
One-step in vivo metabolic labeling as a theranostic approach for overcoming drug-resistant bacterial infections†
Duo
Mao
 a,
Fang
Hu
a,
Kenry
a,
Guobin
Qi
a,
Shenglu
Ji
b,
Wenbo
Wu
a,
Deling
Kong
*b and
Bin
Liu
*a
a,
Fang
Hu
a,
Kenry
a,
Guobin
Qi
a,
Shenglu
Ji
b,
Wenbo
Wu
a,
Deling
Kong
*b and
Bin
Liu
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore, 117585, Singapore. E-mail: cheliub@nus.edu.sg
bState Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials, Ministry of Education and College of Life Sciences, Nankai University, Tianjin, 300071, China. E-mail: kongdeling@nankai.edu.cn
First published on 8th January 2020
Abstract
A bacterial metabolic probe combining D-alanine and a photosensitizer with aggregation-induced emission (AIE) has been developed for in vivo light-up imaging of bacterial walls and efficient antibacterial therapy. This activatable probe could serve as a promising non-antibiotic tool for diagnosis and treatment of biofilms and intracellular bacteria, as well as multi-resistant bacterial infections.
New conceptsCurrently, clinical procedures for the theranostics of bacterial infections are becoming increasingly ineffective due to the emergence of antibiotic-resistant pathogens. We report the development of a bacterial metabolic probe, TPACN-D-Ala, containing D-alanine (D-Ala) and an organic photosensitizer with aggregation-induced emission characteristics, to achieve precise in vivo bacterial diagnosis and photodynamic therapy. TPACN-D-Ala can strongly penetrate biofilms and host cells and specifically incorporate into bacterial walls through metabolic processes, realizing specific light-up imaging of bacteria hidden in biofilms and living cells. More importantly, after intravenous injection of TPACN-D-Ala, the invasive bacteria on the infected tissue can be clearly imaged through metabolic processes and effectively eradicated by light irradiation. This is the first time that ‘one step’ in vivo metabolic labeling is realized, which provides a non-antibiotic strategy for more effective in vivo bacteria diagnosis and treatment as compared to traditional antibiotic therapies. Our simple and intelligent antibacterial probe will have great potential in the clinical diagnosis and treatment of bacteria-associated diseases in future. |
Invasive bacterial infections may lead to numerous severe inflammatory diseases, which are detrimental to human health. To date, clinical procedures for the theranostics of bacterial infections rely mainly on pathological examinations, followed by appropriate antibiotic administrations.1 The conventional tissue biopsies and cultures used in the diagnostic process are time-consuming and inefficient, thus leading to significant delays in treatment. In addition, current anti-bacterial treatments are becoming increasingly ineffective because of the increasing number of multi-resistant bacterial strains.2 Recently, it was found that most of the invasive pathogens can find ways to avoid drug treatments or intrinsic immune system attacks. For example, bacteria can form a biofilm, which can protect them against antimicrobial attacks through a self-secreted protective extracellular matrix, which gives rise to many difficult-to-treat infections, such as periodontitis and diabetic foot ulcers.3 Pathogens can also hide in normal tissues to escape immune attacks and antimicrobial therapies, which render infections latent or recurrent and cause severe diseases such as tuberculosis later on.4 Up to now, numerous studies have been conducted to solve these persistent bacterial-related problems, but with limited success. Therefore, it is essential to develop an alternative method that can overcome biofilm and host cell barriers to specifically identify and kill infection-causing bacterial strains without the concern of antibiotic resistance.
Bio-orthogonal labeling has become a promising tool for bacterial targeting and detection due to its high sensitivity and specificity.5 It is well known that the bacterial wall consists of a peptidoglycan structure, which is a network of oligopeptide cross-linked alternating N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) units.6 The bio-orthogonal labeling technique utilizes the metabolic process of unnatural metabolic precursors (either functionalized D-amino acids or NAM) to insert chemically active groups (either azide or alkyne) onto peptidoglycan, which leads to effective labeling of bacterial walls through the click reaction between the chemically active groups and their corresponding bio-orthogonally modified fluorescent dyes.7 Since D-amino acids typically do not exist naturally in mammalian cells,8 this strategy exhibits unique advantages for bacterial targeting and treatment in vivo. In our previous work, we realized in vivo metabolic labeling of pathogens by the use of an inflammatory responsive carrier to specifically deliver metabolic precursors into infected tissues.9 However, because the in vivo bio-orthogonal labeling strategy involves a two-step manipulation, the carrier design becomes complicated and the labeling process requires repeated administrations, which are not very attractive for clinical translations. Recently, we reported that a positively charged AIE photosensitizer modified with D-alanine (TPEPy-D-Ala) showed excellent bacteria labeling and treatment effects through a simple one-step incubation in vitro.10 Therefore, it is expected that this design of the probe may be advantageous for specific targeting and treatment of bacteria hidden in biofilms or host cells in in vivo systems, which are very difficult to kill using traditional therapeutics. However, becasue cationic substances usually induce strong inflammation reactions and can be rapidly cleared up by the immune system,11 this type of probe is not beneficial for in vivo drug delivery.
To develop a theranostic probe for in vivo applications, several conditions should be considered, which include good water dispersibility and ensuring effective body circulation. Activatable fluorescence and selective antibacterial activity in the infection regions are also ideal. In addition, a theranostic agent with good cellular and tissue penetration capability is beneficial for breaking biofilms and host cell barriers to further reduce antimicrobial resistance. Recently, we have developed a series of organic fluorescent photosensitizers (PSs) with aggregation-induced emission (AIE) properties, some of which have been successfully applied for in vitro bacterial diagnosis and photodynamic therapy (PDT).12 Different from the traditional PSs, AIE PSs show negligible fluorescence in the molecular state, but very bright fluorescence and strong 1O2 generation efficiency in the aggregate state. In this study, we developed a bacterial metabolic probe TPACN-D-Ala, which combines an AIE PS with D-alanine (D-Ala), with activatable fluorescence and strong 1O2 generation capability to realize precise in vivo tracking and efficient eradiation of bacteria in one step. After intravenous injection (i.v.), TPACN-D-Ala exhibits very weak fluorescence under physiological conditions due to the hydrophilic moiety D-Ala, which ensures a low background fluorescence within the normal tissue of a mouse body (Fig. 1A). The good water solubility and very small size of the probe enable TPACN-D-Ala to penetrate the biofilms or host cells with ease. Once reaching the bacteria-infected tissues, TPACN-D-Ala would be metabolically incorporated into the bacterial wall to provide clear in vivo imaging of bacteria because of its AIE property. Furthermore, the hidden bacteria could efficiently be eradicated by the light-induced 1O2 generated by the probe, which showed more superior in vivo bacteria killing as compared to traditional antibiotic therapy (Fig. 1B).
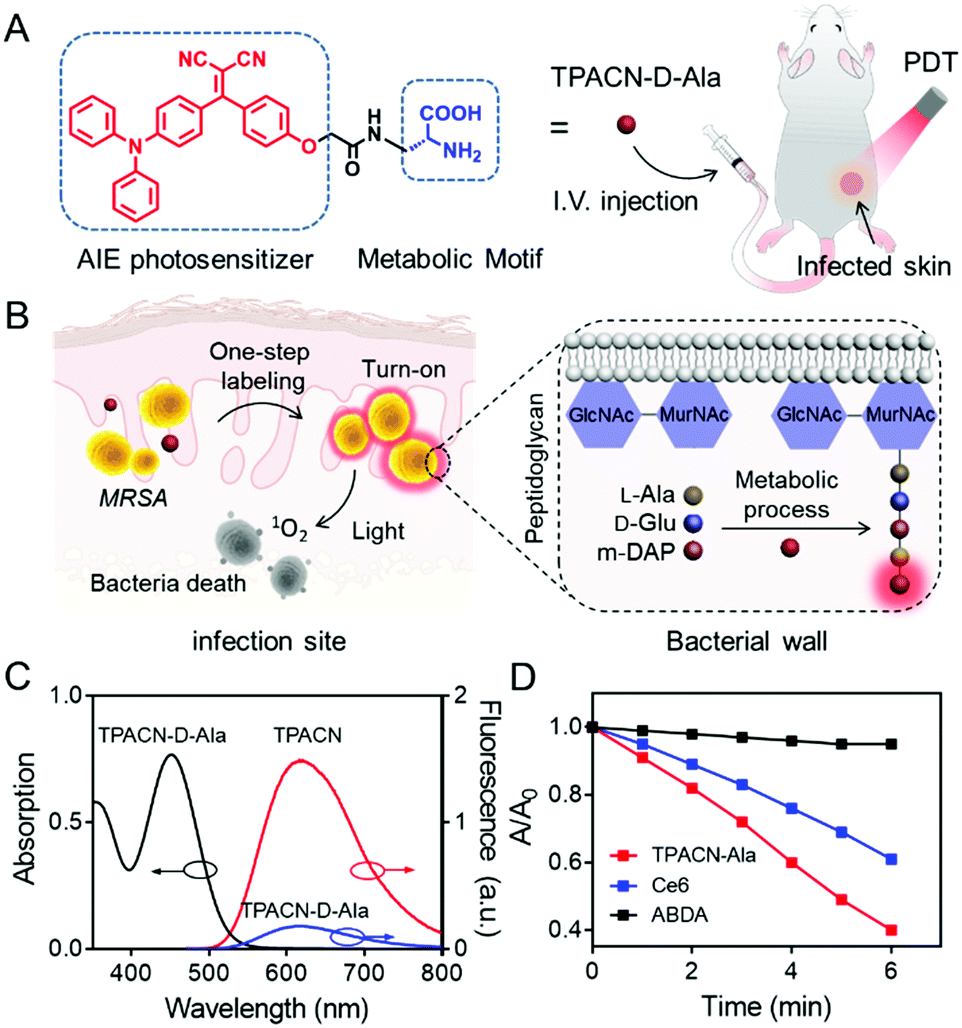 | ||
| Fig. 1 Design and characterization of the developed TPACN-D-Ala probe for bacterial tracking and photodynamic therapy (PDT). (A) TPACN-D-Ala was synthesized by combining D-Ala with the AIE photosensitizer TPACN, which could be intravenously injected into a bacteria-infected mouse to realize specific fluorescence light-up and image-guided antibacterial PDT. (B) Once reaching the infected tissue site through blood circulation, TPACN-D-Ala would be integrated into peptidoglycan to produce intense NIR fluorescence. Furthermore, specific imaging and efficient treatment of invasive bacteria could be achieved in vivo. (C) UV absorption and fluorescence emission of TPACN-D-Ala or TPACN in PBS. (D) Measurement of 1O2 production of TPACN-D-Ala (10 μM) or Ce6 (10 μM) using ADBA under light irradiation (60 mW cm−2). ABDA (black) solutions were used as the control; A0 and A are the absorbance of ABDA at 378 nm. | ||
To enable metabolic labeling of pathogens, the molecular weight of the probe should be as low as possible in order for the probe to pass through the pathogen cell walls. Besides, probes with an electric neutrality are preferred for in vivo study because of the minimal non-specific interaction. In accordance with these requirements, a small-molecular AIE photosensitizer with a molecular weight of 427.2, TPACN, was rationally prepared by conjugating electron-donating diphenylamine groups with electron-accepting dicyano vinyl groups (Scheme S1, ESI†). The D (donor)–A (acceptor) structure gives rise to a visible absorption band with a peak at 450 nm (Fig. S7A, ESI†). Because of the triphenylamine and vinyl benzene units, TPACN has an AIE-active emission property, which enables light-up imaging. As shown in Fig. S7B (ESI†), the emission intensity of TPACN was significantly enhanced when it was in a poor solvent (water) as compared to that in a good solvent (THF). Meanwhile, the donor and acceptor of TPACN are separated by a phenyl unit, which causes a larger torsional angle between the donor and acceptor, endowing TPACN with photosensitizing ability. The photosensitizing ability of TPACN was characterized by the consumption of 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) upon light irradiation (Fig. S8, ESI†). For further study, the pathogen-metabolizable moiety, D-alanine was covalently incorporated with TPACN by the condensation reaction and the deprotection of the fluorenylmethyloxycarbonyl group. The obtained TPACN-D-Ala was purified via high performance liquid chromatography (HPLC) and characterized with NMR and high-resolution mass spectrometry (Fig. S1–S6, ESI†). As shown in Fig. 1C, TPACN-D-Ala inherited the UV absorption of TPACN in the range of 350–550 nm. Compared with TPACN, TPACN-D-Ala exhibits very weak fluorescence and has a smaller hydrodynamic size (around 0.5 nm) in PBS, indicating good water-solubility (Fig. 1C and Fig. S9A, ESI†) of the probe after modification with hydrophilic amino acids. Using (4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran) as the reference, the fluorescence quantum yield of TPACN in water was estimated to be 5.7%. After modification with hydrophilic D-alanine, the water solubility was increased, and the fluorescence quantum yield of the obtained TPACN-D-Ala decreased to 1.6%. When incubated with methicillin-resistant Staphylococcus aureus (MRSA) in PBS, the fluorescence intensity of TPACN-D-Ala was strengthened dramatically, which was much higher than those mixed with other types of bio-macromolecules, such as fetal bovine serum (FBS), phospholipid (1,2-didodecanoyl-sn-glycero-3-phosphocholine), and 3T3 cell lysate (Fig. S9B, ESI†). This result suggests that TPACN-D-Ala could effectively be turned on for specific imaging of bacteria, while it interacts weakly with normal tissues. More importantly, TPACN-D-Ala has an 1O2 quantum yield of 40% measured using Rose Bengal as the reference. It showed stronger 1O2 production than Ce6, a commonly used PS in aqueous media, as demonstrated by the faster consumption of ABDA under light irradiation, indicating its potential bacteria killing capability (Fig. 1D).
Subsequently, we explored whether TPACN-D-Ala could label bacteria in vitro through a D-amino acid-dependent metabolic pathway. As shown in Fig. 2A and Fig. S10 (ESI†), after incubation with TPACN-D-Ala for 10 min, bright red fluorescence could be observed around each MRSA and Van S cell through confocal laser scanning microscopy (CLSM), demonstrating the efficient Gram-positive bacterial wall labeling capability of TPACN-D-Ala. Meanwhile, because L-alanine does not participate in the peptidoglycan synthesis, a competition inhibition test was also performed to further understand the labeling process of TPACN-D-Ala. As shown in Fig. 2B and C, the fluorescent signal of TPACN-D-Ala-treated MRSA decreased in the presence of additional Az-D-Ala in the labeling process, although Az-D-Ala did not affect the light-up imaging of bacteria. Mass spectrometry (MS) analysis was further conducted to support this mechanism. Peptidoglycans of TPACN-D-Ala-treated and normal MRSA cells were isolated by the standard protocol and then analysed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). The results showed that both the TPACN-D-Ala-treated and normal MRSA cells have an m/z (mass-to-charge ratio) peak at 939.3, which belongs to the natural GlcNAc-MurNAc-pentapeptide fragment. However, different from normal MRSA, a peak at 1408.4 can be found in the MS analysis result of TPACN-D-Ala-treated MRSA lysate, which is consistent with the molecular weight of the GlcNAc-MurNAc-pentapeptide-TPACN fragment (Table S1 and Fig. S11, ESI†). In addition, when MRSA was fixed with 4% paraformaldehyde, TPACN-D-Ala cannot label bacterial cell walls (Fig. S12, ESI†). Taken together, these data demonstrate that TPACN-D-Ala can be metabolically integrated into the peptidoglycan of the bacterial wall through a simple incubation step. Due to the protection of host cells, intracellular bacteria are difficult to be visualized and killed through the currently available methods. Since TPACN-D-Ala exhibits a highly specific reaction with bacteria in the presence of various types of biomolecules, we wondered if intracellular bacteria can also be tracked by TPACN-D-Ala. We chose Raw 264.7 as the host cell model, which was first incubated with MRSA. Subsequently, these infected living cells were treated with TPACN-D-Ala. As shown in Fig. S13 (ESI†), the uninfected Raw 264.7 showed low fluorescence background, indicating very weak interaction between TPACN-D-Ala and biomolecules in living cells. However, the bacteria hiding in living cells could clearly be visualized through a red fluorescence turn on, suggesting that invasive bacteria in host cells could be precisely labelled through TPACN-D-Ala treatment (Fig. 2D).
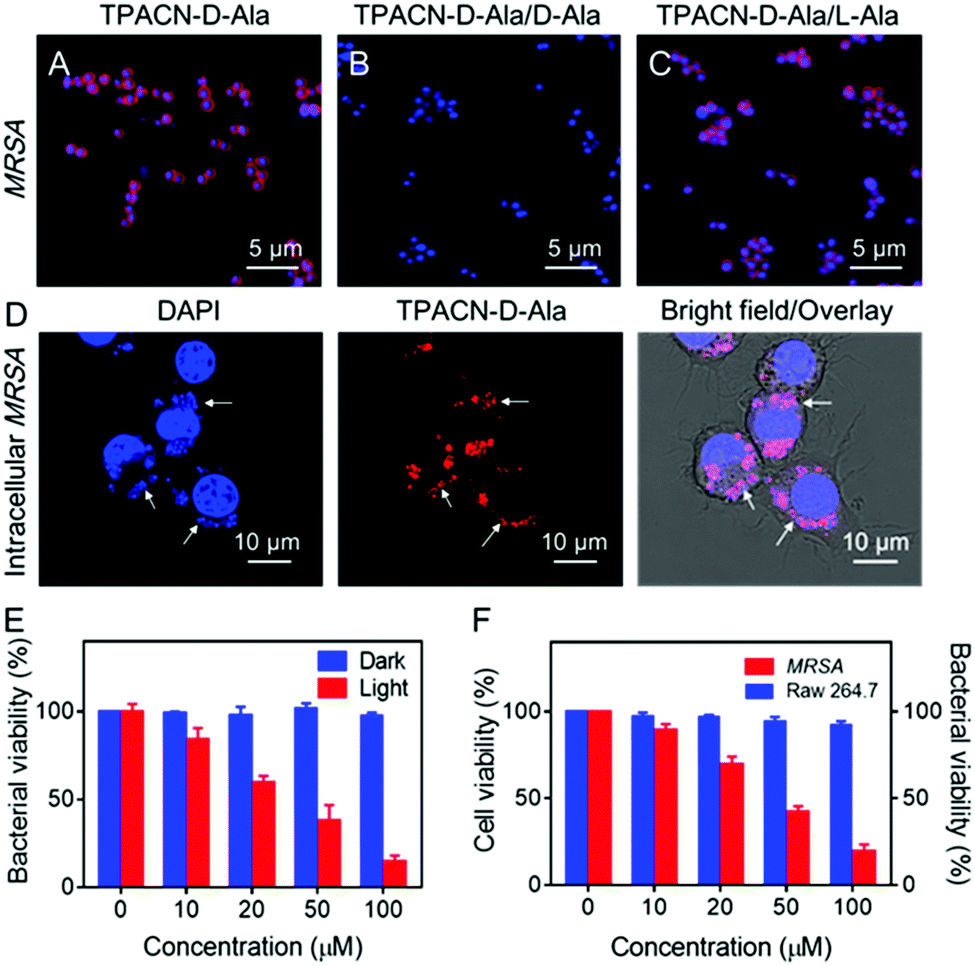 | ||
| Fig. 2 In vitro metabolic labeling ability and antibacterial activity of the TPACN-D-Ala probe. Confocal fluorescence images of MRSA incubated with: (A) TPACN-D-Ala (red color), (B) TPACN-D-Ala plus D-Ala, and (C) TPACN-D-Ala plus L-Ala, followed by staining with Hoechst 33342 (blue color). (D) Confocal fluorescence images of RAW 264.7 cells infected with MRSA, followed by treatment with TPACN-D-Ala (red color) and Hoechst 33342 (blue color). Intracellular bacteria are marked with white arrows. (E) Bacterial viability (CFU%) of MRSA incubated with different concentrations of TPACN-D-Ala under light irradiation (60 mW cm−2) under dark conditions. Bacterial viability of MRSA in PBS (probe concentration 0 μM) under dark conditions was set at 100%. (F) Intracellular bacterial viability of MRSA-infected RAW 264.7 cells incubated with different concentrations of TPACN-D-Ala upon light irradiation (60 mW cm−2) and cell viability of normal RAW 264.7 cells incubated with the same concentrations of TPACN-D-Ala upon light irradiation (60 mW cm−2). The bacterial viability of intracellular MRSA without any treatment and the cell viability of normal RAW 264.7 cells were set at 100%. | ||
Photodynamic therapy (PDT) has been increasingly explored as an alternative antibacterial treatment because the 1O2 used in this therapeutic approach can not only be cytotoxic, but also hardly induce resistance among bacterial strains. Considering its strong photosensitizing capability, TPACN-D-Ala was anticipated to have a good killing effect on bacteria. As shown in Fig. 2E, after incubation of MRSA with different concentrations of TPACN-D-Ala, the bacterial viability was minimally disturbed. However, after light irradiation, TPACN-D-Ala induced severe toxicity on MRSA, with a half-maximal inhibitory concentration (MIC) of 28.7 μM, indicating excellent capability of TPACN-D-Ala to eliminate antibiotic-resistant bacteria. Meanwhile, we also explored the possibility of intracellular bacterial ablation by PDT. Interestingly, when irradiated with light, the MRSA-bearing Raw 264.7 cells treated with TPACN-D-Ala exhibited a significant decrease in the bacterial colony-forming units (CFUs) in a dose-dependent manner (Fig. 2F). It was also found that the cell viability of normal Raw 264.7 cells was not affected with increasing TPACN-D-Ala concentration (0–100 μM) in the presence of light irradiation, suggesting that TPACN-D-Ala had good biocompatibility and negligible cytotoxicity to normal tissues during the photodynamic treatment (Fig. 2F). In addition, after the incubation of MRSA-infected RAW 264.7 cells with TPACN-D-Ala (50 μM) and a reported antibacterial photosensitizer Methylene Blue (MB) (50 μM), respectively, TPACN-D-Ala induced a stronger photoablation effect of intracellular MRSA than Methylene Blue (MB) upon white light treatment (60 mW cm−2), indicating the unique advantage of TPACN-D-Ala in intracellular antibacterial therapy (Fig. S14, ESI†).
A biofilm is a densely packed community of microorganisms, which can act as another obstacle for mitigating the efficacy of antibiotics and host immune systems. Therefore, we sought to test if TPACN-D-Ala could also label the bacteria within the biofilm and then ablate these bacteria through their excellent cell penetration and antibacterial effect, respectively. Herein, the S. aureus WHGFP biofilm was cultured as a model system to visualize accumulation of TPACN-D-Ala on the biofilm. After being exposed to TPACN-D-Ala, the biofilm was examined via CLSM. As shown in Fig. 3A and C, CLSM images and quantitative fluorescence results indicate a rapid diffusion and accumulation of TPACN-D-Ala into the biofilm with time, which corresponds to an increase in the fluorescence intensity of the biofilm, until it reaches saturation within 20 minutes. Moreover, all bacteria within the biofilm could be precisely labeled, as verified by the enlarged CLSM images (Fig. 3B), demonstrating that the infiltrating probe could be metabolically incorporated into bacteria residing in the biofilm. In addition, TPACN-D-Ala showed a negligible effect on the CFUs of these labeled biofilms under dark conditions even at high concentrations. However, once subjected to light irradiation, the biofilm viability could be greatly reduced in a dose-dependent manner, with a minimum inhibitory concentration (MIC) of 51.2 μM, demonstrating the excellent ablation effect of TPACN-D-Ala on the biofilm (Fig. 3D). These results revealed that, due to very small hydrodynamic size, TPACN-D-Ala can easily pass through extracellular polymeric substances of the biofilm. In addition, infiltrating TPACN-D-Ala as a nutrient can metabolically be taken up by the harbored bacteria and incorporated into their bacterial wall, realizing the biofilm bacterial fluorescence light-up and killing by PDT. Taken together, these in vitro experiments indicate the great potential of TPACN-D-Ala for the treatment of biofilm-caused diseases.
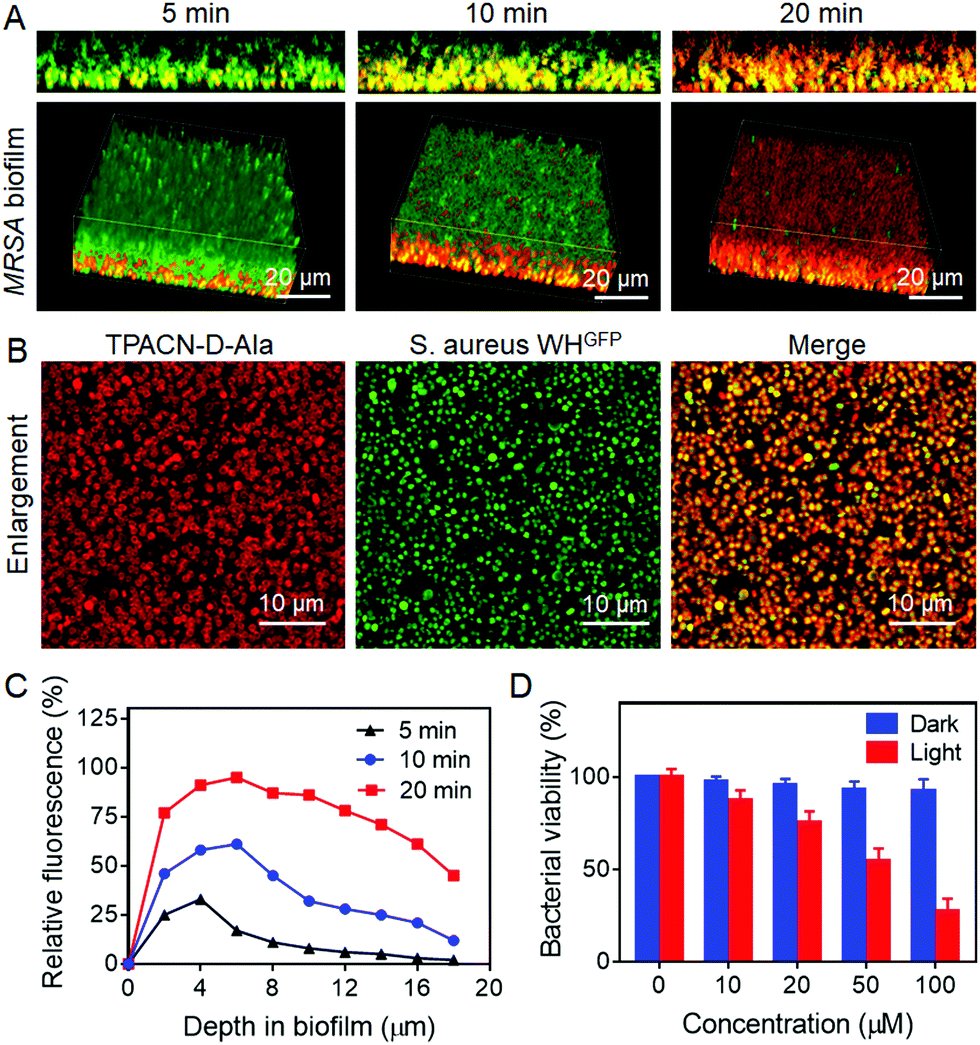 | ||
| Fig. 3 Penetration and accumulation of the TPACN-D-Ala probe into the S. aureus biofilms and the killing efficiency of the probe through PDT. (A) 2D side (upper) and 3D confocal images (bottom) of S. aureus WHGFP biofilms incubated with the TPACN-D-Ala probe at different time intervals. (B) Enlarged CLSM images of S. aureus WHGFP biofilms treated with TPACN-D-Ala. Scale bar = 10 μm. (C) Relative fluorescence intensity (FLI) (%) of the TPACN-D-Ala-treated S. aureus biofilms at different depths of the biofilm at different time intervals. Relative fluorescence intensity represents the number of red fluorescent pixels versus the total number of pixels at a given depth. (D) Bacterial viability of S. aureus WHGFP biofilms incubated with different concentrations of TPACN-D-Ala under light irradiation (60 mW cm−2) and dark conditions. Bacterial viability of biofilms in PBS (probe concentration 0 μM) under dark conditions was set at 100%. | ||
Encouraged by the superior performance of TPACN-D-Ala in in vitro experiments, we further assessed the possibility of its in vivo application. After 14 days intravenous (i.v.) injection of TPACN-D-Ala into healthy mice, negligible in vivo toxicity and rapid body clearance of TPACN-D-Ala were observed through the blood chemistry test for liver/kidney function, body weight measurement, and fluorescence imaging for body clearance (Fig. S15 and S16, ESI†). The bacteria infected mouse model was then built by subcutaneously (s.c.) inoculating S. aureus WHGFP into the right flank skin of Balb/c mice. The in vivo kinetic distribution of TPACN-D-Ala was first evaluated using the CRi imaging system after i.v. injection of TPACN-D-Ala. As shown in Fig. 4A and B, the fluorescent signals of the infected skin continuously increase over time and reach the highest intensity at 6 h post administration, which reflects a preferential accumulation of TPACN-D-Ala within the bacteria-infected tissue. Meanwhile, because of the low background signal of TPACN-D-Ala in a complex biomolecular system, the average fluorescence intensity of the bacteria infected skin was significantly higher than that of the surrounding normal skin, with a bacteria-to-background ratio of ∼3.5, which demonstrates the advantage of the activatable TPACN-D-Ala probe for in vivo bacterial imaging. Similar trends could also be observed from the ex vivo fluorescence distribution of different harvested tissues from MRSA bearing mice treated with TPACN-D-Ala, where the fluorescence intensity of the infected skin was slightly lower than that in the intestines and 2.1–7.5 times higher than those in other tissues (Fig. 4C and Fig. S17, ESI†). Moreover, the infected skin was sectioned and imaged using a confocal microscope. As depicted in Fig. 4D, a large amount of red fluorescent signal could be observed within the GFP-expressing S. aureus aggregates and these labeled bacteria can also be clearly distinguished from the surrounding normal cells, indicating a good tissue penetration capacity of the probe and its precise in vivo bacterial labeling of the infected tissue.
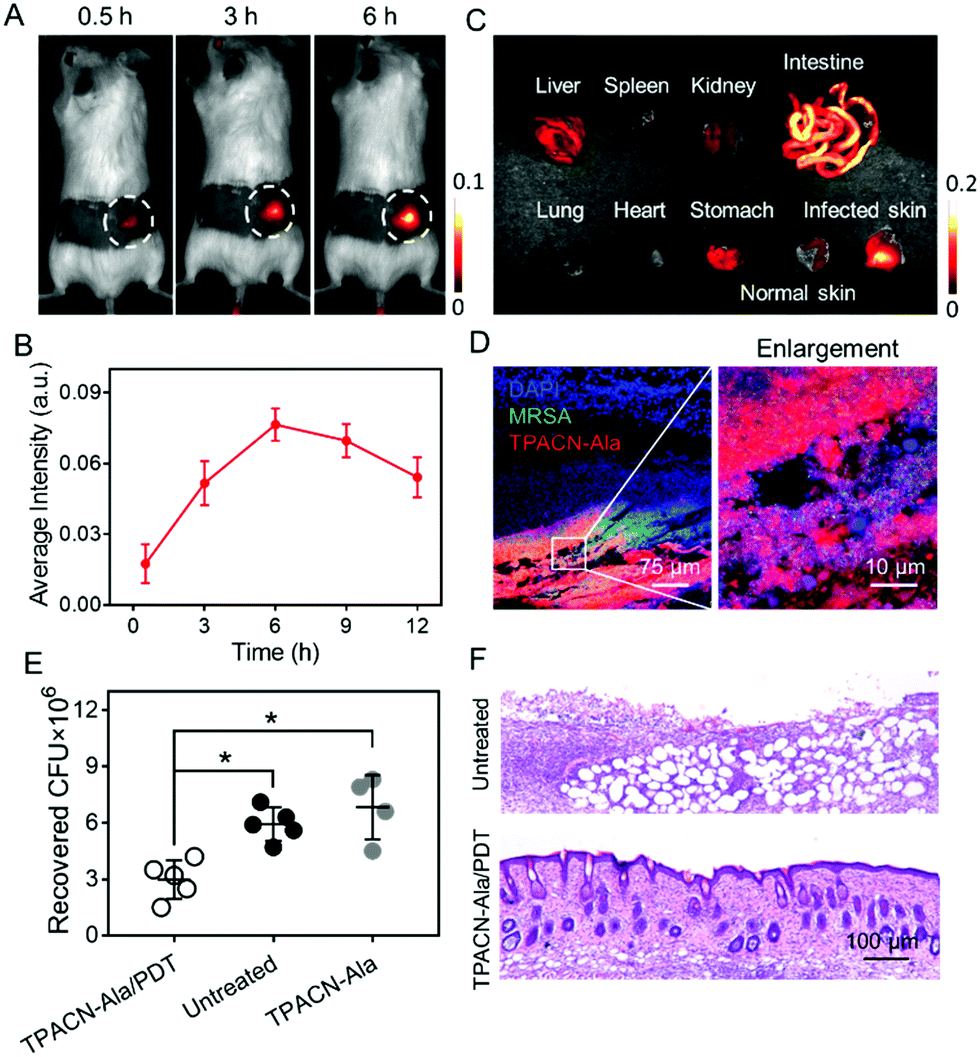 | ||
| Fig. 4 In vivo image-guided bacterial killing using TPACN-D-Ala. (A) Time-dependent in vivo fluorescence images of bacteria-bearing mice after i.v. injection with TPACN-D-Ala. The bacteria-infected regions are labeled with dotted white circles. (B) Average fluorescence intensities of the infected skin of mice treated with TPACN-D-Ala over time. (C) Ex vivo fluorescence pictures of different harvested tissues of mice at 12 h post-injection of TPACN-D-Ala. (D) Confocal images of the infected skin slices of mice treated with TPACN-D-Ala. The right picture is the enlarged region of the white box shown on the left picture. (E) Bacterial colony recovered from the infected skin subjected to different treatments (n = 5, *p < 0.05). (F) H&E staining images of the infected skin slices subjected to different treatments. | ||
Eventually, we tested if the TPACN-D-Ala-labeled bacteria in skin tissue could be ablated through PDT. The bacteria-bearing mice injected with saline were used as controls. The bacteria-infected regions of the mice were exposed to white light irradiation for 10 min. After 7 days, the infected skins were collected for histological analysis and the number of residual bacteria was measured by colony-forming unit (CFU) analysis. As shown in Fig. 4E, the CFU of the infected skin treated with TPACN-D-Ala and light irradiation reduced significantly as compared to those of the control groups. Haemotoxylin and Eosin (H&E) staining images showed that the infected skin treated with TPACN-D-Ala and light irradiation had less inflammatory cell infiltration and normal skin structure as compared to the skin tissues of the control groups (Fig. 4F and Fig. S18, ESI†). These data verify that the direct in vivo labeling method is highly efficient and beneficial for in vivo image-guided antibacterial therapy.
In summary, we developed a bacterial metabolic probe, TPACN-D-Ala, for precise in vivo image-guided antibacterial therapy. TPACN-D-Ala, which combined an AIE photosensitizer with D-Ala, exhibited a very strong 1O2 generation capability and low fluorescent signal in the complex biological environment. In the process of peptidoglycan synthesis of bacteria, five amino acids including D-alanine are successively incorporated onto glycan molecules through enzymatic reactions. Due to excellent water solubility, TPACN-D-Ala has a very small size (∼0.5 nm) under physiological conditions. This ensures that TPACN-D-Ala could successfully be diffused into bacteria and involved in the biosynthesis of peptidoglycan to form peptidoglycan-TPACN, realizing the bio-incorporation of AIE molecules into the bacterial wall with specific light-up imaging as a result of AIE activation. It is more convenient for practical in vivo bacterial labeling than the traditional two-step labeling approach. More importantly, due to its good water solubility, TPACN-D-Ala could diffuse well into the host cells or biofilms to effectively track and ablate the sheltered bacteria through PDT. To the best of our knowledge, this is the first time that simultaneous imaging and eradication of bacteria hidden in biofilms and living cells have been realized, which provides a non-antibiotic strategy for more effective in vivo bacterial ablation. After the i.v. injection of TPACN-D-Ala, the bacteria on infected tissues could be specifically tracked and effectively inhibited through light illumination, realizing precise in vivo image-guided antibacterial therapy. With these unique advantages, we anticipate that the developed probe will have great potential in the treatment of bacteria-associated diseases in clinical settings, which is under investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Singapore NRF Competitive Research Program (R279-000-483-281), the NRF Investigatorship (R279-000-444-281), the National University of Singapore (R279-000-482-133) and the National Science Foundation of China (81830060 and 31771066) for financial support.Notes and references
- J. F. Siqueira Jr and I. N. Rôças, J. Endod., 2008, 34, 1291 CrossRef PubMed.
- A. J. Alanis, Arch. Med. Res., 2005, 36, 697 CrossRef PubMed.
- (a) M. E. Olson, H. Ceri, D. W. Morck, A. G. Buret and R. R. Read, Can. J. Vet. Res., 2002, 66, 86 Search PubMed; (b) R. M. Donlan, Clin. Infect. Dis., 2001, 33, 1387 CrossRef CAS PubMed; (c) H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623 CrossRef CAS PubMed.
- (a) A. P. Bhavsar, J. A. Guttman and B. B. Finlay, Nature, 2007, 449, 827 CrossRef CAS PubMed; (b) R. K. Ernst, T. Guina and S. I. Miller, J. Infect. Dis., 1999, 179, S326 CAS.
- (a) E. Kuru, H. V. Hughes, P. J. Brown, E. Hall, S. Tekkam, F. Cava, M. A. de Pedro, Y. V. Brun and M. S. VanNieuwenhze, Angew. Chem., Int. Ed., 2012, 51, 12519 CrossRef CAS PubMed; (b) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764 CrossRef CAS PubMed.
- (a) I. Melero, A. Rouzaut, G. T. Motz and G. Coukos, Cancer Discovery, 2014, 4, 522 CrossRef CAS; (b) S. Kakarla and S. Gottschalk, Cancer J., 2014, 20, 151 CrossRef CAS PubMed.
- J. M. Fura, D. Kearns and M. M. Pires, J. Biol. Chem., 2015, 290, 30540 CrossRef CAS.
- A. Aliashkevich, L. Alvarez and F. Cava, Front. Microbiol., 2018, 9, 683 CrossRef PubMed.
- D. Mao, F. Hu, S. Ji, W. Wu, D. Ding, D. Kong and B. Liu, Adv. Mater., 2018, 30, 1706831 CrossRef PubMed.
- F. Hu, G. Qi, K. Kenry, D. Mao, S. Zhou, M. Wu, W. Wu and B. Liu, Angew. Chem., Int. Ed., 2019, 58 DOI:10.1002/anie.201910187.
- (a) V. Kononenko, M. Narat and D. Drobne, Arh. Hig. Rada Toksikol., 2015, 66, 97–108 CAS; (b) Y. Cheng, J. Dai, C. Sun, R. Liu, T. Zhai and X. Lou, Angew. Chem., Int. Ed., 2018, 57, 3123–3127 CrossRef CAS PubMed.
- (a) G. Feng, Y. Yuan, H. Fang, R. Zhang, B. Xing, G. Zhang, D. Zhang and B. Liu, Chem. Commun., 2015, 51, 12490 RSC; (b) W. Wu, D. Mao, X. Cai, Y. Duan, F. Hu, D. Kong and B. Liu, Chem. Mater., 2018, 30, 3867 CrossRef CAS; (c) G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404 CrossRef CAS PubMed; (d) E. Zhao, Y. Chen, S. Chen, H. Deng, C. Gui, C. T. Leung, Y. Hong, J. Y. Lam and B. Z. Tang, Adv. Mater., 2015, 27, 4931 CrossRef CAS PubMed; (e) F. Xia, J. Wu, X. Wu, Q. Hu, J. Dai and X. Lou, Acc. Chem. Res., 2019, 52, 3064–3074 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh01675a |
| This journal is © The Royal Society of Chemistry 2020 |