DOI:
10.1039/D0RA07663E
(Review Article)
RSC Adv., 2020,
10, 39304-39322
Synthesis and applications of methyleneaziridines
Received
7th September 2020
, Accepted 19th October 2020
First published on 27th October 2020
Abstract
Methyleneaziridines (MAs) are a special subset of aziridines featuring an exocyclic C–C double bond on the three-membered ring. They have found great potential in organic synthesis. In this review, the structural characterization of MAs, synthetic methods, chemical transformations and mechanisms, especially the advances achieved over the past decade are comprehensively summarized.
 Bin Pan | Bin Pan obtained his M.S. in medicinal chemistry in 2010 from Nanjing Tech University. He obtained his PhD degree in organic chemistry in 2014 from Dalian Institute of Chemical Physics, Chinese Academy of Sciences. The he worked as director of R&D department at Shandong Chengchuang Pharmaceutical R&D Co., ltd for 3 years. In 2018, he joined Weifang University of Science and Technology, where he works on the exploration of transition-metal catalyzed cycloadditions and synthesis of new chemical entities. |
 Feng Li | Feng Li obtained his M.S. degree in medicinal chemistry in 2007 from Nanchang University, China. He obtained his PhD degree in organic chemistry in 2012 from Fudan University, China. After postdoctoral studies (2012–2014) in Zhejiang University, he worked at Ocean University of China for 5 years, then joined Weifang University of Science and Technology in 2019. His current research focuses on medicinal chemistry, chiral chemistry and pharmaceutical process. |
 Yingying Zhao | Yingying Zhao was born in Hebei Province, China, in 1990. She received her B.S. degree from Jiangsu University in 2013 and PhD degree from Dalian Institute of Chemical Physics, Chinese Academy of Sciences, in 2018, under the guidance of Prof. Boshun Wan. Then she began her career at Liaoning Normal University, where she works on the exploration of new catalytic cycloadditions for the synthesis of heterocycles. |
1. Introduction
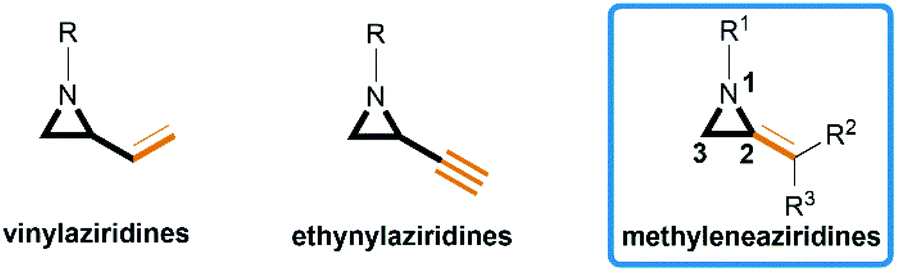
Aziridines are versatile building blocks in organic synthesis because their high strain energy associated with small rings provides a significant driving force for ring-opening functionalizations.1 Besides, their derivatives featuring unsaturated C–C bonds attached to the ring, such as vinylaziridines and ethynylaziridines, have attracted considerable attention as well. Surprisingly, in contrast, methyleneaziridines (MAs) with an exocyclic C–C double bond are much less studied, possibly due to their facile degradation and lack of practical synthetic methods (Fig. 1).
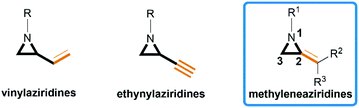 |
| | Fig. 1 Structures of vinylaziridines, ethynylaziridines and methyleneaziridines. | |
One of the most fundamental features of MAs is the introduction of a trigonal sp2-centre into the three – membered ring, which leads to a further increase in the angle strain energy. Computational studies have demonstrated that the strain energy of 2-methyleneaziridine is 12–13 kcal mol−1 (HF/6-31G*) higher than that of aziridine.2 NMR data shows that the 1JC,H of 1-tert-butylmethyleneaziridine is 170 Hz,3 a little higher than the corresponding coupling constants in methylenecyclopropane.4 Additionally, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching frequency in the IR spectra of MAs is raised to around 1770 cm−1.5
C stretching frequency in the IR spectra of MAs is raised to around 1770 cm−1.5
Theoretical studies, together with a number of X-ray analyses,6 indicate that the conjugation between the π-electrons of CC and the lone-pair electrons of nitrogen atom is rather limited.2 Even so, compared with simple aziridines,7 this weak conjugated effect can still shorten the length of the N–C2 bond and cause the faster rate of inversion of the nitrogen, thus leading to the higher reactivity of MAs. Owing to these unique structural properties, the MAs have shown great potential in chemical transformations. In 2006, Shipman and co-workers published an account on their own contributions to the synthesis and reactivity of MAs.8 Tehrani and Kimpe, in 2009, also summarized some representative works on this area.9 Nevertheless, to facilitate the understanding of the principles of MAs and discover new reactions of MAs, it would be highly desirable to provide a comprehensive review of these compounds. Furthermore, given the increasing number of publications in this area over the past decade, there is also an urgent need to give an updated summary on recent advances. This review attempts to cover the literatures of MAs, comprehensively, including the synthesis of these heterocycles and their useful transformations, such as nucleophilic ring-opening reactions, multicomponent reactions, cycloadditions and transition-metal-catalyzed reactions. Additionally, the synthesis of bicyclic MAs and their typical reactions will also be discussed in detail in Section 4.
2. Synthesis of monocyclic MAs
2.1. Base-induced cyclization and dehydrobromination reaction
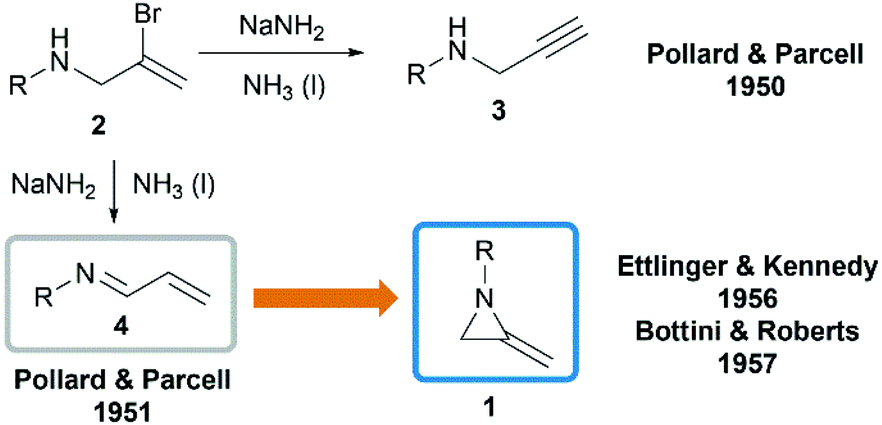
In 1951, when Pollard and Parcell attempted to synthesize propargylamines 3 by reacting N-(2-bromoallyl)ethylamine 2 with sodium amide in liquid ammonia, MAs were first obtained accidentally.10 The results were inconsistent with their previous work on the preparation of propargylamines from the corresponding tertiary amines.11 However, at that time, Pollard and Parcell proposed N-allylidene-alkylamine 4 as the product (Scheme 1), based on the fact that the strong signal detected at 1770 cm−1 in infrared spectrum and absence of the N–H or CC stretching frequencies.
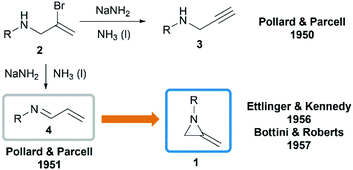 |
| | Scheme 1 Synthesis and structure of MAs. | |
In 1956, Ettlinger and Kennedy noted that the stretching frequency of 1770 cm−1 was similar to exocyclic alkene bond in methylenecyclopropanes and reassigned the structures as methyleneaziridines 1 (Scheme 1),12 which was later confirmed by Bottini and Roberts through NMR spectroscopy and chemical degradation studies.5 It is noteworthy that, up till now, NaNH2-promoted cyclization still remains the most general method for the synthesis of N-alkyl/benzyl substituted monocyclic MAs.13
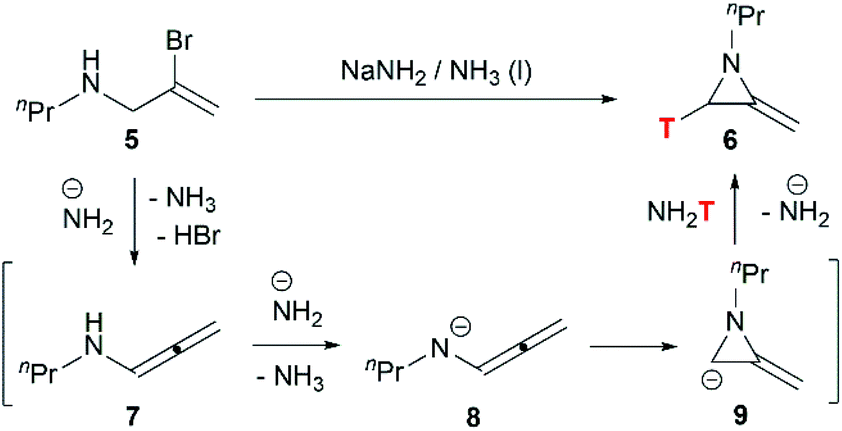
Although this cyclization has been used for nearly 70 years, the mechanism is still controversial. Bottini and Olsen found that when the ring-closing reaction of 5 was performed in tritium-enriched ammonia, C-3 tritium-labelled product 6 was obtained exclusively. Besides, neither 5 nor 6 could undergo hydrogen–tritium exchange with the solvent under the reaction conditions. Therefore, it is suggested that the MAs are formed via an elimination–addition process (Scheme 2).14
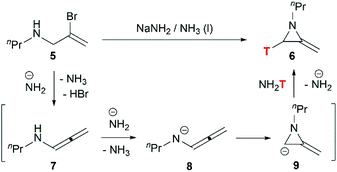 |
| | Scheme 2 Proposed elimination–addition mechanism. | |
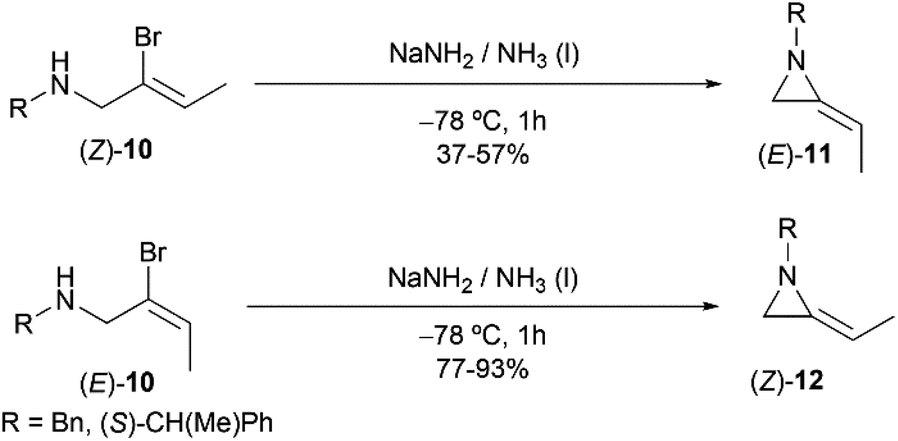
However, this explanation was questioned by Shipman and co-workers. Because they observed that (E)- and (Z)-10 yielded different regioselective products (Scheme 3),15 while if following the elimination–addition mechanism, both substrates would yield the same allene intermediate and lead to the convergence of stereochemistry.
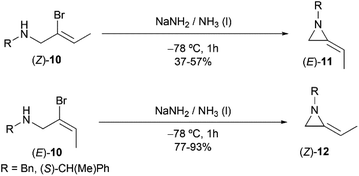 |
| | Scheme 3 Stereocontrolled ring-closing to 2-ethyleneaziridines with inversion at the vinylic carbon atom. | |
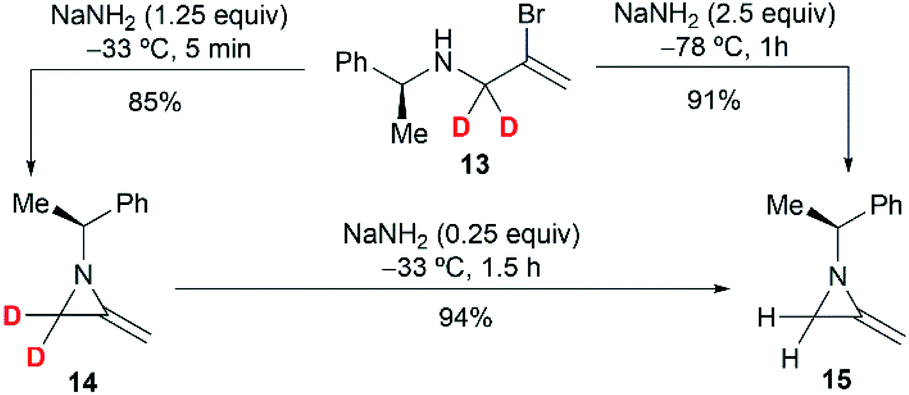
To further investigate the mechanism, the Shipman group studied the ring-closing of deuterated 2-bromoallylamine 13 (Scheme 4). There is no significant deuterium loss in the product 14 (92% D) when treating 13 with NaNH2 for five minutes at −33 °C, indicating that anion 9 could not be intermediate in this cyclization. Treatment of 13 with excess NaNH2 (2.5 equiv.) for a longer time (1 h) yielded normal MA 15 wherein all the deuterium had been lost. Additionally, deuterated MA 14 (92% D) could be completely converted into non-deuterated 15 (2.5% D) under the cyclization conditions. These experimental results, along with the observed stereochemical inversion in the preparation of MAs (Scheme 3) support an ‘SN2-like’ substitution mechanism with inversion by in-plane σ-attack from the backside of the C–Br bond.16
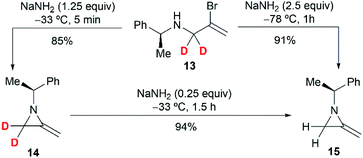 |
| | Scheme 4 Cyclization studies using deuterium labelled substrates. | |
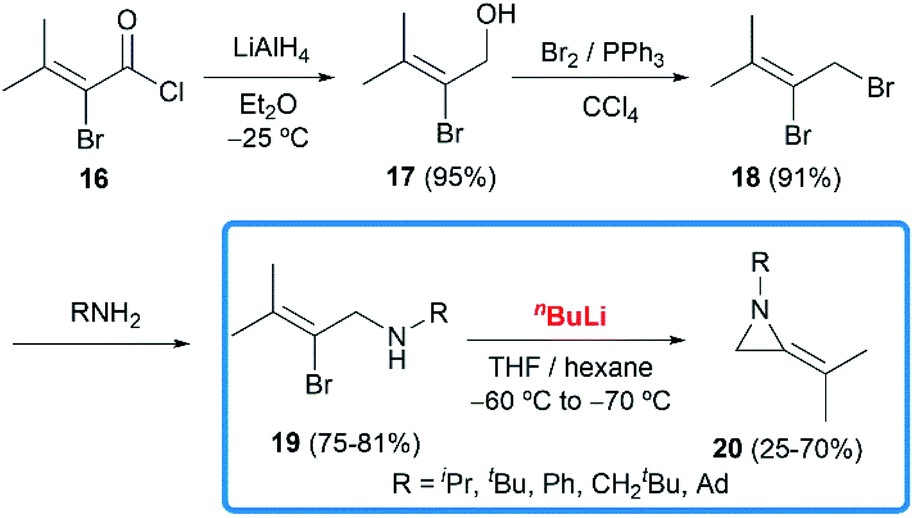
In 1981, Steinberg and co-workers reported the nBuLi-induced cyclization route to obtain MAs.17 Treatment of 19 with nBuLi in THF and hexane at low temperatures gave the disubstituted MAs 20 with acceptable overall yields (16–49% over 4 steps) (Scheme 5).
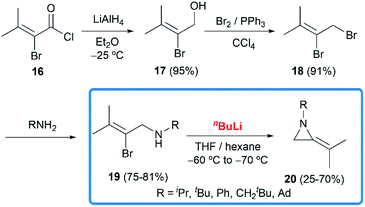 |
| | Scheme 5 nBuLi-induced cyclization to synthesize MAs. | |
De Kimpe and co-workers attempted to build the MA structures by a base-induced, exocyclic β-elimination reaction of 2-(bromomethyl) aziridines.18 A series of substrates were synthesized in good yield, then treated with tBuOK in THF at room temperature. A 1,2-dehydrobromination reaction furnished the expected products 23 in moderate yield, however, the production of an equimolar amount of tert-butyl ether 24 seemed unavoidable (Scheme 6).
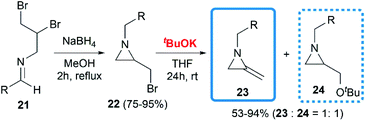 |
| | Scheme 6 tBuOK-induced dehydrohalogenation of aziridines. | |
2.2. Functionalization of monocyclic MAs
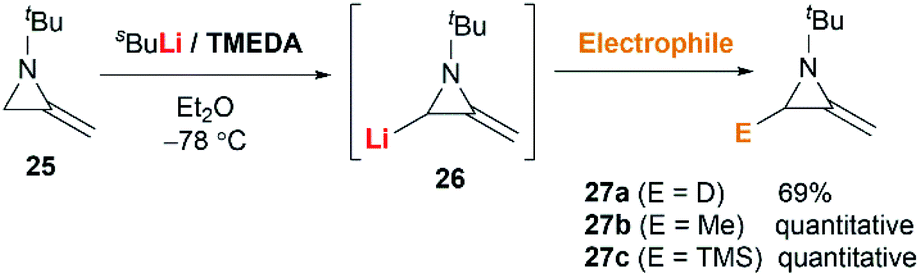
MAs could be lithiated using sec-butyllithium and subsequently alkylated with a range of electrophiles. Quast and Vélez first demonstrated that 1-tert-butyl-2-methyleneaziridine 25 could be converted into 26, quantitatively, by deprotonation with sBuLi in the presence of TMEDA.19 This organolithium species further reacted with a limited range of electrophiles (MeOD, MeI or Me3SiCl) to give MAs 27a–c in good yield (Scheme 7).
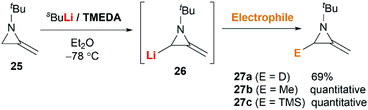 |
| | Scheme 7 Functionalization of MAs by deprotonation–alkylation. | |
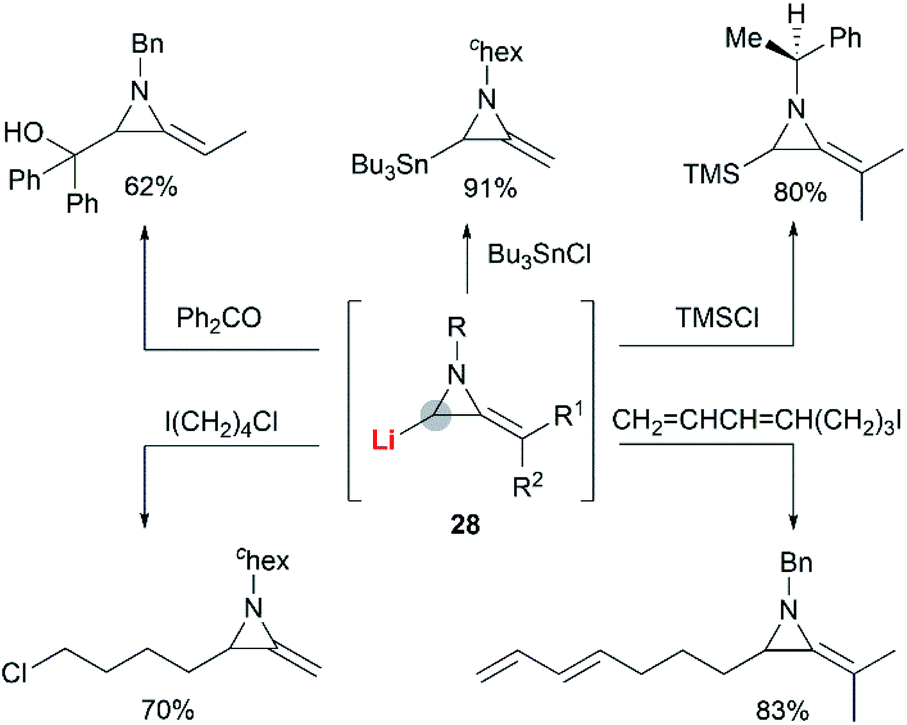
The Shipman group successfully achieved deprotonation–alkylation sequences with MAs bearing different N-substituents and alkene substitution patterns. Many common electrophilic reagents such as MeOD, MeI, PhCH2Br, CH2CHCH2Br, PhCHO, Ph2CO, Me3SiCl and Bu3SnCl could be used successfully (Scheme 8).20 However, the reaction was limited to the formation of monosubstituted products at C-3.
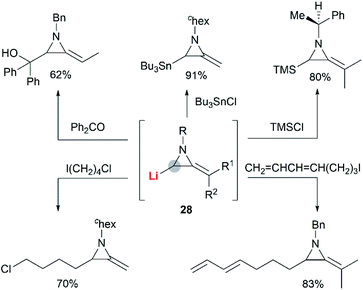 |
| | Scheme 8 Examples of C-3 functionalization using lithiated MAs. | |
Considering a new stereocenter generated in this process, Quast and Vélez had tried adding [(S,S)-(+)-bis(dimethylamino)-2,3-dimethoxybutane] as auxiliary chiral agent to control the stereochemistry of reactions as depicted in Scheme 7.21 Unfortunately, very modest enantioselectivity (12.3% ee) was detected in the deprotonation–alkylation. Shipman et al. reasoned that the introduction of a chiral element into nitrogen atom of the aziridine might improve the enantioselectivity. Gratifyingly, high levels of asymmetric induction have been achieved when isopropylidineaziridine (S)-29 was lithiated and alkylated by a range of electrophiles, including MeI, PhCH2Br, CH2CHCH2Br, Ph2CO and Me3SiCl (Scheme 9).22
 |
| | Scheme 9 Asymmetric induction in the lithiation–alkylation of MAs. | |
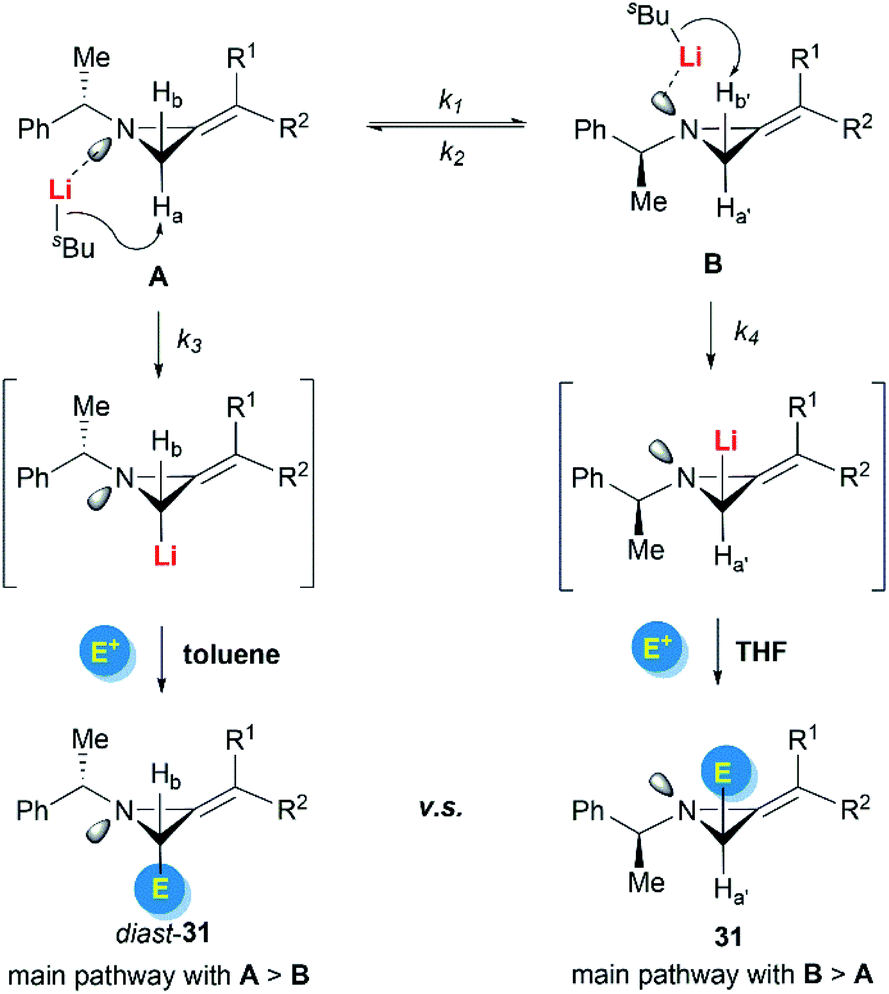
Shipman and Luisi et al. further investigated the stereoselectivity observed in the lithiation–alkylation of MAs.23 Factors such as nitrogen inversion barrier, the stereochemistry at the nitrogen atom, the substitution pattern of the MAs, and the reaction conditions were all taken into consideration. As described in Scheme 10, they concluded that the interplay between nitrogen stereodynamics and complexation phenomena seems to be crucial in determining the stereochemical outcome of the lithiation/trapping sequence. The findings were rationalized by a synergistic use of NMR experiments, run on the lithiated intermediates, alongside computational data.
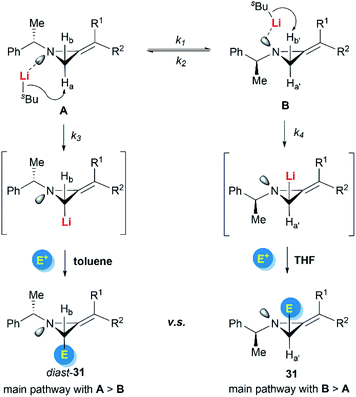 |
| | Scheme 10 Reactivity switch based on nitrogen stereodynamics. | |
2.3. Synthesis of N-aryl and N-electron-withdrawing groups substituted monocyclic MAs
In contrast to the convenient synthesis of MAs bearing alkyl or benzyl groups on nitrogen, construction of aryl or electron-withdrawing groups substituted MAs on the N-atom remains a big challenge.
In 2015, Shipman and co-workers reported their effort on the synthesis of N-aryl MAs.24 Based on the DFT calculations and competition experiments, a vinyl iodide 32 was designed and prepared for ring-closing reaction. Inspiringly, treatment of 32 with NaNH2 successfully provided MAs 33 in a high state of purity after aqueous workup with 84% yield (Scheme 11). Although there was only one case in this research, the methodology did enable the synthesis of N-aryl MA derivatives, that cannot be achieved before.25 Interestingly, MA 33 displayed low thermal stability and rapidly rearranged to cyclopropanimine 34 according to first-order kinetics (t1/2 < 9 min at 88 °C). The similar behaviour has been noted previously for 1-methyl-2-methyleneaziridine but much higher temperature is required (t1/2 = 9 min at 190 °C).26
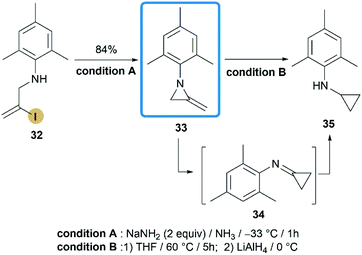 |
| | Scheme 11 Synthesis and rearrangement of 1-mesityl-2-methyleneaziridine. | |
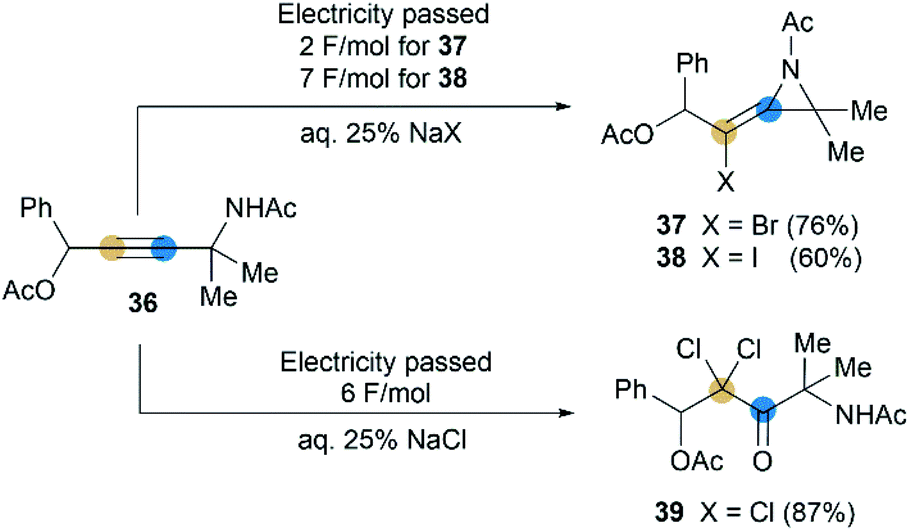
Although rare examples have been reported, intramolecular cyclization of propargyl amides is no doubt a feasible method to synthesize MAs. In 1992, Torii et al. reported that an electrohalogenation of propargyl amides 36 could furnish the corresponding MAs 37 and 38 (Scheme 12),27 which were highly functionalized, bearing halogen atom on exocyclic alkene and acetyl group on N-atom. This reaction was carried out by using CH2Cl2-aqueous 25% NaX (X = I or Br)-Pt system (buffered at pH = 7) under 20 mA cm−2. However, the same electrolytic reaction with NaCl only produced dichloroketone 39 exclusively.
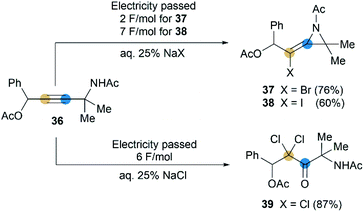 |
| | Scheme 12 Electrohalogenation of propargyl amides. | |
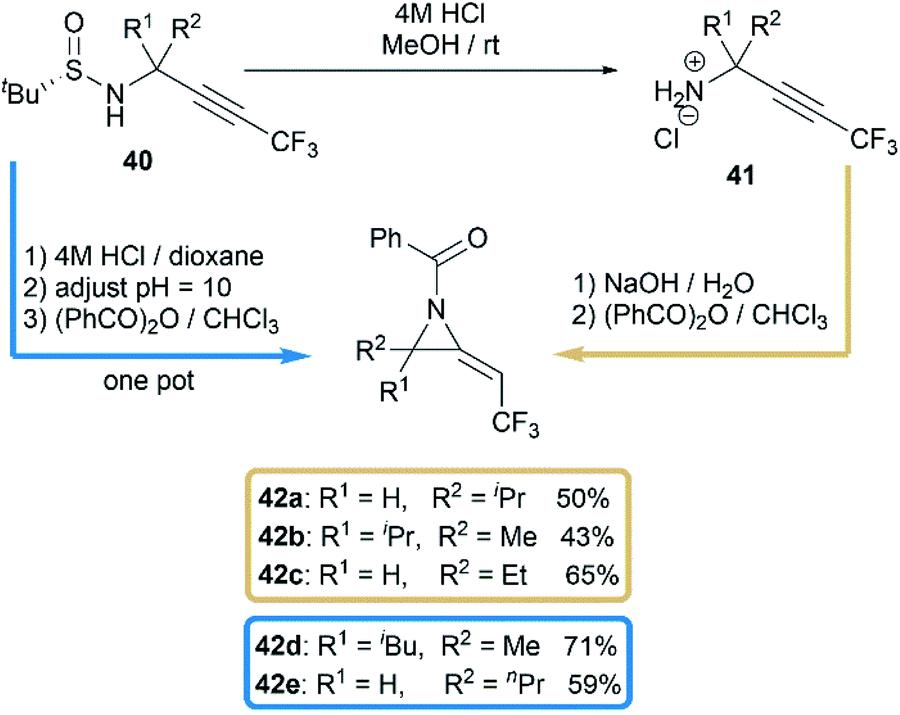
Another interesting research was reported by Qing and co-workers when they studied benzoylation reaction of trifluoromethylated propargylamine substrates 40.28 N-benzoyl-2-trifluoroethylideneaziridines 42a–e were surprisingly obtained in moderate yields when using NaOH as base, as illustrated in Scheme 13. The relatively strong basicity of NaOH, considered as key factor, promoted the deprotonation of N-benzamide intermediates 41, followed by an intramolecular addition of nitrogen anion to alkynyl group to deliver methyleneaziridines.
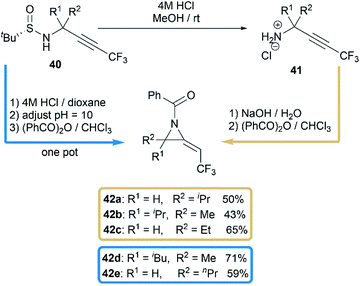 |
| | Scheme 13 Syntheses of N-benzoyl-2-trifluoroethylideneaziridines. | |
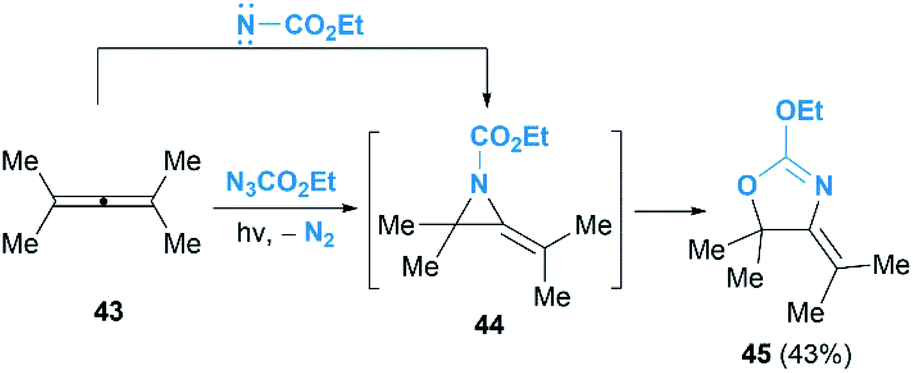
As we known, the allene cyclopropanation is the most common method to access methylenecyclopropanes.29 In contrast, the corresponding aziridination of allenes to construct heteroatom analogues such as MAs have been underutilized. Pioneering research in this area was reported by Bleiholder and Schecter in the reaction of ethyl azidoformate and dimethylallene 43 (Scheme 14).30 They postulated that an addition of singlet nitrenes, in situ generated from ethylazidoformates, to allenes under photolysis condition might deliver MAs. Unfortunately, instead of the MA products, an oxazoline 45 was obtained in 43% yield as the sole product. It is proposed that the oxazoline is produced via further rearrangement of MA 44, but the mechanism involving a direct [3 + 2] cycloaddition of the allene with carbethoxynitrene, N2 extrusion and rearrangement could not be ruled out.
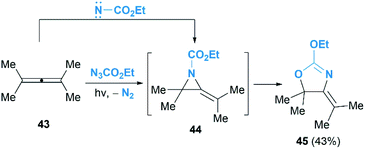 |
| | Scheme 14 Photolysis of ethyl azidoformate in the presence of dimethylallene. | |
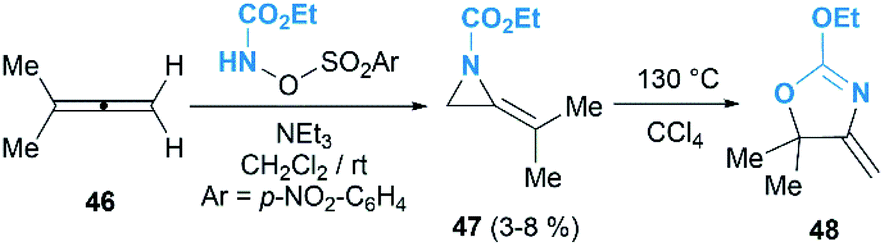
The Gilbert group reinvestigated this work in the reaction of nosyloxycarbamate with allenes.31 Intriguingly, trace amounts of MAs 47 could be isolated, as confirmed by MS and NMR analysis, when using N-(p-nitrobenzenesulfonoxy) urethane as a nitrene precursor (Scheme 15). Thermolysis of the MAs products gave oxazolines 48 indeed, demonstrating the competency of MAs as intermediates in the formation of oxazolines.
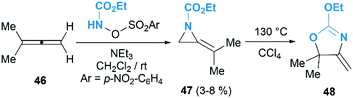 |
| | Scheme 15 Synthesis of MAs by addition of nitrene with allene. | |
2.4. Conversion reactions via aziridine derivatives
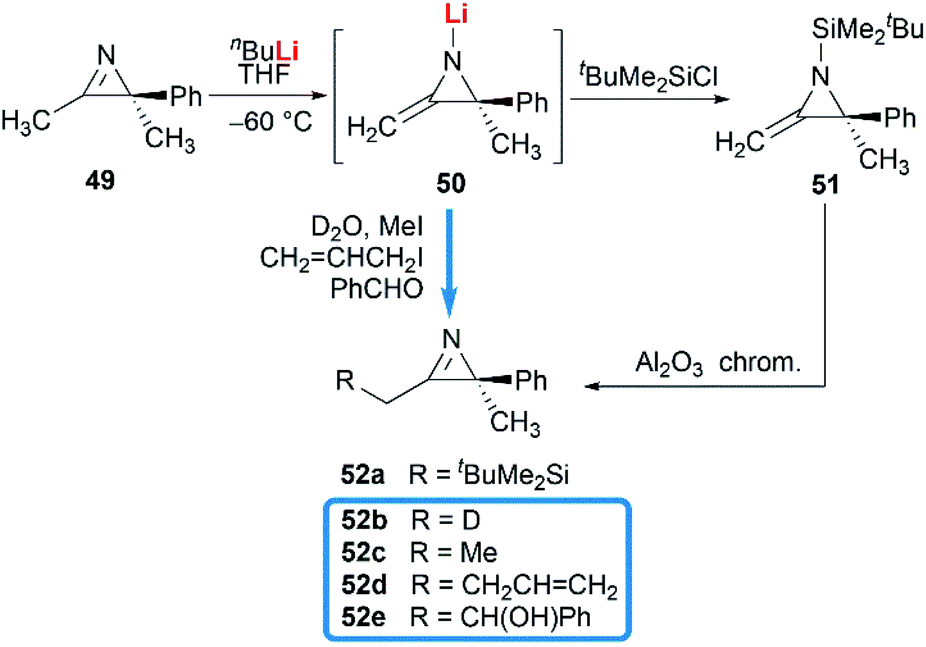
Notably, some aziridine derivatives could be converted into MAs under suitable conditions. A meaningful attempt for building MAs structure via a N-lithiation/silylation of 3-methyl-2H-azirine 49 was reported by Belloir and co-workers.32 Deprotonation of 2H-azirine with butyllithium gave rise to N-lithio-2-methyleneaziridine 50, which could be trapped by tert-butylchlorodimethylsilane to provide N-silyl MA 51 (Scheme 16). Unfortunately, the alkylation of 50 with other electrophiles (MeI, allyl iodide, benzaldehyde, D2O) all failed to give expected MAs but yielded C-alkylation products. In addition, all attempts on the purification of 51 by chromatography on aluminium oxide irreversibly transformed it into the C-silyl isomer 52a.
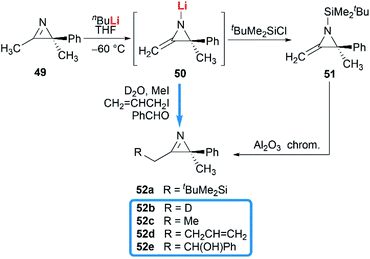 |
| | Scheme 16 N-Lithiation/silylation of 3-methyl-2H-azirine to synthesis of MAs. | |
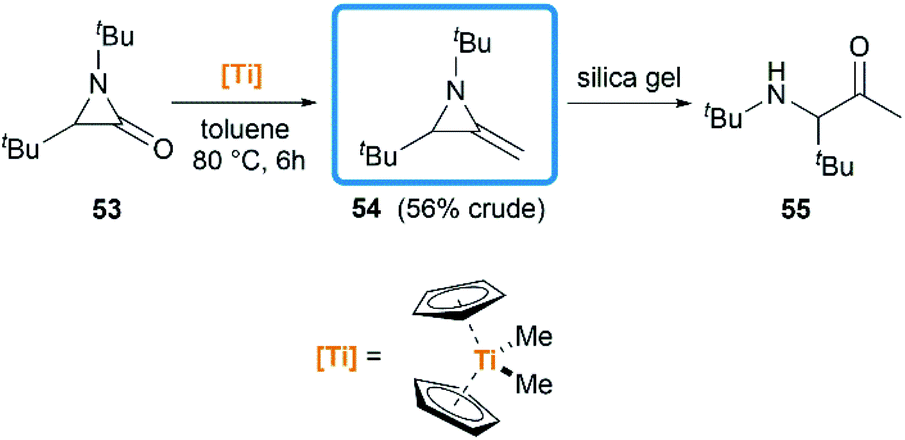
Another conversion involving α-lactam, was reported by De Kimpe et al.33 An olefination reaction of 1,3-di-tert-butyl-2-aziridinone 53 with dimethyltitanocene in toluene gave the MA in 56% crude yield (Scheme 17). Purification of product by column chromatography on neutral alumina or vacuum distillation (24–26 °C/1.3 mm Hg) was achievable. In contrast, the MA 54 was completely hydrolyzed to 3-(tert-butylamino)-4,4-dimethyl-2-pentanone 55 by chromatography on silica gel.
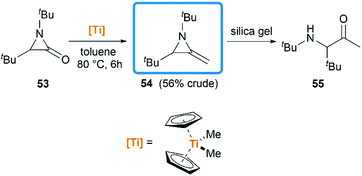 |
| | Scheme 17 Synthesis of MAs by Cp2TiMe2 mediated methylenation of α-lactam. | |
3. Reactions of monocyclic MAs
3.1. Protonations
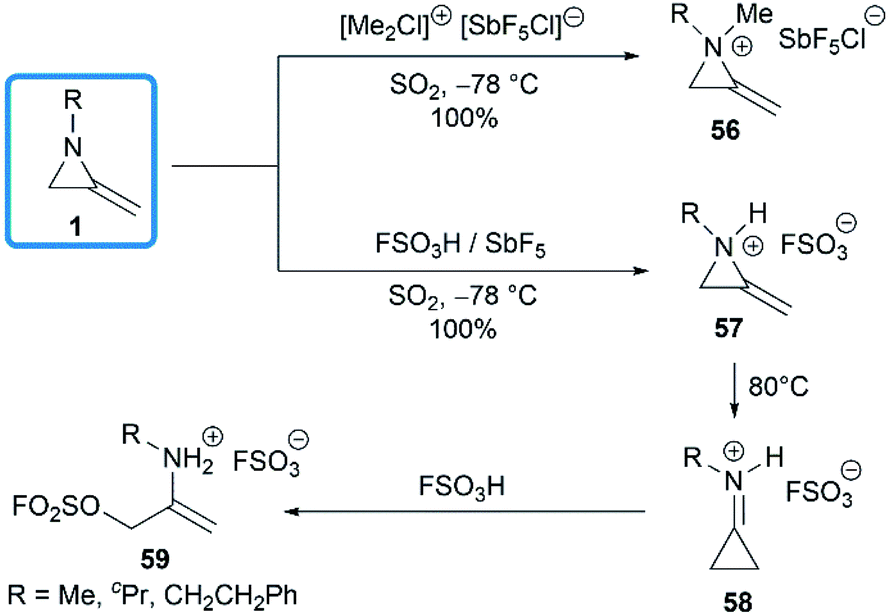
The domino protonation and ring-opening reactions of MAs were investigated in details by Jongejan and co-workers.34 Extraction of pentane solutions of 1 with FSO3H/SbF5 in sulphur dioxide at −78 °C gave the corresponding methyleneaziridinium ions 57 without any side products. Under similar conditions, the nitrogen atom of MA 1 could be methylated by [(Me)2Cl]+[SbF5Cl]− to yield 56. Both methyleneaziridinium salts 56 and 57 showed remarkable thermal stability below 60 °C, no decomposition of either cation being observed by 1H NMR. Thermally induced isomerization and the following ring opening required higher temperature (Scheme 18).13,35
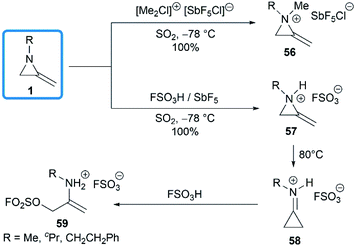 |
| | Scheme 18 Generation and isomerization of methyleneaziridinium cations. | |
3.2. Decompositions and rearrangements
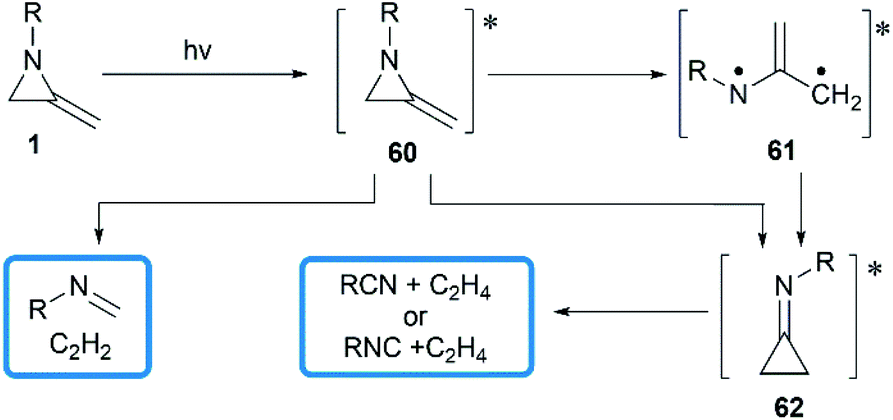
In 1964, Brinton reported the photolysis decomposition of MAs, giving the corresponding alkenes, nitriles and isonitriles as major products.36 The author concluded that the rearrangement mechanism might either proceed by a direct concerted mechanism (60 → 62) or through a short-lived biradical 61 as illustrated in Scheme 19.
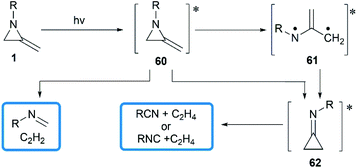 |
| | Scheme 19 Photolysis decomposition of MAs. | |
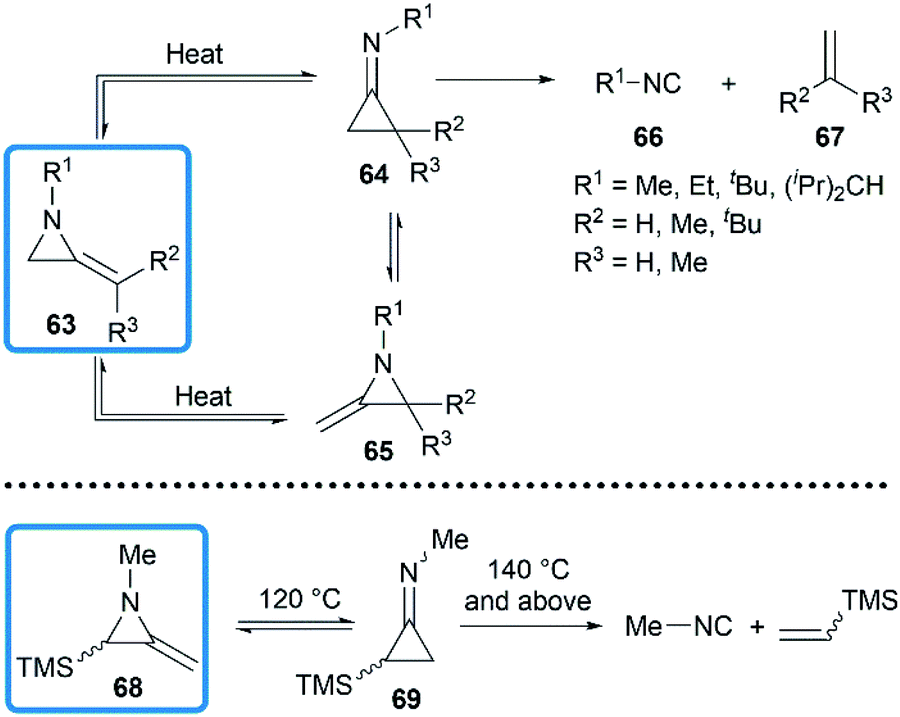
The valence isomerization of methyleneaziridine-cyclopropanimine was also observed under thermal conditions. Quast and Risler demonstrated that a series of MAs primarily rearranged to cyclopropanimine 64 and isomer 65 at 190 °C, which were rapidly converted into isonitriles 66 and alkenes 67 in quantitative yield (Scheme 20).26 Surprisingly, unlike the notorious instability of cyclopropanones,37 cyclopropanimine 64 was relatively stable and readily accessible. The rearrangement of trimethylsilyl MA 68 was also observed at 120 °C, while decomposition did not occur only at 140 °C and above, although to a small extent (Scheme 20).21
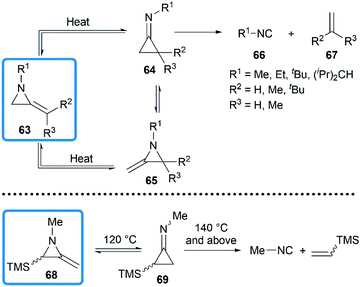 |
| | Scheme 20 Thermal isomerization and decomposition of MAs. | |
3.3. Nucleophilic ring-opening reactions
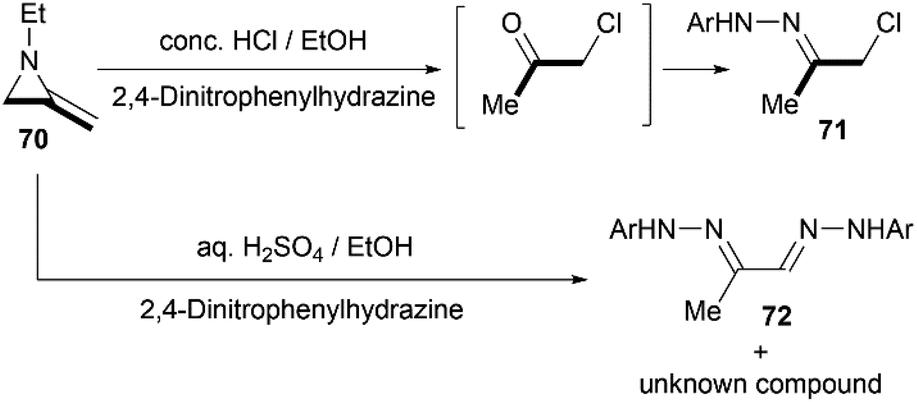
Protonation of the nitrogen atom of MAs and subsequent nucleophilic attack to the aziridine ring lead to ring opening at the N–C3 bond. Bottini and Roberts have reported that a treatment of 1-ethyl-2-methyleneaziridine 70 with hydrochloric acid could provide chloroacetone, which was isolated as its 2,4-dinitrophenylhydrazone derivative 71. However, the hydrolysis with aqueous sulfuric acid in the presence of 2,4-dinitrophenylhydrazine gave osazone 72 and an unknown compound (free of sulfur), as illustrated in Scheme 21.5
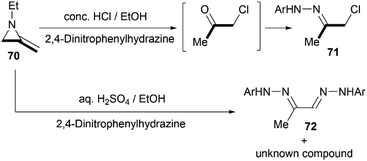 |
| | Scheme 21 Brønsted acid-promoted hydrolysis reaction of MAs. | |
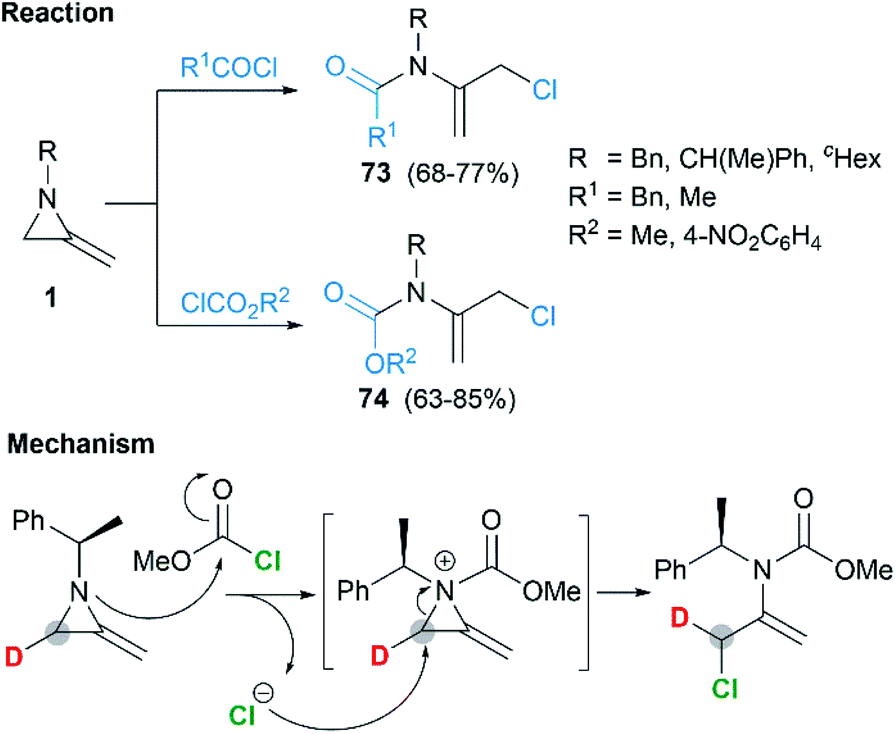
Shipman and co-workers demonstrated that MAs could react with acid chlorides and chloroformates, giving corresponding tertiary amide 73 and carbamates 74 respectively.38 These reactions proceeded well with a series of N-alkyl/benzyl substrates. However, N-trityl MAs, owing to its steric hindrance, could not participate in the process. In addition, these reactions were proved to be less tolerant to more electron-rich acid chlorides such as benzoyl chloride and p-anisoyl chloride. Deuterium labelling studies confirmed that attack of the chloride anion occurred solely at C-3 of the methyleneaziridinium ion (Scheme 22).39
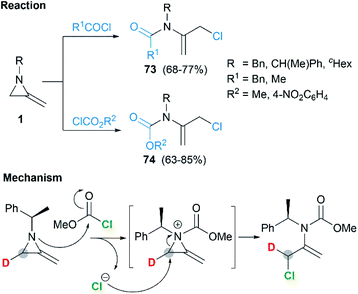 |
| | Scheme 22 Ring-opening and mechanism of MAs with acid chlorides and chloroformates. | |
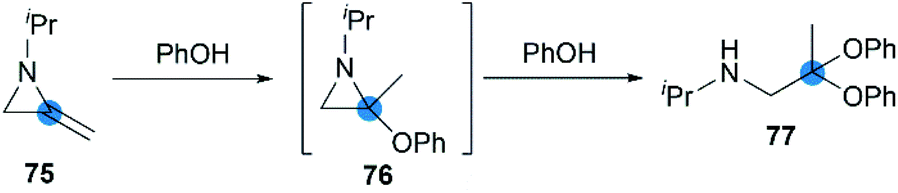
Crandall et al. reported that, N-isopropyl-2,2-diphenoxypropan-1-amine 77 was rapidly and smoothly obtained from the reaction of MA 75 with excess phenol at 25 °C.40 This transformation probably proceeded via initial Markovnikov addition of the first equivalent of the phenol across the exocyclic double bond, followed by ring-opening of the aziridine intermediate by the second equivalent of phenol (Scheme 23).
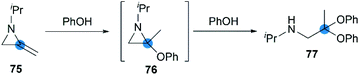 |
| | Scheme 23 Reaction of MA with phenol. | |
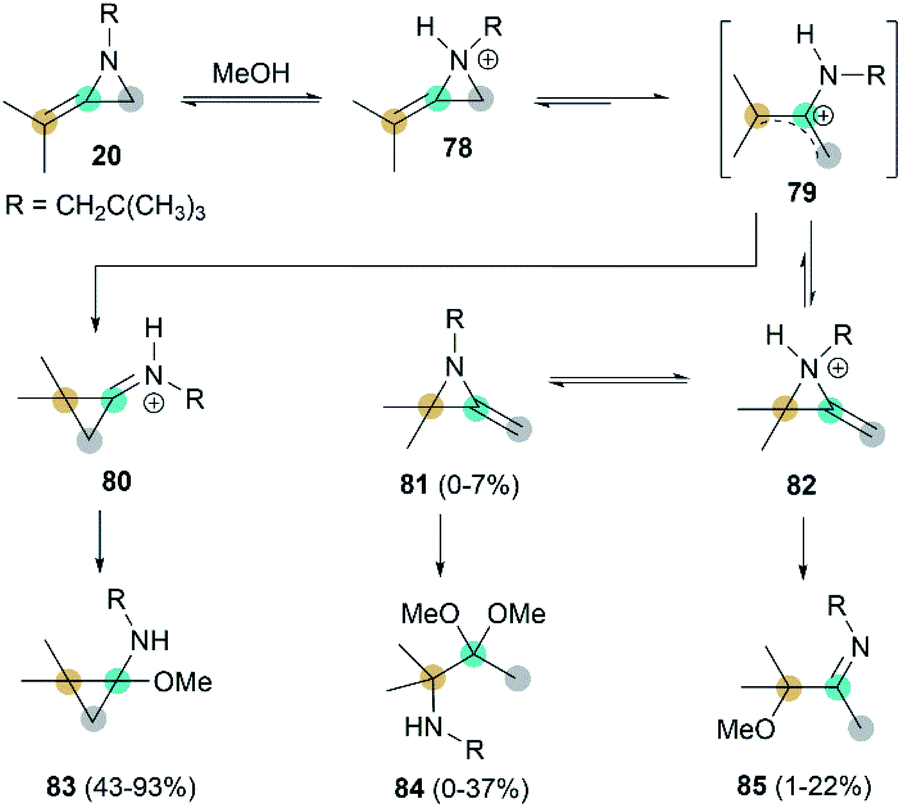
Interestingly, when heating MA 20 and methanol in the presence of benzene, adduct 83 was isolated as the major product.41 This addition begins with rapid N-protonation with methanol, followed by a slow ring opening of 78 to afford the neopentylamino allyl cation 79. Subsequent ring-closure delivers iminium ion 80, which can be easily captured by methanol to generated product 83. The minor ring-opening products methoxyimine 84 and aminoacetal 85 were formed probably via the protonated isomeric MA 82 (Scheme 24).
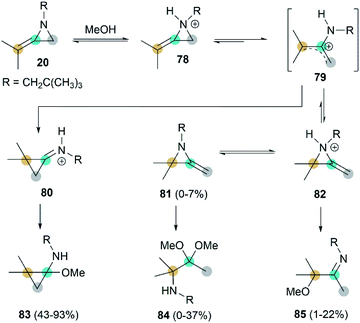 |
| | Scheme 24 Rearrangements of MA in methanol. | |
3.4. Carbon-based nucleophilic ring-opening process and multicomponent reactions (MCR)
In the presence of an organometallic promoter, the ring-opening of MAs 86 with carbon-based nucleophile would produce a metalloenamine 87, which might further undergo C-alkylation with an electrophile to yield ketimine 88 (Scheme 25). Obviously, the structure of the nucleophile, electrophile or the MAs could be changed to prepare a wide range of different ketimines, which displayed diverse chemical reactivities in organic synthesis, as shown in Scheme 25.
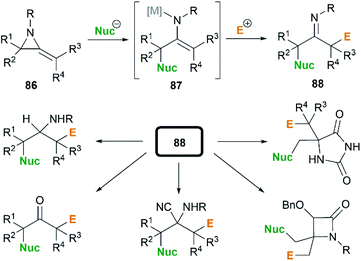 |
| | Scheme 25 Multi-component reactions of MAs. | |
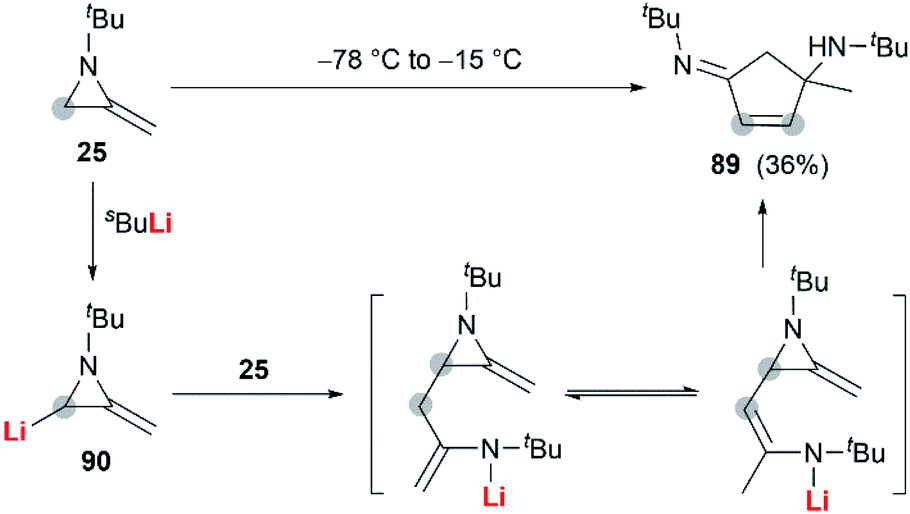
In 1974, Quast and Vélez reported a dimerization reaction of tert-butyl-2-methyleneaziridine 25 (Scheme 26).20 A solution of 2-lithiated MA 90, prepared by the treatment of 25 with butyllithium/TMEDA in ether, was quenched by H2O to give a pale-yellow crystal, which was sensitive to air and moisture. Elemental analysis and mass spectrum confirmed the product structure as a dimer 89. The mechanism involves nucleophilic attack of 25 with lithiated MA 90, ring-opening and intramolecular cyclization.
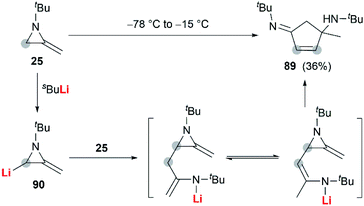 |
| | Scheme 26 Dimerization reaction of MAs. | |
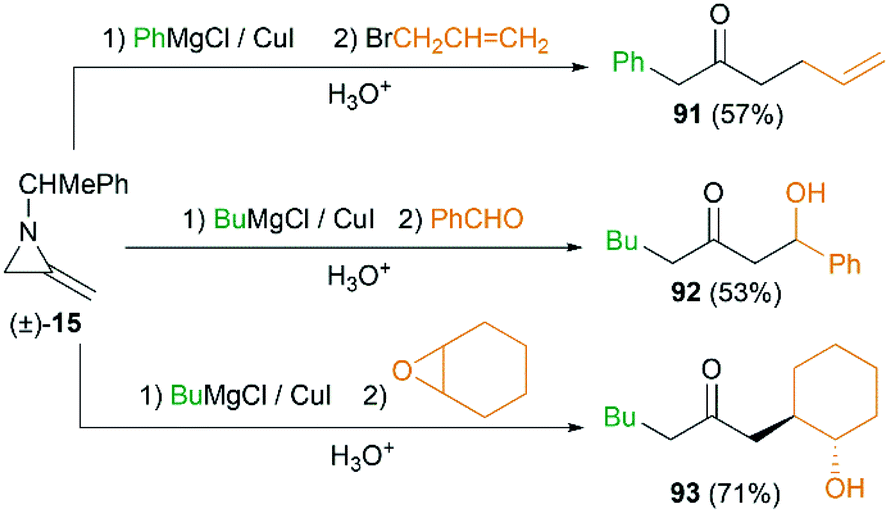
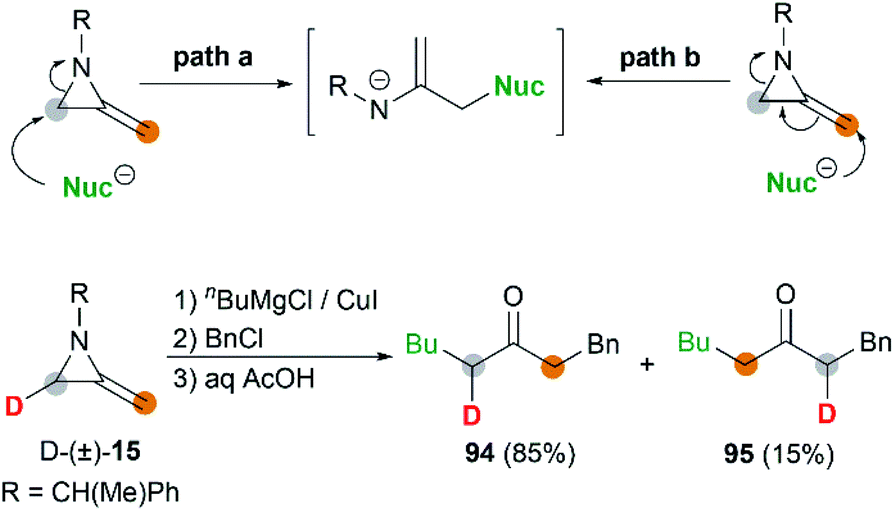
MAs could also be conveniently ring-opened to the metalloenamines using Grignard reagents in the presence of CuI. Subsequent addition of an electrophile facilitates further C-alkylation to yield the ketimines which were then hydrolyzed to ketones. The Shipman group has reported that a range of ketones including 91–93 could be produced using this multicomponent reaction (MCR) (Scheme 27).42 To further elucidate the reaction mechanism, when C-3 deuterated MA 15 was subjected to standard conditions using BuMgCl and BnCl, an inseparable mixture of deuterated ketones 94 and 95 was furnished in a ratio of 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 (Scheme 28). It is clear that ring opening of MA by Grignard reagents occurs predominantly by attack at C-3 of the aziridine ring as illustrated in Scheme 28 (path a), and the path b is also involved as a minor reaction pathway.
:15 (Scheme 28). It is clear that ring opening of MA by Grignard reagents occurs predominantly by attack at C-3 of the aziridine ring as illustrated in Scheme 28 (path a), and the path b is also involved as a minor reaction pathway.
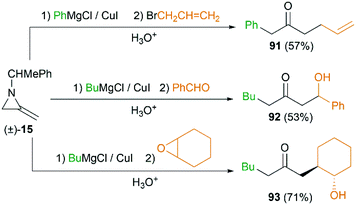 |
| | Scheme 27 Examples of ketone-forming reaction. | |
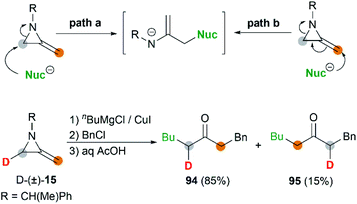 |
| | Scheme 28 Possible machanisms and duterium-labelled study for MAs ring-opening. | |
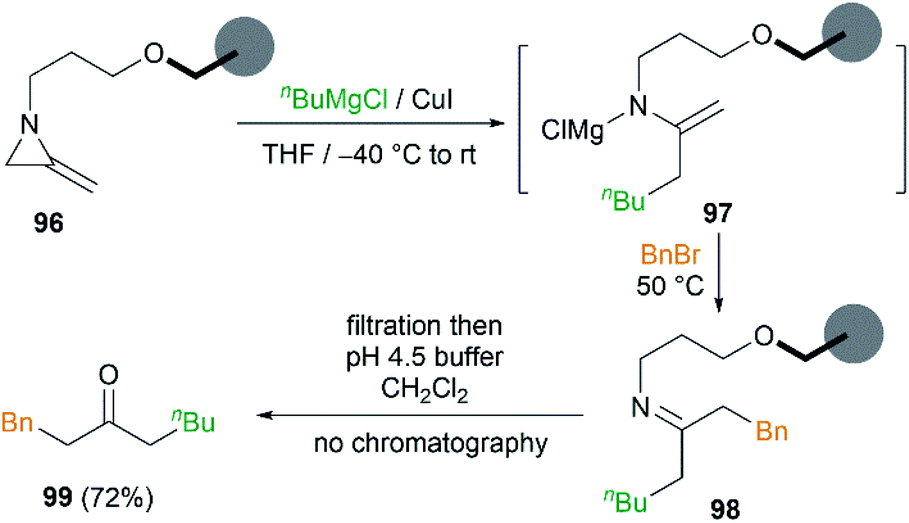
In order to remove excess reagents and simplify the purification of reaction, attachment of the MAs to solid-support via the nitrogen atom was carried out by Shipman et al.43 It was demonstrated that merrifield resin bound MA 96 could be converted into 1-phenyloctan-3-one 99 under optimized conditions in 72% yield (Scheme 29). Although the yield was not better than that obtained in solution, the purification protocol was greatly simplified and no chromatography was required.
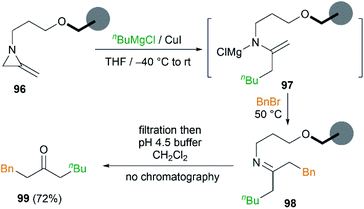 |
| | Scheme 29 Solid-phase ketone-forming reaction. | |
This MCR is also proved to be a useful tool to synthesize amines by way of reduction of the intermediate ketimine (Scheme 30). The stereochemistry of reaction could be well controlled by the substituents on the nitrogen atom of MAs. Isopropylidineaziridine (S)-29 produced homochiral amines 100 in 73% yield and 98% de. Further transformation into amine 101 (≥95% ee) proceeds smoothly by hydrogenation in presence of palladium on carbon.44
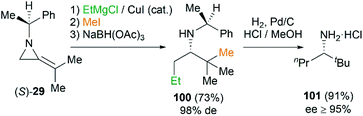 |
| | Scheme 30 Synthesis of homochiral amines using MCR. | |
Interestingly, using difunctionalized electrophiles, the scope of the MCR could be extended to the synthesis of 2-substituted piperidines.45 Shipman group reported a concise asymmetric synthesis of the hemlock alkaloid, (S)-coniine,46 as shown in Scheme 31. Treatment of 15 with EtMgCl, then 1,3-diiodopropane, and finally sodium triacetoxyborohydride gave piperidine 102 in 42% yield as essentially a single diastereomer after chromatography. Hydrogenolysis of 102 furnished (S)-coniine hydrochloride (≥95% ee) in 93% yield.
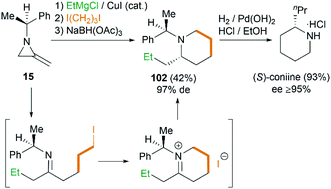 |
| | Scheme 31 Asymmetric synthesis of (S)-coniine using MCR. | |
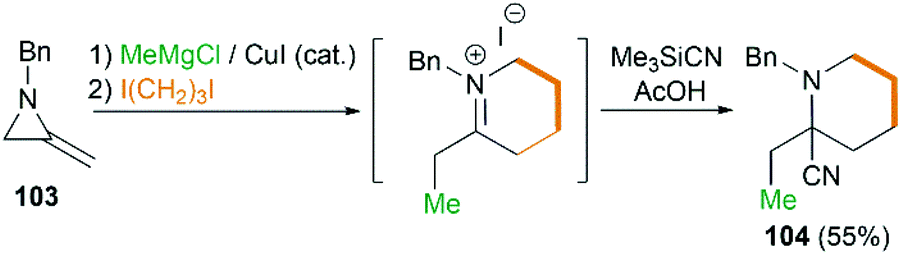
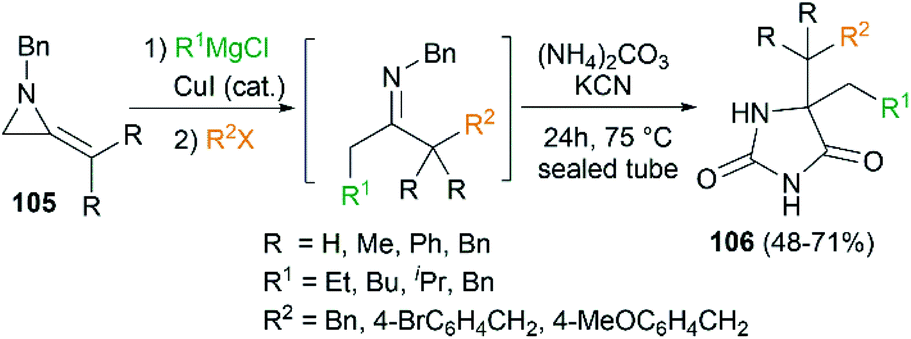
By combination of the MCR strategy with the Strecker reaction, a complicated four-component reaction could be achieved. Treatment of 103 with MeMgCl, 1,3-diiodopropane, then HCN (generated from Me3SiCN and AcOH) provided piperidine 104 in 55% yield (Scheme 32).47 This process is efficient in the production of the total number of new bonds (three C–C bonds and one C–N bond). It's also noteworthy that, the resulting imine in the MCR, without isolation, could be further treated with ammonium carbonate and potassium cyanide to give 5,5′-disubstituted hydantoins 106 via a Bucherer–Bergs protocol (Scheme 33),48 or reacted with (benzyloxy)ketene leading to α-lactams 107 via a Staudinger [2 + 2] cycloaddition (Scheme 34).49
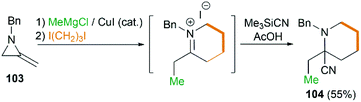 |
| | Scheme 32 Synthesis of cyclic α-amino nitrile. | |
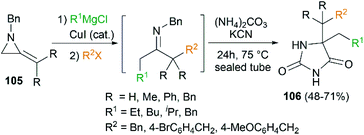 |
| | Scheme 33 Synthesis of 5,5′-disubstituted hydantoins via MCR of MAs. | |
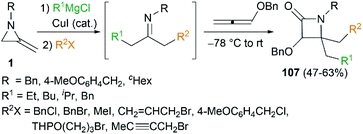 |
| | Scheme 34 Synthesis of α-lactams via MCR of MAs. | |
3.5. Radical cascades
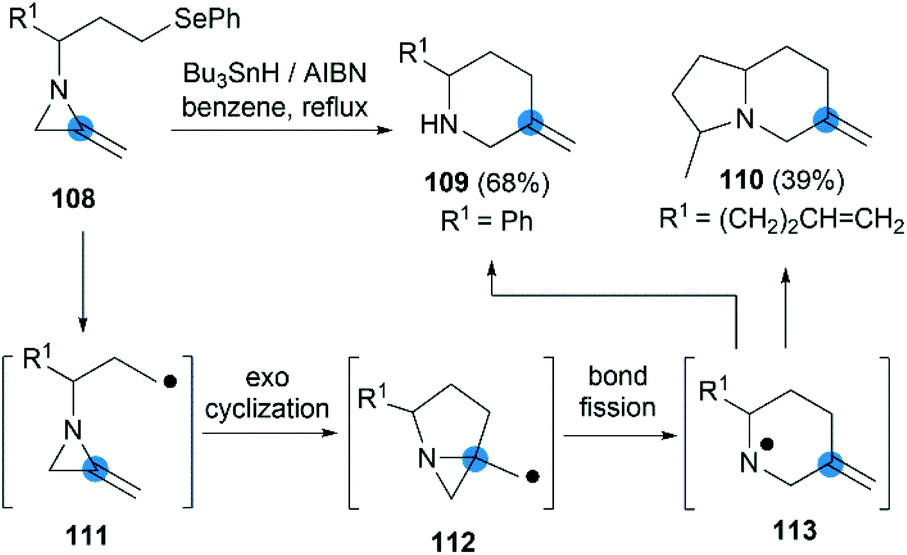
The strain energy of the MAs ring provides a suitable driving force for radical cascades. Shipman et al. first found that the alkyl radical 111, generated from a suitable precursor such as 108, could undergo 5-exo-trig cyclization to form aziridinylcarbinyl radical 112, which was transformed to aminyl radical 113 via N–C2 bond cleavage then provided piperidine derivatives in moderate yield (Scheme 35).50 Using this radical cascade methodology, they also achieved a tandem radical process to form indolizidine 110 in 39% yield (dr = 4:1).51
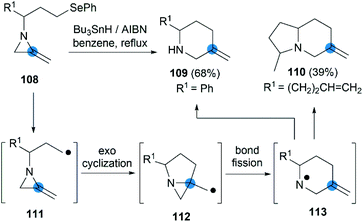 |
| | Scheme 35 Intramolecular radical rearrangement reactions of MAs. | |
3.6. Cycloadditions
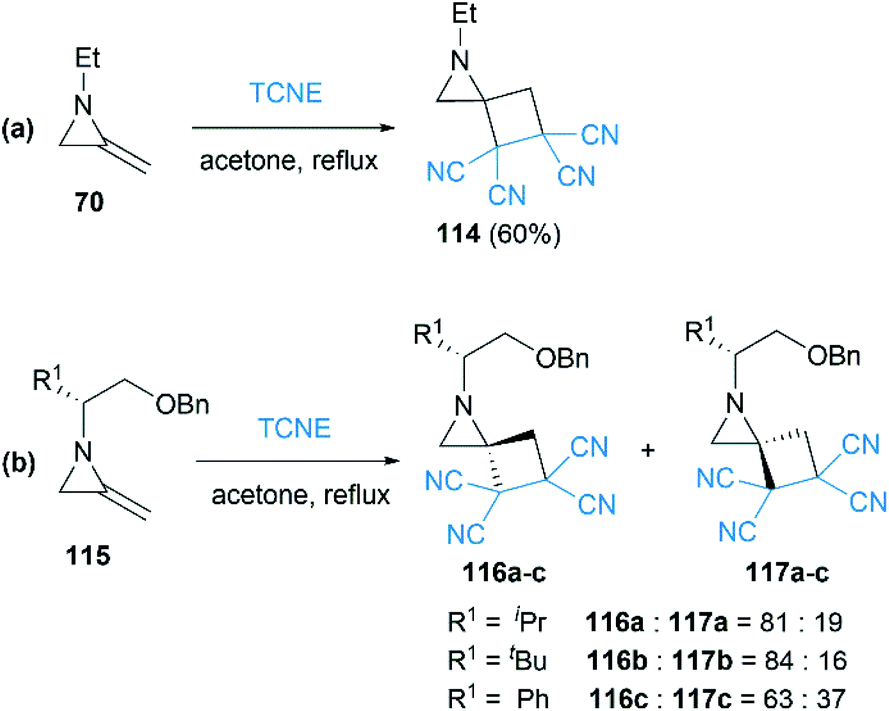
As early as 1967, Cookson and co-workers had shown that the exocyclic double bond of MAs underwent intermolecular [2 + 2] cycloaddition reactions52 with electron deficient alkenes.53 Treatment of 1-ethyl-2-methyleneaziridine 70 with tetracyanoethylene (TCNE) in refluxing acetone resulted in the formation of the corresponding spiroadduct 114 in 60% yield (Scheme 36(a)). Shipman and co-workers employed unique MAs that bearing a chiral motif on the nitrogen atom in this [2 + 2] process.54 The nature of the substituent on the stereocenter exerted an influence on the diastereoselectivity. Increasing the size of the R1 substituent appeared to improve the diastereoselectivity, although low selectivity was observed when R1 = Ph as shown in Scheme 36(b).
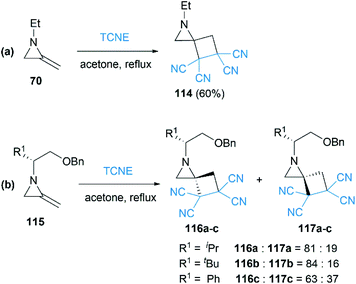 |
| | Scheme 36 [2 + 2] Cycloaddition reactions and diastereoselective control strategy. | |
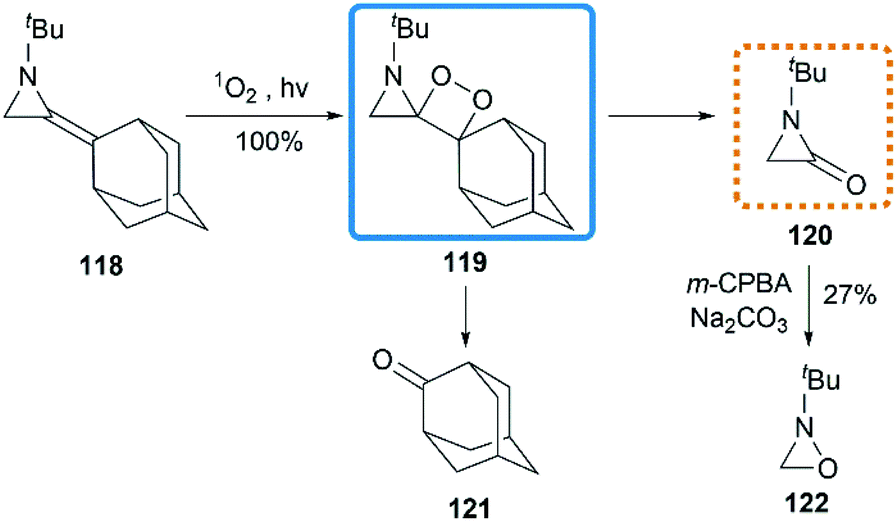
Ando et al. reported that the photooxygenation of 1-tert-butyl-2-adamantylideneaziridine 118 resulted in its oxidative cleavage (Scheme 37).55 A typical [2 + 2] cycloaddition reaction firstly gave dioxetane 119, which was found to be stable below −74 °C and characterized by low temperature NMR. However, the only isolable product in this reaction was adamantanone 121, generated from the rapid decomposition of cycloaddition product 119. While another fragment in this decomposition was α-lactam 120, which was confirmed by the formation of oxaziridine 122 upon in situ oxidation with m-CPBA.
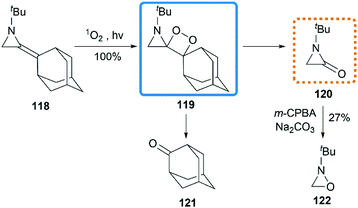 |
| | Scheme 37 Photooxygenation of MAs. | |
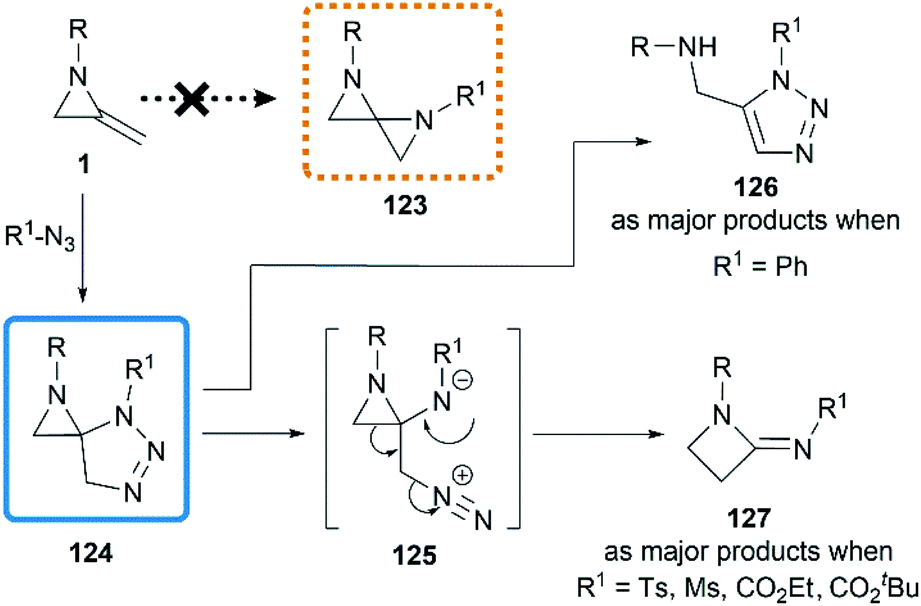
Initial attempts at the [3 + 2] cycloadditon56 were reported by Crandall and co-workers in 1975.40 They expected that a conversion to 1,4-diazospiropentanes 123 might be achieved by treatment of MAs with organic azides following by release of nitrogen (Scheme 38). However, the reaction of 1-isopropyl-2-methyleneaziridine with phenyl azide at 90 °C for 5 days generated a mixture of starting material 1, triazole 126 and β-lactamimide 127 in a ratio of 9:59:32. The triazole 126 probably arose from isomerization of the initial [3 + 2] product 124, driven by relief of ring-strain, and the minor product 127 presumably generated from the rearrangement of 124 via intermediate betaine 125. Notably, the β-lactamimide derivatives became major products when sulfonyl azides or alkyl azidoformates were used, such as p-toluenesulfonyl azide (up to 89% yield), methanesulfonyl azide (up to 82% yield),18 ethyl azidoformate (60%) and tert-butyl azidoformate (sole product, yield was not mentioned).
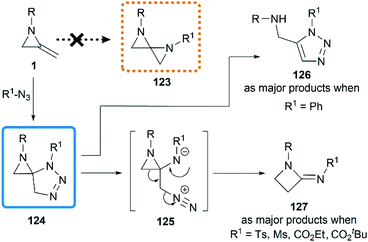 |
| | Scheme 38 Reaction of MAs with azides. | |
In 2013, Shibata group reported a novel [3 + 2] cycloaddition of MAs with isocyanates/isothiocyanate catalyzed by Bu2SnI2/LiI to obtain 4-methylene-2-imidazolidinones.57 As shown in Scheme 39, a pentacoordinate tin Li+[SnBu2I3]− 130 is considered as the active catalytic species in this transformation. Sn–I first attacks at the C-3 position of MA to deliver enamine 131, which is captured by the highly electrophilic carbon of isocyanate to give ureidostannane 132. The following intramolecular N-alkylation affords adducts in moderate to good yields.
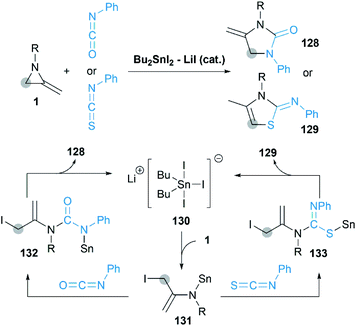 |
| | Scheme 39 [3 + 2] Cycloaddition of MAs with isocyanates/isothiocyanates. | |
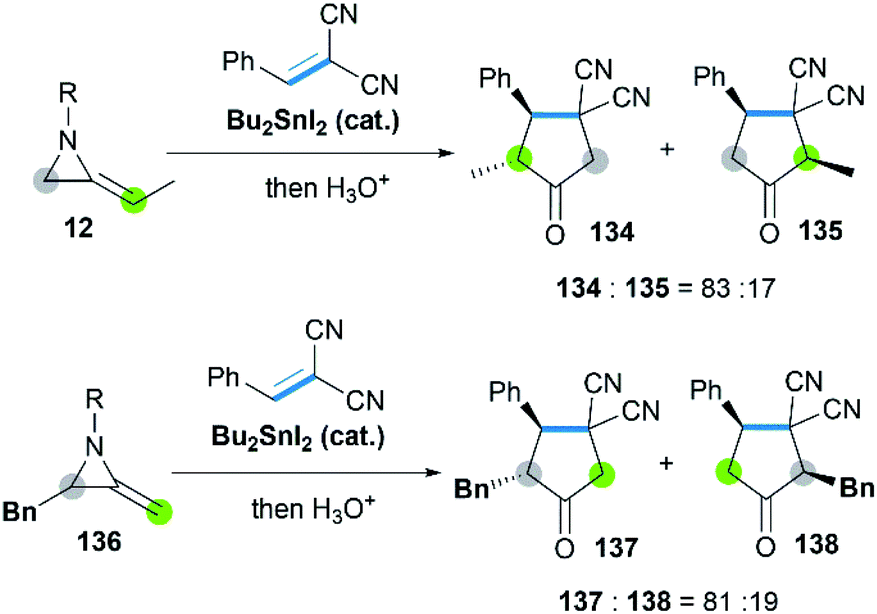
Sn-catalyzed cycloaddition of MAs with activated alkenes such as 1,1-dicyanoalkenes was also proved feasible, giving cyclopentanimines or corresponding cyclopentanone products.58 It's worth noting that both the SN2 attack of Sn–I bond toward C-3 on aziridine ring and CC bond of MAs were observed (Scheme 40), resulting in the different regioselectivity of reactions.
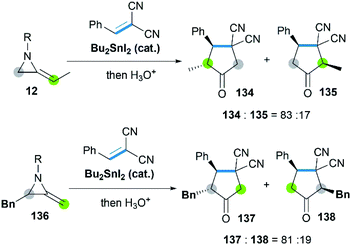 |
| | Scheme 40 Cycloaddition of MAs with 1,1-dicyanoalkenes. | |
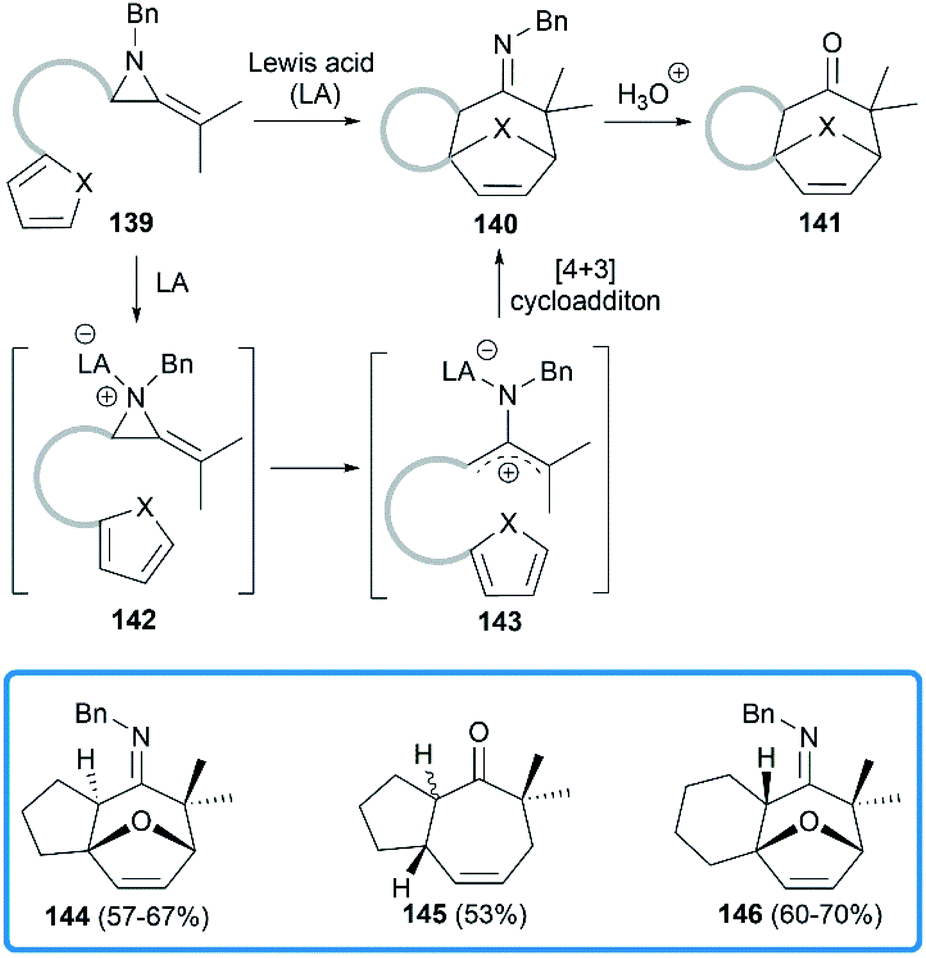
Shipman group focused on the Lewis acids promoted cycloadditions of MAs. In 2004, they reported an efficient approach to access seven membered polycyclic systems via the Lewis acids promoted [4 + 3] cycloadditions59,60 of MAs.61 Treatment of precursor 139 with BF3·Et2O or Sc(OTf)3 provided tricyclic imine in moderate to good yield, or tricyclic ketone after acidic work-up. As shown in Scheme 41, the aziridine 139 is first activated by Lewis acid to generate the intermediate aziridinium ion 142, followed by the formation of 2-aminoallyl cation 143 via C–C bond cleavage. Further intramolecular 1,3-dipolar cycloaddition reaction with the appended 1,3-diene would lead to tricyclic cycloheptenone imine 140.
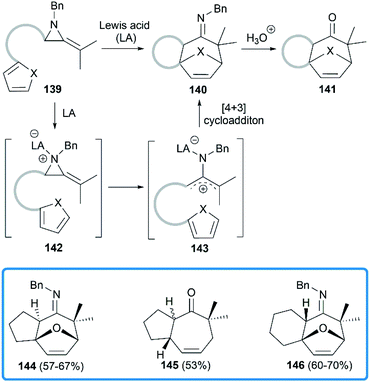 |
| | Scheme 41 Lewis acids promoted [4 + 3] cycloadditions of MAs. | |
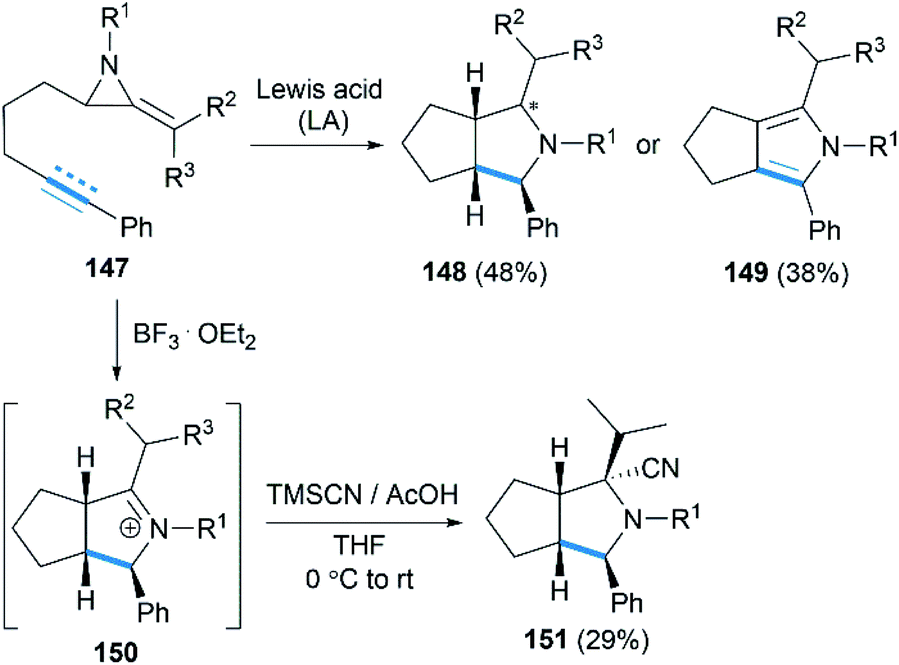
Encouraged by their study on Lewis acid promoted [4 + 3] cycloaddition, Shipman and co-workers reported that intramolecular [3 + 2] cycloadditions of MAs with alkene and alkyne acceptors produced cis-octahydrocyclopenta[c]pyrroles 148 and 2,4,5,6-tetrahydrocyclopenta[c]pyrroles 149, respectively (Scheme 42).62 In addition, the intermediate iminium ions, such as 150, could also be captured by cyanide to generate heterocycles 151 containing fully substituted stereocentres.
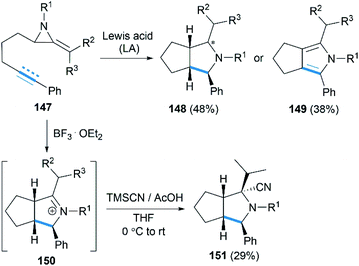 |
| | Scheme 42 Lewis acids promoted [3 + 2] cycloadditions of MAs. | |
The Lewis acid activation strategy was also used in the synthesis of substituted tetrahydro-β-carbolines. In this process, 2-methyleneaziridines 152, bearing an indole nucleus tethered to the aziridine nitrogen, is activated by BF3·OEt2, followed by a nucleophilic attack of alcohol to generate an iminium ion intermediate 155. An eventual Pictet–Spengler cyclization furnishes products in moderate to good yields (Scheme 43).63
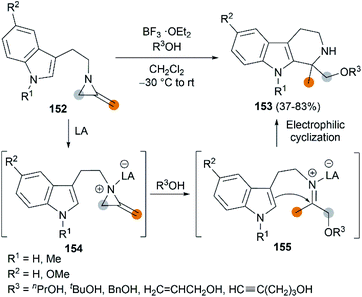 |
| | Scheme 43 Synthesis of 1,1-disubstituted tetrahydro-β-carbolines from MAs. | |
3.7. Transition metal catalyzed reaction
Transition metal catalyzed cycloaddition of MAs was first reported by the Alper group in 1987. Under palladium-catalyzed conditions, ring expansion reactions of these strained heterocycles with carbon monoxide furnished corresponding 3-methyleneazetidin-2-ones 156 in moderate yield, via N–C2 bond cleavage (Scheme 44).64
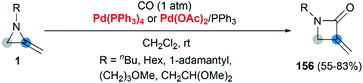 |
| | Scheme 44 Palladium-catalyzed ring expansion reaction of MAs. | |
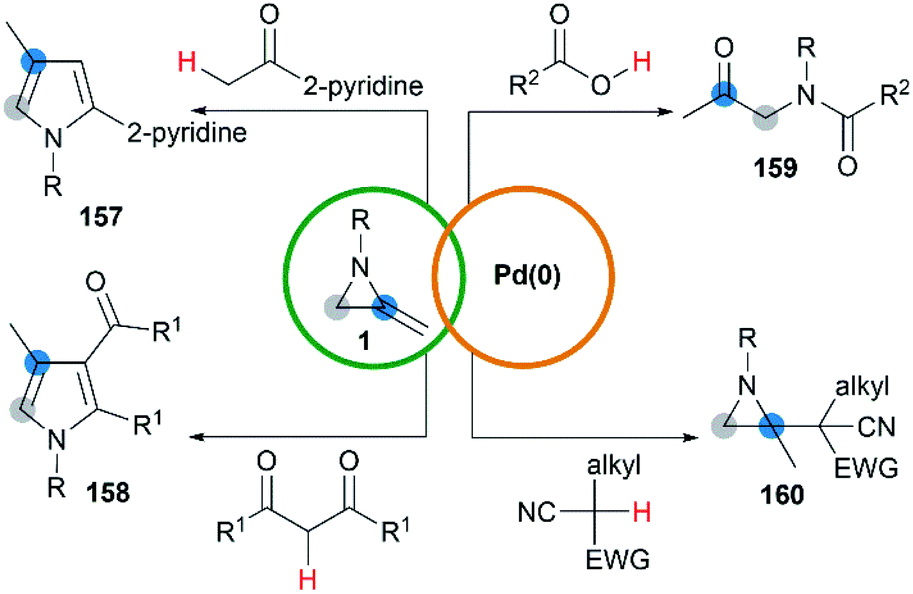
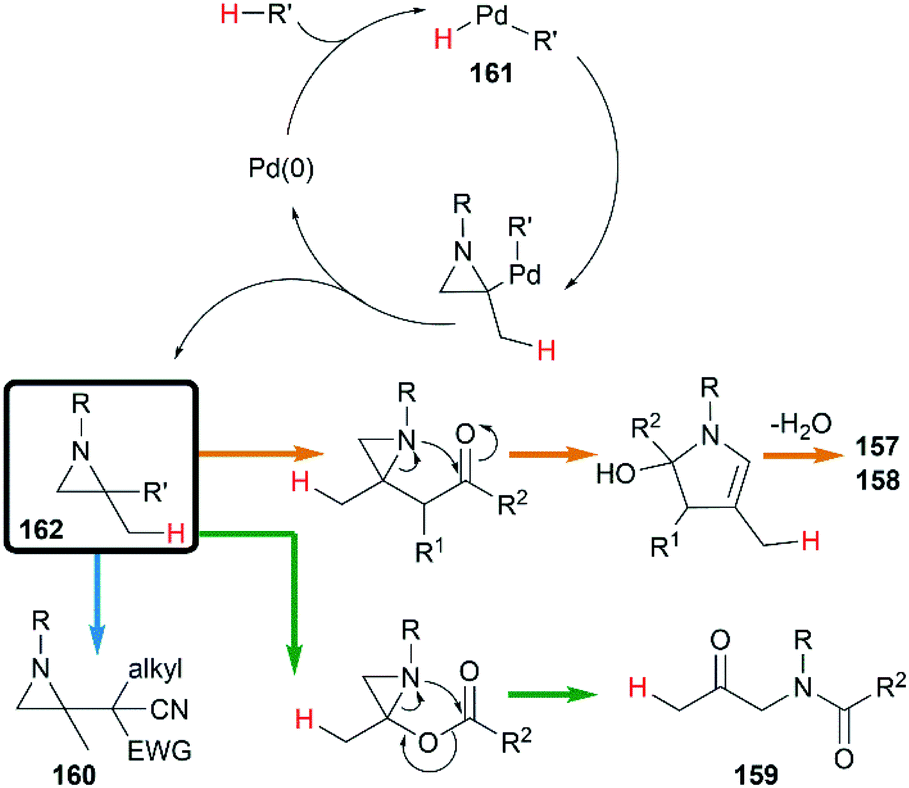
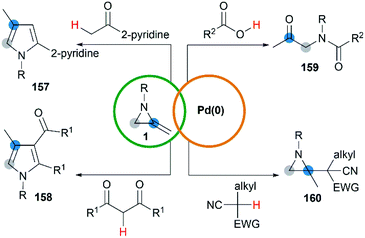
Yamamoto and co-workers found that the hydrocarbonation/hydrocarboxylation reaction of MAs with a range of pronucleophiles proceeded smoothly under palladium-catalyzed conditions (Scheme 45).65 A ring expansion reaction of MAs took place and gave the corresponding pyrroles derivatives 157,65c when acetylpyridine, 2,6-diacetylpyridine or acetylpyrazine were introduced into the reaction. Similar strategy could also be extended to 1,3-diketones, giving tetrasubstituted pyrroles 158 in good yields.65a However, the reaction outcomes were quite different when carboxylic acids or malononitrile derivatives were used as pronucleophiles. The former gave α-amidoketones 159 and the latter gave functionalized aziridines 160 without any ring-opening products.65b,65d,65e
 |
| | Scheme 45 Pd(0)-catalyzed hydrocarbonation reaction of MAs. | |
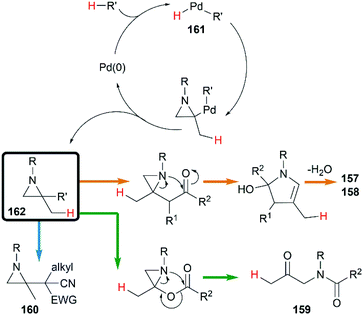
A hydropalladation mechanism was proposed to explain the Pd-catalyzed procedure. Initially, the oxidative addition of Pd(0) with a C–H or O–H bond of pronucleophiles leads to the palladium-hydride species 161. Then a hydropalladation of the exocyclic double bond and reductive elimination provide functionalized aziridines 162 and Pd(0) species. The structural diversities of final products depend on the different thermal rearrangement of disubstituted aziridines 162 as illustrated in Scheme 46.
 |
| | Scheme 46 Plausible mechanism of Pd(0)-catalyzed hydrocarbonation reaction of MAs. | |
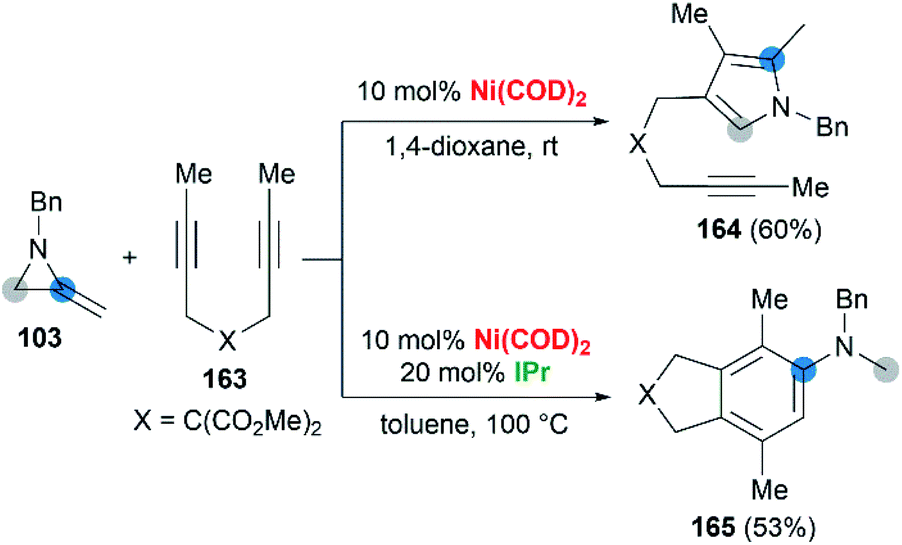
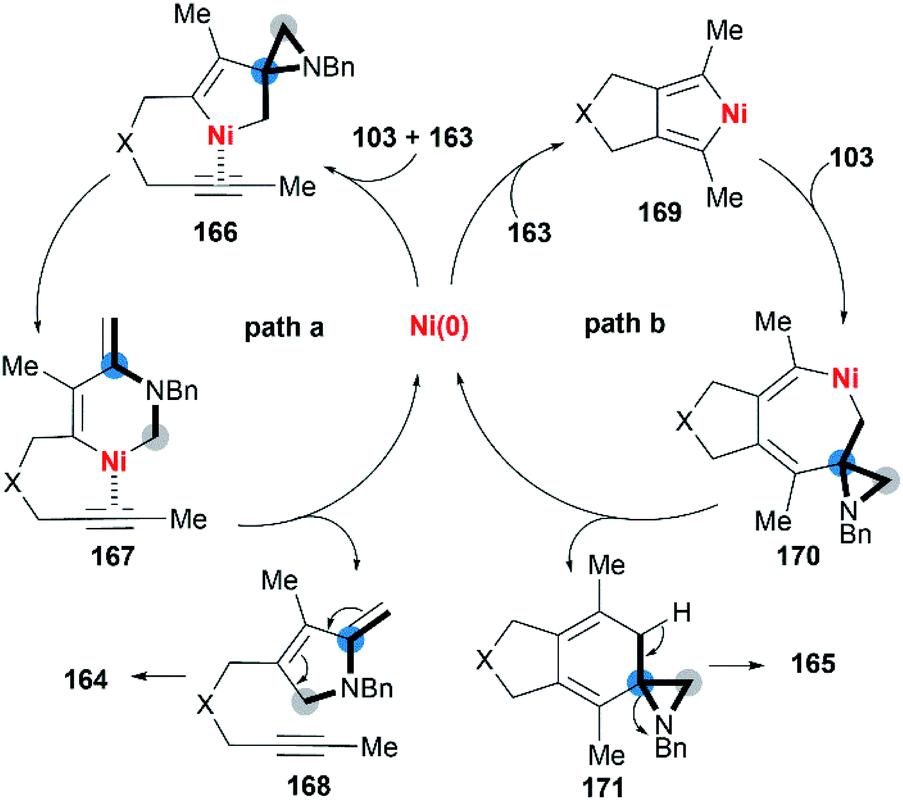
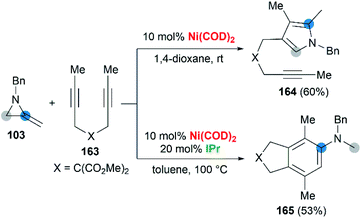
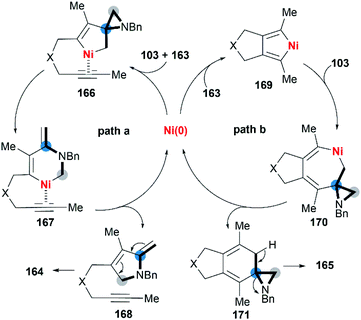
Wan and co-workers first reported the transition-metal-catalyzed cycloaddition reaction66 via C–C bond cleavage67 of MAs (Scheme 47).68 They developed a pyrrole synthesis by combining MAs 103 with diynes 163 in the presence of Ni(COD)2.69 Interestingly, the use of Ni(COD)2/IPr containing a N-heterocyclic carbene (NHC) ligand furnished a fused aniline 165 as the major product. A plausible reaction mechanism is depicted in Scheme 48. In path a (Scheme 48), an oxidative cyclization of MA, alkyne and Ni(0) forms nickelacyclopentene 166, followed by preferential β-carbon elimination (rather than β-nitrogen elimination) to afford 167. Finally, reductive elimination and isomerization provide the products. The spectator alkyne motif in the substrate proves to be essential for the efficiency of the process, likely because its coordination with the nickel center can stabilize the intermediate. In addition, the free alkyne unit of products is advantageous for further derivatization.70 In path b (Scheme 48), alternatively, the catalytic cycle starts with the oxidative cyclization of the diyne 163 on the nickel center to form the nickelacyclopentadiene complex 169. Coordination of the vinyl moiety of MA 103 and insertion into the Ni(II)–C bond gives intermediate 170. Then, reductive elimination of 170 affords the [2 + 2 + 2] cycloadduct 171, which isomerizes to product 165 via C–C bond cleavage.
 |
| | Scheme 47 Ni(0)-catalyzed cycloaddition reactions of MAs with alkyne via C–C bond cleavage. | |
 |
| | Scheme 48 Plausible reaction mechanism of Ni(0)-catalyzed cycloaddition reactions. | |
4. Bicyclic MAs
4.1. Synthesis of bicyclic MAs
As we have discussed in Section 2, intermolecular aziridination of allenes with nitrenes was reported to build MAs structure. But the low yield limited the application of this reaction.71
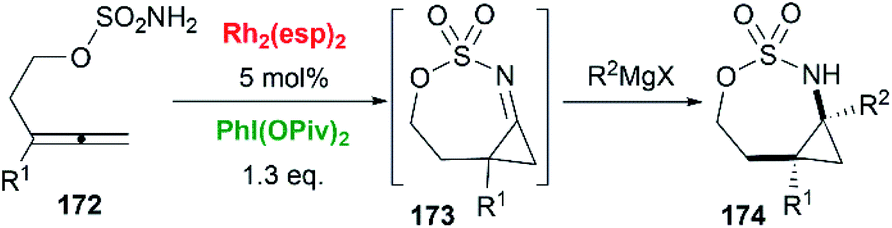
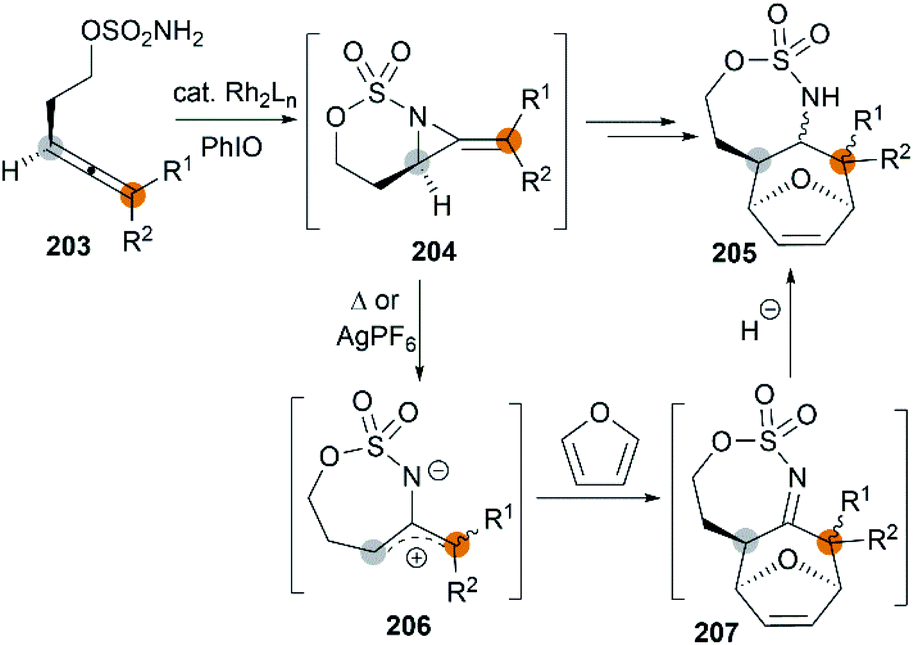
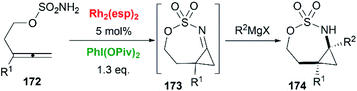
Allenes aziridination was refocused since the popularization of sulfamate-based nitrene precursors by the Du Bois group in the early 2000s.72 The first application of intramolecular Rh-catalyzed allene aziridination was reported by the Blakey group in 2009.73 In the presence of Rh2(esp)2 and PhI(OPiv)2, 1,1-disubstituted allene sulfamates yielded iminocyclopropanes 174, however, Blakey and co-workers did not observe bicyclic MAs formation during the course of their reactions (Scheme 49).
 |
| | Scheme 49 Aminocyclopropane formation from allenes. | |
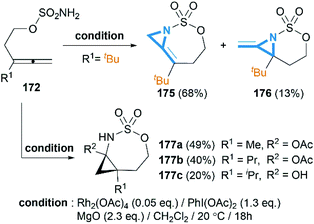
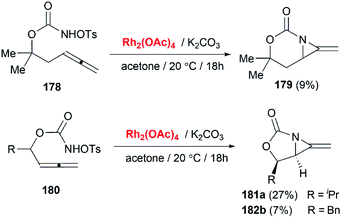
Soon after the publication by Blakey, the Robertson group reported similar work in their experiments with homoallenic sulfamates.74 Interestingly, they found that a more hindered-tBu substrate gave the novel bicyclic MAs as a 5:1 mixture of regioisomers (Scheme 50). Although yields were modest, this work represented the first synthesis of carbamoyl MAs since the work of Gilbert and co-workers in 1975 (Scheme 15).31 Almost at the same time, Robertson group discovered that the corresponding carbamates could also be used as nitrene precursors for allene aziridination. Under the conditions of Rh catalysis, tosyloxycarbamates gave bicyclic MA products in low but reproducible yields (Scheme 51).75
 |
| | Scheme 50 Cyclizations of homoallenic sulfamates. | |
 |
| | Scheme 51 Rh(II)-catalyzed aziridinations of allenyl carbamate derivatives. | |
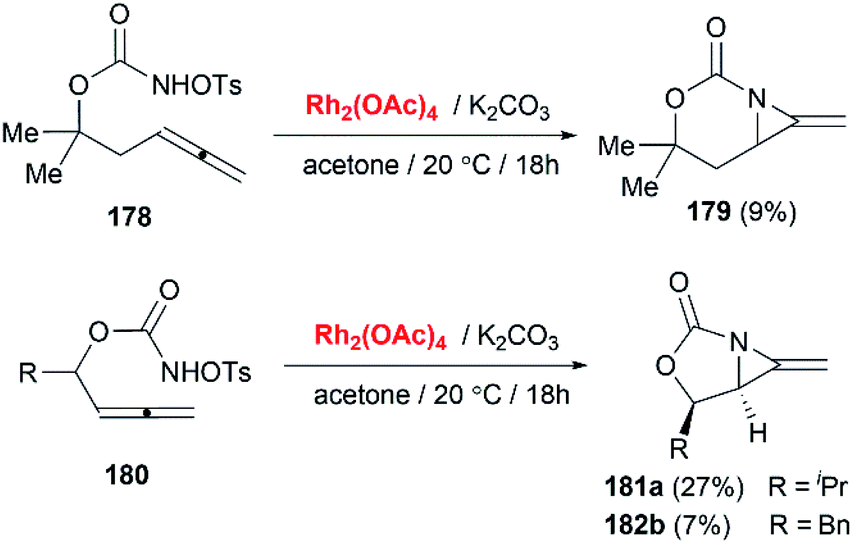
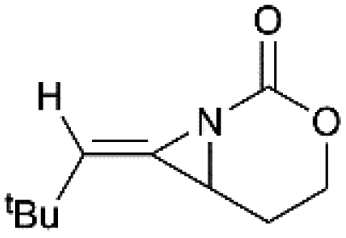
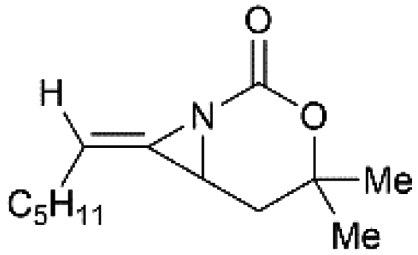
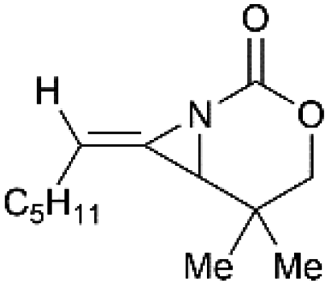
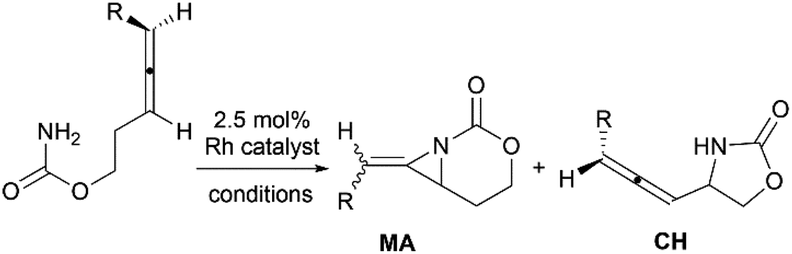
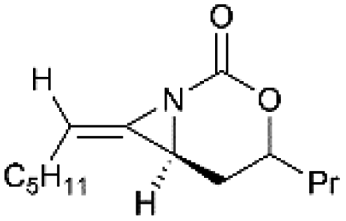
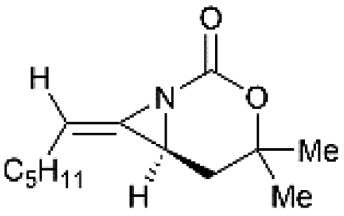
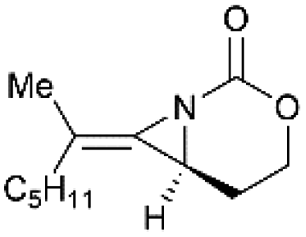
Schomaker and co-workers documented their research on Rh-catalyzed allene aziridination in further detail.76 For the purposes of isolating the target MAs, they found that carbamates provided the best balance between the reactivity of the nitrene precursor and the stability of the product. Rh2esp2 (esp = R,R,R0,R0-tetramethyl-1,3-benzenedipropionate) and Rh2(TPA)4 (TPA = triphenyl acetate) were proved to be more effective catalysts, giving complete conversion of the carbamate in most cases (Table 1). The use of PhIO in the presence of 4 Å molecular sieves minimized the MAs ring-opening and gave 94% yield of the desired MA (Table 1, entry 5). Unfortunately, the chemoselectivity of this aziridination depends heavily on the structural features of the allene substrates.
Table 1 Chemoselectivity in carbamoyl allene aziridination
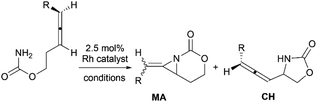
|
| Entry |
Desired product |
Conditions |
MA:CH %:% |
E:Z |
| 1 |
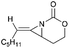 |
Rh2(TPA)4, PhIO, 4 Å MS |
80:15 |
4:1 |
| 2 |
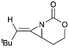 |
Rh2(TPA)4, PhIO, 4 Å MS |
45:23 |
E only |
| 3 |
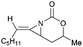 |
Rh2(TPA)4, PhIO, 4 Å MS |
48:31 |
1.5:1 |
| 4 |
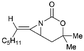 |
Rh2(esp)2, PhI(OPiv)2, MgO |
10:72 |
2.8:1 |
| 5 |
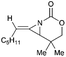 |
Rh2(esp)2, PhI(OPiv)2, MgO |
94 (MA only) |
3:1 |
Competing C–H insertion was exacerbated when allene was trisubstituted or branching in the link between the allene and the N atom.77 To overcome the disadvantage, Schomaker et al. investigated other transition metal catalysts for the aziridination of homoallenic carbamates. Catalysts formed in situ from silver triflate and bipyridyl-type ligands were found to be well-suited to this reaction (Table 2).78 These Ag(I)-based catalysts promote aziridination with superior chemoselectivity over conventional Rh-based catalysts, and greatly increase the yield of bicyclic MAs and the substrate scope of this reaction.
Table 2 Silver vs. Rhodium catalysis in the amination of homoallenic carbamates
4.2. Reactions of bicyclic MAs
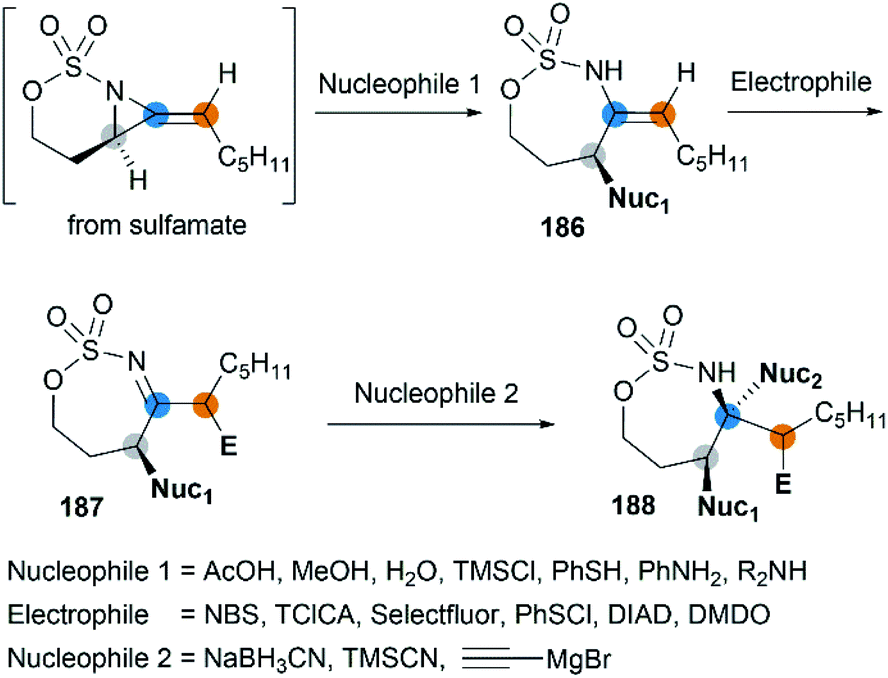
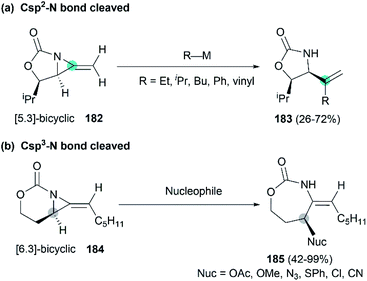
Along with the successful synthesis of bicyclic MAs, research on the chemical transformation of these structurally fascinating molecules was quickly followed. Robertson et al. first investigated the nucleophilic ring-opening reaction of [5.3]-bicyclic MAs with organolithium and Grignard reagents.75 The only identified product was the SNV (nucleophilic vinylic substitution) product, with the cleavage of Csp2–N bond (Scheme 52, (a)). The Schomaker group focused more on the reactivity of the [6.3]-bicyclic MAs. In contrast to [5.3]-bicyclic analogues, the [6.3]-bicyclic MAs were smoothly ring-opened by a series of nucleophiles, with the Csp3-N bond cleaved in preference to the Csp2-N bond (Scheme 52, (b)).76
 |
| | Scheme 52 Cleavage of Csp3–N bond and Csp2–N bond of bicyclic MAs. | |
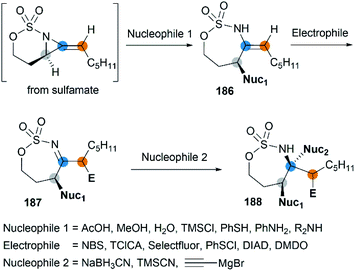
Notably, the resulting ring-opening enecarbamates of [6.3]-bicyclic MAs could serve as building blocks for the synthesis of stereotriad products. Schomaker group developed a strategy to form “all-heteroatom stereotriads” motifs of three contiguous centers containing stereodefined heteroatom substituents.79 (1) Firstly, a series of nucleophiles were proved capable of ring-opening the bicyclic MA in mild conditions. (2) Then, the resulting enesulfamates reacted with a variety of heteroatom-based electrophiles to introduce halogen, oxygen, sulphur, and nitrogen groups at each of the three original allene carbons. (3) Finally, the resulting imines could be trapped with another nucleophile to give stereotriads as shown in Scheme 53.
 |
| | Scheme 53 Conversion of bicyclic MAs to “all-heteroatom stereotriads”. | |
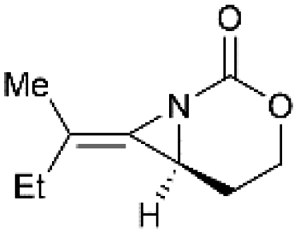
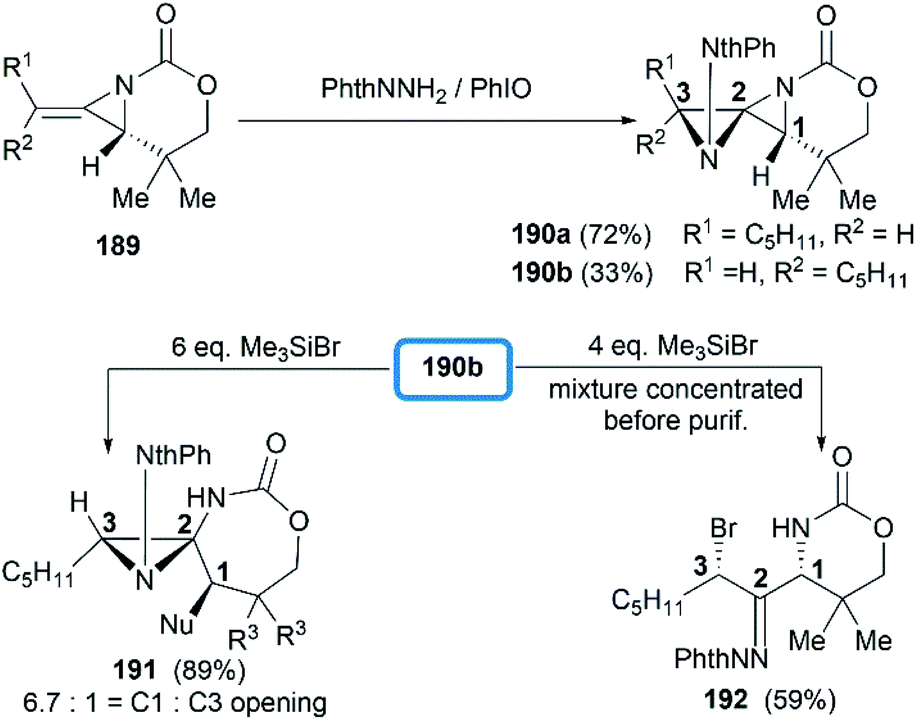
Direct functionalization of the exocyclic olefin of the bicyclic MAs was also proved as an efficient route to stereotriad products. Treatment of bicyclic MAs 189 with N-aminophthalimide in the presence of PhIO gave diazaspiro[2.2]pentane (DASP)80 products 190 with complete facial selectivity (Scheme 54).81
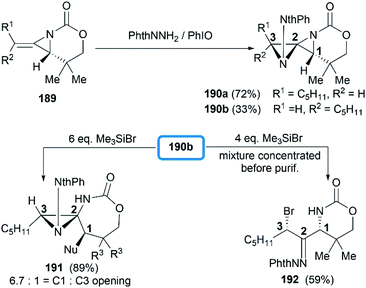 |
| | Scheme 54 Formation of DASP products and ring opening at C1 and C3. | |
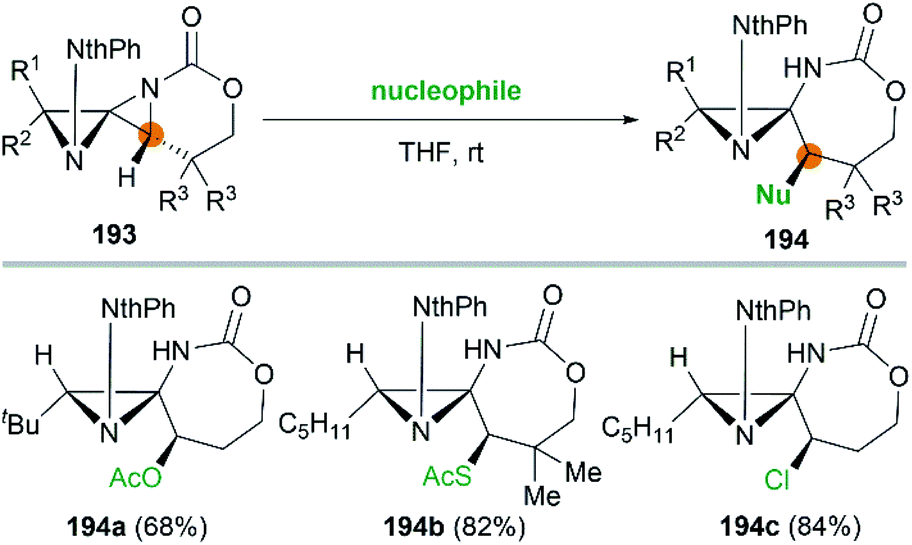
It's worth noting that the ability to electronically differentiate between the two aziridine rings of the DASPs enabled regioselective ring opening at the terminal carbon of either aziridine depending on the reaction conditions. Mostly, the addition of nucleophiles to the DASP typically favored ring-opening at C1 position to give N,N-aminal products 194 (Scheme 55). In certain cases, however, manipulation of the reaction conditions could induce C3 ring-opening to yield α,α′-disubstituted hydrazones (Scheme 54, 192).
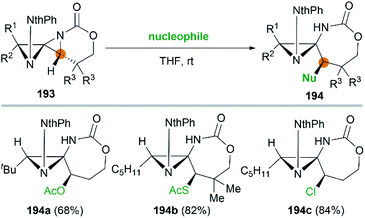 |
| | Scheme 55 Synthesis of N,N-aminals via ring opening at C1 of a DASP. | |
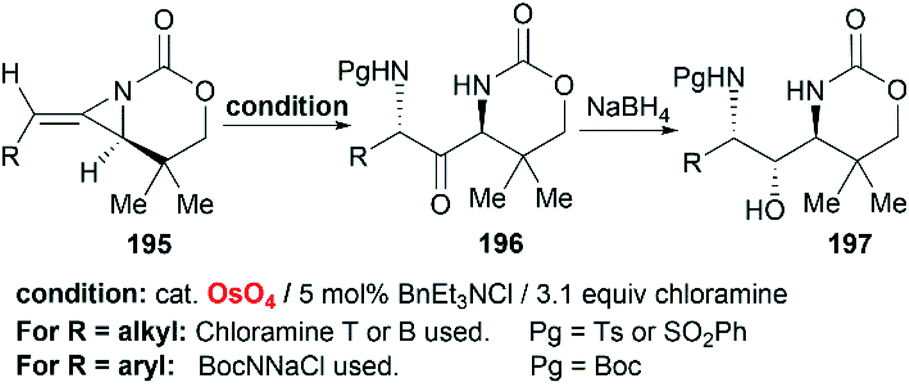
Schomaker et al. also reported the synthesis of 1,3-diaminoketone 197 through Os-catalyzed aminohydroxylation/reduction of bicyclic MAs (Scheme 56).82 This process is general for a variety of alkyl and arylsubstituted MAs with excellent regioselectivity and good yield, which are presumably attributed to the presence of chloramine.
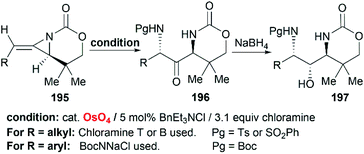 |
| | Scheme 56 Aminohydroxylation/reduction of bicyclic MAs. | |
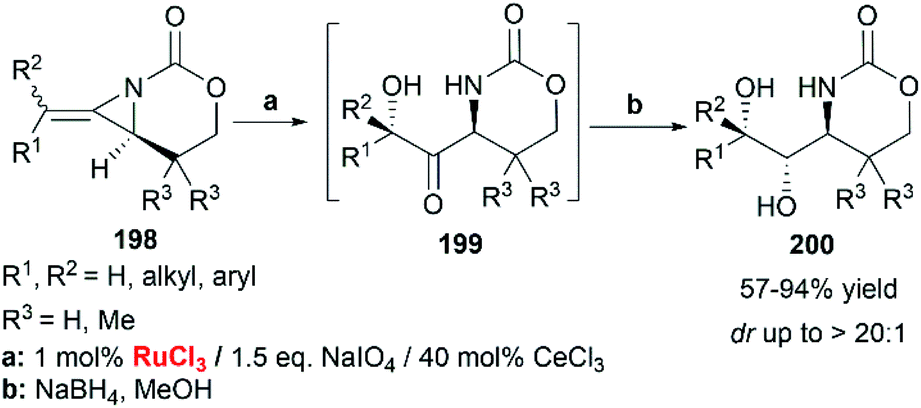
When RuCl3/NaIO4 system was chosen as the terminal oxidant, employing CeCl3 as an additive, bicyclic MAs cleanly provided the ketone 199 as a single diastereomer. Immediate reduction with NaBH4 yielded the 1-amino-2,3-diol 200 in > 20:1 dr (Scheme 57).83
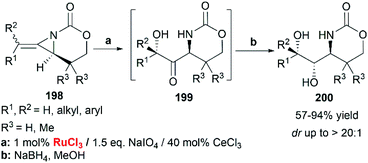 |
| | Scheme 57 Stereocontrolled oxidation of MAs. | |
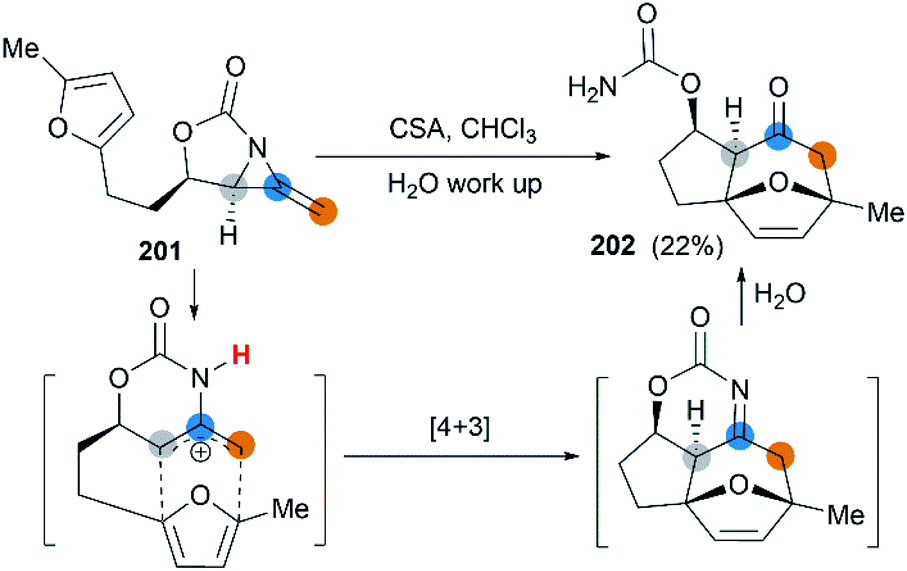
First attempt at cycloaddition reaction of bicyclic MAs was carried out by the Robertson et al.75 Treatment of a furan-tethered [5.3]-bicyclic MA 201 with camphorsulfonic acid in chloroform gave an intramolecular [4 + 3] cycloaddition product in 22% yield (Scheme 58). In this case, the reaction characteristic of bicyclic MAs was analogous to monocyclic MAs, as reported by Shipman.61
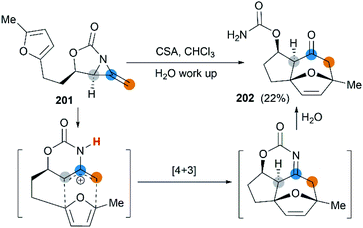 |
| | Scheme 58 Intramolecular [4 + 3] cycloaddition of a bicyclic MA. | |
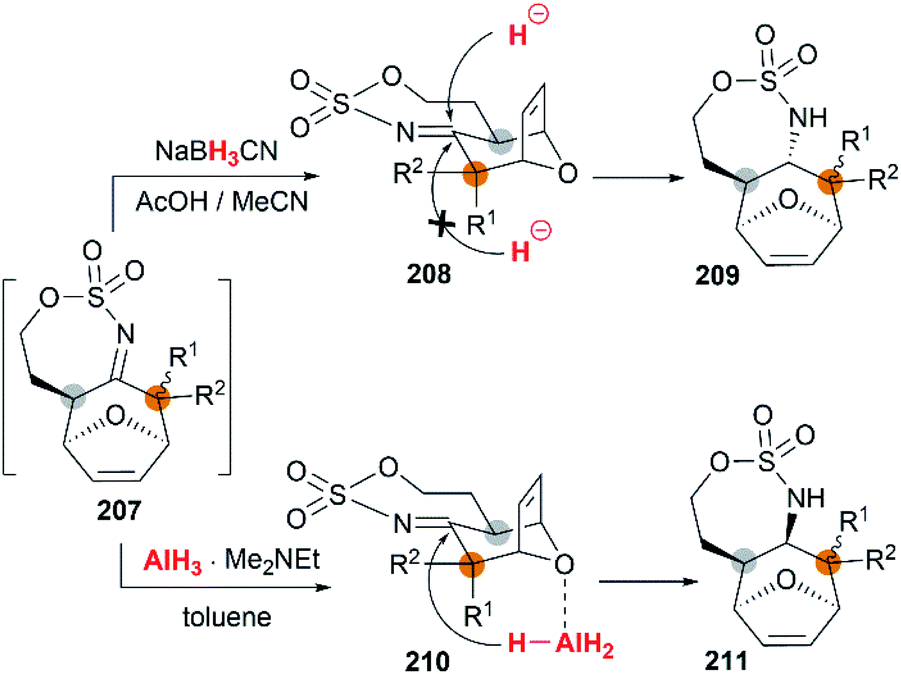
The examples of intermolecular [4 + 3] reactions were reported by the Schomaker group.84 Based on their previous research on bicyclic MAs, a tandem allene aziridination/[4 + 3]/reduction sequence was skillfully designed and successfully utilized to convert homoallenic sulfamates into densely functionalized aminated cycloheptenes. As shown in Scheme 59, this strategy features (1) one-pot allene aziridination/ring opening of 204 giving 2-amidoallyl cations 206. (2) Intermolecular [4 + 3] cycloaddition with furan, where the reaction conditions control the relative stereochemistry at C1 (or C3), and (3) reagent-dependent reduction of the imine 207 provides stereodivergent syntheses of stereoisomers (Scheme 60, 209 and 211). Additionally, the functionality of the products enables access to a series of densely functionalized building blocks in few steps.
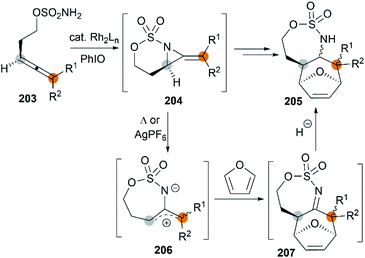 |
| | Scheme 59 Intermolecular [4 + 3] cycloaddition of bicyclic MA intermediates. | |
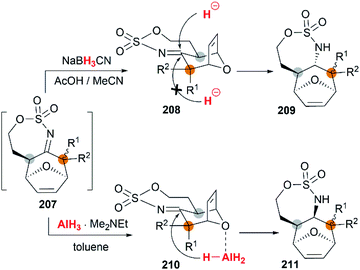 |
| | Scheme 60 Stereodivergent syntheses of stereoisomers. | |
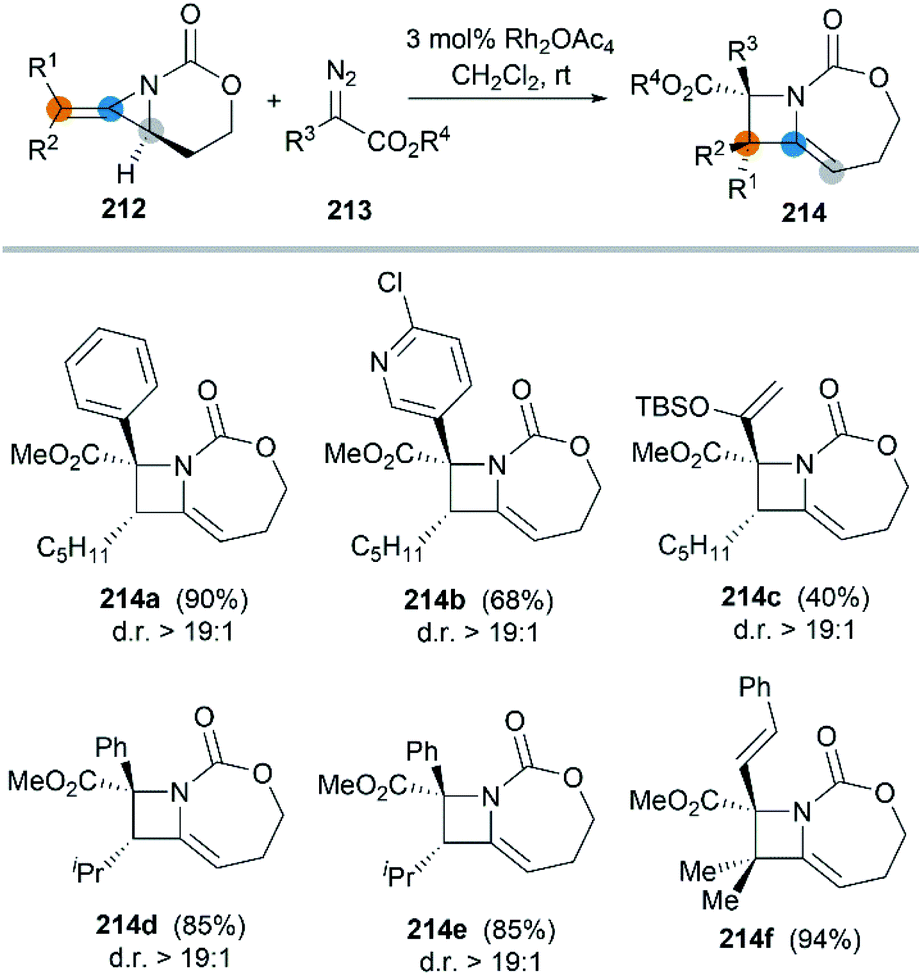
In 2017, the Schomaker group reported an impressive [3 + 1] ring expansion of bicyclic MAs, in combination with Rh-supported carbenes, to deliver tri- and tetra-substituted methylene azetidines.85 The unique strain and structure of the bicyclic MAs promoted this ring-opening/ring-closing cascade to yield highly substituted products with excellent regio- and stereoselectivity (Scheme 61, 214a–f).
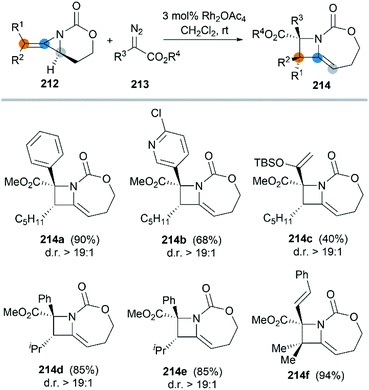 |
| | Scheme 61 Stereoselcetive [3 + 1] ring expansion of bicyclic MAs. | |
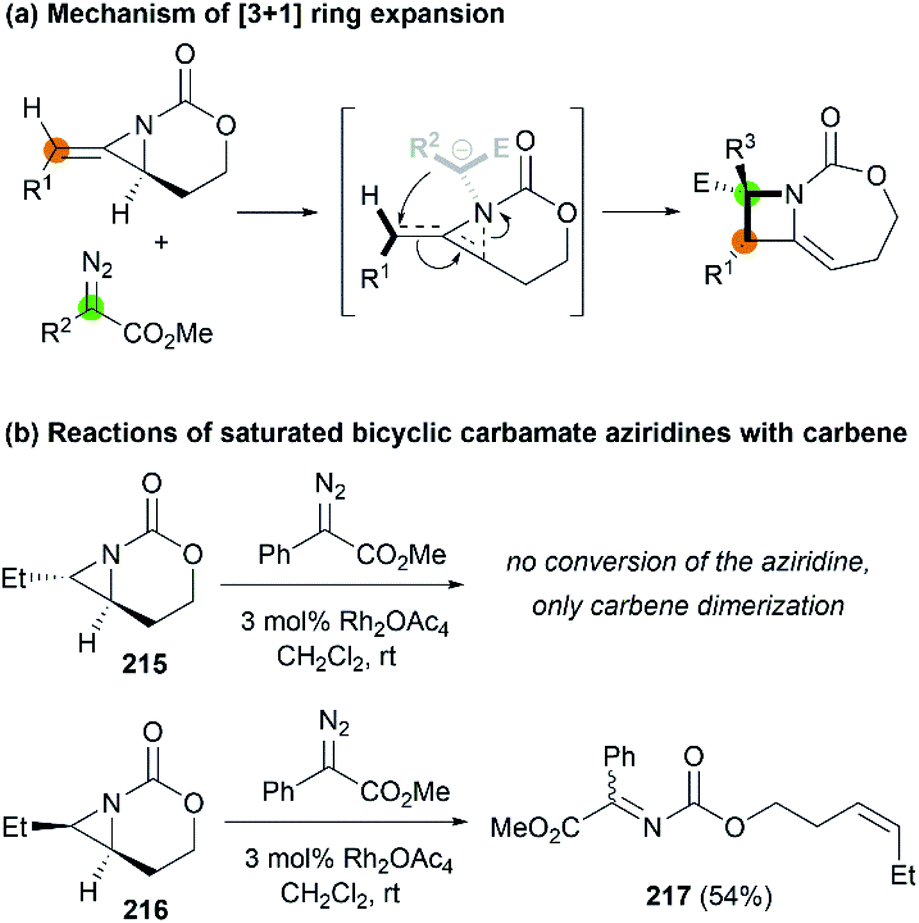
Later, Fernández, Schomaker and co-workers investigated the ring expansion mechanism and confirmed that such reaction proceeds through an aziridinium ylide, which undergoes a concerted, near-barrierless [2,3]-Stevens rearrangement to deliver substituted azetidines (Scheme 62, (a)). The olefin of the MA plays a key role in the success of this chemistry, channeling the aziridinium ylide toward ring expansion processes. In contrast, saturated bicyclic carbamate aziridines could not provide azetidine products (Scheme 62, (b)).86
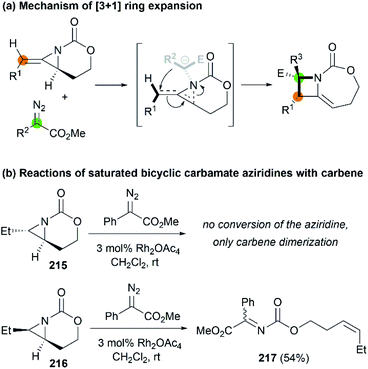 |
| | Scheme 62 Proposed mechanism and stereochemistry of [3 + 1] ring expansion. | |
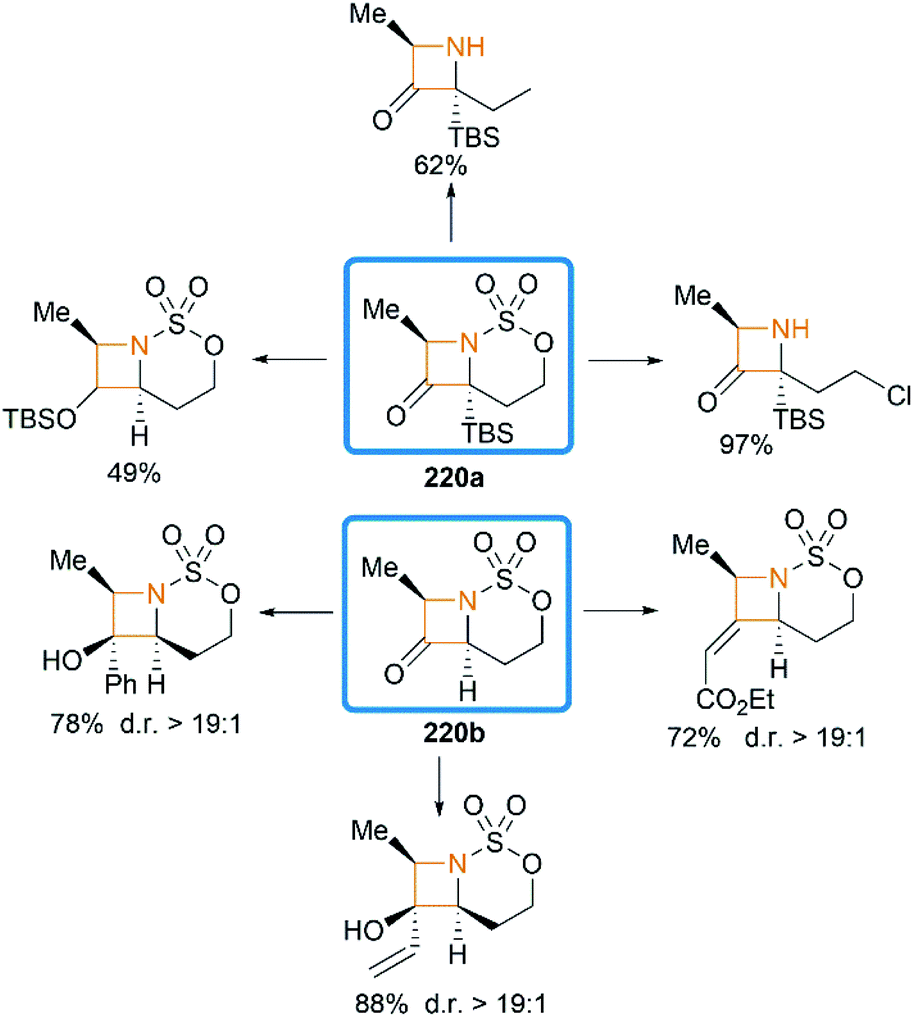
We have so far discussed many conversions involving exocyclic bicyclic MAs. Further studies by Schomaker and co-workers demonstrated that endocyclic bicyclic MAs could also serve as a useful scaffold for the construction of heterocycles. In 2015, they reported a regioselective aziridination of silyl-substituted homoallenic sulfamates for the synthesis of endocyclic bicyclic MAs 218, which could react with m-CPBA to yield azetidin-3-ones 220 (Scheme 63).87 An initial epoxidation of endocyclic bicyclic MAs 218 affords reactive oxazaspiropentanes 219, which rearrange to products in excellent d. r. Another key feature is the flexibility of subsequent transformations which could yield many densely functionalized azetidines (Scheme 64).
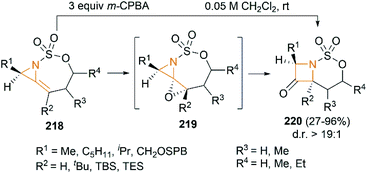 |
| | Scheme 63 Rearrangement of endocyclic bicyclic MAs to fused azetidin-3-ones. | |
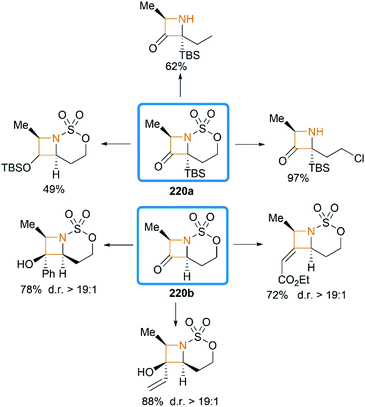 |
| | Scheme 64 Examples of transformation of azetidin-3-ones. | |
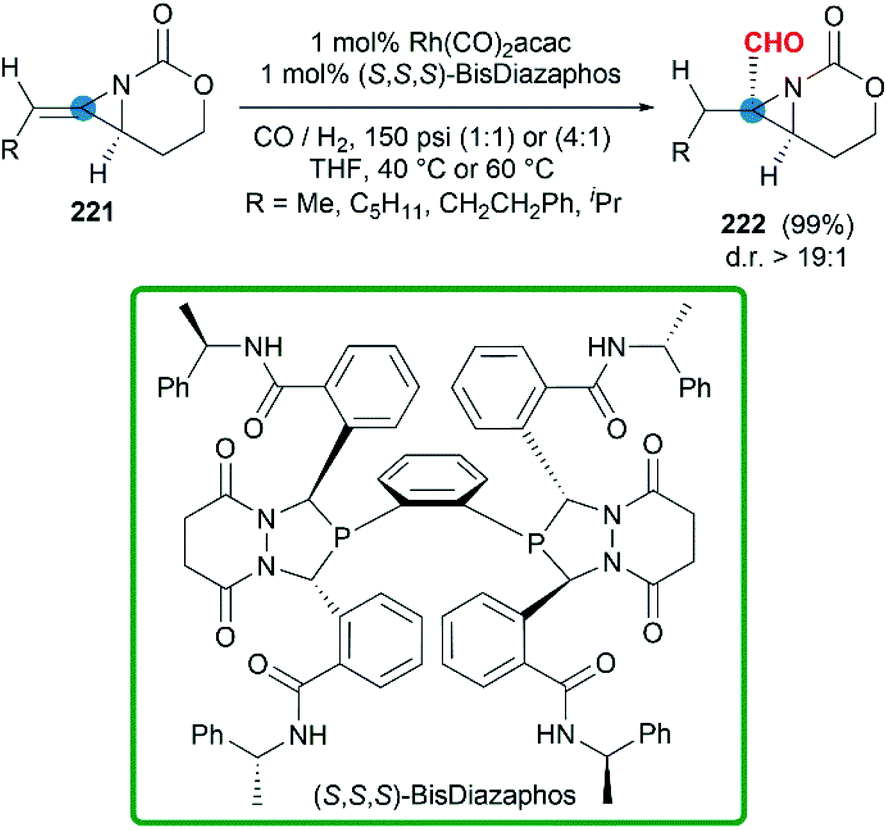
In some cases, bicyclic MAs prefer to show the chemical properties of olefins. More recently, a series of chiral bicyclic MAs were tested in hydroformylation.88 In the presence of Rh(CO)2(acac) and chiral phosphine ligands, the hydroformylation afforded formyl functionalized aziridines in good yield (Scheme 65). Impressively, chiral trisubstituted bicyclic MAs were transformed to products with >99% regioselectivity and >19:1 diastereoselectivity at rates >50 catalyst turnovers per hour.
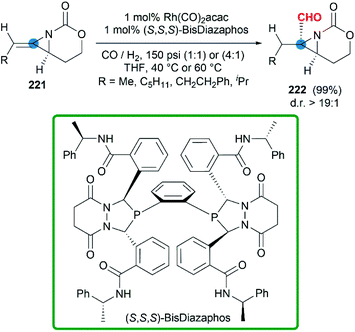 |
| | Scheme 65 Hydroformylation of bicyclic MAs. | |
5. Conclusion
Monocyclic MAs have been synthesized for nearly seventy years. The combination of strained aziridine ring and exocyclic alkene group provides monocyclic MAs diverse reactivity toward a number of useful transformations, including ring-opening reactions, multicomponent reactions, cycloadditions, radical cascades and transition-metal-catalyzed reactions.
However, the methodologies for the synthesis of monocyclic MAs are not well-developed. A major limitation is lack of efficient chemistry to introduce diverse nitrogen substituents, such as aryl groups and electronic-withdrawing groups into its structure.
In the past decade, interest in these attractive scaffolds has led to the development of methods for the synthesis of bicyclic MAs. In addition to the chemical reactivities based on the monocyclic MAs, including nucleophilic ring-opening reactions and Lewis acid catalyzed [4 + 3] reactions, bicyclic MAs could also undergo unique chemical transformations such as diverse oxidation reactions, Rh-catalyzed ring expansion and hydroformylation of exocyclic alkene. Importantly, all of these reactions successfully construct densely functionalized products with structural and stereochemical complexity.
Given the wide occurrence of nitrogen-containing compounds and heterocycles in nature and drugs, we believe that MAs will increasingly attract the interest of synthetic community. Developing new efficient methods for synthesis of MAs with different types of substituents, along with revealing their unknown reactivities, will further expand their synthetic utility. We look forward to the contributions that will be uncovered in the near future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was funded by the National Natural Science Foundation of China (no. 81973540) and the startup research fund of Weifang University of Science and Technology (no. KJRC2019005 and no. KJRC2020002). Dr Zhao acknowledges the startup research fund of Liaoning Normal University and the Department of Science and Technology of Liaoning Province (no. 2019-BS151).
Notes and references
- For reviews on aziridines, see:
(a) H. Pellissier, Tetrahedron, 2010, 66, 1509–1555 CrossRef CAS
 ;
(b) F. Chemla and F. Ferreira, Curr. Org. Chem., 2002, 6, 539–570 CrossRef CAS ;
(c) Q. Y. Wang, H. H. Chang, W. L. Wei, Q. Liu, W. C. Gao, Y. W. Li and X. Li, Chin. J. Org. Chem., 2016, 36, 939–953 CrossRef CAS ;
(d) G. S. Singh, S. Sudheesh and N. Keroletswe, Arkivoc, 2018, i, 50–113 Search PubMed ;
(e) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS ;
(f) H. Ohno, Chem. Rev., 2014, 114, 7784–7814 CrossRef CAS ;
(g) J. J. Feng and J. L. Zhang, ACS Catal., 2016, 6, 6651–6661 CrossRef CAS ;
(h) M. K. Ghorai, A. Bhattacharyya, S. Das and N. Chauhan, Synthesis of 4- to 7-Membered Heterocycles by Ring-Expansion: Ring-Expansions of Activated Aziridines and Azetidines, in Topics in Heterocyclic Chemistry, ed. M. D'hooghe and H.-J. Ha, Springer: Cham, 2016, vol. 41, pp. 49–142 Search PubMed ;
(i) J. Dolfen, N. De Kimpe and M. D'hooghe, Synlett, 2016, 27, 1486–1510 CrossRef CAS ;
(j) S. Tarannum, N. Chauhan and M. K. Ghorai, Aziridines and 2H-Azirines—Monocyclic: Reference Module in Chemistry, in Molecular Sciences and Chemical Engineering, Elsevier 2020, DOI:10.1016/B978-0-12-409547-2.14954-6 .
;
(b) F. Chemla and F. Ferreira, Curr. Org. Chem., 2002, 6, 539–570 CrossRef CAS ;
(c) Q. Y. Wang, H. H. Chang, W. L. Wei, Q. Liu, W. C. Gao, Y. W. Li and X. Li, Chin. J. Org. Chem., 2016, 36, 939–953 CrossRef CAS ;
(d) G. S. Singh, S. Sudheesh and N. Keroletswe, Arkivoc, 2018, i, 50–113 Search PubMed ;
(e) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS ;
(f) H. Ohno, Chem. Rev., 2014, 114, 7784–7814 CrossRef CAS ;
(g) J. J. Feng and J. L. Zhang, ACS Catal., 2016, 6, 6651–6661 CrossRef CAS ;
(h) M. K. Ghorai, A. Bhattacharyya, S. Das and N. Chauhan, Synthesis of 4- to 7-Membered Heterocycles by Ring-Expansion: Ring-Expansions of Activated Aziridines and Azetidines, in Topics in Heterocyclic Chemistry, ed. M. D'hooghe and H.-J. Ha, Springer: Cham, 2016, vol. 41, pp. 49–142 Search PubMed ;
(i) J. Dolfen, N. De Kimpe and M. D'hooghe, Synlett, 2016, 27, 1486–1510 CrossRef CAS ;
(j) S. Tarannum, N. Chauhan and M. K. Ghorai, Aziridines and 2H-Azirines—Monocyclic: Reference Module in Chemistry, in Molecular Sciences and Chemical Engineering, Elsevier 2020, DOI:10.1016/B978-0-12-409547-2.14954-6 . - S. M. Bachrach, J. Phys. Chem., 1993, 97, 4996–5000 CrossRef CAS .
- W. Lwowski, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W Rees, Pergamon, Oxford, UK, 1984, vol. 7. pp. 1–16 Search PubMed .
- H. Quast, R. Frank, A. Heublein and E. Schmitt, Liebigs Ann. Chem., 1980, 11, 1814–1835 CrossRef .
- A. T. Bottini and J. D. Roberts, J. Am. Chem. Soc., 1957, 79, 1462–1464 CrossRef CAS .
-
(a) H. Quast, R. Jakob, K. Peters, E. M. Peters and H. G. von Schnering, Chem. Ber., 1984, 117, 840–849 CrossRef CAS ;
(b) J. Ince, T. M. Ross, M. Shipman, A. M. Z. Slawin and D. S. Ennis, Tetrahedron, 1996, 52, 7037–7044 CrossRef CAS .
-
(a) A. Löwenstein, J. F. Neumer and J. D. Roberts, J. Am. Chem. Soc., 1960, 82, 3599–3601 CrossRef ;
(b) A. T. Bottini and J. D. Roberts, J. Am. Chem. Soc., 1956, 78, 5126 CrossRef CAS ;
(c) A. T. Bottini and J. D. Roberts, J. Am. Chem. Soc., 1958, 80, 5203–5208 CrossRef CAS .
- M. Shipman, Synlett, 2006, 19, 3205–3217 CrossRef .
- K. A. Tehrani and N. De Kimpe, Curr. Org. Chem., 2009, 13, 854–877 CrossRef CAS .
- R. F. Parcell and C. B. Pollard, J. Am. Chem. Soc., 1951, 73, 2925–2927 CrossRef .
- R. F. Parcell and C. B. Pollard, J. Am. Chem. Soc., 1950, 72, 2385–2386 CrossRef CAS .
- M. G. Ettlinger and F. Kennedy, Chem. Ind., 1956, 10, 166–167 Search PubMed .
-
(a) J. Ince, T. M. Ross, M. Shipman and D. S. Ennis, Tetrahedron: Asymmetry, 1996, 7, 3397–3406 CrossRef CAS ;
(b) A. F. Bottini and V. Dev, J. Org. Chem., 1962, 27, 968–973 CrossRef CAS .
-
(a) A. T. Bottini and R. E. Olsen, J. Am. Chem. Soc., 1962, 84, 195–199 CrossRef CAS ;
(b) A. T. Bottini, J. K. Barbara and R. E. Olsen, J. Org. Chem., 1963, 28, 3241–3243 CrossRef CAS .
- J. J. Shiers, M. Shipman, J. F. Hayes and A. M. Z. Slawin, J. Am. Chem. Soc., 2004, 126, 6868–6869 CrossRef CAS .
- V. Lucchini, G. Modena and L. Pasquato, J. Am. Chem. Soc., 1995, 117, 2297–2300 CrossRef CAS .
- J. B. P. A. Wijnberg, P. G. Wiering and H. Steinberg, Synthesis, 1981, 901–903 CrossRef CAS .
- N. De Kimpe, D. De Smaele and Z. Sakonyi, J. Org. Chem., 1997, 62, 2448–2452 CrossRef CAS .
- H. Quast and C. A. Weise Vélez, Angew. Chem., Int. Ed., 1974, 13, 342–343 CrossRef .
- C. Montagne, N. Prévost, G. Prie, S. Rahman, J. Ince, J. F. Hayes and M. Shipman, Tetrahedron, 2006, 62, 8447–8457 CrossRef CAS .
- H. Quast and C. A. Weise Vélez, Angew. Chem., Int. Ed., 1978, 17, 213–214 CrossRef .
- J. F. Hayes, N. Prévost, I. Prokes, M. Shipman, A. M. Z. Slawin and H. Twin, Chem. Commun., 2003, 1344–1345 RSC .
-
(a) L. Degennaro, L. Pisano, G. Parisi, R. Mansueto, G. J. Clarkson, M. Shipman and R. Luisi, J. Org. Chem., 2015, 80, 6411–6418 CrossRef CAS ;
(b) R. Mansueto, L. Degennaro, J.-F. Brière, K. Griffin, M. Shipman, S. Florio and R. Luisi, Org. Biomol. Chem., 2014, 12, 8505–8511 RSC .
- F. H. Bayliffe, A. Steven, K. Ando and M. Shipman, Synlett, 2015, 26, 1371–1374 CrossRef CAS .
- V. M. Ismailov, I. A. Mamedov and N. N. Yusubov, Russ. J. Org. Chem., 2009, 45, 1250–1251 CrossRef CAS , V. M. Ismailov, et al. reported the formation of 1–phenyl–2–methyleneaziridine, which was described as a purple solid and the characterization was very limited [1H NMR (60MHz), microanalysis only]. These findings are inconsistent with observations by M. Shipman et al.
- H. Quast and W. Risler, Angew. Chem., Int. Ed., 1973, 12, 414–415 CrossRef .
- T. Inokuchi, S. Matsumoto, M. Tsuji and S. Torii, J. Org. Chem., 1992, 57, 5023–5027 CrossRef CAS .
- W. Wu, Y. G. Huang and F. L. Qing, Chin. J. Org. Chem., 2009, 29, 1249–1253 CAS .
- Recent reviews of MCP, see:
(a) A. Brandi and A. Goti, Chem. Rev., 1998, 98, 589–635 CrossRef CAS ;
(b) E. Nakamura and S. Yamago, Acc. Chem. Res., 2002, 35, 867–877 CrossRef CAS ;
(c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213–1269 CrossRef CAS ;
(d) G. Audran and H. Pellissier, Adv. Synth. Catal., 2010, 352, 575–608 CrossRef CAS ;
(e) L. Yu and R. Guo, Org. Prep. Proced. Int., 2011, 43, 209–259 CrossRef CAS ;
(f) B. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318–3339 RSC ;
(g) M. Shi, J. M. Lu, Y. Wei and L. X. Shao, Acc. Chem. Res., 2012, 45, 641–652 CrossRef CAS .
- R. F. Bleiholder and H. Schecter, J. Am. Chem. Soc., 1968, 90, 2131–2137 CrossRef CAS .
- E. M. Bingham and J. C. Gilbert, J. Org. Chem., 1975, 40, 224–228 CrossRef CAS .
- P. F. Belloir, A. Laurent, P. Mison, R. Bartnik and S. Lesniak, Tetrahedron Lett., 1985, 26, 2637–2640 CrossRef CAS .
- N. De Kimpe and K. A. Tehrani, Tetrahedron Lett., 2000, 41, 1975–1978 CrossRef .
-
(a) E. Jongejan, H. Steinberg and T. J. De Boer, Recl. Trav. Chim. Pays-Bas Belg., 1979, 98, 66–69 CrossRef CAS ;
(b) E. Jongejan, H. Steinberg and T. J. De Boer, Recl. Trav. Chim. Pays-Bas Belg., 1978, 97, 145–146 CrossRef CAS .
- A. T. Bottini, V. Dev and M. Stewart, J. Org. Chem., 1963, 28, 156–158 CrossRef CAS .
- R. K. Brinton, J. Phys. Chem., 1964, 68, 2652–2656 CrossRef CAS .
- N. J. Turro, Acc. Chem. Res., 1969, 2, 25–32 CrossRef CAS .
- D. S. Ennis, J. Ince and M. Shipman, Tetrahedron Lett., 1997, 38, 5887 CrossRef .
- D. S. Ennis, J. Ince, S. Rahman and M. Shipman, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 2047–2053 RSC .
-
(a) J. K. Crandall, L. C. Crawley and J. B. Komin, J. Org. Chem., 1975, 40, 2045–2047 CrossRef CAS ;
(b) J. K. Crandall and J. B. Komin, J. Chem. Soc., Chem. Commun., 1975, 436 RSC .
- P. G. Wiering and H. Steinberg, Isr. J. Chem., 1982, 22, 56–58 CrossRef CAS .
-
(a) J. F. Hayes, M. Shipman and H. Twin, Chem. Commun., 2000, 1791–1792 RSC ;
(b) J. F. Hayes, M. Shipman and H. Twin, J. Org. Chem., 2002, 67, 935–942 CrossRef CAS .
- J. F. Margathe, M. Shipman and S. C. Smith, Org. Lett., 2005, 7, 4987–4990 CrossRef CAS .
- J. F. Hayes, M. Shipman, A. M. Z. Slawin and H. Twin, Heterocycles, 2002, 58, 243–250 CrossRef CAS .
- For reviews, see:
(a) P. D. Bailey, P. A. Millwood and P. D. Smith, Chem. Commun., 1998, 633–640 RSC ;
(b) S. Laschat and T. Dickner, Synthesis, 2000, 13, 1781–1813 CrossRef .
- J. F. Hayes, M. Shipman and H. Twin, Chem. Commun., 2001, 1784–1785 RSC .
- J. J. Shiers, G. J. Clarkson, M. Shipman and J. F. Hayes, Chem. Commun., 2006, 649–651 RSC .
- C. Montagne, J. J. Shiers and M. Shipman, Tetrahedron Lett., 2006, 47, 9207–9209 CrossRef CAS .
- C. C. A. Cariou, G. J. Clarkson and M. Shipman, J. Org. Chem., 2008, 73, 9762–9764 CrossRef CAS .
- N. Prévost and M. Shipman, Tetrahedron, 2002, 58, 7165–7175 CrossRef .
- N. Prévost and M. Shipman, Org. Lett., 2001, 3, 2383–2385 CrossRef .
- For reviews on [2+2] cycloaddition, see:
(a) N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 10, 1310–1326 Search PubMed ;
(b) T. Michinobu and F. Diederich, Angew. Chem., Int. Ed., 2018, 57, 3552–3577 CrossRef CAS .
- R. C. Cookson, B. Halton, I. D. R. Stevens and C. T Watts, J. Chem. Soc., 1967, 928–931 CAS .
- M. Shipman, T. M. Ross and A. M. Z. Siawin, Tetrahedron Lett., 1999, 40, 6091–6092 CrossRef CAS .
- T. Akasaka, Y. Nomura and W. Ando, J. Org. Chem., 1988, 53, 1670–1672 CrossRef CAS .
- For reviews on [3+2] cycloaddition, see:
(a) M. Rios–Gutierrez and L. R. Domingo, Eur. J. Org. Chem., 2019, 267–282 CrossRef ;
(b) F. Rodier, M. Rajzmann, J. L. Parrain, G. Chouraqui and L. Commeiras, Chem. - Eur. J., 2013, 19, 2467–2477 CrossRef CAS .
- H. Takahashi, S. Yasui, S. Tsunoi and I. Shibata, Eur. J. Org. Chem., 2013, 40–43 CrossRef CAS .
- H. Takahashi, S. Yasui, S. Tsunoi and I. Shibata, Org. Lett., 2014, 16, 1192–1195 CrossRef CAS .
- For reviews on Lewis acids promoted cycloadditions, see:
(a) W. J. Zhu, J. Fang, Y. Liu, J. Ren and Z. W. Wang, Angew. Chem., Int. Ed., 2013, 52, 2032–2037 CrossRef CAS ;
(b) T. Taguchi, A. Saito and H. Yanai, Chem. Rec., 2007, 7, 167–169 CrossRef CAS .
- For reviews on [4+3] cycloaddition, see:
(a) Z. S. Yin, H. Yun and P. Chiu, Chem. Soc. Rev., 2018, 47, 8881–8924 RSC ;
(b) M. Harmata, Adv. Synth. Catal., 2006, 348, 2297–2306 CrossRef CAS .
- G. Prié, N. Prévost, H. Twin, S. A. Fernandes, J. F. Hayes and M. Shipman, Angew. Chem., Int. Ed., 2004, 43, 6517–6519 CrossRef .
- K. Griffin, C. Montagne, C. T. Hoang, G. J. Clarkson and M. Shipman, Org. Biomol. Chem., 2012, 10, 1032–1039 RSC .
- P. M. Mumford, J. J. Shiers, G. J. Tarver, J. F. Hayes and M. Shipman, Tetrahedron Lett., 2008, 49, 3489–3491 CrossRef CAS .
- H. Alper and N. Hamel, Tetrahedron Lett., 1987, 28, 3237–3240 CrossRef CAS .
-
(a) K. K. A. D. S. Kathriarachchi, A. I. Siriwardana, I. Nakamura and Y. Yamamoto, Tetrahedron Lett., 2007, 48, 2267–2270 CrossRef CAS ;
(b) B. H. Oh, I. Nakamura and Y. Yamamoto, Tetrahedron Lett., 2002, 43, 9625–9628 CrossRef CAS ;
(c) A. I. S Siriwardana, K. K. A. D. S. Kathriarachchi, I. Nakamura, I. D. Gridnev and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 13898–13899 CrossRef ;
(d) B. H. Oh, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2004, 69, 2856–2858 CrossRef CAS ;
(e) B. H. Oh, I. Nakamura and Y. Yamamoto, ARKIVOC Online J. Org. Chem., 2003,(viii), 67–78 CAS .
- For recent reviews on transition–metal–catalyzed cycloaddition, see:
(a) B. Kuila, M. Kaur, P. Singh and G. Bhargava, Eur. J. Org. Chem., 2018, 853–868 CrossRef CAS ;
(b) M. Babazadeh, S. Soleimani–Amiri, E. Vessally, A. Hosseiniand and L. Edjlalia, RSC Adv., 2017, 7, 43716–43736 RSC ;
(c) P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS .
- For a recent review on C−C single–bond cleavage of strained ring systems by transition metal complexes, see: G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS .
-
(a) B. Pan, C. X. Wang, D. Wang, F. Wu and B. S. Wan, Chem. Commun., 2013, 5073–5075 RSC ;
(b) X. D. Lu, B. Pan, F. Wu, X. Y. Xin and B. S. Wan, Tetrahedron Lett., 2015, 56, 4753–4755 CrossRef CAS .
- For Ni–catalyzed cycloaddition, see:
(a) A. Thakur and J. Louie, Acc. Chem. Res., 2015, 48, 2354–2365 CrossRef CAS ;
(b) M. Q. Zhou, J. Zhang, X. G. Zhang and X. G. Zhang, Org. Lett., 2019, 21, 671–674 CrossRef CAS ;
(c) S. Pang, X. Yang, Z. H. Cao, Y. L. Zhang, Y. Zhao and Y. Y. Huang, ACS Catal., 2018, 8, 5193–5199 CrossRef CAS .
- B. Pan, X. D. Lu, C. X. Wang, Y. C. Hu, F. Wu and B. S. Wan, Org. Lett., 2014, 16, 2244–2247 CrossRef CAS .
- For a recent review on the conversion of allenes to bicyclic MAs, see: C. S. Adams, C. D. Weatherly, E. G. Burke and J. M. Schomaker, Chem. Soc. Rev., 2014, 43, 3136–3163 RSC .
-
(a) J. D. Bois, Org. Process Res. Dev., 2011, 15, 758–762 CrossRef ;
(b) D. N. Zalatan and J. D. Bois, Top. Curr. Chem., 2010, 292, 347–378 CrossRef CAS .
- A. H. Stoll and S. B. Blakey, J. Am. Chem. Soc., 2010, 132, 2108–2109 CrossRef CAS .
- G. C. Feast, L. W. Page and J. Robertson, Chem. Commun., 2010, 2835–2837 RSC .
- J. Robertson, G. C. Feast, L. V. White, V. A. Steadman, née Doughty and T. D. W. Claridgea, Org. Biomol. Chem., 2010, 8, 3060–3063 RSC .
- L. A. Boralsky, D. Marston, R. D. Grigg, J. C. Hershberger and J. M. Schomaker, Org. Lett., 2011, 13, 1924–1927 CrossRef CAS .
- R. D. Grigg, J. M. Schomaker and V. Timokhin, Tetrahedron, 2011, 67, 4318–4326 CrossRef CAS .
- J. W. Rigoli, C. D. Weatherly, B. T. Vo, S. Neale, A. R. Meis and J. M. Schomaker, Org. Lett., 2013, 15, 290–293 CrossRef CAS .
-
(a) C. S. Adams, L. A. Boralsky, I. A. Guzei and J. M. Schomaker, J. Am. Chem. Soc., 2012, 134, 10807–10810 CrossRef CAS ;
(b) L. Liu, N. C. Gerstner, L. J. Oxtoby, I. A. Guzei and J. M. Schomaker, Org. Lett., 2017, 19, 3239–3242 CrossRef CAS ;
(c) C. S. Adams, R. D. Grigg and J. M. Schomaker, Chem. Sci., 2014, 5, 3046–3056 RSC .
- Pioneer work on the synthesis of DASP, see: R. S. Atkinson and J. R. Malpass, Tetrahedron Lett., 1975, 16, 4305–4306 CrossRef .
- J. W. Rigoli, L. A. Boralsky, J. C. Hershberger, D. Marston, A. R. Meis, I. A. Guzei and J. M. Schomaker, J. Org. Chem., 2012, 77, 2446–2455 CrossRef CAS .
-
(a) C. D. Weatherly, J. W. Rigoli and J. M. Schomaker, Org. Lett., 2012, 14, 1704–1707 CrossRef CAS ;
(b) C. D. Weatherly, I. A. Guzei and J. M. Schomaker, Eur. J. Org. Chem., 2013, 3667–3670 CrossRef CAS .
- J. W. Rigoli, I. A. Guzei and J. M. Schomaker, Org. Lett., 2014, 16, 1696–1699 CrossRef CAS .
- N. C. Gerstner, C. S. Adams, M. Tretbar and J. M. Schomaker, Angew. Chem., Int. Ed., 2016, 55, 13240–13243 CrossRef CAS .
- S. C. Schmid, I. A. Guzei and J. M. Schomaker, Angew. Chem., Int. Ed., 2017, 56, 12229–12233 CrossRef CAS .
- S. C. Schmid, I. A. Guzei, I. Fernández and J. M. Schomaker, ACS Catal., 2018, 8, 7907–7914 CrossRef CAS .
- E. G. Burke and J. M. Schomaker, Angew. Chem., Int. Ed., 2015, 54, 12097–12101 CrossRef CAS .
- J. Eshon, F. Foarta, C. R. Landis and J. M. Schomaker, J. Org. Chem., 2018, 83, 10207–10220 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Feng Li
a and
Yingying Zhao
*a,
Feng Li
a and
Yingying Zhao