
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect sulfonamidation of (hetero)aromatic C–H bonds with sulfonyl azides: a novel and efficient route to N-(hetero)aryl sulfonamides
Zhi Liu
a,
Abdolghaffar Ebadi
 b,
Mohsen Toughani
c,
Nihat Mert
d and
Esmail Vessally
*e
b,
Mohsen Toughani
c,
Nihat Mert
d and
Esmail Vessally
*e
aSchool of Electrical and Automation Engineering, East China Jiaotong University, Nanchang 330013, China. E-mail: liu126lin@gmail.com
bDepartment of Agriculture, Jouybar Branch, Islamic Azad University, Jouybar, Iran. E-mail: dr_ebadi2000@mail.ru
cDepartment of Fishery, Babol Branch, Islamic Azad University, Babol, Iran. E-mail: m.toghani2008@yahoo.com
dDepartment of Biochemistry, Faculty of Veterinary Medicine, University of Yuzuncu Yil, 65080, Van, Turkey. E-mail: mertnihat@hotmail.com
eDepartment of Chemistry, Payame Noor University, Tehran, Iran. E-mail: vessally@yahoo.com; vesali@pnu.ac.ir
First published on 8th October 2020
Abstract
N-Aryl sulfonamides belong to a highly important class of organosulfur compounds which are found in a number of FDA-approved drugs such as dofetilide, dronedarone, ibutilide, sotalol, sulfadiazine, sulfamethizole, vemurafenib, and many more. There is therefore continuing interest in the development of novel and convenient protocols for the preparation of these pharmaceutically important compounds. Recently, direct sulfonamidation of (hetero)aromatic C–H bonds with easily available sulfonyl azides has emerged as an attractive and powerful strategy to access N-(hetero)aryl sulfonamides where non-toxic nitrogen gas forms as the sole by-product. This review highlights recent advances and developments (2012–2020) in this fast growing research area with emphasis on the mechanistic features of the reactions.
 Zhi Liu | Dr Zhi Liu was born in Nanchang, Jiangxi, P. R. China, in 1987. He received the PhD degree in traffic equipment and information engineering from Central South University (CSU), Changsha, China, in 2018. Now, he is currently a Lecturer with East China Jiaotong University. His research interests include intelligent transportation and vehicle material technology. |
 Abdolghaffar Ebadi | Dr Abdol Ghaffar Ebadi finished his doctoral degree in Environmental Biotechnology (Algology) from Tajik Academy of Sciences. Now he is a researcher in TAS in Tajikistan and faculty member at the Islamic Azad University of Jouybar in Mazandaran. Dr Ebadi published more than 400 scientific papers in qualified international journals and attended more than 50 international conferences. He has cooperation with many research project teams around world such as China, Malaysia, and Thailand. His interests are Environmental Biotechnology, Biochemistry, Gene pathways in the phytoremediation processes. |
 Mohsen Toughani | Mr Mohsen Toughani work in Environmental Biology and Chemistry. He is also interested in environmental techniques. Now he is Executive Manager of CAS-Press (http://www.cas-press.com). He is post-graduated in environmental biology from Babol Branch of Islamic University and published more than 30 international papers, and also attended some excellent conferences. His most interest is environmental biology, biotechnology, and pollutions. |
 Nihat Mert | Prof. Nihat Mert was born in Ankara in 1957. He studied for his PhD in Colorado State University in Biochemistry department then finished in Ankara University. He worked in Ankara, Uludag and Yuzuncu Yil Universities. He had more than 200 research mainly biopolymorphism, antioxidants, clinical biochemistry, diabetes mellitus and fitotherapy. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in Organic Chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor of Organic Chemistry. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
Sulfonamides constitute a very important class of drugs which are called sulfa drugs, and display a broad range of pharmacological activities such as antimicrobial, antiretroviral, anticonvulsant, anti-diabetic, antitumor, and anti-depressant.1 Seventy-two currently marketed drugs contain this privileged structural motif in their structure and one-fourth of them are N-(hetero)aryl sulfonamide derivatives.2 For example (Fig. 1), tipranavir 1 with the brand name of Aptivus is a nonpeptidic protease inhibitor marketed worldwide for the treatment of HIV/AIDS infection.3 Sulfamethoxazole 2 is an antibiotic that is used for various bacterial infections and is effective against both Gram negative and positive bacteria.4 Delavirdine 3 with the trade name Rescriptor is an antiretroviral medicine available in many countries worldwide and used as part of highly active antiretroviral therapy for the treatment of HIV type 1 (HIV-1).5 The newer drug dabrafenib 4 (Tafinlar) is a promising anticancer drug that is used to treat certain types of melanoma (a type of skin cancer) that cannot be removed with surgery or that have spread to other parts of the body.6 The drug prevents the formation of dihydrofolic acid, a compound that bacteria must be able to make in order to survive. Consequently, there is continuing interest in the development of expedient and efficient protocols for the preparation of this important class of sulfonamide containing scaffolds.7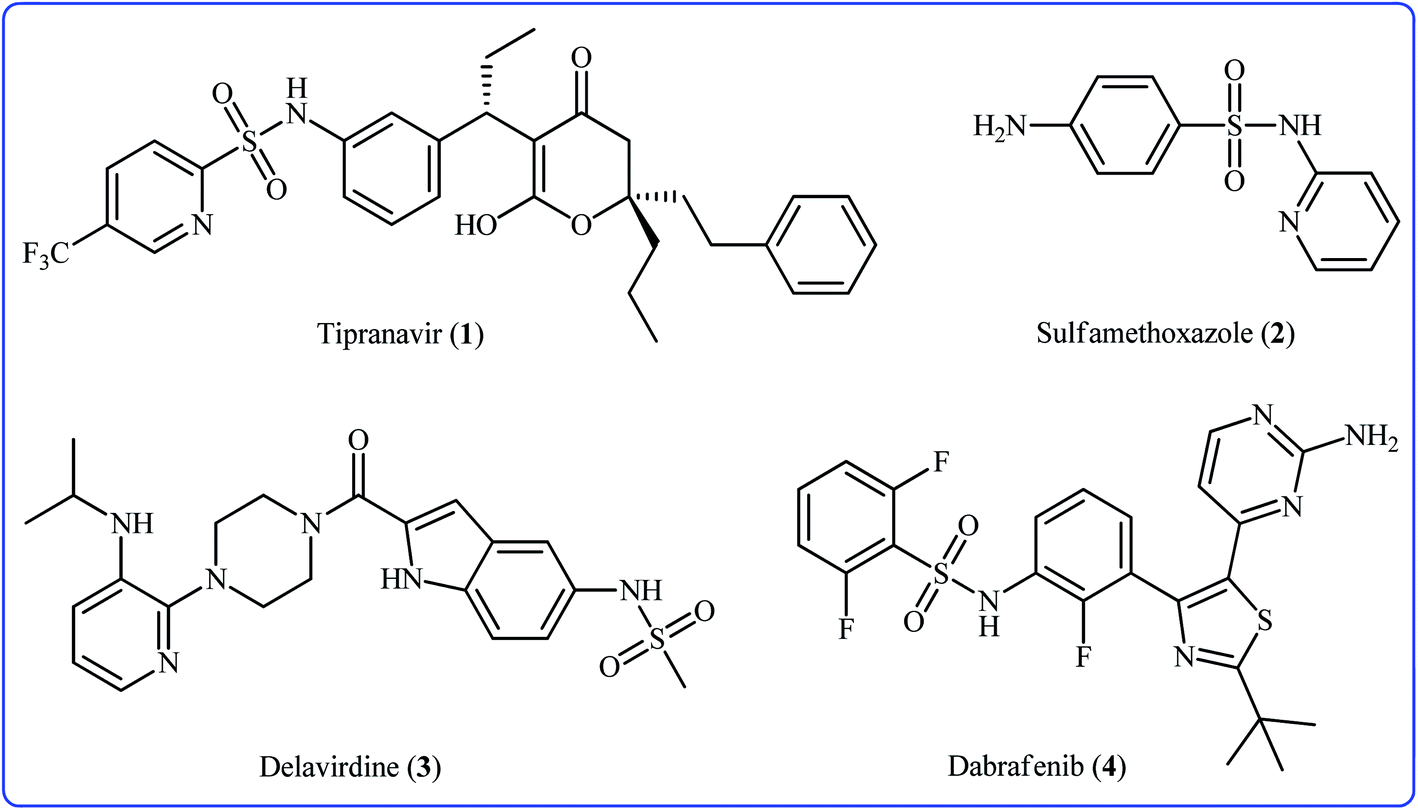 | ||
| Fig. 1 Selected drugs with N-aryl sulfonamide structure motif. | ||
Traditional synthesis of N-(hetero)aryl sulfonamides involves the reaction of sulfonyl chloride derivatives with aromatic amines, which suffers from the necessity for a strong base and generation of halide salts as the by-products.8 Lately, new procedures towards the greener synthesis of the titled compounds have been developed, such as cross-coupling of primary sulfonamides and aryl electrophiles,9 oxidative coupling of anilines with sodium sulfonates,10 reduction coupling of nitroarenes with sodium sulfonates,11 and direct sulfonamidation of aromatic C–H bonds with sulfonyl azides. Among others, transition-metal catalyzed denitrogenative coupling of arenes with sulfonyl azides offers prominent advantages of easily accessible starting materials, high efficiency, and no formation of toxic by-products (Fig. 2).
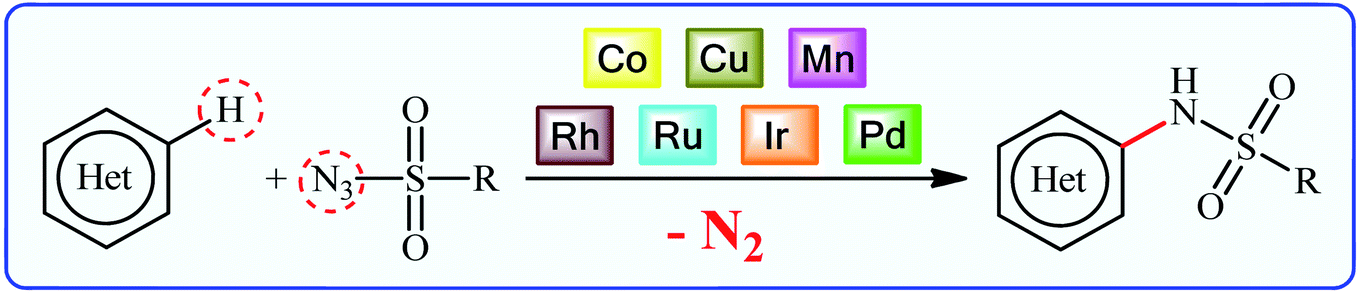 | ||
| Fig. 2 Transition-metal catalyzed direct sulfonamidation of (hetero)aromatic C–H bonds with sulfonyl azides. | ||
Despite the significant progress which has been achieved since 2012 in this research area, a comprehensive review has not appeared on this domain in the literature thus far. In connection with our recent works on the synthesis of organosulfur compounds12 and modern cross-coupling reactions,13 we summarize here a variety of methods for the synthesis of N-(hetero)aryl sulfonamides from the corresponding (hetero)arenes and sulfonyl azides with emphasis on the mechanistic aspects of the reactions. The metal catalysts play main role in the synthesis of organic compounds.14–18 In this regard, the reactions were classified according to the metal center of catalysts.
2. Rhodium-catalyzed reactions
In 2012, Cheng's research team reported one the earliest Rh-catalyzed direct sulfonamidation of aromatic C–H bonds with sulfonyl azides utilizing pyridyl as the ortho-selective coordinating directing group.19 Careful screening of various commercially available rhodium catalysts such as Rh2(O2CCF3)4, [Rh(cod)Cl]2, [RhCp*Cl2]2, [Ru(p-cymene)Cl2]2; and additives like AgBF4, AgSbF6, KPF6 led to [RhCp*Cl2]2/AgSbF6 combination as the most suitable catalytic system for this C–N bond forming reaction and among the various organic solvents (e.g., toluene, 1,2-DCE, t-amylOH); DCE proved to be the most efficient solvent. Under optimized conditions, the C2 position of a wide range of 2-phenylpyridine derivatives 5 was selectively activated and underwent coupling with various types of aryl and alkyl sulfonyl azides 6 in the absence of any external oxidant under atmospheric environment and afforded the target N-aryl sulfonamides 7 in moderate to excellent yields (Scheme 1). It should be noticed that other directing groups, such as pyrazole, pyrimidine, and oxime were also found to effectively mediate this transformation. Based on kinetic isotope effect studies and X-ray crystallographic analysis, the following mechanism is proposed by the authors for this transformation (Scheme 2). Firstly, a cationic Rh(III) species [Rh(III)Cp*][SbF6]2 generated upon treatment of the [RhCp*Cl2]2 precursor with AgSbF6. Next, coordination of nitrogen atom of the phenylpyridine 5 to this species followed by ortho-metalation affords a five-membered rhodacyclic intermediate A. Subsequently, coordination of azide 6 to A leads to the intermediate B that, after insertion of a sulfonamido moiety into the rhodacycle forms a Rh(III) amido complex C. Finally, protonolysis of C produces the desired product 7 and regenerates the cationic Rh(III) species. Later, based on density functional theory (DFT) calculations, the same authors suggested that the liberation of N2 from intermediate B forms a key Rh(V)-nitrenoid species and then a collapse of this intermediate affords the insertion intermediate C.20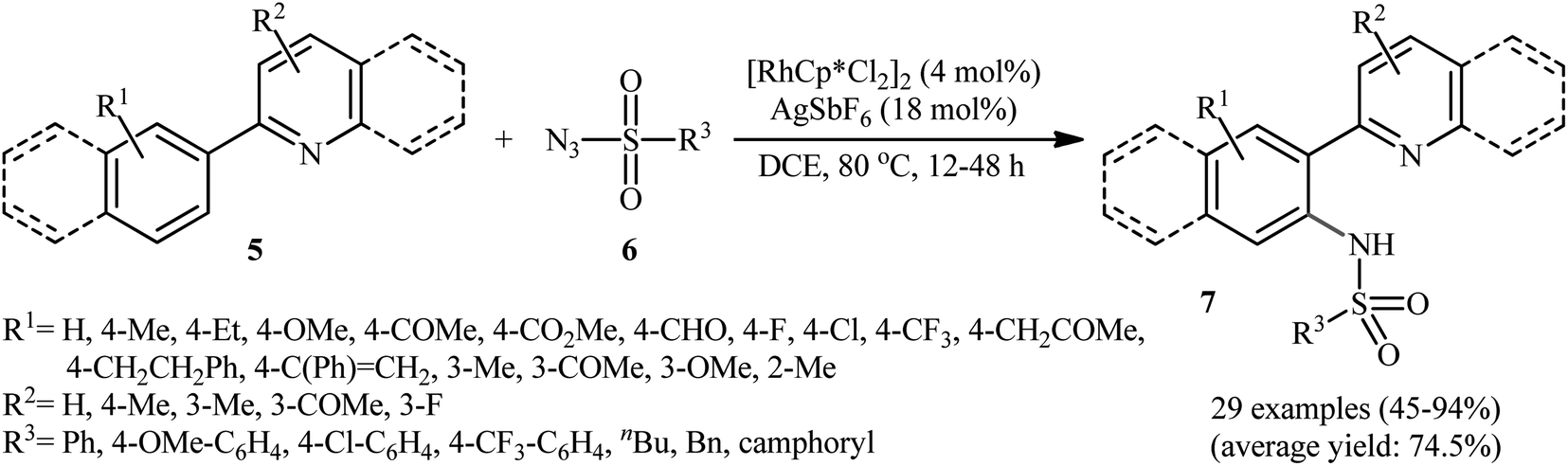 | ||
| Scheme 1 Cheng's synthesis of N-aryl sulfonamides 7. | ||
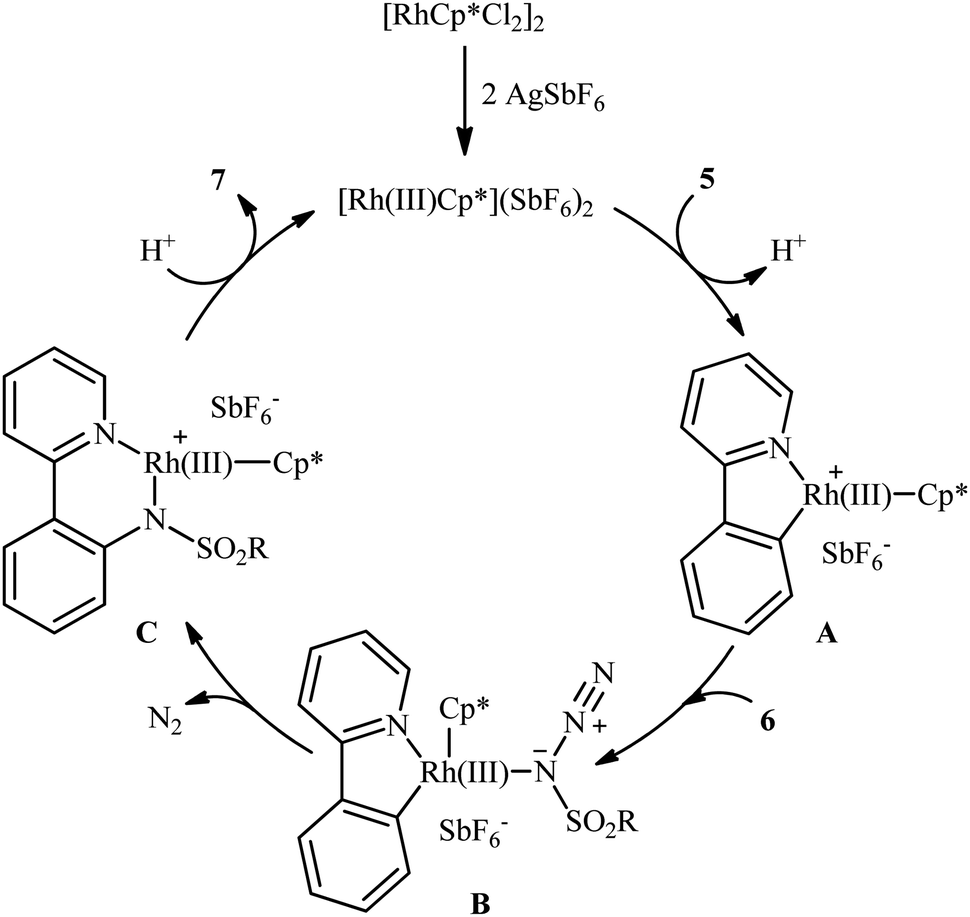 | ||
| Scheme 2 Proposed mechanism for Rh-catalyzed sulfonamidation of phenylpyridines 5 with sulfonyl azides 6. | ||
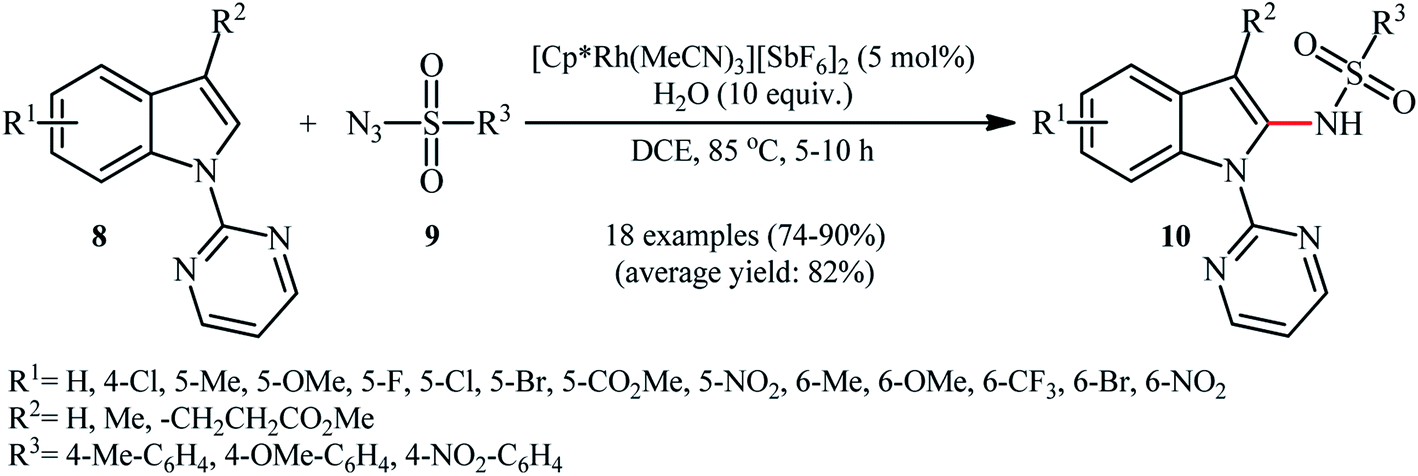
In the same year, Zhou and Li along with their co-workers described an interesting regioselective Rh-catalyzed direct C2-sulfonamidation of indoles 8 bearing a 2-pyrimidyl unit as a directing group through C–H activation by using sulfonyl azides 9 as the sulfonamide source.21 The reactions were carried out in the presence of a catalytic amount of [Cp*Rh(MeCN)3][SbF6]2 and ten equivalents of water as an additive under an open air, tolerated various election-rich and electron-poor functional groups on both coupling partners and provided the expected C2-sulfonamidated indoles 10 in good to excellent yields (Scheme 3). Noteworthy, no product was observed by replacing of pyrimidyl directing group with other groups (e.g., Me, Boc, Ac, Me2NCO). NH-free indoles were also failed to participate in this reaction. It should be mentioned that the 2-pyrimidyl directing group can be easily removed through hydrolysis to yield the corresponding NH-free sulfonamidated indoles under alkaline conditions.
 | ||
| Scheme 3 Rh-catalyzed site-selective sulfonamidation of indoles 8 with sulfonyl azides 9. | ||
In 2014, Jia and Han disclosed that the treatment of azobenzenes 11 with sulfonyl azides 12 in the presence of 5 mol% of [RhCp*Cl2]2 and 20 mol% of AgNTf2 in DCE under an air atmosphere afforded the corresponding ortho-sulfonamidated azobenzene derivatives 13 in moderate to almost quantitative yields, ranging from 35% to 98% (Scheme 4).22 Some of the most important results obtained in this investigation are listed below: (i) the reaction was almost equally efficient for aromatic, heteroaromatic, and aliphatic sulfonyl azides; (ii) electron-rich azobenzenes compare to the electron-poor ones gave higher yield of the expected products; and (iii) unsymmetrical azobenzenes mainly gave the sulfonamidation products on the electron-rich aromatic rings. Concurrently, Xu and colleagues reported a similar strategy to construct ortho-sulfonamidated azobenzenes by using Cheng's standard reaction condition.23 Although this methodology turned out to be highly efficient for the sulfonamidation of ortho- and meta-substituted azobenzenes, its application for the sulfonamidation of unsubstituted and para-substituted azobenzenes is restricted, since the selectivity of mono- and di-sulfonamidation products was generally poor.
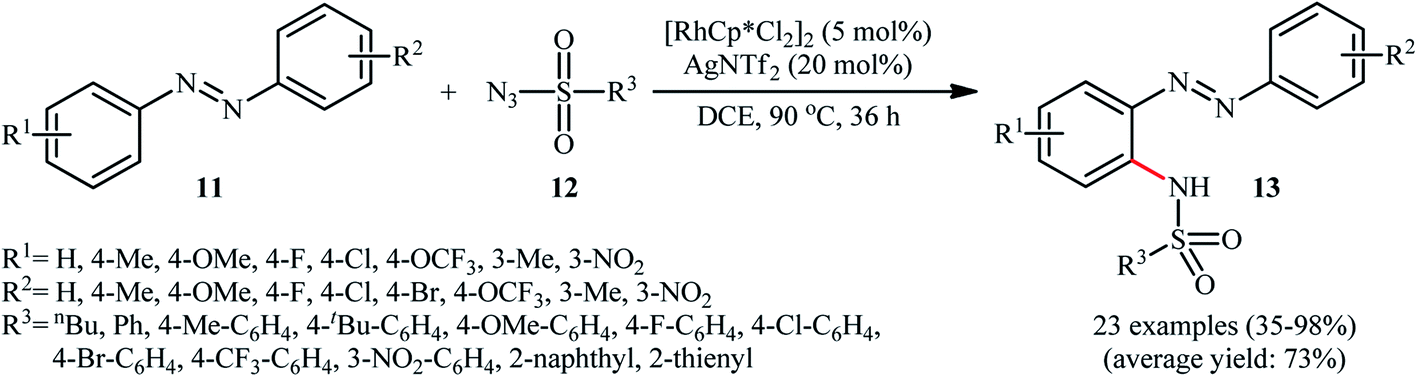 | ||
| Scheme 4 Direct sulfonamidation of azobenzenes 11 with sulfonyl azides 12 catalyzed by [RhCp*Cl2]2. | ||
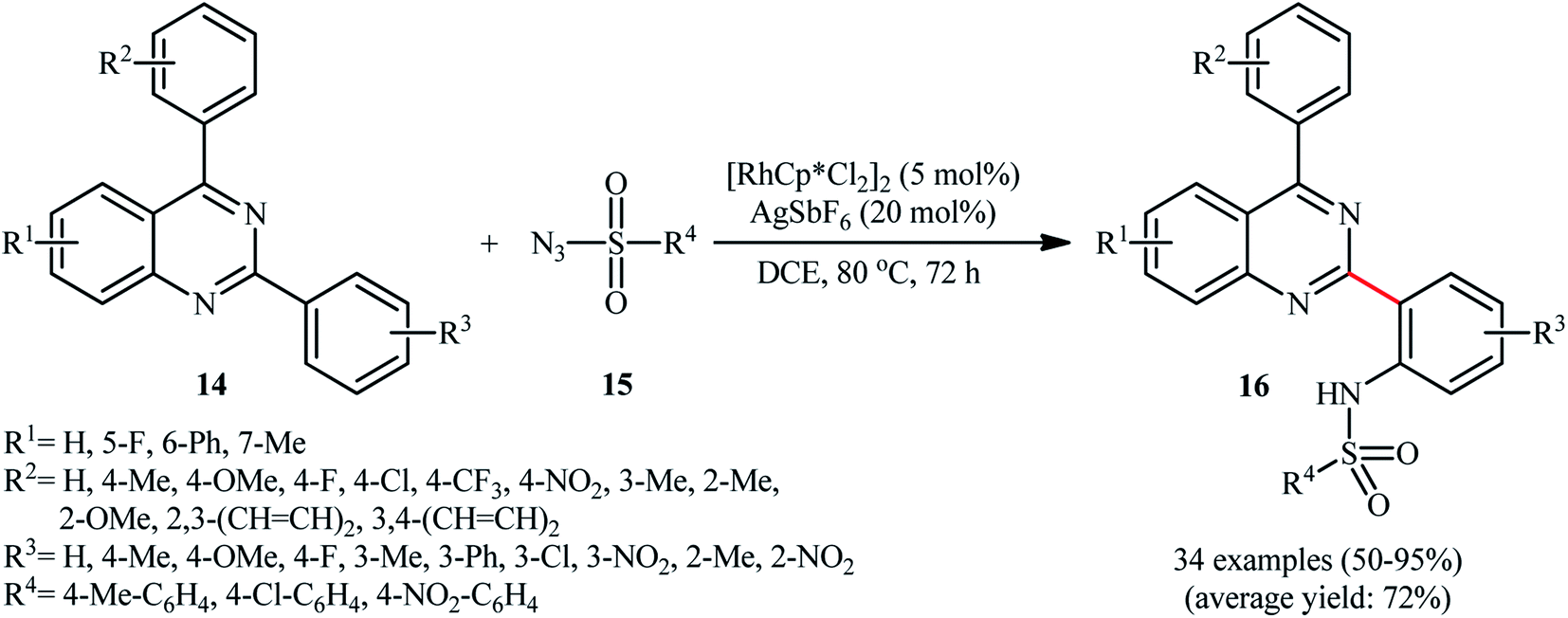
As a continuation of these studies, Peng and co-workers reported the steric hindrance controlled regioselective Rh-catalyzed direct C–H sulfonamidation of 2,4-diarylquinazolines 14 with sulfonyl azides 15 using Cheng's standard reaction condition (Scheme 5).24 They showed that when the meta position of the phenyl ring at the 2-position of quinazolines was blocked by a substituent group, the mono-sulfonamidated products 16 were exclusively obtained without any of di-sulfonamidated product. Likewise, ortho-substituted substrates selectively afforded the corresponding mono-sulfonamidated products. By contrast, the substrates with a substituent at the para position resulted in a mixture of mono- and di-sulfonamidated products. Of note, different substituents on the quinazoline ring failed to enhance the selectivity, albeit total yields were affected by the introduction of various substituents on this ring. Unfortunately, the exact mechanism of this transformation has not been elucidated yet.
 | ||
| Scheme 5 Rh-catalyzed direct C–H sulfonamidation of 2,4-diarylquinazolines 14 with sulfonyl azides 15 developed by Peng. | ||
3. Ruthenium-catalyzed reactions
Ruthenium is a rare transition metal placed in period V of the periodic table, adjacent to rhodium and is much cheaper than it. Therefore, it is reasonable to investigate the catalytic ability of this noble metal in direct C–H sulfonamidation of aromatic compounds with sulfonyl azides. In 2013, Sahoo and co-workers first reported the usefulness of ruthenium catalysts for such transformations.25 They showed that the treatment of ortho- and meta-substituted N-benzoylated sulfoximines 17 with various aromatic and aliphatic sulfonyl azides 18 in the presence of [RuCl2(p-cymene)]2/AgSbF6/KOAc combination as a catalytic system in DCE afforded the corresponding mono-sulfonamidated products 19 in modest to excellent yields and outstanding ortho-selectivity (Scheme 6a). However, para-substituted N-benzoylated sulfoximines provided a mixture of mono- and di-functionalized products with poor selectivity. The authors also successfully applied this strategy to the high yielding synthesis of ataciguat, a drug candidate for the treatment of aortic valve stenosis. They demonstrated that the methylphenyl sulfoximine directing group can be efficiently removed by hydrolysis of the final products under basic conditions (NaOH in MeOH/H2O at 60 °C in 6 h), providing the corresponding anthranilic acid derivatives in excellent yields. Subsequently, the same authors broadened the substrate scope of their methodology to aromatic ketones.26 In this report, thirty-three N-aryl sulfonamides were synthesized in relatively poor to excellent yields by means of 5 mol% of [RuCl2(p-cymene)]2 and 20 mol% of AgSbF6 in DCE at 100 °C in the presence of 50 mol% of Cu(OAc)2·H2O as a base. In contrast to their previous work, para-substituted substrates were also furnished ortho-sulfonamidated products with high mono-selectivity. At the same time, Chang and colleagues reported independently a similar strategy for the preparation of 2-sulfonamidated aromatic ketones 22 from ketones 20 and sulfonyl azides 21 by using a catalytic amount of [RuCl2(p-cymene)]2 in combination with AgNTf2 and NaOAc (Scheme 6b).27 Noteworthy, the authors proposed mechanism for this transformation is analogous to the one depicted for Rh-catalyzed reaction in Scheme 2. Shortly afterwards, by using a slightly modified catalytic system ([RuCl2(p-cymene)]2/AgSbF6/NaOAc/DCE, 60 °C), Ackermann and co-workers described the formation of N-aryl sulfonamides from the corresponding heteroaromatic (pyrimidine, pyridines, and pyrazole) directing-group-containing arenes and sulfonyl azides.28 Following these works, other coordinating directing groups, such as benzo[d]thiazole,29 1,2,3-triazole,30 and azo31 were also successfully utilized in this reaction.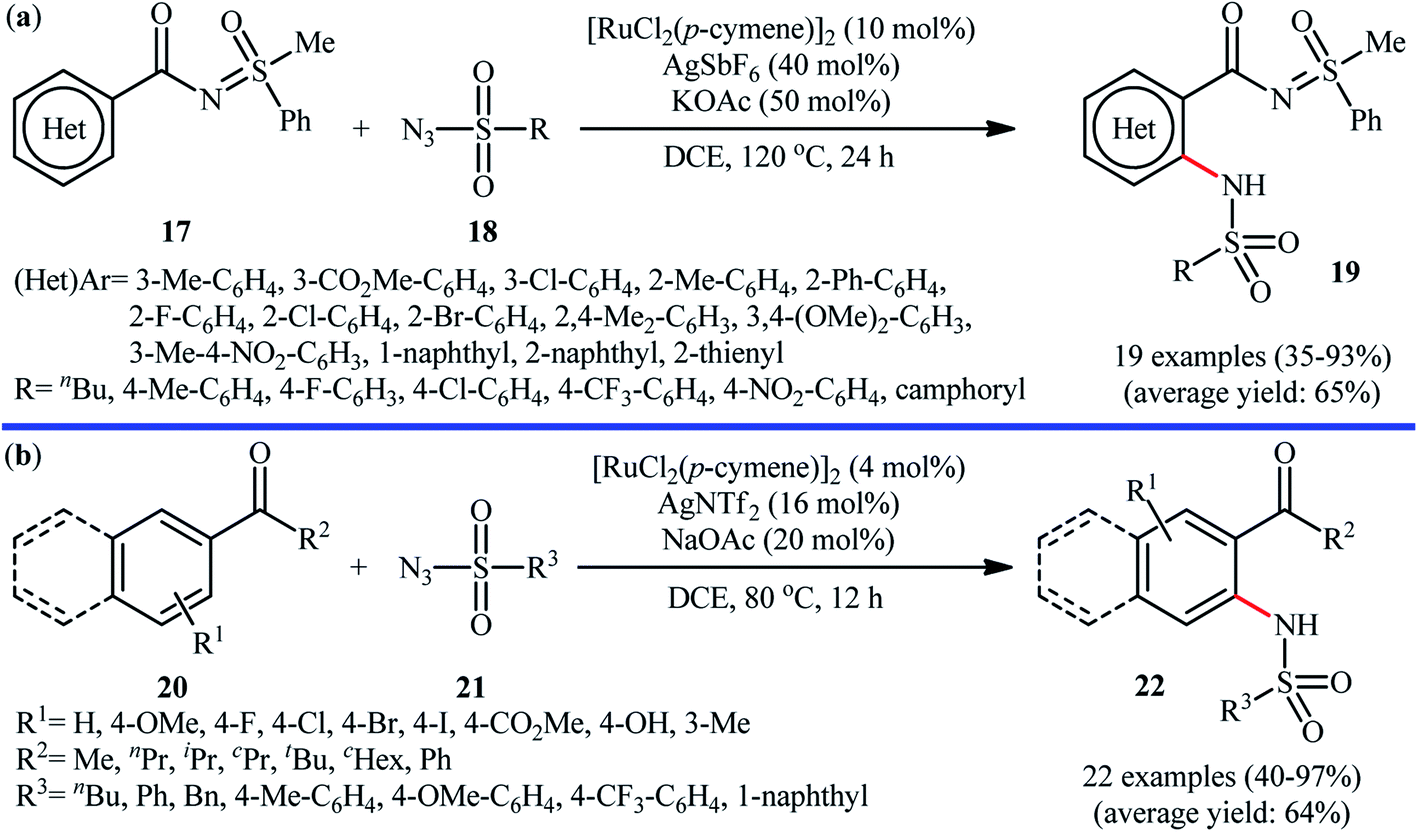 | ||
| Scheme 6 (a) Sulfoximine directed Ru-catalyzed C–H sulfonamidation of arenes 17 with sulfonyl azides 18; (b) Ru-catalyzed synthesis of 2-sulfonamidated aromatic ketones 22 from ketones 20 and sulfonyl azides 21. | ||
Along this line, Zhu and co-workers elaborated a synthetic route to prepare sulfonamidated indolines 25 via a Ru-catalyzed site-selective C–H sulfonamidation of N-carbonylated indolines 23 with sulfonyl azides 24 (Scheme 7).32 The couplings took place in the presence of [RuCl2(p-cymene)]2 (5 mol%), AgSbF6 (20 mol%), AgOAc (50 mol%), at 80 °C in DCE and selectively provided C7-functionalized products in moderate to high yields. Based on preliminary mechanistic investigations involving kinetic isotope effect studies and radical trapping experiments, the authors suggested that this reaction proceeds through the following key steps (Scheme 8): (i) initial formation of active Ru(II) catalyst A via removing of the Cl− ligand from the [{RuCl2(p-cymene)}2] complex by silver salt; (ii) coordination of the carbonyl oxygen of the indoline 23 to the Ru(II) catalyst A to give the metallacycle intermediate B; (iii) coordination of the azide group of the sulfonyl azide 24 to intermediate B to afford the Ru-species C; (iv) insertion of the sulfonamido moiety with evolution of N2 gas into the intermediate C leads to the intermediate D; and (v) protonolysis of D to form the final product 25 and regenerates ruthenium complex A. The above-mentioned catalytic system was then utilized by Reddy's research group in the C8-selective sulfonamidation of 1-tetralones with sulfonyl azides.33 Kim and co-workers also reported a similar strategy to construct 1-sulfonamido-xanthones, 5-sulfonamido-chromones, and 5-sulfonamido-flavonoid derivatives by switching the additive to Cu(OAc)2 and solvent to DCM.34
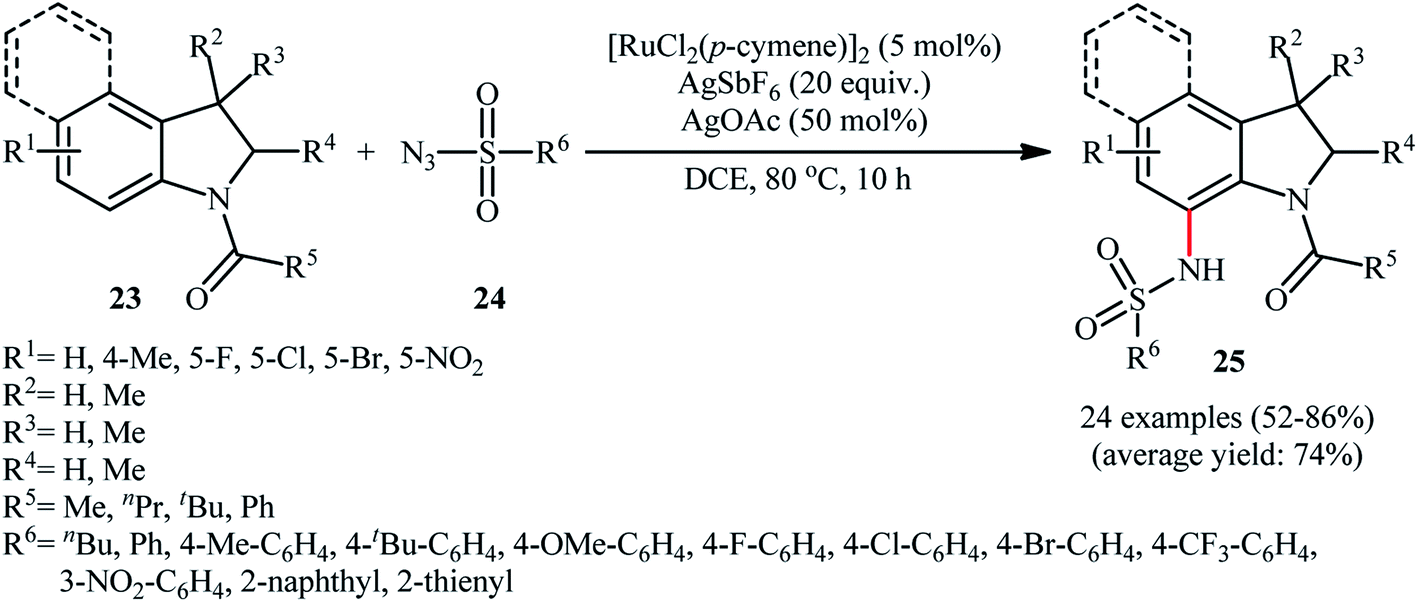 | ||
| Scheme 7 Ru-catalyzed site-selective C–H sulfonamidation of N-carbonylated indolines 23 with sulfonyl azides 24. | ||
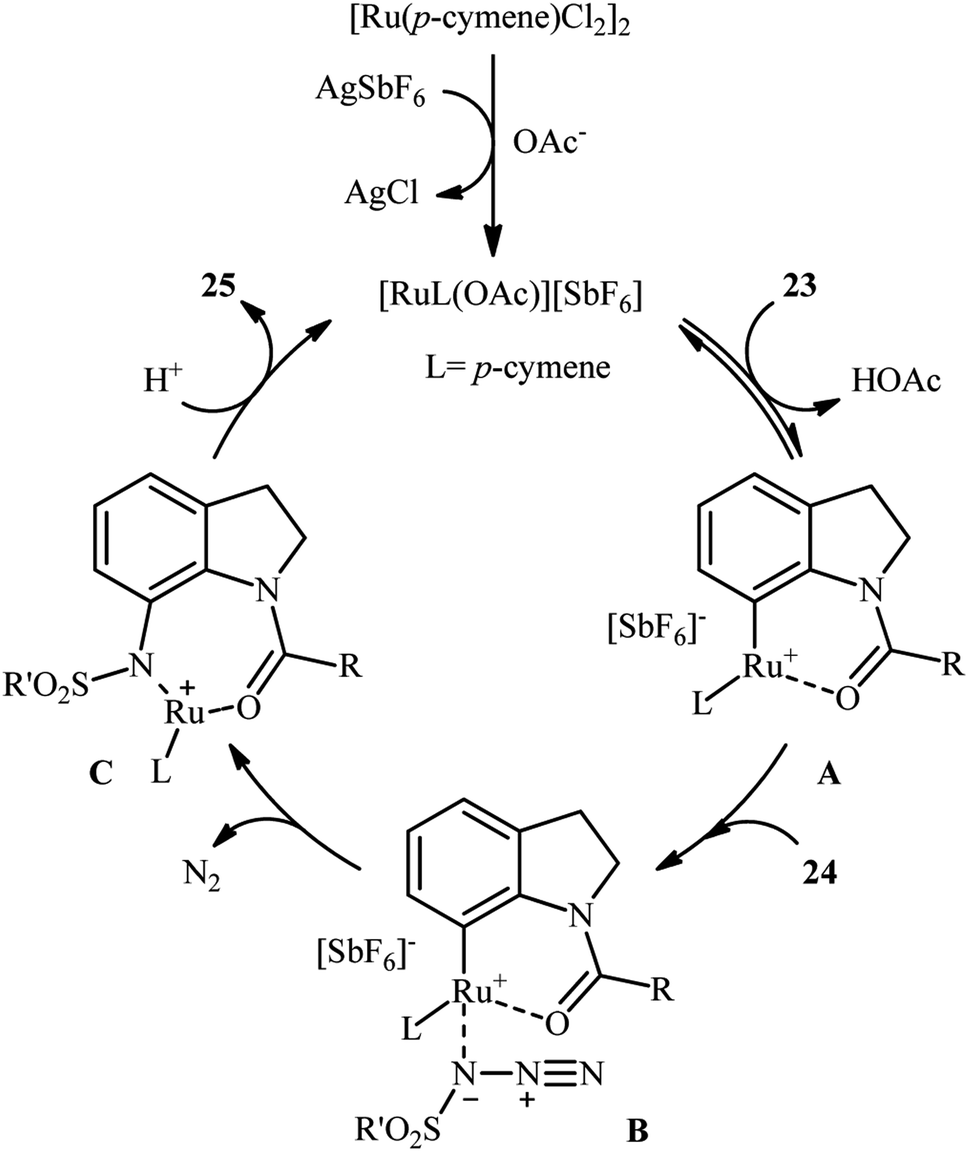 | ||
| Scheme 8 Mechanistic proposal for the reaction in Scheme 7. | ||
Very recently, Bakthadoss and co-workers illustrated a similar site-selective C–H sulfonamidation of oxobenzoxazine derivatives 26 with a range of aromatic sulfonyl azides 27 for the synthesis of ortho-sulfonamido oxobenzoxazine frameworks 28 using [RuCl2(p-cymene)]2 as the catalyst, AgSbF6 an additive and Cu(OAc)2·H2O as an oxidant (Scheme 9).35 The results demonstrated that oxobenzoxazines possessing electron-withdrawing groups afforded higher yields compared to the electron-rich oxobenzoxazines and the reaction was equally efficient for both electron-neutral and electron-rich aryl sulfonyl azides. However, no examples were given with aliphatic and electron-poor aromatic sulfonyl azides as the amide partners. To demonstrate the synthetic application of their methodology, the authors performed further late-stage functional group transformation reactions. They showed that hydrolysis of the synthesized compounds under basic conditions (e.g., NaOH/acetone, KOtBu/tBuOH) or in the presence of HBr in AcOH provided the corresponding 2-(2-sulfonaminobenzamido)benzoic acids in excellent yields, while hydrolysis in the presence of NaOMe in refluxing MeOH gave 2-sulfonaminobenzoic acid derivatives in near quantitative yields.
 | ||
| Scheme 9 Synthesis of ortho-sulfonamido oxobenzoxazine frameworks 28 reported by Bakthadoss.35 | ||
4. Iridium-catalyzed reactions
Of the large amount of published literature, without question, iridium was the most widely used catalyst for the synthesis of N-aryl sulfonamides via denitrogenative coupling of the corresponding aromatic compounds and sulfonyl azides.In 2013, Chang and co-workers communicated the first example of iridium-catalyzed direct sulfonamidation of aromatic C–H bonds with sulfonyl azides.36 They showed that a coordinating directing group (e.g., amide, anilide, ketoxime, hydrazone, carbamate, 2-pyridine, 2-pyrazol, and 2-azoline derivatives) attached to the arene rings leads to sulfonamidated products at the ortho position, adjacent to these groups. Thus, by employing the combination of [IrCp*Cl2]2 with AgNTf2 as an effective catalytic system, mono-selective amidation of aromatic compounds 29 with various aryl and alkyl sulfonyl azides 30 afforded the corresponding ortho-sulfonamidated arenes 31 in moderate to excellent yields (Scheme 10a). The protocol was compatible with various important functional groups such as fluoro, chloro, bromo, nitro, ether, ester, amide and aldehyde functionalities that are useful for further manipulation of products. Noteworthy, the protocol was also applicable for the site-selective amination of arenes with aryl azides. The same authors have next elegantly extended their methodology to access C8-sulfonamidated quinoline N-oxides 34 from the corresponding quinoline N-oxides 32 and sulfonyl azides 33 under a slightly modified condition (Scheme 10b).37 The present synthetic strategy was also successfully applied by the authors for the high yielding synthesis of Zinquin ethyl ester, a derivative of important fluorescent sensors for Zn(II). According to the authors proposed mechanism (Scheme 11), the unique regioselectivity of this synthesis was based on the formation of N-oxide-chelated iridacycle intermediate A, which was isolated and characterized by X-ray crystallographic analysis. Noteworthy, the proposed role of acid additive (AcOH) was to promote the rate-determining protodemetalation step. In a subsequent extension of the substrate scope of the protocol, it was shown that weakly coordinating directing group (ketone and ester) containing arenes could be also selectively amidated with sulfonyl azides at the ortho-position by using the combination of [IrCp*Cl2]2, AgNTf2, AcOH, and Li2CO3.38
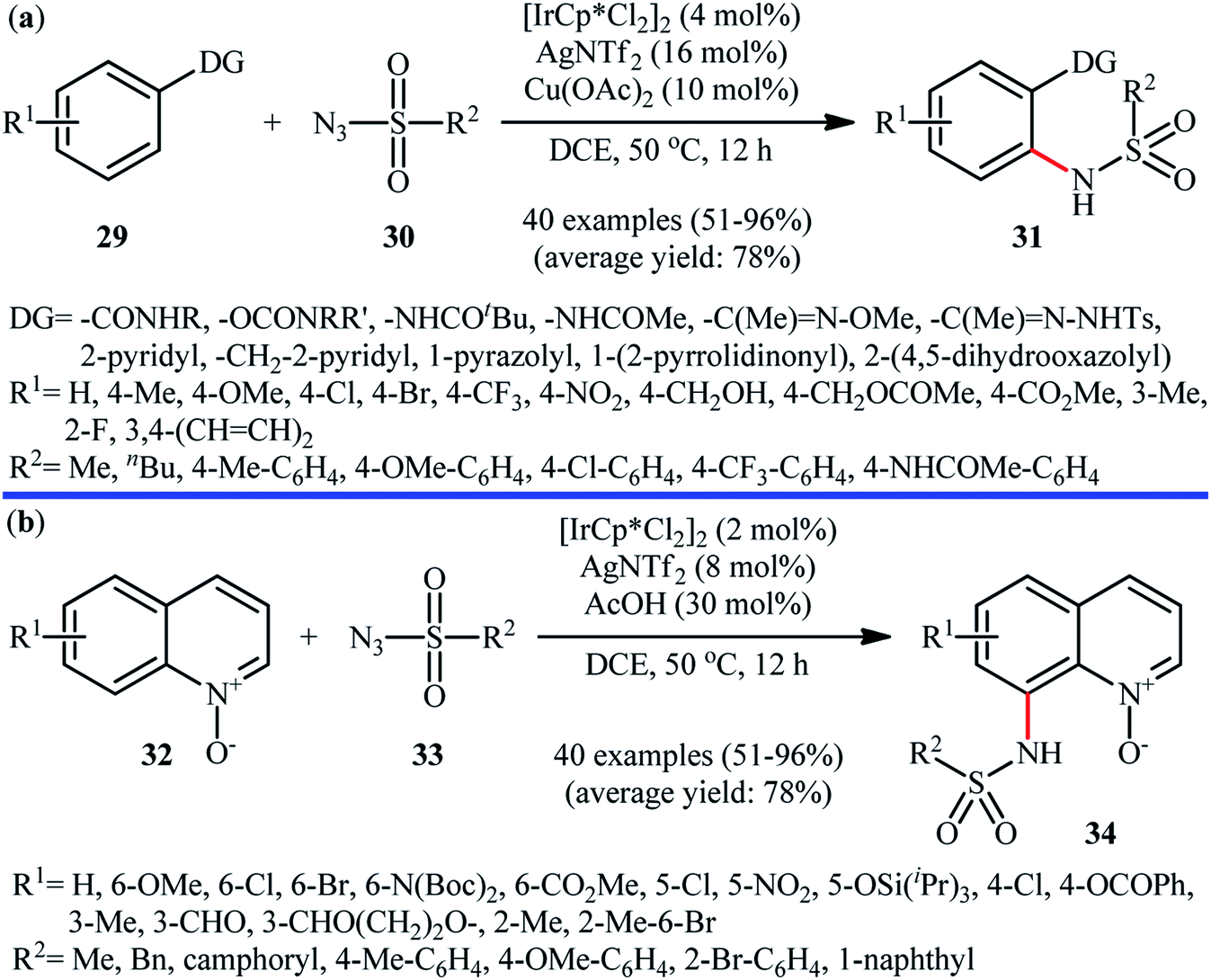 | ||
| Scheme 10 (a) Ir-catalyzed ortho-selective sulfonamidation of aromatic compounds 29 with sulfonyl azides 30; (b) Chang's synthesis of C8-sulfonamidated quinoline N-oxides 34. | ||
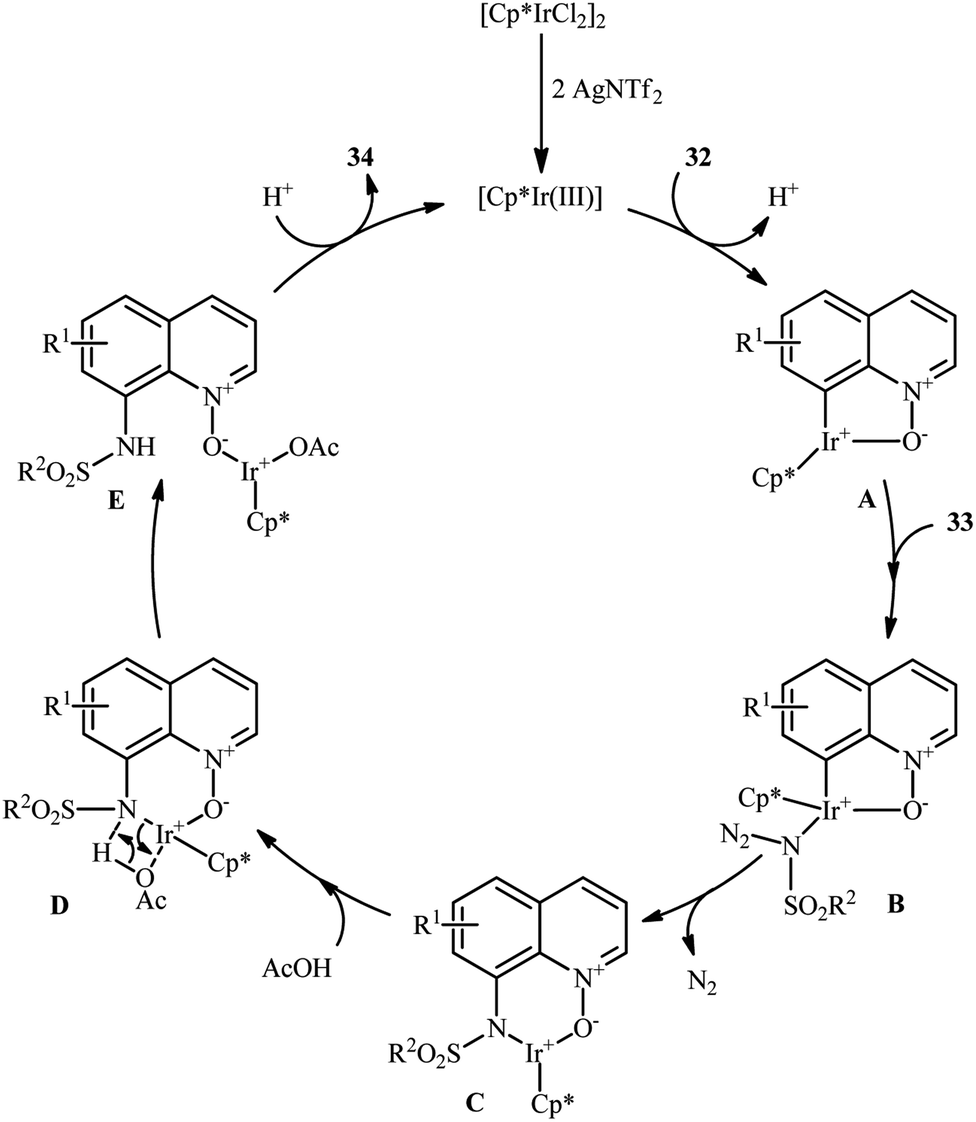 | ||
| Scheme 11 Mechanism for Ir-catalyzed sulfonamidation of quinoline N-oxides 32 and sulfonyl azides 33. | ||
Later, this innovative research group reported further examples of Ir-catalyzed sulfonamidation of aromatic C–H bonds using indolines as the arene reagent.39 Upon treatment with 4 mol% of [IrCp*Cl2]2, 8 mol% of AgNTf2, and 30 mol% of NaOAc in DCE at room temperature, various N-protected indolines 35 underwent regioselective C7-amidation with sulfonyl azides 36 to give C7-sulfonamidated indolines 37 in good to excellent yields, ranging from 66% to 98% (Scheme 12). Shortly thereafter, Zhou and Li along with their co-workers disclosed that this reaction could also successfully performed in the absence of NaOAc additive.40
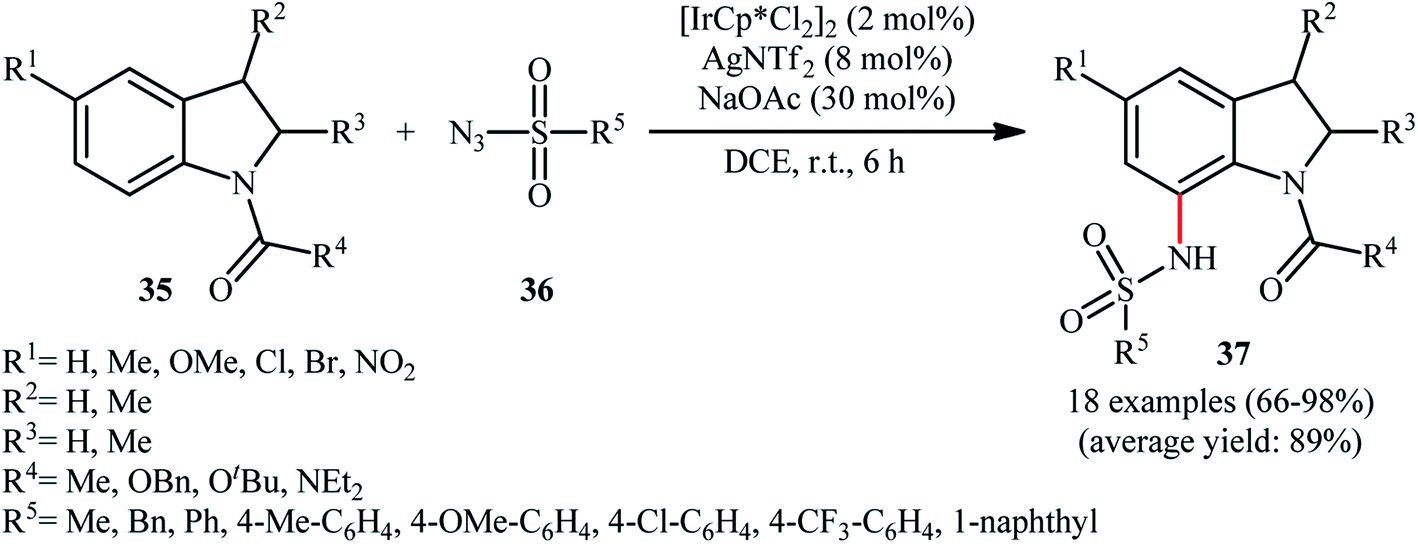 | ||
| Scheme 12 Direct C-7 sulfonamination of indolines 35 with sulfonyl azids 36 catalyzed by [IrCp*Cl2]2. | ||
In 2016, Kim, Park, and Chang employed IrCp*(OAc)2 catalyst, in combination with AgNTf2 for site-selective amidation of N-protected indole derivatives 38 with various aryl, alkyl, and vinyl sulfonyl azides 39.41 These reactions were performed in DCE under air atmosphere, completed within 12 h at room temperature, and provided the expected C7-amidated products 40 in relatively modest to excellent yields (Scheme 13a). The results proved that the efficiency of this C–N bond forming reaction strongly depended on the electronic character of the substituents on the indole ring, clearly in favor of electron-donating groups. Unfortunately, C2-substituted indoles totally failed to enter into this amidation reaction. Notably, the reaction was not operative with Rh or Ru catalyst systems. Drawing inspiration from these works, J. You and colleagues showed that indoles 41 bearing a carbonyl directing group (e.g., aldehyde, ketone, ester, amide) at the C3-position can be selectively sulfonamidated on the C4-position by sulfonyl azides 42 using IrCp*(OAc)2 as a catalyst and AgNTf2 as an additive in DCE under ambient conditions (Scheme 13b).42 The reaction is noteworthy in that both NH-free and N-protected indoles were well tolerated. In addition, the reaction could be scaled up to produce the target secondary sulfonamide in good yield without difficulty. In a closely related investigation, Lanke and Prabhur also described the synthesis of C4-sulfonamidated indoles form the corresponding indole-3-carbaldehydes and sulfonyl azides using Chang's standard condition.43
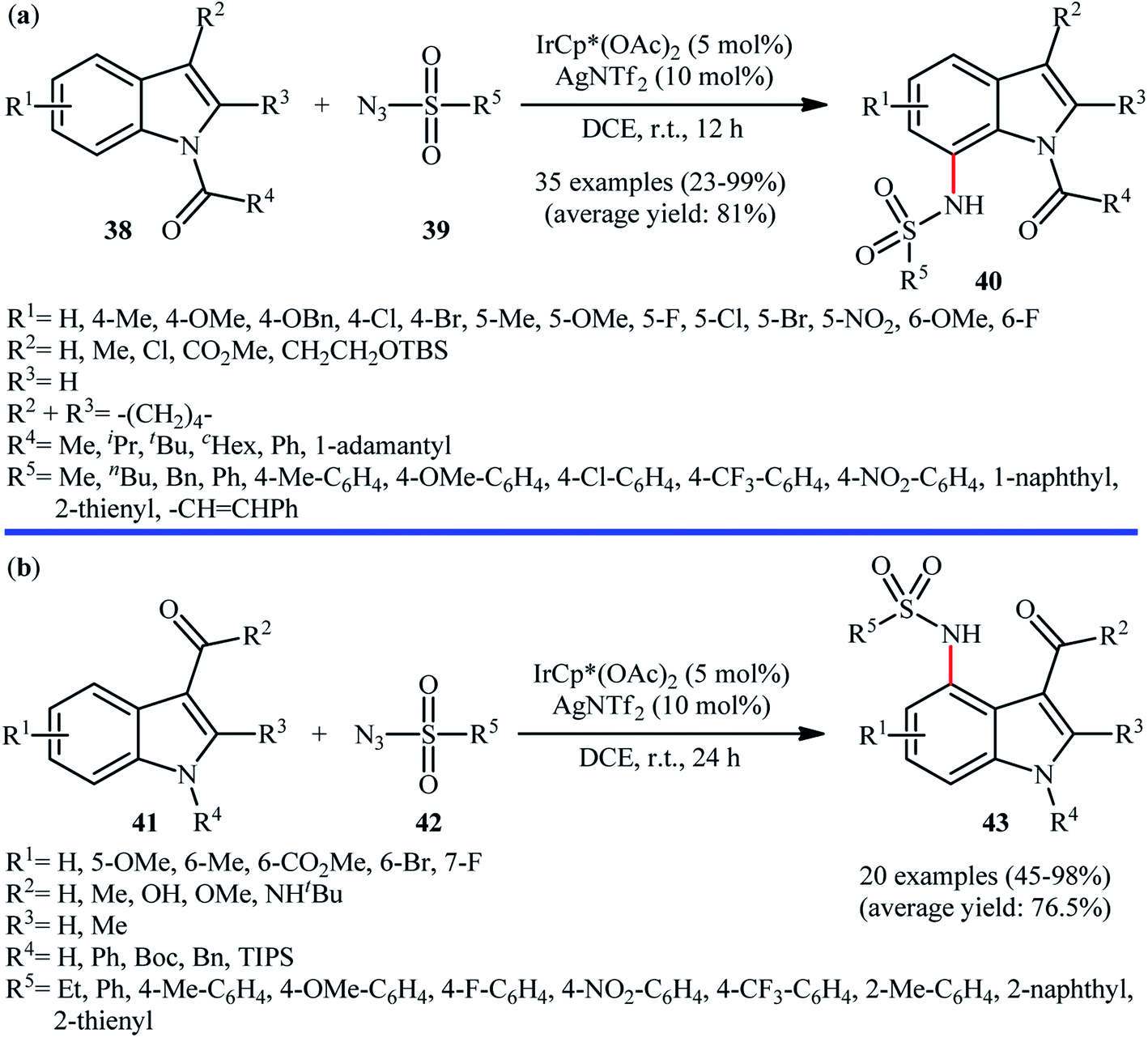 | ||
| Scheme 13 (a) Synthesis of C7-sulfonamidated indoles 40 reported by Chang; (b) You's synthesis of C4-sulfonamidated indoles 43. | ||
In this context, employing the standard [IrCp*Cl2]2 catalyst, different research groups were devised ortho-C–H sulfonamidation reactions of arenes bearing many other directing groups (e.g., carboxylate, amide, sulfonamide, imine, nitrone, triazole N-oxide, benzo[d]isothiazole 1,1-dioxide, pyrimidine, quinazolinone and tetrazine) with sulfonyl azides (Table 1).44–53
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditions | Directing group | N.E.a | Yield (%) | Ref. | |
| Range | Average | |||||
| a Number of examples. | ||||||
| 1 | AgNTf2, HOAc, Li2CO3, DCE, 80 °C | –CO2H | 26 | 40–99 | 81 | 44 |
| 2 | AgBF4, AgOAc, ball milling, 30 Hz | –CONHR | 18 | 42–97 | 76 | 45 |
| 3 | Ag2CO3, HOAc, DCE, 80 °C | –SO2NHR | 21 | 71–95 | 83.5 | 46 |
| 4 | AgNTf2, DCE, r.t. | –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+(O−)R N+(O−)R |
28 | 27–99 | 87 | 47 |
| 5 | AgNTf2, DCE, r.t. | –CHNTs |
21 | 46–95 | 75 | 48 |
| 6 | AgSbF6, DCE, 80 °C | 2-Amino-pyrimidine | 21 | 57–99 | 87.5 | 49 |
| 7 | AgNTf2, PivOH, DCE, 60 °C |  |
29 | 15–97 | 73 | 50 |
| 8 | AgNTf2, CF3CH2OH, 80 °C | 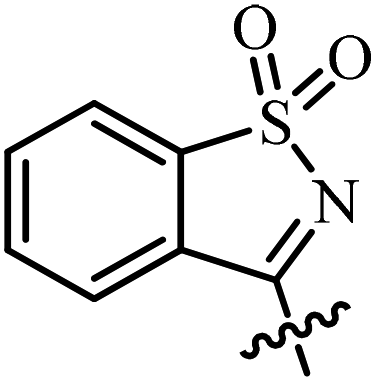 |
27 | 67–99 | 90 | 51 |
| 9 | AgSbF6, AcOH, DCE, 80 °C | 2-Quinazolinonyl | 13 | 45–90 | 78 | 52 |
| 10 | AgNTf2, AgTFA, DCE, 100 °C | 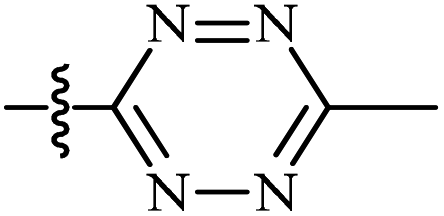 |
21 | 56–98 | 81 | 53 |
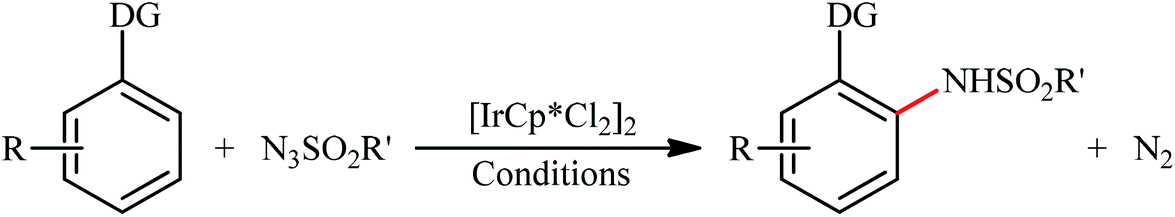
An important contribution to this field was reported by Cui and co-workers in 2017.54 They showed that treatment of N-phenylbenzimidamide derivatives 44 with sulfonyl azides 45 in the presence of [IrCp*Cl2]2/AgNTf2/phenylacetic acid combination in DCE afforded the corresponding 1-(sulfonyl)-2-aryl-1H-benzo[d]imidazoles 46 through C–H activation, sulfonamidation and annulation cascade (Scheme 14). Moderate to high yields, excellent regioselectivity, and broad substrate scope were the advantages, mentioned for this strategy. The suggested reaction mechanism for this transformation is displayed in Scheme 15. The reaction starts with the formation of active catalyst, [Cp*Ir(NTf2)]2, through anion exchange. Next, coordination of N-phenylbenzimidamide 44 to this catalyst and subsequent cyclometalation generates iridacyclic intermediate A, which after coordination with azide 45 affords the intermediate B. Subsequently, elimination of N2 from this intermediate produces iridium carbene species C that, after migratory insertion of the Ir–Ar bond into the carbene unit gives intermediate D. The Ir–N(sulfonamide) bond then undergoes migratory insertion into the C–N(imide) bond to form amide species E. Finally, elimination of the active Ir(III) catalyst and one molecule of NH3 from intermediate E upon protonolysis affords the expected 1-(sulfonyl)-2-aryl-1H-benzo[d]imidazole product 46.
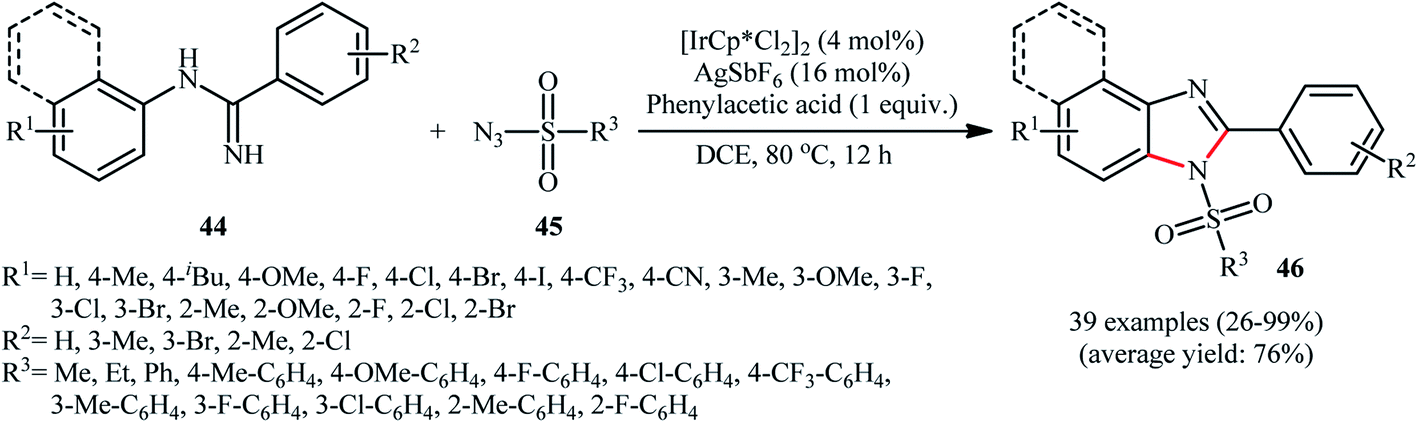 | ||
| Scheme 14 Synthesis of 1-(sulfonyl)-2-aryl-1H-benzo[d]imidazoles 46 from N-phenylbenzimidamides 44 and sulfonyl azides 45 through a C–H activation, sulfonamidation and annulation cascade. | ||
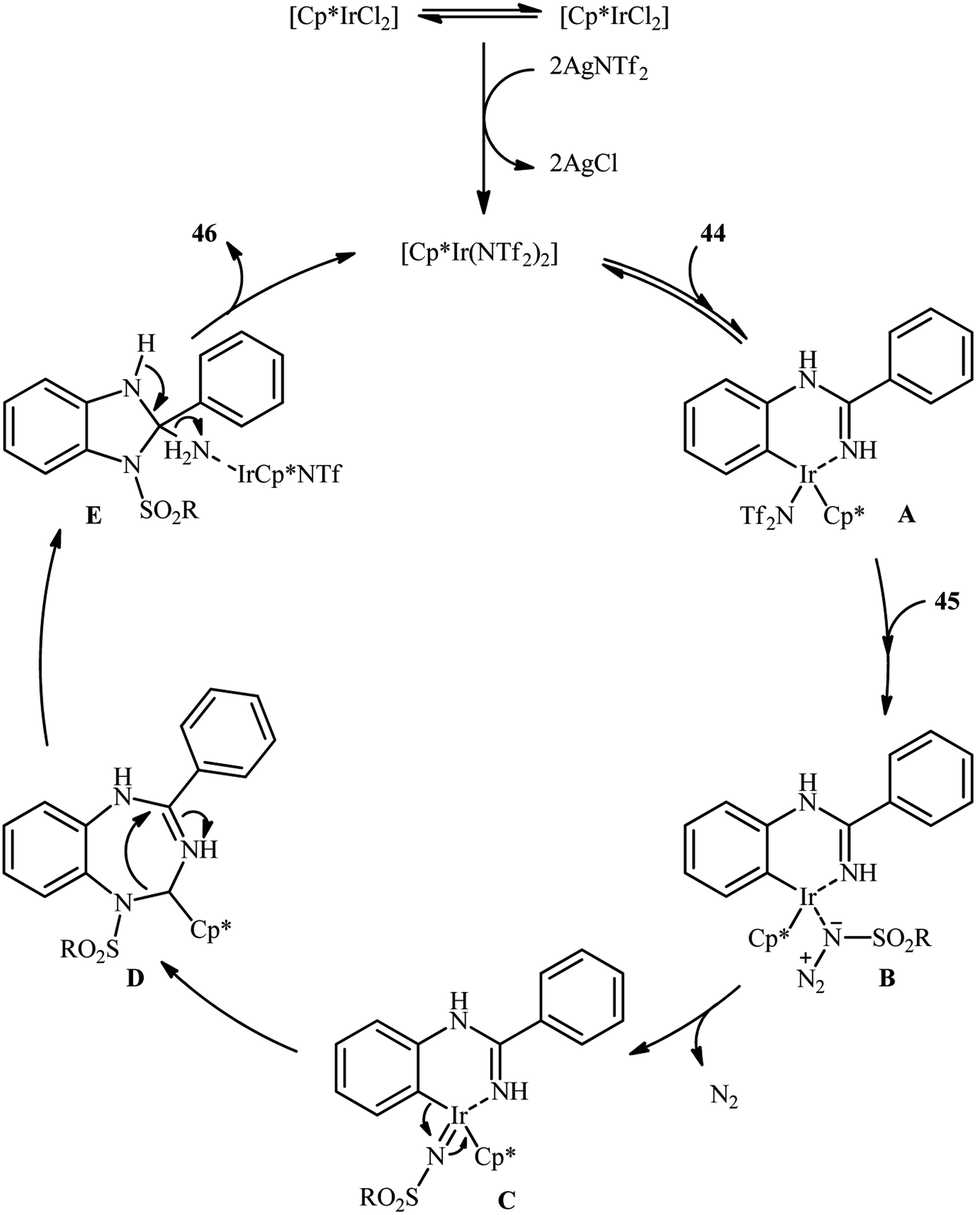 | ||
| Scheme 15 Proposed mechanistic pathway for construction of 1-(sulfonyl)-2-aryl-1H-benzo[d]imidazoles 46. | ||
Recently, Das and Samanta disclosed the synthesis of C3-sulfonamidated isoquinolones 49 in modest to excellent yields and complete regioselectivity from the corresponding 2-pyridyl protected isoquinolones 47 through sulfonamidation with sulfonyl azides 48 in the presence of catalytic quantities of [IrCp*Cl2]2 and AgSbF4, with 50 mol% of NaOAc as an additive (Scheme 16).55 Interestingly, when the pyridyl group was switched to H or methyl group, the C8-sulfonamidated isoquinolones 50 were obtained as the sole products. This interesting directing group regiocontrolled reaction was also worked well with different other azides like benzoyl azide, diphenyl phosphoryl azide, and aziodoformate. However, phenyl azide and benzyl azide did not furnish the desired products.
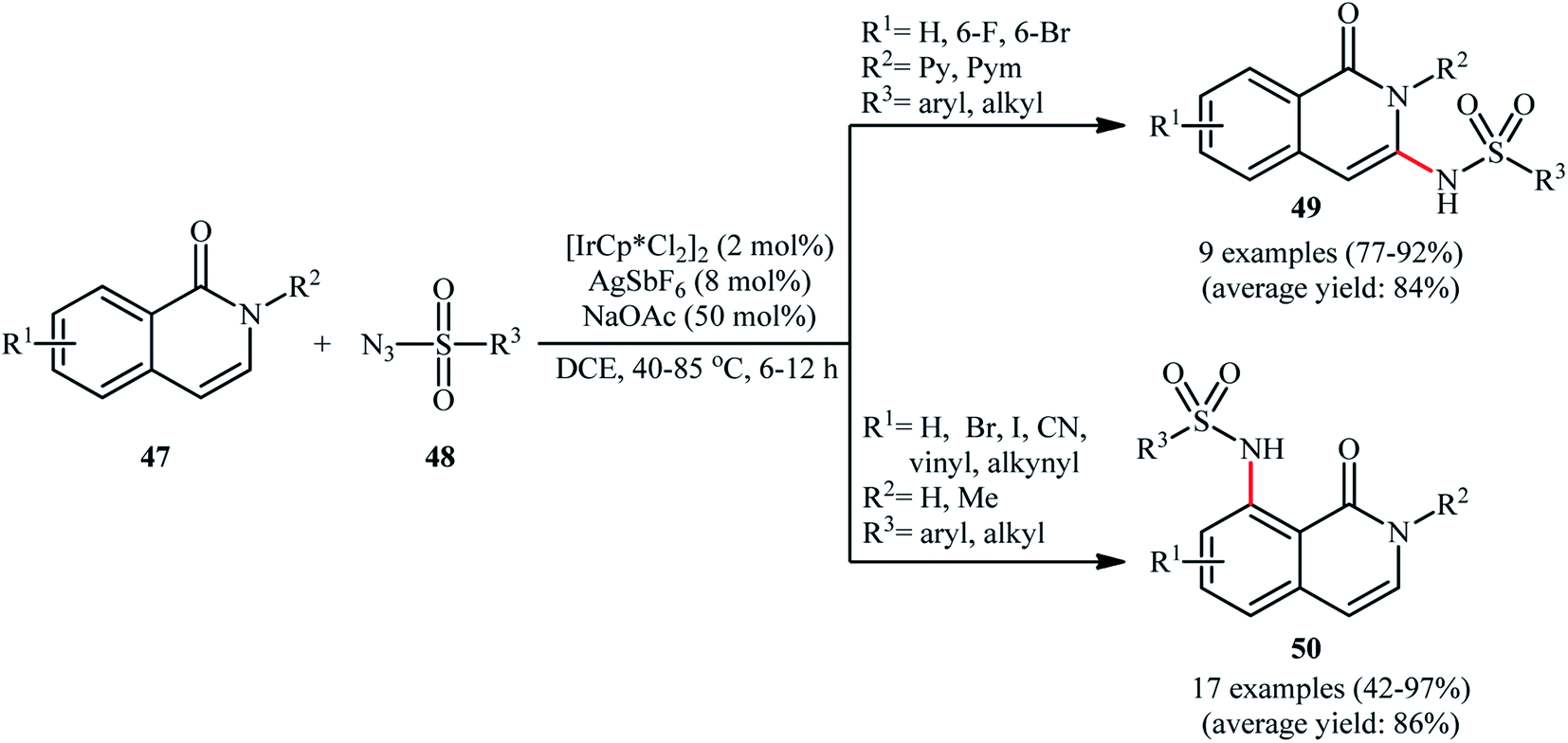 | ||
| Scheme 16 Ir-catalyzed regiocontrolled C3/C8-sulfonamidation of isoquinolones 47 with sulfonyl azides 48. | ||
5. Palladium-catalyzed reactions
Despite wide utility of palladium catalysts in numerous C–H bond functionalization reactions,56 the reported examples of the direct sulfonamidation of C–H bonds with sulfonyl azides using this versatile transition metal are scarce. In fact, only one example of such a reaction was reported in the literature thus far. In this investigation, Hu, Luo, and Zhu described the regioselective introduction of a sulfonamide group at the C-3 position of indoles using Pd(TFA)2 as a catalyst and sulfonyl azides as the nitrogen source.57 A screening of reaction variables indicated that PPh3, H2O, and m-xylene were the most effective ligand, additive, and solvent, respectively. With these optimized reaction conditions, a variety of C3-sulfonamidated indoles 53 were obtained in moderate to excellent yields from the corresponding 2-arylindoles 51 and sulfonyl azides 52 (Scheme 17). Indole itself did not take part in the reaction and therefore no other C2-unsubstituted indoles were examined in the protocol. In order to evaluate the function of ligand, the authors conducted the reaction under the identical conditions by omitting PPh3 and found no product was detected. However, the exact role of the ligand has not been elucidated yet. | ||
| Scheme 17 Pd-catalyzed C–H sulfonamidation of indoles 51 with sulfonyl aides 52. | ||
6. Cobalt-catalyzed reactions
Cobalt-based catalytic systems have played key roles in organic synthesis over decades, due to the high catalytic activity, low-cost and toxicity of this earth-abundant transition metal.58 The first and only example of Co-catalyzed direct sulfonamidation of aromatic C–H bonds with sulfonyl azides was described by Matsunaga and Kanai in 2014.59 The authors reported that indoles 54 bearing a pyrimidine ring as the directing group could undergo a smooth site-selective sulfonamidation with various aryl and alkyl sulfonyl azides 55 in the presence of Cp*Co(CO)I2/AgSbF6/KOAc combination as a catalytic system to give the corresponding C2-amidated products 56 in high to excellent yields (Scheme 18). Of note, Cp*CoI2-dimer was also found to promote this denitrogenative coupling reaction, albeit at lower efficiency. However, other commercially available cobalt-catalysts such as Co(acac)3, Co2(CO)8, Co(NH3)6Cl3, CoI2 and [Cp*Co(C6H6)](PF6)2 proved to be completely ineffective. It should be mentioned that the presence of AgSbF6 was crucial for the success of this C–N bond forming reaction. No product was yielded in the absence of AgSbF6. According to the authors proposed mechanism this reaction proceeds through a five-membered cobaltacyclic intermediate formed by the oxidative cyclometalation of the C2–H bond and a nitrogen atom of pyrimidyl group of the substrates to the active cobalt species.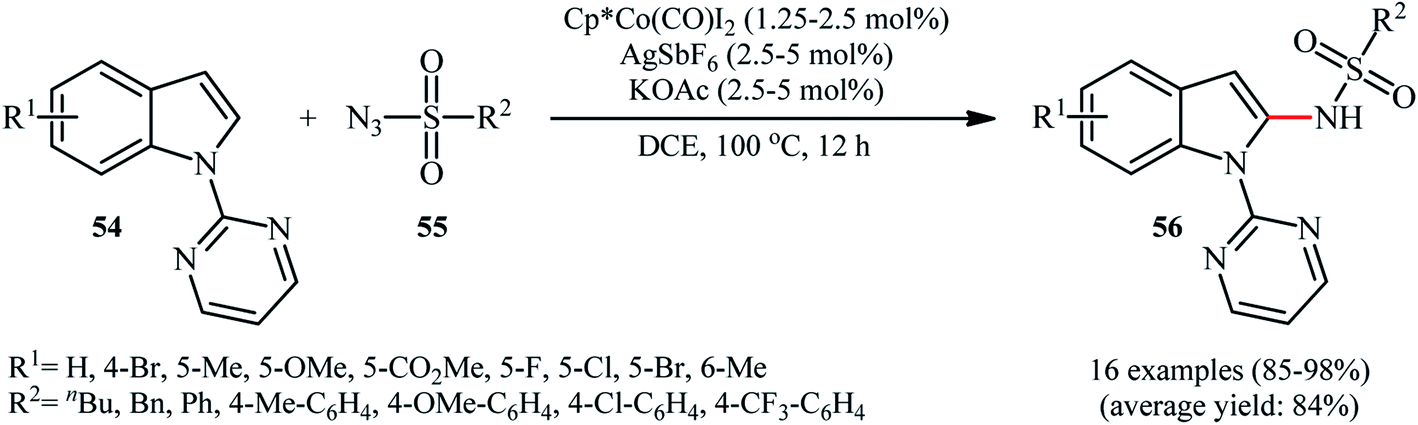 | ||
| Scheme 18 Co(III)-catalyzed C2-selective C–H sulfonamidation of indoles 54 with sulfonyl azides 55. | ||
7. Copper-catalyzed reactions
In 2014, Zhu and co-workers informed for the first time the usefulness of copper catalysts for the direct sulfonamidation of aromatic C–H bonds with sulfonyl azides.60 To evaluate the catalytic activity of different copper salts, 2-phenylpyridine and tosyl azide were chosen as the model substrates. Among the various commercially available Cu-based catalysts [e.g., CuCl, CuCl2, CuTc, Cu(TFA)2, Cu(OAc)2], copper(I) thiophene-2-carboxylate (CuTc) was found to be more effective, which gave a better yield of ortho-sulfonamidated product. In a pursuit to further improve the yield, PivOH was added as an additive to the reaction mixture. The solvents such as 1,4-dioxane, MeOH, DMSO, DMF, DCB, and DCE were examined and a good yield of product was obtained when using DCB as the reaction medium. Under the optimized conditions, various arenes 57 bearing a nitrogen-based directing group reacted efficiently with sulfonyl azides 58 to give the corresponding N-arylsulfonamides 59 in relatively poor to good yields and excellent ortho-selectivity (Scheme 19). The results indicated that arenes possessing electron-donating groups furnished better yields compared to the electron-poor arenes and the relative reaction rates of sulfonyl azides followed the order: aliphatic sulfonyl azides > electron-rich aromatic sulfonyl azides > electron-poor aromatic sulfonyl azides. To the best of our awareness, this is only example dealing with the Cu-catalyzed direct sulfonamidation of aromatic C–H bonds.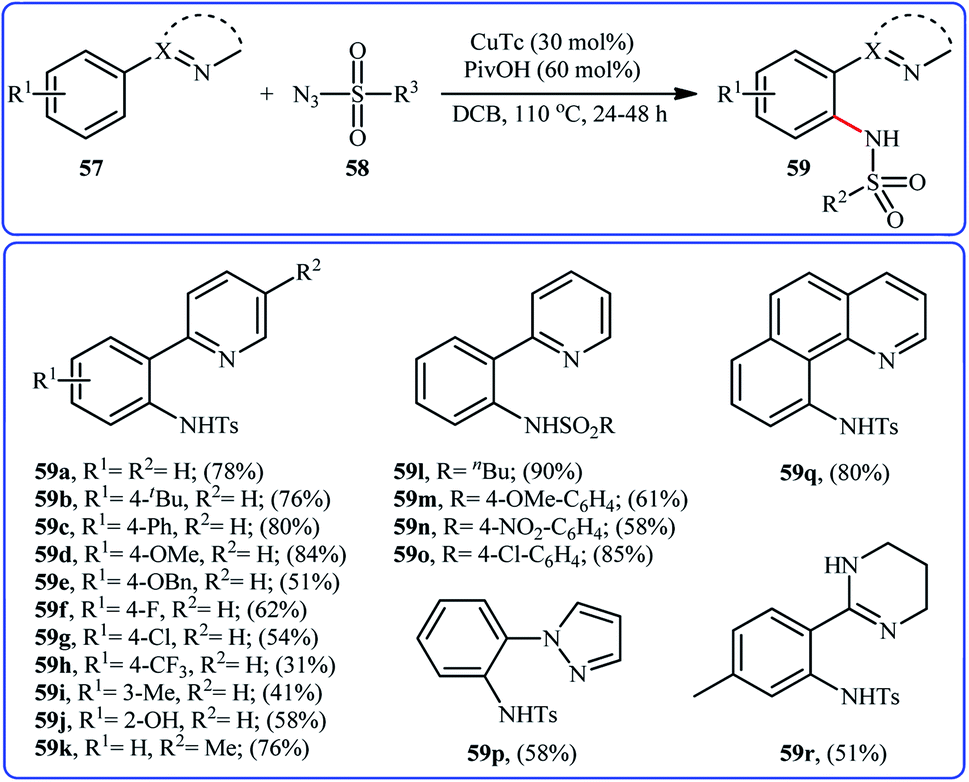 | ||
| Scheme 19 Cu-catalyzed directing group-assisted sulfonamidation of arenes 57 with sulfonyl azides 58. | ||
8. Manganese-catalyzed reactions
Manganese is one of the latest metals that was joined to the story of direct sulfonamidation of aromatic C–H bonds with sulfonyl azides. In 2018, Kong, Ling, Xu disclosed that arene substrates 60 bearing chelating directing groups, can undergo Mn-catalyzed direct ortho-selective sulfonamidation using sulfonyl azides 61 as the nitrogen source (Scheme 20).61 The optimum reaction conditions relied on the use of MnBr(CO)5 as the catalyst and H2O as the solvent, at 120 °C. In addition, the presence of 10 mol% of tetrapropylammonium bromide (TPAB) as an additive proved essential for achieving the expected sulfonamidated products 62 in good yields. The methodology was also useful for C2-selective sulfonamidation of indoles using pyrimidine as the directing group. High to excellent yields, broad substrate scope, and scalability were the main advantages, mentioned of this process. However, the requirement of elevated reaction temperature limited the range of application of this protocol, more or less.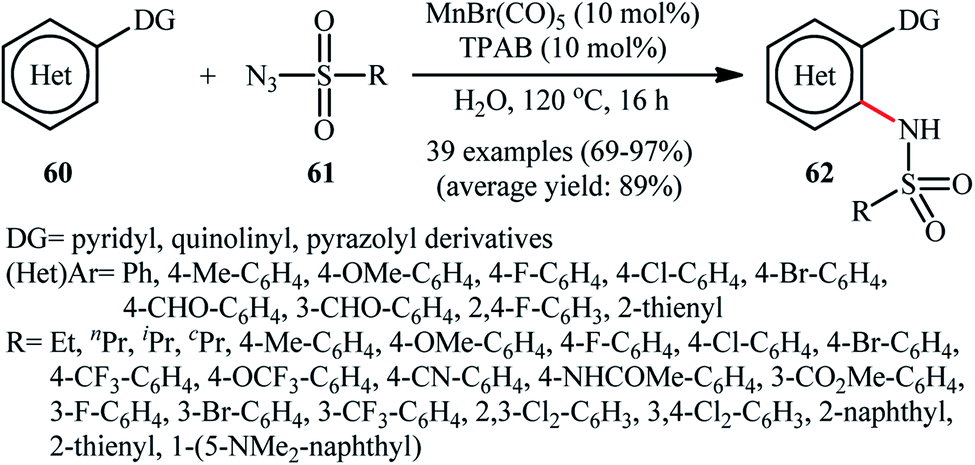 | ||
| Scheme 20 Xu's synthesis of N-(hetero)aryl sulfonamides 62. | ||
The next year, the same authors applied their manganese catalyst for the sulfonamidation of a diverse range of aromatic oximes 63 with sulfonyl azides 64 by using imidazolium ionic liquid [Hmim]OTf as the solvent (Scheme 21).62 This time, no additive was used and the couplings were performed under relatively milder conditions. Importantly, MnBr(CO)5/[Hmim]OTf system could be easily separated from the final reaction mixture and reused for at least five times with tangible decrease in its catalytic activity. In addition, the reaction could be easily scaled up and performed on a multi-gram quantity without further optimization.
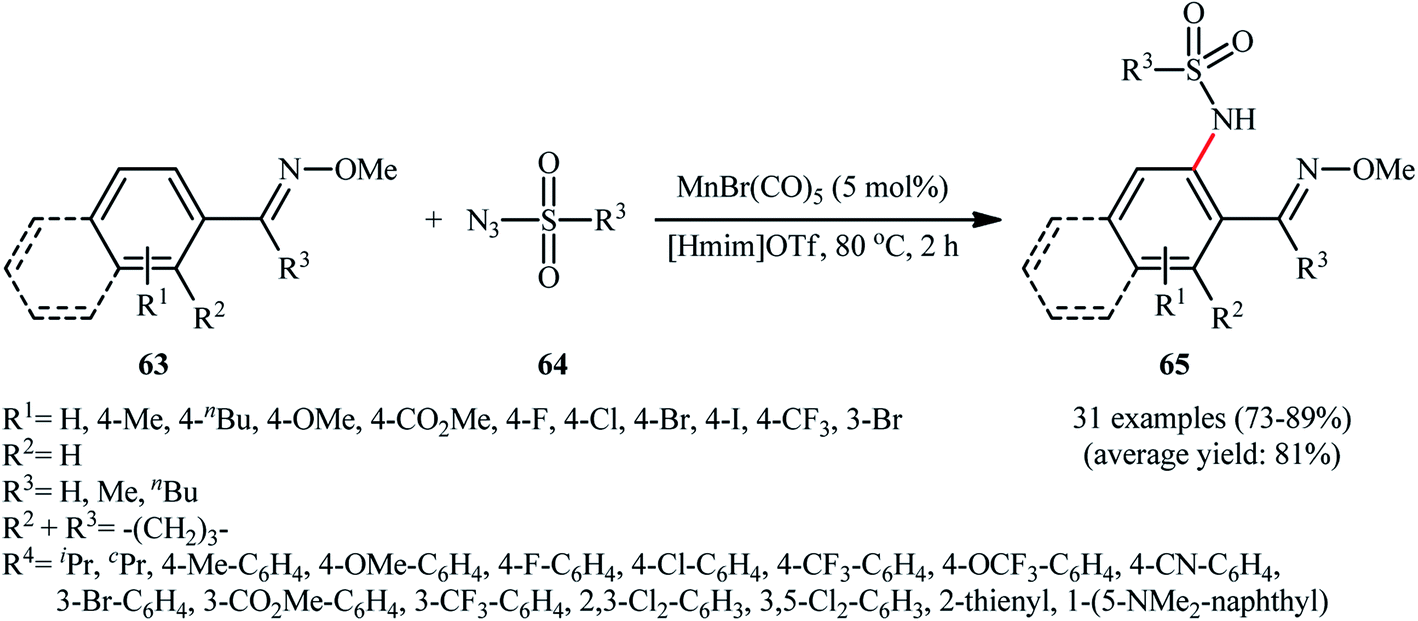 | ||
| Scheme 21 Mn-catalyzed amidation of aromatic oximes 63 with sulfonyl azides 64 in ionic liquid. | ||
9. Summary and outlook
Sulfonamides belong to an important class of organosulfur compounds as they display an array of biological activities including antimicrobial, antiretroviral, anticonvulsant, anti-diabetic, antitumor, and anti-depressant activities. Interestingly, seventy-two FDA-approved drugs contain this privileged structural motif in their structure and up to 25% of them are N-(hetero)arylated derivatives. Therefore, development of facile and efficient procedure for their synthesis from easily available starting materials that meet the objectives of green chemistry is always interesting.The direct functionalization of C–H bonds represents one of the most atom-economical and environmentally friendly approaches to molecular construction. This methodology enables unprecedented, single-step access to a broad range of carbon–carbon and carbon-heteroatom bonds directly from C–H bonds that are unreactive under traditional methods. In this review, recent advances and developments on the synthesis of biologically important N-(hetero)aryl sulfonamides through the transition-metal catalyzed direct sulfonamidation of (hetero)aromatic C–H bonds with easily available sulfonyl azides have been discussed. It is shown that various N-aryl and N-heteroaryl sulfonamides are readily accessible by using this approach in a straightforward modular way where non-toxic nitrogen gas forms as the sole by-product. Of note, majority of the reactions covered in this review were performed under atmospheric environment and some of them were easily scaled up to the gram levels under relatively mild conditions. These results clearly indicate the potential application of this route of N-(hetero)aryl sulfonamide synthesis in industry.
Despite great achievements over the past few years in this fast-growing research area, many challenges remain to be solved. For instance, most of the catalysts used herein are late transition metal catalysts such as Rh, Ru, Ir based-catalysts. Given cost and sustainability concerns, there is a significant need for the development of first-row transition metal-based catalysts. Moreover, despite numerous recent published works on the usefulness of transition metal nanoparticles as highly efficient and reusable catalysts for various C–H functionalization reactions, there have been no reports of the applicability of such catalysts in the titled reactions till date. Thus, the exploration of metallic nanocatalysts for this page of N-aryl sulfonamide are highly desirable in terms of cost and environmental benefits. In addition, almost all of the reactions covered in this review employed directing groups for controlling the site-selectivity. However, the requirement of installing and then removing this groups limited the range of application of this synthetic strategy and therefore, of course, catalyst-controlled site-selective processes should be explored.
Conflicts of interest
There are no conflicts to declare.References
- (a) T. Owa and T. Nagasu, Expert Opin. Ther. Pat., 2000, 10, 1725–1740 CrossRef CAS; (b) A. Scozzafava, T. Owa, A. Mastrolorenzo and C. T. Supuran, Curr. Med. Chem., 2003, 10, 925–953 CrossRef CAS; (c) İ. Gulçin and P. Taslimi, Expert Opin. Ther. Pat., 2018, 28, 541–549 CrossRef; (d) S. Shafiei and S. Davaran, Chem. Rev. Lett., 2020, 3, 19–22 Search PubMed; (e) S. Majedi and S. Majedi, J. Chem. Lett., 2020, 1, 2–8 Search PubMed.
- K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2019, 376, 5 CrossRef.
- L. Doyon, S. Tremblay, L. Bourgon, E. Wardrop and M. G. Cordingley, Antiviral Res., 2005, 68, 27–35 CrossRef CAS.
- (a) R. Gleckman, S. Alvarez and D. W. Joubert, Am. J. Hosp. Pharm., 1979, 36, 893–906 CAS; (b) P. G. Spitzer, S. M. Hammer and A. W. Karchmer, Rev. Infect. Dis., 1986, 8, 427–430 CrossRef CAS.
- (a) L. J. Scott and C. M. Perry, Drugs, 2000, 60, 1411–1444 CrossRef CAS; (b) J. Q. Tran, J. G. Gerber and B. M. Kerr, Clin. Pharmacokinet., 2001, 40, 207–226 CrossRef CAS.
- A. Hauschild, J.-J. Grob, L. V. Demidov, T. Jouary, R. Gutzmer, M. Millward, P. Rutkowski, C. U. Blank, W. H. Miller Jr and E. Kaempgen, Lancet, 2012, 380, 358–365 CrossRef CAS.
- M. Ashfaq, S. S. Shah, T. Najjam, S. Shaheen and G. Rivera, Mini-Rev. Org. Chem., 2013, 10, 160–170 CrossRef CAS.
- T. C. Das, S. A. Quadri and M. Farooqui, Chem. Biol. Interface, 2018, 8, 194–204 CAS.
- (a) W. Deng, L. Liu, C. Zhang, M. Liu and Q.-X. Guo, Tetrahedron Lett., 2005, 46, 7295–7298 CrossRef CAS; (b) M. Nasrollahzadeh, A. Ehsani and M. Maham, Synlett, 2014, 25, 505–508 CrossRef CAS; (c) W. Dong, C. Liu, X. Ma, Y. Zhang, Z. Peng, D. Xie and D. An, Tetrahedron, 2019, 75, 3886–3893 CrossRef CAS.
- (a) X. Tang, L. Huang, C. Qi, X. Wu, W. Wu and H. Jiang, Chem. Commun., 2013, 49, 6102–6104 RSC; (b) W. Wei, C. Liu, D. Yang, J. Wen, J. You and H. Wang, Adv. Synth. Catal., 2015, 357, 987–992 CrossRef CAS; (c) K. Yang, M. Ke, Y. Lin and Q. Song, Green Chem., 2015, 17, 1395–1399 RSC.
- (a) B. Yang, C. Lian, G. Yue, D. Liu, L. Wei, Y. Ding, X. Zheng, K. Lu, D. Qiu and X. Zhao, Org. Biomol. Chem., 2018, 16, 8150–8154 RSC; (b) X. Li, F. Chen and G.-P. Lu, Tetrahedron Lett., 2018, 59, 4226–4230 CrossRef CAS.
- (a) S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638 CrossRef CAS; (b) E. Vessally, K. Didehban, M. Babazadeh, A. Hosseinian and L. Edjlali, J. CO2 Util., 2017, 21, 480–490 CrossRef CAS; (c) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC; (d) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef; (e) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS.
- (a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef; (b) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC; (c) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC; (d) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 20 CrossRef; (e) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS; (f) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 8964–8976 RSC; (g) M. Hamzeloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef; (h) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC; (i) A. Hosseinian, S. Arshadi, S. Sarhandi, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 289–311 CrossRef CAS; (j) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 37 CrossRef CAS; (k) J. Wang, P. Su, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 41684–41702 RSC; (l) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed; (m) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed; (n) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed; (o) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed; (p) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed; (q) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed.
- J. Liu, E. Xu, J. Jiang, Z. Huang, L. Zheng and Z. Q. Liu, Chem. Commun., 2020, 56, 2202–2205 RSC.
- F. Su, Q. Jia, Z. Li, M. Wang, L. He, D. Peng, Y. Song, Z. Zhang and S. Fang, Microporous Mesoporous Mater., 2019, 275, 152–162 CrossRef CAS.
- X. Luo, H. Hu, Z. Pan, F. Pei, H. Qian, K. Miao and G. Feng, J. Hazard. Mater., 2020, 396, 122735 CrossRef CAS.
- M. A. Ashraf, Z. Liu, W. X. Peng, K. Jermsittiparsert, G. Hosseinzadeh and R. Hosseinzadeh, Ceramurgia Int., 2020, 46, 7446–7452 CrossRef CAS.
- L. He, J. Liu, Y. Liu, B. Cui, B. Hu, M. Wang and Z. Peng, Appl. Catal., B, 2019, 248, 366–379 CrossRef CAS.
- J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, J. Am. Chem. Soc., 2012, 134, 9110–9113 CrossRef CAS.
- S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492–2502 CrossRef CAS.
- J. Shi, B. Zhou, Y. Yang and Y. Li, Org. Biomol. Chem., 2012, 10, 8953–8955 RSC.
- X. Jia and J. Han, J. Org. Chem., 2014, 79, 4180–4185 CrossRef CAS.
- H. Wang, Y. Yu, X. Hong, Q. Tan and B. Xu, J. Org. Chem., 2014, 79, 3279–3288 CrossRef CAS.
- C. Zhang, Y. Zhou, Z. Deng, X. Chen and Y. Peng, Eur. J. Org. Chem., 2015, 1735–1744 CrossRef CAS.
- M. R. Yadav, R. K. Rit and A. K. Sahoo, Org. Lett., 2013, 15, 1638–1641 CrossRef CAS.
- M. Bhanuchandra, M. R. Yadav, R. K. Rit, M. R. Kurama and A. K. Sahoo, Chem. Commun., 2013, 49, 5225–5227 RSC.
- J. Kim, J. Kim and S. Chang, Chem.–Eur. J., 2013, 19, 7328–7333 CrossRef CAS.
- V. S. Thirunavukkarasu, K. Raghuvanshi and L. Ackermann, Org. Lett., 2013, 15, 3286–3289 CrossRef CAS.
- X. Zhou, P. Luo, L. Long, M. Ouyang, X. Sang and Q. Ding, Tetrahedron, 2014, 70, 6742–6748 CrossRef CAS.
- X. Wang, C. Zhang, J. Li, C. Jiang, F. Su, Z. Zhan, L. Hai, Z. Chen and Y. Wu, RSC Adv., 2016, 6, 68929–68933 RSC.
- X. Xiao, G. Jia, F. Liu, G. Ou and Y. Xie, J. Org. Chem., 2018, 83, 13811–13820 CrossRef CAS.
- C. Pan, A. Abdukader, J. Han, Y. Cheng and C. Zhu, Chem.–Eur. J., 2014, 20, 3606–3609 CrossRef CAS.
- M. V. Krishna Rao, K. N. Reddy, B. Sridhar and B. V. Subba Reddy, Asian J. Org. Chem., 2017, 6, 1851–1856 CrossRef CAS.
- Y. Shin, S. Han, U. De, J. Park, S. Sharma, N. K. Mishra, E.-K. Lee, Y. Lee, H. S. Kim and I. S. Kim, J. Org. Chem., 2014, 79, 9262–9271 CrossRef CAS.
- M. Bakthadoss, P. V. Kumar, R. Kumar, M. Surender and D. S. Shadra, New J. Chem., 2019, 43, 14190–14195 RSC.
- D. Lee, Y. Kim and S. Chang, J. Org. Chem., 2013, 78, 11102–11109 CrossRef CAS.
- H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS.
- J. Kim and S. Chang, Angew. Chem., Int. Ed., 2014, 53, 2203–2207 CrossRef CAS.
- K. Shin and S. Chang, J. Org. Chem., 2014, 79, 12197–12204 CrossRef CAS.
- W. Hou, Y. Yang, W. Ai, Y. Wu, X. Wang, B. Zhou and Y. Li, Eur. J. Org. Chem., 2015, 395–400 CrossRef CAS.
- Y. Kim, J. Park and S. Chang, Org. Lett., 2016, 18, 1892–1895 CrossRef CAS.
- S. Chen, B. Feng, X. Zheng, J. Yin, S. Yang and J. You, Org. Lett., 2017, 19, 2502–2505 CrossRef CAS.
- V. Lanke and K. R. Prabhu, Chem. Commun., 2017, 53, 5117–5120 RSC.
- M.-E. Wei, L.-H. Wang, Y.-D. Li and X.-L. Cui, Chin. Chem. Lett., 2015, 26, 1336–1340 CrossRef CAS.
- G. N. Hermann, P. Becker and C. Bolm, Angew. Chem., Int. Ed., 2016, 55, 3781–3784 CrossRef CAS.
- H. Hou, Y. Zhao, S. Sheng and J. Chen, Adv. Synth. Catal., 2019, 361, 4393–4398 CrossRef CAS.
- C. Pi, X. Cui and Y. Wu, J. Org. Chem., 2015, 80, 7333–7339 CrossRef CAS.
- Y. Li, Y. Feng, L. Xu, L. Wang and X. Cui, Org. Lett., 2016, 18, 4924–4927 CrossRef CAS.
- L. Wang, Z. Yang, M. Yang, R. Zhang, C. Kuai and X. Cui, Org. Biomol. Chem., 2017, 15, 8302–8307 RSC.
- B. Zhu, X. Cui, C. Pi, D. Chen and Y. Wu, Adv. Synth. Catal., 2016, 358, 326–332 CrossRef CAS.
- M. Maraswami, G. Chen and T. P. Loh, Adv. Synth. Catal., 2018, 360, 416–421 CrossRef CAS.
- Y. Feng, Z. Zhang, Q. Fu, Q. Yao, H. Huang, J. Shen and X. Cui, Chin. Chem. Lett., 2020, 31, 58–60 CrossRef CAS.
- H. Xiong, Y. Gu, S. Zhang, F. Lu, Q. Ji, L. Liu, P. Ma, G. Yang, W. Hou and H. Xu, Chem. Commun., 2020, 56, 4692–4695 RSC.
- L. Xu, L. Wang, Y. Feng, Y. Li, L. Yang and X. Cui, Org. Lett., 2017, 19, 4343–4346 CrossRef CAS.
- D. Das and R. Samanta, Adv. Synth. Catal., 2018, 360, 379–384 CrossRef CAS.
- Y.-l. Yun, J. Yang, Y.-h. Miao, J. Sun and X.-j. Wang, J. Saudi Chem. Soc., 2020, 24, 151–185 CrossRef CAS.
- Z. Hu, S. Luo and Q. Zhu, Sci. China: Chem., 2015, 58, 1349–1353 CrossRef CAS.
- (a) P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS; (b) G. Pototschnig, N. Maulide and M. Schnürch, Chem.–Eur. J., 2017, 23, 9206–9232 CrossRef CAS.
- B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491–1495 CrossRef CAS.
- J. Peng, Z. Xie, M. Chen, J. Wang and Q. Zhu, Org. Lett., 2014, 16, 4702–4705 CrossRef CAS.
- X. Kong, L. Lin and B. Xu, Adv. Synth. Catal., 2018, 360, 2801–2805 CrossRef CAS.
- X. Kong and B. Xu, Asian J. Org. Chem., 2019, 8, 1862–1865 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |