
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the enthalpic contribution of hydrogen bonds by solvent shielding†
Jonathan
Cramer
 a,
Xiaohua
Jiang
a,
Wojciech
Schönemann
a,
Marleen
Silbermann
a,
Pascal
Zihlmann
a,
Stefan
Siegrist
a,
Brigitte
Fiege
a,
Roman Peter
Jakob
b,
Said
Rabbani
a,
Timm
Maier
b and
Beat
Ernst
*a
a,
Xiaohua
Jiang
a,
Wojciech
Schönemann
a,
Marleen
Silbermann
a,
Pascal
Zihlmann
a,
Stefan
Siegrist
a,
Brigitte
Fiege
a,
Roman Peter
Jakob
b,
Said
Rabbani
a,
Timm
Maier
b and
Beat
Ernst
*a
aInstitute of Molecular Pharmacy, University of Basel, Klingelbergstrasse 50, 4056 Basel, Switzerland. E-mail: beat.ernst@unibas.ch
bInstitute of Structural Biology, University of Basel, Klingelbergstrasse 70, 4056 Basel, Switzerland
First published on 28th August 2020
Abstract
In biological systems, polar interactions are heavily burdened by high desolvation penalties resulting from strong solute–solvent interactions. As a consequence thereof, enthalpic contributions of hydrogen bonds to the free energy of binding are severely diminished. However, this effect is strongly attenuated for interactions within solvent-shielded areas of proteins. In microcalorimetric experiments, we show that the bacterial lectin FimH utilizes conformational adaptions to effectively shield its binding site from solvent. The transition into a lower dielectric environment results in an enthalpic benefit of approximately −13 kJ mol−1 for mannoside binding. However, this effect can be abrogated, if the hydrogen bond network within the binding site is disturbed by deoxygenation of the ligand. Conformational adaption leading to reduced local dielectric constants could represent a general mechanism for proteins to enable enthalpy-driven recognition of polar ligands.
Introduction
The energetics of hydrogen bonds in biomolecular recognition are highly affected by severe desolvation penalties imposed by strong interactions with the aqueous solvent in the unbound state. A common conception is that strongly directional hydrogen bond interactions provide selectivity, but do not necessarily augment the overall binding affinity of a ligand because favorable contributions are often canceled out by significant desolvation penalties.1–3 The free energy penalty for the transfer of a hydroxyl group from aqueous solvent to the gas phase was estimated as 26 kJ mol−1, with an associated enthalpy penalty of 36 kJ mol−1 and the transfer of an alcohol function from bulk solvent into a hydrophobic pocket was associated with a free energy penalty of 18–21 kJ mol−1.4,5 Furthermore, in an aqueous system, the contribution of a single hydrogen bond to the free energy of binding has been estimated to equally amount to 18–21 kJ mol−1.6–10 In summary, a hydrogen bond hardly contributes to the binding process, unless additional conditions are fulfilled. It has been proposed that hydrophobic occlusion and solvent shielding are able to advance the strength of hydrogen bonds in biological systems. Thus, enthalpies of formation for solvent-exposed hydrogen bonds on protein surfaces amount to −6 kJ mol−1, whereas buried interactions contribute with up to −25 kJ mol−1.11,12 For the arabinose-binding protein, removal of buried hydrogen bond interactions in deoxy D-galactose derivatives was associated with an enthalpy-driven loss in binding free energy of 30 kJ mol−1.13 Similarly, hydrophobic solvent-shielding has been shown to impact the activity of enzymes, as well as the pKa of buried titratable groups.14–18 The exclusion of polar water molecules effectively creates a low dielectric environment that supports electrostatic interactions and accelerates the reaction of charged intermediates. Thus, solvent shielding of hydrophilic binding sites may contribute to enthalpy-driven recognition of highly polar molecules such as carbohydrates. In this study, we aim to demonstrate that proteins actively harness the modulation of local dielectric properties by conformational transitions and to quantify the thermodynamic consequences on polar hydrogen bond interactions.As a model system to study the impact of the dielectric constant εr on the thermodynamics of binding, we employed FimH, a bacterial adhesin located on type I pili of uropathogenic E. coli.19–21 The functional lectin domain of FimH features a carbohydrate recognition site tailor-made to accommodate mannose epitopes. The mannose binding affinity of the lectin domain is allosterically controlled by the adjacent regulatory pilin domain.22,23 When mechanical forces, such as shear forces during micturition, cause the separation of lectin and pilin domain,23,24 a switch from the low-affinity conformation to the high-affinity conformation of FimH is induced and mannoside ligands are bound with ∼100-fold stronger affinity compared to the domain-associated low affinity state. Here, we compare the properties of the two affinity states by means of two different recombinant FimH constructs.
Results and discussion
The isolated FimH lectin domain (FimHLD) is a common in vitro model for the domain-separated, high-affinity conformation of the protein.25 FimHLD features a rigid structure and a well-defined carbohydrate binding site (Fig. 1). Mannoside ligands engage in an impressive network of ten hydrogen bonds. In the absence of a ligand, water molecules assume the positions of mannose hydroxyl groups, pre-organizing the binding site for carbohydrate recognition (RMSDapo-bound = 0.35 Å). The binding event is a single-step mechanism that proceeds without any major conformational adjustment of the residues in the mannose binding site.26,27 On the other hand, the domain associated full-length FimH construct (FimHFL) has been shown to bind mannoside ligands by a more complex mechanism caused by conformational distortions of the protein due to the presence of the regulatory subunit.22,23 In the absence of a ligand, the protein exists in an ensemble of conformational states that are characterized by a shallow, solvent-exposed binding site, a low affinity to carbohydrate ligands, and a high degree of flexibility (FimHFL-open, Fig. 1).23,28 In the presence of a ligand, the conformational equilibrium is shifted toward a closed state (FimHFL-closed), in which the clamp loop is accommodated close to the carbohydrate binding site and shields the mannose moiety from the solvent. The atomistic features of this mechanism, namely clamp loop mobility in the open and solvent shielding of the binding site in the closed conformation, have been thoroughly characterized in MD simulations.29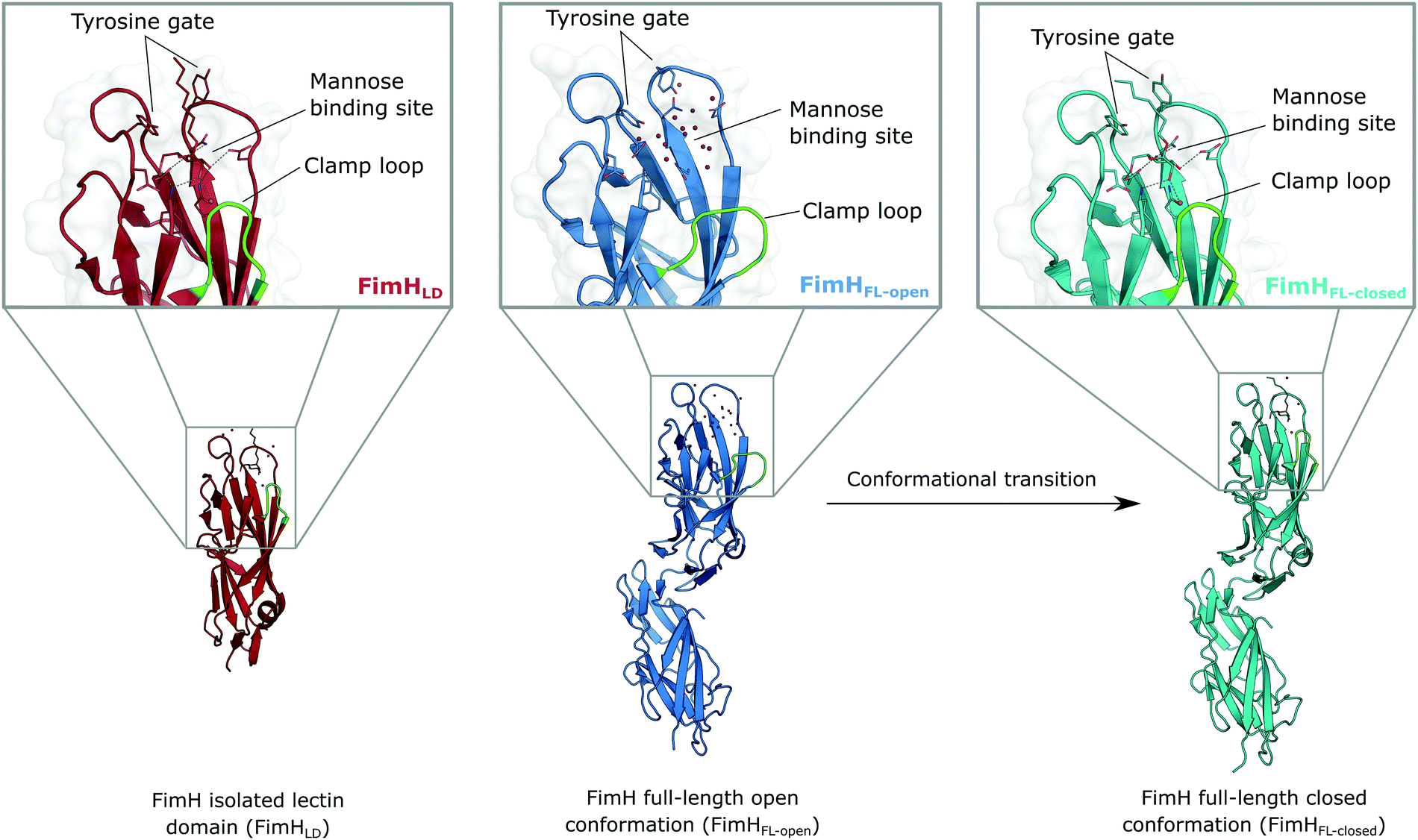 | ||
| Fig. 1 Molecular structure of the bacterial lectin FimH. The isolated lectin domain (FimHLD, PDB 4BUQ) is fairly rigid and pre-organized in the closed conformation even in the absence of a ligand. The recombinant full-length FimH construct (FimHFL) undergoes a conformational transition from an open (FimHFL-open, PDB 4XOD) to a closed (FimHFL-closed, PDB 4XOE) state when a ligand binds to the mannose binding site. | ||
Although the overall structure of this state differs from the high-affinity conformation, the binding site regions are virtually identical (RMSDlectin_domain = 2.0 Å, RMSDbinding_site = 0.2 Å). Fig. 2 depicts a thermodynamic model of ligand binding to FimHLD and FimHFL. The experimentally observed free energy of binding to FimHLD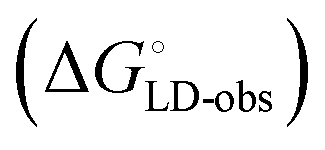 is composed of terms for desolvation
is composed of terms for desolvation 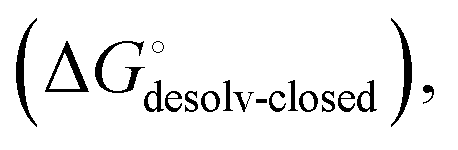 protein–ligand interaction
protein–ligand interaction 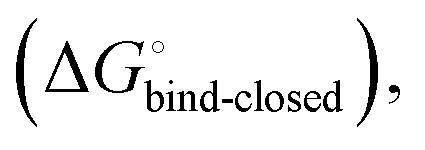 and solvation of the assembled complex
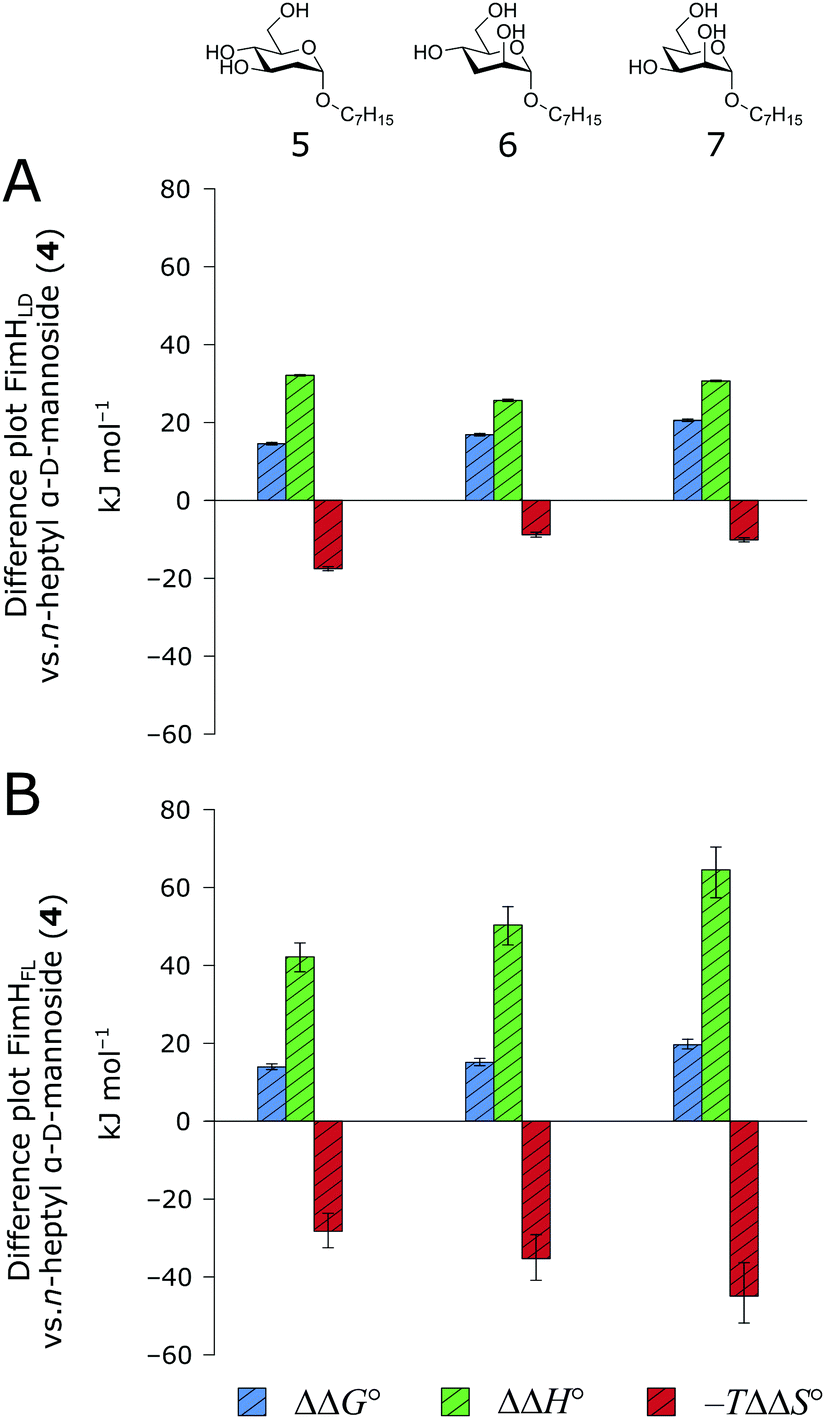
and solvation of the assembled complex 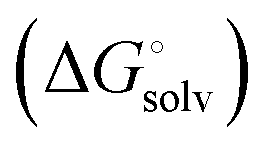 (Fig. 2, pathway A). For FimHFL, the conformational rearrangement associated with ligand binding theoretically proceeds via a conformational selection (Fig. 2, pathway B) or an induced-fit (Fig. 2, pathway C) mechanism. In case of conformational selection, the experimentally observed thermodynamics
(Fig. 2, pathway A). For FimHFL, the conformational rearrangement associated with ligand binding theoretically proceeds via a conformational selection (Fig. 2, pathway B) or an induced-fit (Fig. 2, pathway C) mechanism. In case of conformational selection, the experimentally observed thermodynamics  contain contributions from a conformational transition to the closed state
contain contributions from a conformational transition to the closed state 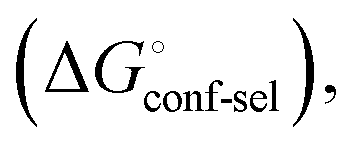 desolvation
desolvation 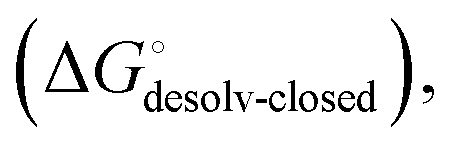 protein–ligand interaction
protein–ligand interaction 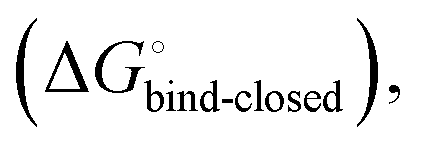 and solvation of the assembled complex
and solvation of the assembled complex  . In the induced-fit pathway C,
. In the induced-fit pathway C, 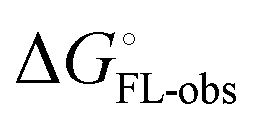 is composed of desolvation
is composed of desolvation 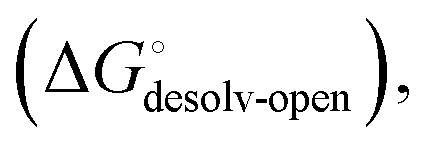 low-affinity binding
low-affinity binding  induced-fit transition
induced-fit transition 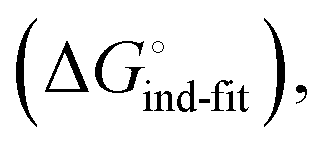 and solvation of the assembled complex
and solvation of the assembled complex 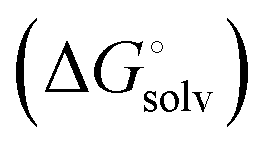 . Thus, the difference in the thermodynamic profiles of ligand binding to FimHFL and FimHLD
. Thus, the difference in the thermodynamic profiles of ligand binding to FimHFL and FimHLD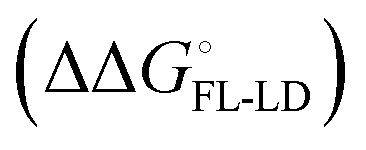 gives access to an experimental approximation of clamp loop rearrangement and hydrophobic occlusion of the polar mannose binding site. In the conformational selection model (Fig. 2, pathway B),
gives access to an experimental approximation of clamp loop rearrangement and hydrophobic occlusion of the polar mannose binding site. In the conformational selection model (Fig. 2, pathway B), 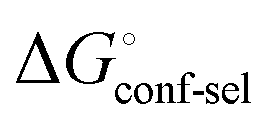 is a constant term, and ligand binding affinity scales entirely with binding affinity to the closed state. In the induced-fit model (Fig. 2, pathway C), however, differences between ligands can scale with the affinity of a low-affinity encounter complex
is a constant term, and ligand binding affinity scales entirely with binding affinity to the closed state. In the induced-fit model (Fig. 2, pathway C), however, differences between ligands can scale with the affinity of a low-affinity encounter complex  and the effect of the conformational transition
and the effect of the conformational transition 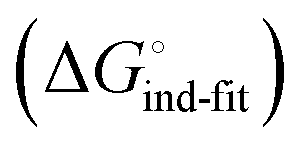 .
.
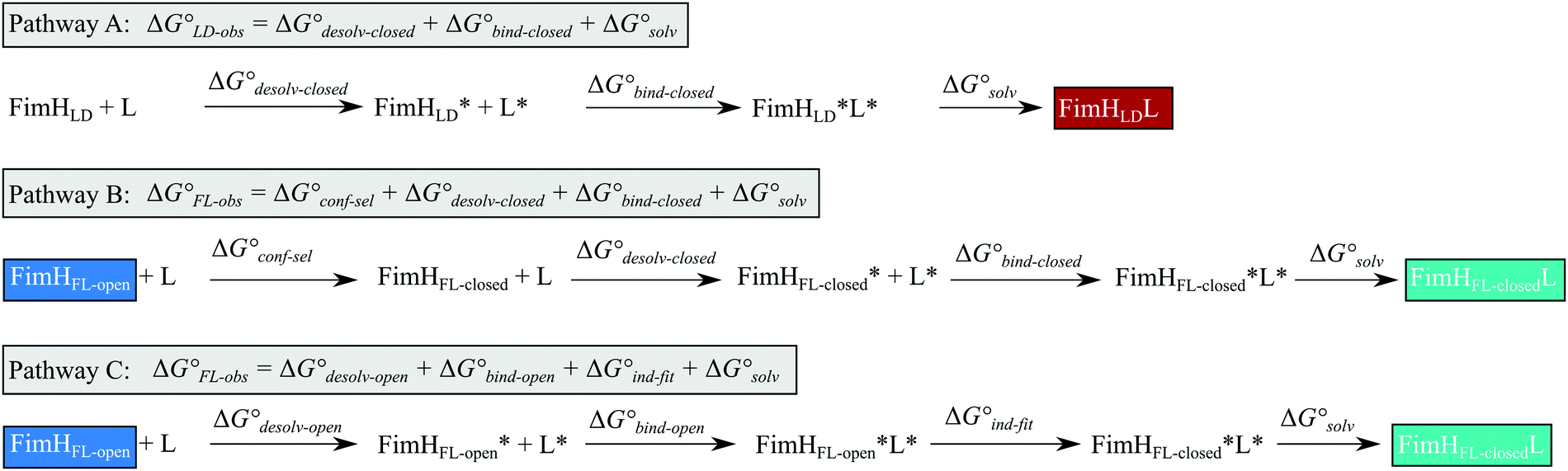 | ||
| Fig. 2 Thermodynamic model of the interaction of FimH with mannoside ligands. FimH states and complexes are colored analogous to available X-ray structures in Fig. 1. Desolvated states are indicated with an asterisk. | ||
As a result of an extensive network of hydrogen bonds between the pre-organized binding site and the mannose moiety, the interaction between mannosides 1–4 and FimHLD (high-affinity state) is characterized by a strongly enthalpic contribution (Fig. 3A). The weak entropic penalty has been linked to the rigidification of the tyrosine gate, an ensemble of hydrophobic residues that control access to the mannose binding site and interact with the aglycones of ligands 1–4.26,27 An improved π–π-stacking and hydrophobic interaction between aglycone and tyrosine gate further accentuates their thermodynamic profile, as the electrostatic nature of the aglycones gives rise to progressively tighter, enthalpy-driven binding from 4 → 3 → 2 → 1.
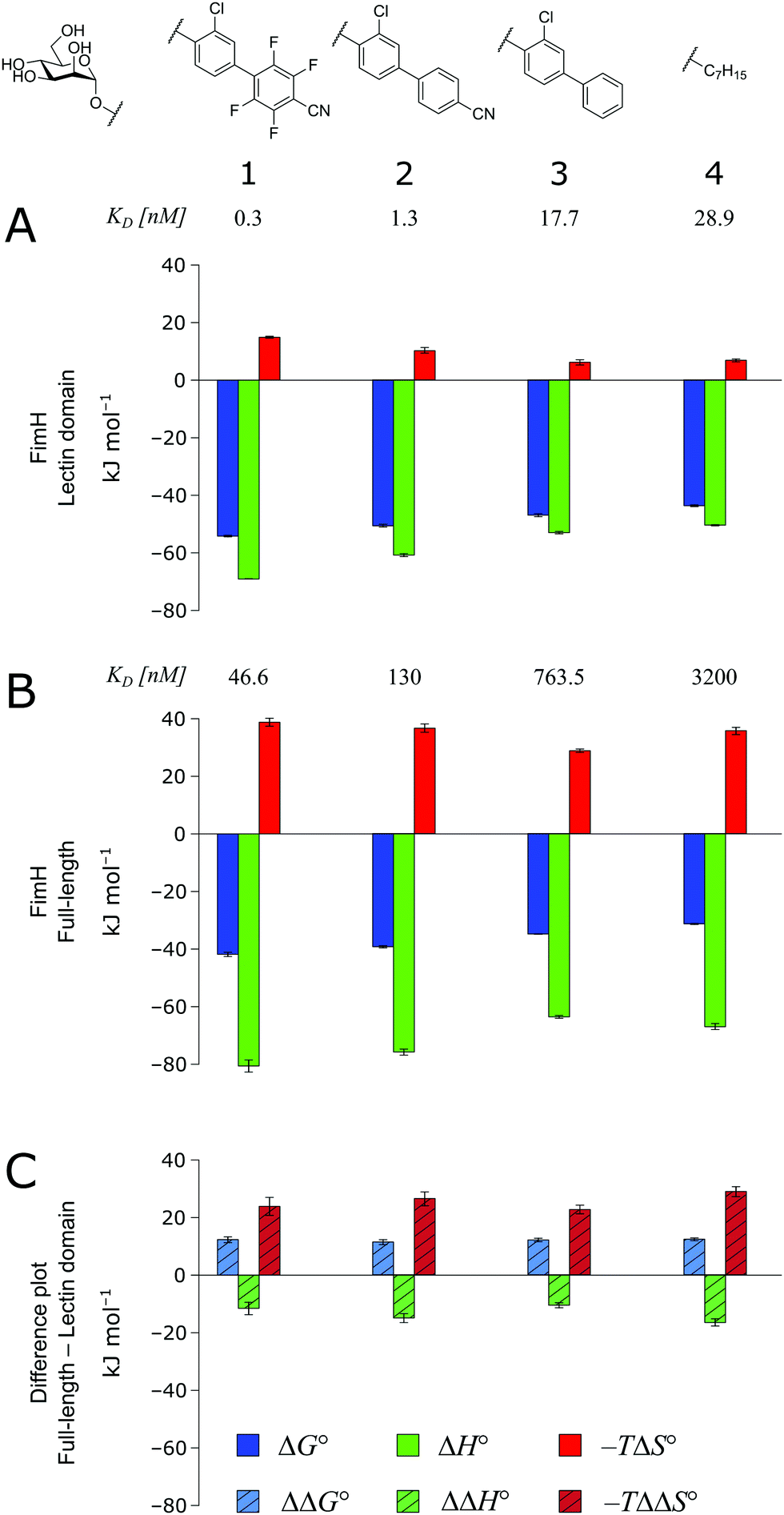 | ||
| Fig. 3 Thermodynamic profiles (see Fig. S1–S4, ESI†) for the interaction of mannosides 1–4 with FimHLD (A) and FimHFL (B). Data for the interactions FimHLD-2/3/4 and FimHFL-2/4 have been published before.24,30,31 (C) Difference in the thermodynamic profiles between ligand binding to FimHFL and FimHLD. Error bars represent (propagated) 68% confidence intervals. Numeric data and further information can be found in the ESI.† | ||
The thermodynamic profiles of mannoside interactions with the FimHFL construct (low-affinity state) show similar characteristics (Fig. 3B). The binding process is equally driven by enthalpy, with a counteracting entropic penalty. However, the magnitude of this enthalpy–entropy compensation is much more substantial. Overall, the binding affinity of the mannosides 1–4 is reduced roughly by 12 kJ mol−1, approximately two orders of magnitude in KD, compared with the same ligands binding to FimHLD. This value represents the experimental approximation of the thermodynamics related to the conformational rearrangement from the open (FimHFL-open) to the closed (FimHFL-closed) state.
The difference in the thermodynamics (Fig. 3C) shows that this fairly constant affinity reduction mainly originates from a severe entropy penalty of roughly 26 kJ mol−1, which can be correlated to a shift of the conformational equilibrium of FimHFL toward a closed state, in which the clamp loop is fixed near the mannose moiety. The high mannose binding affinity of FimHLD thus results from pre-organization of the mannose binding site. This concept has been thoroughly characterized in the study of enzyme catalysis.32,33 Less intuitively, the thermodynamics are also characterized by a favorable enthalpic contribution of approximately −13 kJ mol−1. Since the mannose hydrogen bond networks and any other electrostatic contacts between protein and ligand are identical in the bound states of FimHLD and FimHFL, this difference has to originate from the conformational transition of the clamp loop region upon ligand binding. While the local dielectric constant on protein surfaces, as in the open form of FimHFL, is commonly estimated to be 20, it is reduced to approximately 5 in the shielded binding site of FimHFL-closed.17 Based on these simplified qualitative estimations,34,35 the shift between the open and closed protein states is accompanied by hydrophobic shielding of polar hydrogen bond interactions in the binding site. This leads to reduced local permittivity εr in FimHFL-closed, which is associated with an enhanced enthalpic contribution to binding 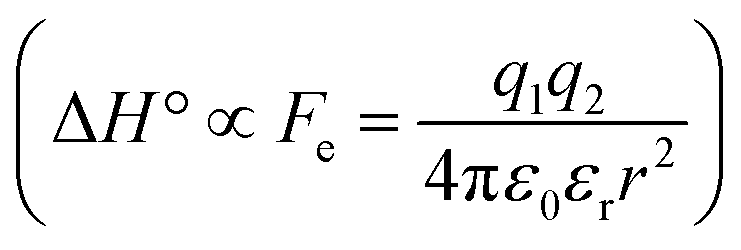 .
.
In order to investigate the outcome of a perturbation of the hydrogen bond network, several deoxygenated derivatives of n-heptyl α-D-mannoside (4) were subjected to thermodynamic and structural analyses. The X-ray crystal structures of 4–7 in complex with FimHLD reveal very little variation (RMSDs ≈ 0.2 Å) upon sequential removal of mannose hydroxyl groups (Fig. 4A), indicating that the loss of hydrogen bonds is not associated with any major conformational adjustments. Next, NMR chemical shift perturbations (CSP) of residues involved in hydrogen bond interactions to mannose hydroxyl groups were studied (Fig. 4B). Gly14, engaged in a water-mediated hydrogen bond to 2-OH, shows a minor CSP upon ligand binding. Variations between different mannosides are negligible, even when the 2-OH group is removed (→5). This indicates that the position of the interacting water molecule W1 is not affected. The backbone amide NH of Asp47 forms a hydrogen bond with 6-OH, leading only to slight CSP upon binding to the ligands 4 and 5 (2-deoxy). In case of the 3-deoxy derivative 6, the CSP is more pronounced, whereas the signal is superimposed to the apo protein for 7 (4-deoxy). CSP of the amide side chain protons of Gln133 report on hydrogen bond formation with 3-OH. For 4, the signal of the amide proton (PDB nomenclature Hε22) is shifted downfield by 0.8 ppm, indicating hydrogen bond formation. A smaller downfield shift of 0.3–0.5 ppm signifies the slightly weaker interaction for 5 and 7, whereas the absence of the interaction for 3-deoxy derivative 6 is obvious from a strong upfield shift of −1.6 ppm.
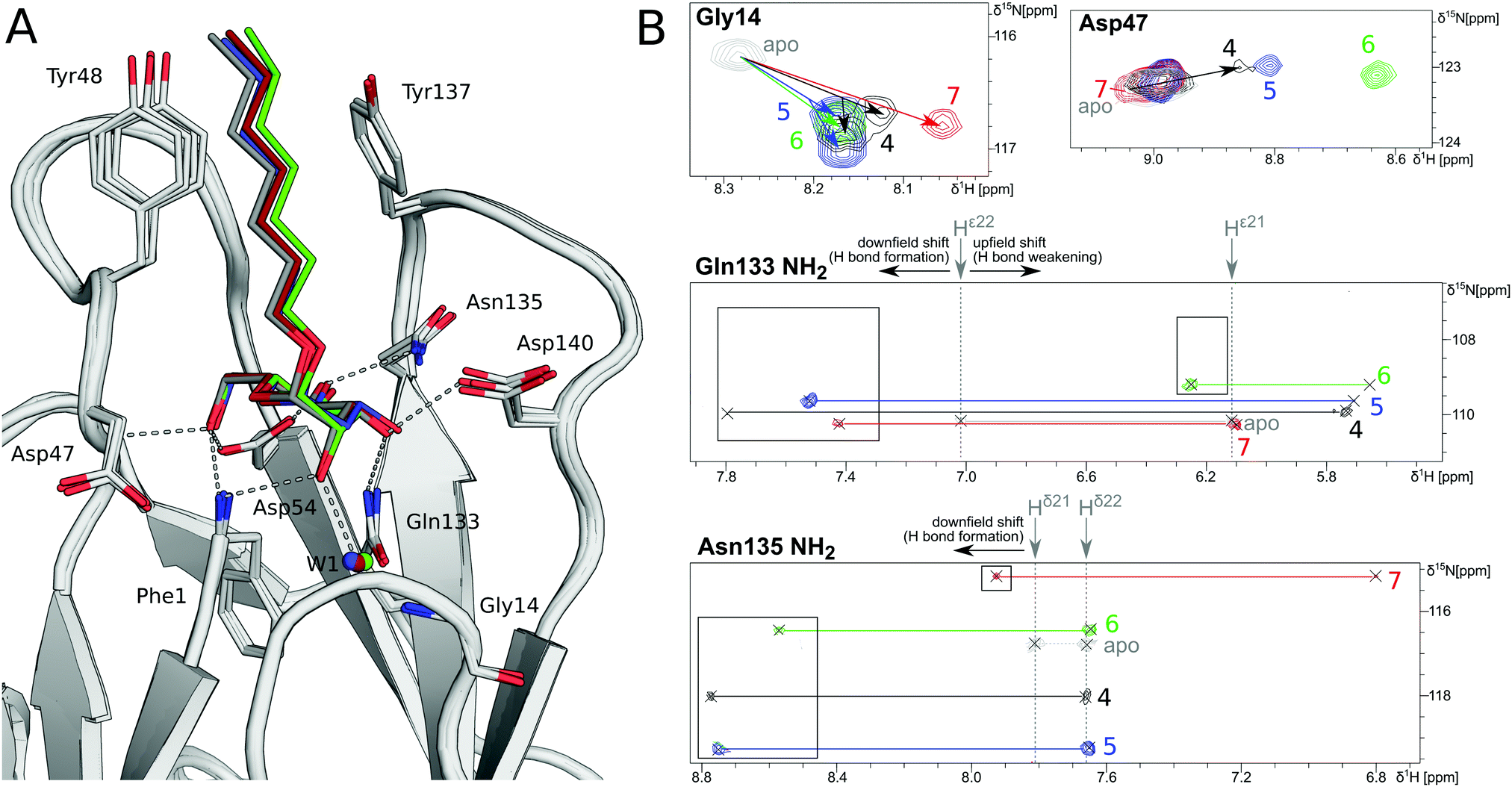 | ||
| Fig. 4 Structural characterization of interactions between mannoside 4 and the deoxygenated compounds 5–7 and FimHLD. (A) X-ray crystal structures of mannoside 4 (black, PDB 4BUQ), 2-deoxy-mannoside 5 (blue, PDB 5L4T), 3-deoxy-mannoside 6 (green, PDB 5L4V), and 4-deoxy-mannoside 7 (red, PDB 5L4X). Hydrogen bond interactions of 4 are indicated as grey dashes. (B) 1H,15N-HSQC NMR experiments reveal chemical shift perturbations for residues in the mannose binding site in the presence of ligands. Color coding according to panel A. Nomenclature of Asn and Gln δ and ε protons according to PDB guidelines. Full spectrum is shown in Fig. S20 (ESI†). | ||
Similarly, CSP of the side chain signals of Asn135 indicate the interaction with 4-OH. The signal is shifted downfield for 4, 5, and 6, whereas a relative upfield shift for the 4-deoxy derivative 7 signifies the absence of the interaction. Importantly, none of the observed residues show a marked shift in the 15N-dimension of the spectra, reporting on changes in dihedral angles of protein backbone and side chains.
In summary, the structural information obtained by NMR and X-ray is in good agreement, and the conformational changes of protein and ligand are remarkably small upon the loss of relevant interactions. In both experiments, low binding affinity prevented a characterization of compound 8.
The thermodynamics of 5–7 binding to FimHLD (Fig. 5A) illustrate that removal of the hydroxyl groups in the 2-, 3-, and 4-position of n-heptyl α-D-mannoside 4 coincide with a sizeable reduction of the enthalpic contribution to binding.
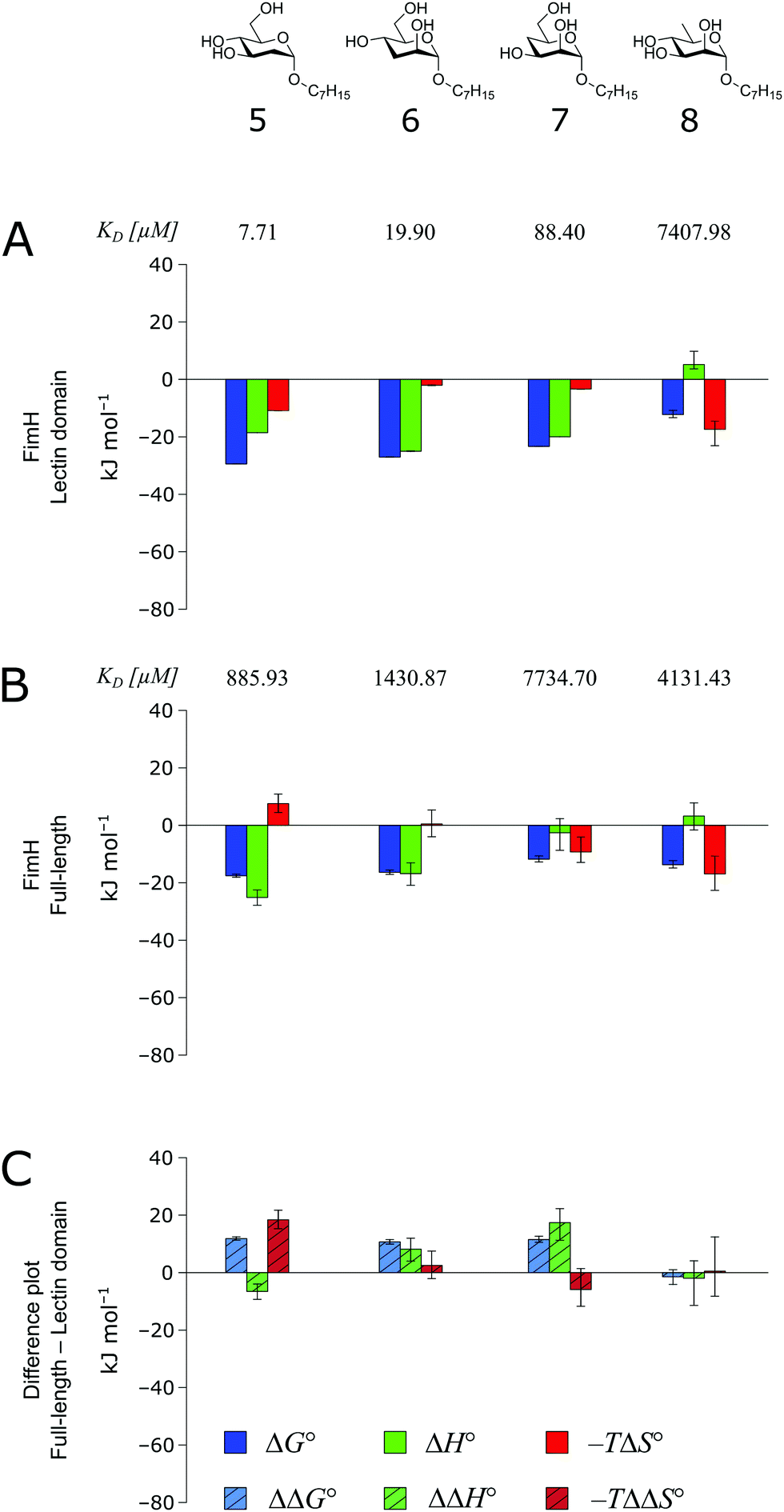 | ||
| Fig. 5 Thermodynamic profiles (see Fig. S5–S8, ESI†) for the interaction of deoxygenated n-heptyl α-D-mannoside derivatives 5–8 with FimHLD (A) and FimHFL (B). Data for the interactions of FimHLD with compounds 5–7 have been published elsewhere.30 (C) Difference in the thermodynamic profiles between ligand binding to FimHFL and FimHLD. Error bars represent (propagated) 68% confidence intervals. | ||
In addition, the negative value for −TΔS° indicates an increased degree of disorder, probably as a consequence of the impaired hydrogen bond network. This is in line with the reduced downfield shifts in NMR experiments. The interaction of 5–7 with FimHFL shows an equivalent trend in the thermodynamic profiles (Fig. 5B). Yet, the magnitude of the enthalpy–entropy compensation effect differs substantially. As a consequence, the fairly constant trend we observed in the differential profiles for 1–4 (Fig. 3C) is lost for 5–7 (Fig. 5C). Here, the enthalpic contribution that was attributed to the modulation of binding site permittivity εr in FimHFL is considerably diminished and even reversed for 6 and 7. Instead, a progressive shift toward entropic binding is observed.
When the thermodynamic profiles of 5–7 are compared with the parent compound 4, notable dissimilarities between the two protein constructs, FimHLD (Fig. 6A) and FimHFL (Fig. 6B) can be observed. For FimHLD, the enthalpic penalty for removal of a hydroxyl group amounts to 26–32 kJ mol−1. The same modification in FimHFL, however, results in an enthalpic penalty that ranges from 42 kJ mol−1 up to 65 kJ mol−1. The discrepancy suggests that the loss of a hydrogen bond in the interaction of 5–7 and FimHFL is superimposed by global effects resulting from a modulation of clamp loop dynamics and the associated solvent shielding mechanism.
 | ||
| Fig. 6 Difference in thermodynamic profiles between 4 and deoxygenated n-heptyl α-D-mannoside derivatives 5–7 with FimHLD (A) or FimHFL (B). Error bars represent propagated 68% confidence intervals. | ||
Thus, the observed thermodynamics are not indicative of the protein–ligand interaction itself, but instead reflect a shift in the conformational equilibrium of the protein. This mechanism has been termed entropy–enthalpy transduction.36 The existence of distinctive enthalpy–entropy compensation profiles for different ligands might also indicate that binding does not proceed via selection of a preexisting FimHFL conformation (Fig. 2, pathway B), but through a ligand-induced fit (Fig. 2, pathway C). The conformational equilibrium in a conformational selection model is an inherent property of the protein and does not depend on the nature of a ligand. Thus, 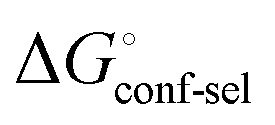 must be a constant term, and differences in ligand thermodynamics originate solely from interaction with the binding-competent conformation
must be a constant term, and differences in ligand thermodynamics originate solely from interaction with the binding-competent conformation  In an induced-fit model, however, formation of an encounter complex
In an induced-fit model, however, formation of an encounter complex 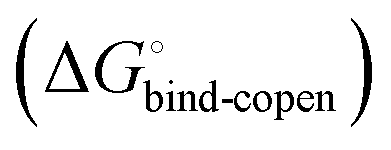 and contrasting consequences of the conformational transition
and contrasting consequences of the conformational transition 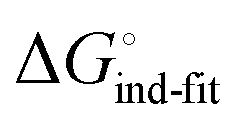 (e.g. loss of enthalpic benefit from solvent shielding) can account for differences between ligands. For ligands with an intact mannoside core (1–4), the induced-fit transition reaches its full effect so that the differential thermodynamic profiles (Fig. 3C) appear constant.
(e.g. loss of enthalpic benefit from solvent shielding) can account for differences between ligands. For ligands with an intact mannoside core (1–4), the induced-fit transition reaches its full effect so that the differential thermodynamic profiles (Fig. 3C) appear constant.
Intriguingly, X-ray and NMR experiments yielded no information about the binding mode of 8 to FimHLD. ITC data suggests that this compound actually binds with very low, but similar affinity to the high- and low-affinity construct of the protein (Fig. 5). These observations could signify a different binding mode that does not proceed via a conformational rearrangement in FimHFL.
Conclusions
In conclusion, we have investigated the binding thermodynamics of two FimH constructs that share a similar binding mode but explore a very different conformational landscape. We could demonstrate that an observed enthalpic contribution of roughly −13 kJ mol−1 for mannoside ligands is a direct consequence of solvent shielding enabled by the rearrangement of the clamp loop in FimHFL and the associated modulation of local dielectric properties. When the hydrogen bond network in the binding site is disrupted, the enthalpic benefit is completely abolished (Fig. 5C). In the case of FimHFL, the effect of the solvent shielding mechanism is superimposed by an entropic penalty for the arrest of the clamp loop dynamics.A similar transition of the binding site into a lower dielectric environment was observed for a number of other carbohydrate binding proteins, such as arabinose-binding protein (ABP), glucose/galactose-binding protein (GGBP) und sialic acid-binding periplasmic protein (SiaP). Their conformational transition upon ligand binding involves a twist of rigid protein domains around a fixed axis (“hinge-bending” motions).37–40 These transitions do not alter the flexibility of individual protein domains to an extent as observed in FimH and can be considered entropically neutral. Thus, modulation of binding site permittivity could represent a general mechanism for carbohydrate binding proteins to enable enthalpy-driven recognition of polar ligands in aqueous solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. E. gratefully acknowledges funding from the Swiss National Science Foundation (PZ) and German Academic Exchange Service (BF).Notes and references
- H.-J. Böhm and G. Klebe, Angew. Chem., Int. Ed. Engl., 1996, 35, 2588–2614 CrossRef
.
- R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359–1472 CrossRef CAS PubMed
-
G. M. Keserü and D. C. Swinney, Thermodynamics and Kinetics of Drug Binding, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed
- S. Cabani, P. Gianni, V. Mollica and L. Lepori, J. Solution Chem., 1981, 10, 563–595 CrossRef CAS
- E. Barratt, A. Bronowska, J. Vondrášek, J. Černý, R. Bingham, S. Phillips and S. W. Homans, J. Mol. Biol., 2006, 362, 994–1003 CrossRef CAS PubMed
-
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS
- C. P. Sager, D. Eriş, M. Smieško, R. Hevey and B. Ernst, Beilstein J. Org. Chem., 2017, 13, 2584–2595 CrossRef CAS
- A. Vedani and J. D. Dunitz, J. Am. Chem. Soc., 1985, 107, 7653–7658 CrossRef CAS
- J. Cramer, C. P. Sager and B. Ernst, J. Med. Chem., 2019, 62, 8915–8930 CrossRef CAS PubMed
- A. V. Efimov and E. V. Brazhnikov, FEBS Lett., 2003, 554, 389–393 CrossRef CAS PubMed
- G. Némethy, I. Z. Steinberg and H. A. Scheraga, Biopolymers, 1963, 1, 43–69 CrossRef
- A. H. Daranas, H. Shimizu and S. W. Homans, J. Am. Chem. Soc., 2004, 126, 11870–11876 CrossRef CAS PubMed
- S.-O. Shan and D. Herschlag, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14474–14479 CrossRef CAS PubMed
- M. M. Malabanan, T. L. Amyes and J. P. Richard, Curr. Opin. Struct. Biol., 2010, 20, 702–710 CrossRef CAS PubMed
- D. G. Isom, C. A. Castañeda, B. R. Cannon, P. D. Velu and E. B. García-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16096–16100 CrossRef CAS PubMed
- C. A. Fitch, D. A. Karp, K. K. Lee, W. E. Stites, E. E. Lattman and E. B. García-Moreno, Biophys. J., 2002, 82, 3289–3304 CrossRef CAS PubMed
- E. L. Mehler, M. Fuxreiter, I. Simon and E. B. Garcia-Moreno, Proteins: Struct., Funct., Genet., 2002, 48, 283–292 CrossRef CAS PubMed
- M. A. Mulvey, J. D. Schilling, J. J. Martinez and S. J. Hultgren, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8829–8835 CrossRef CAS PubMed
- J. D. Schilling, M. A. Mulvey and S. J. Hultgren, J. Infect. Dis., 2001, 183, S36–S40 CrossRef PubMed
- C. H. Jones, J. S. Pinkner, R. Roth, J. Heuser, A. V. Nicholes, S. N. Abraham and S. J. Hultgren, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2081–2085 CrossRef CAS PubMed
- I. Le Trong, P. Aprikian, B. A. Kidd, M. Forero-Shelton, V. Tchesnokova, P. Rajagopal, V. Rodriguez, G. Interlandi, R. Klevit, V. Vogel, R. E. Stenkamp, E. V. Sokurenko and W. E. Thomas, Cell, 2010, 141, 645–655 CrossRef CAS PubMed
- M. M. Sauer, R. P. Jakob, J. Eras, S. Baday, D. Eriş, G. Navarra, S. Bernèche, B. Ernst, T. Maier and R. Glockshuber, Nat. Commun., 2016, 7, 10738 CrossRef CAS PubMed
- K. Mayer, D. Eris, O. Schwardt, C. P. Sager, S. Rabbani, S. Kleeb and B. Ernst, J. Med. Chem., 2017, 60, 5646–5662 CrossRef CAS PubMed
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C.-S. Hung, J. Pinkner, R. Slättegård, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2004, 55, 441–455 CrossRef PubMed
- B. Fiege, S. Rabbani, R. C. Preston, R. P. Jakob, P. Zihlmann, O. Schwardt, X. Jiang, T. Maier and B. Ernst, ChemBioChem, 2015, 16, 1235–1246 CrossRef CAS PubMed
- W. Schönemann, J. Cramer, T. Mühlethaler, B. Fiege, M. Silbermann, S. Rabbani, P. Dätwyler, P. Zihlmann, R. P. Jakob, C. P. Sager, M. Smieško, O. Schwardt, T. Maier and B. Ernst, ChemMedChem, 2019, 14, 749–757 CrossRef PubMed
- V. Kalas, J. S. Pinkner, T. J. Hannan, M. E. Hibbing, K. W. Dodson, A. S. Holehouse, H. Zhang, N. H. Tolia, M. L. Gross, R. V. Pappu, J. Janetka and S. J. Hultgren, Sci. Adv., 2017, 3, e1601944 CrossRef PubMed
- G. Interlandi and W. E. Thomas, Proteins: Struct., Funct., Bioinf., 2016, 84, 990–1008 CrossRef CAS PubMed
- P. Zihlmann, M. Silbermann, T. Sharpe, X. Jiang, T. Mühlethaler, R. P. Jakob, S. Rabbani, C. P. Sager, P. Frei, L. Pang, T. Maier and B. Ernst, Chem. – Eur. J., 2018, 24, 13049–13057 CrossRef CAS PubMed
- S. Kleeb, L. Pang, K. Mayer, D. Eris, A. Sigl, R. C. Preston, P. Zihlmann, T. Sharpe, R. P. Jakob, D. Abgottspon, A. S. Hutter, M. Scharenberg, X. Jiang, G. Navarra, S. Rabbani, M. Smiesko, N. Lüdin, J. Bezençon, O. Schwardt, T. Maier and B. Ernst, J. Med. Chem., 2015, 58, 2221–2239 CrossRef CAS PubMed
- J. Lameira, R. P. Bora, Z. T. Chu and A. Warshel, Proteins: Struct., Funct., Bioinf., 2015, 83, 318–330 CrossRef CAS PubMed
- G. Jindal and A. Warshel, Proteins: Struct., Funct., Bioinf., 2017, 85, 2157–2161 CrossRef CAS PubMed
- C. N. Schutz and A. Warshel, Proteins: Struct., Funct., Genet., 2001, 44, 400–417 CrossRef CAS PubMed
- A. Warshel, P. K. Sharma, M. Kato and W. W. Parson, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 1647–1676 CrossRef CAS PubMed
- A. T. Fenley, H. S. Muddana and M. K. Gilson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20006–20011 CrossRef CAS PubMed
- M. J. Borrok, L. L. Kiessling and K. T. Forest, Protein Sci., 2007, 16, 1032–1041 CrossRef CAS PubMed
- L. Unione, G. Ortega, A. Mallagaray, F. Corzana, J. Pérez-Castells, A. Canales, J. Jiménez-Barbero and O. Millet, ACS Chem. Biol., 2016, 11, 2149–2157 CrossRef CAS PubMed
- A. Müller, E. Severi, C. Mulligan, A. G. Watts, D. J. Kelly, K. S. Wilson, A. J. Wilkinson and G. H. Thomas, J. Biol. Chem., 2006, 281, 22212–22222 CrossRef PubMed
- J. F. Darby, A. P. Hopkins, S. Shimizu, S. M. Roberts, J. A. Brannigan, J. P. Turkenburg, G. H. Thomas, R. E. Hubbard and M. Fischer, J. Am. Chem. Soc., 2019, 141, 15818–15826 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00108b |
| This journal is © The Royal Society of Chemistry 2020 |