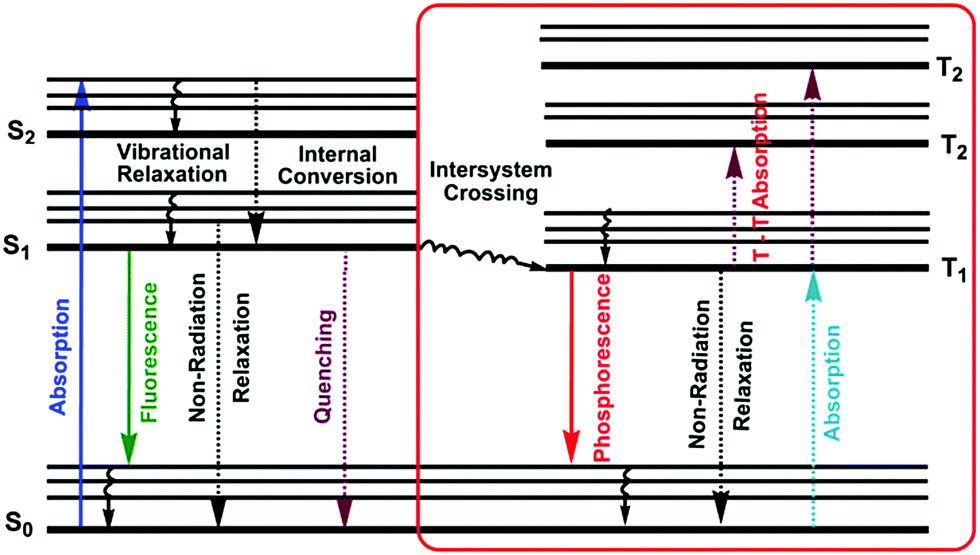 | ||
| Fig. 4 A Jablonski diagram of the relevant photophysical processes in luminescence materials. | ||
By a combination of internal conversions and vibrational relaxations, a molecule in an excited electronic state may return to the ground electronic state without emitting a photon. A final form of radiation-less relaxation is an intersystem crossing in which a molecule in the ground vibrational energy level of an excited electronic state passes into a high vibrational energy level of a lower energy electronic energy state with a different spin state. For example, an intersystem crossing between a singlet excited state (S1), and a triplet excited state (T1). Fluorescence (S1 → S0 + hν) occurs when a molecule in the lowest vibrational energy level of an excited electronic state returns to a lower energy electronic state by emitting a photon.
Phosphorescence (T1 → S0 + hν) is most favorable for molecules that have n → π* transitions, which have a higher probability for an intersystem crossing than do π → π* transitions. Delayed fluorescence results from two intersystem crossings, first from the singlet to the triplet, and then back to the singlet state (S1 → T1 → S1 → S0 + hν).
The intensity of fluorescence or phosphorescence I, is proportional to the amount of radiation from the excitation source that is absorbed and the quantum yield for fluorescence.
| I = kΦ(A0 − A) | (i) |
| A/A0 = 10−εbc | (ii) |
| I = kΦA0(1 − 10−εbc) | (iii) |
| I = 2.303kΦA0εbc | (iv) |
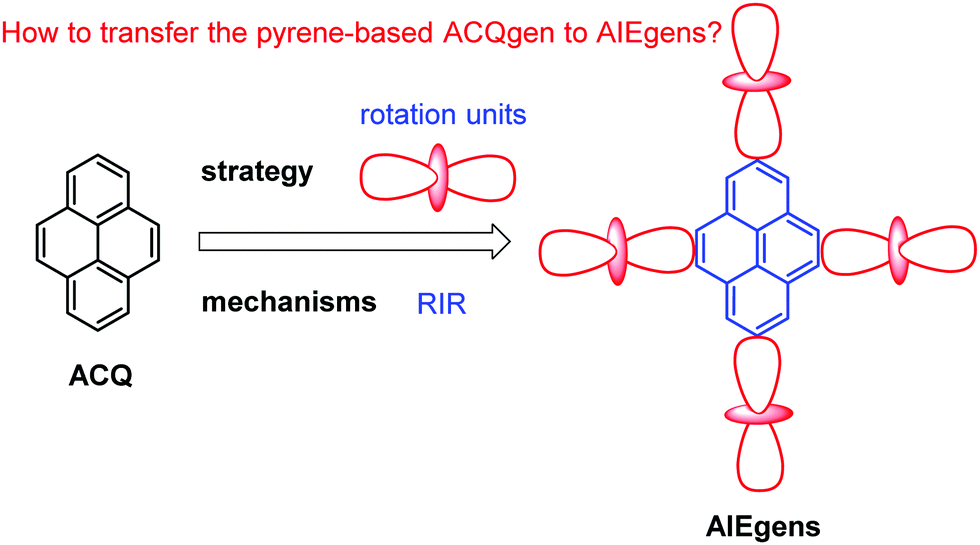
According to the Jablonski diagram, upon aggregate formation, the emission of AIEgen is induced or revitalized by the specific effects of the RIM and the highly twisted molecular conformation that destroys the intermolecular π–π stacking interactions.25,26 We will first carefully elaborate on the working principle of AIE and then demonstrate how to transform traditional ACQgens into new AIEgens, followed by the presentation of a series of new AIEgens derived from the RIM principle (Fig. 5). Finally, we will recap the research efforts devoted toward the exploration of technological applications of the new pyrene-based AIEgens. To understand the mechanisms of AIE phenomena is of great importance in the quest for fundamental knowledge of photophysics, and just as crucial, it will guide our endeavors to design novel pyrene-based AIE luminogens, and to explore practical applications and technological innovations. Since the arrival of the AIE concept in 2001, researchers have shown a persistent thirst for learning what are the real mechanistic causes of the AIE phenomena.25,26 Herein, we will summarize new pyrene luminogens, and more importantly, discuss a feasible method to realize the transformation of pyrene-based ACQ luminogens to AIE. The excellent optical properties of pyrene-based AIEgens for potential applications in organic semiconductor devices in biochemistry will be discussed, as well as recent developments in the area of chemisensors.
 | ||
| Fig. 5 A schematic diagram of pyrene-based AIEgens possessing bright emission. | ||
2. Pyrene-based aggregation-induced emission luminogens
2.1. Pyrene core AIEgens
Since the emission of pyrene 1 is either weak or quenched in the solid state due to its planar structure with a large π-conjugation system, an effective approach to overcome this is the incorporation of bulky substituents or a spiro-type framework at the periphery of the pyrene core in order to inhibit the π–π interactions, leading to a highly emissive solid state. Arylsilyl substituted pyrene derivatives 2 and 3 were synthesized via Suzuki–Miyaura/Sonogashira–Hagihara cross-coupling reactions to afford the corresponding silyl-terminated group at the 1-, 3-, 6- and 8-positions. Triphenylsilylphenyl substituted pyrene 2 exhibited intense emission at 440 nm in the film with a high fluorescence quantum yield (Φf = 0.65); whereas the triphenylsilylphenyl(ethynyl) substituted pyrene 3 showed an excimer emission with low Φf both in THF solution (0.50) and as a film (0.16).39 Despite the introduction of the bulky tetraphenylsilylethynyl groups in 3, the pyrene moieties can clearly still interact in an orthogonal orientation. Density functional theory (DFT) geometry optimization predicts that the incorporation of the coplanar phenylethynyl substituents in the structure of 3 results in the Si atoms being 8.77 Å from the 1-, 3-, 6-, and 8-carbon atom positions, rather than 6.21 Å, as is the case in the structure predicted for 2 (Fig. 6).39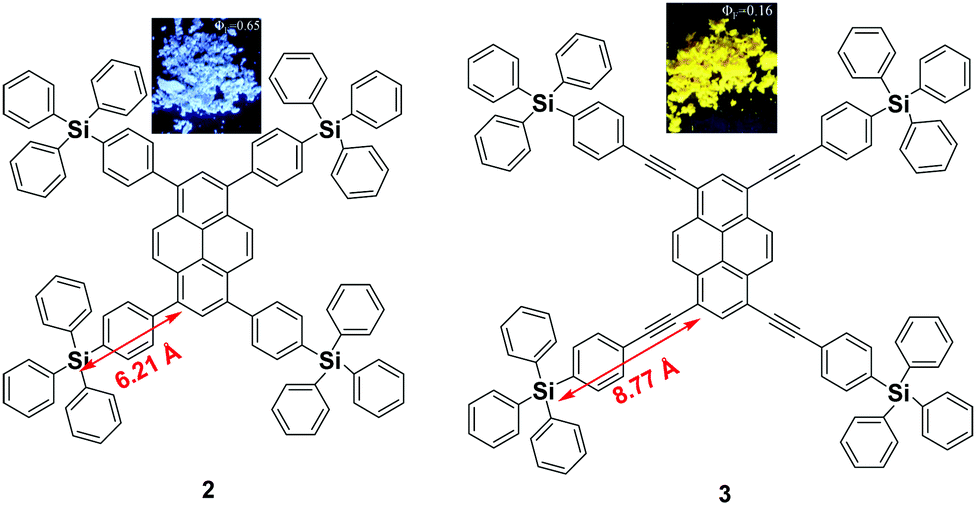 | ||
| Fig. 6 Structures of 2 and 3 calculated via DFT methods. Reproduced from ref. 39. | ||
The longer distance between the Si atoms and the 1-, 3-, 6-, and 8-positions on the outer perimeter of the pyrene π-system, probably enables significant π–π interaction to occur between neighboring pyrenes. Although the pyrene possesses a large π-conjugation planar structure, according to the RIR mechanism, it is possible to convert the pyrene into a highly efficient pyrene-based AIEgen by introducing a twisted fragment, such as TPE or triphenylethylene (TriPE) units etc. Zhao first introduced the TPE units at the pyrene core as peripheral ornaments affording TTPEPy 4 possessing a novel aggregation-induced enhanced emission (AIEE) effect (Fig. 7). It emitted sky-blue emission (483 nm) with efficient PL (Φ = 70%) in the solid state, and excellent thermal stability (Td = 485 °C).40 The temperature-dependent PL spectrum was measured over the range 300 K to 77 K, and the fluorescence lifetime gradually lengthened from 0.25 ns at 300 K to finally 1.29 ns at 77 K. This was due to the fact that the intramolecular rotations of the TPE blades were greatly restricted at the cryogenic temperature and the accompanying RIR process helped enhance the PL of TTPEPy 4 in the frozen solution.
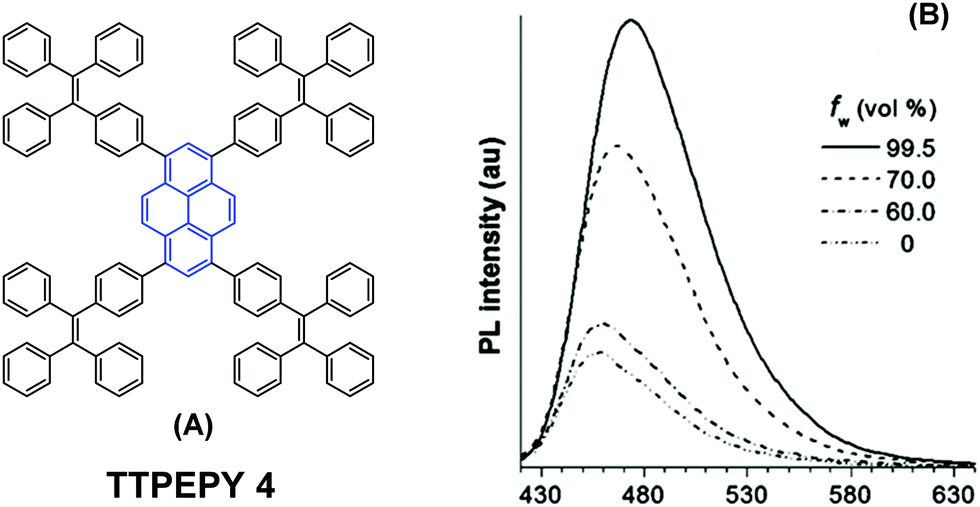 | ||
| Fig. 7 The structure of TTPEPy 4 and emission spectrum of TTPEPy 4 in THF with increasing percentages of water. Reproduced from ref. 40. | ||
In addition, the construction of efficient non-OLEDs using TTPEPy 4 as a blue emitter, which display an excellent current efficiency (ηC), power efficiency (ηP), and maximum external quantum efficiency (ηext) up to 12.3 cd A−1, 7.5 lm W−1 and 4.95%, respectively has been reported.40 Furthermore, to investigate the relationship between the molecular conformation, the emission color and the AIE properties, Li et al. designed and synthesized two pyrene-based luminogens, Py-4MethylTPE (5) and Py-4mTPE (6) (Fig. 8). The differing behavior is due to the linkage mode at the meta-position which can twist the molecular structure and provide a new stereoelectronic effect (Fig. 8). Both compounds exhibit AIE and AEE effects, although they have minor differences in their structures;41 for example the torsion angle of 5 between the pyrene core and the TPE periphery is larger than that of 6 due to the introduction of the methyl groups. On the other hand, use of meta linkages on the TPE moieties shortens the π-conjugation length in comparison with the para linkages and results in the increased rotational barriers. The meta linkage is more effective at controlling the twisted conformation and generating enhanced molecular rotation. Given this, the compound 6 emits a more enhanced blue emission at 445 nm compared with 5, which shows an emission peak at 459 in the aggregation state. Importantly, when using compound 6 as emitter, the OLED device exhibits favorable EL properties with a lower turn-on voltage (3.5 V) and better OLED performance, with values of ηC,max, ηP,max, and ηext,max at 4.02 cd A−1, 3.08 lm W−1, and 2.5%, respectively.41a
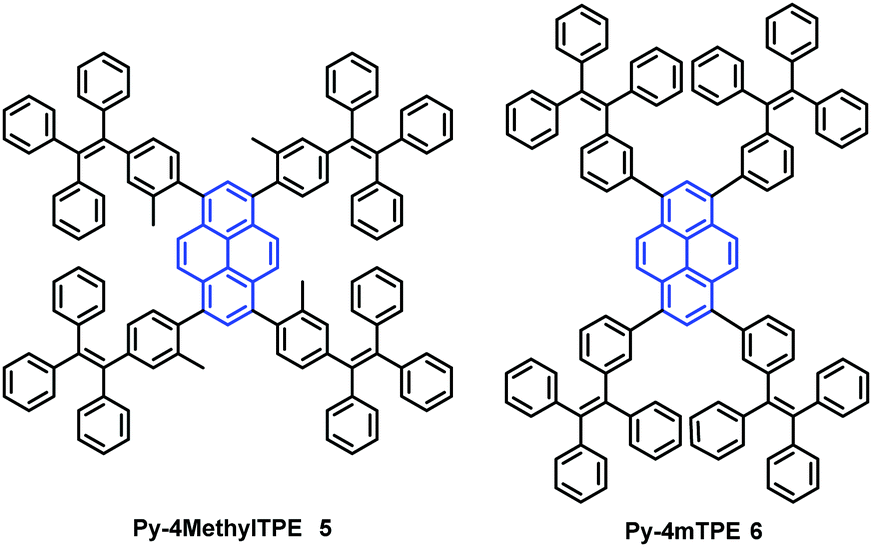 | ||
| Fig. 8 Structures of Py-4MethylTPE 5 and Py-4mTPE 6. | ||
It seems that it is an efficient approach to achieve pyrene-based AIEgens through integrating pyrene with TPE units. Tao's group has presented a pyrene-based fluorescent material, namely 1-[4-(2,2-diphenylvinyl)phenyl]pyrene (PVPP 7), which was synthesized via the Suzuki coupling reaction with TriPE units.20b Interestingly, compound 7 shows a dramatic enhancement of fluorescence intensity with clear AIEE characteristics. The single crystal X-ray diffraction analysis indicated that the two terminal phenyl rings in PVPP 7 are almost perpendicular, with a twist angle of 89.3°, and that several intramolecular C–H⋯π interactions are present which can play a significant role in suppressing the fluorescence quenching of the pyrene units in the solid state (Fig. 9d). The free twisting motion of the 1,1-diphenylvinyl motif is restricted by the intra- and intermolecular C–H⋯π hydrogen bonds. Thus, the non-radioactive decay channel is closed. More importantly, the molecular structure of PVPP 7 is significantly distorted, and the twisted geometry can prevent the strong π–π interactions producing intense fluorescence in the solid state. Li et al. first introduced the twist terminal units at the 2,7-positions of pyrene to access the pyrene-based AIEgens 8–13 (Fig. 10). In this case, the AIE building blocks become more and more twisted, and large blue-shifted emission is achieved on moving from Py-2mTPE 9 (460 nm) to Py-2F 12 (456 nm) and to Py-2TP 13 (444 nm).41b
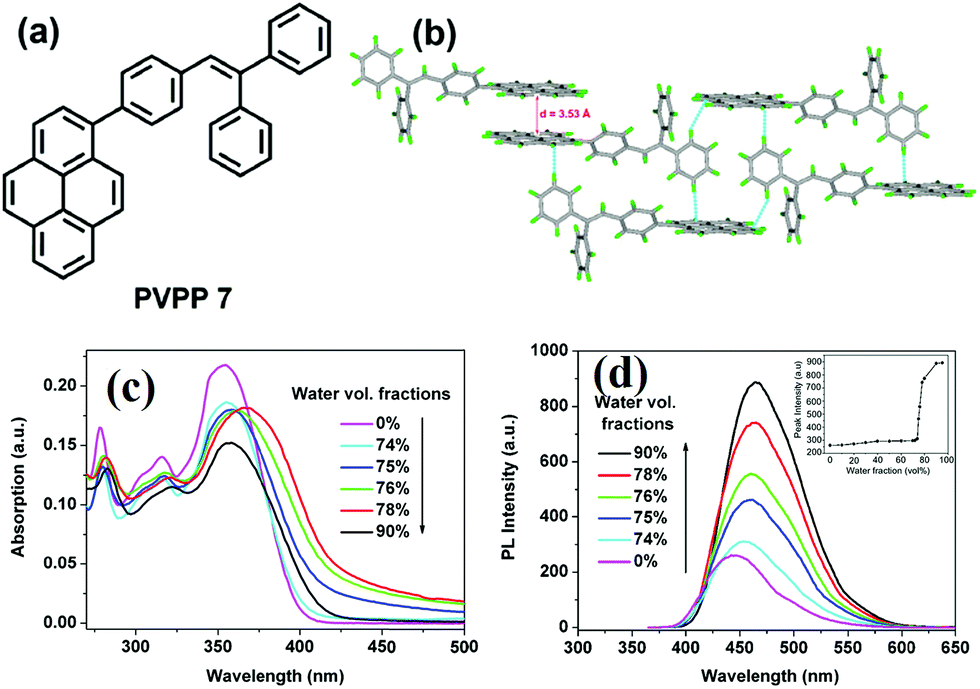 | ||
| Fig. 9 (a) The molecular structure of PVPP 7 and (b) the crystal packing structure PVPP 7. (c) UV-visible absorption spectra of PVPP 7 in THF/water mixtures with different volume fractions of water. (d) PL spectra of PVPP 7 in THF/water mixtures with different volume fractions of water. The inset shows the relationship between the PL peak intensity of PVPP 7 and the water fraction in THF. Reproduced from ref. 20b. | ||
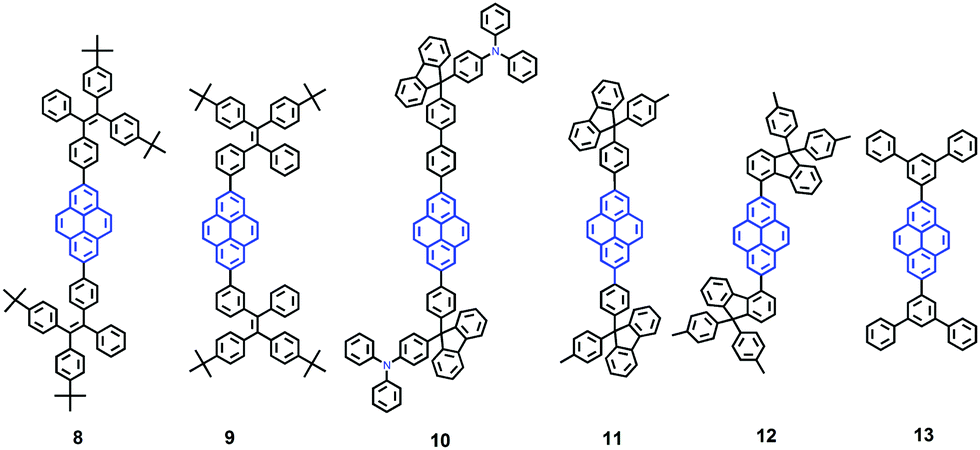 | ||
| Fig. 10 Structures of Py-2pTPE 8, Py-2mTPE 9, Py-2TF 10, Py-2NTF 11, Py-2F 12, and Py-2TP 13. | ||
Py-2 NTF 11 exhibited a blue-shifted absorption compared with Py-2F 12, owing to the weak conjugation at the 9,9′-positions in the fluorene as well as the severely twisted conformation of this linkage mode. There was little difference between Py-2NTF 11 and Py-2TF 10 in the UV absorption spectra due to the non-conjugated 9,9′-positions of fluorene. Py-2pTPE 8 and Py-2mTPE 9, which consisted of typical AIE units of tetraphenylethene, show AIE characteristics (non-emissive in THF solution but a strong fluorescence in the aggregation state).
Interestingly, for Py-2pTPE 8 (Fig. 11), as the water fraction was varied over the range 30–50%, it exhibited a deep blue emission which peaked at about 450 nm. The luminescence intensity increased gradually, and reached a maximum at 50% water fraction. However, when the water fraction increased to 60%, only very weak luminescence was observed, and its emission peak was red-shifted to about 494 nm. This abnormal phenomenon originated from the transition of the crystalline state to an amorphous state.41b
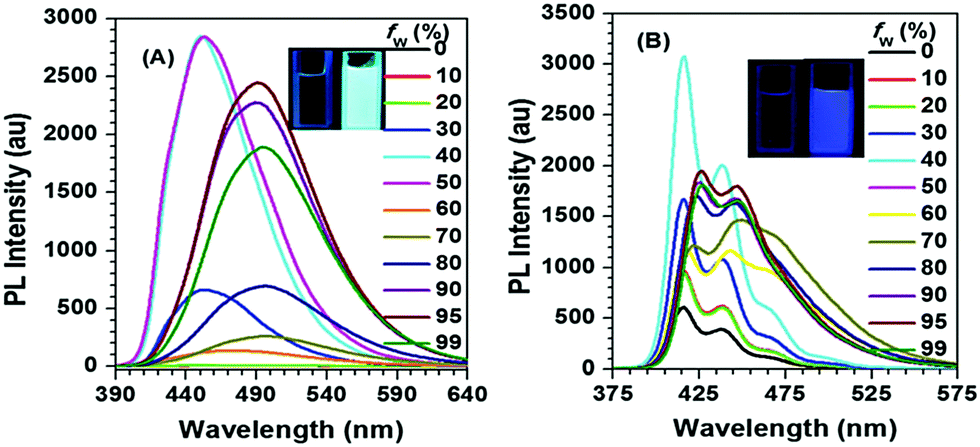 | ||
| Fig. 11 PL spectra in THF/H2O mixtures with different water fractions: (A) Py-2pTPE 8, concentration (mM): 12.0; excitation wavelength 330 nm; (B) Py-2TP 13, concentration (mM): 14.0; excitation wavelength 300 nm. Inset: Photos of Py-2pTPE 8 and Py-2TP 13 in THF/H2O mixtures (fw = 0 and 99%) taken under illumination by a 365 nm UV lamp. Reproduced from ref. 41b. | ||
More recently, the group of Tang reported a new strategy to achieve white-light emission from single tetraphenylethylene substituted pyrenes (TPE-Pys) 14–16 with AIE characteristics (Fig. 12).42 These molecules exhibited tuneable emission from blue (474 nm) to yellow (531 nm) both in solution and in the solid state. Specially, 14c and 16 emitted a dual fluorescence at around 436 nm and 538 nm in a water fraction of 80%, which resulted in white-light with a CIE of x = 0.30, y = 0.41.42 These compounds emit white-colored fluorescence in THF/water mixtures. Indeed, these are rare examples of white-light emission achieved from a single AIE-active molecule via the control of its aggregated state emission by tuning the composition of the solvent mixture.
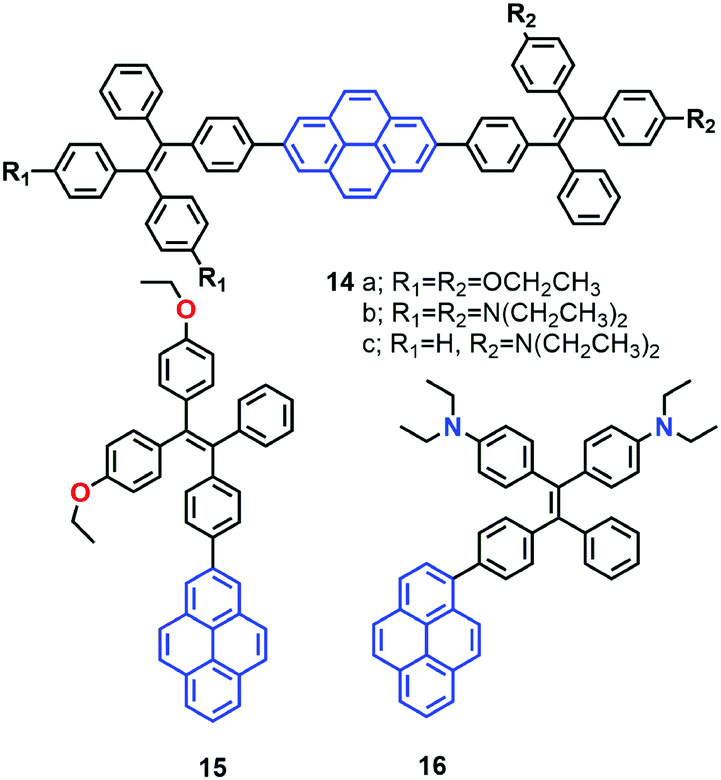 | ||
| Fig. 12 Structures of TPE-Pys 14–16. | ||
A single crystal X-ray diffraction study indicated that compound 16 adopts a twisted conformation and revealed that the torsion angle between the terminal pyrene ring and the benzene ring of the TPE is 48.6° (Fig. 13). The molecular motion of the TPE moiety is suppressed by multiple intramolecular C–H⋯π interactions with distances of 2.75–2.95 Å. The pyrene rings are arranged in a head-to-tail fashion and are separated by the TPE units at a large distance of 8.08 Å. This prevents their π–π stacking which would quench the light emission. Multiple C–H⋯π interactions are formed in the crystal lattice which effectively restrict the phenyl rings of TPE from undergoing intramolecular motions. The non-radiative decay pathways are blocked to allow 16 to emit intense light in the aggregated state.42
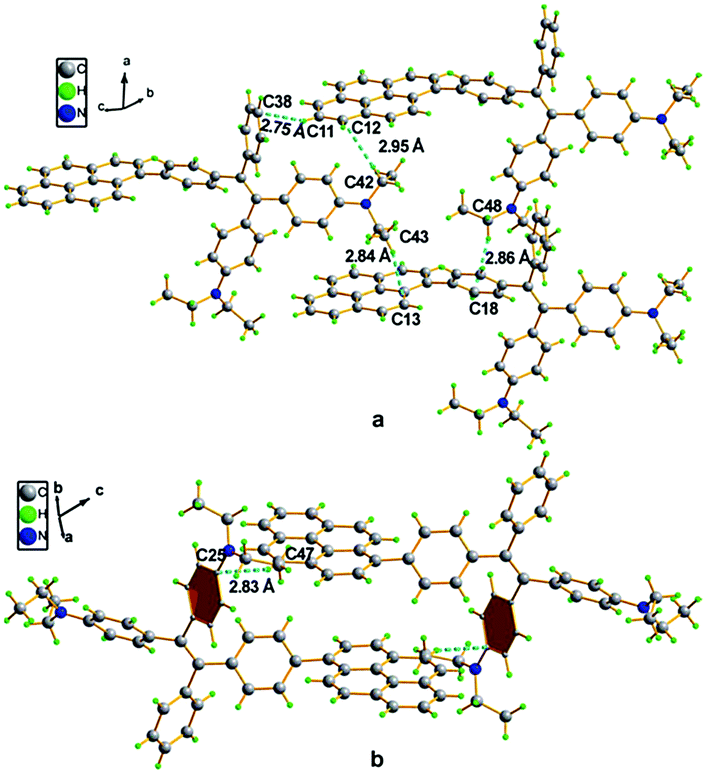 | ||
| Fig. 13 Single crystal structure of 16: (a) face-to-face pattern of pyrene moieties separated by TPE units and (b) head-to-tail packing mode with multiple C–H⋯π interactions with a distance of 2.83 Å. Reproduced from ref. 42. | ||
2.2. TPE-like pyrene-based AIEgens
Tetraphenylethylene (TPE) has attracted much attention because it possesses advantages such as efficient solid-state emission, facile synthesis, and ease of functionalization.43,44 More importantly, TPE is a promising candidate to convert conventional ACQ chromophores into effective AIEgens with excellent photoluminescence properties.45 Given their outstanding AIE properties, much effort has been devoted to developing TPE-like based AIEgens which are expected to have large π-conjugation as well as enhanced emission behavior in the solid state. The main approach to obtaining efficient solid photoluminescence is to replace the phenyl rings of TPE by PAHs which introduce increased π-conjugation into the system. To date, several TPE-based AIEgens with notable solid-state emission have been developed.46 Researchers have speculated whether a tetra(polycyclic aryl)ethene with excellent solid-state emission can be obtained by carefully selecting PAHs as substituent groups. These PAHs not only have an enlarged π-conjugated system, but also have effective steric hindrance to restrict the formation of π-stacking.To verify this assumption, scientists have utilized many PAHs such as pyrene, fluorene, naphthalene, biphenyl etc. to assemble tetra(polycyclic aryl)ethenes.47 Zhao et al. reported two pyrene-substituted ethenes, namely TPPyE 17 and DPDPyE 18 (Fig. 14), where the phenyl ring of TPE was replaced by a pyrene ring. Unexpectedly, both the TPE-like pyrene-based dyes exhibited clear AIE features and were weak emitters in solution, but became strong emitters when aggregated in the condensed phase.46a The crystal packing indicated that the intramolecular rotation was suppressed by π–π stacking and multiple C–H⋯π interactions. These structural features block the non-radiative energy decay channels, and allow for intense emission in the solid state.
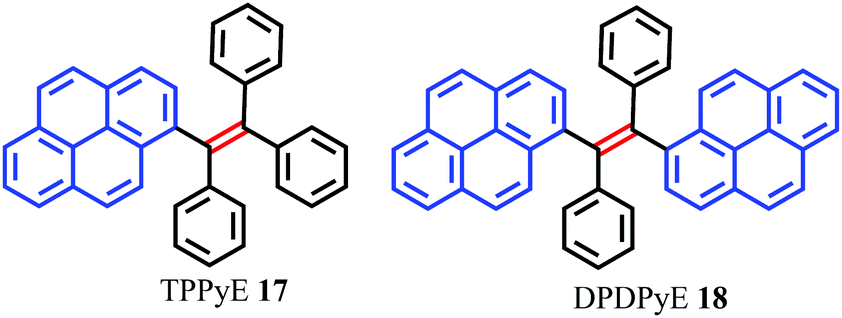 | ||
| Fig. 14 Structures of 17 and 18. | ||
Using TPPyE 17 and DPDPyE 18 as high-efficiency emitters, green OLEDs were fabricated with maximum luminance and power, current, and external quantum efficiencies of 49830 cd m−2, 9.2 lm W−1, 10.2 cd A−1 and 3.3%, respectively. To investigate the effect of substituent size on molecular rotation, Ni et al. reported tetrakis(4,5,9,10-tetrahydropyren-2-yl)ethene 19, which exhibits clear AIE characteristics.47 The fluorescence quantum yield of tetrakis(4,5,9,10-tetrahydropyren-2-yl)ethene 19 in a solid film was up to 74.1%. A crystal structure analysis indicated that no π-stacking interactions are present (Fig. 15). This study reveals that when constructing AIE molecules using substituent groups with expanded π-conjugation, the introduction of appropriate steric hindrance on the substituent groups can effectively restrict the formation of excimers. Meanwhile, hydrogen atoms from the ethylidene groups of compound 19 tend to interact with the π-cloud center of the closest two phenyl rings from the adjacent molecules, and these multiple C–H⋯π interactions rigidify the molecular structure and restrict the rotation of 19, thereby making a valuable contribution in avoiding the formation of excimers. Thus, compound 19 exhibited high emission in the solid state with a high quantum yield (74.1%) compared with that observed in THF solution (1.25%).47
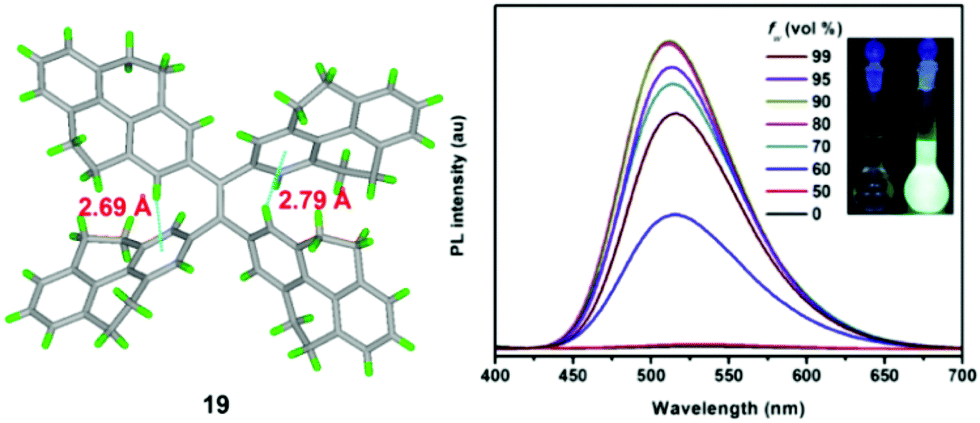 | ||
| Fig. 15 Crystal structure of 19 and PL spectrum of compound 19 in THF/water mixtures with different water fractions (fw). Inset: Photos of compound 19 in THF/water mixtures (fw = 0 and 90%) under UV lamp illumination. Excitation wavelength: 370 nm. Reproduced from ref. 47. | ||
On the other hand, it is an efficient approach to construct AIEgens by expanding the π-conjugation of the ethene derivatives using pyrene units. Li et al. further published a series of efficient TPE-like pyrene-based blue materials 20–25 with an AIE building block acting as the core and pyrene as a peripheral group. These compounds showed enhanced hole-transporting ability, achieving a good interaction between pyrene substituents (Fig. 16).48
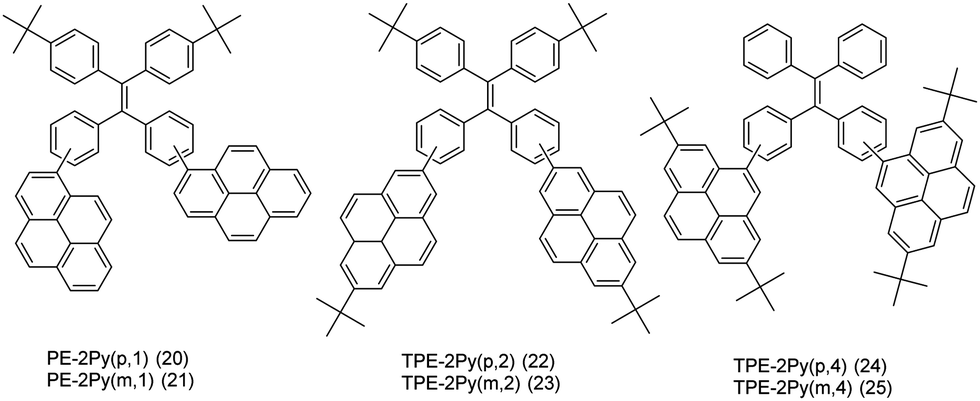 | ||
| Fig. 16 Structures of 20–25. | ||
The effect of substituent position on the emission color and electroluminescence was investigated by modifying the linkage modes of the TPE (para to meta-position) and pyrene units (1- to 2- to 4-position). All of these pyrene derivatives exhibited apparent AIE characteristics and comparable EL performances in their OLED devices. When the pyrene units were attached at the meta-position of TPE, the PL emissions were tuned to deep blue (452 nm) in the solid state,48 with that at the pyrene 4-position producing a better deep blue emitter than the 1- or 2-substituted pyrenes. Using TPE-2Py(p,4) 24 as both a hole-transporting layer and emitting layer, the resulting blue OLED device (CIE coordinate: x = 0.16, y = 0.21) exhibited excellent EL performance, with a current efficiency and external quantum efficiency up to 4.66 cd A−1 and 2.79%, respectively.48 Furthermore, Yanget et al. reported the highly efficient blue emitter TPE-4Py 26 with an AIE effect, which is achieved by a combination of a twisted tetraphenylethene (TPE) core and planar pyrenes on the periphery.49 Its EL efficiency was enhanced in doped OLEDs, at high doping concentration (50%), with a current efficiency up to 4.9 cd A−1 at 484 nm (Fig. 17). The twisted conformation of 26 led to weak conjugation as well as blue emission. Furthermore, the π–π stacking was effectively restricted, which resulted in an abnormal AIE effect. The optimized structure revealed a heavily twisted conformation with a torsion angle of about 50° for the TPE, while the dihedral angle between the pyrene and the adjacent phenyl ring was as wide as 60°.49
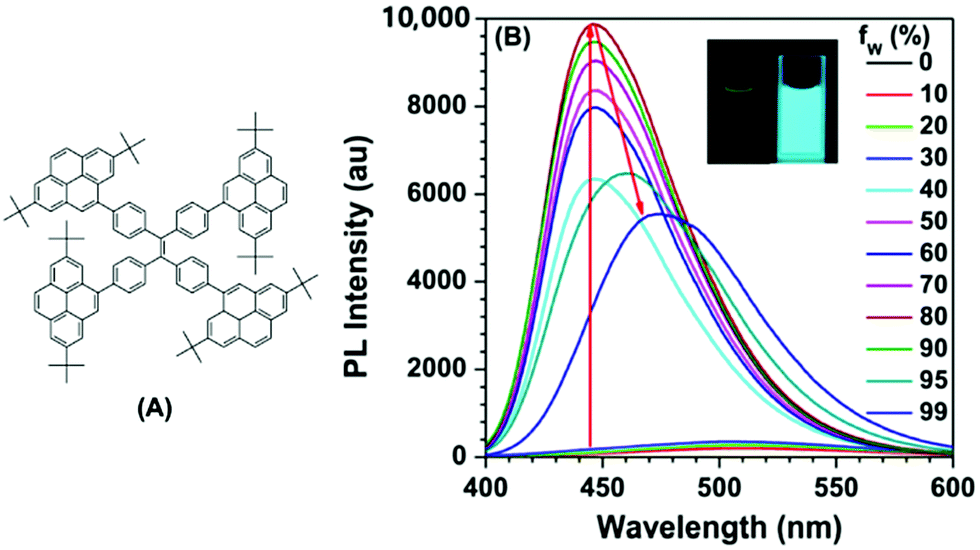 | ||
| Fig. 17 (A) Molecular structure of TPE-4Py 26; (B) PL spectra of TPE-4Py 26 in THF/H2O mixtures with different water fractions. Concentration: 10 μM; excitation wavelength: 330 nm; inset: photographs of TPE-4Py 26 in THF/H2O mixtures (fw = 0% and 90%) taken under illumination by a 365 nm UV lamp. Reproduced from ref. 49. | ||
2.3. Non-typical pyrene-based AIEgens
A study of vinylene-based materials revealed that the vinylene bond geometry plays an important role on the emission.50 For example, the cis-isomer in poly(p-phenylenevinylene) (PPV) molecules generally possesses weak luminescence.51 However, random placement of cis-olefin linkages along the PPV chain significantly increases the luminescence efficiency. For vinylene-based chromophores with an AIE or AIEE property, most tend to adopt the trans-isomer conformation.51 According to the RIR mechanism, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond can also play a significant role when constructing AIE molecules. For example, 1-[(9-anthracenyl)vinyl]pyrene (AVP)-based AIEgens trans-AVP 27 and cis-AVP 28 are presented in Fig. 18, and although the large planar structure of the pyrene and anthracene are both ACQ active, the anthracene ring was connected to a pyrene unit via a vinyl bridge and shows a clear AIEE characteristic with the two isomer conformations in the crystalline state.52
C bond can also play a significant role when constructing AIE molecules. For example, 1-[(9-anthracenyl)vinyl]pyrene (AVP)-based AIEgens trans-AVP 27 and cis-AVP 28 are presented in Fig. 18, and although the large planar structure of the pyrene and anthracene are both ACQ active, the anthracene ring was connected to a pyrene unit via a vinyl bridge and shows a clear AIEE characteristic with the two isomer conformations in the crystalline state.52
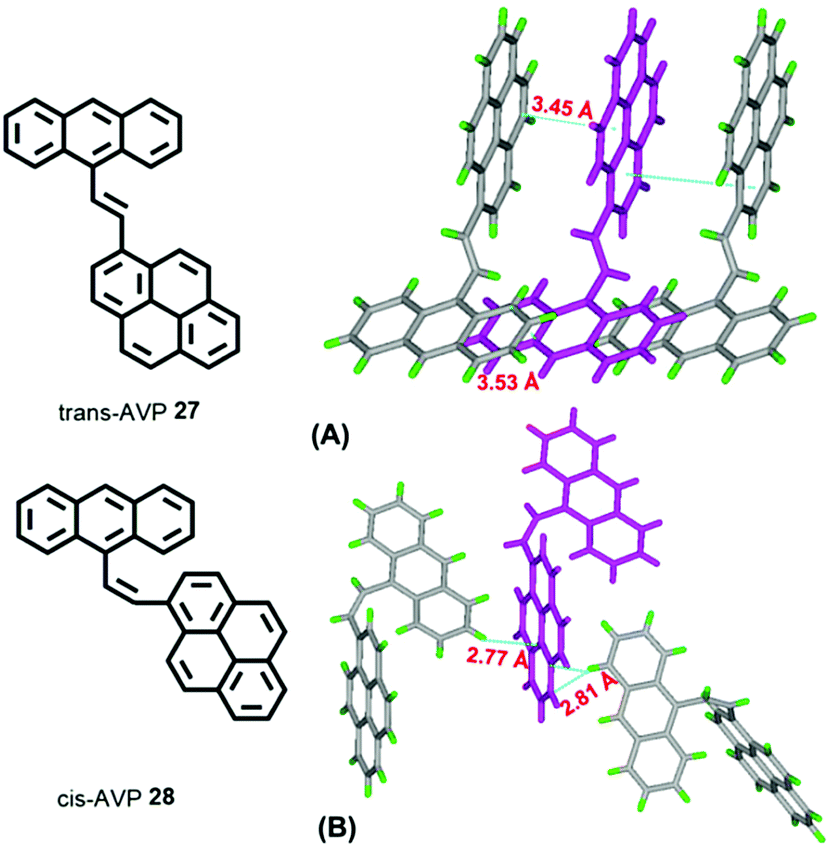 | ||
| Fig. 18 Structures of (A) trans-AVP 27 and (B) cis-AVP 28, and their crystal packing. | ||
The quantum yield of trans-AVP 27 and cis-AVP 28 are 54.0% and 36.0% in the solid state versus in solution 15.0% and 17.0%, respectively. The interacting adjacent molecules in crystals of trans-AVP 27 adopt both edge-to-face and face-to-face arrangements. In contrast, in crystals of cis-AVP 28, the adjacent molecules are inclined to adopt an edge-to-face arrangement and the molecular packing plays an important role in the solid emission efficiency.52 Similarly, Li et al. systematically investigated the substituent position and the vinylene bond geometry on the AIE features and the fluorescence solvatochromism. The structure of the (9-anthryl)vinyl(1-pyrenyl)vinylbenzene isomers 29–32 are presented in Fig. 19.53
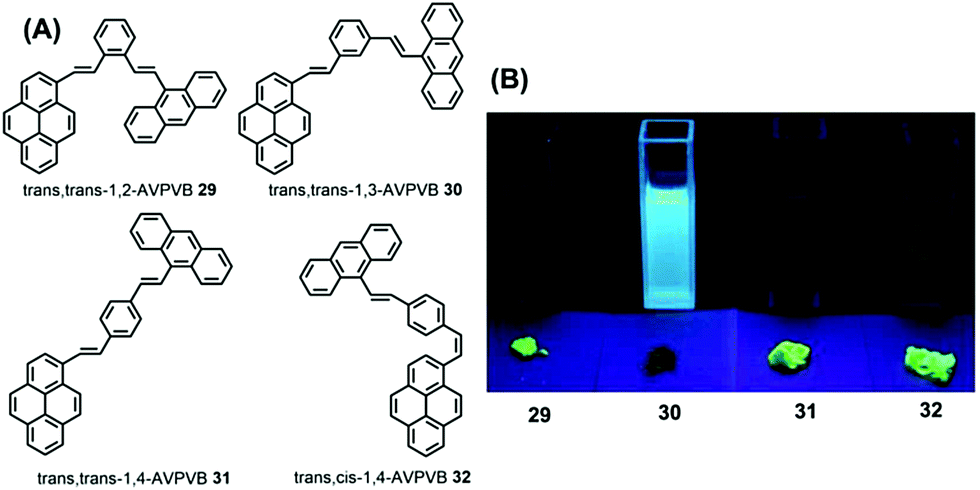 | ||
| Fig. 19 Structures and fluorescence images of 29–32 in CH2Cl2 solution (upper) and as solids (lower) (from left to right) under illumination by a 365.0 nm UV lamp. | ||
Due to differences in the molecular conformations, the series of compounds exhibit different fluorescence properties. For example, trans,trans-1-(9-anthryl)vinyl-4-(1-pyrenyl)-vinylbenzene (trans,trans-1,4-AVPVB 31) shows unusual AIE characteristics with fluorescence solvatochromism and emission shifts of over 80 nm, while trans,trans-1-(9-anthryl)vinyl-3-(1-pyrenyl)-vinylbenzene (trans,trans-1,3-AVPVB 30) exhibits a weak solvatochromic effect due to the meta-substitution which emits most efficiently in solution. In addition, trans,trans-1,2-AVPVB 29 and trans,cis-1,4-AVPVB 32 display moderate fluorescence and solvatochromism.
Theoretical calculations indicate that fluorescence solvatochromism is related to the conformational change from the ground state to the excited states due to the conjugate effect. An X-ray diffraction study revealed that the dihedral angle (53.98°) between the pyrenyl and central benzene ring in trans,cis-1,4-AVPVB 32 is higher than that observed for trans,trans-1,4-AVPVB 31 (22.46°), which is due to the strong intramolecular C–H⋯π steric hindrance between the pyrenyl ring and its adjacent phenyl in trans,cis-1,4-AVPVB 32. Therefore, the trans,trans-1,4-AVPVB 31 molecule is more planar and has better π-conjugation than does trans,cis-1,4-AVPVB 32.53
On the other hand, α-cyanostyrenes of type 33 bearing a pyrene scaffold are weakly emissive in solution but show broad red-shifted emission in water due to aggregation (Fig. 20).54 Methoxy-substituted pyrenyl styrenes (33b and 33c) exhibit a broad emission peak (∼550–560 nm) with a shoulder peak (∼460 nm) in polar acetonitrile. In the solid state, 33a shows a clean emission spectrum with maximum peak at ∼510 nm, while 33b and 33c exhibit a noisy emission spectrum with bands at ∼495 nm and at ∼530–540 nm, respectively. The presence of the methoxy groups led to the formation of uniformly shaped aggregates, and the resulting unique emission behavior.54 On the other hand, the emission of 33c exhibited a bathochromic shift in different water fractions (λem = 516 nm in fw = 60%, λem = 535 nm in fw = 90%, λem = 550 nm in fw = 100%), due to the formation of J-aggregates dominated by head-to-tail interactions.
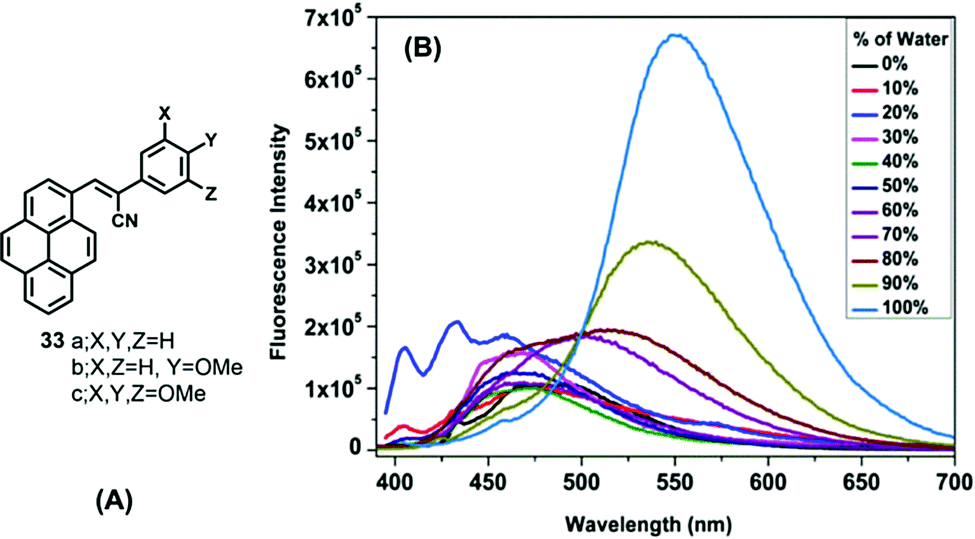 | ||
| Fig. 20 (A) Structures of 33 and (B) emission of 33c in a 1,4 dioxane/water system. Reproduced from ref. 54. | ||
2.4. Pyrene-based chalcone AIEgen
Pyrene-based chalcone derivatives, formed by combining chalcone units with a pyrene core, exhibit special optoelectronic properties, due to a push–pull conjugation effect that improves the luminescent properties and redox activity, while the α,β-unsaturated ketone linkage enhances the non-linearity.55,56 The molecular structure of such pyrenes consists of a donor with an electron-deficient keto vinyl (acceptor) linkage, which helps construct a D–π–A type conformation, which is responsible for the interesting AIEE/AIE features which enhance the luminescence properties of these materials. For instance, the four chalacone dyes 34–35 emitted yellow-green light in the solid state, and showed a large Stokes shifts versus their solution behavior.55 The fluorescence spectra of the methoxy-containing dyes 34b and 35b are found to be blue-shifted by 10 nm relative to the parent ones (34a and 35a), and a beneficial effect of the methoxy function on the degree of aggregation was observed. Quantum yields of 34b (16%) and 35b (19%) were found to be higher in the solid state than in solution, which thus showed a clear AIEE characteristic.The X-ray crystal structure of 35b revealed stacks of head-to-head-oriented chromophores (Fig. 21).55 The case of the dye 35a is different for two reasons; firstly, it forms dimers in which the π-systems of the pyrene styrenyl units in adjacent molecules are parallel, oriented head-to-head, and have no π-overlap.
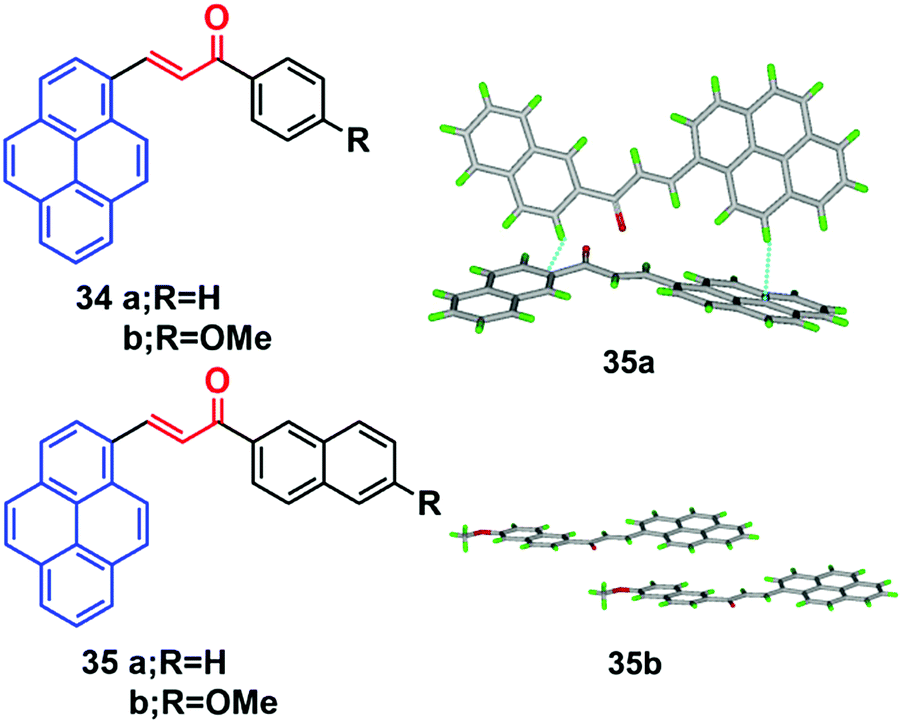 | ||
| Fig. 21 Crystal packing diagrams of 34 and 35 illustrating π–π stacking interactions. Reproduced from ref. 55. | ||
Secondly, the pyrene moieties of adjacent dimers of the former type overlap in an antiparallel fashion with an interplanar distance of 3.489 Å. The X-ray structure of 35a reveals the presence of H-like aggregates, which may explain the quenching of the photoluminescence in these systems as compared with their CH2Cl2 solutions (Fig. 21). The X-ray structure of 35b indicates a J-type aggregation that is not detrimental to solid-state emission.55
On the other hand, pyrene-based chalcone materials containing heterocyclic rings (36–37) were synthesized by a Claisen–Schmidt condensation, and exhibit an AIEE with dual emissive characteristics in solution as well as in the solid.
Specifically, the compounds 36–37 showed a large tuneable color emission from green to orange, depending on the water fraction in the THF/H2O mixtures (Fig. 22).56 In the solid state, three compounds underwent a red-shift of 77 nm, 82 nm, and 81 nm of 36a (PCT), 36b (PCF), and 37 (PCP) compared to their solution state, respectively. In the solid-state emission spectra, 36a exhibits a maximum emission at 558 nm, whereas 36b and 37 display maxima at 569 and 568 nm, respectively,56 which is attributed to intermolecular interactions of the pyrene unit (namely π–π stacking and a J-aggregation arrangement). Indeed, compound 36a (PCT) afforded a good emission quantum yield of 0.74, whereas for 36b (PCF) and 37 (PCP), the quantum yields were 0.68 and 0.53, respectively.56
 | ||
| Fig. 22 Photograph on the AIEE properties of PCT 36a, PCF 36b and PCP 37 in THF/water mixtures with different water fractions (fw = 0–100%) under illumination by long UV-light (365 nm). Reproduced from ref. 56. | ||
2.5. Pyrene-based AIE-active Schiff base molecules
Many scientists are attempting to use the AIE activity of pyrene derivatives to sense molecules by using push–pull or on–off procedures. Schiff base AIE systems have been identified on the basis of intensity turn-on at a specific wavelength.57 Novel pyrene-based Schiff base derivative 38 was synthesized via a condensation reaction and utilized as a turn-on fluorescence sensor toward Cu2+, and showed AIE activity in CH3CN and water mixture up to 80% (Fig. 23).58 The PL intensity of 38 was enhanced ca. 630-fold (Φf = 0.567 in CH3CN:H2O = 20:80) by increasing the water fraction of H2O from 0% to 90% and with a red-shift from 455 to 471 nm. The mechanism possibly arises from RIR. Since the single-bond rotation is mainly responsible for the dominant non-radiative decay, the RIR effect might be the cause of the AIE nature of 38. The hydrogen-bond formation, suppression of the photo-induced electron transfer (PET) process, and suppression of charge transfer (CT) or even intramolecular charge transfer (ICT) are other mechanistic approaches for this category of AIEgens.58 However, the PL spectra for 38 suggested that the red-shift on aggregation probably originated from the suppression of the TICT. Various molecules act as turn-on sensors for a variety of cations and anions based on PET, intramolecular charge transfer (ICT), chelation-enhanced fluorescence (CHEF) and deprotonation mechanisms.59
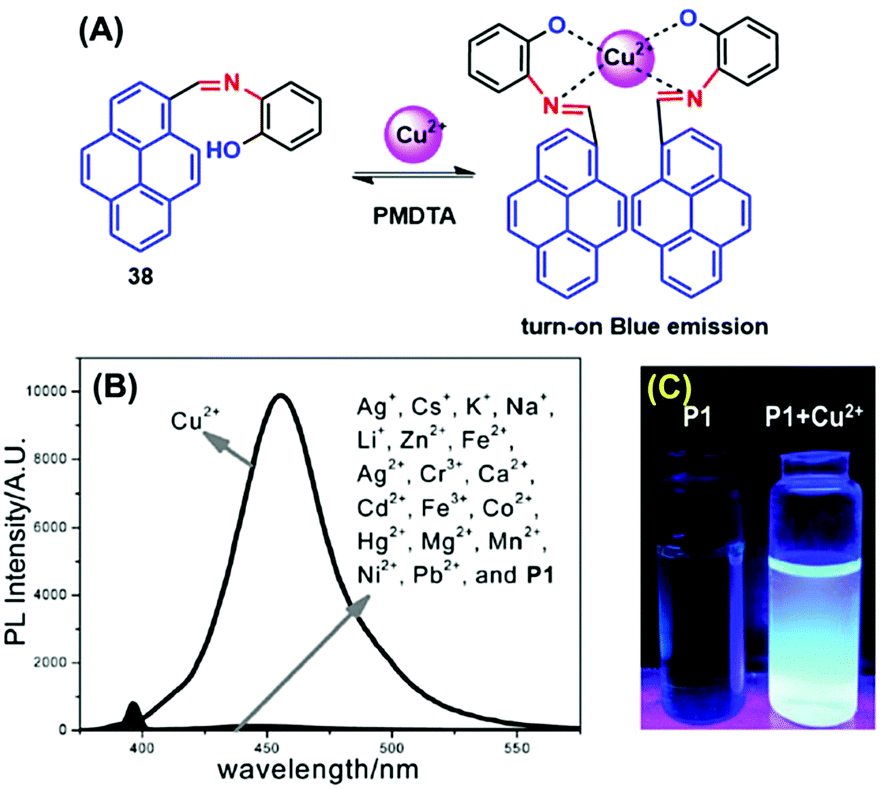 | ||
| Fig. 23 (A) Possible proposed binding mechanism of 38 toward Cu2+ ions. (B) Fluorescence emission spectra of compound 38 (10 μM) upon the addition of various metal ions. (C) Photographs of 38 and 38 + Cu2+ visualized under UV-light irradiation. Reproduced from ref. 58. | ||
The photophysical properties of 39 revealed that the fluorescence of this molecule is completely quenched due to PET processes in solution. The fluorescence intensity remained unchanged over the water fraction range 0–70%. On further addition of water up to 90%, the emission intensity was enhanced with a red-shift (25 nm) from 440 to 465 nm, with a concomitant increase in fluorescence lifetime.60 This clearly signifies that this molecule exhibited an AIE property (Fig. 24).
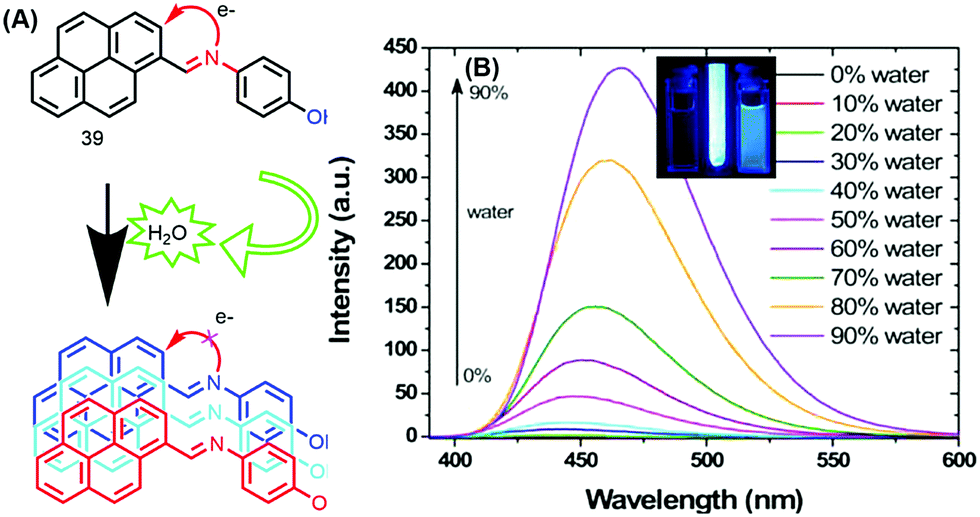 | ||
| Fig. 24 (A) 39 in THF and (B) emission spectra of 39 in THF with increasing percentages of water; inset: photographs taken under illumination by a UV lamp in a THF/water mixture. Reproduced from ref. 60. | ||
The AIE behavior of 39 in the excited state was further investigated by the time-correlated single-photon counting (TCSPC) technique, and the observed longer fluorescence lifetime is due to suppression of the PET process caused by aggregation of the molecules of 39.60 Thus, the mechanism of AIE for this molecule is suppression of PET due to the hydrogen-bonding interactions of the imine donor with water.
Tunable pyrene-based benzohydrazonate molecules of type 40 exhibit drastic aggregation-induced color changes (Δλem,max ∼ 130 nm) with high solid-state quantum yields (∼54.5%). The exciting switch behavior is triggered by intermolecular hydrogen bonding via the carbohydrazone bridge, which leads to restriction of the molecular rotation (Fig. 25).61 Upon increasing the amount of water in the dimethyl formamide (DMF) solution, the absorption did not significantly change until the water fraction reach 90% for (R = H) and 80% for (R = Cl, OMe), whereupon aggregation was initiated. The solid compounds 40 (R = Cl, OMe) revealed red-shifts of 54 nm and 42 nm compared with the spectra of the monomer state.
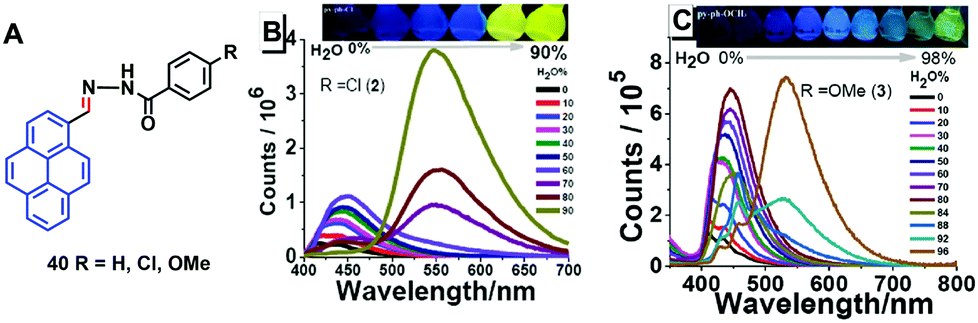 | ||
| Fig. 25 Structures and PL spectra of 40 in DMF/H2O mixtures with different volume fractions of H2O. Inset: Emission color change from blue to yellow in aqueous DMF with fw = 0–90% under 365 nm UV-light. Reproduced from ref. 61. | ||
Furthermore, compound 40 exhibited mechanochromic properties upon exogenic action. The blue-emitting pristine solids were converted to yellowish-green emitting solids, and the emission peaks exhibited varying degrees of red-shift. The pyrenyl-substituted acylhydrazone 41 derivative also exhibited favorable multiple luminescent switching mechano-fluorochromic behavior. In particular, fluorescence color changes were visible by the naked eye, as well as emission that could be reversibly switched between a blue and a green color, as shown in Fig. 26.62 By contrast, xerogel 41 obtained from dimethyl sulfoxide (DMSO) revealed mechanofluorochromic behavior, with the maximum emission peak shifted from 458 nm to 514 nm upon grinding. However, for 41 precipitated from THF, no mechanofluorochromic behavior was observed. The different luminescent properties of 41 are attributed to the switch of the self-assembled structures via intermolecular hydrogen bonding which impacted on the translational and rotational motion of the molecules, and thus the conformational flexibility of the pyrenyl moieties was restricted.62
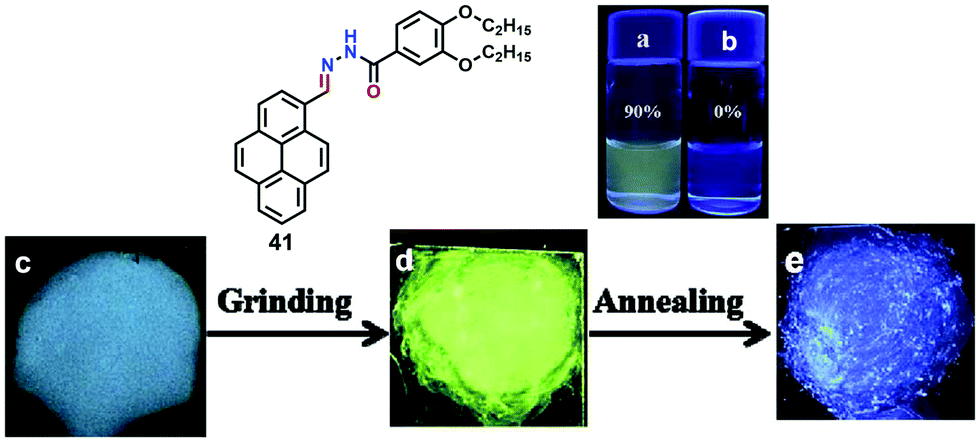 | ||
| Fig. 26 Emission spectra of 41 in 90% water (a) and THF (b). Photographs of emitting samples upon 365 nm UV illumination: (c) xerogel 41, (d) ground xerogel and (e) after annealing treatment. Reproduced from ref. 62. | ||
Two novel pyrene-containing monomeric and dimeric Schiff base derivatives 42 and 43 were reported to show AIEE activity in DMSO upon addition of H2O over the range 0–90% (Fig. 27).63 Both 42 and 43 were also good sensors for trivalent metal ions (Fe3+, Cr3+ and Al3+) in living cells. As an example, when Raw264.7 cells were incubated with 42/43 (10 mM in DMSO), no fluorescence was observed over 30 min. However, bright blue fluorescence images were observed for the Raw264.7 cells after 12 h with 42 and dismal blue fluorescence images with 43 due to intracellular H2O-induced aggregation. The bright cell image of 42 at 12 h is due to the greater intracellular penetration of the free thiol unit, which is superior to the disulfide-containing 43.63 Both 42 and 43 showed “off–on” sensing at highly acidic pH values (1–3) and were employed for live cell applications.63
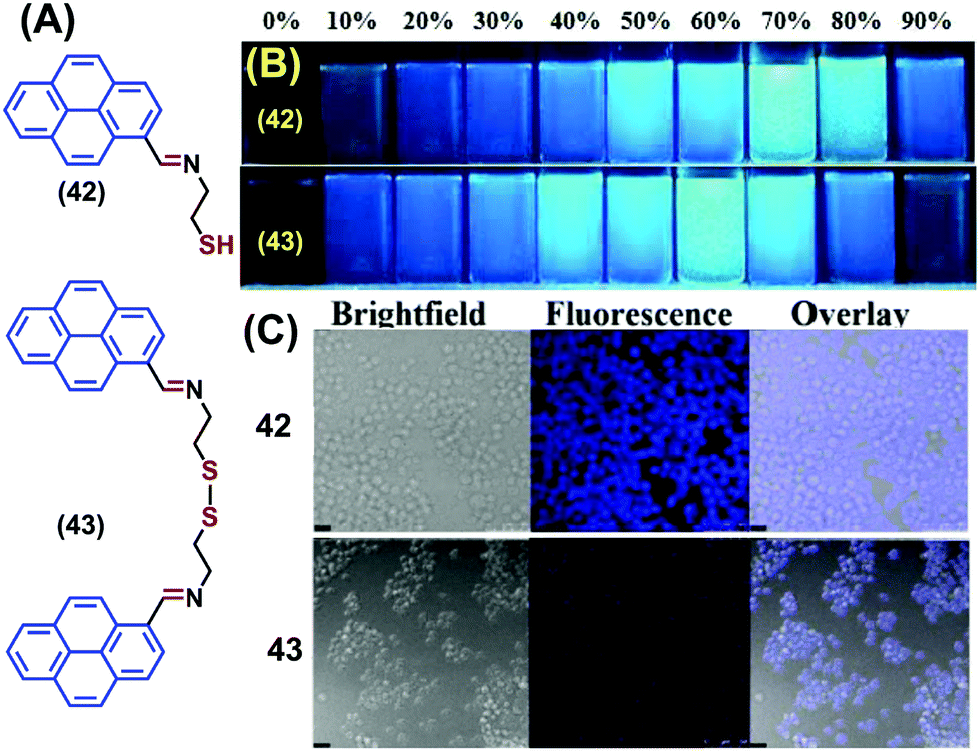 | ||
| Fig. 27 (A) Structures of 42 and 43. (B) Photograph of the AIE of 42 and 43 visualized under UV-irradiation (l = 365 nm). (C) Fluorescence images of Raw264.7 cells treated with 42 and 43 at 12 h. Reproduced from ref. 63. | ||
2.6. ESIPT-based AIEgens
ESIPT compounds have attracted attention due to their interesting and unique emission properties. Essentially, introducing the AIE phenomenon and excimer formation ability into molecular systems has provided the scope for dual emission. By contrast, ESIPT-coupled AIE phenomena operate as an additional handle to adjust the emission wavelength of the corresponding emission peak by varying the solvent polarity.The development of ESIPT molecules as probes is rapidly growing given the recent interest in their photophysical properties, such as unusually large Stokes shifts and outstanding environment-sensitivity that can readily be monitored by spectral changes.64 It should be noted that ESIPT molecules exist exclusively in an enolic form (E) in the ground state. Upon photoexcitation, extremely fast tautomerism from the excited enolic (E*) to the excited keto (K*) form occurs on a sub-picosecond time scale. After decay of K* to the ground state, the K form transforms to the initial E form through reverse proton transfer mediated by intramolecular hydrogen bonds (Fig. 28). The different species between absorbing and emitting in the intrinsic four-level photocycle (E–E*–K*–K) offer a high probability of addressing the disadvantage of fluorescence concentration quenching that is suffered by almost all of the currently available luminescent probes. On the other hand, ESIPT involves a proton-transfer process that is easily affected by changes in the surrounding conditions, leading to sensitive spectral responses. A single and broad emission peak due to complete ESIPT is observed in polar solvents, while in non-polar solvents, conversion of the enol into the keto form is incomplete. The J-type aggregate effects may block the molecular motion and facilitate the ESIPT emission in the solid state/aggregated state.
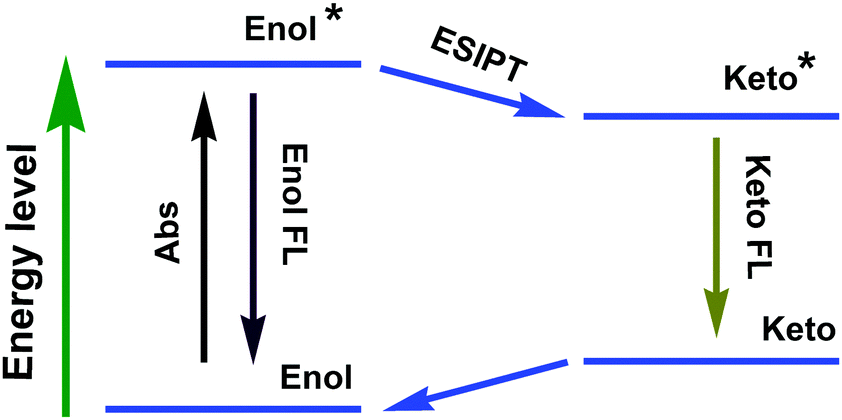 | ||
| Fig. 28 Four-level schematic diagram of ESIPT. | ||
Samanta et al. designed a simple AIE-ESIPT-coupled organic molecule bearing a suitable fluorophore for achieving tuneable white-light emission.65 The designed molecule 44 was found to be very weakly emissive in pure methanol, but this was enhanced in methanol–water mixtures with large water contents (Fig. 29).
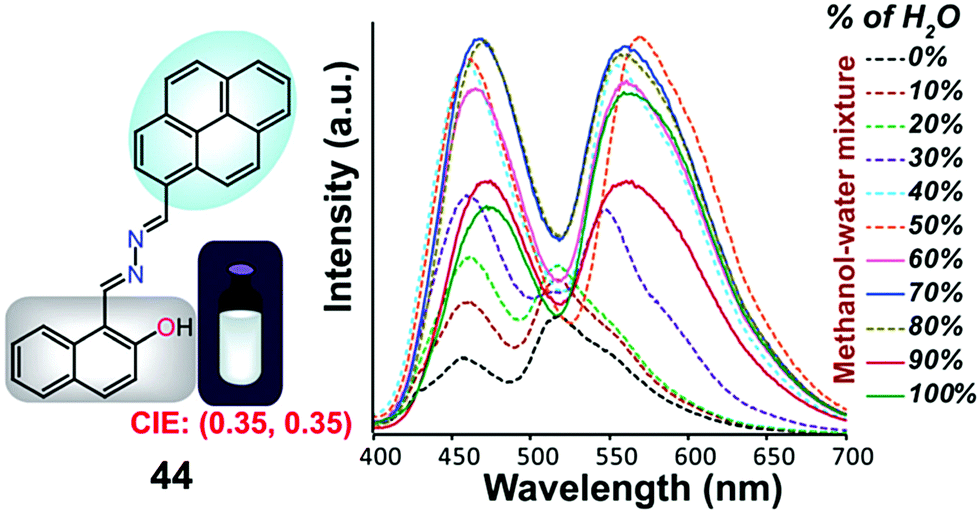 | ||
| Fig. 29 Structure of 44 and fluorescence spectra (10 mM) upon a change in the water fraction of a methanol–water mixed solvent; λex = 380 nm. Reproduced from ref. 65. | ||
A pyrene-based fluorescent probe, 3-methoxy-2-((pyren-2ylimino)methyl)phenol 45 (Fig. 30), was found to exhibit high sensitivity and selectivity toward Al3+ over other relevant metal ions, and also displayed novel AIEE characteristics in its aggregated/solid state.66 This material could sense Al3+ in a 1:1 mode by signaling a significant fluorescence enhancement with a turn-on ratio of over ∼200-fold. Excess addition of Al3+ dramatically enhanced the fluorescence intensity through aggregation. The origin of such an AIEE phenomena for 45 was due to the single-bond rotation, which is mainly responsible for the dominant non-radiative decay.66 Thus, the RIR of 45 plays a crucial role in the AIEE characteristics as well as the PET off in the solid state. The same group also reported the AIEE materials 46 (Fig. 30), which can sense 2,4,6-trinitrophenol (TNP) by PET off and exhibit AIE in the aggregation state.67
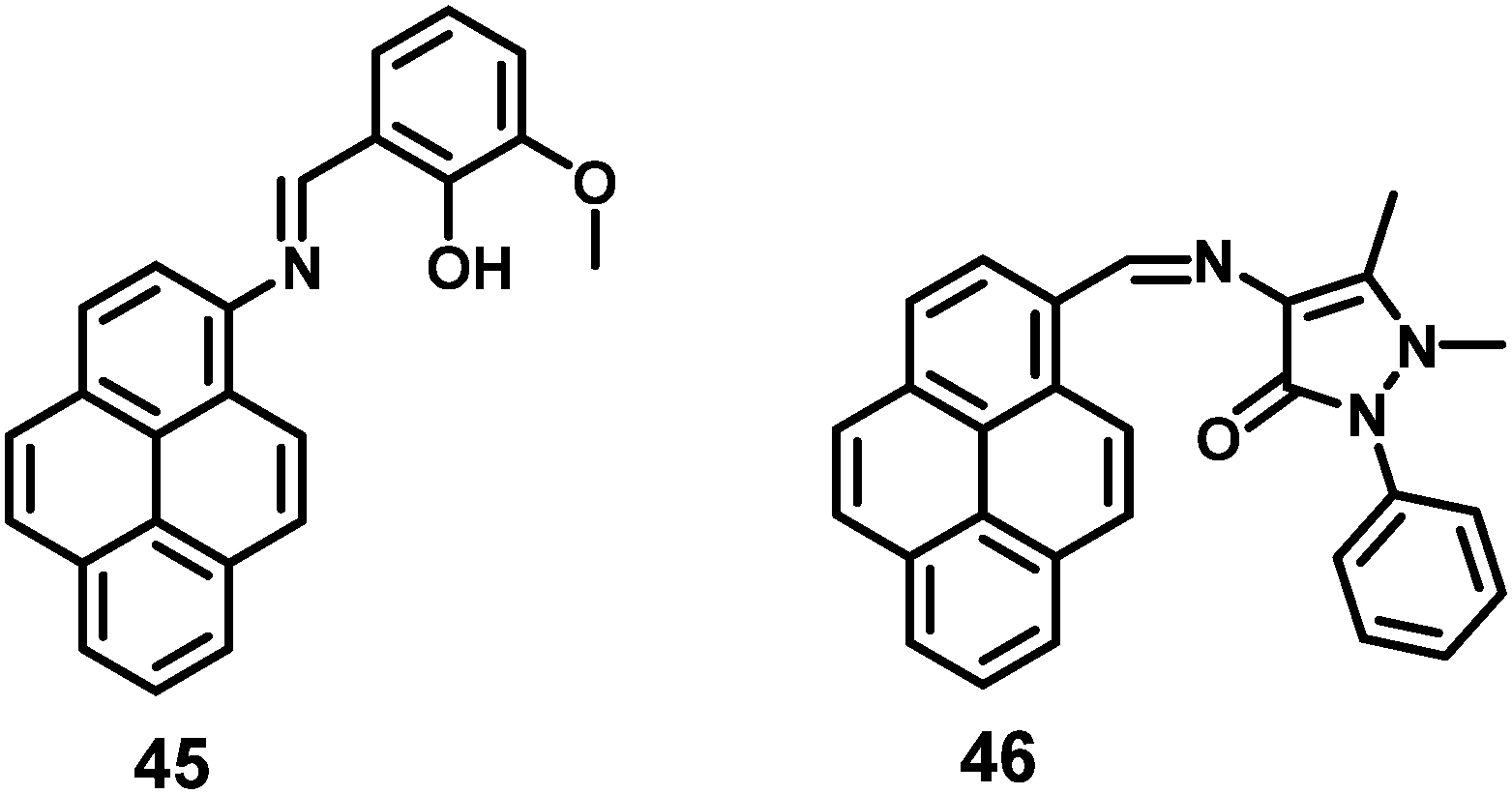 | ||
| Fig. 30 Structures of 45 and 46. | ||
2.7. Non-typical pyrene-based AIEgens: modification at the K-region
The pyrenoimidazoles 47 and 48 are weakly fluorescent in solution, and their AIE behavior has been studied in THF–water mixtures with different water content.68 The poor fluorescence of the pyrenoimidazoles 47 and 48 in THF was enhanced in the aggregated suspension (95% water fraction) by 38- and 9-fold, respectively (Fig. 31). The pristine forms of pyrenoimidazole absorb at 417 nm and 427 nm, respectively, which upon grinding, exhibit bathochromic shifts and absorb at 446 nm and 437 nm, respectively. RIR and X-ray diffraction (XRD) studies reveal a transformation of the twisted crystalline state to the planar amorphous state.68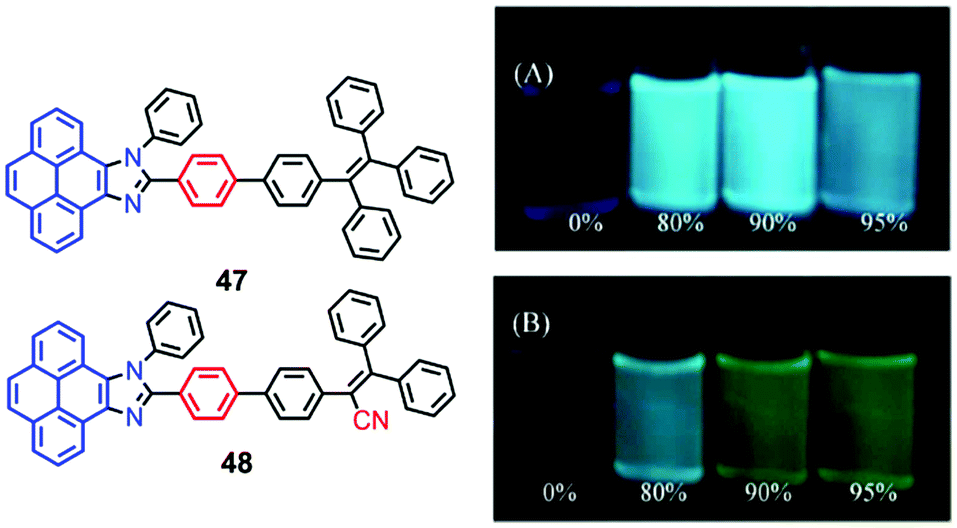 | ||
| Fig. 31 Photographs of 47 (upper) (A) and 48 (lower) (B) in THF–water mixtures with different water fractions (10 mM) under 365 nm UV illumination. Reproduced from ref. 68. | ||
The pyrene-imidazole-based structural isomers, namely axis symmetric syn-PyDTI 49 and centrosymmetric anti-PyDTI 50, are non-emissive in THF but exhibited intense emission in their aggregate states. Crystalline syn-PyDTI 49 exhibited a 2-fold higher quantum efficiency than anti-PyDTI 50, and displayed a much better performance in OLEDs with a ηC,max of 11.4 cd A−1 (8.8 cd A−1 for 50).69
Comparing the crystal emissions revealed a red-shift for the anti-isomer 50; that is, the syn-PyDTI 49 crystal exhibited an emission peak at 465 nm, whereas the anti-isomer 50 crystal displayed an emission peak at 507 nm. High-quality single crystals of syn- and anti-PyDTI emit blue and green, respectively, under UV-light. The blue-shift observed for syn-PyDTI 49 was probably associated with the formation of dimers, which resulted in a flexible geometry and a decreased conjugation length (Fig. 32).69 These observations reveal the dependence of the molecular excited-state properties on distinct structural symmetries. In recent years, pyrene-based, mechanochromism (MC)-active materials have been reported to possess attractive excimer fluorescence in the solid state, which is in contrast to the frequently encountered ACQ phenomena in planar luminogens and offers the possibility to discover mechanoluminescence (ML) luminogens with planar structures.70,71
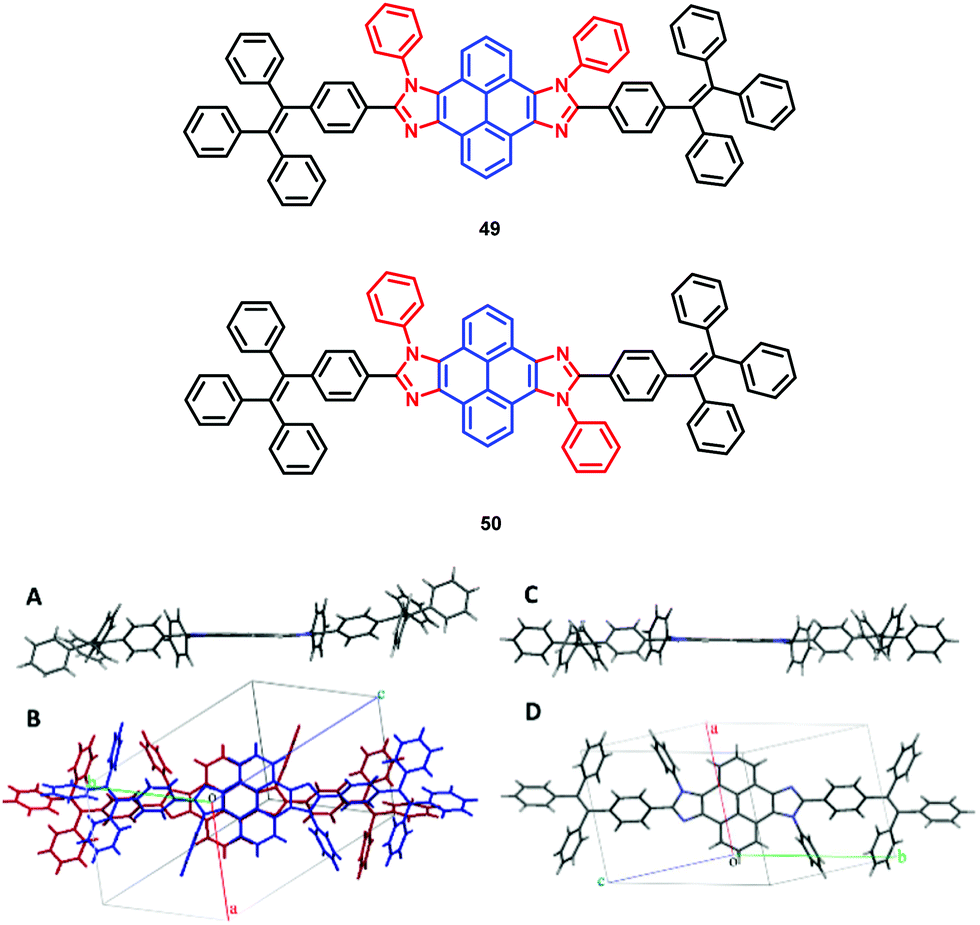 | ||
| Fig. 32 (A and C) Structures of syn-PyDTI 49 and anti-PyDTI 50. (B and D) Unit cell structure top views of crystalline syn- and anti-PyDTI. Reproduced from ref. 69. | ||
An outstanding feature associated with pyrene is its facile and varied structural modification. In addition to its four main points of reactivity (i.e., the 1-, 3-, 6- and 8-positions), recent studies have revealed that the non-typical K-region (4-, 5-, 9-, and 10-positions) is also available for further modification.2,9,72 In contrast to these recent advances at the 1-, 3-, 6,- and 8-positions, pyrene derivatives substituted at the 4-, 5-, 9-, and 10-positions (K-region) have been employed predominantly as synthetic intermediates for fused π-systems.73 However, only a few studies have examined the effects of substitution at the 4-, 5-, 9-, and 10-positions on the photophysical behavior of pyrenes.74
A new design strategy was introduced by the Sasaki group in order to achieve AIE emission in pyrene chromophores. This was accomplished by introducing N,N-dimethylamine substituents at the K-region 4,5-positions of pyrene, and these are likely to stabilize the minimum energy conical intersection (MECI).75 Four pyrene derivatives, which contain highly twisted N,N-dimethylamino groups at the 4- (4-Py), 4,5- (4,5-Py), 1- (1-Py), or 1,6-positions (1,6-Py) are 51–54 (Fig. 33). The non-radiative transitions of 4,5-Py 52 are highly efficient (knr = 57.1 × 107 s−1), so that its fluorescence quantum yield in acetonitrile decreases to Φ = 0.04.75 The solid-state fluorescence of 4,5-Py 52 is also efficient (Φ = 0.49). By contrast, 1,6-Py 54 features strong fluorescence (Φ = 0.48) in solution with a slow non-radiative transition (knr = 11.0 × 107 s−1) that is subject to severe quenching (Φ = 0.03) in the solid state. The introduction of strongly twisted N,N-dimethylamines at the 1- and 1,6-positions resulted in intense fluorescence in solution. These differences are attributed mainly to non-radiative transitions. A comparison of the knr value of 4,5-Py 52 with its fluorescence energetics implies the involvement of a special pathway; e.g. a non-adiabatic relaxation via MECI, for efficient internal conversions.75 The other derivatives 51 and 53 did not show noticeable emission changes in solution or the solid state, although they have AIE activity. The presence of highly twisted N,N-dialkylamino groups, especially at the 4,5-positions, induces fast non-radiative transitions both in polar and non-polar solvents, resulting in faint fluorescence in solution.75
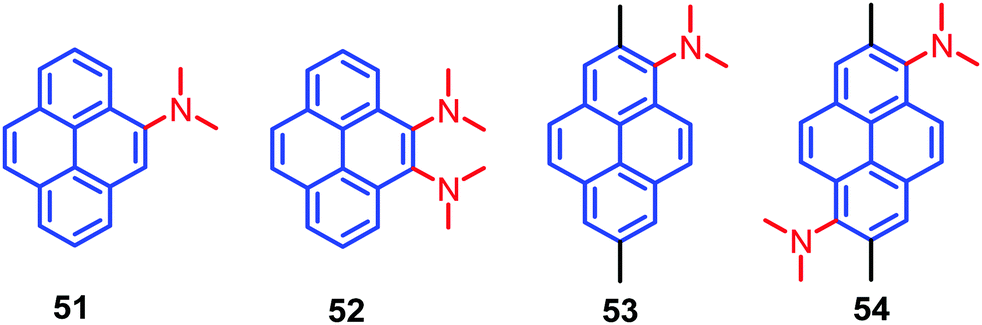 | ||
| Fig. 33 Structures of 51–54. | ||
Interestingly, a pyridine unit was introduced at the 1-position of pyrene, affording 2-(pyren-1-yl)pyridine 55 (Fig. 34).76 This compound exhibits a typical AIEE feature, and mechanisms were proposed by RIR, while the crystal structure of 55 revealed the presence of a C–H⋯π interaction at 2.75 Å between two of the pyridine rings of adjacent molecules which blocks the rotation of the pyridine ring in the solid state (Fig. 34). Furthermore, strong π–π interactions were observed in the range of 3.53–3.59 Å. Such short contacts would suppress the restriction of movement of the pyridine group in the solid state, and block the non-radiative pathways and open up new radiative pathways on the progressive addition of water. On the other hand, the face-to-face π–π interaction also caused an excimer emission in the aggregation state or in the crystal state.
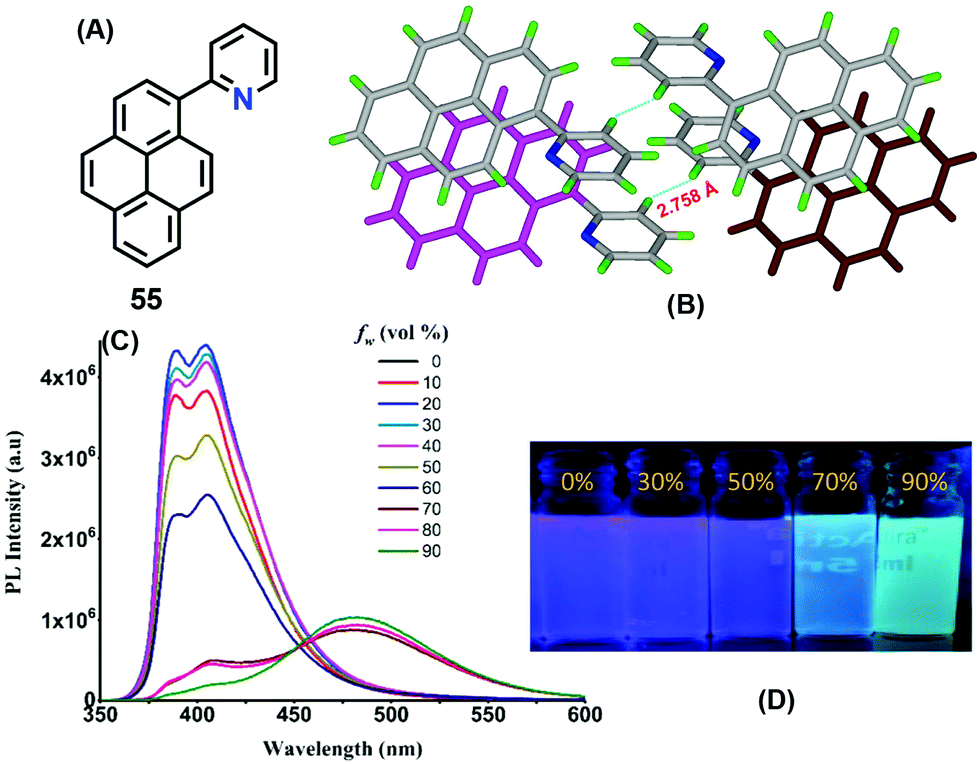 | ||
| Fig. 34 Emission spectra of 55 in different fractions of water in methanol and strong π–π interactions between the centroids of the two pyrene rings in the solid state. Reproduced from ref. 76. | ||
Interestingly, Dastidar et al. first observed the AIE phenomenon in organic salts. The primary ammonium salts of the pyrene-based carboxylic acid and histamine 56 (Fig. 35) exhibited an enhanced fluorescence in the solid state. The N acceptor and N–H donor of the imidazole moiety form a 3D hydrogen bonding network, thereby imposed RIR in the solid state, and triggering AIE activity in the pyrene salt 56.77 The pyrene-substituted o-carborane dyads 57 (Fig. 35) were synthesized via the insertion reaction between decaborane and 1-ethynylpyrene in the presence of a Lewis base using the Sonogashira–Hagihara coupling reaction. O-Carborane dyads emit extremely bright light in the solid state (ΦPL > 0.99).78
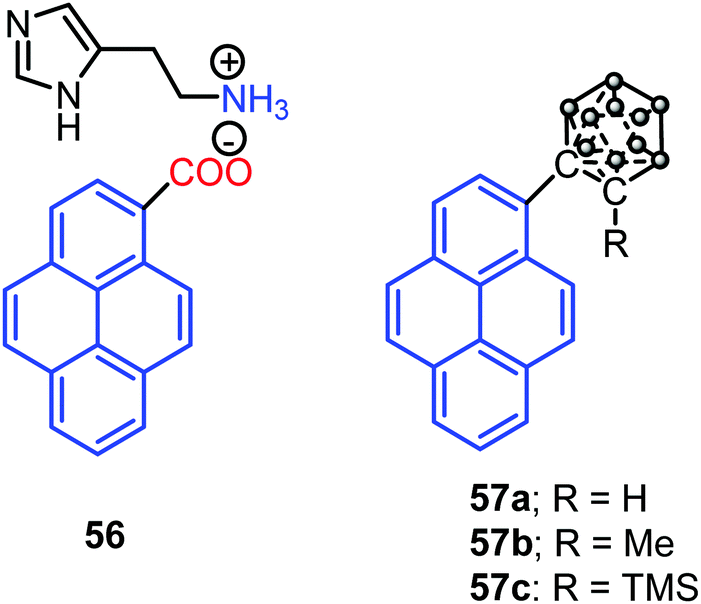 | ||
| Fig. 35 Structures of 56–57. | ||
2.8. Pyrene-based hexaphenylbenzene-like AIEgens
As mentioned above, steric interactions play an important role and can affect molecular configuration, as well as luminescence behavior. For example, the propeller-shaped hexaphenylbenzene (HPB), consists of six rotatable peripheral phenyl units with respect to the central benzene ring, and shows AEE character.79 With this in mind, new pyrene-based hexaphenylbenzene-like AIEgens 58 were explored, as shown in Fig. 36. Two phenyl units were replaced by a pyrene ring, and both anti-58 and syn-58 presented novel AIEE characteristics with a multiple photoluminescence, including monomer, excimer and CT emission. The multiple emission mechanisms originated from the different spatial arrangements, due to the presence of the large planar structure of the pyrene unit.80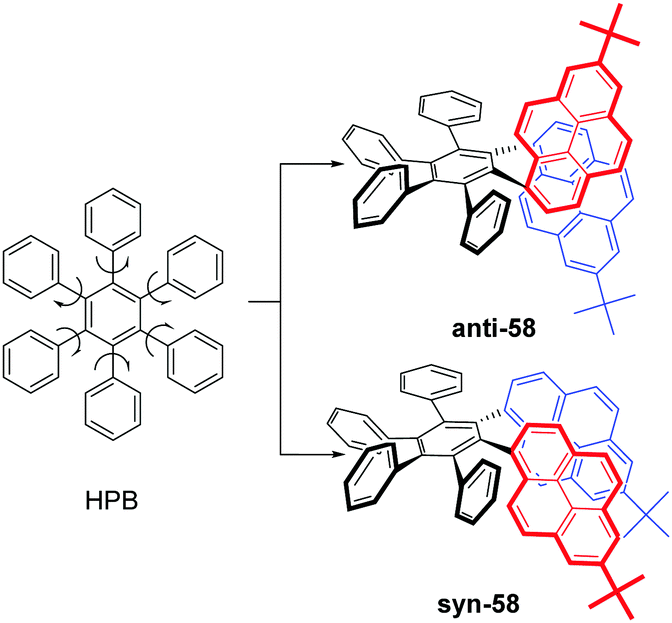 | ||
| Fig. 36 Structure of HPB and the pyrene-based hexaphenylbenzene-like AIEgens 58. | ||
3. Possible applications of pyrene-based AIEgens
“Touch a stone and turn it into gold”: pyrene, a dim luminogen in solution, can be twisted into a bright emitter as a solid by decorating it with suitable groups at different active positions. Solid-state fluorescent organic materials are essential components in optoelectronic devices, such as OLEDs, solid-state lasers fluorescent, sensors, organic solar cells (OSCs).8 However, the emission of most fluorescent organic molecules is effectively suppressed in the solid state, due to aggregation of the planar π-conjugation systems based on π–π interactions or the formation of excimers.7 As a result, there has been considerable interest in preparing pyrene-based AIEgens which retain their photophysical properties in the solid state. Pyrene-containing probes are very topical and remain of widespread interest because of their fast response via PET and excimer/exciplex formation toward specific species of interest.813.1. Organic light-emitting diodes
Pyrene is well known as a good deep blue emitter and is often utilized to construct blue luminogens.22a,82 An OLED, also termed organic electro luminescent device (OELD), is a light-emitting diode (LED) whose emissive electroluminescent layer is composed of an organic film. This layer of organic semiconductor material is formed between two electrodes, where at least one of the electrodes is transparent. An OLED material as a flat-panel display has many unique advantages, such as low driving voltage, high efficiency, excellent flexibility, vivid color, wide viewing angle. As a consequence, this technology has been used successfully for solid-state lighting applications. Pyrene is a promising chromophore with a high fluorescent quantum yield, and has attracted considerable interest in the design of small organic molecules for different applications including OLEDs83 and organic field-effect transistors (OFETs).70,84 For example, the optimization of non-doped OLEDs using pyrene-based 2,7-TPE-substituted AIEgens led to emitters that displayed sky-blue light at 492 nm at a low turn-on voltage (3.1 V) with maximum luminance of 15750 cd m−2, current efficiency of 7.34 cd A−1, and with low efficiency roll-off (Fig. 37).85
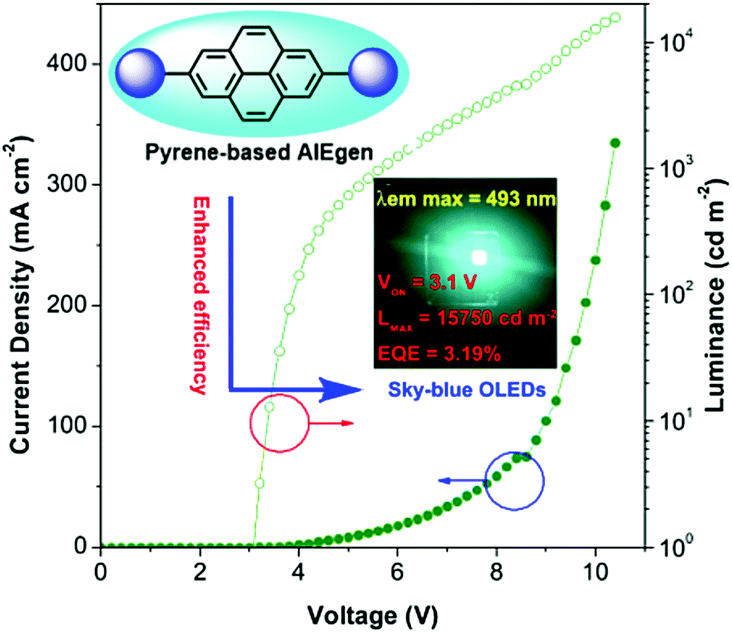 | ||
| Fig. 37 Using 2,7-disubstituted pyrenes with AIE characteristics as an emitter, the sky-blue OLED device exhibited excellent EL properties. Reproduced from ref. 85. | ||
N,N-Di-p-methoxyphenylamine-substituted pyrenes have been successfully used as hole-transporting materials for perovskite-based solar cells with high efficiency.86 Since pyrene has extensive π-electron delocalization, it is a fascinating core for developing fluorescent π-conjugated light-emitting monomers substituted by a variety of functionalized groups or can be deployed themselves as substituents and introduced into others molecules to afford blue EL.
Advantages of pyrene 1 include its excellent thermal and chemical stability, but it is not a good candidate for LEDs alone, due to its excimer emission tendency which lowers the fluorescence efficiency.10 However, this limitation has been overcome by introducing bulky groups with long alkyl chains, which enhances the fluorescence quantum efficiency in OLEDs and also reduces the aggregation of the final material.2
3.2. Chemosensors
As a classic chromophore, pyrene shows interesting fluorescence properties and has been used in chemosensors.23a,87 Pyrene-based AIE/AIEE probe molecules behave as selective and sensitive detectors of metal ions (Cu2+, Fe3+, Al3+), picric acid, and H+ ions etc.58,66,67,783.3. Organic field-effect transistors
The high fluorescence efficiency, electron rich ‘macrocyclic’ conjugation, and high charge carrier mobility all make pyrene a promising candidate for a variety of organic semiconducting materials.88 Pyrene-based organic semiconductors have great opportunities in OFETs as charge carrier channel layer materials possessing transport mobility.8b,87b,89Interestingly, Zhang et al. have reported two trans-β-styryl substituted pyrene-based AIEgens 1,6-PyE 59 and 2,7-PyE 60 which exhibited strong emission with semiconducting properties in the solid state.90 Microrods of 1,6-PyE 59 exhibit intense yellow-green emission (λmax,em: 463 nm), while microplates of 2,7-PyE 60 show brighter blue-emissions (λmax,em: 437 nm) with high quantum yields of 28.8% and 27.4%, respectively. Furthermore, thin films of the pyrene-based AIEgens, 59 and 60 (Fig. 38) exhibited p-type semiconducting properties with hole mobility up to 1.66 cm2 V−1 s−1, which originate from their unique intermolecular interactions and crystal packing. In addition, both compounds exhibited optical waveguiding behavior.
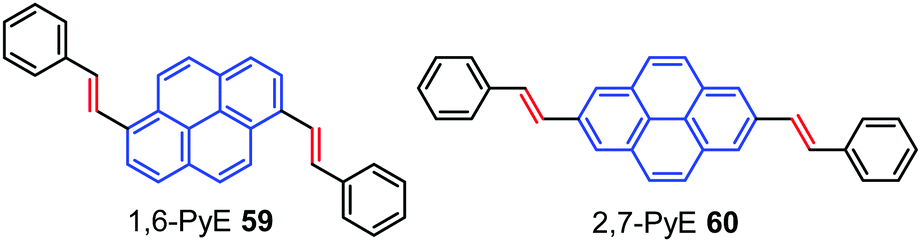 | ||
| Fig. 38 Molecular structures of 1,6-PyE 59 and 2,7-PyE 60. | ||
3.4. Solid-state lasers
Recently, a considerable amount of effort has been devoted to the design of laser materials and devices because of their potential applications.91 Solid-state lasers comprise a host and an active ion doped host material. The active ion should have excellent fluorescent, broad absorption bands, high quantum yield etc., and since pyrene possesses these properties, it is a good candidate for the design and development of new solid-state lasers.3.5. Biological applications
Pyrene-based AIE compounds show good biological activity against both Gram-positive (Rhodococcus rhodochrous and Staphylococcus aureus) and Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacteria and also fungal species (Candida albicans).60 Importantly, AIEE probes can sense M3+ ions, and their highly acidic pH-sensing ability has been successfully applied in cell imaging and cell viability analysis.63 The sensitive and selective detection of nucleic acids is important in biological studies, clinical diagnostics, and bio-defense applications, and pyrene-based AIEgen have potential in these fields.923.6. Probing critical micellar concentration
The critical micellar concentration (cmc) of both ionic and non-ionic surfactants can conveniently be determined from measurement of the UV absorption of pyrene in surfactant solution. Surfactants can assemble in solution, and cmc is an important solution property of surfactants. There are several frequently used methods such as tensiometry, conductometry, fluorimetry, and calorimetry, which can be used for its determination.93 Spectral methods using dyes as probes, and self-absorption of amphiphiles in solution are used for the evaluation of cmc.94 As fluorescent probes, pyrene molecules have been successfully employed to estimate the polarity level of micelles as well as the cmc of amphiphiles.953.7. Mechanoluminescent and mechanochromism agents
The solid-state behavior of organic materials, which exhibit bright emission under mechanical actions such as pressure, grinding, shearing, rubbing, temperature etc., is attracting much attention due to its scientific significance and practical applications in many fields, including uses in deformation detectors, new light sources, and pressure sensing.96For example, Zhen Li et al. reported the unique solid-state packing style of pyrene and the bright dual monomer–excimer-ML and excimer-ML from two pyrene derivatives (Py-Bpin 61b and Py-Br 61c). The simple, planar molecular structure was compared with pyrene and the analog Py-H 61a (Fig. 39).97 These four luminogens display similar optical properties in dilute solution, but different luminescent properties in the solid state, mainly due to their different molecular packing in the crystalline form. For example, pyrene is MC- and ML-inactive, while Py-H 61a is MC-active, Py-Bpin 61b is MC- and ML-active, and Py-Br 61c is ML-active. The MC-active pyrene derivatives (Py-H 61a & Py-Bpin 61b) have monomer arrangements with very few overlaps. The ML-active pyrene derivatives (Py-Bpin 61b & Py-Br 61c) have strong intermolecular interactions in the solid state. Both a monomer arrangement and strong intermolecular interactions are required in MCL-active pyrene derivatives (Py-Bpin 61b). These results demonstrate that planar molecules can also have bright ML if the luminogens possess strong intermolecular interactions and no ACQ in the solid state.
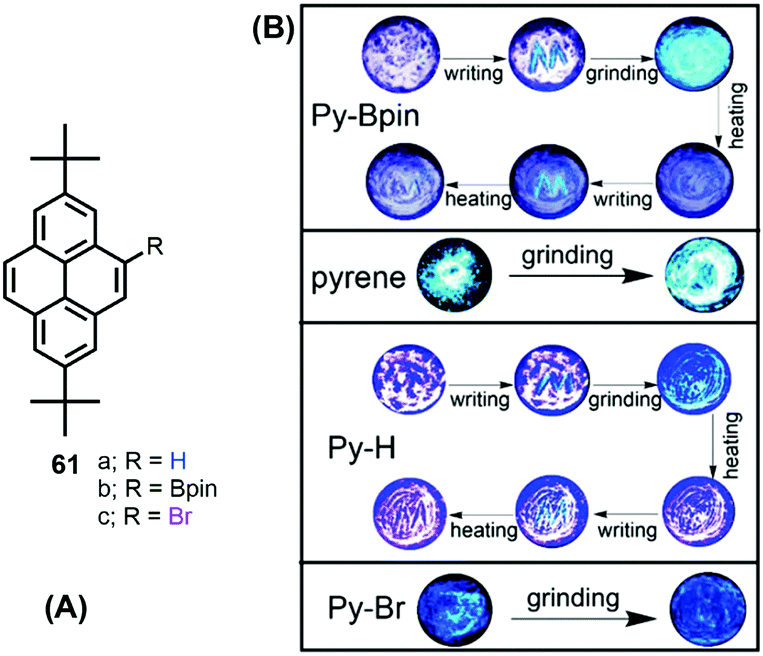 | ||
| Fig. 39 Structures and images of mechanochromic experiments involving pyrene 1, Py-Bpin (61b), Py-H (61a) and Py-Br (61c). Reproduced from ref. 97. | ||
4. Perspectives and conclusions
In conclusion, pyrene is an important member of the PHA family, which has received considerable attention in a variety of applications, and recent reports in the literature reveal that it continues to attract the interest of chemists and those involved in materials science and beyond. To further expand the pyrene chemistry research, the basis of this review is to focus on summarizing the efficient strategies used to develop pyrene-based AIEgens, which are very efficient and promising materials with fantastic optical properties. This includes (1) it is possible to achieve pyrene-based AIEgens by introducing typical AIE units, such as TPE, TriPE, into the active site of pyrenes. (2) It seems that CC or CN bonds play an important role in constructing the pyrene-based AIEgens and this molecular configuration can contribute to the molecular rotation against the CC/CN bond around the single-bond axes which non-radiatively consume the exciton energy. (3) The molecular (crystal) packing can affect the photophysical properties, and can lead to interesting optical phenomena, such as mechanoluminescent/mechanochromism. Thus, the twisted molecular structure bearing a bulky group can suppress intramolecular rotation and enhance the intensity of fluorescence in the aggregated state. (4) In addition, the pyrene-based primary ammonium salts also exhibited an enhanced fluorescence in the solid state, which may be due to the anion–π+ interactions suppressing any π–π stacking. This is useful approach for the design and synthesis of pyrene-based AIEgens.98
The electronic composition at the active sites (1,3,6,8-positions), the nodal plane (2,7-positions), and the K-region (4,5,9,10-positions) of pyrene are all different. When terminal groups are attached at the 1,3,6,8-positions, this leads to a large red-shift in the emission compared to when they are positioned at the nodal plane and the K-region.
By contrast, substitution at the 2,7-positions of pyrene has only a limited effect on the emission shift, due to the weak electronic interaction communication between the pyrene core and the substituted moieties. The K-region of pyrene exhibits a more special electronic structure, the substituent groups at the 4,5,9,10-positions can affect the S1 ← S0, S2 ← S0 and S3 ← S0 transitions.99 According to the Jablonski diagram, 25% of the singlet excitons can be utilized to produce light, while 75% of the radiative decay of triplet excitons is forbidden.100 To improve the quantum yield of luminogens devices, there is needs to harvest both singlet and triplet excitons under electrical excitation for emission. There are two main strategies (i) convert the triplet excitons (75%) into singlet excitons via reverse intersystem crossing (RISC) processes under thermal activation101 and (ii) develop phosphorescent emitters with high efficiency.102 Thus, to achieve highly efficient AIEgens, it is better to design and synthesize novel pyrene-based AIEgens to overcome the ACQ effect. Moreover, the new concepts for molecular design, such as aggregation-induced delayed fluorescence (AIDF),103 thermally activated delayed fluorescence (TADF),104 and room-temperature phosphorimetry (RTP)105 can be introduced into the pyrene-based AIE system to improve the efficient exciton for organic semiconductor applications.
This review will be helpful in understanding and designing new high-efficiency pyrene-based solid emitters for further investigations of different applications, such as OLEDs, biosensors, chemosensors, and chiral recognition. Such studies will also shed further light on the key roles of the AIE or AIEE processes in pyrene chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21602014), and the Youth Fund of Guangdong University of Technology (220413205). MM Islam thanks MOST, PR China for a Talented Young Scientist Program (TYSP) Fellowship; CR thanks the EPSRC for an Overseas Travel Grant.Notes and references
- (a) S. Tao, Z. Peng, X. Zhang, P. Wang, C. S. Lee and S. T. Lee, Adv. Funct. Mater., 2005, 15, 1716 CrossRef CAS; (b) J. Jayabharathi, P. Jeeva, V. Thanikachalam and S. Panimozhi, J. Photochem. Photobiol., A, 2017, 346, 296 CrossRef CAS; (c) J. K. Salunke, F. L. Wong, K. Feron, S. Manzhos, M. F. Lo, D. Shinde, A. Patil, C. S. Lee, V. A. L. Roy, P. Sonar and P. P. Wadgaonkar, J. Mater. Chem. C, 2016, 4, 1009 RSC; (d) D. Chercka, S.-J. Yoo, M. Baumgarten, J.-J. Kim and K. Müllen, J. Mater. Chem. C, 2014, 2, 9083 RSC.
- M. T. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260 CrossRef PubMed.
- S. Karuppannan and J. C. Chambron, Chem. – Asian J., 2011, 6, 964 CrossRef CAS.
- H. Maeda, T. Maeda, K. Mizuno, K. Fujimoto, H. Shimizu and M. Inouye, Chem. – Eur. J., 2006, 12, 824 CrossRef CAS PubMed.
- Z. Wang, C. Xu, X. Wang, X. Dong, B. Zhao and B. Ji, Dyes Pigm., 2011, 92, 732 CrossRef.
- T. Oyamada and H. Uchiuzou, J. Appl. Phys., 2005, 98, 74506 CrossRef.
- (a) Z. Zhang, B. Xu, J. Su, L. Shen, Y. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654 CrossRef CAS; (b) Q. Wang, H. Lu, L. Gai, W. Chen, G. Lai and Z. Li, Dyes Pigm., 2012, 94, 183 CrossRef CAS; (c) M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516 CrossRef CAS PubMed.
- (a) Organic light-emitting materials and devices, ed. Z. Li and H. Meng, CRC Press/Taylor & Francis, Boca Raton, 2007 Search PubMed; (b) F. Cicoira and C. Santato, Adv. Funct. Mater., 2007, 17, 3421 CrossRef CAS; (c) Introduction to organic electronic and optoelectronic materials and devices, ed. S. S. Sun and L. R. Dalton, Taylor & Francis, Boca Raton, 2008 Search PubMed.
- (a) X. Feng, J.-Y. Hu, C. Redshaw and T. Yamato, Chem. – Eur. J., 2016, 22, 11898 CrossRef CAS PubMed; (b) Y. Gong, K. Chang, C. Chen, M. Han, X. Zhan, J. Min, X. Jiao, Q. Li and Z. Li, Mater. Chem. Front., 2019, 3, 93 RSC.
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970 Search PubMed; (b) J. R. Lackowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 2nd edn, 1999, p. 595 CrossRef.
- P. Sonar, M. S. Soh, Y. H. Cheng, J. T. Henssler and A. Sellinger, Org. Lett., 2010, 12, 3292 CrossRef CAS.
- (a) M. J. J. Dewar and R. D. Dennington II, J. Am. Chem. Soc., 1989, 111, 3804 CrossRef CAS; (b) H. Cerfontain, K. Laali and H. J. A. Lambrechts, Recl. Trav. Chim. Pays-Bas, 1983, 102, 210 CrossRef CAS.
- (a) X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261 CrossRef CAS PubMed; (b) H. Tong, Y. Hong, Y. Dong, Y. Ren, M. Haussler, J. W. Y. Lam, K. S. Wong and B. Z. Tang, J. Phys. Chem. B, 2007, 111, 2000 CrossRef CAS PubMed.
- G. Venkataramana and S. Sankararaman, Eur. J. Org. Chem., 2005, 4162 CrossRef CAS.
- (a) C. Lambert, J. Ehbets, D. Rausch and M. Steeger, J. Org. Chem., 2012, 77, 6147 CrossRef CAS PubMed; (b) L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brückner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750 CrossRef CAS PubMed; (c) A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349 CrossRef CAS PubMed.
- C. Wanninger-Weiß and H.-A. Wagenknecht, Eur. J. Org. Chem., 2008, 64 CrossRef.
- (a) J. Wang, Y. Zhao, C. Dou, H. Sun, P. Xu, K. Ye, J. Zhang, S. Jiang, F. Li and Y. Wang, J. Phys. Chem. B, 2007, 111, 5082 CrossRef CAS PubMed; (b) S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS; (c) B. T. Nguyen, J. E. Gautrot, C. Y. Ji, P. L. Brunner, M. T. Nguyen and X. X. Zhu, Langmuir, 2006, 22, 4799 CrossRef CAS PubMed.
- (a) D. Y. Kim, H. N. Cho and C. Y. Kim, Prog. Polym. Sci., 2000, 25, 1089 CrossRef CAS; (b) C. T. Chen, Chem. Mater., 2004, 16, 4389 CrossRef CAS.
- (a) K. A. Zachariasse, Trends Photochem. Photobiol., 1994, 3, 211 CAS; (b) J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. London, Ser. A, 1963, 275, 575 CrossRef CAS.
- (a) J. A. Mikroyannidis, L. Fenenko and C. Adachi, J. Phys. Chem. B, 2006, 110, 20317 CrossRef CAS PubMed; (b) Z.-Q. Liang, Y.-X. Li, J.-X. Yang, Y. Ren and X.-T. Tao, Tetrahedron Lett., 2011, 52, 1329 CrossRef CAS; (c) C. Tang, F. Liu, Y.-J. Xia, L.-H. Xie, A. Wei, S.-B. Li, Q.-L. Fan and W. Huang, J. Mater. Chem., 2006, 16, 4074 RSC.
- W. Z. Yuan, P. Lu, S. M. Chen, J. W. Y. Lam, Z. M. Wang, Y. Liu, H. S. Kwok, Y. G. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159 CrossRef CAS PubMed.
- (a) K. C. Wu, P. J. Ku, C. S. Lin, H. T. Shih, F. I. Wu, M. J. Huang, J. J. Lin, I.-C. Chen and C. H. Cheng, Adv. Funct. Mater., 2008, 18, 67 CrossRef CAS; (b) T. M. Figueira-Duarte, P. G. Del Rosso, R. Trattnig, S. Sax, E. J. W. List and K. Müllen, Adv. Mater., 2009, 21, 1 Search PubMed.
- (a) F. M. Winnik, Chem. Rev., 1993, 93, 587 CrossRef CAS; (b) G. Venkataramana and S. Sankararaman, Org. Lett., 2006, 8, 2739 CrossRef CAS PubMed; (c) V. de Halleux, J.-P. Calbert, P. Brocorens, J. Cornil, J.-P. Declercq, J. L. Bredas and Y. Geerts, Adv. Funct. Mater., 2004, 14, 649 CrossRef CAS; (d) D. Rausch and C. Lambert, Org. Lett., 2006, 8, 5037 CrossRef CAS PubMed; (e) J.-Y. Hu, X. Feng, A. Paudel, H. Tomiyasu, U. Rayhan, P. Thuery, M. R. J. Elsegood, C. Redshaw and T. Yamato, Eur. J. Org. Chem., 2013, 5829 CrossRef CAS; (f) X. Feng, J.-Y. Hu, L. Yi, N. Seto, Z. Tao, C. Redshaw, M. R. J. Elsegood and T. Yamato, Chem. – Asian J., 2012, 7, 2854 CrossRef CAS PubMed; (g) X. Feng, J.-Y. Hu, F. Iwanaga, N. Seto, C. Redshaw and M. R. J. Elsegood, Org. Lett., 2013, 15, 1318 CrossRef CAS PubMed; (h) Y. Qiao, J. Zhang, W. Xua and D. Zhu, Tetrahedron, 2011, 67, 3395 CrossRef CAS.
- I. B. Berlman, J. Phys. Chem., 1970, 74, 3085 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (d) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (e) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC.
- (a) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972 CrossRef CAS PubMed.
- Y. Wang, T. Liu, L. Bu, J. Li, C. Yang, X. Li, Y. Tao and W. Yang, J. Phys. Chem. C, 2012, 116, 15576 CrossRef CAS.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS.
- Y. Li, F. Li, H. Zhang, Z. Xie, W. Xie, H. Xu, B. Li, F. Shen, L. Ye, M. Hanif, D. Ma and Y. Ma, Chem. Commun., 2007, 231 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40 RSC.
- R. Deans, J. Kim, M. R. Machacek and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 8565 CrossRef CAS.
- K. Perumal, J. A. Garg, O. Blacque, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Chem. – Asian J., 2012, 7, 2670 CrossRef CAS PubMed.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956 CrossRef CAS PubMed.
- (a) N. B. Shustova, B. D. McCarthy and M. Dinca, J. Am. Chem. Soc., 2011, 133, 20126 CrossRef CAS PubMed; (b) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- B. Manimaran, P. Thanasekaran, T. Rajendran, R. J. Lin, I. J. Chang, G. H. Lee, S. M. Peng, S. Rajagopal and K. L. Lu, Inorg. Chem., 2002, 41, 5323 CrossRef CAS PubMed.
- B. Pramanik and D. Das, J. Phys. Chem. C, 2018, 122, 3655 CrossRef CAS.
- (a) R. J. Argauer and C. E. White, Anal. Chem., 1964, 36, 368 CrossRef CAS; (b) C. A. Royer, Biophys. J., 1995, 1191 CrossRef CAS; (c) M. R. Eftink, Biophys. J., 1994, 66, 482 CrossRef CAS PubMed; (d) S.-J. Kim, F. N. Chowdhury, W. Stryjewski, E. S. Younathan, P. S. Russo and M. D. Barkley, Biophys. J., 1993, 65, 215 CrossRef CAS PubMed.
- Q. Wang, H. Liu, H. Lu, Z. Xu, G. Lai, Z. Li, J. Mack and Z. Shen, Dyes Pigm., 2013, 99, 771 CrossRef CAS.
- Z. J. Zhao, S. M. Chen, J. W. Y. Lam, P. Lu, Y. C. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221 RSC.
- (a) J. Yang, J. Huang, N. Sun, Q. Peng, Q. Li, D. Ma and Z. Li, Chem. – Eur. J., 2015, 21, 6862 CrossRef CAS PubMed; (b) J. Yang, L. Li, Y. Yu, Z. Ren, Q. Peng, S. Ye, Q. Li and Z. Li, Mater. Chem. Front., 2017, 1, 91 RSC.
- X. Feng, C. Qi, H.-T. Feng, Z. Zhao, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, A. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 5679 RSC.
- (a) C. Y. K. Chan, J. W. Y. Lam, J. Zhao, S. M. Chen, P. Lu, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4320 RSC; (b) T. Jadhav, J. M. Choi, J. Y. Lee, B. Dhokale and R. Misra, Org. Electron., 2016, 37, 448 CrossRef CAS; (c) T. Jadhav, B. Dhokale, Y. Patil, S. M. Mobin and R. Misra, J. Phys. Chem. C, 2016, 120, 24030 CrossRef CAS.
- (a) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed; (b) L. Chen, Y. Jiang, H. Nie, R. Hu, H. S. Kwok, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17215 CrossRef CAS PubMed.
- (a) Y. Liu, S. Chen, J. W. Y. Lam, P. Lu, T. K. Kwok, F. Mahtab, H. S. Kwok and B. Z. Tang, Chem. Mater., 2011, 23, 2536 CrossRef CAS; (b) Z. Zhao, P. Lu, J. W. Y. Lam, Z. Wang, C. Y. K. Chan, H. H. Y. Sung, I. D. Williams, Y. Ma and B. Z. Tang, Chem. Sci., 2011, 2, 672 RSC.
- (a) Z. Zhao, S. Chen, J. W. Y. Lam, Z. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. Ma, H. S. Kwok and B. Z. Tang, J. Mater. Chem., 2011, 21, 7210 RSC; (b) W. Wang, T. Lin, M. Wang, T.-X. Liu, L. Ren, D. Chen and S. Huang, J. Phys. Chem. B, 2010, 114, 5983 CrossRef CAS PubMed; (c) D. Jana, S. Boxi and B. K. Ghorai, Dyes Pigm., 2013, 99, 740 CrossRef CAS.
- Z. Zhang, Y. Zhao, R. Zhang, L. Zhang, W. Cheng and Z. H. Ni, Dyes Pigm., 2015, 118, 95 CrossRef CAS.
- J. Yang, Q. Guo, X. Wen, X. Gao, Q. Peng, Q. Li, D. Ma and Z. Li, J. Mater. Chem. C, 2016, 4, 8506 RSC.
- J. Yang, J. Qin, Z. Ren, Q. Peng, G. Xie and Z. Li, Molecules, 2017, 22, 2144 CrossRef PubMed.
- S. Son, A. Dodabalapur, A. J. Lovinger and M. E. Galvin, Science, 1995, 269, 376 CrossRef CAS PubMed.
- (a) B. Wang, Y. Wang, J. Hua, Y. Jiang, J. Huang, S. Qian and H. Tian, Chem. – Eur. J., 2011, 17, 2647 CrossRef CAS PubMed; (b) B. Gao, H. Wang, Y. Hao, L. Fu, H. Fang, Y. Jiang, L. Wang, Q. Chen, H. Xia, L. Pan, Y. Ma and H. Sun, J. Phys. Chem. B, 2010, 114, 128 CrossRef CAS PubMed; (c) Z. Wang, H. Shao, J. Ye, L. Tang and P. Lu, J. Phys. Chem. B, 2005, 109, 19627 CrossRef CAS PubMed; (d) J. N. Wilson, M. D. Smith, V. Enkelmann and U. H. F. Bunz, Chem. Commun., 2004, 1700 RSC; (e) C. J. Bhongale, C.-S. Chang, C.-S. Lee, E. W.-G. Diau and C.-S. Hsu, J. Phys. Chem. B, 2005, 109, 13472 CrossRef CAS PubMed; (f) H. Tong, Y. Dong, M. Häußler, Y. Hong, J. W. Y. Lam, H. H.-Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Chem. Phys. Lett., 2006, 428, 326 CrossRef CAS; (g) P. An, Z. Shi, W. Dou, X. Cao and H. Zhang, Org. Lett., 2010, 12, 4364 CrossRef CAS PubMed.
- Y.-X. Li, G.-X. Sun, J.-L. Miao, Y. Nie, Z.-W. Zhang and X.-T. Tao, Tetrahedron Lett., 2013, 54, 3263 CrossRef CAS.
- Y.-X. Li, X.-F. Yang, J.-L. Miao and G.-X. Sun, J. Phys. Chem. C, 2016, 120, 21722 CrossRef CAS.
- J. Katla, H. R. Bhat, P. C. Jha, P. S. Ghalsasi and S. Kanvah, ChemistrySelect, 2017, 2, 1902 CrossRef CAS.
- A. D. Aleo, A. Karapetyan, V. Heresanu, M. Giorgi and F. Fages, Tetrahedron, 2015, 71, 2255 CrossRef.
- A. Karuppusamy, T. Vandana and P. Kannan, J. Photochem. Photobiol., A, 2017, 345, 11 CrossRef CAS.
- (a) D. A. Pearce, N. Jotterand, I. S. Carrico and B. Imperiali, J. Am. Chem. Soc., 2001, 123, 5160 CrossRef CAS PubMed; (b) M. Mameli, M. C. Aragoni, M. Arca, C. Caltagirone, F. Demartin, G. Farruggia, G. D. Filippo, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis, S. Murgia, L. Prodi, A. Pintus and N. Zaccheroni, Chem. – Eur. J., 2010, 16, 919 CrossRef CAS PubMed; (c) Y. Bao, B. Liu, H. Wang, F. Du and R. Bai, Anal. Methods, 2011, 3, 1274 RSC; (d) Y. Bao, B. Liu, F. Du, J. Tian, H. Wang and R. Bai, J. Mater. Chem., 2012, 22, 5291 RSC.
- M. Shellaiah, Y.-H. Wu, A. Singh, M. V. R. Raju and H.-C. Lin, J. Mater. Chem. A, 2013, 1, 1310 RSC.
- (a) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS PubMed; (b) G. Aragay, J. Pons and A. Merkoci, Chem. Rev., 2011, 111, 3433 CrossRef CAS PubMed.
- A. Kathiravan, K. Sundaravel, M. Jaccob, G. Dhinagaran, A. Rameshkumar, D. A. Ananth and T. Sivasudha, J. Phys. Chem. B, 2014, 118, 13573 CrossRef CAS PubMed.
- Y. Li, W. Huang, J. Yong, S. Huang, Y. Li, Y. Liu and D. Wu, New J. Chem., 2018, 42, 12644 RSC.
- Q. Chai, J. Wei, B. Bai, H. Wang and M. Li, Dyes Pigm., 2018, 152, 93 CrossRef CAS.
- M. Shellaiah, T. Simon, V. Srinivasadesikan, C.-M. Lin, K. W. Sun, F.-H. Ko, M.-C. Lin and H.-C. Lin, J. Mater. Chem. C, 2016, 4, 2056 RSC.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615 CrossRef CAS PubMed.
- S. Samanta, U. Manna and G. L. Das, New J. Chem., 2017, 41, 1064 RSC.
- M. Shyamal, P. Mazumdar, S. Maity, G. P. Sahoo, G. Salgado-Morán and A. Misra, J. Phys. Chem. A, 2016, 120, 210 CrossRef CAS PubMed.
- M. Shyamal, S. Maity, P. Mazumdar, G. P. Sahoo, R. Maity and A. Misra, J. Photochem. Photobiol., A, 2017, 342, 1 CrossRef CAS.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981 RSC.
- Y. Liu, T. Shan, L. Yao, Q. Bai, Y. Guo, J. Li, X. Han, W. Li, Z. Wang, B. Yang, P. Lu and Y. Ma, Org. Lett., 2015, 17, 6138 CrossRef CAS.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520 CrossRef CAS.
- (a) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS; (b) M. J. Teng, X. R. Jia, S. Yang, X. F. Chen and Y. Wei, Adv. Mater., 2012, 24, 1255 CrossRef CAS PubMed; (c) M. J. Teng, X. R. Jia, X. F. Chen and Y. Wei, Angew. Chem., Int. Ed., 2012, 51, 6398 CrossRef CAS; (d) T. S. Wang, N. Zhang, K. Zhang, J. W. Dai, W. Bai and R. K. Bai, Chem. Commun., 2016, 52, 9679 RSC.
- L. Ji, A. Lorbach, R. M. Edkins and T. B. Marder, J. Org. Chem., 2015, 80, 5658 CrossRef CAS.
- (a) L. Zöphel, D. Beckmann, V. Enkelmann, D. Chercka, R. Rieger and K. Müllen, Chem. Commun., 2011, 47, 6960 RSC; (b) T. Yamato, M. Fuimoto, A. Miyazawa and K. Matsuo, J. Chem. Soc., Perkin Trans. 1, 1997, 1201 RSC; (c) B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051 CrossRef CAS PubMed; (d) H. M. Duong, M. Bendikov, D. Steiger, Q. Zhang, G. Sonmez, J. Yamada and F. Wudl, Org. Lett., 2003, 5, 4433 CrossRef CAS PubMed; (e) K. Baumgärtner, K. A. L. M. Chincha, A. Dreuw, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef.
- (a) L. Zöphel, V. Enkelmann and K. Müllen, Org. Lett., 2013, 15, 804 CrossRef; (b) S. N. Keller, N. L. Veltri and T. C. Sutherland, Org. Lett., 2013, 15, 4798 CrossRef CAS; (c) M. Piotrowicz, J. Zakrzewski, R. Métivier, A. Brosseau, A. Makal and K. Woźniak, J. Org. Chem., 2015, 80, 2573 CrossRef CAS PubMed.
- S. Sasaki, S. Suzuki, K. Igawa, K. Morokuma and G.-I. Konishi, J. Org. Chem., 2017, 82, 6865 CrossRef CAS PubMed.
- V. Kachwal, P. Alam, H. R. Yadav, S. S. Pasha, A. R. Choudhury and I. R. Laskar, New J. Chem., 2018, 42, 1133 RSC.
- U. K. Das and P. Dastidar, J. Chem. Sci., 2014, 126, 1357 CrossRef CAS.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064 CrossRef CAS PubMed.
- V. Vij, V. Bhalla and M. Kumar, Chem. Rev., 2016, 116, 9565 CrossRef CAS PubMed.
- (a) C.-Z. Wang, Y. Noda, C. Wu, X. Feng, P. Venkatesan, H. Cong, M. R. J. Elsegood, T. G. Warwick, S. J. Teat, C. Redshaw and T. Yamato, Asian J. Org. Chem., 2018, 7, 444 CrossRef CAS; (b) C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174 RSC.
- (a) Y. Niko, S. Kawauchi, S. Otsu, K. Tokumaru and G.-I. Konishi, J. Org. Chem., 2013, 78, 3196 CrossRef CAS PubMed; (b) Y. Niko, Y. Cho, S. Kawauchi and G.-I. Konishi, RSC Adv., 2014, 4, 36480 RSC.
- (a) A. Matsumoto, M. Suzuki, D. Kuzuhara, J. Yuasa, T. Kawai, N. Aratani and H. Yamada, Chem. Commun., 2014, 50, 10956 RSC; (b) H. Lee, B. Kim, S. Kim, J. Kim, J. Lee, H. Shin, J. Lee and J. Park, J. Mater. Chem. C, 2014, 2, 4737 RSC.
- (a) J. N. Moorthy, P. Natarajin, P. Venkatakrishnan, D. F. Huang and T. J. Chow, Org. Lett., 2007, 9, 5215 CrossRef CAS PubMed; (b) M. Y. Lo, C. G. Zhen, M. Lauters, G. E. Jabbour and A. Sellinger, J. Am. Chem. Soc., 2007, 129, 5808 CrossRef CAS PubMed; (c) R. D. Xia, W. Y. Lai, P. A. Levermore, W. Huang and D. D. C. Bradley, Adv. Funct. Mater., 2009, 19, 2844 CrossRef CAS; (d) Z. J. Zhao, J. H. Li, X. P. Chen, X. M. Wang, P. Lu and Y. Yang, J. Org. Chem., 2009, 74, 383 CrossRef CAS PubMed.
- (a) H. J. Zhang, Y. Wang, K. Z. Shao, Y. Q. Liu, S. Y. Chen, W. F. Qiu, X. B. Sun, T. Qi, Y. Q. Ma, G. Yu, Z. M. Su and D. B. Zhu, Chem. Commun., 2006, 755 RSC; (b) M. J. Sienkowska, J. M. Farrar, F. Zhang, S. Kusuma, P. A. Heiney and P. Kaszynski, J. Mater. Chem., 2007, 17, 1399 RSC; (c) F. Liu, W. Y. Lai, C. Tang, H. B. Wu, Q. Q. Chen, B. Peng, W. Wei, W. Huang and Y. Cao, Macromol. Rapid Commun., 2008, 29, 659 CrossRef CAS.
- X. Feng, Z. Xu, Z. Hu, C. Qi, D. Luo, X. Zhao, Z. Mu, C. Redshaw, J. W. Y. Lam, D. Ma and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 2283 RSC.
- N. J. Jeon, J. Lee, J. H. Noh, M. K. Nazeeruddin, M. Grätzel and S. Seok, J. Am. Chem. Soc., 2013, 135, 19087 CrossRef CAS PubMed.
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 CrossRef; (b) W. Z. Yuan, S. Chen, J. W. Y. Lam, C. Deng, P. Lu, H. H. Y. Sung, I. D. Williams, H. S. Kwok, Y. Zhang and B. Z. Tang, Chem. Commun., 2011, 47, 11216 RSC; (c) C. Shi, Z. Guo, Y. Yan, S. Zhu, Y. Xie, Y. S. Zhao, W. Zhu and H. Tian, ACS Appl. Mater. Interfaces, 2013, 5, 192 CrossRef CAS PubMed.
- (a) X. Gao and Z. Zhao, Sci. China: Chem., 2015, 58, 947 CrossRef CAS; (b) K. Oniwa, H. Kikuchi, H. Shimotani, S. Ikeda, N. Asao, Y. Yamamoto, K. Tanigaki and T. Jin, Chem. Commun., 2016, 52, 4800 RSC; (c) F. Moggia, C. Videlot-Ackermann., J. Ackermann, P. Raynal, H. Brisset and F. Fages, J. Mater. Chem., 2006, 16, 2380 RSC; (d) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296 CrossRef CAS PubMed.
- H. Ju, K. Wang, J. Zhang, H. Geng, Z. Liu, G. Zhang, Y. Zhao and D. Zhang, Chem. Mater., 2017, 29, 3580 CrossRef CAS.
- (a) X. J. Tang, L. Z. Wu, L. P. Zhang and C. H. Tung, Chem. Phys. Lett., 2002, 356, 573 CrossRef CAS; (b) G. S. He, T. C. Lin, V. K. S. Hsiao, A. N. Cartwright, P. N. Prasad, L. V. Natarajan, V. P. Tondiglia, R. Jakubiak, R. A. Vaia and T. J. Bunning, Appl. Phys. Lett., 2003, 83, 2733 CrossRef CAS.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401 CrossRef CAS PubMed.
- J. H. Clint, Surfactant Aggregation: Blacki, Chapman and Hall, New York, 1991 Search PubMed.
- (a) S. K. Hait, P. R. Majhi, A. Blume and S. P. Moulik, J. Phys. Chem. B, 2003, 107, 3650 CrossRef CAS; (b) M. Prasad, A. MacDonald, R. Palepu and S. P. Moulik, J. Surf. Sci. Technol., 2003, 19, 125 CAS.
- (a) G. B. Ray, I. Chakraborty and S. P. Moulik, J. Colloid Interface Sci., 2006, 294, 248 CrossRef PubMed; (b) K. Kalyansundaran and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039 CrossRef; (c) J. Aguiar, P. Carpana, J. A. Molina-Boliver and C. C. Ruiz, J. Colloid Interface Sci., 2003, 258, 116 CrossRef CAS.
- (a) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS PubMed; (b) Y. Q. Dong, J. W. Y. Lam, B. Z. Tang and J. Phys, Chem. Lett., 2015, 6, 3429 CAS; (c) J. Picard, Science, 1939, 89, 460 CrossRef; (d) N. C. Eddingsaas and K. S. Suslick, Nature, 2006, 444, 163 CrossRef CAS PubMed.
- Y.-B. Gong, P. Zhang, Y.-R. Gu, J.-Q. Wang, M.-M. Han, C. Chen, X.-J. Zhan, Z.-L. Xie, B. Zou, Q. Peng, Z.-G. Chi and Z. Li, Adv. Opt. Mater., 2018, 6, 1800198 CrossRef.
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974 CrossRef CAS.
- (a) M. P. Fernández-Liencres, T. Peña-Ruiz, J. M. Granadino-Roldan, M. Moral, A. Valenzuela-Pereira, A. Garzón-Ruiz and A. Navarro, J. Phys. Chem. C, 2018, 122, 12002 CrossRef; (b) A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS.
- K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 127, 15446 CrossRef.
- (a) W. Zhao, Z. He, Q. Peng, J. W. Y. Lam, H. Ma, Z. Qiu, Y. Chen, Z. Zhao, Z. Shuai, Y. Dong and B. Z. Tang, Nat. Commun., 2018, 9, 3044 CrossRef PubMed; (b) Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181 CrossRef CAS PubMed.
- J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S.-J. Su, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971 CrossRef CAS.
- (a) P. Rajamalli, N. Senthilkumar, P. Gandeepan, P.-Y. Huang, M.-J. Huang, C.-Z. Ren-Wu, C.-Y. Yang, M.-J. Chiu, L.-K. Chu, H.-W. Lin and C.-H. Cheng, J. Am. Chem. Soc., 2016, 138, 628 CrossRef CAS PubMed; (b) G. Xie, J. Luo, M. Huang, T. Chen, K. Wu, S. Gong and C. Yang, Adv. Mater., 2017, 29, 1604223 CrossRef PubMed.
- L. L. Liu, J. Zhou, R. Andrews and D. W. Stephan, J. Am. Chem. Soc., 2018, 14, 7466 CrossRef.
Footnote |
| † This review is dedicated to Emeritus Professor Takehiko Yamato (Saga University, Japan) on the occasion of his 66th birthday. |
| This journal is © the Partner Organisations 2019 |