
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer mechanochemistry-enabled pericyclic reactions
Emilia
Izak-Nau†
 a,
Davide
Campagna†
ab,
Christoph
Baumann†
ab and
Robert
Göstl
*a
a,
Davide
Campagna†
ab,
Christoph
Baumann†
ab and
Robert
Göstl
*a
aDWI – Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, 52056 Aachen, Germany. E-mail: goestl@dwi.rwth-aachen.de
bInstitute for Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 1, 52074 Aachen, Germany
First published on 16th March 2020
Abstract
Over the past decades, it became clear that next to heat and light, pericyclic reactions can be induced mechanochemically when the reacting motifs are embedded as latent force-responsive groups (mechanophores) into polymer architectures. Not only does this enable a variety of functions and applications on a material level, but moreover grants access to symmetry-forbidden reaction products with respect to the Woodward–Hoffmann rules. The latter indicates that polymer mechanochemistry follows its own set of rules that, however, regarding underlying mechanisms and design rationales is far from being holistically understood. Here we review the existing body of literature and identify common structural features and substitution prerequisites to the polymer framework shining light on the differences between polymer mechanochemical pericyclic reactions and their traditional counterparts. By this, we believe to contribute to the major challenge of not only retrospectively describing force-induced reactivity but eventually finding a common molecular design guideline.
Introduction
The use of mechanical force to induce chemical bond transformations in polymers enables reactions that cannot proceed using other, more established physical stimuli, such as light and heat.1 Unlike heat, force is a vector quantity, i.e. it is intrinsically directional with regard to bond orientation. Moreover and contrarily to light, force-controlled reactions proceed on the ground state potential energy surface (PES).2 Reductively, force acting on molecules can be interpreted as alteration of the geometries and distances of the atoms participating in the force-controlled chemical reaction – comparable to Hammond's postulate of transition state theory.3 While force and pressure have been applied to steer chemical reactions for millennia, e.g. in milling processes,4–6 the relatively young field of polymer mechanochemistry allows to exert better control over the applied force to the respective participating reactants. Whereas milling acts on the reactants omnidirectionally by compression and impact, polymer mechanochemistry relies on the extension of macromolecular termini that are substituted to individually defined positions of the reacting molecular motif – the mechanophore. This allows precise localization of the force and hence introduces a sense of directionality by ‘force-coupling’ certain bonds within the mechanophore.7 In consequence, most reactions in polymer mechanochemistry known today rely on the extension-induced selective bond scission as initial step of the mechanochemical reaction. Notably, this paradigm might be challenged in the future with the discovery of ‘molecular anvils’ that can exert compressive forces on molecules in a directional fashion, too.8 However, polymer mechanochemistry is more than a collection of destructive processes as the downstream formation of new bonds can be achieved, e.g. by the force-activation of latent catalysts.9The force-induced bond-scission along the path of hundreds of covalent bonds within the mechanophore-centred polymer is a competition between these bonds. Hence, selectivity regarding the identity of the bond that cleaves can only be achieved by tailor making the mechanophore such that it exhibits a higher force-dependent scission rate than the polymer backbone or other functional moieties. While the electronic structures of the reactants remain widely unaffected by force when compared to light and heat, atomic motion along the reaction coordinate is altered by molecular stress and strain.10 This renders the design process of new mechanophores unintuitive and hence is generally carried out by educated guessing while building on already established motifs.11 Thermal bond dissociation energies (BDEs) are used as a first estimation to assess the propensity of a covalent bond to undergo scission under force.12 This approach is intrinsically flawed as it assumes identical reaction pathways along the PES for thermal and mechanochemical reactions,13 but has yielded reasonable successes in the past. A BDE of about 300 kJ mol−1 (72 kcal mol−1) can be considered mechanochemically promising14 (the BDE of a C–C bond is ca. 360 kJ mol−1 (86 kcal mol−1), for perspective).
A plethora of mechanochemically labile bonds exist that can be cleaved by various forms of force. However, a strikingly large amount of these mechanophores relies on motifs that undergo pericyclic reactions, more precisely those pericyclic reactions that lead to bond dissociation. This is initially surprising as, e.g. cycloeliminations, involve the dissociation of two covalent bonds and intuitively one could assume that this would be mechanochemically more difficult than the dissociation of a single covalent bond elsewhere in the macromolecule. Yet, many papers have been published over the last decades indicating that dissociative pericyclic reactions are indeed selectively accessible by mechanical force, though they might not always follow the thermally or photochemically established mechanisms and thus occasionally oppose the Woodward–Hoffmann (WH) rules.15 While excellent reviews are available focusing on the functions and applications that can be realized by employing mechanophores in materials as well as their interpretation,9,16–23 we here explicitly and comprehensibly review all polymer mechanochemical transformations based on pericyclic and pseudopericyclic reactions on the mechanophore level in relation to their molecular designs.
Cycloelimination of [2 + 2] cycloadducts
Four-membered carbocycles
The formation of strained cyclic structures is an established strategy to reduce the internal stability of molecules. Cyclobutane is one of the simplest strained cycles as a four-membered ring bearing only saturated carbon atoms. As cyclobutane can be formally seen as the [2 + 2] cycloadduct of two alkenes, subjecting it to a force-induced cycloelimination reaction was considered promising and subsequently achieved (Scheme 1).24–26 | ||
| Scheme 1 General scheme of the bonds involved in the cycloelimination of a [2 + 2] cycloadduct. Attachments to polymer framework are represented as dashed bonds, application and direction of force in bold arrows, reacting bonds in red – here and throughout the manuscript. | ||
In 2010, Moore and co-workers synthesized the first mechanophore based on the cyclobutane motif which showed mechanochemical activity in solution exerted by ultrasonication.24 A dicyano-substituted cyclobutane ring (Scheme 2) was integrated into the centre of linear poly(methyl acrylate) (PMA) chains. The mechanochemical activity of the motif was qualitatively confirmed by postfunctionalization with 9-(methylaminomethyl)anthracene (MAMA) that reacted with the mechanochemically generated α,β-unsaturated system via Michael-type addition. UV-Vis absorption spectroscopy revealed the presence of anthryl signals in the polymer underlining the successful formation of the reactive double bonds. From this first encouraging proof of a mechanochemically triggered cycloreversion of a [2 + 2] cycloadduct, further and more detailed studies followed.
 | ||
| Scheme 2 Dicyano-substituted cyclobutane undergoing force-induced cycloelimination.24 | ||
Although the first well-documented experiments were reported in 2010, earlier works had already referred to this mechanically induced cycloelimination. However, this was not investigated in detail, since there the focus was placed on crack formation events in solid state materials.27–30 The first study by Kim and co-workers in 2004 focused on light-triggered healing of the material after crack formation induced by grinding in the solid state.27 Cinnamoyl moieties were used to construct cyclobutane-based mechanophores that in the cleaved state would show high and bathocromically shifted absorptivity to undergo light-induced [2 + 2] cycloaddition (Scheme 3). The cycloelimination was not further investigated as only a small fraction of the cyclobutanes underwent this reaction and the focus was to develop an efficient method for crack healing.
 | ||
| Scheme 3 Cyclobutane as crosslinker in a polymer network designed to heal upon light irradiation after grinding-induced cycloelimination.27 | ||
The reversibility of the mechanochemical scission of cyclobutanes was also demonstrated by heat. For this purpose, Craig and co-workers exploited the well-known heat-induced [2 + 2] cycloaddition of trifluorovinyl ethers (TFVE) yielding perfluorocyclobutanes (PFCB) (Scheme 4).30 Employed as mechanophores in polymers, these could be healed thermally after mechanochemical scission. Notably, a detailed analysis of the PFCB cycloreversion mechanism was performed, which was proposed to involve a 1,4-diradical intermediate. This interpretation was based on two observations: (i) after the sonication a fraction of PFCB showed different stereochemistry compared to the isomer of the starting cyclobutane scaffold implying that a concerted mechanism could be excluded and (ii) a radical trap added during mechanophore activation was incorporated into the polymer backbone indicating the presence of radicals.
 | ||
| Scheme 4 Structure of the PFCB mechanophores capable of thermal healing after mechanical damage.30 | ||
Mechanistic considerations
The first prerogative for validating a mechanophore, concerning the exclusion of factors different from the mechanical force, was proven in detail by Craig and co-workers.26 It was verified that when mechanical force was applied to cyclobutane-based mechanophores within a polymer chain, significant dependencies on the average molar mass of the polymer chain, the dispersity in molar mass and the distribution of the mechanophore along the polymer chain arise. The latter is especially prominent in the purpose of rationalizing the force contribution and it was found that the scission rate dramatically decreased when the mechanophore was not chain-centered. On the other hand, the same activation was completely successful without any ‘chain-centered’ requirement when mechanical force was replaced by light as energy source.To discuss mechanism and stereochemistry of the mechanically-induced cycloelimination of [2 + 2] cycloadducts, a brief recapitulation of the WH rules is essential.15 For the [2 + 2] cycloaddition, the number of π-electrons involved is m + n = 4q, so necessarily q = 1. According to the selection rules this reaction is then thermally allowed in the ‘top–bottom’ (t–b) and ‘bottom–bottom’ (b–b) addition case, i.e. for two reactants with different configuration (Z–E or E–Z), and is photochemically allowed only in the t–b addition case, i.e. for two reactants with equal configuration (Z–Z or E–E). The stereochemistry of the addition process is reflected in the configuration of the cyclic product and due to the principle of microscopic reversibility the cycloreversion follows the same mechanisms and rules. Scheme 5 shows that the requirement for an allowed cycloreversion reaction is that the two reacting σ bonds must have identical configurations (syn–syn or anti–anti).
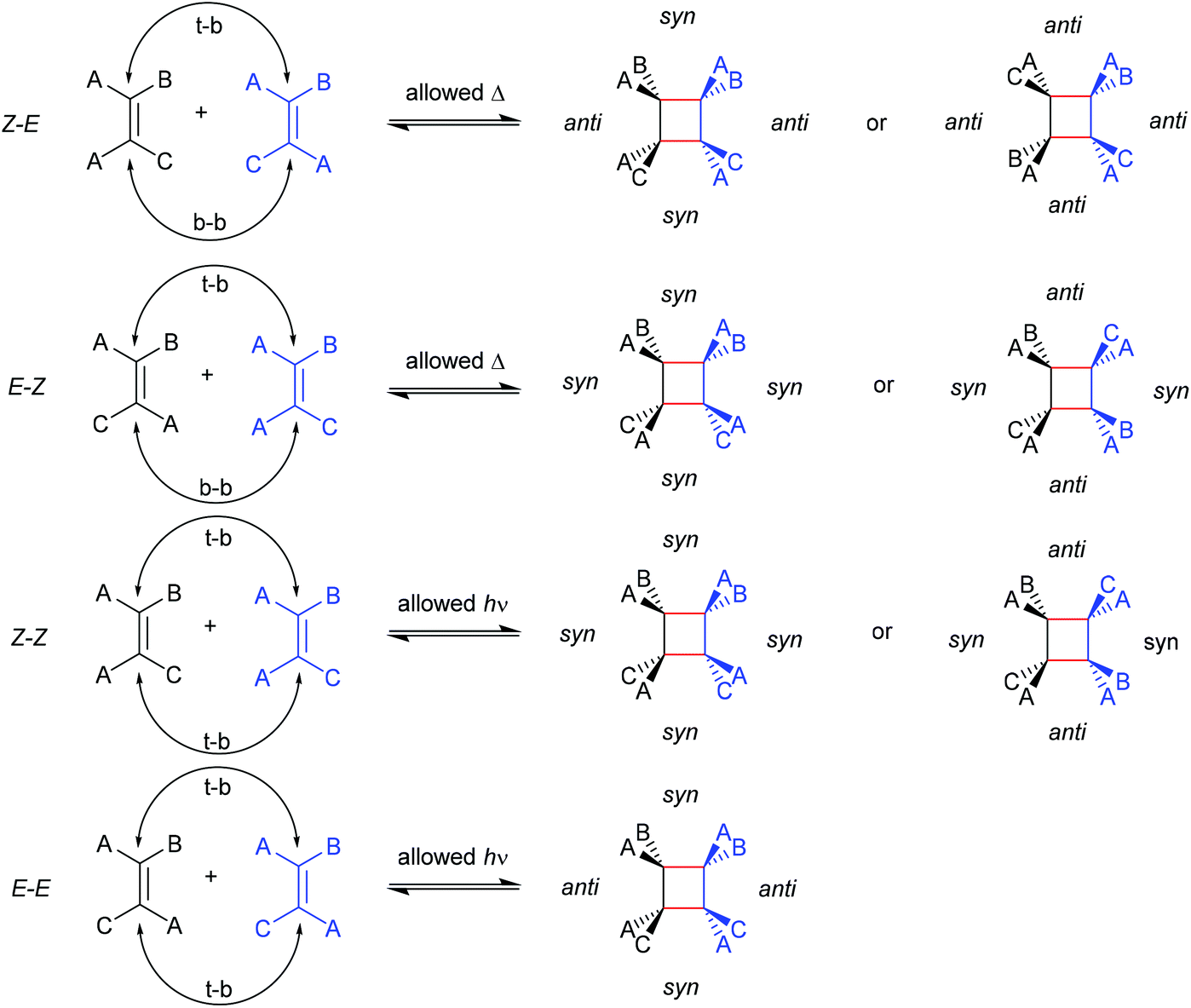 | ||
| Scheme 5 Allowed pathways related to the respective configurations for both thermal and photochemical [2 + 2] cycloaddition and -elimination according to the WH rules. | ||
Although it was strongly suggested that the mechanochemically-induced cycloreversion involved a diradical intermediate,30–32 the mechanism of this process was firstly investigated in more detail in subsequent works.15–19 For this, Craig and co-workers studied the mechanochemical cycloelimination of bicyclo[4.2.0]octane stereoisomers.33 The high symmetry and non-scissile nature of this mechanophore facilitated the comparison of actual and hypothetical products of the cycloelimination reaction. Scheme 6a and b show the mechanophore in syn–syn and anti–syn configuration on the scissile bonds, respectively. While the former syn–syn cycloelimination is photochemically allowed yielding the product with E–E double bonds and thermally allowed yielding the Z–E product, the cycloelimination from the anti–syn reactant is completely symmetry forbidden. However, the experimentally obtained mechanochemical products differ significantly from this prediction and a product mixture of Z–Z, E–E and Z–E was obtained starting from either syn–syn or anti–syn mechanophores (Scheme 6c). In addition, a fraction of 70–80% of the thermodynamically more stable Z–E product was received in all cases. These results strongly underline a diradical intermediate obtained by homolytic bond scission as in this form bond rotation into the thermodynamically more stable pro-configuration can occur. Importantly, this means that through polymer mechanochemistry the non-WH products can be obtained as the reaction pathway can be altered significantly.
 | ||
| Scheme 6 (a), (b) Theoretically allowed thermal products according to the WH rules: (a) starting configuration syn–syn on the scissile bonds, the product is Z–E; (b) starting configuration anti–syn on the scissile bonds, the reaction is symmetry forbidden. (c) Experimentally observed mechanochemically generated products. No significant distinction between the starting configurations concerning the stereochemistry of the products can be made. This suggests formation of a diradical intermediate with three pro-conformations giving three final configurations. | ||
Beyond the mechanistic elucidation, the discovery of a mechanophore class is often accompanied by comprehensive evaluation of the regio- and stereochemical effects on the mechanochemical activity. Structure–activity relationships were investigated by Moore and co-workers starting from the dicyano-substituted cyclobutane structure progressively modifying its scaffold.34 They showed that the number and configuration of the substituents modulated the mechanochemical scission rate, which was confirmed by computational studies. Six different structures were assessed with either no, one, or two cyano-substituents either in E- or Z-configuration (Scheme 7). The mechanochemical scission rate was shown to increase from E- to Z-isomer and with increasing number of cyano substituents.
 | ||
| Scheme 7 Different cyclobutane derivatives studied with regard to their mechanochemical scission rate (increasing with arrow direction).34 | ||
The mechanochemical scission rate in dependence of the mechanophore regioisomerism was moreover investigated by Sijbesma, Doltsinis, Hawker, Leibfarth, and co-workers.35 In this study, four-membered ketene dimers bearing polymer chain substituents in 2,4-position of the cycle (unlike the common 1,2-substitution discussed above) both in Z- and E-form were evaluated (Scheme 8). While the cycloreversion towards the ketenes was observed thermally, force primarily yielded homolytic C–O scission on the polymer attachment moieties. This observation inferred that the favoured scission route is dependent on the position of the application of the mechanical force to the molecular moiety, which was confirmed by computational simulations. The force-coupled potential energy surfaces were calculated for the cycloreversion of both Z- and E-ketene dimer as well as for the homolytic benzylic C–O cleavage and showed that the scission rate of the C–O bond dramatically increased with force application. While the E-ketene dimer simply shows a low decrease in activation energy upon force application, the Z-isomer even revealed an increasing activation energy suggesting a force-induced strengthening effect.
 | ||
| Scheme 8 Application of heat or force to a ketene dimer bearing polymer substituents in 2,4-position.35 | ||
Four-membered heterocycles
The successful mechanochemical activation of four-membered carbocycles inspired the survey of other four-membered rings that exhibit interesting functional groups upon cycloelimination. One of the most commonly occurring four-membered rings is the β-lactam scaffold and its activation by force via cycloelimination was investigated by Moore and Robb (Scheme 9a).36,37 This motif dissociates into a ketene and an imine, i.e. the two reactants used for the synthesis of the β-lactam. Initially, the propensity of the β-lactam to undergo mechanochemical cycloelimination was evaluated by computational simulations. The encouraging results thereof were translated to synthesis and subsequently this motif was proven mechanochemically active, most likely via a diradical intermediate. Following a comparable approach, the mechanical cycloelimination of a 1,2-azetidinone was investigated by Craig and co-workers (Scheme 9b).38 Analogously to the β-lactam, this moiety cleaves into imine and isocyanate, the latter of which is much more useful in downstream synthetic applications due to its longer lifetime compared to ketenes. Importantly, these results demonstrate that the mechanochemical cycloelimination of four-membered rings is not limited to carbocycles alone, granting access to interesting chemical functionalities.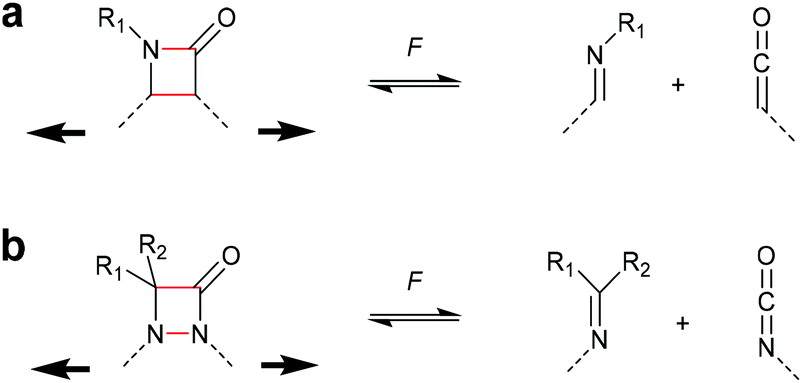 | ||
| Scheme 9 Cycloelimination of (a) β-lactame-based mechanophore into ketene and imine, (b) 1,2-azetidinone-based mechanophore into isocyanate and imine. | ||
Another intriguing [2 + 2] cycloadduct undergoing mechanochemically induced cycloelimination containing heteroatoms is the 1,2-dioxetane. This moiety is particularly interesting as it produces mechanoluminescence and was firstly described by Sijbesma and co-workers (Scheme 10).39 Although no mechanistic evidence was reported yet, the cycloreversion most likely occurs via a diradical intermediate. Prominently, one of the two carbonyl end groups produced after scission remains in the excited state and relaxes by emission of light. This motif was then successfully integrated into a variety of materials for optical stress sensing applications.40–51
 | ||
| Scheme 10 Cycloelimination of a 1,2-dioxetane to the corresponding ketones of which one is in the excited state giving rise to mechanoluminescence. | ||
Applications
 | ||
| Scheme 11 Reversible fluorogenic bicyclic mechanophore based on a cinnamate dimer.53 | ||
 | ||
| Scheme 12 Copolymer of cyclobutane- and gDCC-based mechanophores to achieve a mechanical gating effect.55 | ||
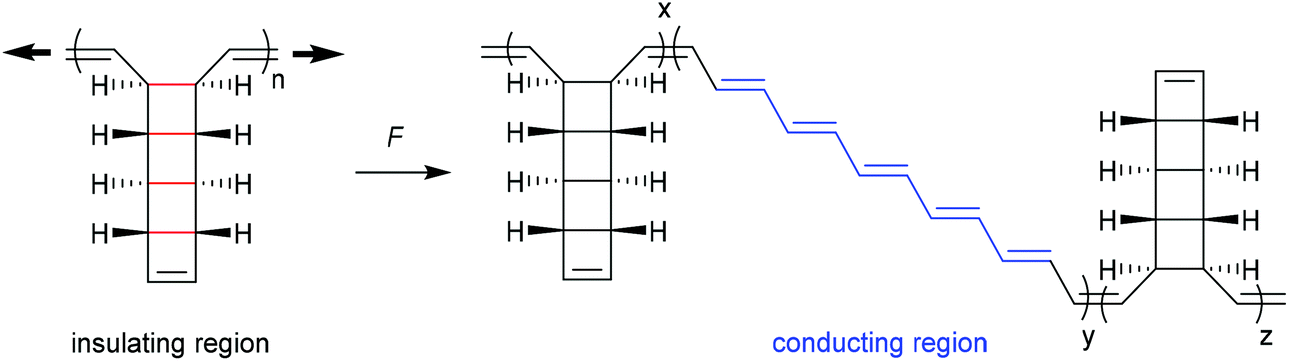 | ||
| Scheme 13 Mechanical unzipping of polyladderenes. | ||
Cycloelimination of [4 + 2] cycloadducts (retro-Diels–Alder reaction)
Scissile activation of [4 + 2] cycloadducts
The formal [4 + 2] cycloelimination or retro-Diels–Alder (rDA) reaction is commonly known as a thermo-reversible reaction in which a [4 + 2] cycloadduct reacts to the corresponding diene and dienophile.62 For polymer mechanochemistry, the rDA was first observed in healable materials.63 As the rDA occurred during crack formation in polymer networks crosslinked with a DA motif, these could be mended at elevated temperatures by the forward DA cycloaddition.64 Two DA motifs studied by Yoshie and co-workers were established in this context: the furan–maleimide (F–M) and the anthracene–maleimide (A–M) cycloadduct (Scheme 14).65,66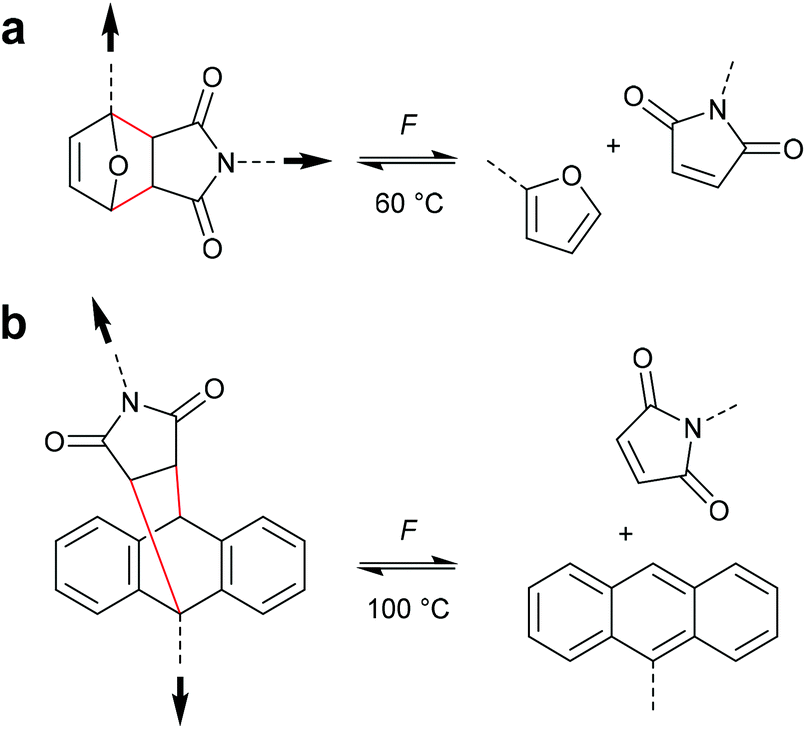 | ||
| Scheme 14 (a) Furan–maleimide and (b) anthracene–maleimide DA adducts and their respective thermoreversible activation by force. | ||
Though it was shown that these healable motifs underwent mechanochemically induced bond scission, their thorough investigation as mechanophores was realized only in subsequent, later works. Up to today, F–M and A–M mechanophores were used in various materials with different properties, including linear and star-shaped polymers,67–70 at heterointerfaces,71–73 hydrophilic and hydrophobic diblock copolymer micelles,74,75 hydrogels,76 and in combination with slide-ring materials.77 The F–M and A–M rDA mechanophore activation was investigated with standard procedures, such as compression of bulk solid state materials78 and ultrasonication of solutions,79 but also by solvent swelling,80 laser-induced stress waves,81 and recently also single-molecule force spectroscopy (SMFS).73
Makarov, Bielawski and co-workers carried out the first systematic investigations on the F–M and A–M cycloadducts as mechanophores in a trendsetting work.82 They used both computational simulations and synthetic experiments to investigate the effect of the substitution pattern of the attached polymer chains. For the F–M cycloadduct, computation suggested that force application in proximal position (Fig. 1a; blue) would mechanochemically accelerate the cycloelimination reaction whereas force application in distal position (Fig. 1a; red) would decelerate mechanochemical reactivity. For the A–M cycloadduct, calculations and experimental data proposed that a substitution in 2-position would completely suppress the mechanochemical cycloelimination (Fig. 1b; red). Confirming this result, no mechanochemical activation was monitored experimentally, whereas the commonly used 9-position remained accessible for the force-induced rDA (Fig. 1b; blue).
 | ||
| Fig. 1 Force-dependent activation energy for the cycloelimination depending on the substitution pattern in DA cycloadducts of (a) F–M and (b) A–M. Adapted with permission from ref. 82. Copyright 2013 American Chemical Society. | ||
Stevenson and De Bo systematically expanded these results for the F–M cycloadducts and additionally differentiated between the endo- and exo-stereoisomers.83 These were investigated with distal and proximal substitution patterns regarding the attachment points of the polymer chains and incorporated within the central region of linear PMA (Fig. 2). Whereas expectedly the endo-stereoisomers thermally cleaved at a higher rate than the exo-variants, the mechanochemical rate was found to depend more on the attachment points of the polymer chains than on the stereochemical identity of the DA adducts. In agreement with the investigations by Makarov and Bielawski discussed above, the proximal isomers reacted at a higher rate than the distal isomers. Importantly, the distal exo-cycloadduct could not be cleaved by force and appeared to be mechanochemically inactive. This was further underlined by computational simulations, in which the distal exo-adduct led to non-selective bond scission.
 | ||
| Fig. 2 Influence of the regio- and stereochemistry in the thermal and mechanochemical cycloelimination of F–M cycloadducts.83 | ||
Notably, Wang and Craig saw room for improvement with regard to the method employed by Stevenson and De Bo to differentiate between endo- and exo-adduct mechanochemical activity.84 By combination of the scissile F–M DA adducts with the non-scissile gDCC (vide infra) motifs, they found that the endo-stereoisomers were mechanochemically more labile than their exo-counterparts.
Otsuka and co-workers combined the F–M DA mechanophore with a photochromic diarylethene (DAE) unit providing a photoregulated lock for mechanochemistry (Scheme 15).85 In the ring-open form DAE the adduct could be cleaved selective through the rDA reaction, but after isomerization to the ring-closed form via UV-irradiation the mechanophore was inert toward elongational forces due to the removal of the crucial double bond from the DA adduct.86–88 In their study they also synthesized and sonicated a control mechanophore with distal geometry without observing any activation. They calculated the required energy and force (Emax = 770 kJ mol−1 and Fmax = 5.19 nN) and confirmed the previously discussed reports regarding the stability of distal isomers.82–84 Notably, Robb and co-workers reported the inverse case, i.e. a mechanoregulated lock for photochemistry, with a cyclopentadiene-bridged DAE in a DA adduct with maleimide.89
 | ||
| Scheme 15 DAE DA mechanophore with a photoregulated lock for mechanochemistry.85 | ||
During their efforts to tailor the optical properties of anthracenes released during cycloelimination of A–M DA cycloadducts, Göstl and co-workers also synthesized the untypical 1,4-cycloadduct of a 9,10-π-extended anthracene (Scheme 16).90,91 Importantly, while polymer substituents coupled the force to the 9- and 10-positions, they were not directly attached to the quaternary carbon atoms participating in the cycloelimination reaction in 1,4-position. This led to the absence of mechanochemical reactivity via the rDA pathway for this derivative highlighting once more the findings of Makarov and Bielawski discussed above. It is likely that the force vector did not align well with the reaction coordinates, yet high thermodynamic stability of the 1,4-cycloadduct could not be excluded since no further investigations on these derivatives were performed.90
 | ||
| Scheme 16 1,4-cycloadduct of 9,10-π-extended anthracene and maleimide resistant to force-induced cycloelimination.90 | ||
Mechanistic considerations
For successful prediction of activation free energies for reactions under force it is necessary to revisit the reaction mechanism. DFT calculations by Makarov, Bielawski and co-workers suggested that the rDA in the absence of force proceeded in a concerted mechanism (Fig. 3a, top).82 Akbulatov and Boulatov compared those computational simulations with other DFT functions (e.g. uMPW1K) and also predicted the existence of an alternative two-step radical dissociation pathway (Fig. 3a, bottom).19 Although the required energy for this path was, compared to the concerted mechanism, considerably higher in a reaction without force, small forces were shown to invert this order (Fig. 3b and c). The stepwise radical mechanism was already dominant at an applied force of ca. 0.1 nN. The concerted mechanism, on the other hand, was disfavoured under force and was abandoned completely at forces of ca. 0.4–0.8 nN, depending on the DFT function used and the substituents attached to the motif. Therefore, dissociation of DA adducts was inhibited at small forces (0.1 nN) but accelerated with increasing force. | ||
| Fig. 3 (a) Concerted (top) and stepwise radical (bottom) mechanism for the dissociation of DA adducts under force. (b) Changes in the free energies of the transition states relative to the reactants as a function of applied force (uMPW1K/6-31+G(d)). (c) A comparison of dissociation barrier calculated by Akbulatov and Boulatov (grey line) with the predicted one by Makarov and co-workers. Adapted from ref. 19 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, copyright 2017. | ||
Flex activation of [4 + 2] cycloadducts
A distinctively alternative way to mechanochemically induce the cycloreversion of [4 + 2] cycloadducts was developed with the so-called ‘flex activation’. In contrast to the activation via the scission of internal bonds that are elongated with the force vector of the attached polymer segments, for flex activation the polymer scaffold is used to distort bond angles releasing small molecules.92 Boydston and co-workers pioneered this method for the release of furan derivatives (Scheme 17).92–94 They converted main chain alkene moieties in oxanorbornadiene DA adducts constructed from furan and dimethyl acetylenedicarboxylate (DMAD) into alkynes in a cycloelimination reaction. Incorporation of the DA adducts as crosslinker into PMA or polyurethane networks followed by compression proved the release of small furan-containing molecules. Calculated potential energies estimate an activation energy of ca. 35 kcal mol−1 for this process. | ||
| Scheme 17 Flex activation via transient bond angle distortion of oxanorbornadiene DA adducts and subsequent cycloelimination without internal polymer bond scission. | ||
Roessler and Zimmerman used the growing string method (GSM) without using the θ value as a constraint coordinate and showed that the reaction mechanism for the flex activation depended on the applied force.95 The activation energy decreased mostly linearly with increasing force and was approximately 24 kcal mol−1 if 4 nN of force were applied relative to the ground state reaction. Transition states calculated with different applied forces showed unique θ values, indicating that the reaction followed a multiparameter pathway and force influenced only one reaction coordinate.
The concept of flex activation was moreover exploited for anthracene DA adducts (Scheme 18). The attachment of polymer chains in 2,6-position of a cycloadduct of anthracene and phenyltriazolinedione, however, only showed 1% mechanochemical cycloelimination in a strained PDMS network at 125 °C (Scheme 18a).96,97 These harsh conditions are not surprising, as triazolinediones are among the most reactive known dienophiles forming cycloadducts with extreme thermodynamic stability. In contrast, no flex activation was observed for a comparable anthracene maleimide cycloadduct (Scheme 18b).78
 | ||
| Scheme 18 Flex activation of anthracene cycloadducts with (a) phenyltriazolinedione and (b) maleimide.78,96,97 | ||
Cycloelimination of [3 + 2] cycloadducts
The debate over 1,2,3-triazoles as mechanophores
One of the most energetically debated motifs regarding its mechanochemical activity is the 1,2,3-triazole undergoing force-induced cycloelimination. The forward reaction is the 1,3-dipolar Huisgen cycloaddition and is counted to the variety of ‘click’ reactions. The scepticism towards this cycloelimination was fuelled by the retraction of the prominent first investigation of a 1,2,3-triazole carrying polymer substituents in 1,4-position claiming to be ‘unclicking the click’ by Bielawski and co-workers (Scheme 19).98,99 | ||
| Scheme 19 Supposed force-induced cycloelimination of the triazole mechanophore yielding azide and alkyne moities.98,99 | ||
In a later study, Bielawski, Makarov and co-workers set out to examine the reliability of their retracted results and firstly discussed the regiochemical effects of this cycloelimination reaction by comparing the mechanochemical scission rate of 1,4-substituted with 1,5-substituted triazoles (Scheme 20).100 There, they found a slightly increased scission rate for the 1,5-substituted triazole derivatives which is a very notable finding hinting towards two distinct scission mechanisms, though the rate difference of ca. 20% was rather small.
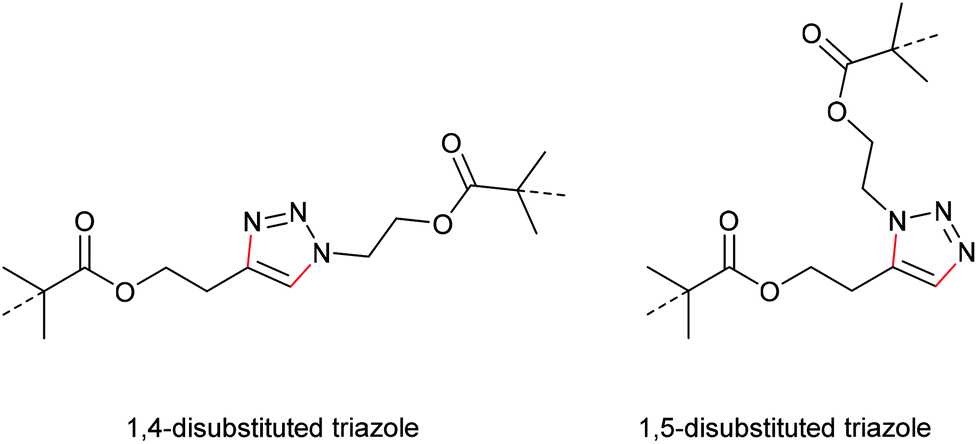 | ||
| Scheme 20 Structures of 1,4- and 1,5-disubstituted triazole mechanophores. | ||
Trying to shine light on this debate, Uggerud and Smalø performed computational simulations in order to mechanistically explain the mechanochemically facilitated cycloreversion of the 1,4-substituted triazoles.101 They found that unspecific chain dissociation, i.e. rupture of the macromolecular backbone, was favoured over the cycloreversion reaction, though the latter was still deemed possible from a theoretical mechanochemical point of view. Hartke, Lüning, Beyer and co-workers even went as far to design and synthesize a 1,4-disubstituted triazole and bridge it with a second cycle turning this previously scissile mechanophore into a non-scissile motif that can be analysed by SMFS employing an atomic force microscope (AFM) (Scheme 21).102 Upon stretching the covalently anchored molecule (between two PEG chains, attached to Si3N4) a characteristic length increase was to be measured in the case of rupture along the triazole branch. While the authors clearly observed and measured a threshold cleaving force on this motif of 1–2 nN, it remained unclear whether these events were indeed attributable to cycloelimination reactions or unspecific chain scission.
 | ||
| Scheme 21 Supposed mechanochemical cycloelimination of the non-scissile bridged 1,4-disubstituteted triazole mechanophore in AFM measurements.102 | ||
Another work raising doubts regarding the mechanochemical activity of the 1,4-disubstituted triazoles was published by Xia and co-workers who prepared amphiphilic block copolymer micelles connecting the blocks with the triazole motif.103 Upon irradiation with high-intensity focused ultrasound (HIFU), they also found that chain scission preferentially occured on the incorporated ester bonds within their polymers and not on the triazole moiety. However, the effects of HIFU on mechanophores are only scarcely investigated and hence comparability of these measurements is limited. Yet, Dreuw and Stauch principally confirmed these results by computational simulation.104 They deemed the cycloreversion of 1,4-substituted triazoles impossible if forces are conducted to aliphatic linkers since the entire mechanochemically robust triazole ring would need to be activated and a rupture event in the aliphatic chain would be energetically much more favourable. However, cycloelimination of 1,5-substituted-triazoles should be possible because of the alignment of the scissile bond with the force vector, although a competition with the rupture of the C–N bond at the pulling point should be observed in stretching experiments due to similar force and energy rupture requirements of these bonds.
Eventually, Marx and co-workers dismissed, also on a computational level, the mechanochemically induced cycloreversion of 1,4-substituted triazoles by CuI-catalysis – analogously to the forward reaction and according to the principle of microscopic reversibility.105 Destructive pathways were favourable when the desired activation free energies were compared. In contrast, a Ru-assisted mechanochemical unclicking of the 1,5-regioisomer could lower the activation free energy for the rate-determining step drastically rendering the reaction feasible.
Blank and co-workers revived the triazoles as mechanophores undergoing cycloelimination in a new context when they investigated the cycloadducts from substituted azides with molecularly strained cyclooctyne derivatives being an important in vitro and in vivo ligation motif in the realm of the biological sciences.106 Employing computational simulations, they compared various 1,4- and 1,5-substituted triazoles and confirmed the previous experiments from Bielawski, Makarov and co-workers100 stating that 1,5-substituted triazoles were more suitable for a force-induced cycloelimination reaction. It was found that 1,5-substituted triazoles opened via an unzipping mechanism in comparison to a shear mechanism in the case of their 1,4-substituted counterparts. Using these cyclooctyne-azide cycloadducts in single molecule force experiments using AFM, Fang, Shahbazian-Yassar and co-workers designed an on-surface reaction cascade that ligated cyclooctyne generated by the mechanochemically induced cycloelimination with gold nanoparticles (AuNPs) to confirm the local character of their experiments.107
The force-induced cycloelimination of 1,4-disubstituted triazoles on unstrained alkynes appears to be an unlikely reaction. In simulations and experiments, the required forces to cleave the triazole ring in a shear mechanism were too high and led to undesired rupture events in the adjacent polymer chains. Conversely, 1,5-substituted triazoles and triazoles with strained cyclooctyne were found to be suitable motifs.
Cycloelimination of [4 + 4] cycloadducts
Anthracene dimer
Very analogously to the mechanophores discussed above, the polymer mechanochemical cycloelimination of [4 + 4] cycloadducts was first observed in the context of photohealable materials. For this purpose, anthracene moieties undergoing a photoreversible [4 + 4] cycloaddition were used.108,109 Dimerization and dissociation of the anthracene and its respective cycloadduct both are accessible by irradiation with light of different wavelengths (Scheme 22). Such scaffolds and their capability of reversible dimerization were studied by Landfester and co-workers, who successfully integrated them into a dendritic, photohealable material.109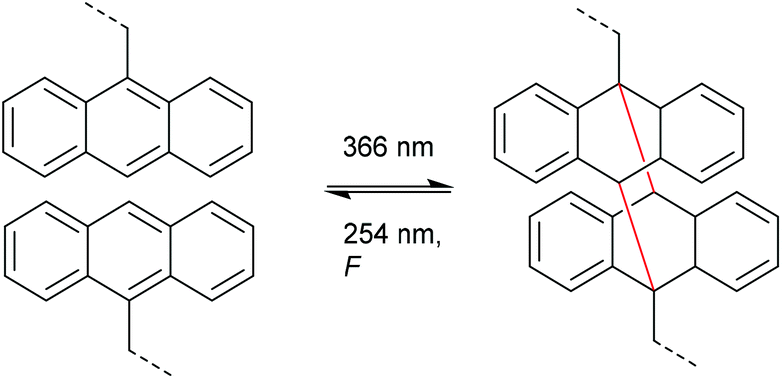 | ||
| Scheme 22 Photoinduced [4 + 4] anthracene dimerization and photo- as well as mechanochemical cycloelimination. | ||
Within healable materials, induced damage is generally of mechanical nature and hence it was reasonable to assume that the corresponding cycloelimination was also accessible via the application of force.110–117 Chung and co-workers investigated a polymer network bearing the anthracene dimer as crosslinker.110 Upon grinding the polymer network, fluorescence emission stemming from the formation of the individual anthracene moieties was recorded thus confirming the successful cycloelimination (Scheme 22). This optical response was exploited as crack sensor with comparably high molar absorptivity and fluorescence almost in the visible region of the spectrum.
The cycloelimination of anthracene dimer induced by mechanical force applied through pressure was also studied more in detail from a kinetic and mechanistic point of view by Chronister, Bardeen and co-workers.111 Macroscopically, an increase of pressure resulted in a dramatic acceleration of the dissociation. This is counterintuitive as dissociative reactions usually exhibit positive activation volumes. These results suggested that pressure acted as catalyst rendering this a purely kinetic effect due to a less voluminous transition state. It was proposed that pressure stabilized the transition state by flattening the anthracene rings, followed by rupture and dissociation – a concept comparable to the ‘flex activation’ discussed above.
Mechanistic considerations
Though the cycloelimination of [4 + 4] cycloadducts was shown to be promoted by mechanical force, no evidence about the mechanistic pathway it follows were presented yet. Computational simulations suggested that the diradical intermediate was energetically favoured compared to the concerted dissociation pathway.118 A radical mechanism would be in agreement with previous reports concerning the mechanochemical cycloelimination of [2 + 2] and [4 + 2] cycloadducts. However, for [4 + 4] cycloadducts no trapping experiments or spectroscopic analyses were performed that could confirm the existence of a metastable diradical intermediate.Yet, a fundamental theoretical study that aimed to elucidate the fairly high rate of the mechanochemical anthracene dimer cycloelimination was performed on the basis of a comparison with the cyclobutane mechanophore.119 Within the anthracene dimer two cyclooctane rings share two covalent bonds connecting the anthracene moieties. Hence, a comparative study between the cyclobutane and a virtual cyclooctane mechanophore were performed. While the cyclobutane showed a ring strain of ca. 26 kcal mol−1 and a BDE of ca. 72 kcal mol−1, cyclooctane reasonably exhibited a lower ring strain of ca. 10 kcal mol−1 and a BDE of ca. 160 kcal mol−1. The ring strain was established as inversely proportional to the BDE and simple cyclooctane was hence shown not to undergo any scission by mechanical force. However, within the anthracene dimer the BDE drastically decreased to 59 kcal mol−1 – even lower than the BDE of cyclobutane. This suggested a high ring strain within the anthracene dimer motif and was invoked to explain the high mechanical sensitivity of this motif.
2-Electron electrocyclic ring-opening reactions
Gem-dihalocyclopropanes (gDHCs)
An electrocyclic reaction is a pericyclic rearrangement in which one π bond is converted to one σ bond or vice versa following either a ring-opening or ring-closing mechanism, respectively. The first proof of a mechanochemically induced 2-electron ring-opening reaction was presented by Craig and co-workers.120 They incorporated multiple gDCCs within a backbone of Z-poly(butadiene) (PB) by reacting the PB with NaOH in CHCl3 under phase-transfer conditions resulting in dichlorocarbene addition. These gDCC polymers underwent mechanochemical ring-opening in conjunction with chlorine migration several hundred times within a single polymer chain, providing a ‘mechanochemical map’ of the stress distributions within linear polymer chains during ultrasonication in solution (Fig. 4). | ||
| Fig. 4 Ultrasonication of gDCC-PB copolymer leads to ring-opened 2,3-dichloroalkenes (○) with simultaneously decreasing Mn (■). Adapted with permission from ref. 120. Copyright 2009 American Chemical Society. | ||
The approach of modifying a PB backbone was consequently extended to other gDHCs.121–128 Notably, Craig and co-workers found by ozonolysis of mechanochemically formed double bonds that those regions of the polymer that did and those that did not undergo force-induced ring-opening were continuous, indicating a smooth and monotonic force reduction from the central chain section towards the termini.125 Moreover, compressive force could be used to perform the ring-opening reaction, but tensile load resulted in no measurable activation, even when applied to the point of failure of the material.124 This was attributed to the minimum domain size for successful mechanochemical activation, which was found to be larger than a single monomer and hence heterogeneous stress distribution within the employed polymer framework needed to be taken into account. In addition, a versatile method for the abundant incorporation of cyclic mechanophores into high molar mass block copolymers with excellent long-term stability was developed.129,130 This allowed access to ABA-type block structures with the mechanophore-rich region in the chain centre allowing high mechanochemical turnover.
Mechanistic considerations
 | ||
| Scheme 23 (a) gDFC E-isomer and (b) Z-isomer thermally ring-open and -close through a disrotatory process via 1,3-diradical transition state. (c) Force leads to the formation and trapping of the 1,3-diradical transition state, which ring-closes to the gDFC Z-isomer once force is removed.123 | ||
Investigating the thermally symmetry-forbidden conrotatory ring-closing after force trapping the diradical transition state, Martinez, Craig and co-workers prepared purely Z-form gDFC-substituted PB (Scheme 24).132 With this, they showed that while mechanochemical activation led to ca. 95% conventional and symmetry-allowed disrotatory ring-closing, ca. 5% conrotatory ring-closing towards the anti-WH product could be observed. This result was confirmed by ab initio molecular dynamics simulations.
 | ||
| Scheme 24 Ring-closing of a force trapped diradical transition state leading i.a. to 5% anti-WH products.132 | ||
In order to quantify the extent of WH rule violation by mechanochemical ring-opening and -closing of gDHCs, SMFS was utilized. In one of the approaches, SMFS of three mechanically induced reactions along both their symmetry-allowed and symmetry-forbidden pathways was performed by Martinez, Craig and co-workers.134 It was found that the symmetry-forbidden ring-opening reactions of benzocyclobutene (BCB, vide infra), gDFC and gDCC required approximately 130 pN less, 560 pN more and 1000 pN more force, respectively, than their corresponding symmetry-allowed counterparts. These results provided the first experimental quantitative benchmarks for mechanically induced anti-WH reactions, while the BCB ring-opening was the first demonstration of a force-coupled symmetry-forbidden reaction that was faster than its force-coupled symmetry-allowed counterpart.
In another work, single-molecule polymer mechanochemistry was utilized to induce the disrotatory outward ring-opening of a Z-dialkyl substituted syn-chloro-gem-chlorofluorocyclopropane (syn-Cl-gCFC).135 The forces required to trigger the anti-WH pathway were ca. 200 pN higher than those involved in the WH compliant process (1290 vs. 1500 pN). The kinetics were complemented by tension trapping experiments, which suggested that the reaction proceeded by the previously established diradical mechanism. Moreover, mechanisms for thermal and mechanical activation of dimethyl-gCFC were proposed (Scheme 25). The methyl groups thermally underwent inward disrotation, during which the electrons of the breaking C–C bond were donated to the antibonding orbital of the C–Cl bond to assist the scission of the C–Cl bond subsequently forming the E-alkene product and following the WH rules. Mechanically pulling on the methyl groups forced the rotation outwards, although this disfavoured the cleavage of the Cl-anion, violating the WH rules. During the mechanically activated ring-opening reaction, syn-Cl-gCFC first formed a diradical, which then underwent ion separation to form the Z-alkene product.
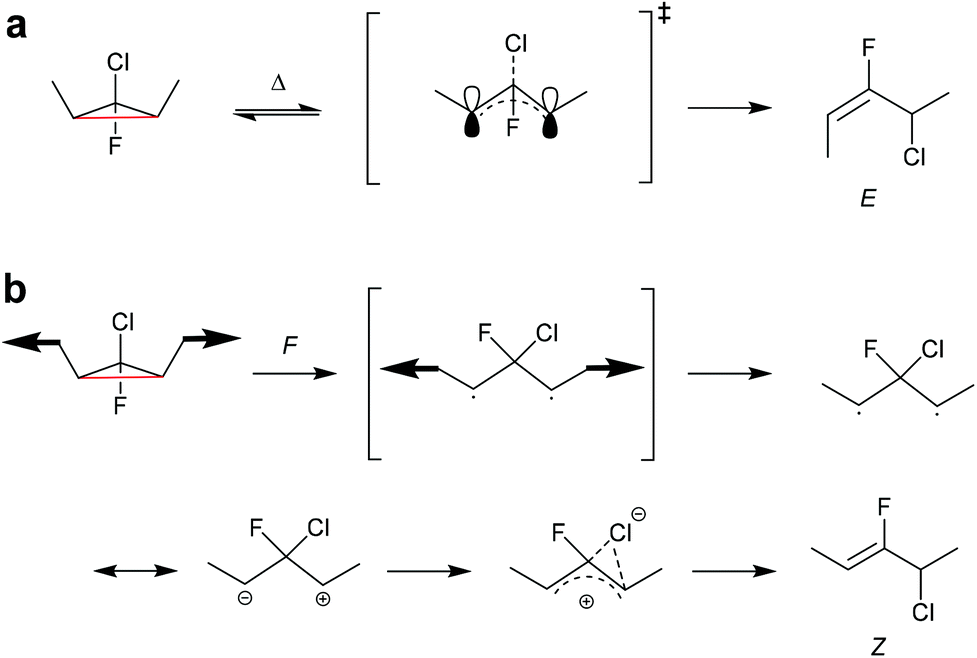 | ||
| Scheme 25 Mechanisms for (a) thermal and (b) mechanochemical activation of dimethyl-gCFC.135 | ||
SMFS was also applied to observe an irreversible extension of a gem-dibromocyclopropane (gDBC)-functionalized PB under tension.122 The extension of close to 28% in contour length of the polymer backbone occurred at roughly 1.2 nN and was attributed to the force-induced isomerization of the gDBCs into 2,3-dibromoalkenes (Fig. 5). This rearrangement is valuable for localized stress relief in polymers and polymer networks under load.
 | ||
| Fig. 5 SMFS indicating the irreversible extension of a gDBC-functionalized PB under tension. Reproduced with permission from ref. 122. Copyright 2010 American Chemical Society. | ||
SMFS investigations further revealed that the force-induced acceleration of the electrocyclic ring opening of gDCC was sensitive to the stereochemistry of an α-alkene substituent on the gDCC.41 The force required to open the E-alkene-substituted gDCC was found to be 0.4 nN lower than that required in the corresponding Z-alkene isomer, despite the effectively identical force-free behaviour of the two isomers as well as the distance between the stereochemical permutation and the scissile bond of the mechanophore. This was termed ‘lever arm effect’ by Craig and co-workers owed to the greater mechanical advantage for a given applied force that could be gained.126,136
Moreover, the mechanochemical activation of gDFC Z-isomer in toluene was investigated by SMFS.137 Unlike previous reports in methyl benzoate (MB),134 two transitions were observed in the force-extension curves of Z-gDFC polymers in toluene. The first and previously observed transition occurred at ∼1300 pN, but a second transition was observed at ∼1800 pN that revealed the partial formation of the E-gDFC isomer (Fig. 6). This was attributed to competing reactions of the Z-gDFC at the 1300 pN plateau: addition of oxygen to a ring-opened diradicaloid intermediate and isomerization of Z-gDFC to its E isomer.
 | ||
| Fig. 6 (a) Representative SMFS force curves of Z-gDFC polymer obtained in MB (blue) and toluene (red). (b) The mechanistic scheme for Z-gDFC when the SMFS experiment is performed in toluene. Adapted with permission from ref. 137. Copyright 2016 American Chemical Society. | ||
Based on computational simulations, Marx and co-workers also confirmed the deviation from the intuitive set of WH rules when biasing the potential energy surfaces with mechanical force instead of heat or light. They examined the mechanochemical reactivity and force-induced stereochemistry of gDCCs based on ab initio simulations.139 By this, they unveiled the mechanisms of force-induced ring-opening of gDCC Z-isomer versus E-isomer, rationalizing the puzzling experimental findings described above.120 Notably, they discovered an unprecedented complex mechano-stereochemical feature, whereby the ring-opening of E-isomers of 2,3-disubstituted gDCCs led to two different diastereomers, with the probabilities of obtaining them depending of the force exerted on the system.
In another study, Marx and co-workers indicated that both the electron-density-based ‘atoms in molecules’ (AIM) analysis and the complementary Lewis-structure-based ‘natural resonance theory’ (NRT) decomposition supported the finding that the electronic structures, underlying the dis- and conrotatory ring-opening mechanisms along the mechanochemically distorted reaction pathways, were very similar to those of the respective (WH-allowed and WH-forbidden) thermal pathways.140 Concomitantly, they claimed that the WH rules, which were based on electronic structure considerations of certain classes of thermally and photochemically activated reactions, should not be considered to be transferable to the same reactions once mechanochemically activated. Instead, the WH rules must be complemented by yet to be formulated ‘mechanochemical rules’.
They further showed the impact of halogen substitution on the thermally activated ring-opening process followed by halogen migration of (2S,3S)-1,1-dihalo-2,3-dimethylcyclopropanes in the case of F, Cl, Br and I distribution.141 Surprisingly, the direction of halogen migration in the case of disrotatory ring-opening was qualitatively different for the difluorinated cyclopropane compared with the homologous Cl, Br and I species. For gDFC E-isomer, the decision was governed by an uphill bifurcation followed by two separate transition states for each migration direction, whereas the disrotatory transition state was first surmounted in the other three cases followed by a downhill bifurcation into the disrotatory products. Thus, chemical substitution was found to qualitatively change the potential energy landscape for the ring-opening of cyclopropanes.
In another attempt, which was based on quantum mechanochemical computations at constant external forces, they discovered that the topology of the energy landscape for mechanochemically activated ring-opening of gDFC E-isomer was qualitatively different in the high-force regime compared with low-forces down to the thermal limit. They showed that both uphill and downhill bifurcations being pre- and post-transition state features, respectively, were shifted continuously by applying external mechanical forces as typically generated by ultrasonication. These findings depicted an approach towards steering the selectivity of chemical reactions in favour of the desired products by mechanochemical activation.
Applications
In further efforts, Craig and co-workers investigated the relative mechanical strengths of polymers bearing comparatively weak scissile covalent bonds.144 Multiple scissile bonds and non-scissile mechanophores were embedded along polymer backbones and stretched by ultrasonication. Thereupon, the non-scissile mechanophores were activated beginning from the centre of the chain radiating outwards until a scissile bond cleaved. The ratio of non-scissile mechanophore activation to bond scission thus provided a measure of the mechanical strength of the weak scissile bond. This methodology was lately used to estimate the mechanical strength of other weak bonds including ferrocene and ruthenocene.145,146
 | ||
| Scheme 26 HCl release from gDCC-substituted indene mechanoacid generator. | ||
4-Electron electrocyclic ring-opening reactions
Benzocyclobutene
Chronologically, one of the first reported mechanophores studied in detail was the benzocyclobutene moiety, which is able to undergo a 4-electron electrocyclic ring-opening reaction upon application of mechanical force (Scheme 27). Up to today, several works were published underlining the general feasibility of this motif as mechanophore.11,149–151 However, the first benzocyclobutene-based mechanophore was designed and synthetized by Moore, White, Sottos and co-workers in 2007.152 This first experimental work did not include any mechanistic study, but rather focussed on the validation of benzocyclobutene mechanochemical activity in solution when integrated in the centre of a linear polymer chain. Benzocyclobutene is non-scissile and hence comparative molar mass analyses were not useful to prove the mechanochemical activity of this motif. However, a maleimide-pyrene conjugate undergoing a DA reaction with the mechanochemically generated diene structure allowed post-functionalization of the force-activated moiety with a chromophore and hence quantitative evaluation. This confirmed the successful mechanochemical activation of this motif and gave rise to further and more detailed studies on this scaffold, especially concerning the stereochemical outcomes in relation to the WH rules. | ||
| Scheme 27 Mechanochemically induced 4-electron electrocyclic ring-opening reaction of benzocyclobutene. | ||
Mechanistic considerations
As for all pericyclic reactions, the WH rules describe the thermal and photochemical 4-electron electrocyclic ring-opening reactions allowing the prediction of the stereochemical outcome based on the configurations of the reactants.15 For ring-opening or ring-closing, the concerted mechanism can follow a conrotatory or a disrotatory pathway, depending on the symmetry of the frontier orbitals involved in the process. The distinction is straightforward and based on the relative position of the orbital phases that can overlap in a constructive manner. Thermal 4-electron electrocyclic reactions follow a conrotatory pathway while photochemically a disrotatory mechanism is favoured. The stereochemical identity of the reaction products is hence a consequence of the symmetry of the involved orbitals and is easy to predict adhering to Scheme 28. | ||
| Scheme 28 Thermally and photochemically allowed pathways of cyclobutene ring-opening according to the respective reactant configurations and the WH rules. | ||
With regard to the WH rules, mechanistic studies on the mechanochemically induced ring-opening of benzocyclobutene were carried out focusing on the configuration of the involved stereoisomers.134,136,152–155 Notably, stereoisomers that were symmetry-forbidden by the WH rules, were accessible by force. For example, both syn- and anti-cyclobutene isomers yield the E,E-diene mechanochemically (Scheme 29). It was argued that elongation of the polymer termini in opposite directions produces a transition state geometry in which the two substituents are as far as possible away from each other, i.e. the E,E-diene. Hence, while employing an anti-reactant a conrotatory pathway in agreement with the WH rules for thermal reactions was followed, employing a syn-reactant a disrotatory pathway conforming to the WH rules for photochemical reactions entailed.15
 | ||
| Scheme 29 Stereochemistry of mechanochemically induced 4-electron electrocyclic ring-opening of benzocyclobutene. | ||
Although no experimental evidence about the concerted (or diradical) character of the underlying ring-opening mechanism exists, computational simulations found that the force-coupled transition state energy levels inverted compared to the thermal pathway and that the disrotatory ring-opening becomes more accessible with the application of force.156
Notably, Craig and co-workers found that structural modification enhanced the mechanochemical activity of the benzocyclobutene mechanophore. They observed that substitution of the motif with an α-alkene lowered the force required for the ring-opening process.136 This is in analogy to the ‘lever arm effect’ discussed above with regard to gDHCs.41,126
Mainly two different computational simulation techniques were used with regard to the mechanical activation of benzocyclobutene: AISMD (ab initio steered molecular dynamics) and CoGEF (constrained geometry to simulate external force).16,156–162 Both examples aimed to characterize the force-modified PES for the ring-opening pathway. Martinez and co-workers were the first to rationalize experimental observations using AISMD simulations.156 They determined the highest energy difference between the force-free and the force-dependent PES for the conrotatory pathway when ring-opening from the anti-isomer and for the disrotatory pathway when ring-opening from the syn-isomer. This results in acceleration of the ring-opening towards the E,E-product from both the syn- and anti-isomers when mechanical force is applied. In addition, Marx and co-workers exploited CoGEF simulations to gain insight into transmission mechanisms of directional forces to the benzocyclobutene scaffold and its associated ring-opening.157 They suggested that a stepwise mechanism involving a diradical intermediate could be excluded and a concerted mechanism would be the most plausible for both syn- and anti-isomers.16,158
6-Electron electrocyclic ring-opening reactions
Spiropyran to merocyanine isomerization
The 6-electron electrocyclic ring-opening of spiropyran (SP) to merocyanine (MC) via temperature, light or pH is among the most studied ‘chromic’ transformations in the literature.163,164 Mechanochromic behaviour of individual small SP molecules was first shown by Tipikin in grinding experiments.165 Based on these initial findings, Moore and co-workers incorporated SP into the centre of linear PMA chains and investigated the polymer mechanochemical activation to MC in ultrasonication experiments (Scheme 30).150 The ring-opening was monitored via UV-Vis spectroscopy and showed absorption changes from a colourless to a pink solution. The activation is reversible by exposure to ambient light or heat. Since then, SP was used as the most successful mechanophore with regard to publication numbers in a variety of materials to investigate the force-related properties of, e.g., polyacrylates,7,150,166–173 polymethacrylates,174,175,176–183,184 polyurethanes,185–193 polydimethylsiloxane (PDMS),194–202 polystyrene,203 polycaprolacton,204,205 polyolefins,206 polycarbonate,207 polyarylenes208 and different copolymers.209–216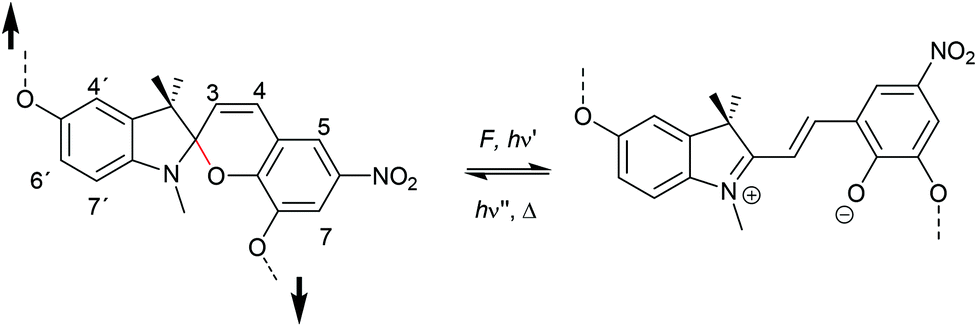 | ||
| Scheme 30 Mechanochemical isomerization of spiropyran to merocyanine. | ||
Regiochemical effects on mechanochemical SP activation were investigated by varying substitution patterns. Based on these patterns, it was possible to distinguish between several SP variants. For mechanochemical activation, it was found that it was critical to attach the polymer chains on opposing sides of the spiro C–O junction.166 If both polymer chain attachment points were located on the indole side, the SP could not be activated by force, yet still functioned as photoswitch (Scheme 31).203 Three widely researched mechanochemically active SPs were described in the literature: in SP1 the attachment points are located at the 5′ position of the indole side and position 8 of the benzopyran side.150,166 In SP2 the polymer chains are attached via a functionalisation of the indole nitrogen and the 8 position of the benzopyran.204 SP3 substitutes the nitro group of the benzopyran side with a polymer chain attachment point.217 SP1 and SP2 were shown to be force- and light-responsive while the absence of the electron withdrawing nitro group was proven to inhibit photoreactivity in SP3. Recovery time after ring-opening for SP3 was quite short (∼30 s) compared to longer thermal ring-closing periods in the cases of SP1 and SP2 (∼40 min).170
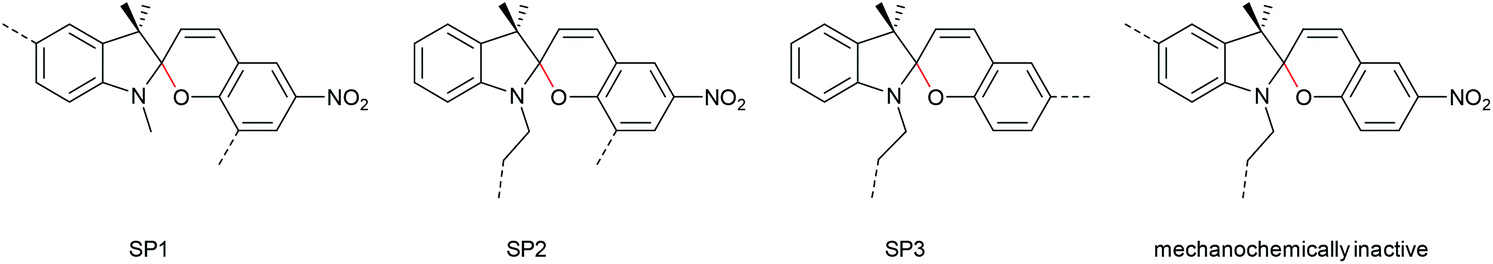 | ||
| Scheme 31 Mechanochemically active and inactive SP derivatives depending on polymer chain substitution patterns. | ||
Craig and co-workers investigated the mechanochemical reaction rate of SP1 and SP2 to their respective MCs by AFM SMFS.218 For this, macrocycles incorporating SP were copolymerized with 9-oxabicyclo[6.1.0]non-4-ene (epoxy-COD) and single molecule force-extension curves were recorded through approach/withdraw cycles at varying velocities. These show the characteristic conversion of SP to MC at ∼260 pN for SP1 and ∼240 pN for SP2. In addition, computational simulations revealed that SP1 undergoes isomerization to MC easier than SP2 in absence of force, but under mechanochemical activation under SMFS conditions the force threshold of SP1 is on average 10% higher (Fig. 7).
 | ||
| Fig. 7 Representative single molecular force–extension curves of macrocycles incorporating SP1 or SP2 copolymerized with epoxy-COD. Adapted with permission from ref. 218. Copyright 2015 American Chemical Society. | ||
Based on this work, Sottos, White, Moore and co-workers investigated the mechanical activation of SP1 and SP2 as crosslinkers in bulk PDMS under mechanical load with the help of computational calculations and in situ fluorescence measurements.219 They could not find a significant difference for the stress or strain required for the activation of the two regioisomeres in the polymer matrix (Fig. 8). Moreover, isomerization rates for SP to MC conversion and vice versa were nearly identical under macroscopic tensile stress. It was hence concluded that the difference in the pulling geometry between SP1 and SP2 had only a negligible influence on the mechanochemical activation in bulk polymeric materials because of the intrinsic complexity of these macroscopic systems.
 | ||
| Fig. 8 Activation behaviour of SP1 or SP2 incorporated in PDMS under tension: comparison of (a) the normalized fluorescence intensity as a function of strain at a strain rate of 0.05 s−1; (b) the onset of activation strain; (c) the activation stress.219 Reprinted with permission from ref. 219. Copyright 2018, American Chemical Society. | ||
Qiao and co-workers systematically investigated the geometric and electronic effects of SPs under compression and tension and determined that geometry dominated the mechanochromic responses in dependence of varying attachment points over electronic effects.220 For this, they synthesized a novel SP mechanophore (SP4) with a combination of all attachment points of SP1 and SP2 (Scheme 32). In conjunction with SP1 and SP2, SP4 was incorporated into PDMS and the colour change in mechanical tests was investigated. They monitored a notable variation in the RGB intensities of the three SP after activation to MC by force followed by relaxation back to the SP form. With the help of DFT calculations they contributed these colour changes towards different MC isomers and hypothesized different path for MC isomers of SP1 and SP2 under force and relaxation (Scheme 33). SP1 reacts and recovers directly to the Z,Z,Z-isomer with a short interatomic distance whereas SP2 reacts to the Z,Z,E-isomer, which relaxes to the Z,Z,Z-isomer, followed by recovery to SP.
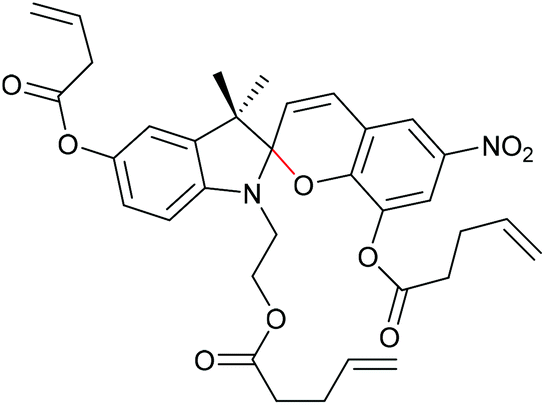 | ||
| Scheme 32 SP4 crosslinker combining the polymer attachment points of SP1 and SP2.220 | ||
 | ||
| Scheme 33 Hypothesized (a) SP1 and (b) SP2 activation and relaxation pathways by Qiao and co-workers.220 | ||
Although SP4 is electronically more related to SP1 because of the slightly electron donating vinyl ester, SP4 behaves comparable to SP2 showing a hypsochromic shift during and after mechanical loading, which is most likely contributed to by the attachment point in the 1′ N-position.
Roessler and Zimmerman previously used GSM computations to locate reaction paths and products for SP1.95 In their study, they identified two merocyanine products, the Z- and E-isomer (Scheme 34). At forces <2 nN the Z-product was found, but at higher forces the ring-open structure isomerized to the E-structure without an energy barrier. With increased applied force the activation barrier for the ring opening decreased roughly linearly (slope of −5.9 kcal mol−1 nN−1) as Z- and E-products had similar transition states (TS) and the Z–E-isomerisation took place after a rate limiting ring-opening TS.
 | ||
| Scheme 34 Z- and E-isomers of merocyanine for the force induced SP1 reaction, according to computational simulations by Roessler and Zimmermann. | ||
Craig and co-workers investigated the effect of the polymer attachment point on the benzopyran-ring for three SP regioisomers.221 SP derivatives with one attachment point at the indoline fragment and the other one in ortho- (SP(o)), meta- (SP(m)) or para- (SP(p)) position of the spirocyclic C–O bond were incorporated in PDMS films and their colour change was studied under tensile load. RGB images were analysed and the intensity in blue and green channels (B/G ratio) was used to evaluate the mechanophore activity. The extrapolated strain onset for detectable activation of all three regioisomeres occurred at ca. 90% uniaxial strain in bulk PDMS. At higher strains (90–135%) the relative activation of SP(o)/SP(m)/SP(p) was estimated through the normalized increase in B/G value as 3.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6:1, which was consistent with the expected relative response of the isolated isomers (Fig. 9). Although their results suggested that the mechanochemical activation was determined foremost by the network structure or was too small to detect, they discussed that in case of individual mechanophores the onset activation should follow the same trend (SP(o) > SP(m) > SP(p)), because electronic effects are modest and therefore the geometric effect should dominate.
:1.6:1, which was consistent with the expected relative response of the isolated isomers (Fig. 9). Although their results suggested that the mechanochemical activation was determined foremost by the network structure or was too small to detect, they discussed that in case of individual mechanophores the onset activation should follow the same trend (SP(o) > SP(m) > SP(p)), because electronic effects are modest and therefore the geometric effect should dominate.
 | ||
| Fig. 9 (a) Spiropyran regioisomers with force handles in ortho-, meta- and para-position towards the spiro C–O bond. (b) Alteration of B/G intensity. (c) Alteration of B/G intensity normalized to the value at 135% strain.221 Adapted with permission from ref. 221 Copyright 2018 American Chemical Society. | ||
The effect of substituents with different electronegativity in the para-position to the spirocyclic C–O bond in SP2 was recently studied employing SMFS.222 SP2 derivatives with para H, Br or NO2 were subjected to force and the change in contour length was measured (Fig. 10). Each SP derivative showed a different critical force for the conversion of SP to MC dependent on the electronegativity of the substituent. Lower electronegativity of the substituent translated to a higher necessary transition force f*: H (410 pN) > Br (360 pN) > NO2 (240 pN) because a largely heterolytic and polar transition state in the mechanochemical reaction was stabilized by electron withdrawing groups in the para C–O position.
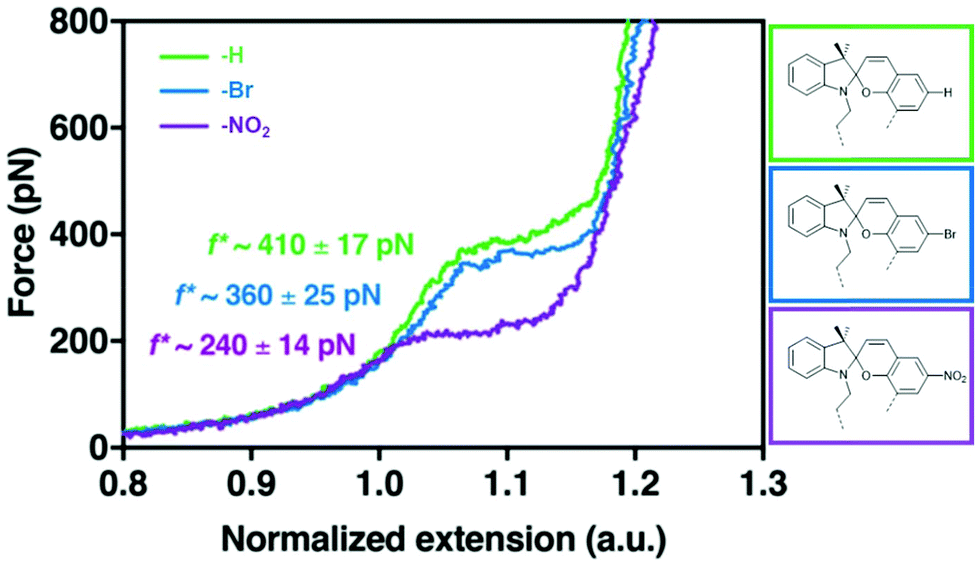 | ||
| Fig. 10 SMFS curves for SP2 derivatives with para-H, -Br or -NO2 and their transition force f*.222 Adapted with permission from ref. 222. Copyright 2018 American Chemical Society. | ||
Spirothiopyran to thiomerocyanine isomerization
The sulphur derivative to SP spirothiopyran (STP) was also shown to undergo thermo- and photoactivated isomerization to thiomerocyanine (TMC), which is reversible by visible light.223,224 Boulatov and co-workers studied the mechanochemistry of STP.225 They incorporated multiple STP moieties into the backbone of polyester and polyurethane and showed the STP activation via sonication or irradiation at 365 nm (Scheme 35). The light-yellow polymer solution turned green and a new absorption band at 635 nm indicated the isomerization to TMC. | ||
| Scheme 35 Mechanochemical isomerization of spirothiopyran to thiomerocyanine. | ||
Naphthopyran to merocyanine isomerization
Analogously to SP, naphthopyran (NP) mechanophores react to MC via a 6-electron electrocyclic ring-opening reaction.48,226–228 Concomitantly to SP, the polymer attachment point for the mechanochemical activation of NP is crucial. Moore and co-workers investigated three different NP derivatives with varying polymer attachment points and determined that only the attachment of polymer chains to the 5-positon led to mechanochemical activity (Fig. 11).226 Regioisomers with substituents in 8- and 9-position were mechanochemically inactive because of disadvantageous alignment of the C–O pyran bond with the direction of the externally applied force. | ||
| Fig. 11 6-Electron electrocyclic ring-opening of NP under mechanical force. (a) Reaction scheme and (b) DFT calculations for three NP regioisomers. Cleavage of C–O pyran bond is only predicted for the NP with polymer attachment at the 5-position.226 Adapted with permission from ref. 226. Copyright 2016 American Chemical Society. | ||
Rhodamine
The activation of rhodamine (Rh) derivatives is another well-researched electrocyclic ring-opening reaction in which the non-fluorescent spirolactam opens to the strongly fluorescent amide.229 Mechanochromic behavior of single organic molecule Rh was first shown in shearing and grinding experiments.230,231 Based on these results, Jia and co-workers synthesized a Rh-mechanophore, incorporated it into polyurethane (PU) films and sensitivity of the material towards mechanical force and UV light was shown (Scheme 36).232 The ring opening reaction from the twisted spirolactam towards the planarized zwitterion was monitored via UV-Vis and fluorescence spectroscopy and showed a distinct color change from dark blue to reddish, as well as a new fluorescent band at 550 nm. Similar to SP, the mechanophore activation was reversible at ambient temperature or by heating. In following studies the pH responsiveness of Rh mechanophores was demonstrated.233 An acidic pH activates the mechanophore, whereas basic pH promotes the cyclization reaction. | ||
| Scheme 36 Mechanochemical isomerization of rhodamine to the planarized zwitterionic amide.232 | ||
Bai and co-workers synthesized a three armed Rh-mechanophore crosslinker for the incorporation in elastomeric networks (Scheme 37, left).234 In their experiments, they showed a yellow fluorescence after the release of force. Theoretical calculations revealed that a large dipole moment in the zwitterionic Rh leads to the formation of a bent geometry with a C–C–O bond angle of 134°. Upon breaking of the C–N bond in the twisted spirolactam this C–C bond was bent and the conformation of the zwitterion was planarized to facilitate the delocalization of the π electrons in the phenyl ring to that of the xanthene resulting in a bathochromic shift. After relaxation, the zwitterion immediately changed back to the bent geometry resulting in a hypsochromic shift.
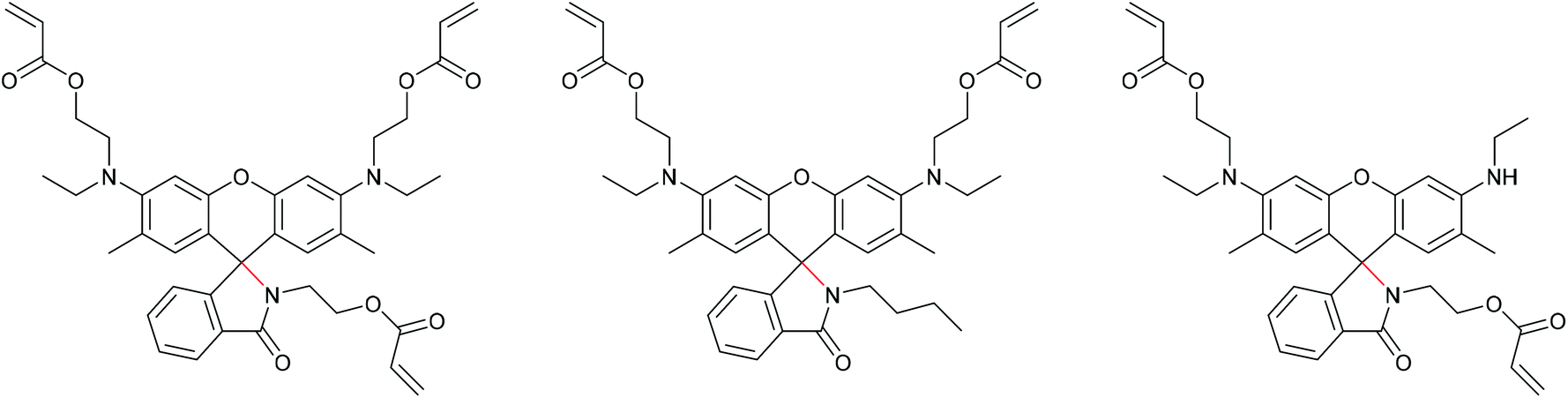 | ||
| Scheme 37 Three-armed Rh-mechanophore (left) and control Rh-mechanophores (middle and right). | ||
In this work, they further introduced a control Rh mechanophore with both polymer attachment points located on the xanthenes which is mechanochemically inactive because force cannot be transferred across the spiro C–O bond (Scheme 37, middle). Another control two-armed Rh mechanophore with attachment points across the C–O junction (Scheme 37, right) showed expected activation but the bathochromic shift and the activated fraction were lower compared to the three armed mechanophore.234,235
Conclusions and outlook
Dissociative pericyclic reactions have proven themselves excellent candidates for their force-activation in the context of polymer mechanochemistry. While plenty of uncertainties about the underlying mechanisms and design rationales remain to be resolved, some overarching conclusions can be drawn from the existing body of literature:1. Polymer mechanochemical pericyclic reactions mostly do not follow the established concerted mechanisms their small-molecular counterparts do. In many cases it was shown, both experimentally and by computational simulations, that diradical or rarely ionic transition states dominate the reaction pathway. While it is arguable whether these reactions thus formally can be considered pericyclic, discussing them in line with their heat- and light-induced counterparts is reasonable as they offer access to reaction products that are unobtainable according to the Woodward–Hoffmann rules.
2. As force is a directional vector quantity, the attachment points, identity and size of the substituted polymer chains transmitting the force to the molecular motif are of paramount importance. Empirically, polymer mechanochemical pericyclic reactions require the polymer chains to be attached in the vicinity or at the position of the atoms that are directly involved in the bond rearrangement. More generally expressed, this means that the force vector must be aligned with the reaction coordinates.
3. Care must be taken when investigating new motifs, as the debate over the 1,2,3-triazoles as mechanophores impressively demonstrates. Inferring mechanochemical rates from GPC elugrams, which can rarely differentiate selective from non-selective scission, cannot be considered sufficient proof for a successful polymer mechanochemical pericyclic reaction. The reaction products need to be identified unequivocally or transition states derived indirectly by trapping. This holds particularly true for non-scissile motifs where GPC analyses become progressively impractical.
While we thus begin to understand the polymer mechanochemistry of pericyclic reactions and can retrospectively rationalize their outcomes, we still lack a powerful and intuitive set of rules to predict the behaviour of novel motifs within a confined set of reaction conditions. Although theoretical frameworks for examining force-dependent reaction mechanisms exist, their application and development needs to be furthered alongside their force-free counterparts. Next to the realization of exciting new applications and functions using force in materials, this will be a major challenge and obligation for researchers involved in this exhilarating field in the future.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
R. G., D. C. and C. B. are grateful for support by a Freigeist-Fellowship of the Volkswagen Foundation (92888). E. I.-N. acknowledges financial support from the European Commission (EUSMI, 731019).References
- S. L. Craig, Nat. Chem., 2017, 9, 1154–1155 CrossRef CAS PubMed.
- W. Kauzman and H. Eyring, J. Am. Chem. Soc., 1940, 62, 3113–3125 CrossRef.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- G. Kaupp, CrystEngComm, 2009, 11, 388–403 RSC.
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- B. A. Beiermann, S. L. B. Kramer, J. S. Moore, S. R. White and N. R. Sottos, ACS Macro Lett., 2012, 1, 163–166 CrossRef CAS.
- H. Yan, F. Yang, D. Pan, Y. Lin, J. N. Hohman, D. Solis-Ibarra, F. H. Li, J. E. P. Dahl, R. M. K. Carlson, B. A. Tkachenko, A. A. Fokin, P. R. Schreiner, G. Galli, W. L. Mao, Z.-X. Shen and N. A. Melosh, Nature, 2018, 554, 505–510 CrossRef CAS PubMed.
- R. Groote, R. T. M. Jakobs and R. P. Sijbesma, Polym. Chem., 2013, 4, 4846–4859 RSC.
- G. S. Kochhar, A. Bailey and N. J. Mosey, Angew. Chem., Int. Ed., 2010, 49, 7452–7455 CrossRef CAS PubMed.
- C. L. Brown and S. L. Craig, Chem. Sci., 2015, 6, 2158–2165 RSC.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- T. J. Kucharski and R. Boulatov, J. Mater. Chem., 2011, 21, 8237–8255 RSC.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- R. Hoffmann and R. B. Woodward, Acc. Chem. Res., 1968, 1, 17–22 CrossRef CAS.
- J. Ribas-Arino, M. Shiga and D. Marx, Angew. Chem., Int. Ed., 2009, 48, 4190–4193 CrossRef CAS PubMed.
- N. De Souza, Nat. Methods, 2012, 9, 873–877 CrossRef CAS PubMed.
- M. Dal Molin, Q. Verolet, S. Soleimanpour and S. Matile, Chem. – Eur. J., 2015, 21, 6012–6021 CrossRef CAS PubMed.
- S. Akbulatov and R. Boulatov, ChemPhysChem, 2017, 18, 1418 CrossRef CAS.
- S. Garcia-Manyes and A. E. M. Beedle, Nat. Rev. Chem., 2017, 1, 2397–3358 Search PubMed.
- G. De Bo, Chem. Sci., 2017, 9, 15–21 RSC.
- N. Willis-Fox, E. Rognin, T. A. Aljohani and R. Daly, Chem, 2018, 4, 2499–2537 CAS.
- M. Stratigaki and R. Göstl, ChemPlusChem, 2020 DOI:10.1002/cplu.201900737.
- M. J. Kryger, M. T. Ong, S. A. Odom, N. R. Sottos, S. R. White, T. J. Martinez and J. S. Moore, J. Am. Chem. Soc., 2010, 132, 4558–4559 CrossRef CAS PubMed.
- Z. S. Kean, A. L. Black Ramirez, Y. Yan and S. L. Craig, J. Am. Chem. Soc., 2012, 134, 12939–12942 CrossRef CAS PubMed.
- Z. S. Kean, G. R. Gossweiler, T. B. Kouznetsova, G. B. Hewage and S. L. Craig, Chem. Commun., 2015, 51, 9157–9160 RSC.
- C. Chung, Y. Roh, S. Cho and J. Kim, Chem. Mater., 2004, 16, 3982–3984 CrossRef CAS.
- S. Y. Cho, J. G. Kim and C. M. Chung, Sens. Actuators, B, 2008, 134, 822–825 CrossRef CAS.
- S.-Y. Cho, J.-G. Kim, S.-Y. Oh and C.-M. Chung, Macromol. Res., 2010, 18, 212–214 CrossRef CAS.
- H. M. Klukovich, Z. S. Kean, S. T. Iacono and S. L. Craig, J. Am. Chem. Soc., 2011, 133, 17882–17888 CrossRef CAS PubMed.
- B. Koo, A. Chattopadhyay and L. Dai, Comput. Mater. Sci., 2016, 120, 135–141 CrossRef CAS.
- M. F. Pill, K. Holz, N. Preußke, F. Berger, H. Clausen-Schaumann, U. Lüning and M. K. Beyer, Chem. – Eur. J., 2016, 22, 12034–12039 CrossRef CAS PubMed.
- Z. S. Kean, Z. Niu, G. B. Hewage, A. L. Rheingold and S. L. Craig, J. Am. Chem. Soc., 2013, 135, 13598–13604 CrossRef CAS PubMed.
- M. J. Kryger, A. M. Munaretto and J. S. Moore, J. Am. Chem. Soc., 2011, 133, 18992–18998 CrossRef CAS PubMed.
- R. Groote, B. M. Szyja, F. A. Leibfarth, C. J. Hawker, N. L. Doltsinis and R. P. Sijbesma, Macromolecules, 2014, 47, 1187–1192 CrossRef CAS.
- M. J. Robb and J. S. Moore, J. Am. Chem. Soc., 2015, 137, 10946–10949 CrossRef CAS PubMed.
- D. Marx and M. I. Menéndez, J. Org. Chem., 2018, 83, 2438–2441 CrossRef CAS PubMed.
- Y. Lin, C. C. Chang and S. L. Craig, Org. Chem. Front., 2019, 6, 1052–1057 RSC.
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559–562 CrossRef CAS PubMed.
- J. M. Clough and R. P. Sijbesma, ChemPhysChem, 2014, 15, 3565–3571 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. S. Kean, L. Fan, B. D. Mar, T. J. Martinez and S. L. Craig, J. Am. Chem. Soc., 2014, 136, 15162–15165 CrossRef CAS PubMed.
- Y. Chen and R. P. Sijbesma, Macromolecules, 2014, 47, 3797–3805 CrossRef CAS.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- J. M. Clough, A. Balan, T. L. J. van Daal and R. P. Sijbesma, Angew. Chem., Int. Ed., 2016, 55, 1445–1449 CrossRef CAS PubMed.
- J. M. Clough, J. van der Gucht and R. P. Sijbesma, Macromolecules, 2017, 50, 2043–2053 CrossRef CAS PubMed.
- V. N. Khiêm and M. Itskov, Int. J. Plast., 2018, 102, 1–15 CrossRef.
- W. Yuan, Y. Yuan, F. Yang, M. Wu and Y. Chen, Macromolecules, 2018, 51, 9019–9026 CrossRef CAS.
- G. Kim, V. M. Lau, A. J. Halmes, M. L. Oelze, J. S. Moore and K. C. Li, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10214–10222 CrossRef CAS PubMed.
- C. Yan, F. Yang, M. Wu, Y. Yuan, F. Chen and Y. Chen, Macromolecules, 2019, 52, 9376–9382 CrossRef CAS.
- Y. Yuan, M. Li, W. Yuan, F. Yang and Y. Chen, Macromol. Mater. Eng., 2019, 304, 1900056 CrossRef.
- Y. Yuan, W. Chen, Z. Ma, Y. Deng, Y. Chen, Y. Chen and W. Hu, Chem. Sci., 2019, 10, 2206–2211 RSC.
- E. M. Nofen, N. Zimmer, A. Dasgupta, R. Gunckel, B. Koo, A. Chattopadhyay and L. L. Dai, Polym. Chem., 2016, 7, 7249–7259 RSC.
- H. Zhang, X. Li, Y. Lin, F. Gao, Z. Tang, P. Su, W. Zhang, Y. Xu, W. Weng and R. Boulatov, Nat. Commun., 2017, 8, 1147 CrossRef PubMed.
- M. Li, H. Zhang, F. Gao, Z. Tang, D. Zeng, Y. Pan, P. Su, Y. Ruan, Y. Xu and W. Weng, Polym. Chem., 2019, 10, 905–910 RSC.
- J. Wang, T. B. Kouznetsova, R. Boulatov and S. L. Craig, Nat. Commun., 2016, 7, 13433 CrossRef PubMed.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS PubMed.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS PubMed.
- J. K. Su, J. D. Feist, J. Yang, J. A. M. Mercer, J. A. H. Romaniuk, Z. Chen, L. Cegelski, N. Z. Burns and Y. Xia, J. Am. Chem. Soc., 2018, 140, 12388–12391 CrossRef CAS PubMed.
- Z. Chen, X. Zhu, J. Yang, J. A. M. Mercer, N. Z. Burns, T. J. Martinez and Y. Xia, Nat. Chem., 2020 DOI:10.1038/s41557-019-0396-5.
- B. H. Bowser, C. H. Ho and S. L. Craig, Macromolecules, 2019, 52, 9032–9038 CrossRef CAS.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 2105–2109 CrossRef CAS PubMed.
- B. Rickborn, in Organic Reactions, ed. L. A. Paquette, John Wiley & Sons, Inc., New York, 1987, vol. 52, pp. 1–393 Search PubMed.
- L. P. Engle and K. B. Wagener, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1993, C33, 239–257 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- N. Yoshie, M. Watanabe, H. Araki and K. Ishida, Polym. Degrad. Stab., 2010, 95, 826–829 CrossRef CAS.
- N. Yoshie, S. Saito and N. Oya, Polymer, 2011, 52, 6074–6079 CrossRef CAS.
- D. C. Church, G. I. Peterson and A. J. Boydston, ACS Macro Lett., 2014, 3, 648–651 CrossRef CAS.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- G. I. Peterson, J. Lee and T. Choi, Macromolecules, 2019, 52, 9561–9568 CrossRef CAS.
- Y. Fang, J. Li, X. Du, Z. Du, X. Cheng and H. Wang, Polymer, 2018, 158, 166–175 CrossRef CAS.
- J. Li, B. Hu, K. Yang, B. Zhao and J. S. Moore, ACS Macro Lett., 2016, 5, 819–822 CrossRef CAS.
- J. Li, T. Shiraki, B. Hu, R. A. E. Wright, B. Zhao and J. S. Moore, J. Am. Chem. Soc., 2014, 136, 15925–15928 CrossRef CAS PubMed.
- A. R. Sulkanen, J. Sung, M. J. Robb, J. S. Moore, N. R. Sottos and G. Y. Liu, J. Am. Chem. Soc., 2019, 141, 4080–4085 CrossRef CAS PubMed.
- H. Li, R. Göstl, M. Delgove, J. Sweeck, Q. Zhang, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2016, 5, 995–998 CrossRef CAS.
- H. Li, Y. Zhang, Y. Liu, R. P. Sijbesma, J. P. A. Heuts and Q. Zhang, Polym. Chem., 2017, 8, 3971–3976 RSC.
- M. Stratigaki, C. Baumann, L. C. A. van Breemen, J. P. A. Heuts, R. P. Sijbesma and R. Göstl, Polym. Chem., 2020, 11, 358–366 RSC.
- M. Zhang and G. De Bo, J. Am. Chem. Soc., 2018, 140, 12724–12727 CrossRef CAS PubMed.
- C. P. Kabb, C. S. O'Bryan, C. D. Morley, T. E. Angelini and B. S. Sumerlin, Chem. Sci., 2019, 10, 7702–7708 RSC.
- H. Y. Duan, Y. X. Wang, L. J. Wang, Y. Q. Min, X. H. Zhang and B. Y. Du, Macromolecules, 2017, 50, 1353–1361 CrossRef CAS.
- B. Lyu, W. Cha, T. Mao, Y. Wu, H. Qian, Y. Zhou, X. Chen, S. Zhang, L. Liu, G. Yang, Z. Lu, Q. Zhu and H. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 6254–6259 CrossRef CAS PubMed.
- J. Sung, M. J. Robb, S. R. White, J. S. Moore and N. R. Sottos, J. Am. Chem. Soc., 2018, 140, 5000–5003 CrossRef CAS PubMed.
- S. S. M. Konda, J. N. Brantley, B. T. Varghese, K. M. Wiggins, C. W. Bielawski and D. E. Makarov, J. Am. Chem. Soc., 2013, 135, 12722–12729 CrossRef CAS PubMed.
- R. Stevenson and G. De Bo, J. Am. Chem. Soc., 2017, 139, 16768–16771 CrossRef CAS PubMed.
- Z. Wang and S. L. Craig, Chem. Commun., 2019, 55, 12263–12266 RSC.
- J. Kida, K. Imato, R. Goseki, D. Aoki, M. Morimoto and H. Otsuka, Nat. Commun., 2018, 9, 3504 CrossRef PubMed.
- R. Göstl and S. Hecht, Angew. Chem., Int. Ed., 2014, 53, 8784–8787 CrossRef PubMed.
- R. Göstl and S. Hecht, Chem. – Eur. J., 2015, 21, 4422–4427 CrossRef PubMed.
- A. Fuhrmann, R. Göstl, R. Wendt, J. Kötteritzsch, M. D. Hager, U. S. Schubert, K. Brademann-Jock, A. F. Thünemann, U. Nöchel, M. Behl and S. Hecht, Nat. Commun., 2016, 7, 13623 CrossRef PubMed.
- X. Hu, M. E. McFadden, R. W. Barber and M. J. Robb, J. Am. Chem. Soc., 2018, 140, 14073–14077 CrossRef CAS PubMed.
- R. Göstl and R. P. Sijbesma, Chem. Sci., 2016, 7, 370–375 RSC.
- D. Yildiz, C. Baumann, A. Mikosch, A. J. C. Kuehne, A. Herrmann and R. Göstl, Angew. Chem., Int. Ed., 2019, 58, 12919–12923 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189–8192 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2014, 136, 1276–1279 CrossRef CAS PubMed.
- B. Cao, N. Boechler and A. J. Boydston, Polymer, 2018, 152, 4–8 CrossRef CAS.
- R. Roessler and A. Zimmerman, J. Phys. Chem. C, 2018, 122, 6996–7004 CrossRef.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS.
- N. R. Sottos, Nat. Chem., 2014, 6, 381–383 CrossRef CAS PubMed.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Science, 2011, 333, 1606–1609 CrossRef CAS PubMed.
- M. McNutt, Editorial retraction, Science, 2015, 347, 834 CrossRef CAS PubMed.
- J. N. Brantley, S. S. M. Konda, D. E. Makarov and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 9882–9885 CrossRef CAS PubMed.
- H. S. Smalø and E. Uggerud, Chem. Commun., 2012, 48, 10443–10445 RSC.
- D. Schütze, K. Holz, J. Müller, M. K. Beyer, U. Lüning and B. Hartke, Angew. Chem., Int. Ed., 2015, 54, 2556–2559 CrossRef PubMed.
- F. Li, C. Xie, Z. Cheng and H. Xia, Ultrason. Sonochem., 2016, 30, 9–17 CrossRef CAS PubMed.
- T. Stauch and A. Dreuw, Chem. Sci., 2017, 8, 5567–5575 RSC.
- M. Krupička, P. Dopieralski and D. Marx, Angew. Chem., Int. Ed., 2017, 56, 7745–7749 CrossRef PubMed.
- M. J. Jacobs, G. Schneider and K. G. Blank, Angew. Chem., Int. Ed., 2016, 55, 2899–2902 CrossRef CAS PubMed.
- A. Khanal, F. Long, B. Cao, R. Shahbazian-Yassar and S. Fang, Chem. – Eur. J., 2016, 22, 9760–9767 CrossRef CAS PubMed.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468–473 CrossRef CAS PubMed.
- Y. K. Song, K. H. Lee, W. S. Hong, S. Y. Cho, H. C. Yu and C. M. Chung, J. Mater. Chem., 2012, 22, 1380–1386 RSC.
- S. R. Jezowski, L. Zhu, Y. Wang, A. P. Rice, G. W. Scott, C. J. Bardeen and E. L. Chronister, J. Am. Chem. Soc., 2012, 134, 7459–7466 CrossRef CAS PubMed.
- F. Tong, C. D. Cruz, S. R. Jezowski, X. Zhou, L. Zhu, R. O. Al-Kaysi, E. L. Chronister and C. J. Bardeen, J. Phys. Chem. A, 2014, 118, 5349–5354 CrossRef CAS PubMed.
- E. M. Nofen, J. Wickham, B. Koo, A. Chattopadhyay and L. L. Dai, Mater. Res. Express, 2016, 3, 035701 CrossRef.
- E. M. Nofen, A. Dasgupta, N. Zimmer, R. Gunckel, B. Koo, A. Chattopadhyay and L. L. Dai, Polym. Eng. Sci., 2017, 57, 901–909 CrossRef CAS.
- L. Y. Hsu, S. Maity, Y. Matsunaga, Y. F. Hsu, Y. H. Liu, S. M. Peng, T. Shinmyozu and J. S. Yang, Chem. Sci., 2018, 9, 8990–9001 RSC.
- B. Koo, J. Miller, R. Gunckel, A. Hall, L. Dai and A. Chattopadhyay, Smart Mater. Struct., 2019, 28, 115035 CrossRef.
- L. Kan, H. Cheng, B. Li, X. Zhang, Q. Wang, H. Wei and N. Ma, New J. Chem., 2019, 43, 2658–2664 RSC.
- T. Nishiuchi, S. Y. Uno, Y. Hirao and T. Kubo, J. Org. Chem., 2016, 81, 2106–2112 CrossRef CAS PubMed.
- B. Koo, E. Nofen, A. Chattopadhyay and L. Dai, Comput. Mater. Sci., 2017, 133, 167–174 CrossRef CAS.
- J. M. Lenhardt, A. L. Black and S. L. Craig, J. Am. Chem. Soc., 2009, 131, 10818–10819 CrossRef CAS PubMed.
- J. M. Lenhardt, M. T. Ong, R. Choe, C. R. Evenhuis, T. J. Martinez and S. L. Craig, Science, 2010, 329, 1057–1060 CrossRef CAS PubMed.
- D. Wu, J. M. Lenhardt, A. L. Black, B. B. Akhremitchev and S. L. Craig, J. Am. Chem. Soc., 2010, 132, 15936–15938 CrossRef CAS PubMed.
- J. M. Lenhardt, J. W. Ogle, M. T. Ong, R. Choe, T. J. Martinez and S. L. Craig, J. Am. Chem. Soc., 2011, 133, 3222–3225 CrossRef CAS PubMed.
- J. M. Lenhardt, A. L. Black, B. A. Beiermann, B. D. Steinberg, F. Rahman, T. Samborski, J. Elsakr, J. S. Moore, N. R. Sottos and S. L. Craig, J. Mater. Chem., 2011, 21, 8454–8459 RSC.
- A. L. Black Ramirez, J. W. Ogle, A. L. Schmitt, J. M. Lenhardt, M. P. Cashion, M. K. Mahanthappa and S. L. Craig, ACS Macro Lett., 2012, 1, 23–27 CrossRef CAS.
- H. M. Klukovich, T. B. Kouznetsova, Z. S. Kean, J. M. Lenhardt and S. L. Craig, Nat. Chem., 2013, 5, 110–114 CrossRef CAS PubMed.
- A. L. B. Ramirez, Z. S. Kean, J. A. Orlicki, M. Champhekar, S. M. Elsakr, W. E. Krause and S. L. Craig, Nat. Chem., 2013, 5, 757–761 CrossRef CAS PubMed.
- A. L. Black Ramirez, A. K. Schmitt, M. K. Mahanthappa and S. L. Craig, Faraday Discuss., 2014, 170, 337–344 RSC.
- Z. S. Kean, A. L. B. Ramirez and S. L. Craig, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3481–3484 CrossRef CAS PubMed.
- B. Lee, Z. Niu and S. L. Craig, Angew. Chem., Int. Ed., 2016, 55, 13086–13089 CrossRef CAS PubMed.
- R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395–397 CrossRef CAS.
- J. Wang, M. T. Ong, T. B. Kouznetsova, J. M. Lenhardt, T. J. Martinez and S. L. Craig, J. Org. Chem., 2015, 80, 11773–11778 CrossRef CAS PubMed.
- K. C. Neuman and A. Nagy, Nat. Methods, 2008, 5, 491–505 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. Niu, M. T. Ong, H. M. Klukovich, A. L. Rheingold, T. J. Martinez and S. L. Craig, Nat. Chem., 2015, 7, 323–327 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 11554–11557 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. Niu, A. L. Rheingold and S. L. Craig, J. Org. Chem., 2015, 80, 11895–11898 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2016, 138, 10410–10412 CrossRef CAS PubMed.
- J. Ribas-Arino and D. Marx, Chem. Rev., 2012, 112, 5412–5487 CrossRef CAS PubMed.
- P. Dopieralski, J. Ribas-Arino and D. Marx, Angew. Chem., Int. Ed., 2011, 50, 7105–7108 CrossRef CAS PubMed.
- M. Wollenhaupt, M. Krupička and D. Marx, ChemPhysChem, 2015, 16, 1593–1597 CrossRef CAS PubMed.
- M. Wollenhaupt, C. Schran, M. Krupička and D. Marx, ChemPhysChem, 2018, 19, 837–847 CrossRef CAS PubMed.
- A. L. Black, J. A. Orlicki and S. L. Craig, J. Mater. Chem., 2011, 21, 8460–8465 RSC.
- J. M. Lenhardt, A. L. Black Ramirez, B. Lee, T. B. Kouznetsova and S. L. Craig, Macromolecules, 2015, 48, 6396–6403 CrossRef CAS.
- B. Lee, Z. Niu, J. Wang, C. Slebodnick and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 10826–10832 CrossRef CAS PubMed.
- Y. Sha, Y. Zhang, E. Xu, Z. Wang, T. Zhu, S. L. Craig and C. Tang, ACS Macro Lett., 2018, 7, 1174–1179 CrossRef CAS PubMed.
- Y. Sha, Y. Zhang, E. Xu, C. W. McAlister, T. Zhu, S. L. Craig and C. Tang, Chem. Sci., 2019, 10, 4959–4965 RSC.
- C. E. Diesendruck, B. D. Steinberg, N. Sugai, M. N. Silberstein, N. R. Sottos, S. R. White, P. V. Braun and J. S. Moore, J. Am. Chem. Soc., 2012, 134, 12446–12449 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 99–103 CrossRef CAS PubMed.
- G. Cravotto and P. Cintas, Angew. Chem., Int. Ed., 2007, 46, 5476–5478 CrossRef CAS PubMed.
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS PubMed.
- J. Wang, I. Piskun and S. L. Craig, ACS Macro Lett., 2015, 4, 834–837 CrossRef CAS.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- I. Frank and J. Friedrichs, Nat. Chem., 2009, 1, 264–265 CrossRef CAS PubMed.
- K. M. Wiggins, J. N. Brantley and C. W. Bielawski, ACS Macro Lett., 2012, 1, 623–626 CrossRef CAS.
- T. B. Kouznetsova, J. Wang and S. L. Craig, ChemPhysChem, 2017, 18, 1486–1489 CrossRef CAS PubMed.
- M. T. Ong, J. Leiding, H. Tao, A. M. Virshup and T. J. Martinez, J. Am. Chem. Soc., 2009, 131, 6377–6379 CrossRef CAS PubMed.
- J. Ribas-Arino, M. Shiga and D. Marx, Chem. – Eur. J., 2009, 15, 13331–13335 CrossRef CAS PubMed.
- J. Ribas-Arino, M. Shiga and D. Marx, J. Am. Chem. Soc., 2010, 132, 10609–10614 CrossRef CAS PubMed.
- S. S. M. Konda, J. N. Brantley, C. W. Bielawski and D. E. Makarov, J. Chem. Phys., 2011, 135, 164103 CrossRef PubMed.
- P. Dopieralski, P. Anjukandi, M. Rückert, M. Shiga, J. Ribas-Arino and D. Marx, J. Mater. Chem., 2011, 21, 8309–8316 RSC.
- Y. Tian and R. Boulatov, Chem. Commun., 2013, 49, 4187–4189 RSC.
- Y. Tian and R. Boulatov, ChemPhysChem, 2012, 13, 2277–2281 CrossRef CAS PubMed.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- D. S. Tipikin, Russ. J. Phys. Chem. A, 2001, 75, 1720–1722 Search PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- B. A. Beiermann, S. L. B. Kramer, P. A. May, J. S. Moore, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2014, 24, 1529–1537 CrossRef CAS.
- M. N. Silberstein, L. D. Cremar, B. A. Beiermann, S. B. Kramer, T. J. Martinez, S. R. White and N. R. Sottos, J. Mech. Phys. Solids, 2014, 63, 141–153 CrossRef CAS.
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177–180 CrossRef CAS.
- M. Li, Q. Zhang and S. Zhu, Polymer, 2016, 99, 521–528 CrossRef CAS.
- M. Li, L. Lei, Q. Zhang and S. Zhu, Macromol. Rapid Commun., 2016, 37, 957–962 CrossRef CAS PubMed.
- W. Qiu, P. A. Gurr and G. G. Qiao, ACS Appl. Mater. Interfaces, 2019, 11, 29268–29275 CrossRef CAS PubMed.
- Y. Zhang, B. Ren, F. Yang, Y. Cai, H. Chen, T. Wang, Z. Feng, J. Tang, J. Xu and J. Zheng, J. Mater. Chem. C, 2018, 6, 11536–11551 RSC.
- B. A. Beiermann, D. A. Davis, S. L. B. Kramer, J. S. Moore, N. R. Sottos and S. R. White, J. Mater. Chem., 2011, 21, 8443–8447 RSC.
- C. M. Kingsbury, P. A. May, D. A. Davis, S. R. White, J. S. Moore and N. R. Sottos, J. Mater. Chem., 2011, 21, 8381–8388 RSC.
- C. M. Degen, P. A. May, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2013, 46, 8917–8921 CrossRef CAS.
- M. N. Silberstein, K. Min, L. D. Cremar, C. M. Degen, T. J. Martinez, N. R. Aluru, S. R. White and N. R. Sottos, J. Appl. Phys., 2013, 114, 023504 CrossRef.
- J. R. Hemmer, P. D. Smith, M. Van Horn, S. Alnemrat, B. P. Mason, J. R. De Alaniz, S. Osswald and J. P. Hooper, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1347–1356 CrossRef CAS.
- C. K. Lee, C. E. Diesendruck, E. Lu, A. N. Pickett, P. A. May, J. S. Moore and P. V. Braun, Macromolecules, 2014, 47, 2690–2694 CrossRef CAS.
- A. D. N. Celestine, B. A. Beiermann, P. A. May, J. S. Moore, N. R. Sottos and S. R. White, Polymer, 2014, 55, 4164–4171 CrossRef CAS.
- J. W. Kim, Y. Jung, G. W. Coates and M. N. Silberstein, Macromolecules, 2015, 48, 1335–1342 CrossRef CAS.
- G. Kister, M. Moniruzzaman, M. Khan and S. Debnath, Dyes Pigm., 2019, 162, 309–314 CrossRef CAS.
- J. Y. Jo, H. G. Jang, Y. C. Jung, D. C. Lee and J. Kim, ACS Macro Lett., 2019, 8, 882–887 CrossRef CAS.
- A. D. N. Celestine, N. R. Sottos and S. R. White, Strain, 2019, 55, 1–17 CrossRef.
- S. R. White, C. K. Lee, N. R. Sottos, D. A. Davis, P. V. Braun and J. S. Moore, J. Am. Chem. Soc., 2010, 132, 16107–16111 CrossRef PubMed.
- C. K. Lee, B. A. Beiermann, M. N. Silberstein, J. Wang, J. S. Moore, N. R. Sottos and P. V. Braun, Macromolecules, 2013, 46, 3746–3752 CrossRef CAS.
- T. A. Kim, B. A. Beiermann, S. R. White and N. R. Sottos, ACS Macro Lett., 2016, 5, 1312–1316 CrossRef CAS.
- G. Hong, H. Zhang, Y. Lin, Y. Chen, Y. Xu, W. Weng and H. Xia, Macromolecules, 2013, 46, 8649–8656 CrossRef CAS.
- X. Fang, H. Zhang, Y. Chen, Y. Lin, Y. Xu and W. Weng, Macromolecules, 2013, 46, 6566–6574 CrossRef CAS.
- H. Zhang, Y. Chen, Y. Lin, X. Fang, Y. Xu, Y. Ruan and W. Weng, Macromolecules, 2014, 47, 6783–6790 CrossRef CAS.
- Y. Chen, H. Zhang, X. Fang, Y. Lin, Y. Xu and W. Weng, ACS Macro Lett., 2014, 3, 141–145 CrossRef CAS.
- Q. Zhang, Y. Wang, C. Xing, Y. Cai, K. Xi and X. Jia, RSC Adv., 2017, 7, 12682–12689 RSC.
- C. Xing, L. Wang, L. Xian, Y. Wang, L. Zhang, K. Xi, Q. Zhang and X. Jia, Macromol. Chem. Phys., 2018, 219, 1800042 CrossRef.
- G. R. Gossweiler, C. L. Brown, G. B. Hewage, E. Sapiro-Gheiler, W. J. Trautman, G. W. Welshofer and S. L. Craig, ACS Appl. Mater. Interfaces, 2015, 7, 22431–22435 CrossRef CAS PubMed.
- S. Cho, S. Kang, A. Pandya, R. Shanker, Z. Khan, Y. Lee, J. Park, S. L. Craig and H. Ko, ACS Nano, 2017, 11, 4346–4357 CrossRef CAS PubMed.
- C. L. Brown, M. H. Barbee, J. H. Ko, H. D. Maynard and S. L. Craig, J. Chem. Educ., 2017, 94, 1752–1755 CrossRef CAS.
- R. C. Rohde, A. Basu, L. B. Okello, M. H. Barbee, Y. Zhang, O. D. Velev, A. Nelson and S. L. Craig, Polym. Chem., 2019, 10, 5985–5991 RSC.
- J. Park, Y. Lee, M. H. Barbee, S. Cho, S. Cho, R. Shanker, J. Kim, J. Myoung, M. P. Kim, C. Baig, S. L. Craig and H. Ko, Adv. Mater., 2019, 31, 1–10 Search PubMed.
- Z. Xia, V. D. Alphonse, D. B. Trigg, T. P. Harrigan, J. M. Paulson, Q. T. Luong, E. P. Lloyd, M. H. Barbee and S. L. Craig, Molecules, 2019, 24, 542 CrossRef PubMed.
- M. H. Barbee, K. Mondal, J. Z. Deng, V. Bharambe, T. V. Neumann, J. J. Adams, N. Boechler, M. D. Dickey and S. L. Craig, ACS Appl. Mater. Interfaces, 2018, 10, 29918–29924 CrossRef CAS PubMed.
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, Nat. Commun., 2014, 5, 4899 CrossRef PubMed.
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, J. Mech. Phys. Solids, 2015, 82, 320–344 CrossRef CAS.
- M. E. Grady, B. A. Beiermann, J. S. Moore and N. R. Sottos, ACS Appl. Mater. Interfaces, 2014, 6, 5350–5355 CrossRef CAS PubMed.
- G. O'Bryan, B. M. Wong and J. R. McElhanon, ACS Appl. Mater. Interfaces, 2010, 2, 1594–1600 CrossRef PubMed.
- N. Yenpech, V. Intasanta, K. Tashiro and S. Chirachanchai, Polym. Chem., 2020, 11, 91–101 RSC.
- M. Li, W. Liu and S. Zhu, Polymer, 2017, 112, 219–227 CrossRef CAS.
- Y. Vidavsky, S. J. Yang, B. A. Abel, I. Agami, C. E. Diesendruck, G. W. Coates and M. N. Silberstein, J. Am. Chem. Soc., 2019, 141, 10060–10067 CrossRef CAS PubMed.
- F. Kempe, O. Brügner, H. Buchheit, S. N. Momm, F. Riehle, S. Hameury, M. Walter and M. Sommer, Angew. Chem., Int. Ed., 2018, 57, 997–1000 CrossRef CAS PubMed.
- L. J. Wang, X. J. Zhou, X. H. Zhang and B. Y. Du, Macromolecules, 2016, 49, 98–104 CrossRef CAS.
- H. Chen, F. Yang, Q. Chen and J. Zheng, Adv. Mater., 2017, 29, 1606900 CrossRef PubMed.
- Y. Jia, W. J. Wang, B. G. Li and S. Zhu, Macromol. Mater. Eng., 2018, 303, 1800154 CrossRef.
- S. Jiang, L. Zhang, T. Xie, Y. Lin, H. Zhang, Y. Xu, W. Weng and L. Dai, ACS Macro Lett., 2013, 2, 705–709 CrossRef CAS.
- M. Li, W. Liu, Q. Zhang and S. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 15156–15163 CrossRef CAS PubMed.
- Y. Jia, S. Wang, W. J. Wang, B. G. Li and S. Zhu, Macromolecules, 2019, 52, 7920–7928 CrossRef CAS.
- M. Raisch, D. Genovese, N. Zaccheroni, S. B. Schmidt, M. L. Focarete, M. Sommer and C. Gualandi, Adv. Mater., 2018, 30, 1802813 CrossRef PubMed.
- M. Takaffoli, T. Zhang, D. Parks and X. Zhao, J. Appl. Mech., 2016, 83, 071007 CrossRef.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CrossRef CAS PubMed.
- G. R. Gossweiler, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 6148–6151 CrossRef CAS PubMed.
- T. A. Kim, M. J. Robb, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2018, 51, 9177–9183 CrossRef CAS.
- W. Qiu, P. A. Gurr, G. Da Silva and G. G. Qiao, Polym. Chem., 2019, 10, 1650–1659 RSC.
- Y. Lin, M. H. Barbee, C. C. Chang and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 15969–15975 CrossRef CAS PubMed.
- M. H. Barbee, T. Kouznetsova, S. L. Barrett, G. R. Gossweiler, Y. Lin, S. K. Rastogi, W. J. Brittain and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 12746–12750 CrossRef CAS PubMed.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed.
- Y. Shiraishi, K. Tanaka, E. Shirakawa, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2013, 52, 8304–8308 CrossRef CAS PubMed.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed.
- T. Kosuge, X. Zhu, V. M. Lau, D. Aoki, T. J. Martinez, J. S. Moore and H. Otsuka, J. Am. Chem. Soc., 2019, 141, 1898–1902 CrossRef CAS PubMed.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- M. J. Teng, X. R. Jia, X. F. Chen and Y. Wei, Angew. Chem., Int. Ed., 2012, 51, 6398–6401 CrossRef CAS PubMed.
- Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 52, 12268–12272 CrossRef CAS PubMed.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS PubMed.
- L. Wang, W. Zhou, Q. Tang, H. Yang, Q. Zhou and X. Zhang, Polymers, 2018, 10, 994 CrossRef PubMed.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- L. J. Wang, K. X. Yang, Q. Zhou, H. Y. Yang, J. Q. He and X. Y. Zhang, Chin. J. Polym. Sci., 2019, 38, 24–36 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |