
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of the key steps in the self-assembly of homogeneous gold metal nanoparticles produced using inverse micelles†
Donato
Decarolis
ab,
Yaroslav
Odarchenko
 ab,
Jennifer J.
Herbert
abc,
Chengwu
Qiu
ab,
Alessandro
Longo
cd and
Andrew M.
Beale
*ab
ab,
Jennifer J.
Herbert
abc,
Chengwu
Qiu
ab,
Alessandro
Longo
cd and
Andrew M.
Beale
*ab
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: andrew.beale@ucl.ac.uk
bResearch Complex at Harwell (RCaH), Harwell, Didcot, Oxfordshire OX11 0FA, UK
cNetherlands Organisation for Scientific Research (NWO), DUBBLE CRG@ESRF, Grenoble 38042, France
dIstituto per lo Studio dei Materiali Nanostrutturati, Sezione di Palermo, Consiglio Nazionale delle Ricerche, Via La Malfa, 153, I-90146 Palermo, Italy
First published on 23rd August 2019
Abstract
The self-assembly of gold nanoparticles (Au NPs) using polymer-encapsulated inverse micelles was studied using a set of advanced X-ray techniques (i.e. XAFS, SAXS) in addition to DLS, UV-vis spectroscopy and TEM. Importantly the combination of these techniques with the inverse micelle approach affords us detailed insight and to rationalize the evolving molecular chemistry and how this drives the formation of the Au NPs. We observe that the mechanism comprises three key steps: an initial fast reduction of molecular Au(III) species to molecular Au(I)Cl; the latter species are often very unstable during the self-assembly process. This is followed by a gradual reduction of these molecular Au(I) species and the formation of sub-nanometric Au clusters which coalesce into nanoparticles. It was also found that addition of small amounts of HCl can accelerate the formation of the Au clusters (the second phase) without affecting the final particle size or its particle size distribution. These findings would help us to understand the reaction mechanism of Au NP formation as well as providing insights into how NP properties could be further tailored for a wide range of practical applications.
1. Introduction
Metal nanoparticles (NPs) can be used in a variety of applications ranging from catalysis to sensors, antibacterials and drug delivery.1–4 In order to prepare a metal nanoparticle, various methods have been developed.5,6 Synthesis methods can be divided into top-down methods (NPs are removed from ‘bulk’ material) and bottom-up (atoms assemble NPs).5,6 Whereas top-down approaches, such as e-beam lithography7 or thermal evaporation,8 allow NP production with regular physical attributes (e.g. size and shape). It is generally not viable for large scale applications, for example, to manufacture heterogeneous catalysts, due to the time required to produce the material, expensive instrumentation and the fact that it only allows for the formation of 1D and 2D supported NP arrays9 and therefore is mainly suitable to fabricate model systems. The bottom-up approach, more commonly reported as a method to prepare NPs, involves the reduction of metal ions in solution and a subsequent agglomeration.5,10–13 Bottom-up methods include templating,5 chemical,10 electrochemical11 and sonochemical12 reduction, with a common occurrence in these types of synthesis being the presence of a capping agent, often a surfactant, in order to restrict the NPs growth to the desired size and shape. The main disadvantage of these bottom-up approaches is that they rarely produce homogeneous NPs, often resulting in a broad particle size distribution and the average particle size may vary from batch to batch. This uncertainty in the NP size makes it difficult to truly understand the impact of NPs size on their properties e.g. catalytic activity14,15 or for other applications such as colorimetric sensing.3 Despite these drawbacks, bottom-up methods are used for industrial applications, such as production of the fixed bed heterogeneous catalysts, mainly for being relatively fast and straightforward and resulting in lower cost compared to top-down methods. Therefore, improving the synthesis methods in order to produce NPs with a particle size distribution as narrow as possible is paramount for the improvement in the understanding of NP properties.The fabrication of monodisperse NPs using bottom-up approaches, however, is not trivial and is hampered by the lack of detailed knowledge of all the steps in the self-assembly process.5 Various studies have been performed in order to understand the evolution of colloidal solution from the metal ions to NPs in different systems, such as Au,10 Fe2O3,16 Ag,17 and several NPs’ growth modes including Ostwald ripening, fusion, coalescence, condensation and coagulation, and combinations of these modes, thoroughly described in a review article from Thanh et al.18 Despite similarities in the fabrication mechanisms, the NP growth shows some dependence on the synthesis method.10,19 Perhaps the best known self-assembly study concerns the work of Polte et al. who studied the mechanism of Au NPs formation in the commonly used citrate method and showed that the combination of Small-angle X-ray Scattering (SAXS) and X-ray Absorption Near Edge Structure (XANES) analysis is able to reveal insight into the growth of metal nanoparticles during synthesis.10 The authors concluded that NP growth goes through four different phases:
– fast initial formation of small gold nuclei comprising a few atoms;
– coalescence of these nuclei into a nanoparticle;
– slow growth of the nanoparticle sustained by an ongoing reduction of the precursor;
– fast reduction ending with the complete consumption of the precursor species.
The second step (coalescence of the nuclei) has been identified as a key step influencing the polydispersity of the nanoparticles.10 However it is challenging to control multiple factors such as reagents mixing and pH of the citrate solution acting as reducing agent and stabiliser. On the other hand, a study from Sakai et al. shows that in the case of a block copolymer (poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide), also known as PEO–PPO–PEO) the NPs can be grown in a single step, due to the presence of the polymer acting both as a stabilizer as well as a reductant.19 Here the presumed mechanism of gold NPs formation is proposed as below:
– reduction of the metal ions facilitated by the polymer to form clusters (i.e. structures containing a small number of atoms);
– adsorption of the polymer onto the cluster surface and a (further) reduction of [AuCl4]− ions adsorbed on the surface of the clusters;
– growth of the NP through a repeat of step 2 until stabilisation.
In this process the polymer plays an important role in determining the particle size resulting from the synthesis. This is due to the dual attribute of the polymer as reductant and stabilizer, which varies the reaction activity of [AuCl4]− based on the polymer molecular weight.19 By comparing the citrate10 and copolymer-assisted19 methods one can conclude that the reduction of metal ions into a cluster and the coalescence of the clusters are two common steps during metal NP synthesis. However, there are also key differences in the mechanism for gold NP growth, the reasons for which could be attributable to the different stabilizer (citrate in one case and block copolymer in the other) that could alter the pathway of the reduction process. Ideally, a detailed investigation is required to determine the suggested mechanism for Au NP synthesis using the PEO–PPO–PEO based method, however considering it is a “single pot” synthesis procedure it could be extremely challenging to unravel the many chemical processes that lead to the formation of the Au NPs.19
Among the bottom-up methods, polymer-encapsulated inverse micelle synthesis, introduced by Spatz et al., can be used to prepare, in a relatively quick and reproducible way, uniform monodisperse metal NPs which can be conveniently loaded onto a variety of supports, e.g. refractive oxide powders.20–22 This advanced synthesis method mimics water-in-oil systems by employing precursor-filled micelles formed by PS-b-P2VP diblock-copolymer with low polydispersity index (PDI) in organic solvent, that allows for full encapsulation of all metal ions and inhibits the formation of sub-species.22 Another advantage of this approach includes self-assembly into regular arrays with controlled inter-particle distance which, when deposited on a flat substrate, can improve the properties of the final material e.g. minimises the interactions between NPs and prevents sintering during heat treatment.22,23 The size of the deposited NPs and their density (e.g. inter-particle distance) can be conveniently controlled by altering the length of shell- (PS) or core-forming (P2VP) copolymer blocks.23 Despite the multiple reports where the polymer-based method has been shown to produce well-defined metal NPs with suitably small particle size distributions,20,22–25 to the best of our knowledge no detailed studies have been performed to understand the mechanism of metal NP formation. As such important questions remain regarding the time required to complete the self-assembly and reduction processes, and the step-by-step pathway to self-assemble metal ions into Au NPs and whether/how such methods can be exploited to prepare multicomponent/multi-layered structures such as core–shell multi-metallics. Although the use of inverse micelles consisting in the encapsulation of Au species inside AOT (dioctyl sodium sulfosuccinate) liquid crystal has been explored in the literature,8,26 this approach would not have made possible the in situ observation of the formation mechanism of the gold NPs, compared to the solvent based method here employed.
In situ X-ray Absorption Fine Structure (XAFS) is a powerful method to follow the changes in the electronic structure of metals during the reduction process.27,28 In particular, XANES is used to observe changes in oxidation state of the Au species, whereas Extended X-ray Absorption Fine Structure (EXAFS) technique can monitor the formation of Au clusters through detection of the Au–Au scattering pair evolution.10,27 Whereas XAFS reveals information on the local environment of the gold atoms (i.e. the short-range order) in the micelles, SAXS is sensitive (in a size range of 1 to 100 nm) to the changes in the electron density fluctuations and is more suitable to probe the micelles’ core where gold clusters formation occurs.10,17 Thus, a combination of these two X-ray techniques with the use of other more commonly available experimental methods such as dynamic light scattering (DLS), ultraviolet-visible spectroscopy (UV-vis) and transmission electron microscopy (TEM), allows to understand the chemical and physical processes happening during the formation of the metal NPs.
This work is focused on the investigation of gold NP formation during polymer-encapsulated inverse micelle synthesis using in situ X-ray spectroscopy and scattering. The inverse micelle method, by offering tight control over the NPs’ size, allows a more detailed mechanistic insight into the process of homogeneous NP formation. Key to this study is the avoidance of ‘fast’ reducing agents such as NaBH429 or citrate,10 that can often result in uncontrolled particle growth and a large particle size distribution, hindering our attempts at a detailed understanding of the self-assembly process.30 By modifying the inverse micelle method developed by Spatz et al., using a slower reductant compared to the commonly used hydrazine, it was possible to observe the presence of Au(I) species, previously supposed to be an intermediate in the Au nanoparticle formation but hardly detected due to their inherent instability. Furthermore, it was observed that the addition of small quantities of HCl during the synthesis appear to catalyse the formation of Au nanoclusters, most probably due to the formation of in situ N2H4. The insight obtained using these advanced characterization methods in combination with the inverse micelle synthesis allowed us to identify the key stages in the self-assembly process that are useful when trying to optimize NPs for a wide range of practical applications.
2. Experimental section
2.1 Nanoparticles synthesis
Gold nanoparticle solutions were prepared using the polymer-encapsulated inverse micelles method.20 Poly(styrene-b-2-vinyl pyridine) (PS-b-P2VP) block-copolymer P18226-S2VP was purchased from the Polymer Source Inc (electronic grade purity). The metal salt used was chloroauric acid (HAuCl4) (Aldrich 99.99999% trace metal basis). p-Toluene sulfonyl hydrazide (p-tosyl hydrazide) (Aldrich 97%) was used as the reducing agent, as an alternative to the more commonly used toxic and explosive hydrazine.31 All reagents were used as received. 0.5 wt% polymer (c = 5 mg ml−1) solution was prepared in 25 ml of toluene (Aldrich 99.9%) and left to stir for 3 days in order to allow the polymer to dissolve and homogenize. After 3 days, 0.021 g of the metal precursor was added to the solution and left to allow the metal salt to incorporate inside the micelles. The metal atoms to pyridine ratio (metal loading) was fixed at 0.3. Before the measurements 10 ml of metal loaded polymer solution was put in a glass vial with a 10 ml of a solution of p-tosyl hydrazide. To compare the effect of the HCl, used in literature to remove the excess of hydrazide,20 on the metal reduction inside the polymer micelle, 50 μl of HCl (ACS reagent 37%, Fluka) was added to the solution after ∼30 s and left to stir. Table 1 summarises a list of the samples used in this work.2.2 In situ small-angle X-ray scattering (SAXS)
In situ Small-angle X-ray scattering (SAXS) measurements were conducted on the DUBBLE beamline (BM26A) of the European Synchrotron Radiation Facility (ESRF) using X-ray photons of 11.850 keV. The 2D SAXS images were recorded in transmission mode over a period of 3 min using the Mar CCD 165 detector with a pixel size of 80 μm. The modulus of the scattering vector q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ, where θ is the Bragg angle and λ is the wavelength, was calibrated using silver behenate. The sample to detector distance was approximately 2 m, resulting in a q range spanning 0.016 and 0.4 nm−1.
sinθ/λ, where θ is the Bragg angle and λ is the wavelength, was calibrated using silver behenate. The sample to detector distance was approximately 2 m, resulting in a q range spanning 0.016 and 0.4 nm−1.
For the sample measurement, an aliquot of each solution, taken at different reduction times (0, 0.3, 1.25, 2.25, 6.25 h), was loaded in 1 ml quartz capillaries of 1 mm diameter. The data were corrected for the detector dark field and background scattering from the capillary with polymer and solvent only. The data reduction was performed using Fit2D and 1D curves were fitted using SANS Analysis package for the Igor Pro software.32
 | (1) |
The Schultz–Zimm distribution function was chosen, that can account for the asymmetrical NPs size distribution. Also this model was successfully used in the past to reveal the mechanism of the Au NPs formation in the classical citrate method.10 The Schultz–Zimm distribution can be written as33
 | (2) |
NP (P2VP core of the core–shell polymer micelle) is considered to be a hard (uniform density) sphere with the radius R and volume V. This approximation is supported by the electron microscopy data that is discussed later in the paper. NP's form factor is given by the Rayleigh formula34
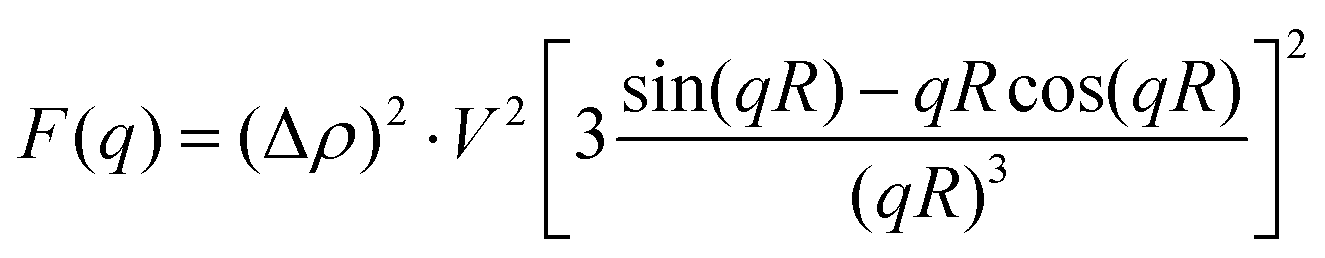 | (3) |
2.3 In situ X-ray absorption fine structure (XAFS)
Extended X-ray absorption fine structure (EXAFS) and X-ray Absorption Near Edge Structure (XANES) were measured on the DUBBLE beamline (BM26A) of the European Synchrotron Radiation Facility (ESRF) using Au LIII edge (11.91 keV) in fluorescence mode using a 9-element monolithic Ge detector (Ortec), with the sample holder mounted at 45° with the respect to the incident beam in order to maximize the fluorescence yield. An aliquot of each solution, taken at different times during the reduction process (0 and 2.25 h), was loaded in 1 ml quartz capillary and mounted on the sample holder. Each XAFS spectrum was acquired over a period of ∼10 min and repeated a further 3 times for each sample measured. The reference data used for XANES analysis were obtained by the author during previous XAFS experiment at the B18 beamline of Diamond Light Source (DLS) using Au LIII edge in transmission mode on a solution of AuCl3 in water and on an Au foil for Au(III) and Au0 reference, respectively.2.4 Transmission electron microscopy (TEM)
Transmission Electron Microscopy (TEM) was measured using JEOL: JEM-2100 operating at 200 keV using a LaB6 filament, after deposition through drop-casting on a carbon film supported by a 300 mesh copper TEM grid. Sample A-HCL solution before and after the reduction treatment were analysed. Due to a small scattering contrast of the block copolymer compared to the carbon film renders it impossible to obtain information regarding the state of the micelles before the addition of the Au precursor.2.5 UV-vis spectroscopy
UV-vis spectroscopy measurements were taken in transmission mode, using a Shimadzu UV-1800 spectrometer for sample A and an Agilent Cary series for sample B, with the samples loaded into a cuvette with 5 mm pathlength to determine the position of the Au plasmon peak. A toluene solution with the polymer micelles was used as a background in order to remove any interference to the plasmon peak. Each spectrum was acquired using a total of 5 min per scan.2.6 Dynamic light scattering (DLS)
Dynamic light scattering (DLS) measurements were performed on samples reduced over different times using Zetasizer nano ZS from Malvern. The incident laser wavelength used in the setup was 532 nm and the refractive index was derived from polystyrene (η = 1.5). Micelles containing solutions were loaded into a cuvette with 5 mm pathlength.3. Results and discussion
3.1 DLS
With the help of DLS one can observe how the micellar system in its entirety responds during the reduction of metal ions through the impact this has on the micelles’ hydrodynamic diameter. DLS plots, including the weighted average micelle radius obtained from the DLS measurement for sample A during reduction are shown in Fig. 1. The mean size of the micelles before reduction appears to be around 60 nm which increases to 70 nm in 0.1 h after addition of the reducing agent. As can be seen on Fig. 1 the swelling of the metal-containing polymer particles continues and results in almost doubling of the mean micelle size. A similar effect is observed in sample A-HCl when HCl is added during the reduction process, where the micelles size increases from 60 to 100 nm with the reduction time.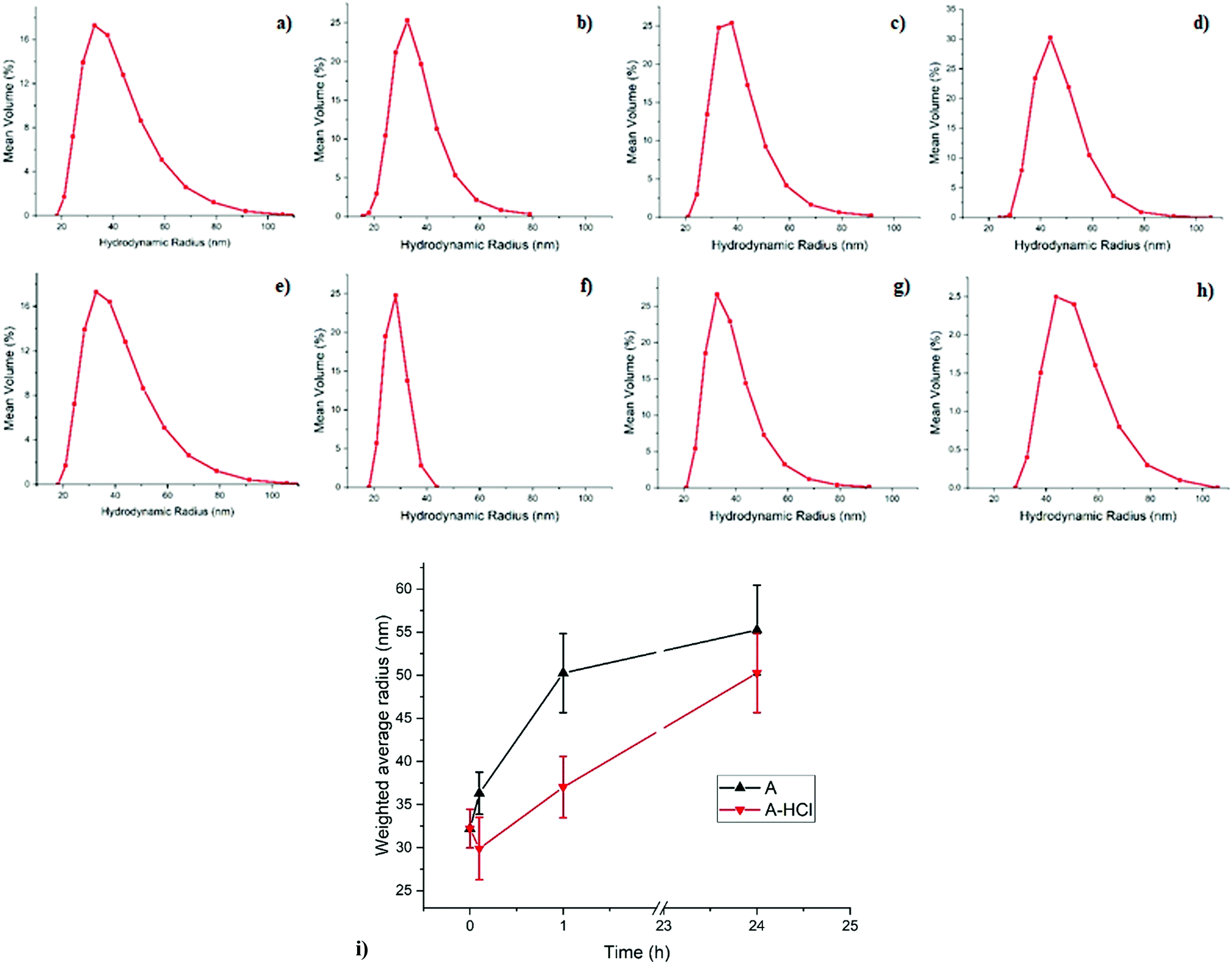 | ||
| Fig. 1 DLS plots of samples A (a–d) and A-HCl (e–h). Sample A before reduction (a) and after 0.1 h (b), 1 h (c) and 24 h (d) of the reduction process. Sample A-HCl before reduction (e) and after 0.1 h (f), 1 h (g) and 24 h (h) of the reduction process. In (i) are shown the weighted average radius obtained from the DLS measurements as function of time for sample A and A-HCl. The average diameter of the polymer micelle increases from ∼60 to ∼110 nm after the reduction for both samples. | ||
We propose that the observed increase of the micelle size is a consequence of the reduction of the metal salt. The reduction reaction of [AuCl4]− with H2, produced by the p-tosyl hydrazide decomposition, to Au(I) is thought to occur as follows:
| H[AuCl4]− + H2 → AuCl + 3HCl | (4) |
| 2AuCl + H2 → 2Au + 2HCl | (5) |
| 2H[AuCl4]− + 3H2 → 2Au0 + 8HCl | (6) |
DLS studies on the similar systems have been reported before including copolymer- and surfactant-based inverse micelles,37–39 colloidal gold and polystyrene nanoparticles,40,41 and inverse micelles filled with Au NPs.42,43 In the most relevant study Mössmer et al.,42 using PS-b-P2VP with block lengths comparable to sample A, observed a mean radius of 28 nm (56 nm diameter) for the micelles filled with H[AuCl4], which is similar to the size of 60 nm measured for sample A before reduction. In their study, the micelles’ size for fully reduced Au NPs was reported to increase to 30 nm (60 nm diameter), compared to the 110 nm diameter observed here for sample A at the end of the reduction process.42 This discrepancy could be due to the different reductant used: hydrazine42versus the much bulkier p-tosyl hydrazide used here. Also the authors did not perform a time-resolved study and thus the intermediate stages of the reduction process were not seen,42 whereas our DLS measurements as a function of time allows us to follow the swelling of the micelles as the reduction process is taking place (Fig. 1). It has to be noted that DLS is only capable of measuring the total diameter of the core–shell polymer micelle, consisting of the PS core and P2VP shell based on the miscibility of copolymer blocks, and does not allow for probing the local structural changes concerning the Au NPs of primary interest.42 Therefore, the combination of microscopic and spectroscopic methods have also been employed to obtain information regarding the local environment of the gold loaded P2VP core and the role of HCl in the reduction process.
3.2 UV-vis spectroscopy
UV-vis spectroscopy was employed to follow Au species evolution in solution during the reduction process. The UV-vis spectra shown in Fig. 2a for sample A contain a plasmon peak at 550 nm characteristic of Au0 clusters formed by the reduction of HAuCl4.44 The increase in intensity with time suggests continuous oxidic Au species reduction and formation of Au NPs; interestingly the band does not change position suggesting continued growth of the Au nanoparticles without any dramatic change in size. In the case of sample A-HCl, the red shift of the plasmon peak from 550 to 600 nm in Fig. 2b could be explained by nanoparticles aggregation possibly caused by the presence of “water pockets” induced by the addition of HCl, by the unlikely possibility that particles larger than the micelles themselves (≫100 nm) are forming or else by the presence of anisotropic particles.45 It can be also noted that characteristic plasmon band at 6.5 h in Fig. 2b has lower intensity compared to spectrum at 3.5 h. This could be possibly due to the precipitation of some of the already formed Au nanoparticles, causing a decrease in the amount of Au species which are observed by the beam. After 24 h of reduction since the micelles containing reduced Au NPs were observed to precipitate as can be seen in Fig. S1 (ESI†) and hence no UV-vis data could be reported. Additional band located at ∼330 nm (Fig. 2b) decreases in intensity. This peak could be attributed to gold(III) chloride species that are slowly consumed during the reduction.46 To follow the changes in the gold clusters’ size and oxidation state of the Au species during the reduction process a combination of XANES and EXAFS was utilised.10,27 | ||
| Fig. 2 UV-vis spectra of samples A (a) and A-HCl (b) during reduction. The dotted line, added as an eye guide, represents the maximum of the plasmon peak that is 550 nm for sample A and 600 nm for sample A-HCl.44 | ||
3.3 XAFS
In situ XAFS was used to determine further the mechanism of the Au reduction and aggregation of the gold clusters. The results of the EXAFS fit for all four samples after 0 and 2.25 h of the reduction process are summarised in Table 2. Fig. 3 contains XANES spectra and EXAFS Fourier Transforms (FTs) for samples A and A-HCl. A reference AuCl3 solution which contains a strong white line at 11.920 keV due to an electronic (dipole) transition from the 2p orbitals to the partially unoccupied 5d orbitals is also shown in Fig. 3a and c.47 After the reductant is added there is a drop in the intensity in the white line (reduced transition probability) as a consequence of increased filling of the Au 5d orbitals. This change is observed in conjunction with a slight shift of the edge position towards higher energies (from 11.197 to 11.199 keV). Both changes are consistent with the reduction of Au(III) to Au(I).48 However, no further changes were observed in the XANES between 0 and 2.25 h suggesting the Au(I) species formed are stable.| Sample | Time (h) | CNAu–Cl | R Au–Cl (Å) | σ Au–Cl 2 (Å−2) | CNAu–Au | R Au–Au (Å) | σ Au–Au 2 (Å−2) | ΔE | R factor |
|---|---|---|---|---|---|---|---|---|---|
| CN, the coordination number; R, the radial distance; σ2, the Debye–Waller factor. | |||||||||
| Au foil | — | — | — | — | 12.0 ± 0.5 | 2.861 ± 0.002 | 0.008 ± 0.0002 | 4.23 ± 0.25 | 0.014 |
| AuCl4 in solution | — | 4.29 ± 0.18 | 2.280 ± 0.003 | 0.0020 ± 0.0003 | 4.29 ± 0.18 | 0.007 | |||
| A | 2.25 | 2.0 ± 0.2 | 2.248 ± 0.008 | 0.003 ± 0.001 | — | — | — | 7.3 ± 1.1 | 0.02 |
| A-HCl | 0 | 2.9 ± 0.4 | 2.266 ± 0.009 | 0.0025 ± 0.0013 | — | — | — | 8.1 ± 1.3 | 0.03 |
| 2.25 | 0.7 ± 0.4 | 2.25 ± 0.02 | 0.0027 ± 0.0018 | 8.2 ± 1.8 | 2.86 ± 0.01 | 0.007 ± 0.002 | 6.9 ± 1.3 | 0.05 | |
 | ||
| Fig. 3 XAFS spectra of sample A (a and b) at 2.25 h of reduction time and A-HCl (c and d) at 0 and 2.25 h of reduction time. XANES spectra include Au0 and AuCl3 references for sample A (a) and A-HCl (c). EXAFS Fourier Transform for sample A (b) and A-HCl (d). | ||
The EXAFS data for sample A in Fig. 3b are clearly different to that of the reference compounds but do possess a peak at ∼2.24 Å which can be assigned to a Au–Cl scattering contribution, although the reduced amplitude (when compared with AuCl4 in solution) suggests the presence of Au(I)Clx species (see eqn (4)). These values (and FTs) are similar to those observed previously by Salama et al.49 in Na-Y zeolites and are consistent with the formation of AuCl species. Au(I) species in the form of a Au2Cl2 closed square dimer configuration previously reported by Hargittai et al.50 can be ruled out on the basis that the Au–Cl distance reported there (2.567 Å) is too long. Interestingly, the Debye–Waller factors obtained for both samples are comparatively low suggesting low static/thermal disorder in the AuCl species. No sign of any metallic Au NPs were observed after 2.25 h of reaction (see Fig. 3b).
For sample A-HCl, the higher Au–Cl coordination number (>2) at the start of the reduction (0 h) suggests that initially the reduction process is slightly less advanced in comparison to the non-HCl samples. However, after 2.25 h the formation of Au NPs can be seen by the presence of the Au–Au scattering contribution at 2.86 Å in the second shell of the FT (Fig. 3d). Assuming an isotropic shape, one can estimate the size of the Au NPs from the CNAu–Au value to be of the order of ∼2 nm.51 However, reduction is not complete at this stage as a clearly discernible, if low, CN value is obtained for the Au–Cl first shell. XANES spectra also confirm the presence of metallic Au by the increase intensity of the feature located at 11.945 keV which can be attributed to a multiple scattering path of metallic Au52 (Fig. 3c). However, the higher edge energy and the intense white line of sample A-HCl compared to Au0 also suggest that Au(I)Clx species are still present after 2.25 h.
3.4 SAXS
In order to investigate the structural transformation, in particular in terms of size and polydispersity, of the NPs formed in the micelle's core during the reduction process in situ SAXS studies were conducted. 1D SAXS profiles for samples A and A-HCl during the reduction process are shown in Fig. 4. According to the fit (for the details see Experimental section and Fig. S2 and S3) there is a slight increase in the scattering component size from 13.4 to 13.8 nm (Fig. 4a, c and Fig. S2, ESI†) for sample A after 6.25 h of reduction. For sample A-HCl a shift of the scattering component towards smaller q-values can be seen which corresponds to sizes from 16.2 to 18.2 nm depending on reduction time, with 1.25 h being the time when the size stabilizes (Fig. 4b, c and Fig. S3, ESI†). | ||
| Fig. 4 Structural changes during reduction as followed by in situ SAXS. 1D SAXS profiles for the sample A (a), A-HCl (b). Evolution of the micelles core diameter with time for the samples A and A-HCl (c). | ||
The polydispersity of the system53 obtained from the fit results is relatively low (≤13%). The calculated value of polydispersity is stable throughout the NP formation process (see Fig. S2 and S3, ESI†), indicating that as the NPs grow the homogeneity of the system is not affected.
The in situ SAXS fit results summarised in Fig. 4c show a growth of the NPs for both samples during the reduction process that is in a good agreement with the DLS data (Fig. 1) whereas the difference in size obtained for sample A (∼14 nm from SAXS, ∼60 nm from DLS) is due to the PS shell which is not detected by the SAXS due the low electron density contrast between toluene and polystyrene (Fig. S4a, ESI†). The increase of the P2VP core size from ∼16 to ∼18 nm for sample A-HCL, when HCl is included into the solution can be attributed to an increment in the speed of NPs’ growth. This was also suggested from the XAFS analysis, indicating that HCl acts as a catalyst for the reduction of the Au species.
3.5 TEM
TEM has been used to characterize the morphology (e.g. shape and size) of the polymer micelles before and after adding reductant. As an example, Fig. 5a and b shows the PS-b-P2VP micelles loaded initially only with HAuCl4 for sample A-HCl. It can be seen that the metal ions are preferentially located in the micelle cores (Fig. 5a and b). Although, for sample A-HCl, the gold appears as sub-nanometre clusters inside the core of the micelle, which we propose is caused by the reduction of the precursor caused by the electron beam (Fig. 5b). The loaded micelles’ core size for sample A-HCl is ∼11 nm (Fig. 5b). The fact that the micelle's core size observed in TEM compares well with the SAXS data further supports the assumption that Au species are confined and self-assemble from within the polymer micelle core. | ||
| Fig. 5 TEM micrographs of sample A-HCl before (a and b) and after reduction (c and d). Polymer A loaded with HAuCl4 before the addition of reductant (a) and higher magnification image (b). Average micelle core size is ∼11 nm. Sample A-HCl after addition of reducing agent and HCl (c) and higher magnification image (d) with the average NP size of 4.5 ± 0.8 nm. | ||
After p-tosyl hydrazide is added to the micelle-containing solution, reduction of Au ions occurs and leads to the formation of nanoparticles, as shown for example in Fig. 5c and d for the reduced sample A-HCl after 24 h. The calculated NP size from the TEM data is 4.5 ± 0.8 nm that is considerably smaller than the value of ∼11 nm for the micelle core of sample A-HCl before the reduction.
3.6 Rationalisation for self-assembly process
From the results obtained by the combination of time-resolved DLS, UV-vis, XAFS, SAXS and TEM it was possible to propose a model for the self-assembly of the homogeneous metal nanoparticles in the inverse micelles system shown in Scheme 1, that can be delineated into the following steps: | ||
| Scheme 1 Proposed schematic model of the Au nanoparticle formation during the polymer inverse micelle synthesis, including the time scale and techniques used to identify each stage. (1) the [AuCl4]− ions attach to the pyridine group; (2) the [AuCl4]− ions get reduced to Au(I) species; (3) the Au(I) species are reduced to form Au(0) species that start to form Au nanoclusters; (4) resulted Au nanoparticle encapsulated in the PS-P2VP micelle. Characteristic sizes are shown using an example of sample A. The addition of HCl increase the rate of formation of Au clusters. | ||
– an initial fast reduction of [AuCl4]− to Au(I)Clx species;
– further slow reduction of the Au(I) and formation of Au(0) clusters;
– agglomeration of the small clusters to form nanoparticles confined inside the core of the micelle.
The rational for our scheme is given as follows. Firstly, according to TEM data (Fig. 5), the micelle core is loaded with the gold precursor at the beginning of synthesis (stage 1 in Scheme 1). This in combination with the XAFS data dominated by Au–Cl species leads us to conclude that Au must be initially homogeneously dispersed all over the micelle cores as a complex between the nitrogen from pyridine and [AuCl4]−, in a similar way to that previously observed for iron and molybdenum precursors.54 After the reductant is added (stage 2 in Scheme 1) the [AuCl4]− ions reduce almost immediately to Au(I) species, namely [AuCl2]−, this again concluded mainly on the basis of the XAFS results (Table 2 and XANES data shown in Fig. 3). Furthermore, we observe no immediate change in the core diameter from the SAXS data with reaction time (Fig. 4). The core diameter obtained from the fit of the SAXS data, 13.4 nm for sample A and 16.2 nm for sample A-HCl (Fig. S2 and S3, ESI†), in addition to the TEM data, suggests that the [AuCl2]− species are well dispersed within the micelle core; this we conclude based on the good agreement obtained for SAXS and TEM measurements for the size of the micelle core when filled with Au species (Scheme 1).
After 2.25 h (stage 3 in Scheme 1) the formation of Au0 species can be seen for sample A-HCl (Fig. 3d), likely comprised of small clusters based on the low EXAFS-derived values of CNAu–Au shown in Table 2. An increase of the NP diameter at this time during the reduction process measured using SAXS indicates self-assembly of gold clusters into the NPs (Fig. 4). The swelling of the micelle core continues (Fig. 4c) until Au reduction is complete. Interestingly for sample A whereas XAFS does not show any presence of Au0 species, the UV-vis data display a, albeit very small, plasmon peak (Fig. 2a). This could be attributed by the large amount of Au(I) species which dominate the XAFS spectra, whereas UV-vis is only sensible to the few Au0 species present in solution.
Further agglomeration of the gold clusters to form NPs (stage 4 in Scheme 1) was confirmed by the UV-vis (Fig. 2) and TEM data (Fig. 5c and d), the latter showing that once the reduction process is completed, the formed NPs are positioned at the centre of the micelles. Moreover, the nanoparticles show a defined spherical shape, that is clearly different from the sub-nanometric gold cluster formed due to electron beam exposure (Fig. 5a and b). We note however that the DLS data revealed that the whole micelle, and not just the core, is affected during the reduction of the gold precursor to the metal NP (Fig. 1).
3.7 The role of HCl in the self-assembly process
The XAFS, UV-vis and SAXS data suggest that the presence of HCl catalyses the reduction process of Au(I) to Au0 and its agglomeration. We rationalise that HCl helps with the in situ formation of hydrazine through the following reaction sequence:| NH2–NH–p-toluenesulfonyl + HCl ↔ NH2NH2 + Cl–p-toluenesulfonyl | (7) |
| 2H[AuCl4] + N2H4 → N2 + 6HCl + 2AuCl | (8) |
| 4AuCl + N2H4 → N2 + 4HCl + 4Au | (9) |
| 4H[AuCl4] + 3N2H4 → 4Au + 3N2 + 16HCl | (10) |
The low polydispersity values calculated from the SAXS data are consistent with the presence of micelles containing evenly loaded and highly dispersed Au ions attached as molecular species to the pyridine groups of the P2VP chain. We propose that this interaction is important as it slows the reduction of Au(I) to Au0 and agglomeration process, compared to more commonly used Au NP synthesis procedures such as the Turkevich method.10,55 This retardation then seems to realise a more controlled Au NP nucleation and growth process resulting in a much narrower particle size distribution. This also demonstrates the protective effect of the polymer micelles, preventing undesired coalescence of metal NPs. Our results can be interpreted as being more widely reflective of how Au NP assembly occurs in the presence of species that can coordinate and/or complex with Au species (i.e. block co-polymers, capping agents such as PVP,56 PVA,57etc.). For example, Sakai et al. using block PEO–PPO–PEO co-polymers observed simultaneously the reduction of [AuCl4]− precursor to Au0, formation of Au clusters and finally the growth of the NPs.19 Due to the comparative slowness the inverse polymer micelle encapsulation method allows to separate different stages of Au NP growth and to follow the formation of homogeneous particles step by step. Moreover time-resolved X-ray techniques help to unequivocally identify the presence of a stable Au(I) species as intermediates in the formation process. Indeed, such Au(I) species have been reported only for the short time (≤30 min) during Au NP self-assembly,58,59 partially due to the fast disproportionation to Au0 and Au(III).
3.8 Effects of X-ray irradiation on stability of Au species
Shown in Fig. S4b (ESI†) are two XANES spectra acquired in succession for sample A, 5 min after addition of the reductant. The similarity of the two XANES spectra are testament of the stability of the samples in the X-ray beam and indicate that the mixed Au metal/ionic environments observed are a direct consequence of a slow reduction process.4. Conclusions
Although the formation of Au NPs within polymer-encapsulated inverse micelles is a dynamic process, by obtaining multi-technique ‘snap-shots’ during this process, the key stages associated with their self-assembly could be identified. This is the first study, to the best of our knowledge, where the combination of SAXS/EXAFS methods and slow reducing agent allowed to obtain the comprehensive pathway on the mechanism of gold NPs formation. We speculate that whereas this mechanism has been explored for the inverse micelle method, it is highly likely that the formation process would be similar if not identical when using similar synthetic methods i.e. those which employ polymers or liquid crystals as capping agents, or complexes, etc. Importantly, when considering the application of such bottom-up synthetic procedures the detailed insight obtained here concerning the rate and mechanism of Au NP formation allows us to identify the variables, i.e. time or presence of additional reagents, that can be altered to aid production or else to tune the nanoparticle’ properties, i.e. composition and morphology, thus allowing a better control over NP size and particle size distribution and ultimately to a more optimal performance in their intended application.Abbreviations
| NP | Nanoparticle |
| SAXS | Small-angle X-ray scattering |
| XAFS | X-ray absorption fine structure |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC for funding (grant EP/K007467/1 & EP/L015862/1). The authors also thank NWO for part sponsorship of a studentship for JH and for DUBBLE@ESRF for the (in-house) beamtime.References
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Gold nanoparticles in delivery applications, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- F. Tao and J. Spivey, Metal Nanoparticles for Catalysis, The Royal Society of Chemistry, Cambridge, 2014 Search PubMed.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Selective Colorimetric Detection of Polynucleotides Based on the Distance-Dependent Optical Properties of Gold Nanoparticles, Science, 1997, 277, 1078LP–1081LP CrossRef PubMed.
- M. Moritz and M. Geszke-Moritz, The newest achievements in synthesis, immobilization and practical applications of antibacterial nanoparticles, Chem. Eng. J., 2013, 228, 596–613 CrossRef CAS.
- G. Cao, Nanastructures and Nanomaterials – Synthesis, Properties and Applications, Imperial College Press, London, 2004, vol. 2 Search PubMed.
- A. S. Edelstein and R. C. Cammarata, Nanomaterials: synthesis, properties, and applications, Taylor & Francis, Abingdon, 1998 Search PubMed.
- P. A. Schaal, A. Besmehn, E. Maynicke, M. Noyong, B. Beschoten and U. Simon, Electrically Conducting Nanopatterns Formed by Chemical e-Beam Lithography via Gold Nanoparticle Seeds, Langmuir, 2012, 28, 2448–2454 CrossRef CAS PubMed.
- A. Longo, P. Calandra, M. P. Casaletto, C. Giordano, A. M. Venezia and V. T. Liveri, Synthesis and physico-chemical characterization of gold nanoparticles softly coated by AOT, Mater. Chem. Phys., 2006, 96, 66–72 CrossRef CAS.
- W. Karim, A. Kleibert, U. Hartfelder, A. Balan, J. Gobrecht, J. A. van Bokhoven and Y. Ekinci, Size-dependent redox behavior of iron observed by in-situ single nanoparticle spectro-microscopy on well-defined model systems, Sci. Rep., 2016, 6, 18818 CrossRef CAS PubMed.
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thünemann and R. Kraehnert, Mechanism of Gold Nanoparticle Formation in the Classical Citrate Synthesis Method Derived from Coupled In Situ XANES and SAXS Evaluation, J. Am. Chem. Soc., 2010, 132, 1296–1301 CrossRef CAS PubMed.
- B. Yin, H. Ma, S. Wang and S. Chen, Electrochemical Synthesis of Silver Nanoparticles under Protection of Poly(N-vinylpyrrolidone), J. Phys. Chem. B, 2003, 107, 8898–8904 CrossRef CAS.
- A. Nemamcha, J.-L. Rehspringer and D. Khatmi, Synthesis of Palladium Nanoparticles by Sonochemical Reduction of Palladium(II) Nitrate in Aqueous Solution, J. Phys. Chem. B, 2006, 110, 383–387 CrossRef CAS.
- H. Zhang, W. Wang, M. Akinc, S. Mallapragada, A. Travesset, D. Vaknin, S. Mallapragada, D. Vaknin, T. Xu and A. P. Alivisatos, Assembling and ordering polymer-grafted nanoparticles in three dimensions, Nanoscale, 2017, 9, 8710–8715 RSC.
- M. C. Saint-Lager, I. Laoufi, A. Bailly, O. Robach, S. Garaudee and P. Dolle, Catalytic properties of supported gold nanoparticles: new insights into the size-activity relationship gained from in operando measurements, Faraday Discuss., 2011, 152, 253–265 RSC.
- I. Laoufi, M. C. Saint-Lager, R. Lazzari, J. Jupille, O. Robach, S. Garaudée, G. Cabailh, P. Dolle, H. Cruguel and A. Bailly, Size and catalytic activity of supported gold nanoparticles: An in operando study during CO oxidation, J. Phys. Chem. C, 2011, 115, 4673–4679 CrossRef CAS.
- A. Kabelitz, A. Guilherme, M. Joester, U. Reinholz, M. Radtke, R. Bienert, K. Schulz, R. Schmack, R. Kraehnert and F. Emmerling, Time-resolved in situ studies on the formation mechanism of iron oxide nanoparticles using combined fast-XANES and SAXS, CrystEngComm, 2015, 17, 8463–8470 RSC.
- J. Polte, X. Tuaev, M. Wuithschick, A. Fischer, A. F. Thuenemann, K. Rademann, R. Kraehnert and F. Emmerling, Formation Mechanism of Colloidal Silver Nanoparticles: Analogies and Differences to the Growth of Gold Nanoparticles, ACS Nano, 2012, 7, 5791–5802 CrossRef.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Mechanisms of Nucleation and Growth of Nanoparticles in Solution, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- T. Sakai and P. Alexandridis, Single-step synthesis and stabilization of metal nanoparticles in aqueous pluronic block copolymer solutions at ambient temperature, Langmuir, 2004, 20, 8426–8430 CrossRef CAS PubMed.
- J. P. Spatz, S. Mössmer, C. Hartmann, M. Möller, T. Herzog, M. Krieger, H. G. Boyen, P. Ziemann and B. Kabius, Ordered deposition of inorganic clusters from micellar block copolymer films, Langmuir, 2000, 16, 407–415 CrossRef CAS.
- D. James Martin, D. Decarolis, Y. I. Odarchenko, J. J. Herbert, T. Arnold, J. Rawle, C. Nicklin, H.-G. Boyen and A. M. Beale, Reversible restructuring of supported Au nanoparticles during butadiene hydrogenation revealed by operando GISAXS/GIWAXS, Chem. Commun., 2017, 53, 5159–5162 RSC.
- D. J. Martin, D. Decarolis, R. Tucoulou, G. Martínez-Criado and A. M. Beale, Towards the interrogation of the behaviour of a single nanoparticle under realistic catalytic reaction conditions, Catal., Struct. React., 2017, 3, 63–70 CrossRef CAS.
- U. Wiedwald, L. Han, J. Biskupek, U. Kaiser and P. Ziemann, Preparation and characterization of supported magnetic nanoparticles prepared by reverse micelles, Beilstein J. Nanotechnol., 2010, 1, 24–47 CrossRef CAS.
- B. R. Cuenya, Metal nanoparticle catalysts beginning to shape-up, Acc. Chem. Res., 2013, 46, 1682–1691 CrossRef.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, Exceptional Size-Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- P. Calandra, C. Giordano, A. Longo and V. T. Liveri, Physicochemical investigation of surfactant-coated gold nanoparticles synthesized in the confined space of dry reversed micelles, Mater. Chem. Phys., 2006, 98, 494–499 CrossRef CAS.
- M. Harada and Y. Kamigaito, Nucleation and Aggregative Growth Process of Platinum Nanoparticles Studied by in Situ Quick XAFS Spectroscopy, Langmuir, 2012, 28, 2415–2428 CrossRef CAS.
- G. F. Tierney, D. Decarolis, N. Abdullah, S. M. Rogers, S. Hayama, M. Briceno de Gutierrez, A. Villa, C. R. A. Catlow, P. Collier, N. Dimitratos and P. P. Wells, Extracting structural information of Au colloids at ultra-dilute concentrations: identification of growth during nanoparticle immobilization, Nanoscale Adv., 2019, 1, 2546–2552 RSC.
- Z. Zhang and Y. Wu, Investigation of the NaBH4-Induced Aggregation of Au Nanoparticles, Langmuir, 2010, 26, 9214–9223 CrossRef CAS.
- L. Bronstein, M. Antonietti and P. Valetsky, Nanoparticles and Nanostructured Films, Wiley-VCH Verlag GmbH, 1998, pp. 145–171 Search PubMed.
- J.-M. Yun, J.-S. Yeo, J. Kim, H.-G. Jeong, D.-Y. Kim, Y.-J. Noh, S.-S. Kim, B.-C. Ku and S.-I. Na, Solution-Processable Reduced Graphene Oxide as a Novel Alternative to PEDOT:PSS Hole Transport Layers for Highly Efficient and Stable Polymer Solar Cells, Adv. Mater., 2011, 23, 4923–4928 CrossRef CAS.
- S. R. Kline, Reduction and analysis of SANS and USANS data using IGOR Pro, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- M. Kotlarchyk, R. B. Stephens and J. S. Huang, Study of Schultz distribution to model polydispersity of microemulsion droplets, J. Phys. Chem., 1988, 92, 1533–1538 CrossRef CAS.
- O. Glatter and O. Kratky, Small Angle X-ray Scattering, Academic Press, London, 1982 Search PubMed.
- B. Ravel and M. Newville, Athena, artemis, hephaestus: data analysis for x-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS.
- M. Newville, IFEFFIT: interactive XAFS analysis and FEFF fitting, J. Synchrotron Radiat., 2001, 8, 322 CrossRef CAS.
- H. B. Bohidar and M. Behboudnia, Characterization of reverse micelles by dynamic light scattering, Colloids Surf., A, 2001, 178, 313–323 CrossRef CAS.
- R. D. Falcone, J. J. Silber and N. M. Correa, What are the factors that control non-aqueous/AOT/n-heptane reverse micelle sizes? A dynamic light scattering study, Phys. Chem. Chem. Phys., 2009, 11, 11096 RSC.
- D. Voulgaris, C. Tsitsilianis, V. Grayer, F. J. Esselink and G. Hadziioannou, Amphiphile micelles formed by polystyrene/poly(2-vinyl pyridine) heteroarm star copolymers in toluene, Polymer, 1999, 40, 5879–5889 CrossRef CAS.
- B. N. Khlebtsov and N. G. Khlebtsov, On the measurement of gold nanoparticle sizes by the dynamic light scattering method, Colloid J., 2011, 73, 118–127 CrossRef CAS.
- C. M. Hoo, N. Starostin, P. West and M. L. Mecartney, A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions, J. Nanopart. Res., 2008, 10, 89–96 CrossRef CAS.
- S. Mössmer, J. P. Spatz, M. Möller, T. Aberle, J. J. Schmidt, W. Burchard, S. Moessner, J. P. Spatz, M. Moeller, T. Aberle, J. J. Schmidt, W. Burchard, S. Mössmer, J. P. Spatz, M. Möller, T. Aberle, J. J. Schmidt, W. Burchard, S. Moessner, J. P. Spatz, M. Moeller, T. Aberle, J. J. Schmidt, W. Burchard, S. Mössmer, J. P. Spatz, M. Möller, T. Aberle, J. J. Schmidt, W. Burchard, S. Moessner, J. P. Spatz, M. Moeller, T. Aberle, J. J. Schmidt and W. Burchard, Solution Behavior of Poly(styrene)- b lock -poly(2-vinylpyridine) Micelles Containing Gold Nanoparticles, Macromolecules, 2000, 33, 4791–4798 CrossRef.
- J. P. Spatz, S. Sheiko and M. Möller, Ion-stabilized block copolymer micelles: film formation and intermicellar interaction, Macromolecules, 1996, 29, 3220–3226 CrossRef CAS.
- S. Link and M. A. El-Sayed, Size and Temperature Dependence of the Plasmon Absorption of Colloidal Gold Nanoparticles, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Determination of Size and Concentration of Gold Nanoparticles from UV-vis Spectra, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS.
- P. Kwolek and M. Wojnicki, The kinetic study of photoreduction of tetrachloroaurate acid by methanol in acidic media, J. Photochem. Photobiol., A, 2014, 286, 47–54 CrossRef CAS.
- P. Zhang and T. K. Sham, Tuning the electronic behavior of Au nanoparticles with capping molecules, Appl. Phys. Lett., 2002, 81, 736–738 CrossRef CAS.
- G. S. Pokrovski, B. R. Tagirov, J. Schott, E. F. Bazarkina, J.-L. Hazemann and O. Proux, An in situ X-ray absorption spectroscopy study of gold-chloride complexing in hydrothermal fluids, Chem. Geol., 2009, 259, 17–29 CrossRef CAS.
- T. M. Salama, T. Shido, R. Ohnishi and M. Ichikawa, EXAFS/XANES, XRD, and UV-vis Characterization of Intrazeolitic Gold(I) Prepared by Monolayer Dispersion of AuCl3 inside Na−Y Zeolite, J. Phys. Chem., 1996, 100, 3688–3694 CrossRef CAS.
- M. Hargittai, A. Schulz, B. Réffy and M. Kolonits, Molecular Structure, Bonding, and Jahn−Teller Effect in Gold Chlorides: Quantum Chemical Study of AuCl3, Au2Cl6, AuCl4-, AuCl, and Au2Cl2 and Electron Diffraction Study of Au2Cl6, J. Am. Chem. Soc., 2001, 123, 1449–1458 CrossRef CAS.
- A. M. Beale and B. M. Weckhuysen, EXAFS as a tool to interrogate the size and shape of mono and bimetallic catalyst nanoparticles, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- S. N. Reifsnyder, H. H. Lamb and N. Carolina, Characterization of Silica-Supported Pd–Au Clusters by X-ray Absorption Spectroscopy, J. Phys. Chem. B, 1999, 2, 321–329 CrossRef.
- R. Hilfiker, H.-F. Eicke, W. Sager, C. Steeb, U. Hofmeier and R. Gehrke, Form and Structure Factors of Water/AOT/Oil Microemulsions from Synchrotron SAXS, Ber. Bunsen-Ges., 1990, 94, 677–683 CrossRef CAS.
- A. Riskin, A. M. Beale, H.-G. Boyen, A. Vantomme, A. Hardy and M. K. Van Bael, The use of XAFS to determine the nature of interaction of iron and molybdenum metal salts within PS-b-P2VP micelles, Phys. Chem. Chem. Phys., 2013, 15, 1675–1681 RSC.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, Turkevich Method for Gold Nanoparticle Synthesis Revisited, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, D. Gianolio, E. K. Gibson, A. L. Gould, N. Jian, A. J. Logsdail, R. E. Palmer, L. Prati, N. Dimitratos, A. Villa and P. P. Wells, Tailoring Gold Nanoparticle Characteristics and the Impact on Aqueous-Phase Oxidation of Glycerol, ACS Catal., 2015, 5, 4377–4384 CrossRef CAS.
- H. Shi, N. Xu, D. Zhao and B.-Q. Xu, Immobilized PVA-stabilized gold nanoparticles on silica show an unusual selectivity in the hydrogenation of cinnamaldehyde, Catal. Commun., 2008, 9, 1949–1954 CrossRef CAS.
- B. Abécassis, F. Testard, Q. Kong, B. Francois and O. Spalla, Influence of Monomer Feeding on a Fast Gold Nanoparticles Synthesis: Time-Resolved XANES and SAXS Experiments, Langmuir, 2010, 26, 13847–13854 CrossRef PubMed.
- F. Hubert, F. Testard, A. Thill, Q. Kong, O. Tache and O. Spalla, Growth and Overgrowth of Concentrated Gold Nanorods: Time Resolved SAXS and XANES, Cryst. Growth Des., 2012, 12, 1548–1555 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp03473k |
| This journal is © the Owner Societies 2020 |