
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal-free catalytic reduction of primary amides using an abnormal NHC based potassium complex: integrating nucleophilicity with Lewis acidic activation†
Mrinal
Bhunia‡
 ,
Sumeet Ranjan
Sahoo‡
,
Arpan
Das
,
Jasimuddin
Ahmed
,
Sreejyothi
P.
and
Swadhin K.
Mandal
*
,
Sumeet Ranjan
Sahoo‡
,
Arpan
Das
,
Jasimuddin
Ahmed
,
Sreejyothi
P.
and
Swadhin K.
Mandal
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur-741246, India. E-mail: swadhin.mandal@iiserkol.ac.in
First published on 27th December 2019
Abstract
An abnormal N-heterocyclic carbene (aNHC) based potassium complex was used as a transition metal-free catalyst for reduction of primary amides to corresponding primary amines under ambient conditions. Only 2 mol% loading of the catalyst exhibits a broad substrate scope including aromatic, aliphatic and heterocyclic primary amides with excellent functional group tolerance. This method was applicable for reduction of chiral amides and utilized for the synthesis of pharmaceutically valuable precursors on a gram scale. During mechanistic investigation, several intermediates were isolated and characterized through spectroscopic techniques and one of the catalytic intermediates was characterized through single-crystal XRD. A well-defined catalyst and isolable intermediate along with several stoichiometric experiments, in situ NMR experiments and the DFT study helped us to sketch the mechanistic pathway for this reduction process unravelling the dual role of the catalyst involving nucleophilic activation by aNHC along with Lewis acidic activation by K ions.
Introduction
Amines belong to a privileged class of compounds, routinely present in many fine and bulk chemicals, drugs, agrochemicals, dyes, surfactants, detergents and organic materials.1–6 One of the most attractive methods for synthesis of amines is the deoxygenative reduction of amides.7 However, the reduction of amides is the most challenging among all carboxylic derivatives because of their higher thermodynamic stability. The reduction of primary amides to primary amines is more stringent compared to secondary and tertiary amides.8Among various synthetic methods to prepare primary amines such as reductive amination of carbonyls,9–15 borrowing hydrogenation methodologies,16–18 hydroamination of alkynes,19,20 reduction of nitriles,21–24 primary amides,25 and carboxylic acids in the presence of ammonia,26 reduction of primary amides is a straightforward route as amides are stable, naturally abundant, and inexpensive. Common methods for reduction of primary amides employing stoichiometric amounts of reactive metal hydride reagents such as lithium aluminum hydride or sodium borohydrides are unattractive in terms of atom economy and environmental issues. In addition, they suffer from safety and selectivity of the process generating a stoichiometric amount of inorganic waste.7 In contrast, atom economical catalytic hydrogenation of primary amides is highly desirable and to date no example of catalytic hydrogenation of primary amides has been reported (Scheme 1).26
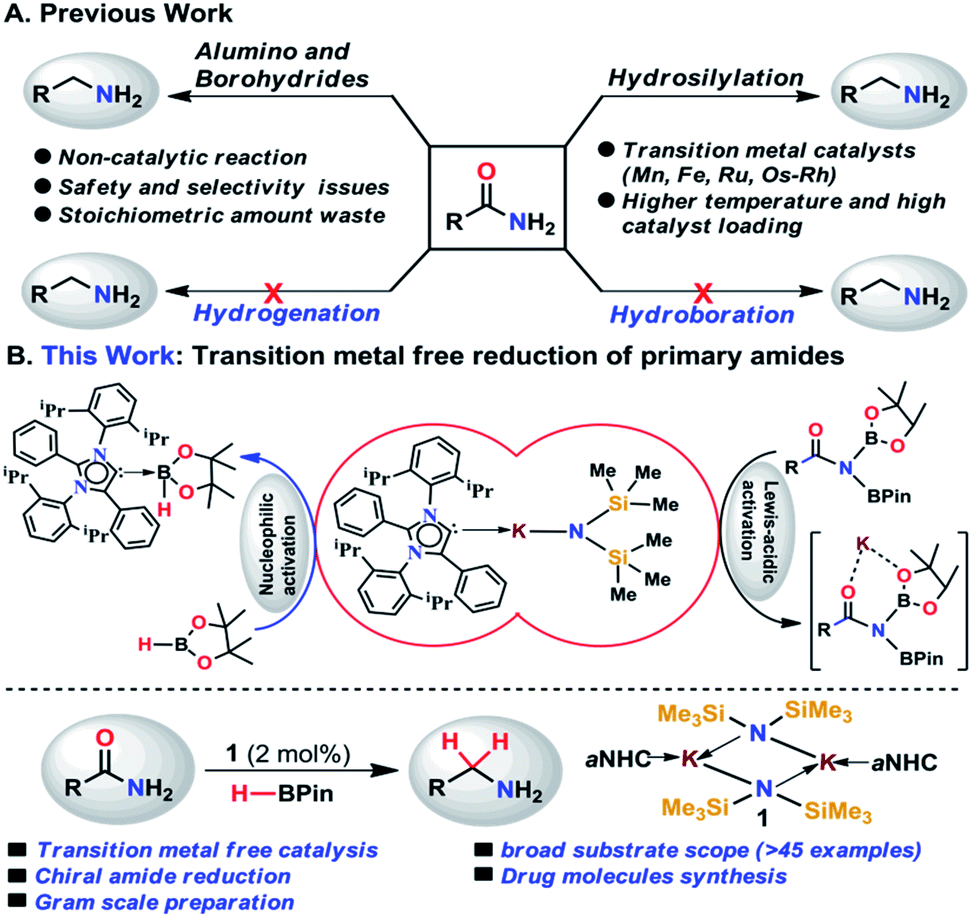 | ||
| Scheme 1 (A) Summary of the reported methods for reduction of primary amides. (B) Overview of the present work where integration of nucleophilicity and Lewis-acidic activation enables the primary amide reduction avoiding transition metals. | ||
As an alternative, catalytic hydrosilylation is an attractive approach for facile and efficient reduction of primary amides to amines.27–30 Earlier, Beller and coworkers utilized two different iron catalysts for efficient reduction of primary amides to primary amines in which the [NHEt3][Fe3H(CO)11] catalyst dehydrates primary amides to nitriles and the second catalyst Fe(OAc)2 was used for reduction of nitriles to the corresponding primary amines.25 Very recently, our group has developed a single catalyst [MnIII(O,N,N,O-PLY)Cl] for hydrosilylation of primary amides into amines.31 Only a few examples of catalytic hydrosilylation of primary amides have been reported to date,25,31–33 while catalytic deoxygenative hydroboration of primary amides to amines has not been reported to date.
In the present study, we have achieved the reduction of primary amides to amines through deoxygenative hydroboration exploiting an abnormal N-heterocyclic carbene (aNHC) based potassium complex34 (1) under ambient conditions in which the catalyst acts by integrating the nucleophilicity of weakly bound aNHC and Lewis acidic activation imparted by the K ions. It may be noted that to date a very few NHC–potassium complexes have been known in the literature35–37 and in these complexes, NHCs are found to be weakly bound (average bond length ∼ 3.00 Å) primarily by ionic interaction with K ions. Thus, we posed a question, whether a weakly bound N-heterocyclic carbene (NHC) in the form of its potassium complex is able to utilize its σ-donating (nucleophilic) properties in activating a hydride transfer reagent (such as borane) to reduce primary amides in a catalytic fashion. It is interesting to note that very recently we have utilized the superior nucleophilicity of aNHC for the reduction of thermodynamically stable CO2 from cylinder38 as well as from the atmosphere39 including use of CO2 for formylation of primary amides.40 These findings motivated us to check the catalytic efficacy of the aNHC based potassium complex (1) for selective reduction of primary amides under ambient conditions.
Results and discussion
At first, the catalytic reduction of primary amides was investigated using 4-nitrobenzamide as a model substrate and pinacolborane (HBPin = 4,4,5,5-tetramethyl-1,3,2-dioxaborolane) as the reducing agent. Reaction of 4-nitrobenzamide (0.5 mmol) and HBPin (2.0 mmol) in the presence of catalyst 1 (5 mol%) at 40 °C in toluene afforded 85% yield of 4-nitrobenzyl amine hydrochloride salt within 24 h (see entry 3, Table S1 in the ESI†). Notably, further lowering the catalyst loading and optimizing various solvents, time and equivalents of HBPin used, the optimized condition was realized (83% yield) in the presence of 2 mol% catalyst (1) loading in toluene at 40 °C for 12 h when 4 equivalents of HBPin were used (entries 4–11, Table S1, ESI†). Control experiments clearly demonstrated that the deoxygenative hydroboration of 4-nitrobenzamide could not occur in the absence of either catalyst 1 (entries 8, and 12–14, Table S1, ESI†) or combination of aNHC and KN(SiMe3)2 (entries 15 and 16, Table S1, ESI†).Having optimized the reaction conditions (entry 8, Table S1, ESI†) in hand, further the scope of the reduction was investigated using various aromatic and aliphatic primary amides to hydroborylated products which essentially upon hydrolysis resulted in the corresponding primary amines and were isolated as their hydrochloride salts (Tables 1 and 2). Under the standardized conditions, benzamide and the diverse range of benzamides bearing electron-donating (Me, OMe, OEt, and tBu) as well as electron-withdrawing functional groups (Cl, NO2, and CN) at the para position were reduced in good to excellent yields (61–97%) to their corresponding amine hydrochlorides (3a–3h, Table 1). Interestingly, in the presence of highly reducible functional groups such as nitro and cyano groups at the para position of benzamides, only amine hydrochloride salts were obtained without reduction of these functional groups (3g–3h, Table 1). Under similar conditions, a varied range of primary benzamides containing electron-donating and electron-withdrawing groups at the meta position (Me, OMe, Cl, CF3, and NO2) and the ortho position (Me, OMe, OEt, F, Cl, and Br) was reduced to the corresponding amines in moderate to excellent yields (50–98%) (3i–3s, Table 1). Next, we probed reduction of primary amides in the presence of sterically encumbering groups such as 2,6-dimethoxybenzamide, 2,6-difluorobenzamide, 2-methyl-3-chlorobenzamide, and 2-methyl-5-fluorobenzamide which rendered very good yields (89–93%) of their corresponding benzylamine hydrochloride (3t–3w, Table 1), whereas sterically less bulky 3,5-dimethoxybenzamide, 3,4-dimethylbenzamide and 3-methyl-4-bromobenzamide afforded 90%, 91% and 97% yields of the reduced products, respectively (3x–3z, Table 1).
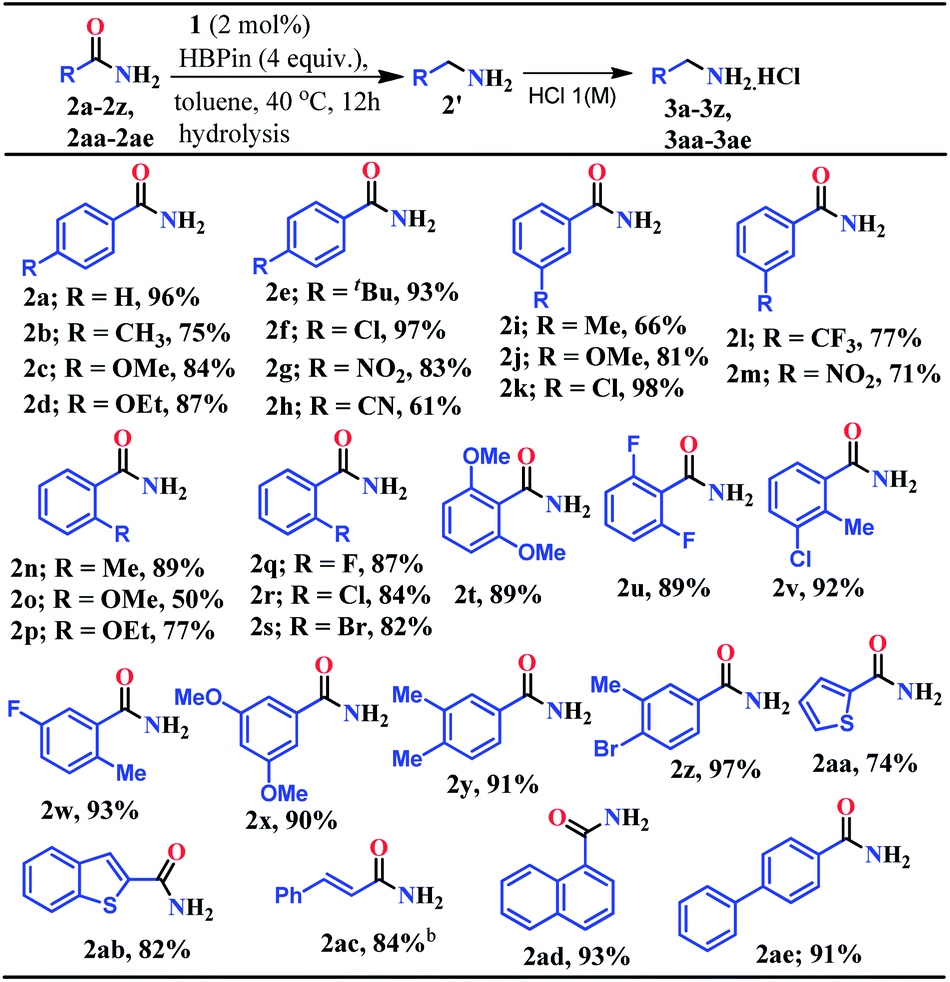
Given the importance of the preparation of heterocyclic primary amines for the production of pharmaceutically and agriculturally valuable products, we attempted to reduce thiophene-2-carboxamide and benzothiophene-2-carboxamide which rendered 74% and 82% yields of the corresponding amine hydrochloride, respectively (3aa–3ab, Table 1). Interestingly, trans-cinnamamide bearing internal alkene afforded 84% yield of the corresponding amine hydrochloride (3ac) without reduction of the internal olefin moiety. These results unarguably established that our reduction methodology is appreciably chemoselective in nature where highly reducible groups such as –NO2, –CN, and alkene units remained intact (3g, 3h, 3m, and 3ac, Table 1). Furthermore, 1-naphthamide and [1,1′-biphenyl]-4-carboxamide also afforded the reduction products in 93% and 91% yields, respectively (3ad and 3ae, Table 1).
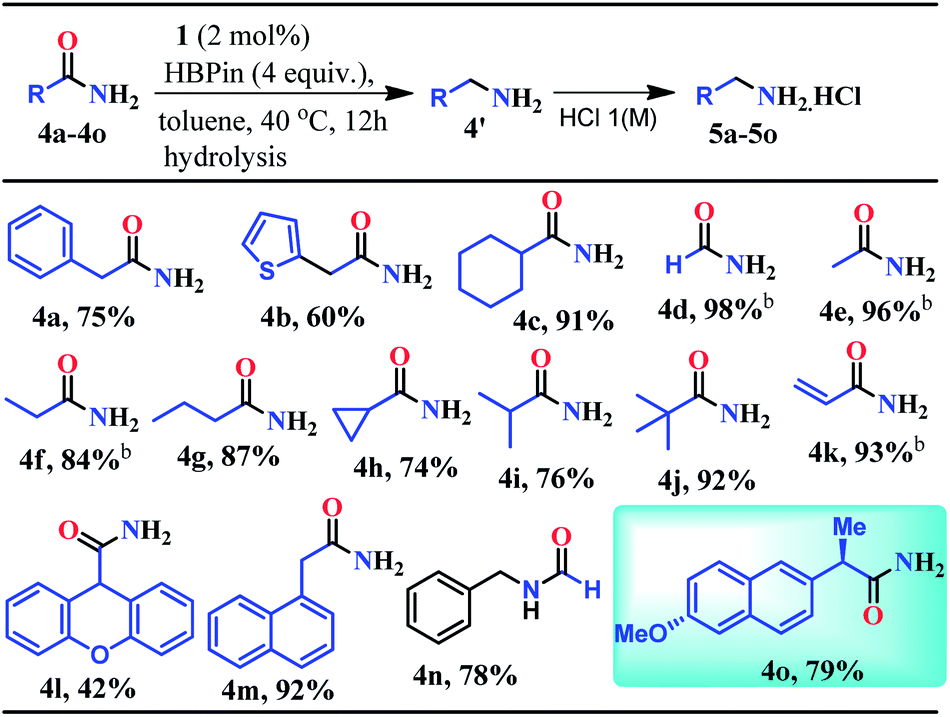
Given the fact that many drug molecules contain an aliphatic amine moiety in their structural unit,41–43 we employed this optimized reaction condition for the reduction of various aliphatic primary amides to their corresponding primary amines which were also isolated as hydrochloride salts (Table 2). Under the standard reaction conditions, aliphatic amides such as 2-phenylacetamide and 2-(thiophen-2-yl)acetamide furnished good yields (75% and 60%) of their corresponding reduced products (5a–5b, Table 2). Moreover, aliphatic primary amides such as cyclohexanecarboxamide, formamide, acetamide, propionamide, butyramide, cyclopropanecarboxamide, isobutyramide, and pivalamide when subjected to reduction with HBPin under the optimized reaction conditions, the corresponding anilinium salts were realized in excellent yield 74–98% (5c–5j, Table 2). Notably, selective reduction products of acrylamides and allylamines are ubiquitously present in various biologically active compounds.44–47 Interestingly, acrylamides are also well-tolerated by the current catalytic system and rendered 93% yield of the corresponding reduced borylated amines which can produce allyl amines upon hydrolysis and considered as valuable intermediates for a wide range of organic compounds.48,49 Interestingly, 2-(naphthalen-1-yl)acetamide, N-benzylformamide, heterocyclic primary amide, and 9H-xanthene-9-carboxamide afforded very good to excellent yields (42–92%) of their reduced products (5l–5m, Table 2). Moreover, N-benzylformamide under optimized reaction conditions afforded 78% yield of the corresponding amine hydrochloride (5n, Table 2). It may be noted that free amine of 5n is considered as an intermediate for the preparation of biologically active compounds. Additionally, this reduction methodology was also applicable for chiral amides. Under the standard reaction conditions, (R)-2-(6-methoxynaphthalen-2-yl)propanamide rendered 79% yield of the amine which was isolated as the hydrochloride salt (5o, Table 2).
Furthermore, to demonstrate the practical applicability of this transition metal-free reduction protocol, 2-phenylethylamine, a monoaminergic neuromodulator and a neurotransmitter15 in the human central nervous system (CNS), was prepared in gram-scale (1.1 g) and isolated as 2-phenylethanamine hydrochloride salt (5a, Scheme 2). In addition, N-methyl-1-phenylmethanamine can be easily transformed into an antifungal drug naftifine analog, 6, which was prepared on a gram-scale (76% yield, 3.5 g, Scheme 2) using this transition metal-free protocol.50
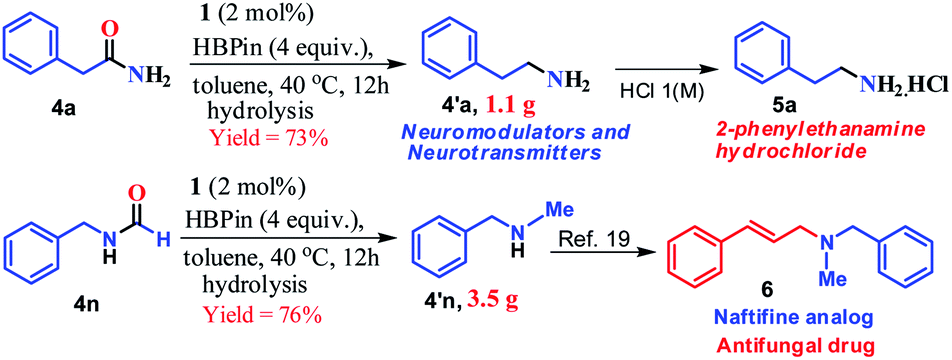 | ||
| Scheme 2 Synthetic route to phenylethylamine and the naftifine analog on a gram scale. | ||
The phenomenal catalytic activity of the potassium aNHC complex, 1, for the reduction of primary amides prompted us to uncover the mechanistic details on the basis of a series of stoichiometric reactions. At first, to find out the role of 1, we performed the stoichiometric reaction of 1 with pinacolborane in toluene at room temperature which afforded colorless crystals of the aNHC–borane adduct (1a) at −30 °C along with formation of KN(SiMe3)2. Formation of KN(SiMe3)2 in the reaction mixture was supported by the 1H NMR spectrum displaying a resonance at δ 0.06 ppm. 1a was isolated and further characterized using NMR spectroscopy and mass spectrometry as well as using single-crystal X-ray diffraction studies (Scheme 3a). Compound 1a crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a single molecule in the asymmetric unit where the carbene–borane bond length was determined as 1.668 (2) Å.51 Formation of 1a in the reaction mixture in the presence of a borane establishes the role of 1 during catalysis. The weak ionic bond between K ion and aNHC in 1 as evident from the bond length (2.973 (2) Å)34 makes the free aNHC available for adduct formation with borane. Thus 1 primarily acts as a carrier of aNHC during the catalytic reaction. Furthermore, to corroborate whether 1a is an active catalyst or not, we accomplished the catalytic reduction of benzamide and 4-nitrobenzamide using the aNHC–borane adduct in the presence of 2 mol% 1a, when benzamide and 4-nitrobenzamide offered only 22% and 7% yields of the corresponding phenylmethanamine hydrochloride salt, respectively, under the standard reaction conditions (Scheme 3b). On the other hand, the same reaction when performed with catalytic amounts of 1a (2 mol%) and KN(SiMe3)2 (2 mol%), an excellent yield (94% for benzamide and 78% for 4-nitrobenzamide) of the reduction product was realized (Scheme 3c). However, only KN(SiMe3)2 failed to afford any reduction product (vide supra). These results clearly suggest that combination of KN(SiMe3)2 and aNHC is accountable for reduction of primary amides. Next, we performed the catalytic reduction of 4-nitrobenzamide in the presence of aNHC and different MN(SiMe3)2 (M = Na, and K), and scrutinized the relative yield. When KN(SiMe3)2 was used, it resulted in 78% yield while NaN(SiMe3)2 led to only 56% yield (Table S4, ESI†). This clearly indicates the role of alkali metal ions in the catalytic outcome of this reaction. To further investigate the reactivity difference between KN(SiMe3)2 and NaN(SiMe3)2, we undertook a density functional theory (B3LYP/6-31+g(2d,p)) analysis (Fig. 1). Optimized geometries clearly exhibited both the HOMO and HOMO−1 are primarily aNHC based (Fig. 1b), and a more destabilized HOMO (−8.3190 eV) and HOMO−1 (−9.0364 eV) for aNHC–KN(SiMe3)2 infers its better nucleophilicity towards borane (HBPin) in comparison with the sodium analogue which in turn makes the B–H bond more nucleophilic upon adduct formation. Such adduct mediated nucleophilic activation of borane by aNHC has been reported in earlier studies.38 Furthermore, when a catalytic amount of less nucleophilic normal NHC (2.0 mol% IPr carbene) along with KN(SiMe3)2 (2.0 mol%) was used to reduce 4-nitrobenzamide, only 9% yield of the reduction product was realized (78% yield for 1), suggesting the importance of higher nucleophilicity of aNHC in increasing the electron density on the boron center which promotes the nucleophilic addition of the B–H bond towards the carbonyl group of borylated amides (vide supra).52 Moreover, a stoichiometric reaction between benzamide and pinacolborane generates N-borylated amide (2a′), which was characterized through 1H, 13C and 11B NMR spectroscopic studies as well as mass spectrometry (Fig. S6–S8, ESI†). Notably, during the formation of N-borylated amide (2a′), evolution of hydrogen gas was monitored which was unambiguously supported by 1H NMR resonance at δ 4.49 ppm (Scheme 3d).53 Furthermore, a stoichiometric reaction was performed between N-borylated amide (2a′) and KN(SiMe3)2 to substantiate the role of KN(SiMe3)2. The sharp downfield shift of the carbonyl group (by 9.9 ppm) in the 13C NMR spectra (δ 179.1 ppm) compared to its borylated amide, 2a′ (δ 169.2 ppm) (Fig. S10, ESI†), clearly implies a Lewis acidic interaction between the K ion and carbonyl oxygen which was further supported by the DFT study (vide infra, Fig. 2b).54 Such a Lewis acidic interaction may enhance the electrophilicity of amide carbonyl functionality. Furthermore, on treatment of catalyst 1 with borylated amide, 2a′, in the presence of HBPin and stopping the reaction prematurely after 2 h, partial formation of N-borylated imine was detected in the 1H NMR spectrum at δ 10.34 ppm and in the 13C NMR spectrum at δ 172.7 ppm in benzene-d6 for benzamide (Fig. S12 and S13, ESI†). A similar reaction with 4-chlorobenzamide after 2 h exhibited the 1H NMR resonance of the corresponding N-borylated imine (9) at δ 9.78 ppm (Fig. S14, ESI†) in toluene-d8 (Scheme 3e).55 Next, N-borylated imine, 9, reacts with HBPin in the presence of catalyst 1 to accomplish the reduced product N-diborylated amine (10) which was also characterized through 1H, 13C and 11B NMR spectroscopy.23
with a single molecule in the asymmetric unit where the carbene–borane bond length was determined as 1.668 (2) Å.51 Formation of 1a in the reaction mixture in the presence of a borane establishes the role of 1 during catalysis. The weak ionic bond between K ion and aNHC in 1 as evident from the bond length (2.973 (2) Å)34 makes the free aNHC available for adduct formation with borane. Thus 1 primarily acts as a carrier of aNHC during the catalytic reaction. Furthermore, to corroborate whether 1a is an active catalyst or not, we accomplished the catalytic reduction of benzamide and 4-nitrobenzamide using the aNHC–borane adduct in the presence of 2 mol% 1a, when benzamide and 4-nitrobenzamide offered only 22% and 7% yields of the corresponding phenylmethanamine hydrochloride salt, respectively, under the standard reaction conditions (Scheme 3b). On the other hand, the same reaction when performed with catalytic amounts of 1a (2 mol%) and KN(SiMe3)2 (2 mol%), an excellent yield (94% for benzamide and 78% for 4-nitrobenzamide) of the reduction product was realized (Scheme 3c). However, only KN(SiMe3)2 failed to afford any reduction product (vide supra). These results clearly suggest that combination of KN(SiMe3)2 and aNHC is accountable for reduction of primary amides. Next, we performed the catalytic reduction of 4-nitrobenzamide in the presence of aNHC and different MN(SiMe3)2 (M = Na, and K), and scrutinized the relative yield. When KN(SiMe3)2 was used, it resulted in 78% yield while NaN(SiMe3)2 led to only 56% yield (Table S4, ESI†). This clearly indicates the role of alkali metal ions in the catalytic outcome of this reaction. To further investigate the reactivity difference between KN(SiMe3)2 and NaN(SiMe3)2, we undertook a density functional theory (B3LYP/6-31+g(2d,p)) analysis (Fig. 1). Optimized geometries clearly exhibited both the HOMO and HOMO−1 are primarily aNHC based (Fig. 1b), and a more destabilized HOMO (−8.3190 eV) and HOMO−1 (−9.0364 eV) for aNHC–KN(SiMe3)2 infers its better nucleophilicity towards borane (HBPin) in comparison with the sodium analogue which in turn makes the B–H bond more nucleophilic upon adduct formation. Such adduct mediated nucleophilic activation of borane by aNHC has been reported in earlier studies.38 Furthermore, when a catalytic amount of less nucleophilic normal NHC (2.0 mol% IPr carbene) along with KN(SiMe3)2 (2.0 mol%) was used to reduce 4-nitrobenzamide, only 9% yield of the reduction product was realized (78% yield for 1), suggesting the importance of higher nucleophilicity of aNHC in increasing the electron density on the boron center which promotes the nucleophilic addition of the B–H bond towards the carbonyl group of borylated amides (vide supra).52 Moreover, a stoichiometric reaction between benzamide and pinacolborane generates N-borylated amide (2a′), which was characterized through 1H, 13C and 11B NMR spectroscopic studies as well as mass spectrometry (Fig. S6–S8, ESI†). Notably, during the formation of N-borylated amide (2a′), evolution of hydrogen gas was monitored which was unambiguously supported by 1H NMR resonance at δ 4.49 ppm (Scheme 3d).53 Furthermore, a stoichiometric reaction was performed between N-borylated amide (2a′) and KN(SiMe3)2 to substantiate the role of KN(SiMe3)2. The sharp downfield shift of the carbonyl group (by 9.9 ppm) in the 13C NMR spectra (δ 179.1 ppm) compared to its borylated amide, 2a′ (δ 169.2 ppm) (Fig. S10, ESI†), clearly implies a Lewis acidic interaction between the K ion and carbonyl oxygen which was further supported by the DFT study (vide infra, Fig. 2b).54 Such a Lewis acidic interaction may enhance the electrophilicity of amide carbonyl functionality. Furthermore, on treatment of catalyst 1 with borylated amide, 2a′, in the presence of HBPin and stopping the reaction prematurely after 2 h, partial formation of N-borylated imine was detected in the 1H NMR spectrum at δ 10.34 ppm and in the 13C NMR spectrum at δ 172.7 ppm in benzene-d6 for benzamide (Fig. S12 and S13, ESI†). A similar reaction with 4-chlorobenzamide after 2 h exhibited the 1H NMR resonance of the corresponding N-borylated imine (9) at δ 9.78 ppm (Fig. S14, ESI†) in toluene-d8 (Scheme 3e).55 Next, N-borylated imine, 9, reacts with HBPin in the presence of catalyst 1 to accomplish the reduced product N-diborylated amine (10) which was also characterized through 1H, 13C and 11B NMR spectroscopy.23
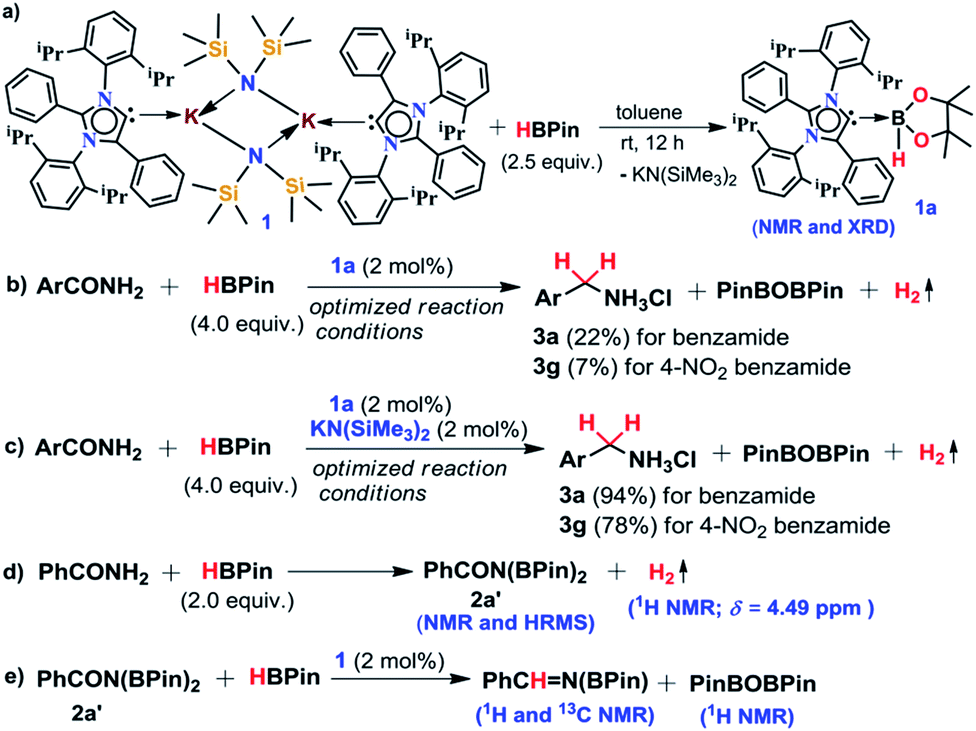 | ||
| Scheme 3 A series of control experiments (a–e) conducted to understand the mechanistic pathway. | ||
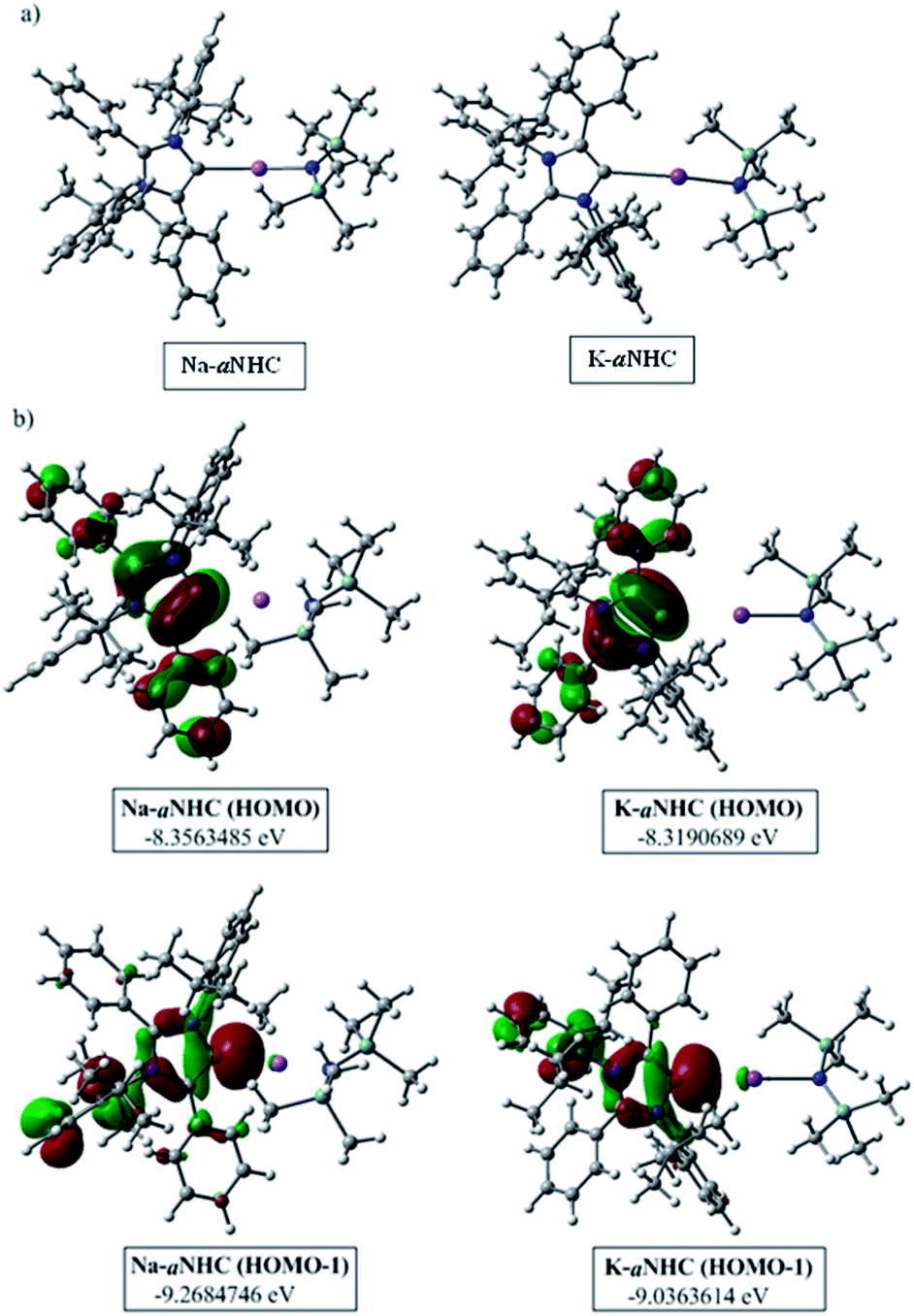 | ||
| Fig. 1 (a) Optimized structures of aNHC–NaN(SiMe3)2 and aNHC–KN(SiMe3)2 complexes. (b) HOMO and HOMO−1 of the corresponding optimized species. | ||
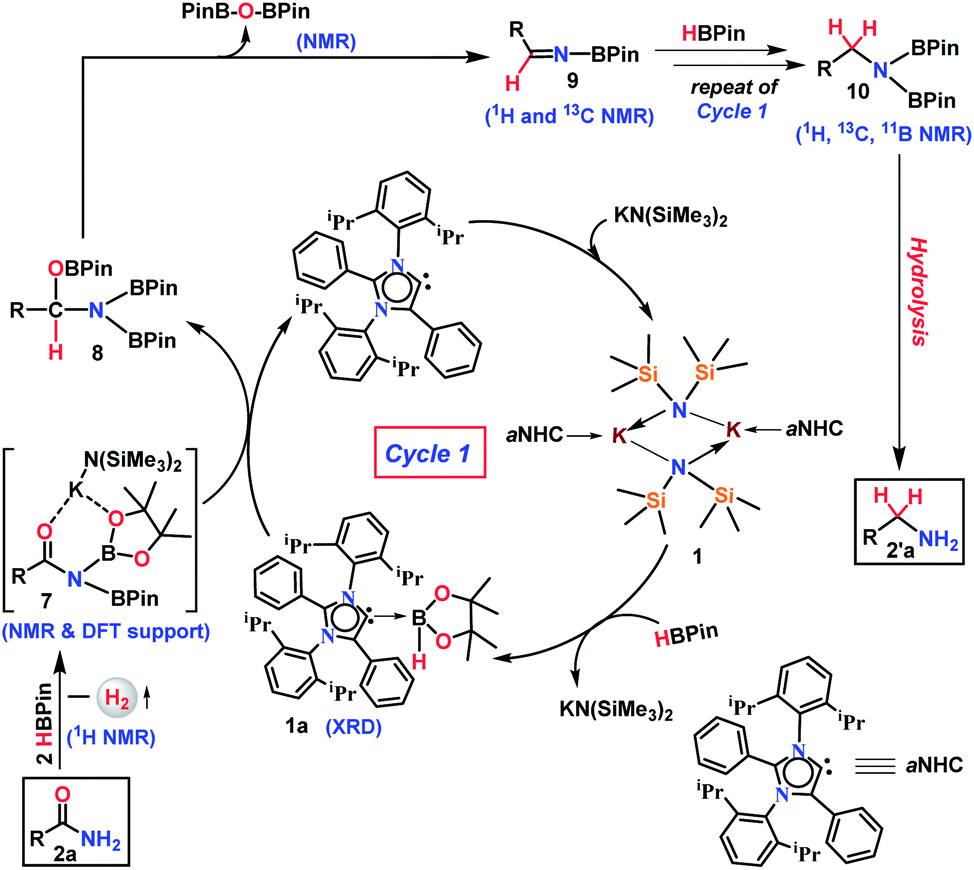 | ||
| Scheme 4 Proposed catalytic cycle based on stoichiometric reactions. | ||
Based on these stoichiometric control experiments described above, a plausible mechanism for this transition metal-free reduction of primary amides is outlined in Scheme 4. In the first step, 1 reacts with pinacolborane to form 1a (X-ray) where the strong σ-donating property of aNHC increases the nucleophilicity to the B–H bond of borane (Fig. 2a). The organometallic K–aNHC complex acts as the carrier of aNHC which transfers the weakly bound aNHC to borane and generates KN(SiMe3)2. Next, KN(SiMe3)2 increases the electrophilicity of borylated amide, through Lewis-acidic interactions (evinced by NMR spectroscopy and the DFT calculated structure of 7, Fig. 2b).
 | ||
| Fig. 2 (a) View of the molecular structure of 1a. Ellipsoids are set at the 50% probability level; hydrogen atoms except H1 of 1a have been omitted for the sake of clarity. (b) Computationally optimized structure of 7 at the B3LYP/6-31G* level of theory, showing interaction between the K ions and carbonyl oxygen. | ||
On the other hand, the aNHC–borane adduct, 1a, reacts with activated borylated amide, 7, to form an intermediate borylated amine ester, 8. Strikingly, deoxygenative reduction of the carbonyl groups in isocyanate and formylated amines was carried out through hydroboration using alkaline earth metals which involved a similar hydride attack on carbonyl carbon to produce methylamine derivatives.56,57 Also, it may be noted that the borylated amide can be formed by the dehydrogenation reaction of benzamide and pinacolborane (as evidenced by stoichiometric experiments, see Scheme 3d) which is further activated by the in situ generated KN(SiMe3)2 through formation of 7 (DFT supported and 13C NMR evidence). Next, elimination of a diboryl ether (authenticated by 1H and 11B NMR spectroscopies) leads to the formation of corresponding N-borylated imine, 9, which was also characterized by 1H and 13C NMR spectroscopies. The N-borylated imine next can undergo further reduction via another catalytic cycle (likewise cycle 1) to generate deoxygenative hydroborylated amine, 10, which was characterized through 1H, 13C and 11B NMR spectroscopy and subsequent hydrolysis produces the corresponding primary amine.
Conclusions
In conclusion, we have demonstrated a transition metal-free catalytic reduction of primary amides to the corresponding primary amines using a well-defined aNHC based potassium complex through hydroboration. Notably, our reduction protocol exhibited a wide range of substrate scope including aromatic, heteroaromatic, and aliphatic primary amides. Moreover, this reduction methodology is considerably chemo-selective and applicable for chiral amides. Gram-scale production of phenylethylamine, a neuromodulator and neurotransmitter in the human CNS was also executed with this reduction strategy. Furthermore, with the help of a series of stoichiometric control experiments, in situ NMR experiments, X-ray crystallography and DFT study, we propose a mechanistic pathway for this challenging transformation where strong σ-donation of aNHC and labile character of organometallic K-aNHC were integrated with Lewis acidity imparted by in situ generated KN(SiMe3)2. Conceivably, the strong σ-donating properties of aNHC activated HBPin for nucleophilic addition of the B–H bond and the Lewis-acidic properties of KN(SiMe3)2 activated in situ generated borylated amide. Such a strategy of merging both nucleophilic and Lewis-acidic activation enables a transition metal free approach towards reduction of various primary amides.Experimental section
General considerations
All manipulations were carried out using standard Schlenk techniques, high vacuum and also glovebox techniques below 0.1 ppm of O2 and H2O. All glassware were oven-dried at 130 °C and evacuated while hot prior to use. All solvents were distilled from Na/benzophenone prior to use. All other chemicals were purchased from Sigma Aldrich and used as received. Elemental analyses were carried out using a PerkinElmer 2400 CHN analyzer and samples were prepared by keeping under a reduced pressure (10−2 mbar) overnight. The FT-IR spectra were recorded by transmission measurements of thin films with a PerkinElmer FT-IR spectrometer Spectrum RXI. The NMR spectra were recorded on a JEOL ECS 400 MHz spectrometer and on a Bruker Avance III 500 MHz spectrometer. All chemical shifts were reported in ppm using tetramethylsilane as a reference. Crystallographic data for structural analysis of 1a (aNHC–HBPin adduct) were deposited at the Cambridge Crystallographic Data Center, CCDC number 1900619.General method for reduction of primary amides
An oven dried 20 mL reaction tube was charged with [aNHC·KN(SiMe3)2]2, 1 (14.8 mg, 2 mol%), and pinacolborane (290 μL, 2.0 mmol, 4 equivalents) along with 1 mL toluene inside a N2 filled glovebox. Next, primary amides (0.5 mmol, 1 equivalent) were added to the reaction mixture and stirred for 12 h at 40 °C. After completion of the reaction, 1.0 mL 2.0 (M) NaOH solution was added to the reaction mixture along with 1.0 mL Et2O and stirred for another 1 h. Next, the reaction mixture was worked up with an Et2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixture (1:1) and the corresponding reduced amines were concentrated in a vacuum. Consequently, 1.0 mL 1.0 (M) HCl was added to the concentrated amines followed by addition of 1.0 mL Et2O and the corresponding amine hydrochloride salt was purified by washing with Et2O. The isolated amine hydrochloride salts were characterized through NMR spectroscopy in DMSO-d6.
:H2O mixture (1:1) and the corresponding reduced amines were concentrated in a vacuum. Consequently, 1.0 mL 1.0 (M) HCl was added to the concentrated amines followed by addition of 1.0 mL Et2O and the corresponding amine hydrochloride salt was purified by washing with Et2O. The isolated amine hydrochloride salts were characterized through NMR spectroscopy in DMSO-d6.
Computational details
All theoretical calculations for geometry optimization and Natural Bonding Orbital (NBO) analysis of all the complexes were carried out with the help of Gaussian 16 (ref. 58) at the B3LYP level of theory by using the 6-31+g(2d,p) basis set.59X-ray crystallographic details
A single crystal of compound 1a was mounted on a glass tip. Intensity data were collected on a SuperNova, Dual, Mo at zero, Eos diffractometer. The crystals were kept at 100 K during data collection. Atomic coordinates, isotropic and anisotropic displacement parameters of all the non-hydrogen atoms of two compounds were refined using Olex2,60 and the structure was solved with the Superflip61 structure solution program using charge flipping and refined with the ShelXL62 refinement package using least squares minimization. Structure graphics shown in the figures were created using Olex2 and X-Seed software (version 2.0) packages.63 Crystallographic data for structural analysis of 1a were deposited at the Cambridge Crystallographic Data Center, CCDC number 1900619.Conflicts of interest
There is no conflict to declare.Acknowledgements
S. K. M. thanks SERB (DST), India (Grant No. EMR/2017/000772) for financial support. M. B. thanks Akamara Biomedicine Pvt. Ltd. for a research fellowship. S. R. S., A. D., and J. A. thank IISER Kolkata for a research fellowship. S. P. thanks DST, Inspire for a fellowship.References
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed.
- R. S. Vardanyan and V. J. Hruby, Synthesis of Best-Seller Drugs, Academic Press, Amsterdam, 2016 Search PubMed.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge Univ. Press, Cambridge, 2004 Search PubMed.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS.
- B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. L. Rotgerink, K. D. Möbus, J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, Appl. Catal., A, 2005, 280, 17–46 CrossRef CAS.
- J. Seyden-Penne, Reductions by the Alumino- and Boro-hydrides in Organic Synthesis, John Wiley & Sons, Inc, New York, 1997 Search PubMed.
- L. Hie, N. F. F. Nathel, T. K. Shah, E. L. Baker, X. Hong, Y. F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed.
- S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS.
- J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86–95 CrossRef CAS PubMed.
- G. Hahn, P. Kunnas, N. de Jonge and R. Kempe, Nat. Catal., 2019, 2, 71–77 CrossRef CAS.
- H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021–1049 CrossRef CAS.
- L. Legnani, B. Bhawal and B. Morandi, Synthesis, 2017, 49, 776–789 CAS.
- A. Ruffoni, F. Juliá, T. D. Svejstrup, A. J. McMillan, J. J. Douglas and D. Leonori, Nat. Chem., 2019, 11, 426–433 CrossRef CAS PubMed.
- T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumansn, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Nat. Commun., 2018, 9, 4123–4134 CrossRef.
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS.
- D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS.
- S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126–8129 CrossRef CAS PubMed.
- W. A. Herrmann, J. A. Kulpe, J. Kellner, H. Riepl, H. Bahrmann and W. Konkol, Angew. Chem., Int. Ed. Engl., 1990, 29, 391–393 CrossRef.
- B. Zimmermann, J. Herwig and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 2372–2375 CrossRef CAS.
- S. Enthaler, D. Addis, K. Junge, G. Erre and M. Beller, Chem.–Eur. J., 2008, 14, 9491–9494 CrossRef CAS PubMed.
- D. V. Gutsulyak and G. I. Nikonov, Angew. Chem., Int. Ed., 2010, 49, 7553–7556 CrossRef CAS.
- C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC.
- S. Saha and M. S. Eisen, ACS Catal., 2019, 9, 5947–5956 CrossRef CAS.
- S. Das, B. Wendt, K. Möller, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 1662–1666 CrossRef CAS PubMed.
- A. A. Núñez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC.
- Y. Motoyama, K. Mitsui, T. Ishihda and H. Nagashima, J. Am. Chem. Soc., 2005, 127, 13150–13151 CrossRef CAS PubMed.
- S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032–15040 CrossRef CAS PubMed.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770–1771 CrossRef CAS PubMed.
- Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama and H. Nagashima, Angew. Chem., Int. Ed., 2009, 48, 9511–9514 CrossRef CAS PubMed.
- H. S. Das, S. Das, K. Dey, B. Singh, R. K. Haridasan, A. Das, J. Ahmed and S. K. Mandal, Chem. Commun., 2019, 55, 11868–11871 RSC.
- M. Igarashi and T. Fuchikami, Tetrahedron Lett., 2001, 42, 1945–1947 CrossRef CAS.
- W. Yao, H. Fang, Q. He, D. Peng, G. Liu and Z. Huang, J. Org. Chem., 2019, 84, 6084–6093 CrossRef CAS PubMed.
- M. Bhunia, G. Vijaykumar, D. Adhikari and S. K. Mandal, Inorg. Chem., 2017, 56, 14459–14466 CrossRef CAS PubMed.
- P. L. Arnold, M. Rodden and C. Wilson, Chem. Commun., 2005, 1743–1745 RSC.
- R. W. Alder, M. E. Blake, C. Bortolotti, S. Bufali, C. P. Butts, E. Linehan, J. M. Oliva, A. G. Orpen and M. J. Quayle, Chem. Commun., 1999, 241–242 RSC.
- M. S. Hill, G. Kociok-Köhn and D. J. MacDougall, Inorg. Chem., 2011, 50, 5234–5241 CrossRef CAS PubMed.
- S. C. Sau, R. Bhattacharjee, P. K. Vardhanapu, G. Vijaykumar, A. Datta and S. K. Mandal, Angew. Chem., Int. Ed., 2016, 55, 15147–15151 CrossRef CAS PubMed.
- S. C. Sau, R. Bhattacharjee, P. K. Hota, P. K. Vardhanapu, G. Vijaykumar, R. Govindarajan, A. Datta and S. K. Mandal, Chem. Sci., 2019, 10, 1879–1884 RSC.
- P. K. Hota, S. C. Sau and S. K. Mandal, ACS Catal., 2018, 8, 11999–12003 CrossRef CAS.
- A. Allen and R. Ely, Synthetic methods for amphetamine, www.nwafs.org/newsletters/SyntheticAmphetamine.pdf Search PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- G. Petranyi, N. S. Ryder and A. Stütz, Science, 1984, 224, 1239–1241 CrossRef CAS PubMed.
- S. M. Nanavati and R. B. Silverman, J. Am. Chem. Soc., 1991, 113, 9341–9349 CrossRef CAS.
- M. Johannsen and K. Jørgensen, Chem. Rev., 1998, 98, 1689–1708 CrossRef CAS PubMed.
- K. Das, R. Shibuya, Y. Nakahara, N. Germain, T. Ohshima and K. Mashima, Angew. Chem., Int. Ed., 2012, 51, 150–154 CrossRef CAS PubMed.
- A. Ricci, in Modern Amination Method, Wiley-VCH, Weinheim, 2000 Search PubMed.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed.
- T. Ohshima, Y. Miyamoto, J. Ipposhi, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317–14328 CrossRef CAS PubMed.
- Crystal data for 1a: C45H57BN2O2, Mr= 668.73, triclinic, space group P, a = 9.6381(5) Å, b = 11.4850(6), c = 19.0369(9) Å, α = 85.238(4)°, β = 75.946(4)°, γ = 75.838(5)°, V = 1981.44(18) nm3, Z = 4, ρcalc = 1.121 g cm−3, μ (MoKα) = 0.513 mm−1, T = 100(2) K, θ range for data collection = 4.786–131.826°, R1 = 0.0529 (I > 2σ(I)), wR2 = 0.1208 (all data).
- M. Bhunia, P. K. Hota, G. Vijaykumar, D. Adhikari and S. K. Mandal, Organometallics, 2016, 35, 2930–2937 CrossRef CAS.
- M. Bhunia, S. R. Sahoo, B. K. Shaw, S. Vaidya, A. Pariyar, G. Vijaykumar, D. Adhikari and S. K. Mandal, Chem. Sci., 2019, 10, 7433–7441 RSC.
- D. Mukherjee, S. Shirase, K. Mashima and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 13326–13329 CrossRef CAS PubMed.
- C. Boone, I. Korobkov and G. I. Nikonov, ACS Catal., 2013, 3, 2336–2340 CrossRef CAS.
- Y. Yang, M. D. Anker, J. Fang, M. F. Mahon, L. Maron, C. Weetman and M. S. Hill, Chem. Sci., 2017, 8, 3529–3537 RSC.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahonand and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Rev. D.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. Palatinus and G. Chapuis, SUPERFLIP, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, SHELXL, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- L. J. Barbour and X-Seed: Graphical Interface to SHELX97 and POV-Ray, University of Missouri-Columbia, Columbia, MO, 1999 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, NMR and HRMS spectra, crystallographic details, and coordinates of the computed structures. CCDC 1900619. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05953a |
| ‡ M. Bhunia and S. R. Sahoo contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |