
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChan–Lam coupling reaction of sulfamoyl azides with arylboronic acids for synthesis of unsymmetrical N-arylsulfamides†
Suk-Young
Won
a,
Seo-Eun
Kim
a,
Yong-Ju
Kwon
a,
Inji
Shin
bc,
Jungyeob
Ham
d and
Won-Suk
Kim
 *a
*a
aDepartment of Chemistry and Nanoscience, Ewha Womans University, Seoul 03760, South Korea. E-mail: wonsukk@ewha.ac.kr
bTherapeutics & Biotechnology Division/Innovative Therapeutics Research Center, Korea Research Institute of Chemical Technology (KRICT), Daejeon 34602, South Korea
cDepartment of Medicinal and Pharmaceutical Chemistry, University of Science and Technology, Daejeon, 34101, South Korea
dNatural Product Research Institute, Korea Institute of Science and Technology (KIST), Gangneung 25451, South Korea
First published on 18th January 2019
Abstract
An efficient method was developed for the synthesis of unsymmetrical N-arylsulfamides using sulfamoyl azides and arylboronic acids in the presence of 10 mol% of copper chloride as the catalyst. The reaction was facilitated in MeOH in an open flask at room temperature. Unlike the coupling of sulfamides and boronic acids, the use of sulfamoyl azides was found to be beneficial with respect to the yield and reaction time.
Introduction
N-Arylsulfamides can be found in various therapeutic agents, drugs, and bioactive compounds, such as c-Met inhibitors and β3-adrenergic agonists (Fig. 1).1 Selective inhibition of c-Met signalling is known to have a connection with human cancer was proposed in the case of c-Met kinase inhibitors.1d In particular, the sulfamide structural motif is the defining characteristic of the distinct inhibitory effect. Moreover, compounds containing unsymmetrical sulfamides are favorable for applications in medicinal chemistry.2 Therefore, numerous methods were developed to synthesize these important compounds.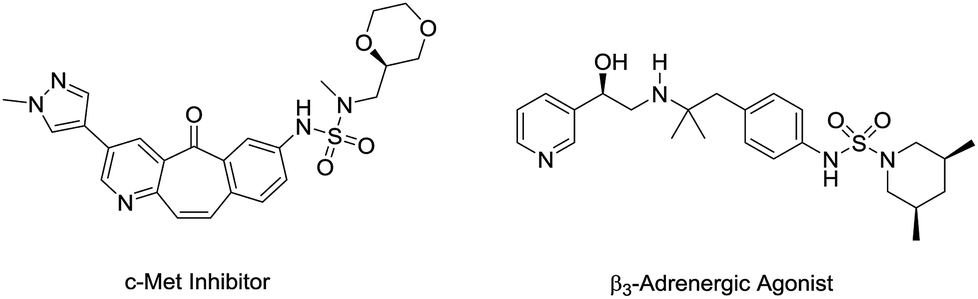 | ||
| Fig. 1 N-Arylsulfamide-containing bioactive compounds. | ||
N-Arylsulfamides were conventionally prepared by the reaction involving sulfamoyl chloride derivatives and an amine with a base (Scheme 1, route A).3 Despite being an effective approach to produce unsymmetrical N-arylsulfamides, it provides low yield as amines exhibit steric hindrance or deficient reactivity. In 2004, a novel intermolecular cross coupling process between N,N-disubstituted sulfamides and aryl halides using a palladium catalyst was first reported to produce unsymmetrical N-arylsulfamides by L. Alcaraz et al. (Scheme 1, route B).4 However, high temperatures to activate the starting reagents, sulfamides, and aryl halides are unavoidable in this approach. A Lossen-like rearrangement was demonstrated and this reaction carried out at room temperature, however, over 12 h were required to synthesize the sulfamides (Scheme 1, route C).5 Therefore, it is still necessary to develop a more efficient and less stringent method to obtain the desired product.
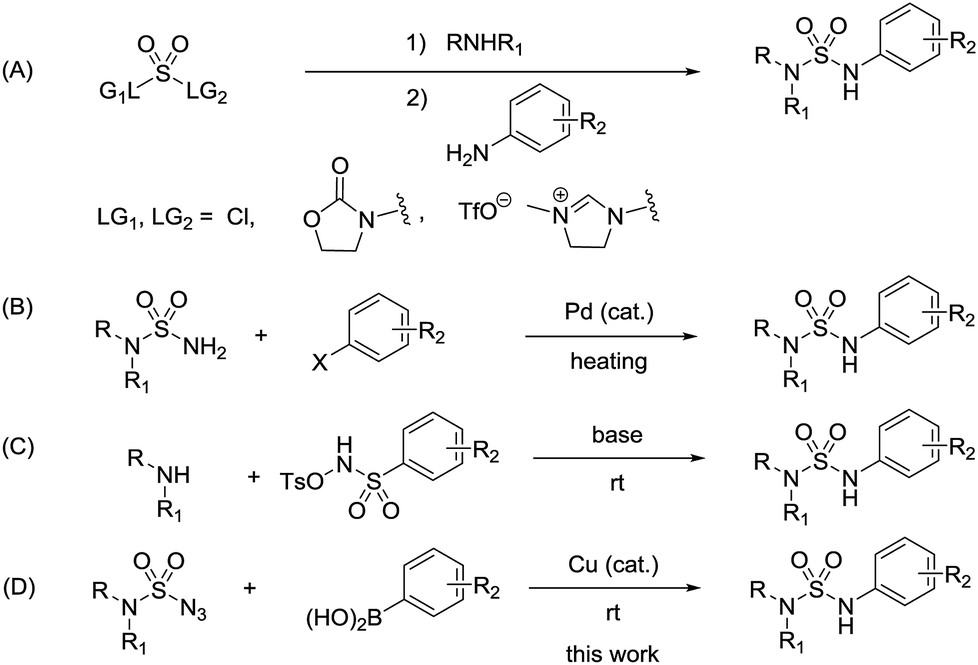 | ||
| Scheme 1 (A) General routes (B) Pd-catalyzed cross-coupling reaction (C) Lossen-like rearrangement (D) The reaction in the present work, towards the production of unsymmetrical N-arylsulfamides. | ||
Recently, an improved approach for the synthesis of N-arylsulfonamides and N-arylcarbamates using sulfonyl azides and azidoformates respectively at room temperature in the presence of a copper catalyst and boronic acids has been successfully reported by our group (Scheme 2).6,7 We thus envisioned an alternative synthetic method in order to contribute to the existing unsymmetrical N-arylsulfamides synthesis. Unlike previous synthetic methods, we expected our process would proceed rapidly at room temperature in high yields without the use of any additives and would not require relatively expensive palladium catalyst (Scheme 1, route D). Furthermore, to the best of our knowledge, the formation of N-arylsulfamides by copper-catalyzed processes has not yet been reported. Therefore, this study aims to present the first copper-catalyzed process for the synthesis of unsymmetrical N-arylsulfamides by employing N,N-disubstituted sulfamoyl azides and aryl boronic acids at room temperature. However, in general, organic azides are widely used in organic synthesis, but special precautions are necessary when using sulfamoyl azides.8
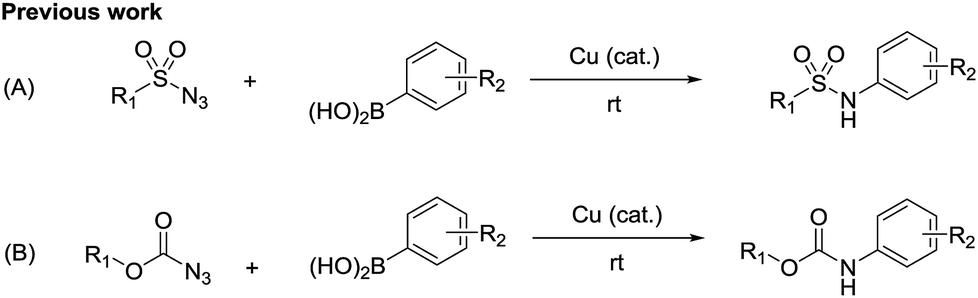 | ||
| Scheme 2 (A) Synthesis of N-arylsulfonamides (B) synthesis of N-arylcarbamates. | ||
Results and discussion
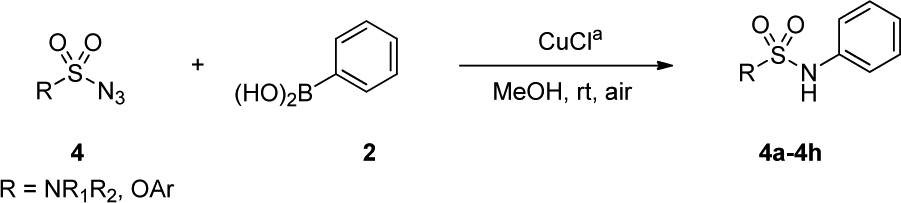
An optimization of the Chan–Lam coupling reaction was initially conducted with N-benzyl-N-methylsulfamoyl azide 19 with phenylboronic acid 2 (Table 1). The reaction was first investigated with a CuCl catalyst and 1.2 equiv. of phenylboronic acid 2 in the solvent MeOH in an open flask. The desired product 3a was generated in 72% yield with 83% conversion in 12 h (Table 1, entry 1). However, when 1.5 equiv. of boronic acid was used, the product was obtained with a slightly higher yield of 81% with 92% conversion in 16 h (Table 1, entry 2).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cu cat. | Equiv. of 2 | Oxidant | Solvent | Time | Yield of 3ab |
| a Reaction conditions: 1.0 equiv. of 1, solvent (0.5 M), rt. b Isolated yield. c Reaction time based on complete consumption of boronic acid as determined by TLC analysis. d Conversion. e 5 mol% of CuCl. | ||||||
| 1 | CuCl | 1.2 | Air | MeOH | 12 hc | 72%, (83%)d |
| 2 | CuCl | 1.5 | Air | MeOH | 16 hc | 81%, (92%)d |
| 3 | CuCl | 2.0 | Air | MeOH | 45 min | 91% |
| 4c | CuCl | 2.0 | Air | MeOH | 70 min | 78% |
| 5 | CuBr | 2.0 | Air | MeOH | 2 h | 87% |
| 6 | Cu(OAc)2 | 2.0 | Air | MeOH | 4 h | 77% |
| 7 | CuCl2 | 2.0 | Air | MeOH | 16 h | 74% |
| 8 | CuCl | 2.0 | Ag2CO3 | MeOH | 16 hc | 3%, (12%)d |
| 9 | CuCl | 2.0 | O2 | MeOH | 55 min | 78% |
| 10e | CuCl | 2.0 | None | MeOH | 85 min | 88% |
| 11 | None | 2.0 | Air | MeOH | 24 h | 0% |
| 12 | CuCl | 2.0 | Air | CH2Cl2 | 16 hc | 0% |
| 13 | CuCl | 2.0 | Air | THF | 24 hc | 17%, (45%)d |
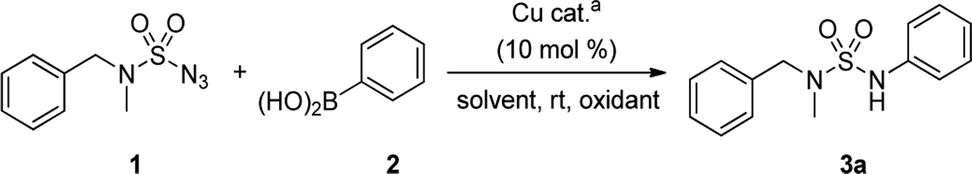
Finally, the use of 2.0 equiv. of phenylboronic acid 2 produced the highest yield of 91% of N-arylsulfamide 3a (Table 1, entry 3). When the catalyst loading was reduced from 10 mol% to 5 mol%, 3a was isolated at a lower yield (Table 1, entry 4). Moreover, the use of other copper catalysts or addition of Ag2CO3 and pure oxygen instead of air produced a lower yield (Table 1, entries 5–9). The use of argon was also furnished the desired product in a slightly lower 88% yield in 85 min (Table 1, entry 10). The absence of the catalyst resulted in 0% yield without conversion, thus demonstrating the importance of the Cu catalyst in this method (Table 1, entry 11). Furthermore, when different solvents having less polarity were used, the yield of 3a was poor (Table 1, entries 12 and 13).
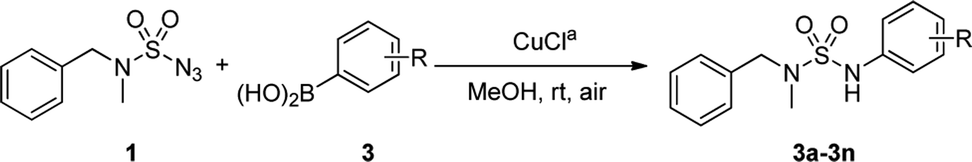
The scope of the copper-catalyzed coupling reactions using N-benzyl-N-methyl-sulfamoyl azide 1 and various boronic acids 3 was investigated under this optimized condition. As illustrated in Table 2, most of the boronic acids that were applied in the optimal conditions produced the desired products with good to excellent yields. However, in the case of 3f, yield of only 30% with 46% conversion in 3 h was achieved due to steric hindrance. In particular, halo-substituted aryl boronic acids exhibited less reactivity and produced 3j and 3k with yields of 80% and 71%, respectively, after 12 h. In addition, the use of 3-thienylboronic acid yielded 82% of the desired 3n with 92% conversion after prolonged reaction time of 12 h. The competing homocoupling of arylboronic acids in the presence of CuCl and MeOH completely consumed the boronic acids when 2-methoxyphenyl boronic acid or 3-thienylboronic acid was used.10
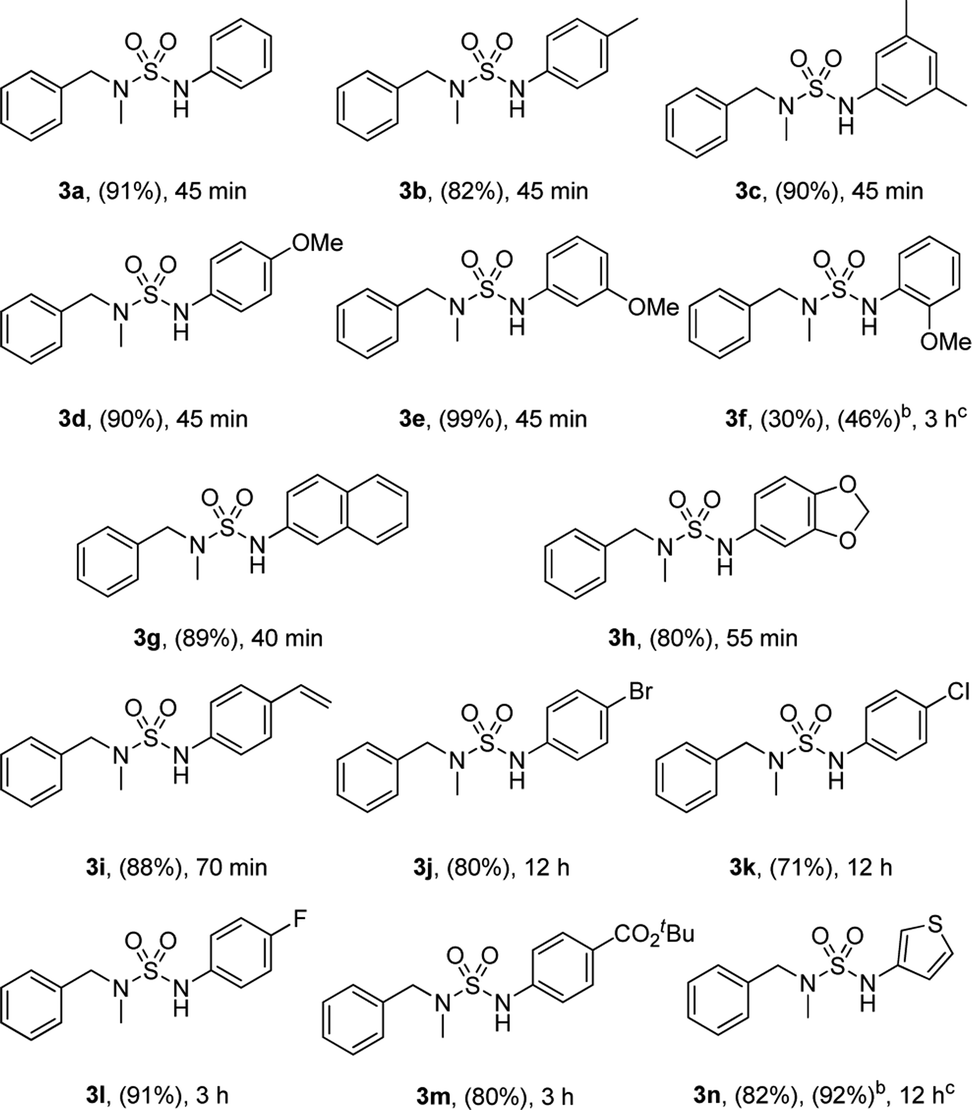
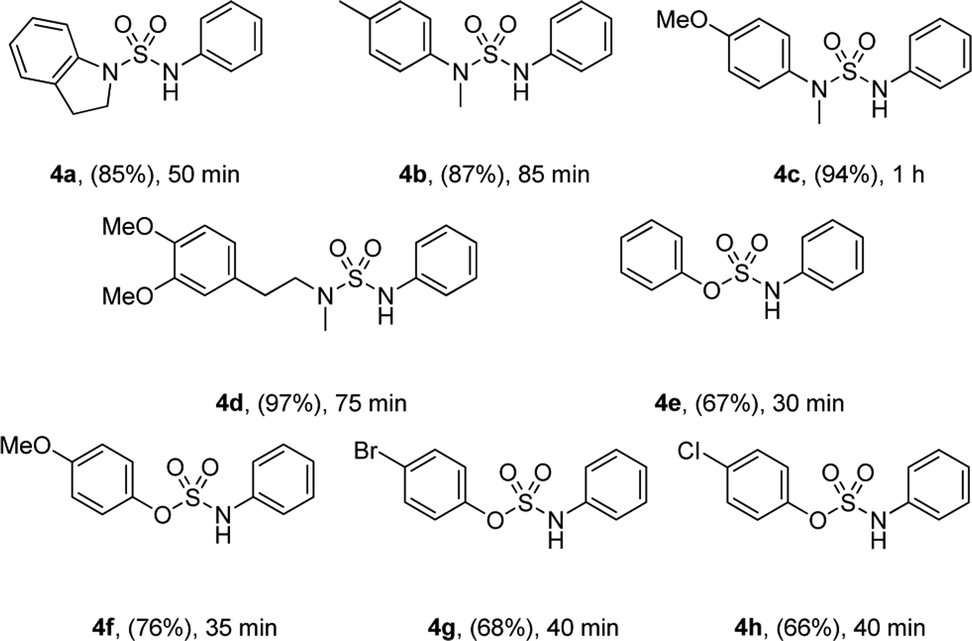
Moreover, the reactions of various sulfamoyl azides 4 with phenylboronic acid 2 were investigated. As shown in Table 3, the desired 4a–d were obtained at a good to excellent yield within 90 min. In addition, by using aryloxysulfonyl azides,11 the formation of N-arylsulfamic esters (Table 3, 4e–h)11c was expected, and their moderate to good yield suggests that the result depends on the reactivity of the azides. The yield gradually improved when an electron-donating group was substituted for the aryloxysulfonyl azides.
|
|
|---|
| a Reaction conditions: 1.0 equiv. of 4, 2.0 equiv. of 2, 10 mol% of CuCl, MeOH (0.5 M), air, rt. |
|
Next, the application of various phenylboronic acid derivatives as coupling partners in the optimized condition was investigated (Scheme 3). The use of phenyltrifluoroborate 5 yielded 42% of 3a with 86% conversion, and with pinacol phenylboronate 6, 31% of 3a was obtained with 60% conversion (Scheme 3, eqn (1) and (2)). Lastly, when dimethyl phenylboronate 7 was used, the desired sulfamide 3a was synthesized at 92% yield in 30 min (Scheme 3, eqn (3)).
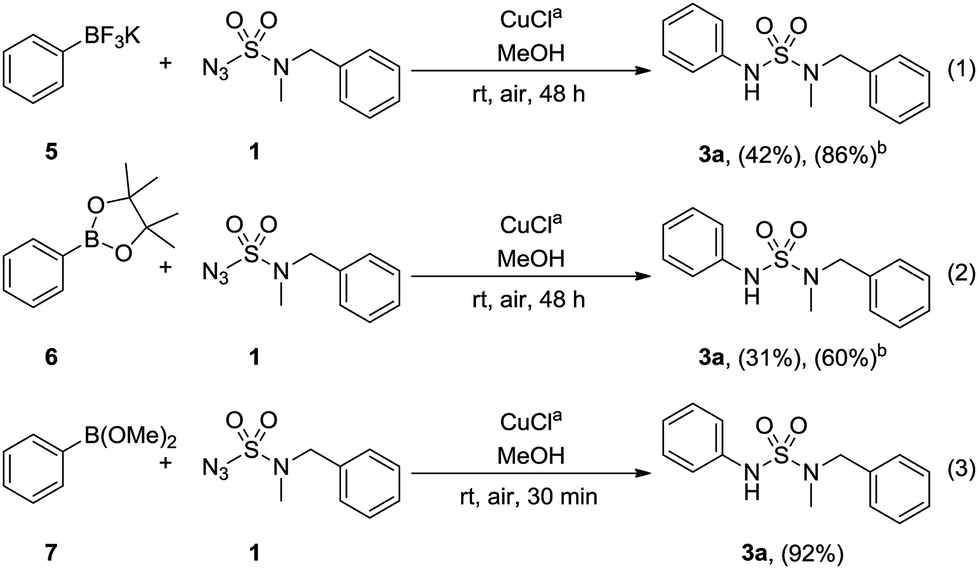 | ||
| Scheme 3 Cu-catalyzed N-arylation of N-benzyl-N-methyl-sulfamoyl azide 1 with boronic acid derivatives. aReaction conditions: 1.0 equiv. of 1, 2.0 equiv. of boronic acid derivative, 10 mol% of CuCl, MeOH (0.5 M), air, rt. bConversion. | ||
A control experiment was then carried out by using N-benzyl-N-methylsulfamide 8 and phenylboronic acid 2 under conventional Chan–Lam coupling conditions12 (Scheme 4, eqn (1)). Despite the extended reaction time, the desired N-arylsulfamide 3a was obtained at a yield of only 28% with 49% conversion. Next, sulfamide 8 was used in the optimized condition to investigate the influence of sulfamoyl azide 1 (Scheme 4, eqn (3)). Only a yield of 12% was obtained with a conversion of 35% over 16 h compared to the yield of 91% produced by sulfamoyl azide over 45 min (Scheme 4, eqn (2)). In view of these results, it could be hypothesized that sulfamoyl azide has a major impact on the synthesis of N-arylsulfamides in this method.
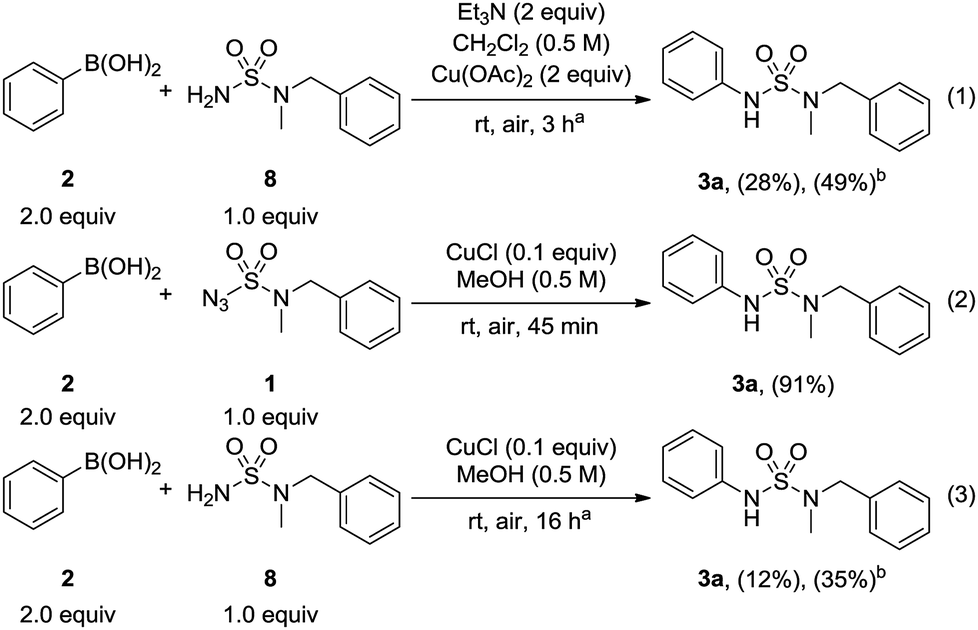 | ||
| Scheme 4 Control experiments. aReaction time based on complete consumption of boronic acid as determined by TLC analysis. bConversion. | ||
The possible mechanism for the Chan–Lam coupling between sulfamoyl azides and arylboronic acids is presented below (Scheme 5).7,13 At first, the sulfamoyl azides coordinate with the Cu(I) salt A that provides copper nitrene and are transformed to Cu(II) complex B.14 Subsequently, the aryl boronic acid is in equilibrium with the methyl boronate ester,15 which undergoes transmetalation with B to produce Cu(II) complex C. After air oxidation of C, the Cu(III) complex D is obtained and E is derived with the regeneration of the Cu(I) catalyst A through reductive elimination.
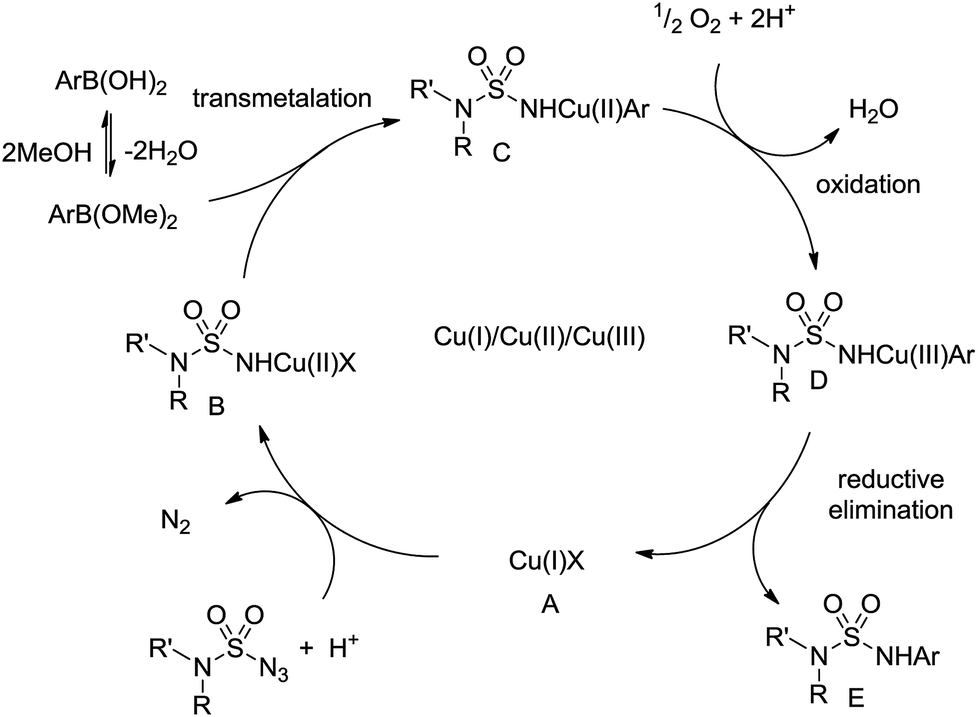 | ||
| Scheme 5 Possible mechanism. | ||
Conclusions
In summary, this study explained the Chan–Lam coupling reaction between sulfamoyl azides and arylboronic acids for synthesizing unsymmetrical N-arylsulfamides. The reaction was carried out in the presence of 10 mol% of the copper chloride catalyst at room temperature in an open flask without any additional additives. The scope of this method employing various sulfamoyl azides and boronic acids was demonstrated. The sulfamoyl azides were established as efficient precursor of the reaction with considerable effect on the yield and reaction time. The application of other organic azides for this method is under investigation.Experimental
General procedure for synthesis of unsymmetrical N-arylsulfamides
A sealed tube was charged with an arylboronic acid (2.0 mmol), CuCl (10 mol%) and the corresponding sulfamoyl azide (1.0 mmol). MeOH (1.0 mL) was then added to the flask. The reaction mixture was stirred at room temperature in an open flask. After completion of the reaction, the reaction mixture was filtered through celite and washed with EtOAc. The filtrate was collected and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel to obtain the desired product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF) (2018R1A2B6005533), the Korea Institute of Science and Technology (KIST) (No. 2Z05310) and the New & Renewable Energy of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant (No. 20163030013900).Notes and references
-
(a) R. Kuang, J. B. Epp, S. Ruan, L. S. Chong, R. Venkataraman, J. Tu, S. He, T. M. Truong and W. C. Groutas, Bioorg. Med. Chem., 2000, 8, 1005 CrossRef CAS
; (b) S. He, R. Kuang, R. Venkataraman, J. Tu, T. M. Truong, H.-K. Chan and W. C. Groutas, Bioorg. Med. Chem., 2000, 8, 1713 CrossRef CAS
-
(a) C. H. Lee and H. J. Kohn, J. Pharm. Sci., 1990, 79, 716 CrossRef CAS
- For selected examples:
(a) J.-L. Montero, G. Dewynter, B. Agoh, B. Delaunay and J.-L. Imbach, Tetrahedron Lett., 1983, 24, 3091 CrossRef CAS
- L. Alcaraz, C. Bennion, J. Morris, P. Meghani and S. M. Thom, Org. Lett., 2004, 6, 2705 CrossRef CAS PubMed
- L. Pantaine, F. Richard, J. Marrot, X. Moreau, V. Coeffard and C. Greck, Adv. Synth. Catal., 2016, 358, 2012 CrossRef CAS
- S.-Y. Moon, J. Nam, K. Rathwell and W.-S. Kim, Org. Lett., 2014, 16, 338 CrossRef CAS
- S.-Y. Moon, U. B. Kim, D.-B. Sung and W.-S. Kim, J. Org. Chem., 2015, 80, 1856 CrossRef CAS
- S. Bräse, C. Gil, K. Knepper and V. Zimmerman, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef
- J. C. Culhane and V. V. Fokin, Org. Lett., 2011, 13, 4578 CrossRef CAS
- Homocoupling of arylboronic acid in the presence of CuCl in MeOH has been reported by Luo and Cheng. See: G. Cheng and M. Luo, Eur. J. Org. Chem., 2011, 2519 CrossRef CAS
-
(a) T. Yang, H. Cui, C. Zhang, Li. Zhang and C.-Y. Su, ChemCatChem, 2013, 5, 3131 CrossRef CAS
- D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933 CrossRef CAS
-
(a) P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, K. M. Averill, D. M. T. Chan and A. Combs, Synlett, 2000, 674 CAS
-
(a) E. Haldón, E. Álvarez, M. C. Nicasio and P. J. Pérez, Chem. Commun., 2014, 50, 8978 RSC
-
D. G. Hall, Boronic Acids, Wiley-VCH, Weinheim, 2005 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09219b |
| This journal is © The Royal Society of Chemistry 2019 |