
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium based antimicrobial theranostics – using nanoscopy to identify therapeutic targets and resistance mechanisms in Staphylococcus aureus†
Kirsty L.
Smitten
 *ab,
Simon D.
Fairbanks
a,
Craig C.
Robertson
a,
Jorge
Bernardino de la Serna
cd,
Simon J.
Foster
b and
Jim A.
Thomas
*a
*ab,
Simon D.
Fairbanks
a,
Craig C.
Robertson
a,
Jorge
Bernardino de la Serna
cd,
Simon J.
Foster
b and
Jim A.
Thomas
*a
aDepartment of Chemistry, University of Sheffield, Sheffield S10 2TN, UK. E-mail: ksmitten1@sheffield.ac.uk; james.thomas@sheffield.ac.uk
bThe Florey Institute and Department of Molecular Biology and Biotechnology, University of Sheffield, S10 2TN, UK
cNational Heart and Lung Institute, Faculty of Medicine, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
dResearch Complex at Harwell, Rutherford Appleton Laboratory, Central Laser Facility, United Kingdom Research and Innovation, OX11 0FA, UK
First published on 29th October 2019
Abstract
In previous studies we reported that specific dinuclear RuII complexes are particularly active against pathogenic Gram-negative bacteria and, unusually for this class of compounds, appeared to display lowered activity against Gram-positive bacteria. With the aim of identifying resistance mechanisms specific to Gram-positive bacteria, the uptake and antimicrobial activity of the lead complex against Staphylococcus aureus SH1000 and other isolates, including MRSA was investigated. This revealed differential, strain specific, sensitivity to the complex. Exploiting the inherent luminescent properties of the RuII complex, super-resolution STED nanoscopy was used to image its initial interaction with S. aureus and confirm its cellular internalization. Membrane damage assays and transmission electron microscopy confirm that the complex disrupts the bacterial membrane structure before internalization, which ultimately results in a small amount of DNA damage. A known resistance mechanism against cationic antimicrobials in Gram-positive bacteria involves increased expression of the mprF gene as this results in an accumulation of positively charged lysyl-phosphatidylglycerol on the outer leaflet of the cytoplasmic membrane that electrostatically repel cationic species. Consistent with this model, it was found that an mprF deficient strain was particularly susceptible to treatment with the lead complex. More detailed co-staining studies also revealed that the complex was more active in S. aureus strains missing, or with altered, wall teichoic acids.
Introduction
Over the last few decades it has become increasingly clear that antimicrobial resistance, AMR, is evolving into a global crisis.1–5 Although reports of resistance to antibiotics emerged quite soon after their rollout, the current extent and breadth of resistant infections is unprecedented and continues to rapidly increase.6–8 Due to a combination of scientific and commercial reasons, this problem is being exacerbated by a dearth of new therapeutic leads.9–17 Consequently, in the last few years, a series of national and international initiatives have been launched to provide the required economic pull and push to incentivize new drug discovery programs.2,18 In terms of chemistry, the need for leads with novel mechanisms of action to established antibiotics has driven searches into new chemical space.19,20 In this context, the potential of metal complexes has begun to be revisited.Even though ground-breaking studies by the Dwyer group in the mid-twentieth century had established that polypyridyl complexes of mid-transition metal ions display antimicrobial activity21 – particularly against Gram-positive organisms22 – research into metal-based leads stagnated for almost fifty years. Now, with the rise in concerns over AMR, interest in such systems is undergoing a renaissance.23–26
Whilst a number of reports have demonstrated that the activity of conventional antibiotics can be modulated or potentiated by connection to metal–organic fragments,27 molecular architectures that are very different to natural systems have also been investigated.28,29 A range of metals have been used in these studies, but – inspired by Dwyer's early work – many reports have involved RuII complexes.30–32
In 2011, Aldrich-Wright, Bolhuis and co-workers showed that mononuclear RuII complexes incorporating the DNA intercalating ligand dppz (dppz = dipyridophenazine) display promising in vivo activity against strains of the Gram-positive bacterium, Staphylococcus aureus.33 Subsequent reports on other intercalating34,35 and non-intercalating36 mononuclear RuII complexes have revealed comparable activities and such studies have also been extended to oligonuclear compounds.
Inspired by work from the Kelly group,37,38 Keene and Collins began investigating dinuclear RuII complexes with flexible linkers as probes for non-canonical DNA structures.39,40 However, on discovering the interesting antibacterial activity of these systems,41 their work switched focus. A series of studies on oligonuclear complexes revealed that longer, flexible linkers between metal centres produces higher activity against therapeutically resistant Gram-positive pathogens, although activity against Gram-negative pathogens is still lower.42
The mechanism for the selective therapeutic action of these compounds has been investigated but is still not fully established. Studies indicate that active complexes bind ribosomal RNA in prokaryotes, but it is also true that they bind RNA in the eukaryotic nucleus43 A recent combined NMR and computational study using membrane models suggested that – due to the increased anionic charge of bacterial membranes – the complexes selectively bridge bacterial walls, but not eukaryote membranes.44 Interestingly, these studies also showed that the bound complexes do not structurally compromise the membrane as leakage assays revealed no lytic activity.
As part of a program to identify oligonuclear complexes that recognize biologically relevant ions and biomolecules, the Thomas group has been investigating the biological properties of RuII, OsII, and IrIII complexes containing the rigidly planar poly-aromatic ligand tetrapyridophenazine, tpphz.45–50 Most of this work has centred on their bio-imaging and therapeutic potential with eukaryotic cells. But – as our original report on cell imaging showed that the prototype system, [{Ru(phen)2}2(tpphz)]4+, 14+, was readily taken up by S. aureus cells46 – we set out to develop new dual imaging/antimicrobial theranostics based on this architecture.
In a recent paper we described the first results of these studies, identifying derivatives of 14+, Fig. 1, containing more lipophilic ancillary ligands that are active against Gram-positive and Gram-negative bacteria, including members of the multidrug resistant ESKAPE group of pathogens.51 Experiments on the most promising lead suggested that its therapeutic action was, at least partly, due to disruption of bacterial membrane structure. Given this distinctive spectrum of activity, we set out to explore whether 14+ and its analogues are active against the pathogen S. aureus. Herein, we report the X-ray crystal structure of the lead system, explore its mechanism of action and identify resistance mechanisms that explain why it is less active against Gram-positive bacteria compared to Gram-negative bacteria.
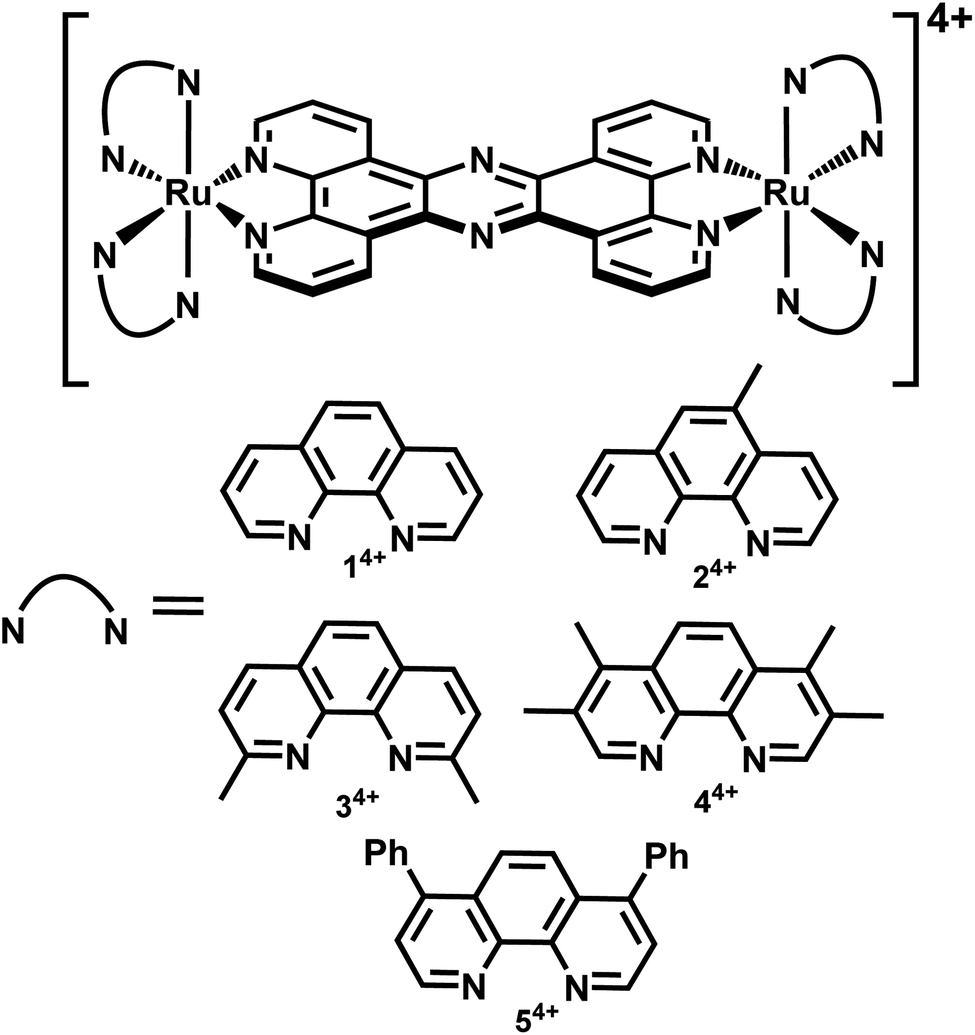 | ||
| Fig. 1 Complexes investigated in this study. | ||
Results and discussion
Crystallographic studies
Crystals grown from rac-44+ suitable for X-ray structure determination were successfully obtained by vapour diffusion of diethyl ether into a nitromethane solution of its hexafluoride salt. A summary of the crystallographic data and thermal ellipsoid plots can be found in the ESI.†As shown in Fig. 2, coordination ligands around the ruthenium centres 44+ define the expected octahedral geometry, distorted by accommodation of three chelating ligands. The resultant Ru–N bond lengths (2.036–2.069 Å) and bite angles (79.48–80.84°) are consistent with other reported dinuclear ruthenium polypyridyl complexes, including the closely related complex [{Ru(bpy)2}2(tpphz)]4+ (bpy = 2′2-bipyridine)50,52 Although crystals were grown from rac-44+ – a mixture of the Δ,Δ, Λ,Λ and Λ,Λ isomers of 44+ – the crystals shows cations packed in layers of offset coplanar stacks, where each alternating layer is solely comprised of either the Δ,Δ or Λ,Λ enantiomer. Interestingly, when [{Ru(bpy)2}2(tpphz)]4+ was similarly crystallized from an unresolved mixture of its stereoisomers the resulting structure also only contained the Δ,Δ and Λ,Λ forms and it was suggested that Λ,Δ form is more soluble than the enantiomeric pair.52 Alternatively, this phenomenon may be due to crystal packing effects.
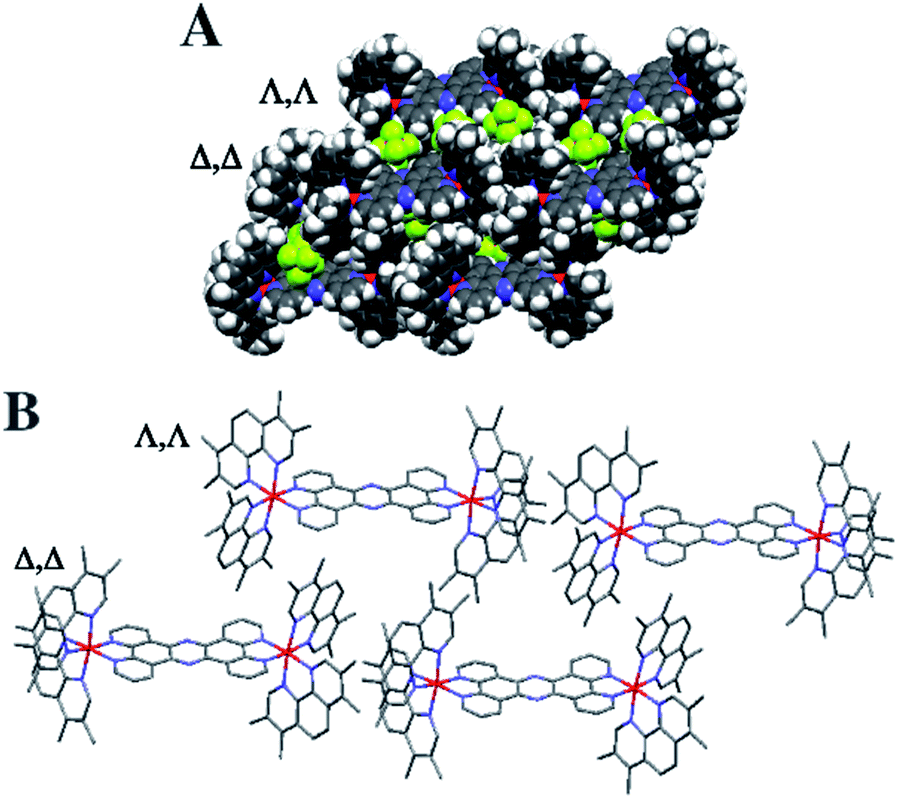 | ||
| Fig. 2 Crystal structure grown from rac-44+. Crystal structure showing Δ,Δ and Λ,Λ layers connected by interaction. (A) Space filling representation with PF6 counter ions. (B) Stick structure of 44+ without counter ions. | ||
Biological studies
As discussed above, in our recent report, we investigated the uptake and therapeutic activity of 14+, 34+–54+ against two examples of the pathogenic ESKAPE group of bacteria; the EC958 ST131 strain of Gram-negative Escherichia coli and the V583 strain of Gram-positive Enterococcus faecalis.51 and found they were more active against the Gram-negative organism. Further studies showed that the most promising lead, 44+, appears to be therapeutically active through a number of mechanisms, which include structural disruption of the Gram-negative cell membranes.These intriguing results prompted us to investigate the therapeutic action of this class of compounds against Gram-positive bacteria in more detail. For our exemplar of a typical Gram-positive microorganism we chose S. aureus.
Partially, this choice was prompted by the fact that we had already established 14+ is readily internalized by S. aureus,46 but this bacterium also provides an ideal “test-bed” to investigate potential resistance mechanisms. Apart from the fact that pathogenic S. aureus strains – particularly methicillin-resistant variants (MRSA), cause pernicious infections associated with treatment failure, higher morbidity/mortality rates, and prolonged hospitalizations greatly increasing health care costs53–55 – the mechanisms of therapeutic resistance in specific strains are well studied56–59 and so they can be used to probe the molecular mechanism of new antimicrobials. Therefore, in this study we investigate the potency of 14+–54+ against a range of S. aureus strains. Compounds used in biological studies were the mixture of stereoisomers, not the crystallised material.
Initially, to identify the most active lead against a representative wild type strain of S. aureus (SH1000),60 the minimum inhibitory concentration, MIC, of the complexes in chemically defined minimal media, CDM, and the commonly used Mueller-Hinton-II media, MH-II was obtained – Table 1. Although nutrient-rich growth media, like MH-II, are used as a standard in clinical assessments of antimicrobials, the nutrient limited environment provided by CDM also represents relevant biological conditions during different stages of infection in vivo.61 As with previous studies, the complexes show increased activity in the CDM, the lower potencies in MH-II relative to the CDM is likely due to the complexes interacting with components in the complex media.
| Complex | MH-II | CDM |
|---|---|---|
| 1 4+ | 66.7 | 21.3 |
| 2 4+ | 100 | 64 |
| 3 4+ | 50 | 58.7 |
| 4 4+ | 40 | 4 |
| 5 4+ | 20 | 2 |
| Oxacillin control | 2.5 | 2.5 |
Minimum bactericidal concentrations, MBC, were also calculated for the compounds in minimal media to determined bactericidal effects – Table 2. As a compound is seen to be bactericidal if it possesses an MBC that is ≤4 [MIC],62 a comparison of data indicated that all the tested compounds are bactericidal.
| Complex | MH-II | CDM | MIC/MBC (CDM) |
|---|---|---|---|
| 1 4+ | 114 | 58.7 | 2.8 |
| 2 4+ | 170 | 106.7 | 1.7 |
| 3 4+ | 100 | 85.3 | 1.5 |
| 4 4+ | 80 | 13.3 | 3.3 |
| 5 4+ | 40 | 6.7 | 3.3 |
| Oxacillin control | 2.5 | 2.5 | 2.5 |
To confirm that these compounds are bactericidal, cell death curves were conducted at five times the MIC concentration for the two most potent compounds, 44+ and 54+ – see ESI.† A 3Log10 decrease (99.9%) in CFU mL−1 is observed, verifying their bactericidal activity.
Previous research indicated that increasing the lipophilicity of the compounds increases the activity.41,51 This effect is apparent here: the most potent compound appears to be the only true lipophilic compound. However, such direct comparisons are misleading, as 54+ precipitated at high concentrations and 5% DMSO was required to improve its solubility – see ESI† for growth curves. As 44+ displays comparable potency, but without addition of DMSO, it was taken as the more promising lead and investigated in more detail.
In time-kill assays the majority of cells exposed to 44+ die within 30 min. This is much faster than the oxacillin control, suggesting that the complex displays a different mechanism of action to this beta-lactam antibiotic. To investigate this rapid bactericidal effect, ICP-AES-based uptake experiments with wild-type SH1000 cells exposed to 44+ at time points up to an hour were carried out – Fig. 3.
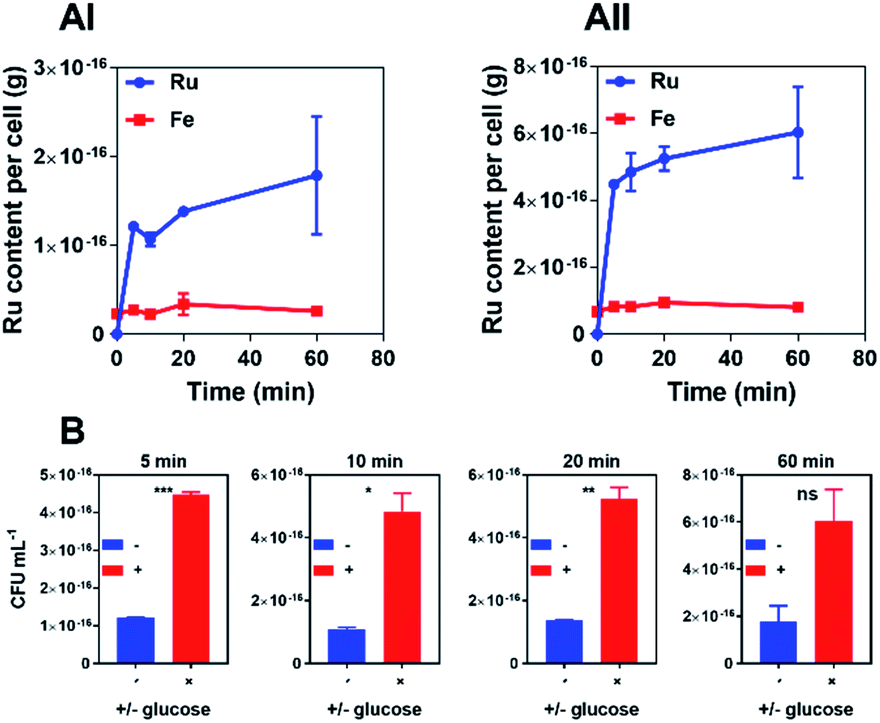 | ||
| Fig. 3 Accumulation experiments. ICP-AES data for the uptake of ruthenium by S. aureus, SH1000, after exposure to 44+ in the absence (AI) and presence (AII) of glucose. Ru (blue) and Fe (red) levels per cell are exposed as metal (g) per cell. T-test stats are given as a comparison at each time-point (B). Fe levels were calculated as a control. Conditions: concentration of 44+ = 4 μM. Cells were washed with 0.5% (v/v) nitric acid to remove unbound complex. Error bars represent three independent biological repeats ±SD. | ||
In these experiments, measurement of iron content (a trace element in all cells) was used as a control and, as expected, a negligible change in [Fe] was observed. CFU/mL readings were also taken to monitor whether cells were lysing/growing during the experiment – see ESI.†
Consistent with the rapid cell death observed in the time-kill assays, accumulation of RuII was also found to be rapid. As previous work demonstrated that 44+ is taken up into Gram-negative bacteria through an active transport mechanism,51 uptake by S. aureus was studied in the presence and absence of glucose over a 60 minute time course and the rates of uptake were determined (μM min−1) for 44+ – see ESI† – at each time point. In the absence of glucose, accumulation over 5 minutes is followed by gradual uptake to a final figure of around 2 × 10−16 g per cell; assuming an average cell volume63,64 of 1 μm3 this is equivalent to an intracellular concentration of >1 mM per cell.
Although the uptake profile of the complex in the presence of glucose displays a similar profile in these conditions there is significantly greater intracellular accumulation of ruthenium (∼3 mM per cell), suggesting that an energy-dependent active transport mechanism contributes to the uptake of 44+ into SH1000 cells. However, in contrast to previously reported experiments with E. coli,51 ruthenium accumulation within S. aureus in the absence of glucose does not take place over two phases, suggesting that the mechanism of uptake for 44+ may be different in Gram-negative and Gram-positive bacteria.
Our previous microscopy experiments suggested that the compound initially binds to lipopolysaccharides on the outer membrane of E. coli, however S. aureus does not possess an outer membrane. Therefore, to study the target of 44+ at the molecular scale, its cellular uptake was monitored by a super resolution technique, structure illumination microscopy, SIM, allowing for sub-diffraction resolutions of >100 nm (ref. 65 and 66) – Fig. 4.
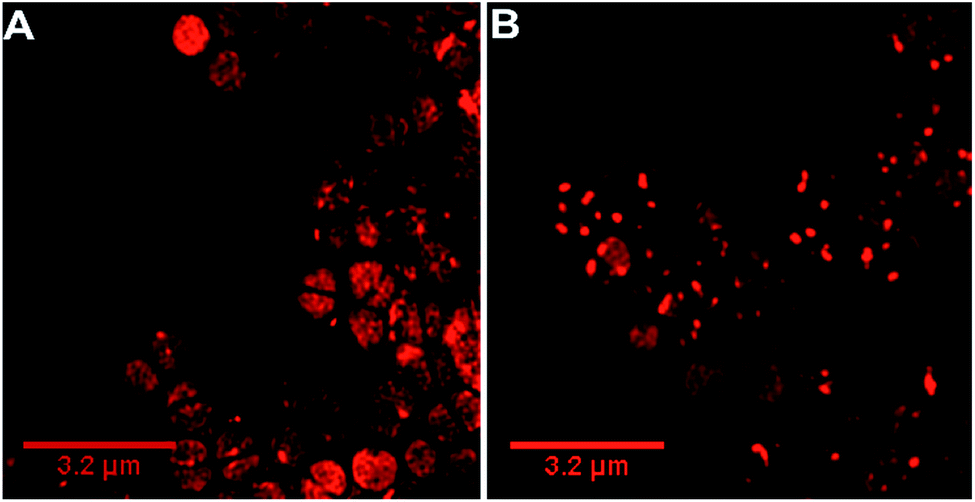 | ||
| Fig. 4 Super-resolution SIM imaging revealing localization of 44+ in S. aureus SH1000 cells at (A) 5 min and (B) 60 min. Cells images using the emission of 44+ on excitation at 450 nm using the A568 filter. After treatment with 4 μM 44+, cells were washed with PBS before fixing with paraformaldehyde (4%). | ||
Within 5 minutes, the majority of cells show that 44+ localizes at cell membranes and within the cell cytoplasm. Accumulation of 44+ is uneven, with punctate patterns. By 60 minutes the compound shows distinct foci within cells. The observation of intracellular accumulation is similar to that for E. coli when exposed to 44+.51
In recent studies we have demonstrated how complexes like 14+ and its analogues can be employed as optical probes for STED, a powerful technique that can, potentially, produce resolutions equal to those of stochastic techniques such as STORM.65,67–69 In our 3D-STED experiments, the resolution of images probing localization of 44+ on the membrane of SH1000 cells was improved up to 43 nm and we regularly resolved structural patterns below 80 nm in the XY plane and above 150 nm in the axial plane – Fig. 5. The direct imaging of the cell septum indicated that the compound is not bound to the peptidoglycan within the cell wall; instead 44+ is localized within or on the membrane. At the improved resolutions provided by 3D-STED the uneven accumulation observed using SIM is revealed to be a distinctive patterning over the entire surface of the S. aureus membrane.
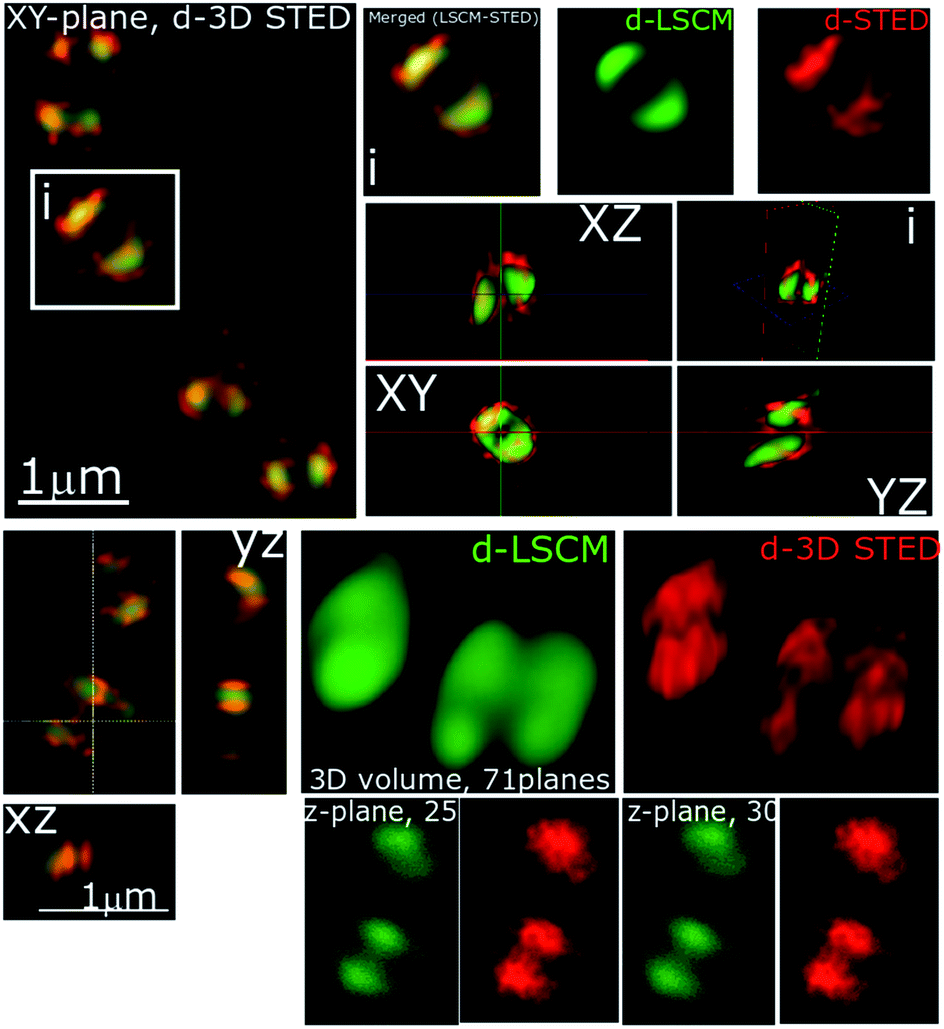 | ||
| Fig. 5 Stimulated emission depletion (STED) nanoscopy. Representative data showing section planes of full-volume cells deconvoluted diffraction-limited (d-LCSM, green) and 3D super-resolution images (Red, d-3D STED). Zoomed in areas of the white square are shown in adjacent images, as well as the orthogonal representation of every axis (XY, XZ and YZ), where the increased resolution and better localization of 44+ is shown in red. This reveals accumulation at specific locations within the cell membrane. Conditions: after 60 minute treatment with 4 μM 44+, cells were washed with PBS before fixing with paraformaldehyde (4%). | ||
As complexes 14+–54+ are all known to bind nucleic acids45,51 and the parent compound localizes in the nucleus of eukaryotic cells,46 the commonly used prokaryotic and eukaryotic DNA stain DAPI was also used as a co-stain to determine whether DNA is targeted once 44+ internalizes with cells imaged using SIM – Fig. 6.
 | ||
| Fig. 6 DAPI co-localization SIM experiments. Cells imaged using the emission of 44+ on excitation at 450 nm using the 488 nm laser and the A568 filter (A), emission of DAPI using the 405 nm laser and DAPI filter (B), overlay image (C). After treatment with 4 μM 44+, cells were incubated with DAPI for 5 minutes and washed with PBS before fixing with paraformaldehyde (4%). | ||
Visual inspection of images recorded after 10 minutes of exposure indicated that the intracellular accumulation of 44+ and DAPI do not strongly overlap, which is confirmed by a relatively low Pearson's correlation coefficient of 0.28. At 60 minutes there is an increase in co-localization with the Pearson coefficient rising to 0.49. As these data suggest that the complex binds to DNA within SH1000 cells this could result in increased mutational rates. Therefore, a mutagenesis assay (Ames test – fluctuation method) was performed; see ESI.†
These tests reveal that, at concentrations above the MIC, exposure to 44+ results in bacterial DNA damage, providing further evidence that the complex does bind intracellular DNA. However, this effect is less pronounced than that caused by exposure to mutagenic UV-irradiation, suggesting that, above the MIC concentration, DNA damage is only a contributing cause of cell death and is part of a multi-target mechanism of action.
Further co-staining studies with a second imaging probe – the commonly used membrane and cell wall stain NHS-ester 405 – resulted in some revealing results – see ESI.† Initially, co-localization tests between NHS-ester and 44+ show a high correlation; however, co-localization decreases over 60 minutes when there is a concomitant increase in DAPI co-localization. These data suggest that 44+ initially binds to bacterial cell surface regions, then consequently internalizes and increasingly binds to DNA.
Revealingly, when the NHS-ester is added to cells after 44+, within 20 minutes NHS-ester staining around the periphery of cells becomes discontinuous. After 60 minutes of exposure this discontinuity becomes increasingly prominent – see ESI,† confirming the induction of cell damage.
Many of the observations described above are consistent with our previous research on Gram-negative bacteria, which showed that cell membrane damage occurs on exposure to 44+. To confirm an analogous mechanism operates in Gram-positive bacteria, the integrity of the S. aureus membranes on treatment with 44+ was tested through an ATP release assay. In this assay, cells were exposed to 44+ at its MIC and 0.5 MIC – see ESI.† These experiments showed that treated cells release more ATP than untreated controls. Furthermore, at the MIC concentration a build-up of cellular debris was observed, indicating that cell lysis was occurring.
The effect of 44+ on the membrane potential of S. aureus cells was also measured using the membrane potential dye DiOC2(3) – Fig. 7. In intact bacteria cells DiOC2(3) emits in the green; however once a membrane is polarized and its potential increases the emission of DiOC2(3) undergoes a red shift, due to dye self-association at higher cytosolic concentrations.70 A diagnostic shift from green to red (red-green) is observed when cells are treated with 44+ at its MIC concentration, signifying an increase in membrane potential. Together, these two experiments provide definitive proof that exposure to 44+ does lead to membrane damage in S. aureus.71,72
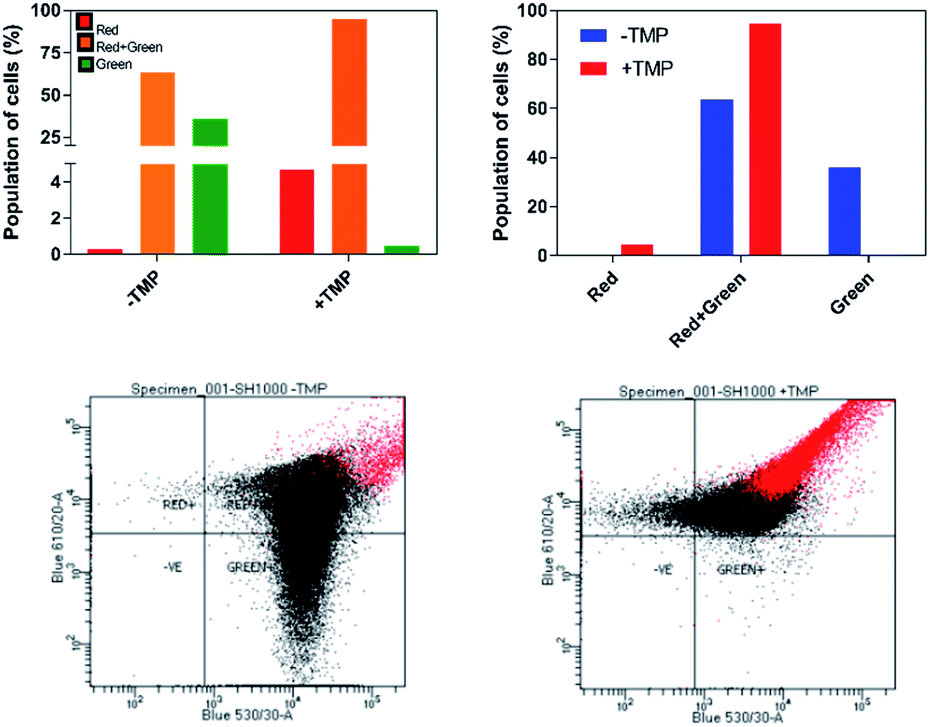 | ||
| Fig. 7 Detection of membrane potential in S. aureus SH100 cells. Red/green cell populations were calculated using population mean fluorescence intensities for S. aureus cells incubated with 30 μM DiOC2(3) for 30 minutes, in the presence or absence of μM of 44+ for 10 minutes. Top: Percentage population is shown in two representation styles. Analysis was conducted using red and green fluorescence parameters. Bottom: The red-versus green fluorescence dot plots. Flow cytometer data were collected with log amplification. | ||
As 44+ contains two electron dense RuII-centres, it is an excellent contrast stain for Transmission Electron Microscopy (TEM). Therefore, damage to the cell wall and membrane was directly imaged using TEM – Fig. 8.
 | ||
| Fig. 8 Detection of membrane damage using transmission electron microscopy (TEM). Images were taken at (A) in the absence of compound and at 20 (B), 60 (C) and 120 (D) minutes after exposure to 44+. In the healthy control the cell wall (CW), plasma membrane (PM) and septum (S) are labelled. Cells were fixed using 2.5% glutaraldehyde in sodium cacodylate buffer (pH 7.4) and stained solely with 44+ (40 μM). The 0 minute control was stained with osmium tetroxide, uranyl acetate and lead citrate. | ||
From these images it is clear the compound causes disruption to both the cell wall and the cell membrane within 20 minutes. At concentrations above its MIC, complex 44+ causes rapid cell death; cell walls appear swollen, and blebbing/channelling is evident. These effects account for the ATP leakage and membrane potential changes seen in the membrane damage assays.
As complex 44+ displays high activity against a wild-type strain and appeared to function through multi-target mechanisms that are analogous to those observed in Gram-negative pathogens, its activity against other S. aureus strains was then investigated. This screen involved a clinical isolate strain taken from a patient with septic arthritis and septicaemia and a confirmed MRSA strain BH1CC – Table 3. BH1CC is a mecA positive, methicillin resistant strain of S. aureus.
| Strain | MH-II | CDM |
|---|---|---|
| SH1000 | 40 | 4 |
| BH1CC | 76.5 | 38.3 |
| Clinical isolate | 51.0 | 19.1 |
In contrast to our work on pathogenic drug resistant Gram-negative bacteria, a significant decrease in activity is observed across both strains in comparison to SH1000. This suggests some strain specific susceptibility against 44+. The molecular basis for resistance was therefore investigated.
Although the cellular responses of S. aureus are similar to those observed for E. coli, one noticeable difference is the distinctive punctated, cell-associated pattern of 44+ – in S. aureus cell walls, shown in Fig. 4 and 5. This suggests that the initial interaction of the complex with Gram-positive and Gram-negative bacteria is different. This is not surprising – a major difference between these two classes of microorganisms is the composition and structure of their cell walls. To investigate this issue, a series of more specialized experiments were carried out.
One obvious difference between the two classes is that the cell wall of Gram-positive bacteria contains a much thicker outer peptidoglycan layer compared to Gram-negative bacteria. Indeed, the biosynthesis of this structure is the established therapeutic target of both beta-lactam and glycopeptide antibiotics.73 To ascertain whether this component of the cell wall is a binding target for 44+, co-staining with a blue emitting peptidoglycan stain – the D-amino acid, HCC-amino-D-alanine (HADA) – was carried out and imaged through SIM, Fig. 9.74 These experiments revealed that, although some co-localization between the two dyes emerged after 60 minutes exposure, the initial distinctive distribution of 44+ shows little co-localization with HADA; therefore, specific peptidoglycan binding by the complex can be discounted.
 | ||
| Fig. 9 Localization of 44+ and HADA in S. aureus SH1000 cells visualized through SIM at 10, 20 and 60 minutes. The emission of 44+ upon excitation with the 488 nm laser was collected using the A568 filter (A), emission of HADA from excitation with the 405 nm laser was collected using the DAPI filter (B), overlay image (C). Cells were stained with HADA for 5 minutes prior to incubation with 44+. After treatment with 4 μM 44+ cells were washed with PBS and fixed with PFA (4%). Co-localisation scatter plots are shown (D). See ESI† for Pearson's correlation coefficient. | ||
A second characteristic feature of cell walls in Gram-positive bacteria is that it is densely functionalized with glycopolymers. The two most common glycopolymers, wall teichoic acid and lipoteichoic acid, contain anionic residues and also moieties that protrude from the cell wall.75–78 As these structures are known to bind extracellular metal cations, the possibility that they interact with 44+ was then investigated.
To address this question, experiments on two mutant strains of S. aureus were carried out. The tarO strain is deficient in wall teichoic acids,79 whilst the dltA strain is specifically deficient in D-alanylated teichoic acids.80
First, MICs for these two mutants were quantified and compared to the data obtained for the SH1000 wild-type strain – Table 4. These data revealed that both mutant strains possess lower MICs for 44+ than SH1000, indicating that binding to the anionic residues of teichoic acid in the wild-type strain lowers susceptibility to 44+ and prevents the compound from reaching other cellular targets. This hypothesis is consistent with previous studies showing increased synthesis of wall teichoic acid is a known resistance mechanism in S. aureus.
| Strain | MIC |
|---|---|
| SH1000 | 40 |
| dltA | 20 |
| tarO | 15 |
| MprF | 1.5 |
Imaging experiments involving the dltA mutant were also revealing. Using SIM, the uptake and subsequent localization pattern produced on exposure to 44+ were compared to SH1000, Fig. 10. In contrast to the wild-type, staining of the dltA mutant did not produce a punctated accumulation pattern over the membrane and the septa of dividing cells are not significantly stained by 44+. Furthermore, although the mutant was exposed to the same concentration of 44+ as the wild-type strain, dltA displays brighter luminescence from the internalized complex, suggesting a higher concentration of compound enters the cell. This provides further evidence that binding to teichoic acids within the cell wall of Gram-positive bacteria inhibits cellular internalization and lowers the therapeutic potency of 44+.
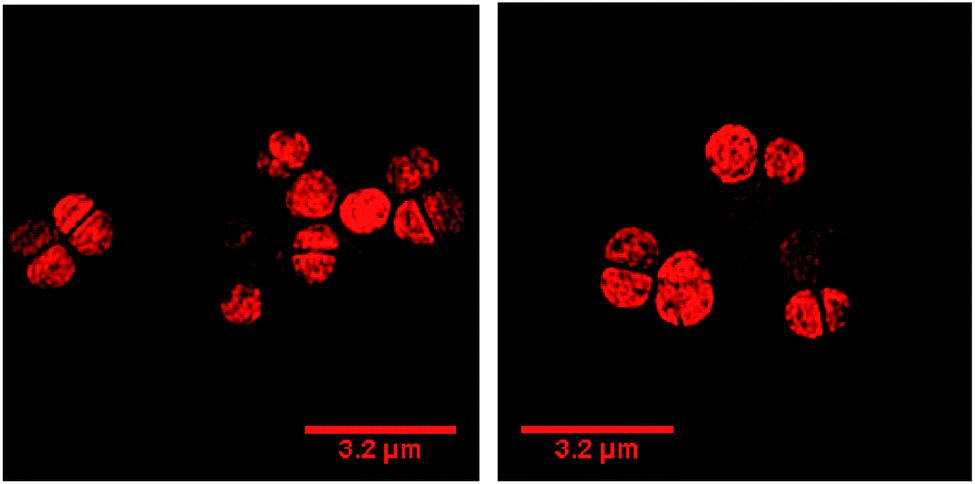 | ||
| Fig. 10 Localization of 44+ in S. aureus dltA cells visualized through SIM at 20 min (left) and 60 min (right). Cells imaged using the emission of 44+ on excitation at 450 nm using the A568 filter. After treatment with 4 μM 44+, cells were washed with PBS before fixing with paraformaldehyde (4%). | ||
The uptake of 44+ by dltA was monitored by measuring Ru content (g) per cell using ICP-AES – see ESI† – Fe content was also recorded. The experiment was conducted in the absence of glucose to monitor the differences in compound uptake between dltA and SH1000. The different profiles suggest a different mechanism of uptake. The intracellular concentration of 44+ at the earlier time-points (5, 10 and 20 min) is higher in the dltA; however, the final intracellular compound concentration is similar across both strains. This supports the hypothesis that 44+ binds teichoic acids on the cell membrane preventing optimal intracellular concentrations, thus lowering the activity. In addition, this observation is consistent with the time-kill assays where significant cell death occurs within the first 30 minutes of exposure to 44+, showing that the higher initial intracellular concentration accounts for the lower MIC in dltA cells. Cell viability was monitored through the course of the experiment by CFU mL−1 counts and the cells did not lyse.
Along with our previous work on Gram-negative bacteria, the experiments described above demonstrate that exposure of S. aureus cells to 44+ causes cell membrane damage and lysis. To confirm this hypothesis and explore another known resistance mechanism available to S. aureus we carried out experiments with a third mutant strain, associated with membrane charge.
An established resistance mechanism to membrane active agents in S. aureus involves up-regulation of mprF.80 This gene codes for an enzyme that links lysine to negatively charged phosphatidylglycerol and translocates the resulting positively charged lysyl-phosphatidylglycerol to the outer leaflet of the cytoplasmic membrane, a process that increases positive charge on the outer surface of the S. aureus membrane and thus reduces susceptibility to cationic antibiotics. Knockout of mprF necessitates that the bacterial membrane outer leaflet retains a higher negative charge compared to the wild-type.81,82 Revealingly, 44+ is considerably more active against this strain than any other tested – Table 4.
Another known mechanism of resistance in MRSA strains is upregulation of teichoic and lipoteichoic acids.83 To investigate whether this may cause the decrease in activity of 44+ any binding interactions were probed by STED nanoscopy – Fig. 11. It is clear that in the MRSA strain, 44+ does not penetrate the cell wall and membrane. Indeed, the complex is clearly retained on the cell–wall and the previously described spotting accumulation is noticeably more prevalent. Strikingly in these 3D-STED images the septa of the cell are no longer visible confirming that the complex is not internalized. This supports the hypothesis that the compound is electrostatically binding teichoic and lipoteichoic acids, preventing high concentrations of 44+ within the cell, thus lowering the activity.
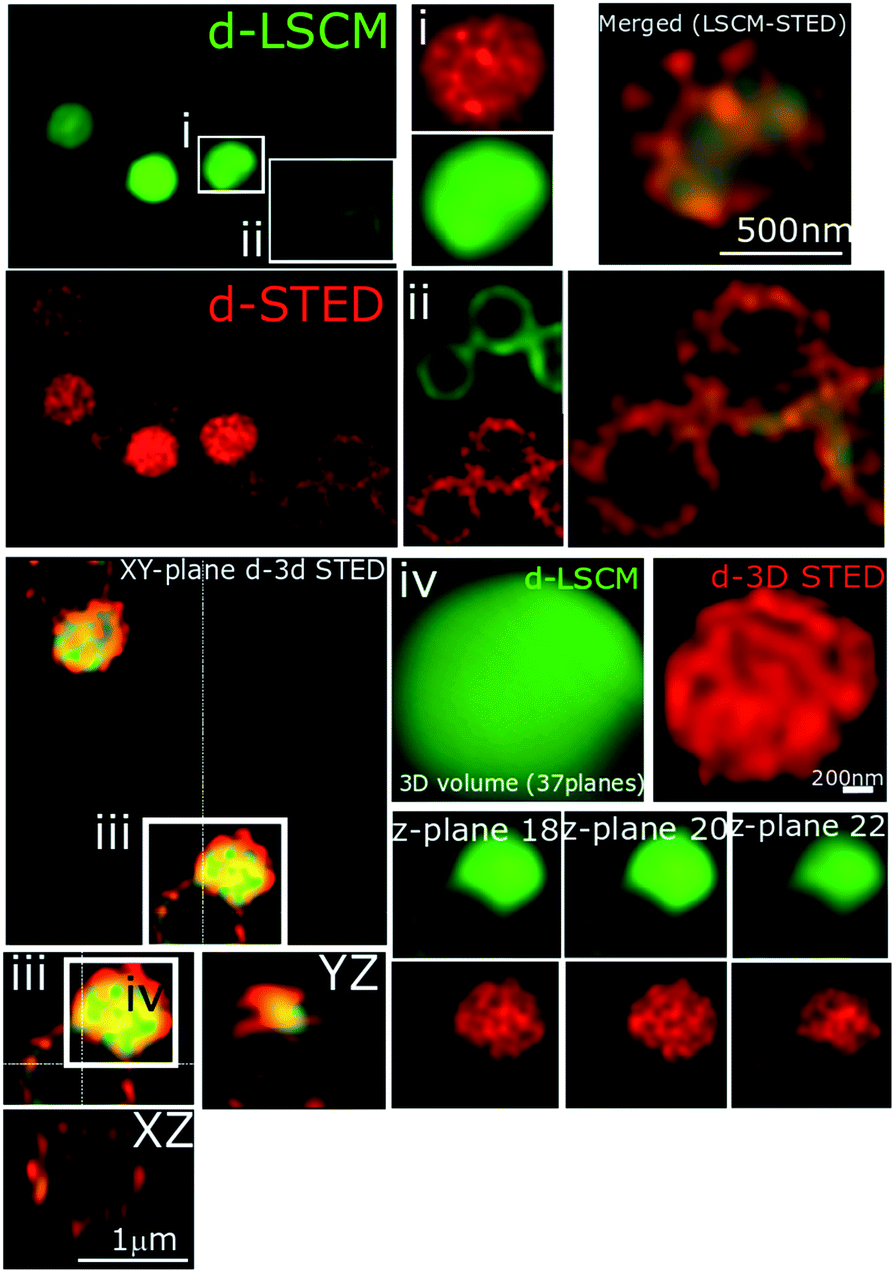 | ||
| Fig. 11 STED nanoscopy of S. aureus BH1CC cells stained with 44+. Representative images showing section planes of full-volume deconvoluted diffraction-limited cells and super-resolution images (d-LCSM and d-3D STED). Zoomed in areas of the white squares are shown adjacent to the images, as well as the orthogonal representation of every axis (XY, XZ and YZ), where the increased resolution confirms accumulation of 44+ within the cell wall and membrane. Images (i and ii) show the probe distribution at two different sections of the cell, where the higher structural resolution provided by STED at the equatorial and the apical section of the cells is apparent. Inset (iii) shows two adjacent cells, and to right-hand side, 3 different Z-planes (87 nm separation between them) again, increased resolution of structural features can be delineated in 3D dSTED images (red). Conditions: after treatment with 4 μM 44+ for 60 minutes, cells were washed with PBS before fixing with paraformaldehyde (4%). | ||
Conclusions
Taken together, the studies in this report confirm that two of the therapeutic targets of 44+ in bacteria are their cell membranes and DNA content. Furthermore, the experiments on the mutant strains reveal that the reason the complex is more active against Gram-negative bacteria compared to Gram-positive bacteria is due to intrinsic differences in the molecular structures of these microorganisms.As discussed above, as has been observed with other antibacterials accretion of cationic lysyl-phosphatidylglycerol residues on the outside of the cytoplasmic membrane inhibits internalization of the complex. Indeed a very recent report on helicates, published as we compiled this study, showed that bacterial resistance to such systems also involves reduction of net negative surface charge.84 Cell walls densely functionalized with the glycopolymers teichoic acid and lipoteichoic acids also provides an additional barrier to uptake toward 44+.
Identification of these mechanisms provide new therapeutic opportunities. For example, studies have shown that combinations such as a small molecule tarO inhibitor that blocks the production of WTA and a conventional antimicrobial are synthetically lethal to therapeutically resistant bacterial strains.85,86 Such a strategy will be explored in future work. Furthermore, the identification of resistance mechanisms in S. aureus strains has afforded insights into the on-going construction of new molecular architectures, based on 44+, possessing structural features designed to mitigate the effect of these mechanisms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to our reviewers, whose insightful comments on our original submission have considerably improved the quality of this study.Notes and references
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS.
- World Health Organization, Antimicrobial resistance: global report on surveillance, 2014 Search PubMed.
- E. Toner, A. Adalja, G. K. Gronvall, A. Cicero and T. V. Inglesby, Health Security, 2015, 13, 153–155 CrossRef.
- R. Kelly and S. Davies, Nat. Microbiol., 2016, 1, 16187 CrossRef.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, F. R. Burkert, O. Cars, G. Cornaglia, S. Gandra, C. G. Giske, D. A. Goff, M. Guzman Blanco, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, K. Leder, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, A. Pan, D. L. Paterson, M. Paul, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, G. E. Thwaites, J. W. van der Meer, S. Vega, A. Wechsler-Fördös, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef.
- A. J. Alanis, Arch. Med. Res., 2005, 36, 697–705 CrossRef.
- A. H. Holmes, L. S. P. Moore, A. Sundsfjord, M. Steinbakk, S. Regmi, A. Karkey, P. J. Guerin and L. J. V. Piddock, Lancet, 2016, 387, 176–187 CrossRef CAS.
- A. R. Brochado, A. Telzerow, J. Bobonis, M. Banzhaf, A. Mateus, J. Selkrig, E. Huth, S. Bassler, J. Zamarreño Beas, M. Zietek, N. Ng, S. Foerster, B. Ezraty, B. Py, F. Barras, M. M. Savitski, P. Bork, S. Göttig and A. Typas, Nature, 2018, 559, 259–263 CrossRef CAS.
- S. J. Projan, Curr. Opin. Microbiol., 2003, 6, 427–430 CrossRef.
- K. M. Overbye and J. F. Barrett, Drug Discovery Today, 2005, 10, 45–52 CrossRef.
- M. N. Gwynn, A. Portnoy, S. F. Rittenhouse and D. J. Payne, Ann. N. Y. Acad. Sci., 2010, 1213, 5–19 CrossRef.
- A. R. White, J. Antimicrob. Chemother., 2011, 66, 1948–1953 CrossRef CAS.
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS.
- M. S. Butler, M. A. Blaskovich and M. A. Cooper, J. Antibiot., 2017, 70, 3–24 CrossRef CAS.
- M. J. Renwick, D. M. Brogan and E. Mossialos, J. Antibiot., 2016, 69, 73–88 CrossRef CAS.
- M. F. Richter and P. J. Hergenrother, Chem, 2017, 3, 10–13 CAS.
- J. Jampilek, Curr. Med. Chem., 2019, 25, 4972–5006 CrossRef.
- C. O. Årdal, D. Findlay, M. Savic, Y. Carmeli and I. Gyssens, Drive-AB Report: Revitalizing the antibiotic pipeline: Stimulating innovation while driving sustainable use and global access, 2018 Search PubMed.
- R. Tommasi, D. G. Brown, G. K. Walkup, J. I. Manchester and A. A. Miller, Nat. Rev. Drug Discovery, 2015, 14, 529–542 CrossRef CAS PubMed.
- M. Fridman, Chem, 2017, 3, 8–10 CAS.
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190–191 CrossRef CAS.
- F. P. Dwyer, I. K. Reid, A. Shulman, G. M. Laycock and S. Dixson, Aust. J. Exp. Biol. Med. Sci., 1969, 47, 203–218 CrossRef CAS.
- M. Patra, G. Gasser and N. Metzler-Nolte, Dalton Trans., 2012, 41, 6350–6410 RSC.
- N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef.
- L. Viganor, O. Howe, P. McCarron, M. McCann and M. Devereux, Curr. Top. Med. Chem., 2017, 17, 1280–1302 CrossRef CAS.
- A. Regiel-Futyra, J. M. Dąbrowski, O. Mazuryk, K. Śpiewak, A. Kyzioł, B. Pucelik, M. Brindell and G. Stochel, Coord. Chem. Rev., 2017, 351, 76–117 CrossRef CAS.
- M. A. Sierra, L. Casarrubios and M. C. de la Torre, Chem.–Eur. J., 2019, 25, 7232–7242 CrossRef CAS.
- S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2011, 4, 31–36 CrossRef.
- M. Wenzel, M. Patra, C. H. R. Senges, I. Ott, J. J. Stepanek, A. Pinto, P. Prochnow, C. Vuong, S. Langklotz, N. Metzler-Nolte and J. E. Bandow, ACS Chem. Biol., 2013, 8, 1442–1450 CrossRef CAS.
- A. I. Ramos, T. M. Braga and S. S. Braga, Mini-Rev. Med. Chem., 2012, 12, 227–235 CrossRef CAS.
- H. M. Southam, J. A. Butler, J. A. Chapman and R. K. Poole, Adv. Microb. Physiol., 2017, 71, 1–96 CrossRef.
- Y. Yang, G. Liao and C. Fu, Polymers, 2018, 10, 650–712 CrossRef.
- A. Bolhuis, L. Hand, J. E. Marshall, A. D. Richards, A. Rodger and J. Aldrich-Wright, Eur. J. Pharm. Sci., 2011, 42, 313–317 CrossRef CAS.
- X. Liu, B. Sun, R. E. M. Kell, H. M. Southam, J. A. Butler, X. Li, R. K. Poole, F. R. Keene and J. G. Collins, ChemPlusChem, 2018, 83, 643–650 CrossRef CAS.
- A. K. F. Mårtensson, M. Bergentall, V. Tremaroli and P. Lincoln, Chirality, 2016, 28, 713–720 CrossRef.
- S. V. Kumar, S. Ø. Scottwell, E. Waugh, C. J. McAdam, L. R. Hanton, H. J. L. Brooks and J. D. Crowley, Inorg. Chem., 2016, 55, 9767–9777 CrossRef CAS.
- F. O'Reilly, J. Kelly and A. Kirsch-DeMesmaeker, Chem. Commun., 1996, 1013–1014 RSC.
- F. M. O'Reilly and J. M. Kelly, New J. Chem., 1998, 22, 215–217 RSC.
- J. L. Morgan, C. B. Spillane, J. A. Smith, D. P. Buck, J. G. Collins and F. R. Keene, Dalton Trans., 2007, 4333 RSC.
- Y. Mulyana, D. K. Weber, D. P. Buck, C. A. Motti, J. G. Collins and F. R. Keene, Dalton Trans., 2011, 40, 1510 RSC.
- F. Li, Y. Mulyana, M. Feterl, J. M. Warner, J. G. Collins and F. R. Keene, Dalton Trans., 2011, 40, 5032 RSC.
- X. Li, A. K. Gorle, M. K. Sundaraneedi, F. R. Keene and J. G. Collins, Coord. Chem. Rev., 2018, 375, 134–147 CrossRef CAS.
- F. Li, E. J. Harry, A. L. Bottomley, M. D. Edstein, G. W. Birrell, C. E. Woodward, F. R. Keene and J. G. Collins, Chem. Sci., 2014, 5, 685–693 RSC.
- D. K. Weber, M.-A. Sani, M. T. Downton, F. Separovic, F. R. Keene and J. G. Collins, J. Am. Chem. Soc., 2016, 138, 15267–15277 CrossRef CAS PubMed.
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem.–Eur. J., 2006, 12, 4611–4619 CrossRef CAS.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- A. Wragg, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem.–Eur. J., 2014, 20, 14004–14011 CrossRef CAS PubMed.
- A. Wragg, M. R. Gill, C. J. Hill, X. Su, A. J. H. M. Meijer, C. Smythe and J. A. Thomas, Chem. Commun., 2014, 50, 14494–14497 RSC.
- A. Wragg, M. R. Gill, L. McKenzie, C. Glover, R. Mowll, J. A. Weinstein, X. Su, C. Smythe and J. A. Thomas, Chem.–Eur. J., 2015, 21, 11865–11871 CrossRef CAS.
- S. D. Fairbanks, C. C. Robertson, F. R. Keene, J. A. Thomas and M. P. Williamson, J. Am. Chem. Soc., 2019, 141, 4644–4652 CrossRef CAS PubMed.
- K. L. Smitten, H. M. Southam, J. Bernardino de la Serna, M. R. Gill, P. J. Jarman, C. G. W. Smythe, R. K. Poole and J. A. Thomas, ACS Nano, 2019, 13, 5133–5146 CrossRef CAS.
- J. Bolger, A. Gourdon, E. N. Ishow and J.-P. Launay, J. Chem. Soc., Chem. Commun., 1995, 1799 RSC.
- K. Sieradzki, R. B. Roberts, S. W. Haber and A. Tomasz, N. Engl. J. Med., 1999, 340, 517–523 CrossRef CAS.
- M. C. Enright, D. A. Robinson, G. Randle, E. J. Feil, H. Grundmann and B. G. Spratt, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7687–7692 CrossRef CAS.
- S. Deresinski, Clin. Infect. Dis., 2005, 40, 562–573 CrossRef CAS PubMed.
- C. Walsh, Nature, 2000, 406, 775–781 CrossRef CAS.
- K. Hiramatsu, Lancet Infect. Dis., 2001, 1, 147–155 CrossRef CAS PubMed.
- F. D. Lowy, J. Clin. Invest., 2003, 111, 1265–1273 CrossRef CAS PubMed.
- M. N. Alekshun and S. B. Levy, Cell, 2007, 128, 1037–1050 CrossRef CAS PubMed.
- A. J. O'Neill, Lett. Appl. Microbiol., 2010, 51, 358–361 CrossRef.
- J. Flatley, J. Barrett, S. T. Pullan, M. N. Hughes, J. Green and R. K. Poole, J. Biol. Chem., 2005, 280, 10065–10072 CrossRef CAS.
- G. A. Pankey and L. D. Sabath, Clin. Infect. Dis., 2004, 38, 864–870 CrossRef CAS PubMed.
- H. E. Kubitschek and J. A. Friske, J. Bacteriol., 1986, 168, 1466–1467 CrossRef CAS PubMed.
- M. Loferer-Krossbacher, J. Klima and R. Psenner, Appl. Environ. Microbiol., 1998, 64, 688–694 CAS.
- J. Z. Rappoport, in Fluorescence Microscopy: Super-Resolution and other Novel Techniques, Elsevier, 2014, pp. 199–212 Search PubMed.
- L. Schermelleh, A. Ferrand, T. Huser, C. Eggeling, M. Sauer, O. Biehlmaier and G. P. C. Drummen, Nat. Cell Biol., 2018, 21, 1–13 Search PubMed.
- L. Meyer, D. Wildanger, R. Medda, A. Punge, S. O. Rizzoli, G. Donnert and S. W. Hell, Small, 2008, 4, 1095–1100 CrossRef CAS.
- G. Vicidomini, G. Moneron, K. Y. Han, V. Westphal, H. Ta, M. Reuss, J. Engelhardt, C. Eggeling and S. W. Hell, Nat. Methods, 2011, 8, 571–573 CrossRef CAS PubMed.
- H. Blom and J. Widengren, Chem. Rev., 2017, 117, 7377–7427 CrossRef CAS PubMed.
- L. V. Johnson, J. Cell Biol., 1981, 88, 526–535 CrossRef CAS PubMed.
- A. J. O'Neill and B. Oliva, J. Antimicrob. Chemother., 2004, 54, 1127–1129 CrossRef PubMed.
- Y. Zhang, X. Chen, C. Gueydan and J. Han, Cell Res., 2018, 28, 9–21 CrossRef CAS PubMed.
- N. Malanovic and K. Lohner, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 936–946 CrossRef CAS PubMed.
- E. Kuru, S. Tekkam, E. Hall, Y. V. Brun and M. S. Van Nieuwenhze, Nat. Protoc., 2014, 10, 33–52 CrossRef.
- F. C. Neuhaus and J. Baddiley, Microbiol. Mol. Biol. Rev., 2003, 67, 686–723 CrossRef CAS PubMed.
- C. Weidenmaier and A. Peschel, Nat. Rev. Microbiol., 2008, 6, 276–287 CrossRef CAS.
- S. Brown, J. P. Santa Maria Jr and S. Walker, Annu. Rev. Microbiol., 2013, 67, 313–336 CrossRef CAS PubMed.
- M. G. Percy and A. Gründling, Annu. Rev. Microbiol., 2014, 68, 81–100 CrossRef CAS.
- C. Weidenmaier, C. Weidenmaier, A. Peschel, A. Peschel, Y.-Q. Xiong, Y. Q. Xiong, S. A. Kristian, S. A. Kristian, K. Dietz, K. Dietz, M. R. Yeaman, M. R. Yeaman, A. S. Bayer and A. S. Bayer, J. Infect. Dis., 2005, 191, 1771–1777 CrossRef CAS PubMed.
- M. Gross, S. E. Cramton, F. Gotz and A. Peschel, Infect. Immun., 2001, 69, 3423–3426 CrossRef CAS PubMed.
- Y. Oku, Microbiology, 2004, 150, 45–51 CrossRef CAS PubMed.
- J. Andrä, T. Goldmann, C. M. Ernst, A. Peschel and T. Gutsmann, J. Biol. Chem., 2011, 286, 18692–18700 CrossRef PubMed.
- U. Bertsche, C. Weidenmaier, D. Kuehner, S.-J. Yang, S. Baur, S. Wanner, P. Francois, J. Schrenzel, M. R. Yeaman and A. S. Bayer, Antimicrob. Agents Chemother., 2011, 55, 3922–3928 CrossRef CAS PubMed.
- D. H. Simpson, A. Hapeshi, N. J. Rogers, V. Brabec, G. J. Clarkson, D. J. Fox, O. Hrabina, G. L. Kay, A. K. King, J. Malina, A. D. Millard, J. Moat, D. I. Roper, H. Song, N. R. Waterfield and P. Scott, Chem. Sci., 2019, 8, 402–413 Search PubMed.
- J. Campbell, A. K. Singh, J. P. Santa Maria Jr, Y. Kim, S. Brown, J. G. Swoboda, E. Mylonakis, B. J. Wilkinson and S. Walker, ACS Chem. Biol., 2010, 6, 106–116 CrossRef PubMed.
- L. Pasquina, J. P. Santa Maria, B. McKay Wood, S. H. Moussa, L. M. Matano, M. Santiago, S. E. S. Martin, W. Lee, T. C. Meredith and S. Walker, Nat. Chem. Biol., 2015, 12, 40–45 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1952088. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04710g |
| This journal is © The Royal Society of Chemistry 2020 |