
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic, transannular carbonyl-olefin metathesis reactions†
Paul S.
Riehl
 ,
Daniel J.
Nasrallah
and
Corinna S.
Schindler
*
,
Daniel J.
Nasrallah
and
Corinna S.
Schindler
*
Willard Henry Dow Laboratory, Department of Chemistry, University of Michigan, 930 North University Avenue, Ann Arbor, Michigan 48109, USA. E-mail: corinnas@umich.edu
First published on 25th September 2019
Abstract
Transannular carbonyl–olefin metathesis reactions complement existing procedures for related ring-closing, ring-opening, and intermolecular carbonyl-olefin metathesis. We herein report the development and mechanistic investigation of FeCl3-catalyzed transannular carbonyl-olefin metathesis reactions that proceed via a distinct reaction path compared to previously reported ring-closing and ring-opening protocols. Specifically, carbonyl-ene and carbonyl-olefin metathesis reaction pathways are competing under FeCl3-catalysis to ultimately favor metathesis as the thermodynamic product. Importantly, we show that distinct Lewis acid catalysts are able to distinguish between these pathways to enable the selective formation of either transannular carbonyl-ene or carbonyl-olefin metathesis products. These insights are expected to enable further advances in catalyst design to efficiently differentiate between these two competing reaction paths of carbonyl and olefin functionalities to further expand the synthetic generality of carbonyl-olefin metathesis.
Introduction
Carbonyl-olefin metathesis reactions represent desirable strategies for catalytic carbonyl olefinations as a result of their potential for direct carbon–carbon bond formation between carbonyl and alkene functionalities.1 In recent years, distinct classes of catalytic carbonyl-olefin metathesis have been developed to effect ring-closing2 (3), ring-opening3 (6) and intermolecular4 (8) transformations5 (Fig. 1A). We herein report the development of FeCl3-catalyzed transannular carbonyl-olefin metathesis (11) that complements existing classes as a fourth, mechanistically distinct reaction category (Fig. 1B). Specifically, in transannular carbonyl-olefin metathesis reactions, carbonyl-ene6 and carbonyl-olefin metathesis reaction paths initially compete to ultimately favour the formation of the thermodynamic metathesis product. However, our studies described herein show that distinct Lewis acid catalysts can differentiate between these divergent reaction paths to enable selective access to both products. Furthermore, transannular carbonyl-olefin metatheses are ring-contraction reactions that result in the rapid formation of new and distinct skeletal frameworks. Consequently, these transformations hold great potential for the molecular editing of biologically important natural products and allow access to structurally diverse carbon scaffolds in a single synthetic transformation. Importantly, the concept of molecular editing, “whereby one could selectively insert, delete, or exchange atoms in highly elaborated molecules”7 was recently described as an area of opportunity and high potential impact that is predicted to drive innovation in pharmaceutical drug discovery for the next 50 years. While important advances have already been made that enable the insertion and deletion of atoms in complex molecules via C–H activation approaches,8 strategies that allow for a selective exchange of atoms remain rare and represent a challenge for current synthetic chemistry.9 We herein show that catalytic, transannular carbonyl-olefin metathesis reactions can contribute to this emerging area of research as transformations that facilitate such a specific exchange. Moreover, distinct Lewis acids enable a divergent and catalyst-controlled strategy for the molecular editing of natural product scaffolds10 through either carbonyl-olefin metathesis or carbonyl-ene reactions.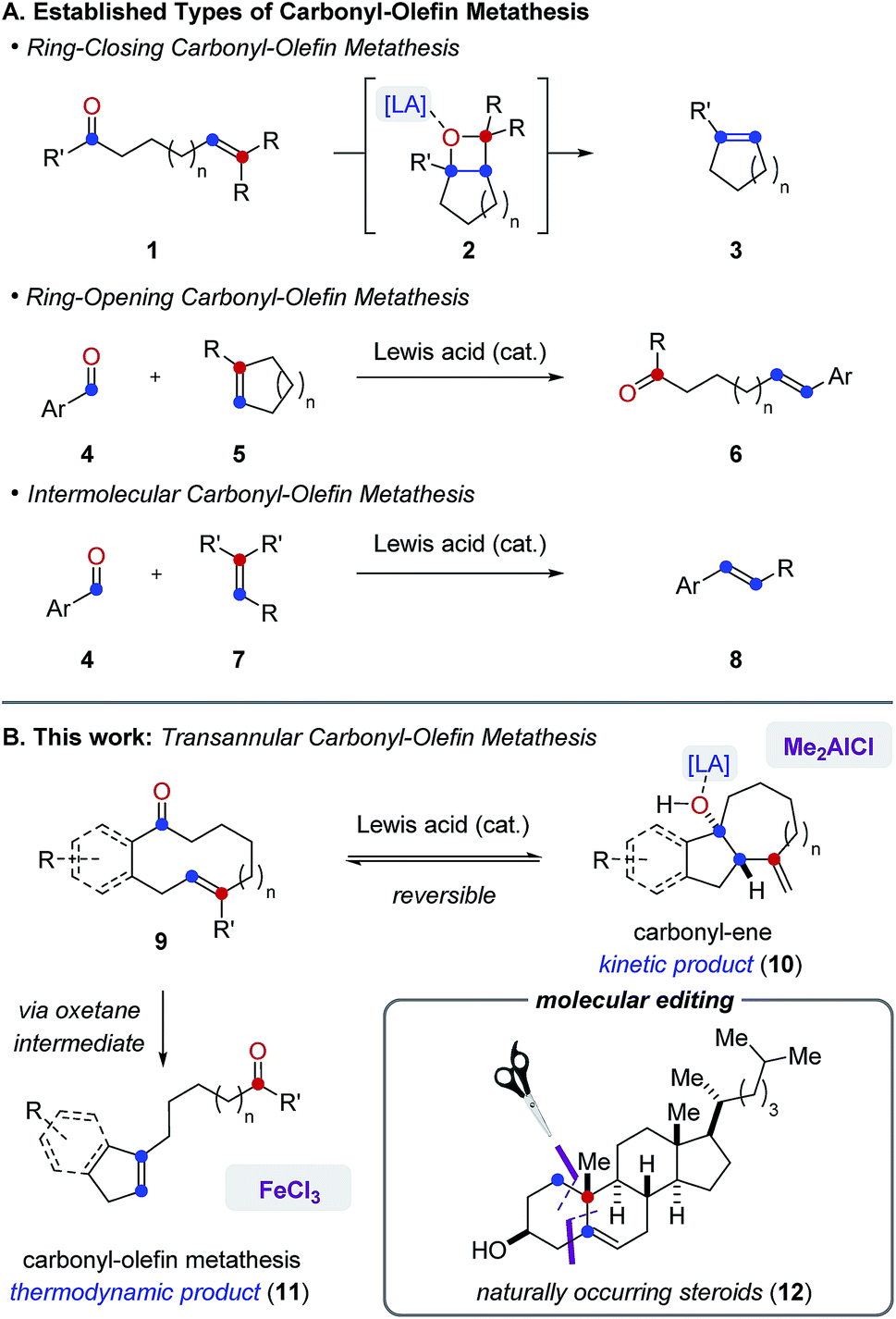 | ||
| Fig. 1 (A) Established categories of catalytic carbonyl-olefin metathesis reactions. (B) Divergent reactivity of Lewis acids enables transannular carbonyl-olefin metathesis. | ||
Results and discussion
Our studies towards catalytic, transannular carbonyl-olefin metathesis reactions initially required efficient synthetic access to functionalized decalin derivatives (9).11 To assess the potential of this transformation for molecular editing, we developed a short sequence to convert naturally occurring steroids into highly functionalized cyclodecenone systems (Scheme 1, see ESI for details†).12 Specifically, epoxidation of cholesterol (12), reduction and selective acetylation of the secondary alcohol gave rise to acetate 13. Subsequent Suárez oxidation13 with (diacetoxy-iodo)benzene and iodine initiates an alkoxy radical fragmentation to form 14 in 58% yield.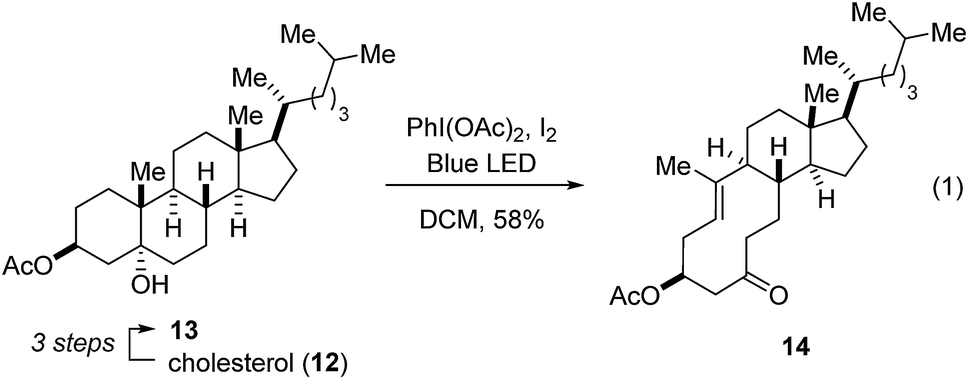 | ||
| Scheme 1 Synthesis of cyclodecenone derivative 14. | ||
We next focused on the evaluation of distinct Lewis acids to promote transannular metathesis between carbonyl and olefin functionalities (Fig. 2A). When cyclodecenone 14 was converted with Sc(OTf)3 as catalyst, the desired metathesis product 17 was isolated in 33% yield together with two additional compounds, subsequently identified as the carbonyl-ene product 16 in 15% yield and tetrahydrofuran 19 in 20% yield (entry 1, Fig. 2A). However, when treated with catalytic amounts of BF3·Et2O, the formation of only two distinct products was observed, including metathesis product 17 and tetrahydrofuran 19 in 30% and 23% yield, respectively (entry 2, Fig. 2A). Importantly, stoichiometric amounts of BF3·Et2O14 also resulted in the formation of both products, albeit in diminished yields of 11% and 25%, respectively (entry 3, Fig. 2A). Although GaCl3 was previously identified as a superior Lewis acid for ring-opening carbonyl-olefin metathesis3, catalytic amounts of GaCl3 failed to promote transannular reactivity of cyclodecenone 14 (entry 4, Fig. 2A). In comparison, 10 mol% SnCl4 resulted in increased reactivity providing metathesis product 17 in 22% yield and tetrahydrofuran 19 in 44% yield (entry 5, Fig. 2A). Interestingly, when cyclodecenone 15 incorporating a free secondary alcohol was subjected to otherwise identical reaction conditions, the metathesis product 18 was formed exclusively in 65% yield (entry 6, Fig. 2A). The increased selectivity observed with the free alcohol 15 led us to evaluate additional Lewis acid catalysts. When acetate 14 was converted with catalytic amounts of FeCl3, a mixture of both carbonyl-olefin metathesis product 17 and tetrahydrofuran 19 was observed, albeit with increased yields of 39% and 31%, respectively (entry 7, Fig. 2A). However, exclusive formation of the desired carbonyl-olefin metathesis product 18 was observed in superior yields of 75% when cyclodecenone 15 was converted under otherwise identical conditions (entry 8, Fig. 2A).
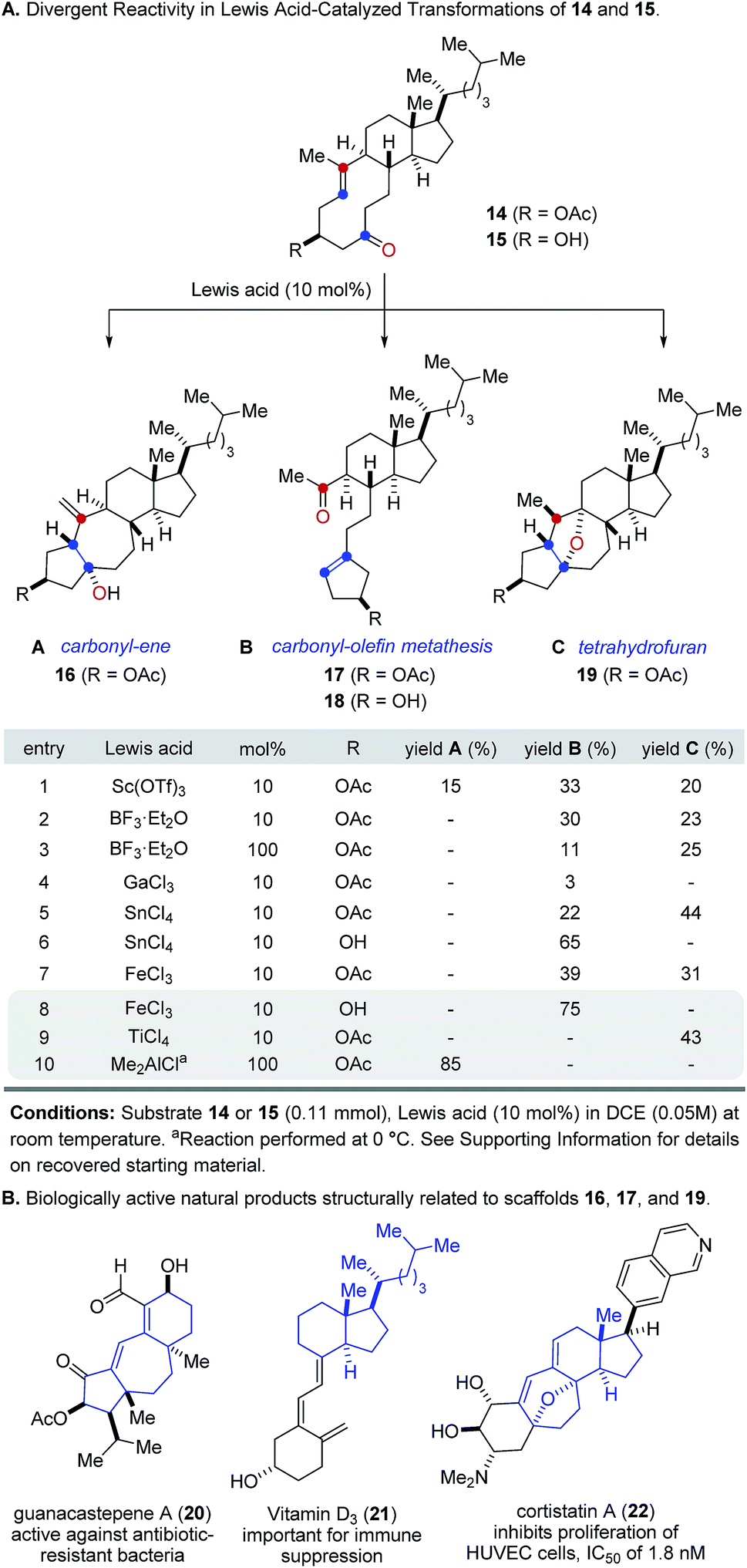 | ||
| Fig. 2 (A) Reaction optimization for transannular carbonyl-olefin metathesis. (B) Distinct molecular scaffolds obtained from steroid precursors resemble biologically active molecules. | ||
Subsequent investigations showed that distinct Lewis acids are also capable of promoting the selective formation of either the carbonyl-ene product 16 or tetrahydrofuran 19. Specifically, 10 mol% TiCl4 resulted in exclusive formation of tetrahydrofuran 19 in 43% yield, while Me2AlCl formed the corresponding carbonyl-ene product 18 in 85% yield (entries 9 and 10, Fig. 2A). These results suggest that the appropriate choice of Lewis acid now enables selective access to distinct molecular frameworks and consequently represents an approach for divergent molecular editing via transannular reactions between carbonyl and olefin functionalities. Importantly, the resulting diverse molecular scaffolds of the products closely resemble naturally occurring compounds of biological importance, including guanacastepene A (20)15, vitamin D3 (21)16, and cortistatin A (22)17 (Fig. 2B).
The optimal reaction conditions developed for FeCl3-catalyzed, transannular carbonyl-olefin metathesis proved general for a variety of 9- and 10-membered ring systems (Table 1). Unfunctionalized medium-sized rings proved viable substrates for transannular carbonyl-olefin metathesis and resulted in good overall yields. Specifically, cyclodecenone 23 formed the desired metathesis product 24 in 42% yield while differently substituted cyclononenones 25a, 25b, and 25c bearing methyl and bromide substituents resulted in yields of 40–64% (entries 1 and 2, Table 1). To further evaluate the potential of Lewis acid-catalyzed transannular carbonyl-olefin metathesis for the molecular editing of natural products, we subsequently investigated additional naturally occurring steroids. Specifically, cholesterol, stigmasterol, pregnenolone, and dehydroepiandrosterone derived cyclodecenones readily available via our synthetic strategy (Scheme 1). Acetate 14 underwent the desired transannular carbonyl-olefin metathesis reaction in 39% yield while the corresponding alcohol 15 resulted in increased yields of 75%. Similarly, stigmasterol-derived cyclodecenones 27 and 29 resulted in 26% and 51% yield, respectively, when converted with catalytic amounts of FeCl3 (entries 5 and 6, Table 1). Silylether 31 obtained from pregnenolone formed the desired metathesis product in 69% yield while the corresponding alcohol 33 resulted in 84% yield. Furthermore, cyclodecenones 35 and 37 obtained from naturally occurring dehydroepiandrosterone formed the desired metathesis products 36 and 38 in 46% and 44% yield, respectively (entries 9 and 10, Table 1).
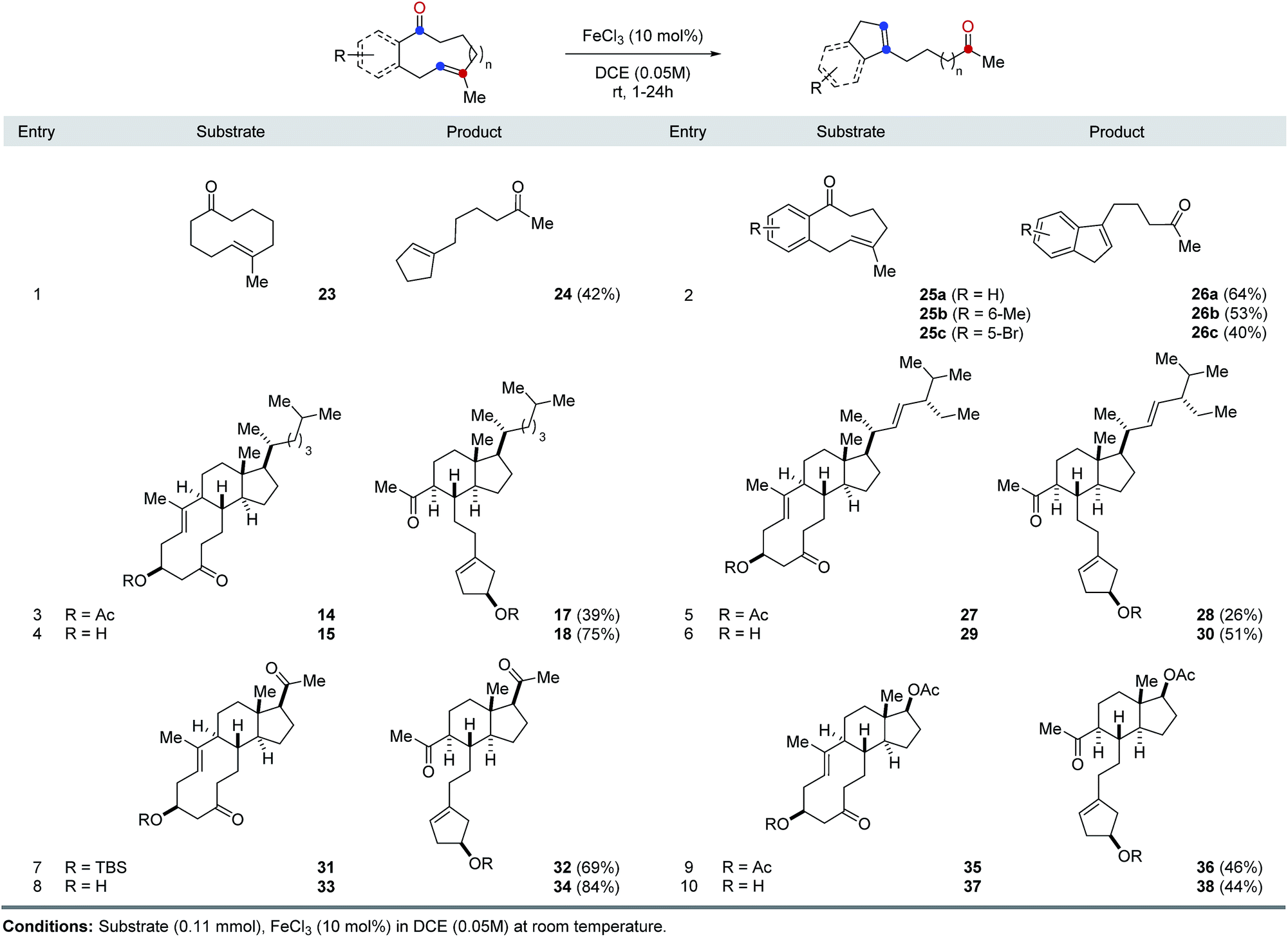
|
To obtain insights into the controlling features of FeCl3-catalyzed transannular carbonyl-olefin metathesis reactions, we conducted additional experimental investigations. We first evaluated the temperature dependence of this transformation (Scheme 2). Surprisingly, when cyclodecenone 14 was converted with 10 mol% FeCl3 at 0 °C, neither metathesis product 17 nor tetrahydrofuran 19 were obtained; instead, the carbonyl-ene product 16 was isolated in 41% yield along with recovered starting material in 11% yield (Scheme 2). However, when carbonyl-ene product 16 was subjected to the conditions optimized for transannular carbonyl-olefin metathesis (10 mol% FeCl3 in DCE at room temperature), the formation of cyclodecenone 14 was observed in 41% yield together with recovered starting material 16 in 45% yield, suggesting the reversibility of the carbonyl-ene reaction (Scheme 2). These observations are in stark contrast to results previously obtained in GaCl3-catalyzed ring-opening carbonyl-olefin metathesis in which a competing, irreversible carbonyl-ene reaction path is responsible for diminished yields of the metathesis product.3
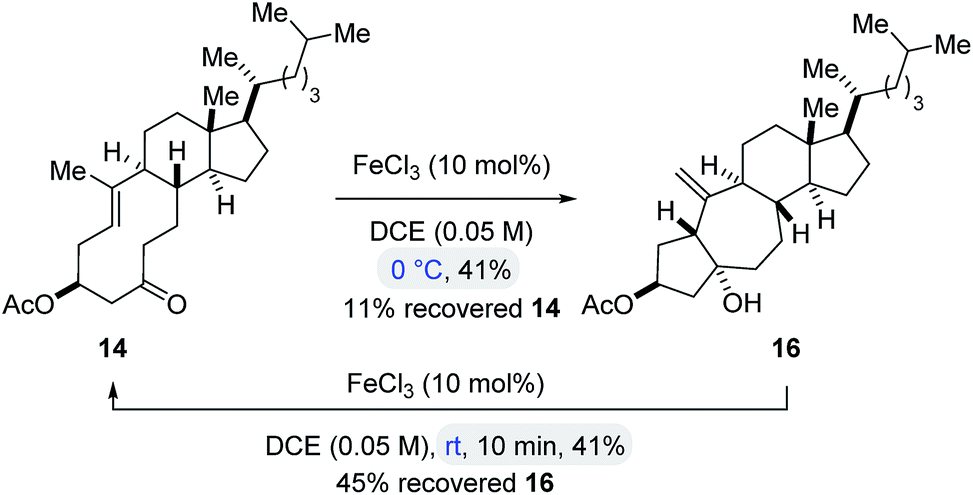 | ||
| Scheme 2 FeCl3 also catalyses a reversible transannular carbonyl-ene reaction. | ||
To further probe this hypothesis suggesting a reaction path for transannular carbonyl-olefin metathesis that is distinct from ring-closing, ring-opening, and intermolecular carbonyl-olefin metathesis, we conducted computational investigations (unrestricted B97-D density functional and 6-31G* basis set).18 Our studies show that cyclodecenone 14 (ref. 19) can undergo either of two reversible reactions upon binding to FeCl3 (14 + Fe, Fig. 3A) leading to oxetane formation via an asynchronous, concerted [2 + 2]-cycloaddition that proceeds with an enthalpic barrier of 5.9 kcal mol−1 (39 + Fe via TS-I) or carbonyl-ene reaction that proceeds with a barrier of 14.1 kcal mol−1 (16 + Fe via TS-II) above the substrate complex (14 + Fe; Fig. 3A). Additionally, our previous experimental results have shown that in comparison to oxetane 39, only the carbonyl-ene product 16 is stable upon isolation. These observations are supported by the relative enthalpies of the uncoordinated oxetane 39 (0.2 kcal mol−1), which was found to be higher than the uncoordinated carbonyl-ene product 16 by 13.6 kcal mol−1 and is therefore less stable (Fig. 3). Subsequent efforts focused on investigating the fragmentation of oxetane 39 computationally. Importantly, our investigations revealed two possible paths for oxetane fragmentation; (1) an asynchronous, concerted retro [2 + 2]-cycloaddition proceeds with an enthalpic barrier of 15.1 kcal mol−1 above the iron-substrate complex (14 + Fe) to form the transannular carbonyl-olefin metathesis product (17 + Fe, Fig. 4). (2) Alternatively, oxetane 39 can fragment upon elimination to result in cycloheptene (40 + Fe). This elementary step proceeds with a barrier of 17.9 kcal mol−1 above the substrate complex (14 + Fe). Notably, this elimination reaction represents an unprecedented mode of Lewis acid-catalyzed oxetane fragmentation. Intramolecular addition of the tertiary alcohol to the alkene moiety results in the formation of the furan byproduct observed under FeCl3-catalysis (19 + Fe, Fig. 4).
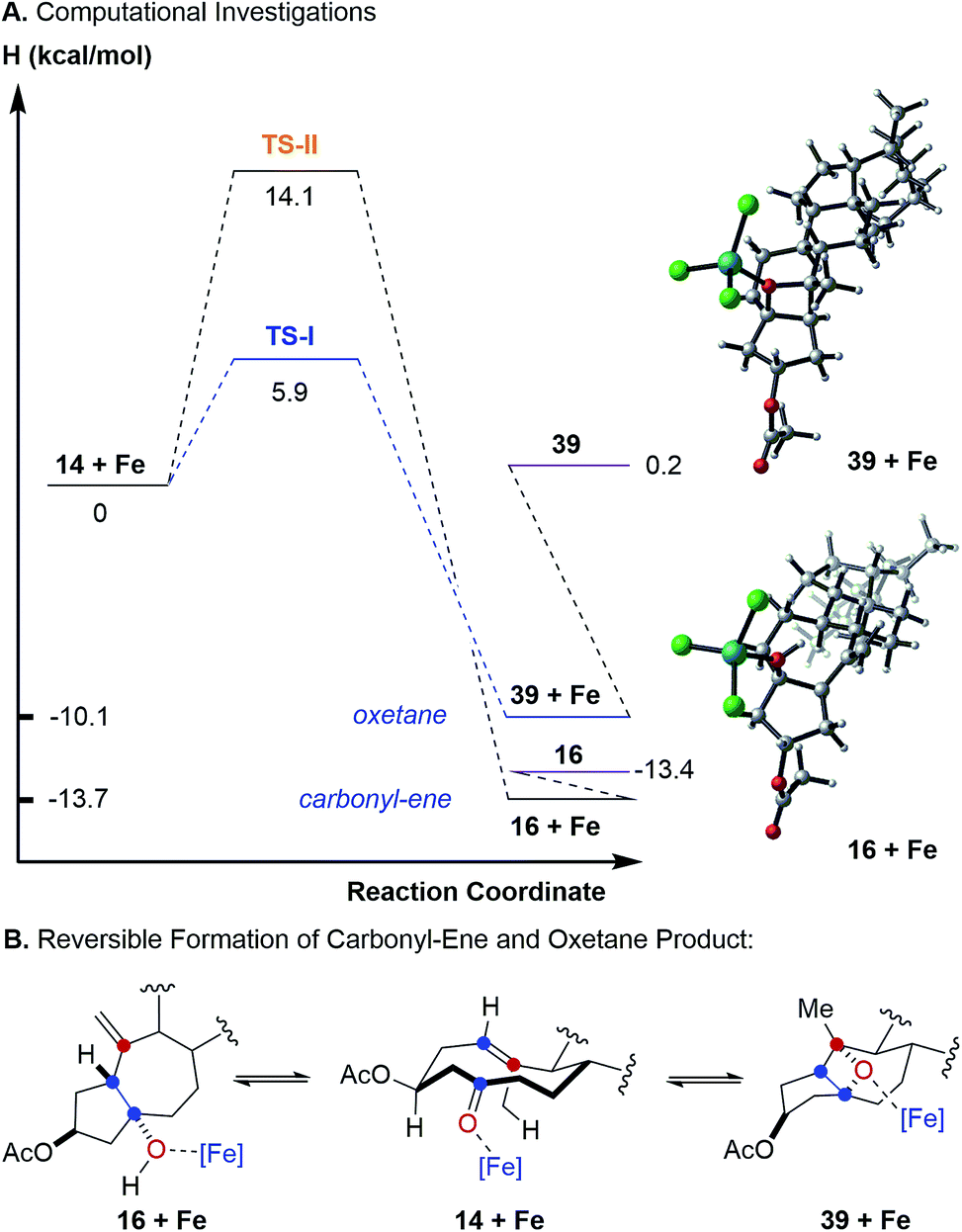 | ||
| Fig. 3 (A) Computed energies comparing carbonyl-ene and oxetane-forming pathways. (B) Carbonyl-ene reaction and oxetane formation proceed reversibly under FeCl3-catalysis in transannular carbonyl-olefin metathesis. | ||
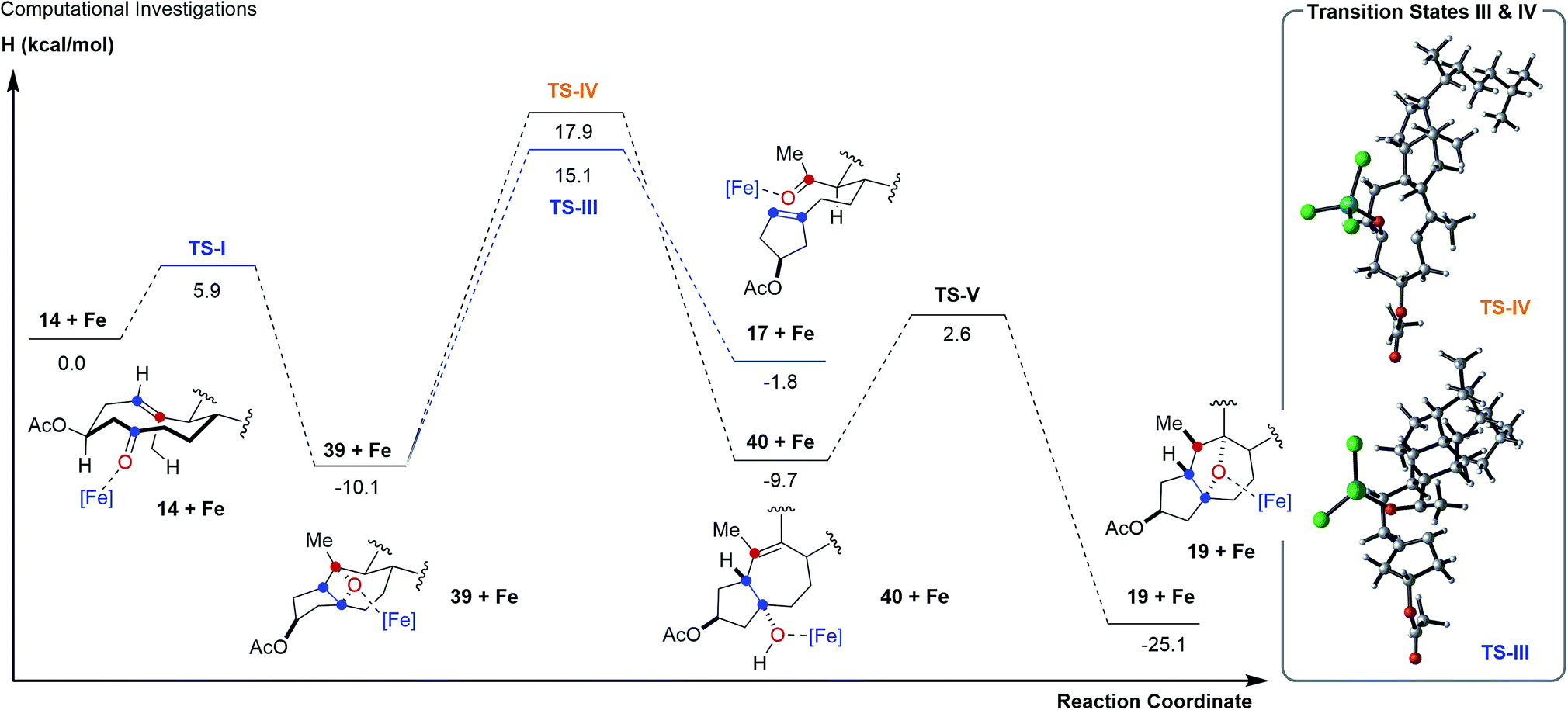 | ||
| Fig. 4 Reaction path for transannular carbonyl-olefin metathesis and the formation of furans from intermediate oxetanes 39. | ||
Our computational investigations suggest oxetane fragmentation as the rate-limiting steps of both transannular carbonyl-olefin metathesis and furan formation. These results are consistent with our experimental observations as temperatures greater than 0 °C are necessary to afford carbonyl-olefin metathesis product 17 (TS-III, 15.1 kcal mol−1) or furan product 19 (TS-IV, 17.9 kcal mol−1) (Fig. 4).
Based on our computational and experimental results, we postulate a reaction mechanism for FeCl3-catalyzed transannular carbonyl-olefin metathesis that differs from ring-closing, ring-opening, and intermolecular carbonyl-olefin metathesis (Fig. 5). Upon activation with the Lewis acid catalyst, the cyclodecenone substrate 14 can undergo either of two reversible transformations. Specifically, a reversible carbonyl-ene reaction forms the kinetic product 16 while a reversible asynchronous, concerted [2 + 2]-cycloaddition results in oxetane 39 as the thermodynamic product. Importantly, the carbonyl-ene product 16 can be isolated at lower temperatures under otherwise identical reaction conditions as a stable product. In comparison, our attempts to isolate oxetane 39 were unsuccessful which is corroborated by our theoretical investigations that found the uncoordinated species to be significantly less stable than 16 by 13.6 kcal mol−1. The FeCl3-coordinated oxetane can subsequently undergo two distinct fragmentation pathways. Asynchronous, concerted retro [2 + 2]-cycloaddition results in the formation of the desired transannular carbonyl-olefin metathesis product 17. Alternatively, oxetane 39 can undergo a competing FeCl3-catalyzed fragmentation via elimination to result in the formation of a cycloheptene intermediate (40 + Fe, Fig. 5).20 Subsequent addition of the activated tertiary alcohol to the alkene subunit in 40 gives rise to tetrahydrofuran 19 as the product resulting via a distinct mechanism for oxetane fragmentation. Importantly, other Lewis acids can differentiate selectively between these competing reaction paths (see Fig. 2). Specifically, when cyclodecenone 14 is converted with Me2AlCl as Lewis acid, carbonyl-ene product 16 is isolated exclusively, suggesting that under Me2AlCl-mediated conditions the carbonyl-ene reaction is not reversible and 16 is formed as the thermodynamic product. Furthermore, when cyclodecenone 14 is reacted with catalytic amounts of TiCl4, tetrahydrofuran 19 is obtained as the sole product, suggesting that the fragmentation of intermediate oxetane 39 proceeds exclusively via elimination under these reaction conditions.
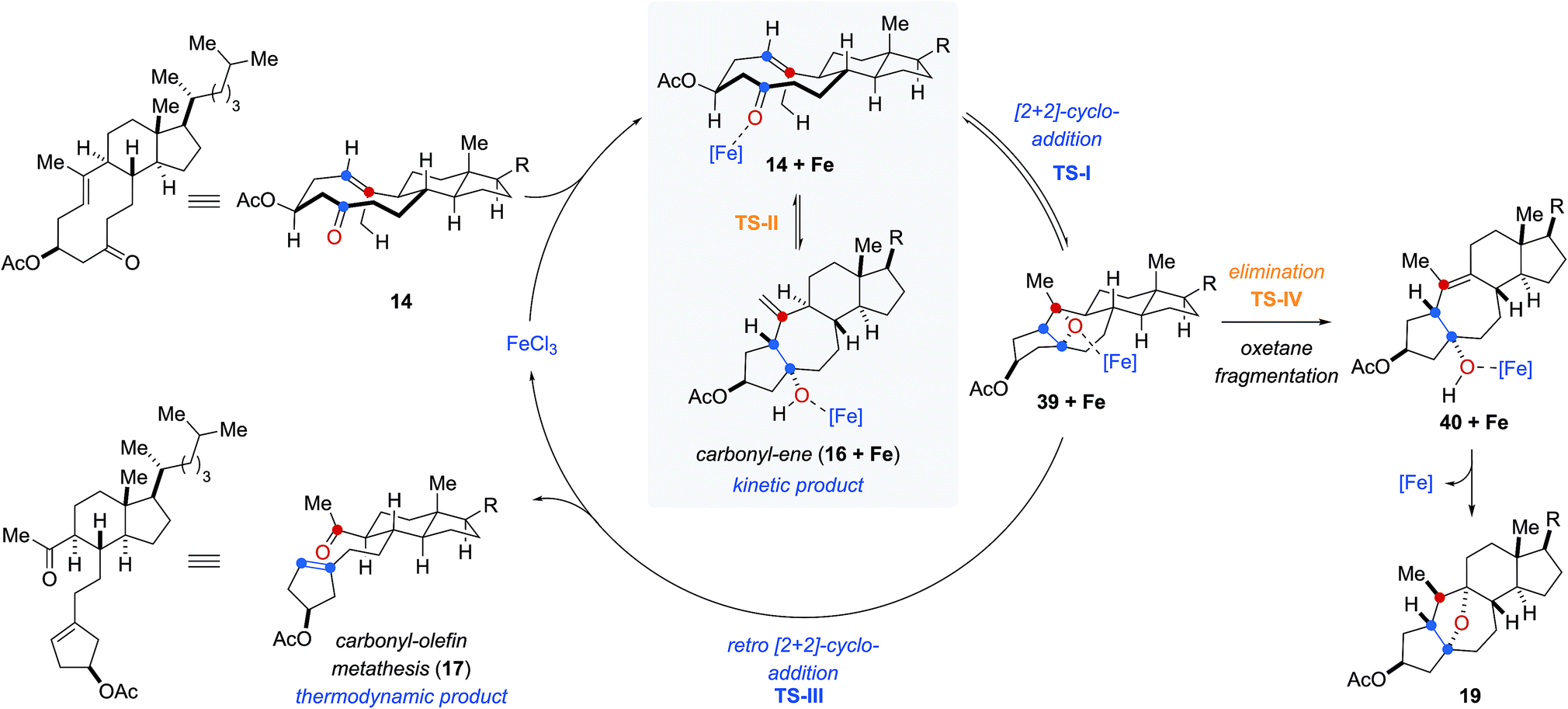 | ||
| Fig. 5 Mechanistic hypothesis for FeCl3-catalyzed transannular carbonyl-olefin metathesis reactions. | ||
Additionally, we observed that the substituents in the 2-position to the reactive carbonyl can affect yield and selectivity of the transannular carbonyl-olefin metathesis reaction (Table 1). We initially hypothesized that hydroxyl functionalities in this position could lead to a reactive conformation that enables bidentate binding of the FeCl3 catalyst and ultimately favours transannular carbonyl-olefin metathesis. However, our subsequent computational investigations do not provide support for this hypothesis. Based on these results, we favour a revised postulate to explain the origin of this selectivity. Specifically, the Lewis basic acetate functionality is capable of coordinating FeCl3 bound to the intermediate oxetane (39 + Fe, Fig. 5), resulting in elongation of one C–O bond and disruption of the retro [2 + 2] pathway (TS-III) to ultimately facilitate the elimination pathway (TS-IV). The inability of free hydroxyl (15, 29, 33, 37) or silyl ether (31) substrates to afford the tetrahydrofuran product is consistent with this mechanistic proposal. This notable control over selectivity for carbonyl-olefin metathesis is an important observation that we expect to have valuable implications for future catalyst design and development in carbonyl-olefin metathesis reactions, specifically those that are hampered by competing carbonyl-ene and/or oxetane fragmentation paths that do not favour metathesis.
Additionally, the reactivity of the analogous Z isomer Z-14 was investigated (Fig. 6). However, treatment of Z-14 with catalytic amounts of FeCl3 under optimal reaction conditions resulted in the exclusive isolation of starting material. Importantly, no formation of carbonyl-ene or carbonyl-olefin metathesis product was observed which was subsequently supported by computational investigations (Fig. 6). Specifically, we were unable to identify a reactive pathway leading to carbonyl-ene or carbonyl-olefin metathesis formation with resulting energy barriers being prohibitively high at 135.6 kcal mol−1 (see ESI for details†). This lack of reactivity is consistent with high strain being introduced when forming an oxetane intermediate from starting material Z-14.
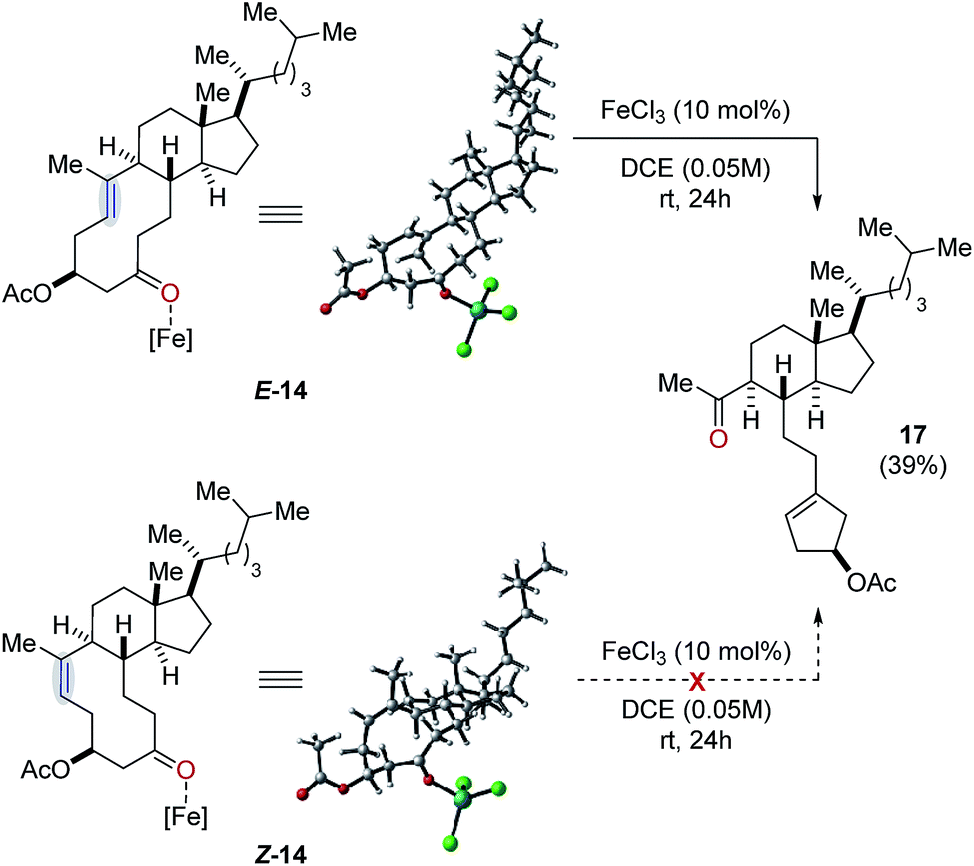 | ||
| Fig. 6 Comparison of reactivity between E and Z isomers of cholesterol-derived substrate 14. | ||
Our investigations next focused on investigating the generality of Me2AlCl in preferentially promoting transannular carbonyl-ene21 over carbonyl-olefin metathesis reaction paths (Table 2). Importantly, when various cyclodecenone substrates were treated with stoichiometric amounts of Me2AlCl, the formation of the corresponding transannular carbonyl-ene products is observed in all cases in good to excellent yields as the exclusive product. Specifically, cyclodecenone 14 resulted in tetracycle 16 in 85% yield, while stigmasterol-derived substrate 27 resulted in 76% of carbonyl-ene product 39 (entries 1 and 2, Table 2). Similarly, exclusive formation of the corresponding carbonyl-ene products was observed with pregnenolone- and androsterone-derived cyclodecenones 31 and 35 in 73% and 59% yield, respectively (entries 3 and 4, Table 2).
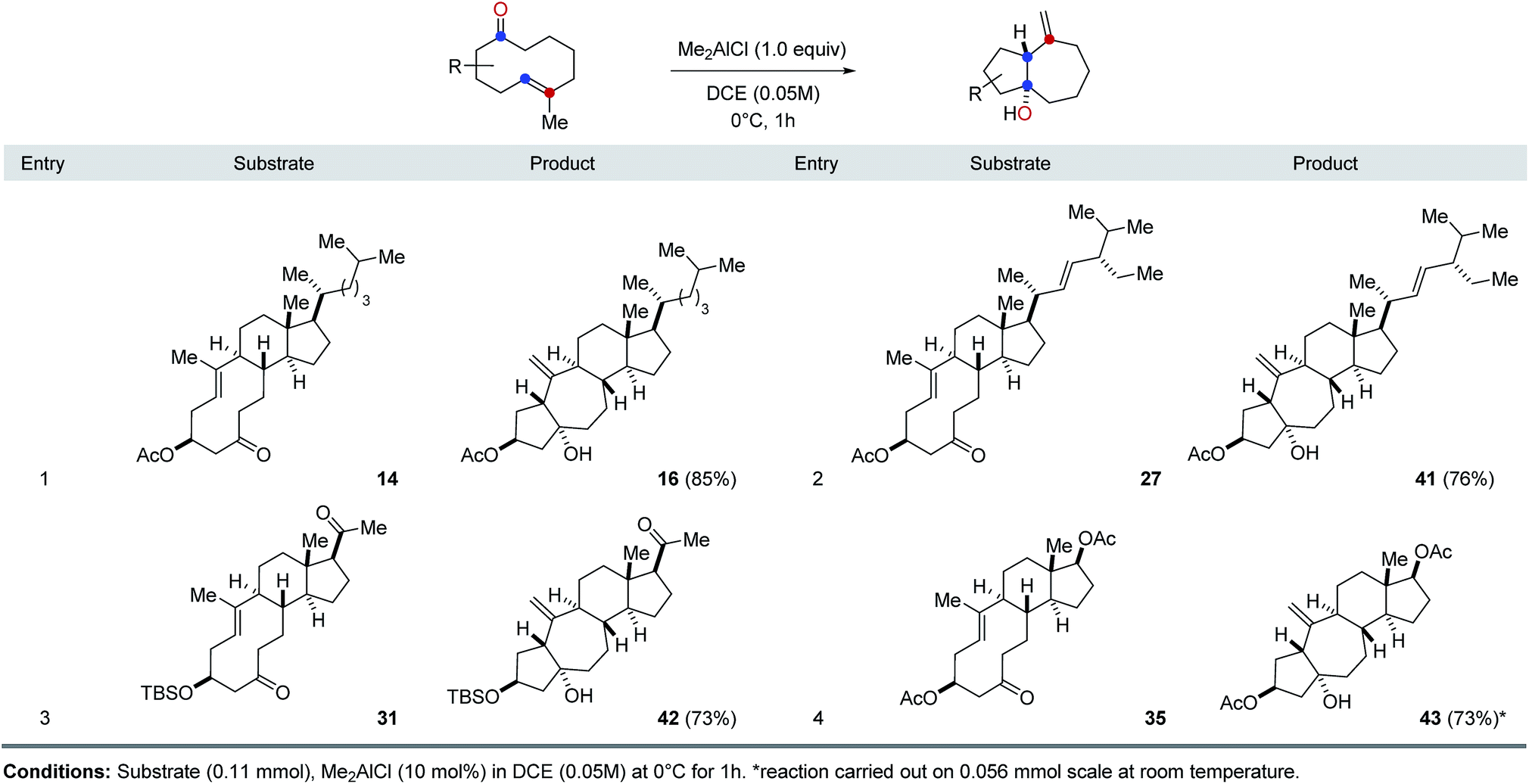
|
Conclusions
This report describes the development and mechanistic investigation of catalytic, transannular carbonyl-olefin metathesis reactions as a fourth category of these transformations. The mechanism for transannular carbonyl-olefin metathesis is distinct and relies on competing carbonyl-ene and carbonyl-olefin metathesis reaction paths that ultimately favour metathesis as the thermodynamic product. Importantly, distinct Lewis acids are shown to be capable of differentiating between these pathways, providing a new opportunity for the molecular editing of naturally occurring complex molecules. The feasibility of this approach is demonstrated on several complex cyclodecenone systems readily derived from naturally occurring steroids that provide rapid access to three new and distinct molecular scaffolds simply based on the choice of Lewis acid.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the NIH (R01-GM118644) for financial support. P. S. R. thanks the Rackham Graduate School of the University of Michigan for a dissertation fellowship. C. S. S. thanks the Alfred P. Sloan Foundation, the David and Lucile Packard Foundation, and the Camille Dreyfus Foundation for fellowships. We thank Duncan Ellinwood and Yuan Lu for help with preliminary experiments. We thank Ren Wiscons and Jeff Kampf for X-ray crystallography. We are grateful to Paul Zimmerman and Michael Robo for helpful discussions regarding computational studies.Notes and references
- For early approaches to carbonyl-olefin metathesis, see:
(a) I. Schopov and C. Jossifov, Makromol. Chem., Rapid Commun., 1983, 4, 659–662 CrossRef
; (b) G. C. Fu and R. H. Grubbs, J. Am. Chem. Soc., 1993, 115, 3800–3801 CrossRef CAS
-
(a) J. R. Ludwig, P. M. Zimmerman, J. B. Gianino and C. S. Schindler, Nature, 2016, 533, 374–379 CrossRef CAS PubMed
- H. Albright, H. L. Vonesh, M. R. Becker, B. W. Alexander, J. R. Ludwig, R. A. Wiscons and C. S. Schindler, Org. Lett., 2018, 20, 4954–4958 CrossRef CAS PubMed
-
(a) U. P. N. Tran, G. Oss, D. P. Pace, J. Ho and T. V. Nguyen, Chem. Sci., 2018, 9, 5145–5151 RSC
- B. N. Bhawal and B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 10074–10103 CrossRef CAS PubMed
- For reviews on carbonyl-ene and ene reactions, see:
(a) M. L. Clarke and M. B. France, Tetrahedron, 2008, 64, 9003–9031 CrossRef CAS
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, DOI:10.1126/science.aat0805
- For selected recent reviews on transition metal catalyzed C–H activation, see:
(a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed
-
(a) A. M. Szpilman and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9592–9628 CrossRef CAS PubMed
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80, DOI:10.1038/ncomms1081
- For detailed studies on the reactivity of steroids and steroid derivatives including acid-catalyzed transformations, see:
(a) H. Fuhrer, Lj. Lorenc, V. Pavlović, G. Rihs, G. Rist, J. Kavloda and M. L. Mihailović, Helv. Chim. Acta, 1979, 62, 1770–1784 CrossRef CAS
- For preparation of compound 14 using Pb(OAc)4, see:
(a)
Ref. 11a. For preparation of 14 using I2 and HgO, see ;
(b) L. Lorenc, L. Bondarenko, M. Rajković, A. Milavanović and M. Lj. Mihailović, Tetrahedron, 1983, 39, 3609–3614 CrossRef CAS
- For hypervalent iodine mediated opening of steroid frameworks, see:
(a) A. Boto, C. Betancor, T. Prangé and E. Suárez, Tetrahedron Lett., 1992, 33, 6687–6690 CrossRef CAS
- Efforts undertaken by Khirpach and coworkers to protect a carbonyl in a seco-steroid as a dithioketal upon treatment with 3 equivalents of BF3·Et2O was reported to form a transannular carbonyl–olefin metathesis product: V. A. Khripach, V. N. Zhabinskii, A. I. Kuchto, V. V. Gromak, M. B. Goen, J. van der Louw and A. de Groot, Tetrahedron Lett., 2006, 47, 6715–6718 CrossRef CAS
- M. P. Singh, J. E. Janso, S. W. Luckman, S. F. Brady, J. Clardy, M. Greenstein and M. W. Maiese, J. Antibiot., 2000, 53, 256–261 CrossRef CAS PubMed
- D. D. Bikle, Chem. Biol., 2014, 21, 319–329 CrossRef CAS PubMed
- S. Aoki, Y. Watanabe, M. Sanagawa, A. Setiawan, N. Kotoku and M. Kobayashi, J. Am. Chem. Soc., 2006, 128, 3148–3149 CrossRef CAS PubMed
- See ESI for details.†.
- Detailed previous reports have identified distinct low energy conformations of steroid 14 based in spectroscopic and theoretical investigations, see:
(a) H. Fuhrer, Lj. Lorenc, V. Pavlović, G. Rihs, G. Rist, J. Kavloda and M. L. Mihailović, Helv. Chim. Acta, 1979, 62, 1770–1784 CrossRef CAS
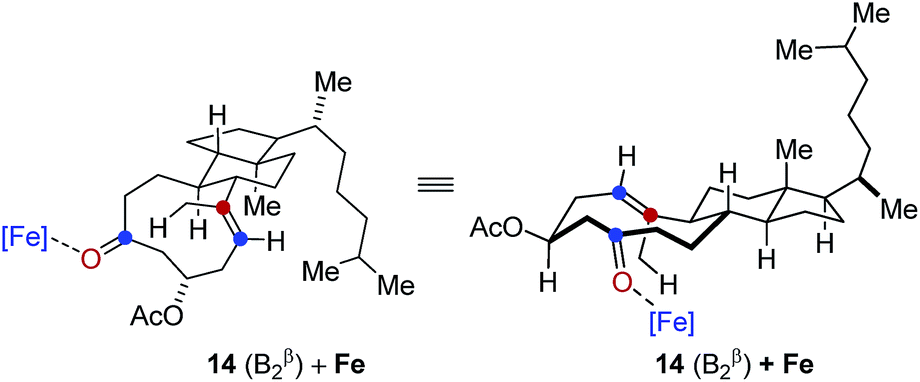
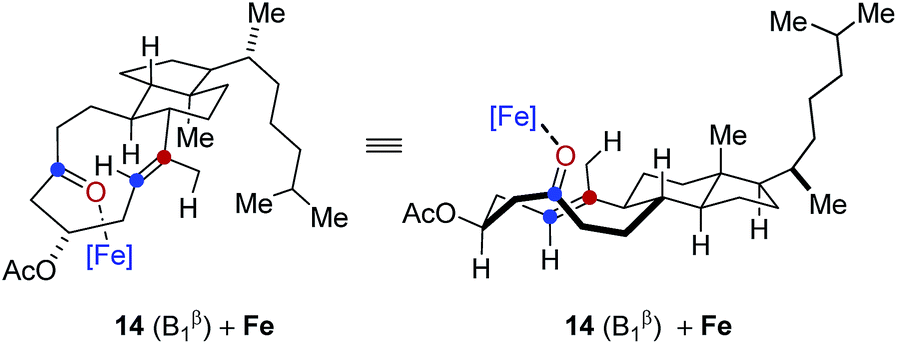
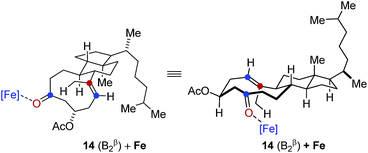 In comparison, conformation 14 (B1β) was found to lead to the preferential formation of diastereomeric product epi-16 that is not observed experimentally under our optimal reaction conditions. See ESI for details.†
In comparison, conformation 14 (B1β) was found to lead to the preferential formation of diastereomeric product epi-16 that is not observed experimentally under our optimal reaction conditions. See ESI for details.†
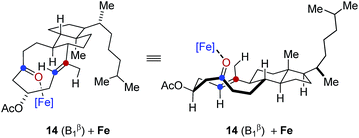 .
. - For a Brønsted acid-catalyzed oxetane fragmentation proceeding via intermediate carbocations, see: J. R. Ludwig, R. B. Watson, D. J. Nasrallah, J. B. Gianino, P. M. Zimmerman, R. A. Wiscons and C. S. Schindler, Science, 2018, 361, 1363–1369 CrossRef CAS PubMed
- Brønsted acid-catalyzed transannular carbonyl-ene reactions have been previously reported on steroid scaffolds. For details:
(a) See ref. 11a;
(b) M. Lj. Mihailović, Lj. Lorenc, J. Foršek, H. Nešović, G. Snatzke and P. Trška, Tetrahedron, 1970, 26, 557–573 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03716k |
| This journal is © The Royal Society of Chemistry 2019 |