
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triple the fun: tris(ferrocenyl)arene-based gold(I) complexes for redox-switchable catalysis†
Axel
Straube
 a,
Peter
Coburger‡
a,
Luis
Dütsch
b and
Evamarie
Hey-Hawkins
*a
a,
Peter
Coburger‡
a,
Luis
Dütsch
b and
Evamarie
Hey-Hawkins
*a
aInstitute of Inorganic Chemistry, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de; Web: http://https://anorganik.chemie.unileipzig.de/de/anorganik/ak-hey-hawkins/
bInstitute of Inorganic Chemistry, Universität Regensburg, Universitätsstr. 31, D-93053 Regensburg, Germany
First published on 3rd August 2020
Abstract
The modular syntheses of C3-symmetric tris(ferrocenyl)arene-based tris-phosphanes and their homotrinuclear gold(I) complexes are reported. Choosing the arene core allows fine-tuning of the exact oxidation potentials and thus tailoring of the electrochemical response. The tris[chloridogold(I)] complexes were investigated in the catalytic ring-closing isomerisation of N-(2-propyn-1-yl)benzamide, showing cooperative behaviour vs. a mononuclear chloridogold(I) complex. Adding one, two, or three equivalents of 1,1′-diacetylferrocenium[tetrakis(perfluoro-tert-butoxy)aluminate] as an oxidant during the catalytic reaction (in situ) resulted in a distinct, stepwise influence on the resulting catalytic rates. Isolation of the oxidised species is possible, and using them as (pre-)catalysts (ex situ oxidation) confirmed the activity trend. Proving the intactness of the P–Au–Cl motif during oxidation, the tri-oxidised benzene-based complex has been structurally characterised.
Introduction
Attaining control over catalytic reactions by external stimuli, preferably with as tight a grip as demonstrated in Nature's complex regulation pathways, has long been at the centre of interest for the chemical community.1–3 It has been 25 years since the seminal work by the groups of Wrighton and Rebek, the former demonstrating a cobaltocene-based rhodium complex A (Chart 1) to function as either a good hydrogenation (CoII) or hydrosilylation (CoIII) catalyst, the activity for the corresponding reaction being significantly lower in the respective other oxidation state.4 Rebek and co-workers used light to reversibly isomerise an azobenzene-derived organo-catalyst B and thus tuning it towards an amide-forming reaction.5 A vast body of work concerning artificially switchable catalysis has since been assembled,6–9 with a major focus on redox-switchable catalysis (RSC).10–13 Even though switching between oxidation states is usually achieved through the addition of chemical redox agents, RSC holds great potential for applications using electrodes and thus greatly reducing chemical waste.14–18 Conceptually, both ligand and metal can form the centres of (reversible) electron transfer for changing the activity state of the catalyst.11 For ligand-based switching, ferrocene has proven and remains a cornerstone for ligand design owing to its amenability to synthetic modification and favourable, while modifiable, redox properties.19,20 | ||
| Chart 1 Examples of redox- (A [activity indicated for hydrogenation of alkenes],4C,21D29) and light-/thermo-switchable (B,5F42) catalyst systems. The switching moiety is highlighted in blue and the catalytic sites are highlighted in purple. The most active (ON) state is labelled in green and underlined (bold), the intermediate state in yellow and underlined (dashed), and the least active state (OFF) is labelled in red. | ||
Even though one of the first redox-switchable catalysts by Long and co-workers used for the ring-opening polymerisation of rac-lactide, C (Chart 1),21 already contained two pendant ferrocenyl groups, and a plethora of compounds featuring multiple ferrocenyl groups is available,22–28 only one report by Zhao and Chen deals with exploiting the possibility of addressing more than just two catalytic states. Employing an α-diimine palladium catalyst D with two pendant ferrocenyl groups, the three activity states resulting from sequential two-step oxidation were found to differ with respect to polymerisation activity, resulting in tuneable polymer molecular weight, topology, and polydispersity.29 The scarcity of this concept in RSC is surprising given how multi-state switchable molecules feature prominently in molecular machines30–33 and molecular electronics and logic.34–41 Combining molecular machines and catalysis, Wang and Feringa have impressively demonstrated a unidirectionally light- and thermo-switchable rotor F to display three different activity states in an organo-catalysed asymmetric Michael addition, in turn also leading to different enantioselectivities (P,P-trans: racemic; M,M-cis: S enantiomer; M,M-trans: R enantiomer).42
Expanding on this idea, we sought to prepare a system with four accessible oxidation states by making use of the C3-symmetric s-tris(ferrocenyl)arene motif recently first exploited for ligand design.43 So far, we have focused on the monotopic use of these tris-phosphanes 1 (Scheme 1); however, put to use as tritopic ligands renders them miniaturised and thus easier-to-study analogues of the ferrocenyl-based dendrimers we could recently show to function as redox-switchable ruthenium(II) catalysts for the isomerisation of an allylic alcohol44,45 and for transfer hydrogenation of a prochiral ketone with two distinct catalytic activity states (neutral and fully oxidised).46
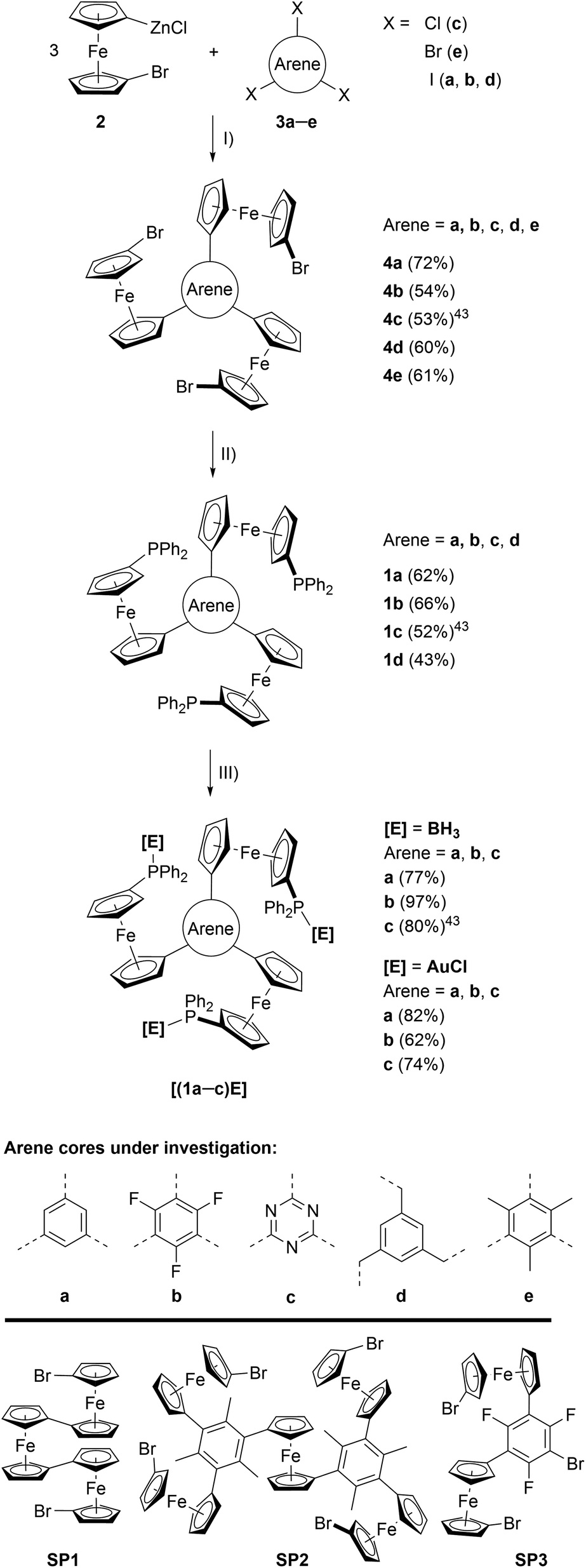 | ||
| Scheme 1 (Top) Preparation of tris-phosphanes 1a–d and complexes [(1a–c)E]. (I) Pd precatalyst, THF, r. t.; (II) nBuLi, Ph2PCl, THF, −80 °C to r. t.; (III) [E]·X ([E]·X = BH3·SMe2 or [AuCl(tht)]), CH2Cl2, r. t.; (Bottom) crystallographically characterised side products obtained during the syntheses of 4d (SP1), 4e (SP2), and 4b (SP3). For their solid-state structures, see Fig. S3.† | ||
C 3 symmetry in general has been recognised as a promising ligand design principle,47,48 and adorning one ligand fragment with multiple metal centres can furthermore allow for cooperative effects to occur.49–54 The underlying key feature of the catalytically active sites in close proximity has been also found crucial in the design of single-molecule magnets55,56 and for molecular recognition and supramolecular assemblies.57–60
Results and discussion
Preparation and electrochemical characterisation of tris-phosphanes and trinuclear gold(I) complexes
The preparation of tris-phosphanes 1 (Scheme 1, top), as described for 1c,43 starts with a triple Negishi coupling of the in situ-generated 1-bromo-1′-ferrocenylene zinc halide 2, prepared from 1,1′-dibromoferrocene,61n-butyllithium and anhydrous zinc chloride in THF,25,62 with C3-symmetric arenes 3a–e. Introducing electron-withdrawing (b, c) and electron-donating (e) arenes as well as a tris-benzylic arene core (d), preventing conjugation between the three ferrocenylene groups, allows for modularly fine-tuning the system's electrochemical response (vide infra). Tris(1-bromo-1′-ferrocenylene)arenes 4a–e are obtained in moderate to good yields as crystalline solids and the potential to incorporate more and highly functionalised cores is only limited by the functional group tolerance of the Negishi protocol.634a, b, d, e were analysed by single crystal X-ray diffraction (XRD) analysis (Fig. S1 and Table S3†), their structural parameters falling within the expected standard ranges for C3-symmetric tris(ferrocenyl)arenes.25,64–68 In solution, 4a–e are characterised by unhindered rotation about the Carene–CCp bonds on the NMR timescale, thus displaying C3v symmetry in their 1H and 13C{1H} NMR spectra.Depending on the choice of PdII precatalyst, formation of bi- and triferrocenes has also been observed. Among them, triferrocene SP1 (Scheme 1, bottom) had not been crystallographically characterised until now69 and is most likely formed from hindered reductive elimination followed by a second transmetallation step of 2 onto the Pd catalyst.70 The formation of pentanuclear SP2 during the preparation of 4e can be similarly rationalised. Together with the isolation of only di-ferrocenylated bromotrifluorobenzene SP3, these findings point towards the use of our synthetic protocol to access ever more complex and functionalisable redox-active structures.
Tris-phosphanes 1a–d are obtained following the established protocol and purified by column chromatography.43 Attempting the synthesis of a mesitylene-based tris-phosphane from 4e has only resulted in the isolation of impure trace amounts, potentially due to side reactions involving the methyl protons of 4e. Crystals suitable for XRD analysis have been obtained for 1a and 1d (Fig. 1 and S4†). Their structural parameters are in agreement with those previously reported for 1c43 and other diphenylphosphanyl ferrocenes.71–74 Similar to their precursors 4, no rotamers are observed in solution, while small changes in the 31P{1H} NMR chemical shifts of 1a–d reflect the electronic nature of the arene core (Table 1).
 | ||
| Fig. 1 Molecular structures of tris-phosphane 1a and borane adduct [1a(BH3)3]. Thermal ellipsoids are set at the 50% probability level. For clarity, the phenyl rings are depicted in wireframe style, disorder in the phenyl rings of 1a has been omitted, and hydrogen atoms except for those of the BH3 groups in [1a(BH3)3] are not depicted. | ||
Borane adducts [1a–c(BH3)3] have been prepared ([1c(BH3)3] has been reported before)43 to study the electrochemistry of the phosphanes (vide infra), since direct cyclopentadienyl–phosphorus bonds usually render the oxidations irreversible due to the lone pair of electrons on phosphorus.75–77 Adduct [1a(BH3)3] was found to crystallise with crystallographic C3 symmetry (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , Fig. 1) and compares well (Table S5†) to structural parameters of similar ferrocenyl-phosphane boranes described by us78 as well as by Štěpnička and co-workers.79
, Fig. 1) and compares well (Table S5†) to structural parameters of similar ferrocenyl-phosphane boranes described by us78 as well as by Štěpnička and co-workers.79
Recently we reported the capability of 1c to bind coinage metal ions in a tridentate trigonal planar coordination mode;43 now we have turned our focus to potential trinuclear gold(I) complexes of 1a–c. Indeed, reacting the tris-phosphanes with the common gold(I) precursor [AuCl(tht)] (tht = tetrahydrothiophene) in slight stoichiometric excess afforded, after simple precipitation, homotrinuclear metal complexes [1a–c(Au)3] in good yields. Their trinuclear composition is confirmed by CHN analyses, multinuclear NMR spectroscopy, and they remain homotrinuclear in the gas phase as assessed by high-resolution electrospray-ionisation mass spectrometry (HR-ESI MS).
Representative for all three gold(I) chloride complexes, the molecular structure of [1c(Au)3] was determined from suitable crystals (Fig. 2) and confirms the trinuclearity in the solid state as well. In line with the experimentally determined non-centrosymmetric space group P1, [1c(Au)3] crystallises as an enantiopure compound (xFlack = −0.008(7)); however, in solution, there is no indication of a corresponding hindered rotation as assessed from NMR spectroscopy.
 | ||
| Fig. 2 Molecular structure of homotrinuclear [1c(Au)3] with partial atom numbering scheme. Thermal ellipsoids are set at the 50% probability level. For clarity, the phenyl rings are depicted in wireframe style, and co-crystallised solvent, disorder in the P(2)–Au(2)–Cl(2) fragment, and hydrogen atoms have been omitted. | ||
The P–Au bond lengths (2.225(5)–2.231(6) Å, Tables 2 and S6†) are similar to mononuclear analogues featuring an N-heterocyclic substituent at the 1′-position of the 1-diphenylphosphanyl-ferrocenylene moiety described by the groups of Siemeling (d(P–Au) = 2.242 Å [2-pyridyl], 2.234 Å [3-pyridyl])80 and Lang (d(P–Au) = 2.215 Å [2,2′:6′,2′′-terpyridin-4′-yl]),81 yet [1c(Au)3] is the first such complex containing ferrocene in the tris-phosphane backbone and the second to incorporate three ferrocene moieties into a trinuclear gold complex.82
The spectral data of both [1a(Au)3] and [1b(Au)3] are in line with that of [1c(Au)3] and all complexes are thus presumed to have similar structures. Notably, [1c(Au)3] does not show aurophilic interactions83–85 or close ferrocene⋯metal contacts86 in the solid state (for M⋯M distances see Table 2), setting it apart from related structures regularly displaying aurophilic interactions (Table S7†). In the context of cooperative effects operating in multimetallic catalysis,49,52,87–90[1c(Au)3] thus falls short of the proximity criterion formulated by Feringa as the gold⋯gold separation exceeds 6 Å.49 Yet, owing to the flexibility of the ligand backbone, gold⋯gold distances in solution might well become much closer.
| a Intramolecular distances; the shortest intermolecular distance is shown italicised. | |
|---|---|
| Au(m)–P(m) | 2.231(6)/2.286(7)/2.225(5) |
| Au(m)–Cl(m) | 2.288(6)/2.229(4)/2.281(5) |
| P(m)–M(m)–Cl(m) | 179.0(2)/177.0(2)/176.9(2) |
| Au(1,2,3)⋯Au(2,3,1)a | 14.187(1)/8.012(1)/9.100(1)/6.4204(9) |
| Au(m)⋯Fe(m) | 4.302(3)/4.080(3)/4.544(2) |
Regarding our vision of using 1-derived complexes as “rotary” or “dimmable” switches for multi-redox-state applications, electrochemical characterisation by cyclic voltammetry (CV) was of utmost relevance. 1,3,5-Tris(ferrocenyl)arenes have previously been shown91–93 to display reversible redox activity and, in supporting electrolytes (SE) containing weakly coordinating anions such as [B(C6F5)4]− or [B{3,5-(CF3)2C6H3}4]− (BArF4−),94,95 to furthermore be oxidisable in three separate, resolved steps.25
Triazine-based 4c and [1c(BH3)3] displayed a similar behaviour,43 while 1c, as expected, showed an irreversible electrochemical oxidation due to the direct cyclopentadienyl–phosphorus linkage.75–77,96 Their analogues reported herein share these redox features (s. Fig. 3, left, for 4a, 1a, and [1a(BH3)] and Fig. S8–S11†). Gratifyingly, the arene core determines the exact oxidation potentials (Tables 3 and S9–S12†), in line with substitution by electron-donating or -withdrawing groups. The choice of arene thus allows for electrochemically fine-tuning the whole system. The arene trend also holds true for [1a–c(Au)3] (Fig. 3, right). In a BF4−-based SE, the three gold(I) complexes show partly irreversible behaviour (Fig. S12†), but in all cases, their tris(ferrocenyl)arene cores are oxidised in one (quasi)reversible event. In a BArF4−-based SE, the tris(ferrocenyl)arene core is again oxidised in three resolved steps. In our setup (0.1 mol L−1 (nBu4N)BF4 or (nBu4N)BArF4 in CH2Cl2, −1.75 to 1.2 V vs. FcH/[FcH]+),97 no oxidation of gold(I) in [1a–c(Au)3] was observed.
 | ||
| Fig. 3 Cyclic voltammograms of 4a, 1a (the grey trace was recorded with a vertex potential of 245 mV vs. FcH/[FcH]+), [1a(BH3)3], and [1a–c(Au)3] at 1 mmol L−1 in 0.1 mol L−1 (nBu4N)BArF4/CH2Cl2 (scan rate: 100 mV s−1, working electrode: glassy carbon, counter electrode: platinum wire). The 2nd of 3 cycles is shown for all compounds (for full voltammograms, s. Fig. S8–S12†). | ||
| E 01 (ΔEp)a [mV] | |||||
|---|---|---|---|---|---|
| a Determined on 1 mmol L−1 samples in anhydrous 0.1 mol L−1 (nBu4N)BArF4/CH2Cl2 (working electrode: glassy carbon) at 100 mV s−1. The difference between oxidation and reduction potential, ΔEp, is given in brackets. b Determined from square-wave voltammetry due to close peak-to-peak separation, leaving ΔEp inaccessible. | |||||
| 4d | 195 (85) | 1d | 113b | [1a(BH3)3] | 270 (91) |
| 4e | 212 (87) | 1a | 138 (98) | [1b(BH3)3] | 353 (87) |
| 4a | 247 (100) | 1b | 206 (116) | [1c(BH3)3] 43 | 421 (100) |
| 4b | 293 (96) | 1c 43 | 275 (160) | [1a(Au)3] | 376 (101) |
| 4c 43 | 386 (127) | [1b(Au)3] | 419 (93) | ||
| [1c(Au)3] | 535 (108) | ||||
This is in line with results from DFT calculations locating the HOMO at the iron centres; in the mono-oxidised model complex [1a(Au)3]+, the spin density solely resides at the three iron centres, too (Fig. S14–S19†). Among the three tris-phosphanes, 1c yields the most anodically shifted redox potentials in its complexes (Fig. 3, right, and Table 3). In line with our previous findings, complexes of 1c were also found to display the least straightforward electrochemistry such as cathodically shifted reductions connected to electron transfer-induced chemical transformations (EC mechanism; cf. Fig. S13†).43
Redox-switchable gold(I) catalysis
Seeking to demonstrate the applicability of the stepwise oxidation of the s-tris(ferrocenyl)arene core, our choice fell on the gold-catalysed 5-exo-dig ring-closing isomerisation of N-(2-propyn-1-yl)benzamide (5) to 5-methylene-2-phenyl-4,5-dihydrooxazole (6) (Scheme 2) as a read-out. Uncovered by Hashmi and co-workers in 2004,98 the catalytic synthesis of oxazolines has quickly developed into a standard reaction for gold(I) complexes.99–104 The groups of Sarkar and Heinze, among others, have established the transformation of 5 to 6 as a platform to perform and study RSC using gold(I) catalysts.105–109 Rendering the gold(I) centres reversibly more Lewis-acidic and hence more catalytically active110 through oxidation of a connected ferrocenyl moiety is one way of obviating the sometimes problematic use of silver salts for activation of gold(I) pre-catalysts by halide abstraction and enables temporal control over the activity of the catalyst.111–115 Since we aimed for a detailed understanding of the switching process, the reaction was performed on the NMR scale in CD2Cl2, allowing for a time-resolved study of the reaction through protons Ho (reaction involving oxidised species) and Hm (reactions not involving oxidised species) of oxazoline 6 (Fig. S21 and S22†) vs. 1,3,5-trimethoxybenzene as an internal standard.116–118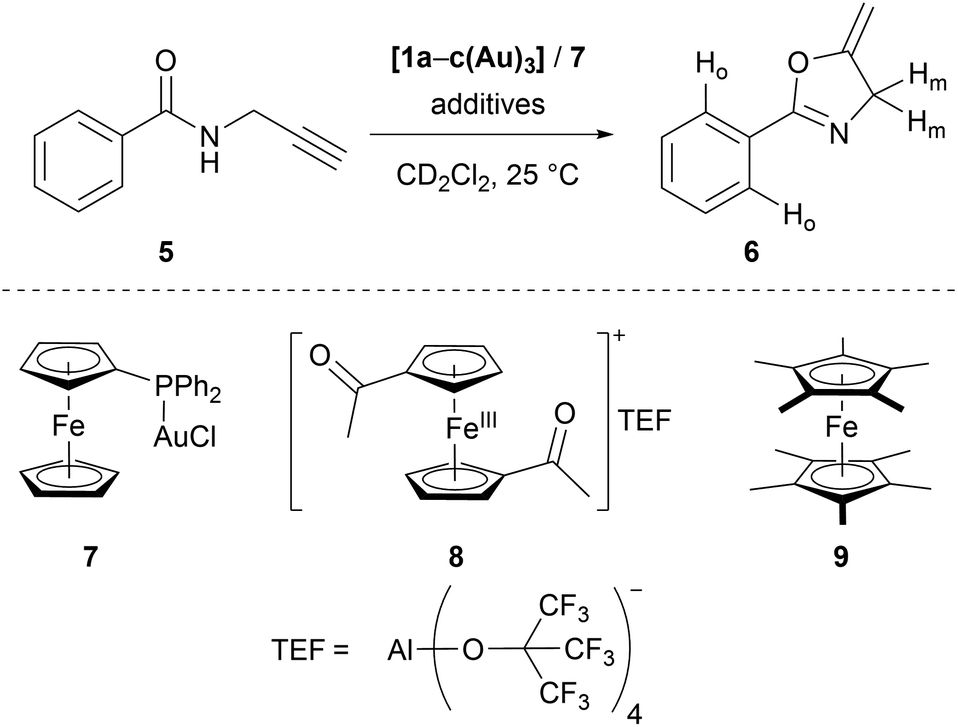 | ||
| Scheme 2 Gold(I)-catalysed ring-closing isomerisation of N-(2-propyn-1-yl)benzamide 5 to oxazoline 6 (top, labelled protons used for following the reaction), including alternative mononuclear (pre-)catalyst 7, oxidant 8, and reductant 9. | ||
Native [1a(Au)3] (crossed light blue circles; Fig. 4), employed in a 3 mol% Au loading (referring to substrate 5), performed with low activity (TOF = 2.0 ± 0.2 h−1; TOF fits, s. Fig. S39–S53†). A control experiment without any added gold(I) complex yielded no product. While [1b(Au)3] (crossed navy squares, TOF = 2.5 ± 0.1 h−1) performed slightly better than [1a(Au)3], potentially due to the slightly more electron-withdrawing 1,3,5-trifluorobenzene core, [1c(Au)3] (crossed turquoise triangles) did, reproducibly, not show any catalytic activity under the same conditions. Given that the only difference is in the s-triazine core, these nitrogen atoms might interact with the amide protons of substrate 5, in turn preventing the completion of the catalytic cycle by protodeauration.98 This hypothesis is supported comparing the 1H NMR spectra of [1a(Au)3] and [1c(Au)3] with 3 equivalents of 5, respectively, at −60 °C in CD2Cl2, resulting in a signal splitting for the amide proton signal of 5 in presence of [1c(Au)3] (Fig. S23†).
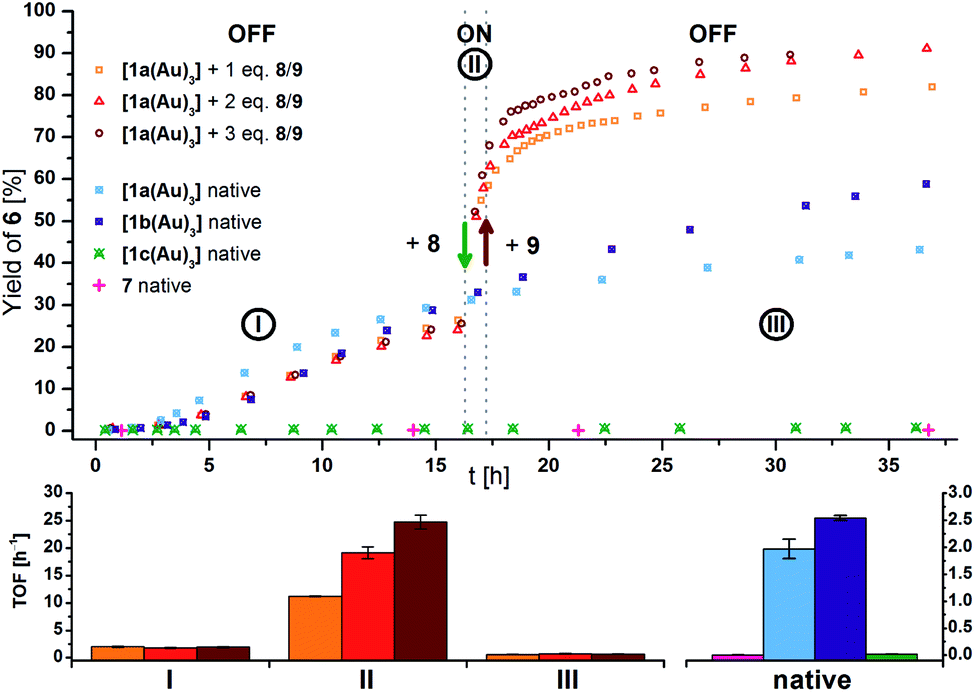 | ||
| Fig. 4 (Top) Yield-over-time graphs for native (crossed symbols) and redox-switchable (hollow symbols) gold(I)-catalysed cyclisation of 5 to 6 (3 mol% Au as [1(Au)3] and 7, [5]0 = 60 mmol L−1, CD2Cl2, 25 °C). Arrows indicate the addition of additives. I: initial OFF phase with little catalytic activity; II: ON phase after addition of 1.0–3.0 eq. oxidant 8; III: 2nd OFF phase of little catalytic activity after addition of 1.1–3.3 eq. reductant 9. (Bottom) Comparison of turnover frequencies (TOF), determined from linear fits of fixed time frames (s. Fig. S39–S45†) for the switched reaction phases shown above (left) and for the native complexes (right). | ||
In order to test for potential cooperative effects due to its three gold centres, [AuCl(FcPPh2)] (7) was prepared as a mononuclear gold complex analogue of [1a(Au)3].119 Quite surprisingly, a catalytic evaluation of 7 has not yet been reported. As judged from 31P{1H} NMR spectroscopy ([1a(Au)3]: δ(CD2Cl2) = 28.3 ppm; 7: δ(CDCl3) = 28.4 ppm) and CV ([1a(Au)3]: E0((nBu4N)BF4) = 340 mV; 7: E0((nBu4N)PF6) = 320 mV),1197 is a well-suited model compound with respect to the electronic properties of both gold(I) and the ferrocene unit.
At the same concentration of gold(I), [1a(Au)3] clearly outperformed the catalytically silent 7 (pink pluses, Fig. 4). This mirrors results from Mendoza-Espinosa and co-workers who have observed a similar effect in comparing tetranuclear mesoionic carbene gold(I) halide complexes and their mononuclear analogues in the hydroamination and hydrohydrazination of terminal alkynes.90 Peris and co-workers found a less prominent cooperative effect for the gold(I)-catalysed hydroamination of phenylacetylene using a trinuclear gold(I) chloride complex with a triphenylene-based tris(N-heterocyclic carbene) ligand.53
Contrastingly, an anti-cooperative effect was found when [1a(Au)3] and 7 were activated by halide abstraction (Fig. S24†) using NaBArF4.117,120 At a 1 mol% gold(I) loading, the catalytic activity of 7/NaBArF4 (TOF = 20.1 ± 0.6 h−1) surpassed that of [1a(Au)3]/NaBArF4 (TOF = 3.2 ± 0.1 h−1) greatly. Following the chloride abstraction of [1a(Au)3] by 1H and 31P{1H} NMR spectroscopy proved inconclusive but hinted at the slow formation of a P,P′-dicoordinated gold(I) complex (δP = 42.9 ppm, Fig. S25†).121–123 An HR-ESI mass spectrum showed signals corresponding to [M–Au–2Cl]+, [M–Au–3Cl]2+, and [M–2Cl]2+ species. The inferior performance of [1a(Au)3]/NaBArF4vs.7/NaBArF4 might thus relate to the formation of chelated and therefore less substrate-accessible gold(I) species.
Given its favourable redox properties – the lowest oxidation potentials and three fully reversible and well-separated oxidation events in the BArF4−-based SE (cf.Fig. 3) – [1a(Au)3] was chosen as a model (pre-)catalyst for initial tests concerning RSC. For the oxidation, 1,1′-diacetylferrocenium tetrakis(perfluoro-tert-butoxy)aluminate (= teflonate) (8) was chosen. Next to the ready synthetic availability from silver(I) teflonate124 and 1,1′-diacetylferrocene and the sufficiently high oxidation potential (E0 = 490 mV vs. FcH/[FcH]+ in CH2Cl2),125 the highly inert non-coordinating anion informed this choice. Smaller anions such as BF4− and SbCl6− have been shown to form tight ion pairs with ferrocenium,105,126 and anion effects in general have been found crucial for the understanding and tailoring of gold catalysts.127
After the reaction shown in Fig. 4 had reached a 25% yield (phase I), 1.0 (orange squares), 2.0 (red triangles), or 3.0 (maroon circles) equivalents of 8 were added. The catalytic activity increased considerably (phase II; cf.Fig. 4, bottom left), in line with findings for similar systems.105–109 Gratifyingly, the activity differed according to the amount of oxidant added: one equivalent of oxidant resulted in a 5.5-fold, two in a 10-fold, and three in a 13-fold increase of the experimentally determined TOF (Fig. 4, bottom). Upon addition of decamethylferrocene (9) as reductant (phase III), less-than-initial activity (TOFØ = 0.67 ± 0.06 h−1) was restored after some delay. In control experiments, employing only 8 or 8 + 9 did not trigger product formation (Fig. S20 and S21†).
The amplified activity is usually explained by an increase in Lewis acidity at the gold(I) site,105–109 and DFT calculations for mono-oxidised [1a(Au)3]+ showed a decrease of energy of unoccupied gold-centred orbitals by over 1.9 eV (Fig. S14–S19†). Such a decrease in LUMO energy can cautiously be understood in terms of increased electrophilicity.128
The different activities, which might correspond to distinct activity states in line with findings of Zhao and Chen,29 were found to be easier to distinguish at 1 mol% Au catalyst loading (Fig. 5, top), decelerating the reaction in so much as to allow for an OFF–ON–OFF–ON switching sequence. Moreover, adding two equivalents of 8 to a catalytic run previously oxidised by one equivalent of 8 (VII to VIII, rose squares), a distinct increase of activity was found. Due to the negligible initial activity (TOF < 0.05 h−1), the great increase in activity is difficult to quantify by numbers (Fig. 5, centre left). It is, however, evident that the TOF effected by the re-oxidised species (VI to VII) are slightly larger than those originating from the first oxidation for one (rose squares) and two (pink triangles), yet not for the second oxidation with three equivalents of 8 (purple circles). Oxidising what we assume to be the mono-oxidised species [1a(Au)3]+ to presumably tri-oxidised [1a(Au)3]3+ (VII to VIII) leads to an even larger reaction rate (TOF = 4.3 ± 0.2 h−1). The ON-switching can also be used on a macroscopic level, to which end two 5 mL-scale reactions (57 mg 5, 2.2 mg [1a(Au)3], i.e., 1 mol% Au, [5]0 = 60 mmol L−1) were compared. One run was oxidised using 3.0 eq. 8 at 12.5 h after the start and led to a quantitative yield vs. only 20% conversion for the non-switched case after 42.0 h (29.5 h after the ON-switch).129
 | ||
| Fig. 5 (Top) Yield-over-time graphs for redox-switchable gold(I)-catalysed cyclisation of 5 to 6 using [1a(Au)3] (1 mol% Au, [5]0 = 60 mmol L−1, CD2Cl2, 25 °C). Arrows indicate the addition of additives. IV: initial OFF phase; V: 1st ON phase after addition of 1.0–3.0 eq. oxidant 8; VI: 2nd OFF phase after addition of 1.1–3.3 eq. reductant 9; VII: 2nd ON phase after addition of 1.1–3.3 eq. oxidant 8; VIII: addition of 2.2 eq. 8 to 1 eq.-switched reaction (rose squares). (Centre) Comparison of turn-over frequencies (TOF) determined from linear fits (cf. Fig. S40–S48† for regression plots) of reaction phases IV to VIII (top), IX (bottom left), and X (bottom right). (Bottom) Left: yield-over-time graphs for gold(I)-catalysed cyclisation of 5 to 6 using [1b(Au)3] (red) and [1c(Au)3] (yellow; conditions as above), oxidised in situ with 2 eq. of 8. Right: yield-over-time graphs for gold(I)-catalysed cyclisation of 5 to 6 using isolated oxidised complexes [1a(Au)3](TEF)n (n = 1–3, conditions as above). Dashed lines represent the chosen timeframe for linear fitting. | ||
These findings are invariant to the order of addition, that is, the presence of substrate during oxidation;130 when [1a(Au)3] is oxidised with 1–3 equivalents of 8 before the addition of 5, similar reaction profiles and TOF result (Fig. S26, Table S13†). In all cases, the first ON-switch is accompanied by an induction period which is absent for the re-oxidation (e.g., IV to Vvs.VI to VII in Fig. 5). When [1b(Au)3] (red triangles) is used in the cyclisation (Fig. 5, bottom left) and oxidised using two equivalents of 8, the same behaviour, including a similar TOF (2.3 ± 0.1 h−1) as for [1a(Au)3], is found. Oxidising [1c(Au)3] (yellow triangles) with two equivalents of 8 converted the otherwise silent complex (vide supra) into an active catalyst with higher TOF (3.3 ± 0.1 h−1). Even though other redox-switchable gold(I) catalysts perform with sometimes significantly higher activity, complexes [1a–c(Au)3] were intended as models demonstrating the feasibility of tailorable multi-oxidation-state applications. Lower observed activities for [1a–c(Au)3] are hence an acceptable trade-off prior to further optimisation.105,109 In the same way, the significantly higher reaction rate of using oxidised 7 (1 mol% Au, TOF = 10.6 ± 0.4 h−1, Fig. S24†) comes at the expense of not being principally able to address separate states with differing catalytic activity.
In order to gain more insight into the switching process, the oxidation of [1a(Au)3] in the absence of substrate was first followed by 1H and 31P{1H} NMR spectroscopy (Fig. S27 and S28†). The 31P{1H} NMR resonance shifted upfield and broadened with each added equivalent of 8, while the subsequent addition of 9 reversed this trend. In line with the findings from the catalytic runs, the reduction took quite long to take its full effect.
Given these encouraging results, we sought to isolate the individually switched complexes [1a(Au)3](TEF)n (n = 1–3), attempting to dismiss the possibility that oxidising [1a(Au)3] during the catalytic reaction (in situ) might lead to mixtures of different oxidation states (e.g. by disproportionation). Furthermore, we wanted to ascertain the integrity of the oxidised species, since Nataro and co-workers observed loss of a chlorido ligand (G, Chart 2) from dinuclear gold(I) complexes based on 1,1′-bis(phosphanyl)ferrocenes after chemical oxidation using tris(p-bromophenyl)ammoniumyl tetrakis(pentafluorophenyl)-borate (“Magic Blue”).131 While the group of Peris found oxidation-induced protonation (H, Chart 2) of the ferrocenyl-imidazolylidene backbone in their gold(I) chloride complexes upon addition of acetylferrocenium tetrafluoroborate,126 Heinze and co-workers described valence isomerisation from, initially, FeIII/AuI to FeII/AuII (J, Chart 2) assisted by both the SbCl6− anion of their oxidant and propargylic amide 5.105 In the last two instances, the respective Au–Cl fragment was found to remain intact.
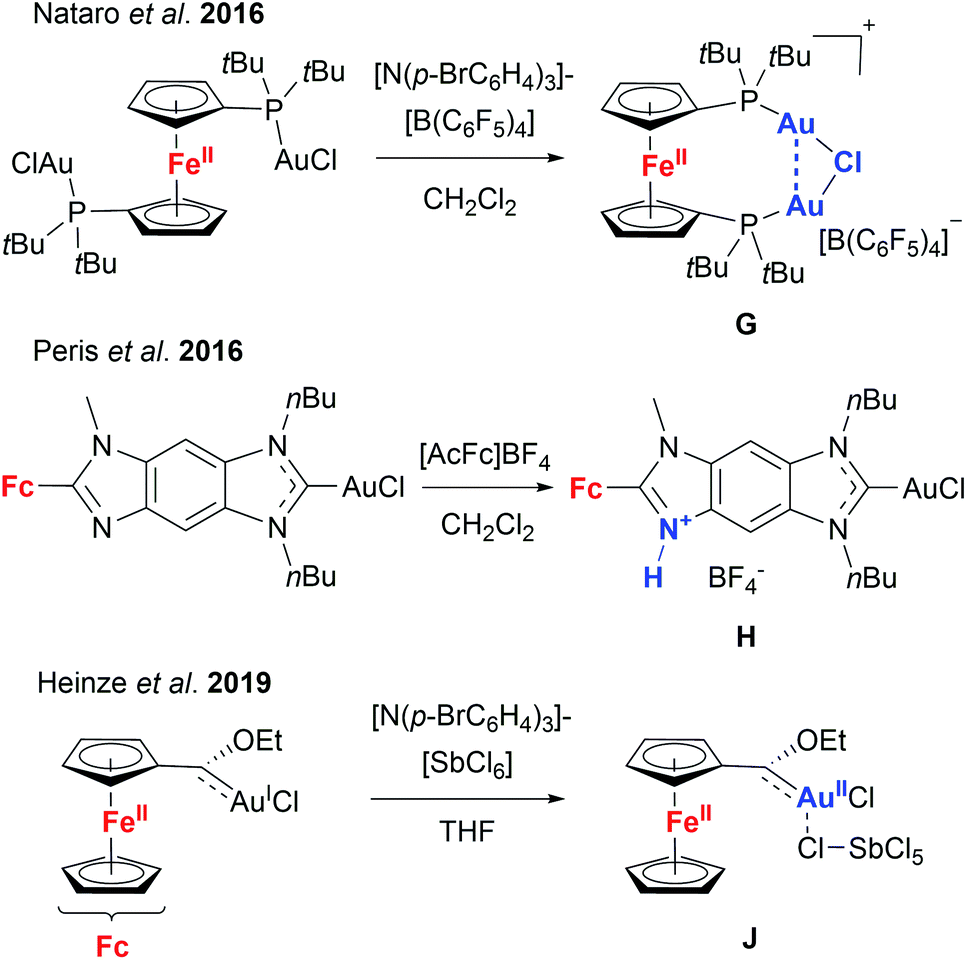 | ||
| Chart 2 Behaviour of ferrocene-derived chloridogold(I) complexes upon chemical oxidation (compounds G and H have been characterised by XRD in the solid state, while information about the structure of J has been gathered from DFT calculations and various analytical techniques). | ||
Gratifyingly, [1a(Au)3](TEF)n can indeed by obtained as analytically pure compounds from the reaction of [1a(Au)3] with 1, 2, or 3 equivalents of 8 as evidenced by CHN analyses. While [1a(Au)3](TEF) is a pale green powder, both [1a(Au)3](TEF)2 and [1a(Au)3](TEF)3 are dark-green microcrystalline solids. In contrast to [1a(Au)3], they are readily soluble in diethyl ether which allows for their purification by precipitation with pentanes, as 1,1′-diacetylferrocene is soluble under these conditions and can hence be extracted. The effective magnetic moments μeff of [1a(Au)3](TEF)n in solution were determined using Evans' method (Fig. S99, S103 and S107†) and match the expected spin-only values for one (2.03 μB, expected: 1.73 μB), two (2.98 μB, expected: 2.83 μB), and three (3.52 μB, expected: 3.82 μB) unpaired electrons reasonably well.132,133 The 1H and 31P{1H} NMR-spectroscopic features (Fig. S98–S109†) match those of the previously mentioned stepwise oxidation (Fig. S27 and S28†). HR-ESI mass spectra show peaks for the three different cations respectively, the tri-cation [1a(Au)3]3+ apparently stabilised by a tight contact to the teflonate anion as {[1a(Au)3](TEF)}2+ under these conditions; similarly, the 19F resonance of [1a(Au)3](TEF)3 (ω1/2 = 17 Hz) is slightly broadened with respect to that of the mono- and dioxidised species (ω1/2 = 4 Hz). IR spectra (Fig. S29†) of [1a(Au)3](TEF)n display both the signature of the teflonate anion and a band at 860 cm−1, characteristic for the δ(C–H) vibration of ferrocenium.134,135 In line with previous reports,136 the ferrocenylene ν(C–H) stretches shift to higher wavenumbers upon increasing degree of oxidation and do not indicate the presence of native or lower oxidation states as expected for disproportionation. Neither can this be inferred from UV/Vis spectra of [1a(Au)3](TEF)n (Fig. 6 top right, Fig. S30†), as they differ in the position of their bands at long wavelength, most likely related to LMCT, inner ferrocenyl, and potentially even to AuI–FeIII MMCT transitions,106,137,138 between 500 and 1000 nm.
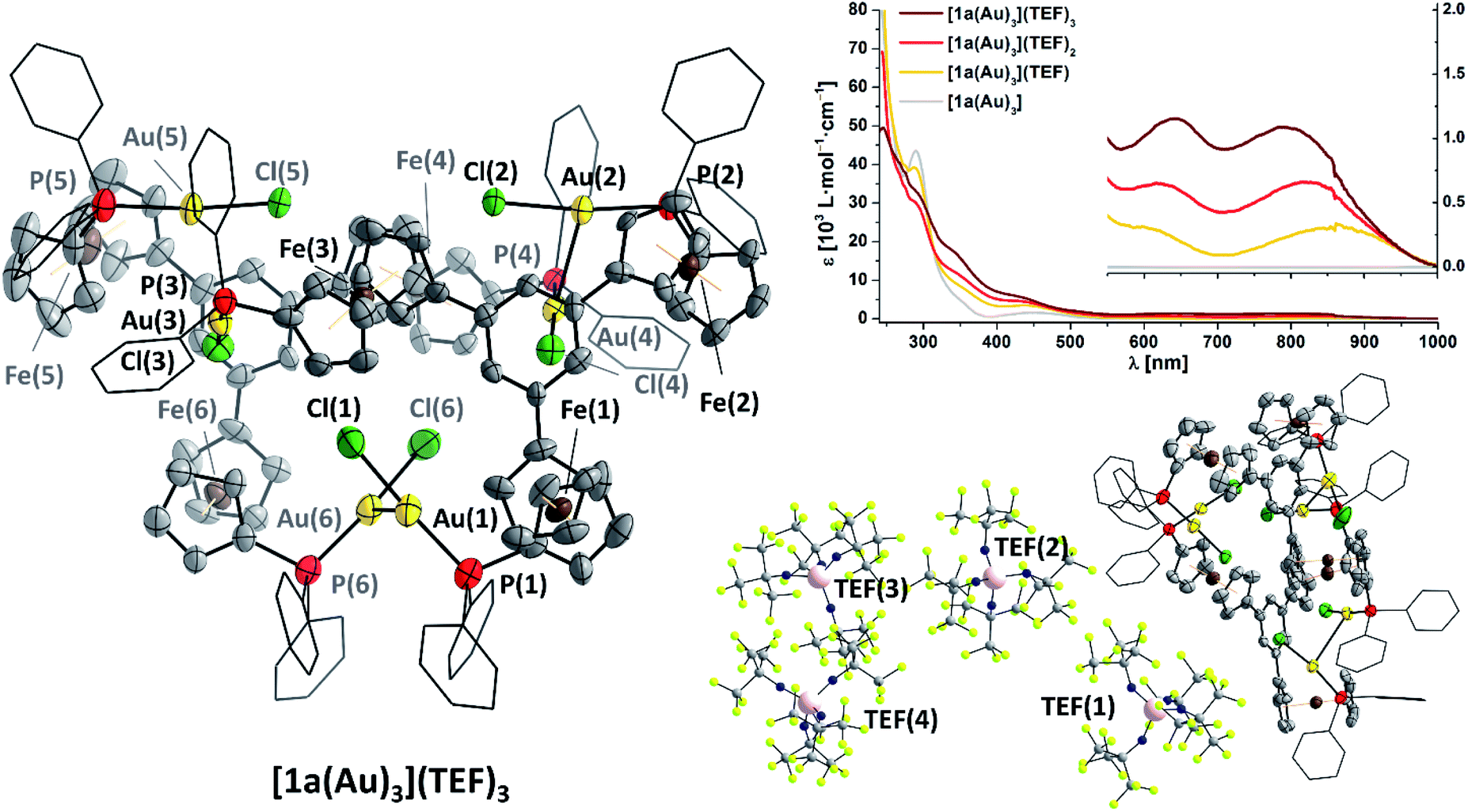 | ||
| Fig. 6 (Left) Molecular structure of {[1a(Au)3]}2(TEF)6 with partial atom numbering scheme. Thermal ellipsoids are set at the 50% probability level. For clarity, P-bound phenyl rings are depicted in wireframe style, and co-crystallised solvent, hydrogen atoms, and anions have been omitted. (Bottom right) Asymmetric unit of {[1a(Au)3]}2(TEF)6 with the four localisable teflonate anions in ball-and-stick style. The presence of severely disordered teflonate anions TEF(5) and TEF(6) has been confirmed through their electron density and the unoccupied volume. (Top right) Stacked UV/Vis spectra of [1a(Au)3](TEF)n (n = 0–3) in CH2Cl2. The inset shows a magnification for the absorptions at long wavelength. | ||
As final proof for the site of oxidation, we were able to isolate single crystals of the tri-oxidised complex as {[1a(Au)3]}2(TEF)6 (Fig. 6, left and bottom right), thus unambiguously confirming the oxidation state of three times FeIII through the presence of six, albeit only partly crystallographically describable, teflonate anions per asymmetric unit. Notably, all three P–Au–Cl fragments are still intact and form dimers of an overall sixfold positive charge through a set of two aurophilic interactions (d(Au(1)–Au(6)) = 2.989(1) Å, d(Au(2)–Au(4)) = 3.188(1) Å) and a less-close Au⋯Au contact (d(Au(3)⋯Au(5)) = 3.397(1) Å).139 While the poor crystal quality precludes detailed metric analyses, generally larger C5H4(centroid)–Fe distances of over 1.68 Å and, accordingly, Fe–C bond lengths of up to 2.15(2) Å, typical for ferrocenium species,136,140 are discernible (Table S8†). Other structural parameters, particularly with respect to the P–Au–Cl moieties are, within error, comparable to the molecular structure of [1c(Au)3]. While ferrocenium cations are frequently encountered as counter cations for metallate anions and several homo- and heteromultinuclear bridged metallocenes featuring ferrocenium units have been listed in the CSD, {[1a(Au)3]}2(TEF)6 is a very rare example of a crystallographically characterised metal complex containing ferrocenium in its ligand backbone. To the best of our knowledge, a rhenium(0) carbonyl complex by Nataro and co-workers is the only other example to contain both ferrocenium and a phosphane-bound metal complex fragment,141 while the +III oxidation state of iron in reported bis[1,1′-bis(diphenylphosphanyl)ferrocenium]hexadecachlorotetraantimonate(III) by the group of Kasim142 has been questioned by both Nataro and Connick.143 Not backed up by a solid-state molecular structure, Grandberg and co-workers also reported a phosphanyl ferrocenium-based gold(III) bromide complex in 1977.144 Last but not least, a very recent report by the Lapinte group details a related tetranuclear iron(II/III) half-sandwich array with potential use in molecular electronics.145
[1a(Au)3](TEF)n appear to be air- and moisture-insensitive in the solid state for short periods. They can be weighed out in ambient conditions but quickly turn from green to yellow solutions in wet solvents. Their stability in dry CD2Cl2 was monitored by 1H and 31P{1H} NMR spectroscopy (Fig. S31–S33†). Surprisingly, a solution of [1a(Au)3](TEF)3 did show little change over three days while [1a(Au)3](TEF) and [1a(Au)3](TEF)2 slowly decomposed under the same conditions (room temperature, protected from light). 31P{1H} resonances attributable to P,P′-dicoordinate species at around 40 ppm appeared, and [1a(Au)3](TEF)2 produced a metallic mirror of presumably elemental gold on the wall of the NMR tube. Furthermore, the 1H NMR spectral evolution of [1a(Au)3](TEF)2 (Fig. S32†) indicates at least some degree of disproportionation to the mono- and trioxidised species, making it the least solution-stable of the three isolated oxidation products. Although a mass spectrum of the crystals of [1a(Au)3](TEF)3 does not contain signals for dimeric species, aurophilic interactions in solution might play a role in stabilising the trication. When fresh solutions of [1a(Au)3](TEF)n (n = 1–3) are reacted with one, two, or three equivalents of reductant 9, native [1a(Au)3] is re-obtained respectively (Fig. S34–S36†).
The isolated oxidised species [1a(Au)3](TEF)n have finally also been tested for their catalytic performance (ex situ; Fig. 5, bottom right). Their TOF show the previously noted distinct dependence on the degree of oxidation, with tri-oxidised [1a(Au)3](TEF)3 (light blue circles, 5.8 ± 0.1 h−1) performing with about threefold activity than mono-oxidised [1a(Au)3](TEF) (navy squares, 1.8 ± 0.1 h−1). All of them outperform the in situ-oxidised species, most notably [1a(Au)3](TEF)3. This points towards redox equilibria between 8 and the complexes in higher oxidation states which might be overcome using stronger oxidants, thus ensuring full conversion upon addition. Even though we cannot fully exclude potential disproportionation or the presence of mixtures under these catalytic conditions, the distinctly different activities in the read-out catalytic conversion strongly suggest that [1a(Au)3] and its analogues can function as molecular switches with four addressable states.
Similar to the in situ-generated species, induction periods are observed for [1a(Au)3](TEF)n. Notably, the addition of a fresh batch of 5 to an almost-completed reaction after 24 h (Fig. S37†) using [1a(Au)3](TEF)3 did not result in another induction period but led to a slight loss of activity (3.2 ± 0.1 vs. 5.0 ± 0.1 h−1). Mixing isolated [1a(Au)3](TEF)3 with 5 (Fig. S38†) in CH2Cl2 led to an appreciably slow colour change which was followed by time-resolved UV/Vis spectroscopy. We thus speculate that the catalytically active species are, in general, formed from a chemical process involving 5. Bearing in mind the aforementioned results from Heinze and co-workers and in accordance with loss of the FeIII-associated absorptions of [1a(Au)3](TEF)3 at long wavelength,105 a coordination-assisted valence isomerisation from FeIII/AuI to FeII/AuII might be at the heart of this behaviour.
Conclusions
In summary, we have demonstrated the modular syntheses of a new class of tris(ferrocenyl)arene-based tris-phosphanes 1 which can be used to form well-defined, C3-symmetric homotrinuclear gold(I) complexes. Four oxidation states relating to the tris(ferrocenyl)arene backbone – non-, mono-, di-, and tri-oxidised – have been identified by cyclic voltammetry. Stoichiometric oxidation of [1a(Au)3] produces isolable products [1a(Au)3](TEF)n. This redox behaviour can be advantageously used in redox-switchable catalysis ex and in situ, as we were able to show for the proof-of-principle ring-closing isomerisation of N-(2-propyn-1-yl)benzamide (5) forming oxazoline 6. The arene cores determine the exact redox potential and were also found to influence the catalytic performance of the native and oxidised species, a feature which we are currently investigating in more detail. Metal complexes of 1 and its analogues thus hold great promise for applications in molecular electronics and logic, possibly extending binary to quaternary signal processing, as four different oxidation states can be addressed and isolated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Studienstiftung des deutschen Volkes (doctoral fellowships to P. C. and A. S.), the DFG (He 1376/51-1) and the Graduate School BuildMoNa is gratefully acknowledged. L. D. acknowledges funding from DFG Sche 384/36-1. The authors thank Dr Andy Schmied for the XRD measurement of 4a, Dr Peter Lönnecke for help with the structure solution of {[1a(Au)3]}2(TEF)6, and Ramona Oehme (MS Core Facility of Leipzig University) for HR-ESI MS measurements.Notes and references
- M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2018, 2, 12236 CrossRef.
- H. H. Kung and M. C. Kung, Catal. Lett., 2014, 144, 1643 CrossRef CAS.
- M. Desage-El Murr, ChemCatChem, 2019, 12, 53 CrossRef.
- I. M. Lorkovic, R. R. Duff and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617 CrossRef CAS.
- F. Würthner and J. Rebek, Angew. Chem., Int. Ed., 1995, 34, 446 CrossRef.
- J. Choudhury, Tetrahedron Lett., 2018, 59, 487 CrossRef CAS.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969 CrossRef CAS PubMed.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC.
- E. Peris, Chem. Rev., 2018, 118, 9988 CrossRef CAS PubMed.
- J. Wei and P. L. Diaconescu, Acc. Chem. Res., 2019, 52, 415 CrossRef CAS PubMed.
- A. M. Allgeier and C. A. Mirkin, Angew. Chem., Int. Ed., 1998, 37, 894 CrossRef PubMed.
- Y. Ryu, G. Ahumada and C. W. Bielawski, Chem. Commun., 2019, 55, 4451 RSC.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 149, S21 Search PubMed.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 2017, 6 CrossRef CAS.
- Ferrocenes. Ligands, Materials and Biomolecules, ed. P. Štěpnička, J. Wiley, Chichester, England, Hoboken, NJ, 2008 Search PubMed.
- C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410 CrossRef CAS PubMed.
- M. S. Inkpen, S. Scheerer, M. Linseis, A. J. P. White, R. F. Winter, T. Albrecht and N. J. Long, Nat. Chem., 2016, 8, 825 CrossRef CAS PubMed.
- D. Astruc, C. Ornelas and J. Ruiz, Acc. Chem. Res., 2008, 41, 841 CrossRef CAS PubMed.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629 CrossRef CAS PubMed.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Hahn, S. Liebing, J. Kortus and H. Lang, Organometallics, 2012, 31, 6761 CrossRef CAS.
- U. Pfaff, G. Filipczyk, A. Hildebrandt, M. Korb and H. Lang, Dalton Trans., 2014, 43, 16310 RSC.
- B. Topolinski, B. M. Schmidt, S. Schwagerus, M. Kathan and D. Lentz, Eur. J. Inorg. Chem., 2014, 2014, 5391 CrossRef CAS.
- Y. Yu, A. D. Bond, P. W. Leonard, U. J. Lorenz, T. V. Timofeeva, K. P. C. Vollhardt, G. D. Whitener and A. A. Yakovenko, Chem. Commun., 2006, 2572 RSC.
- M. Zhao and C. Chen, ACS Catal., 2017, 7, 7490 CrossRef CAS.
- G. Haberhauer, Angew. Chem., Int. Ed., 2011, 50, 6415 CrossRef CAS PubMed.
- L. van Dijk, M. J. Tilby, R. Szpera, O. A. Smith, H. A. P. Bunce and S. P. Fletcher, Nat. Rev. Chem., 2018, 2, 22 Search PubMed.
- M. Baroncini, L. Casimiro, C. de Vet, J. Groppi, S. Silvi and A. Credi, ChemistryOpen, 2018, 7, 169 CrossRef CAS PubMed.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200 CrossRef CAS PubMed.
- A. P. de Silva, T. P. Vance, B. Wannalerse and M. E. S. West, in Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd edn, 2011, pp. 669–696 Search PubMed.
- H. Keisar, M. Lahav and M. E. van der Boom, ChemPhysChem, 2019, 20, 2403 CrossRef CAS PubMed.
- K. Szaciłowski, Chem.–Eur. J., 2004, 10, 2520 CrossRef PubMed.
- C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266 CrossRef CAS PubMed.
- F. Pina, A. Roque, M. J. Melo, M. Maestri, L. Belladelli and V. Balzani, Chem.–Eur. J., 1998, 4, 1184 CrossRef CAS.
- H. J. Chandler, M. Stefanou, E. E. B. Campbell and R. Schaub, Nat. Commun., 2019, 10, 1 CrossRef CAS PubMed.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, Nat. Nanotechnol., 2012, 7, 41 CrossRef PubMed.
- S. Silvi, E. C. Constable, C. E. Housecroft, J. E. Beves, E. L. Dunphy, M. Tomasulo, F. M. Raymo and A. Credi, Chem.–Eur. J., 2009, 15, 178 CrossRef CAS PubMed.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429 CrossRef CAS PubMed.
- A. Straube, P. Coburger, M. R. Ringenberg and E. Hey-Hawkins, Chem.–Eur. J., 2020, 26, 5758 CrossRef CAS PubMed.
- P. Neumann, H. Dib, A.-M. Caminade and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2015, 54, 311 CrossRef CAS PubMed.
- P. Neumann, H. Dib, A. Sournia-Saquet, T. Grell, M. Handke, A.-M. Caminade and E. Hey-Hawkins, Chem.–Eur. J., 2015, 21, 6590 CrossRef CAS PubMed.
- J. Popp, A.-M. Caminade and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2020, 17, 1654 CrossRef.
- S. E. Gibson and M. P. Castaldi, Angew. Chem., Int. Ed., 2006, 45, 4718 CrossRef CAS PubMed.
- C. Moberg, Angew. Chem., Int. Ed., 1998, 37, 248 CrossRef CAS PubMed.
- E. K. van den Beuken and B. L. Feringa, Tetrahedron, 1998, 54, 12985 CrossRef CAS.
- R. Peters, Cooperative Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed.
- X. Liu, P. Du and R. Cao, Nat. Commun., 2013, 4, 2375 CrossRef PubMed.
- L.-I. Rodríguez, T. Roth, J. Lloret Fillol, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2012, 18, 3721 CrossRef PubMed.
- S. Gonell, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2013, 52, 7009 CrossRef CAS PubMed.
- R. Maity, A. Mekic, M. van der Meer, A. Verma and B. Sarkar, Chem. Commun., 2015, 51, 15106 RSC.
- T. Glaser, M. Heidemeier, T. Weyhermüller, R.-D. Hoffmann, H. Rupp and P. Müller, Angew. Chem., Int. Ed., 2006, 45, 6033 CrossRef CAS PubMed.
- C.-G. Freiherr von Richthofen, A. Stammler, H. Bögge, M. W. DeGroot, J. R. Long and T. Glaser, Inorg. Chem., 2009, 48, 10165 CrossRef PubMed.
- S. Naik, M. Kumaravel, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2014, 53, 1370 CrossRef CAS PubMed.
- G. S. Ananthnag, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2015, 54, 10985 CrossRef CAS PubMed.
- J. Zhang, P. W. Miller, M. Nieuwenhuyzen and S. L. James, Chem.–Eur. J., 2006, 12, 2448 CrossRef CAS PubMed.
- C. S. A. Fraser, M. C. Jennings and R. J. Puddephatt, Chem. Commun., 2001, 1310 RSC.
- M. S. Inkpen, S. Du, M. Driver, T. Albrecht and N. J. Long, Dalton Trans., 2013, 42, 2813 RSC.
- S. Steffens, M. H. Prosenc, J. Heck, I. Asselberghs and K. Clays, Eur. J. Inorg. Chem., 2008, 1999 CrossRef CAS.
- D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540 CrossRef CAS.
- K. Schlögl and H. Soukup, Tetrahedron Lett., 1967, 8, 1181 CrossRef.
- V. Mamane, I. Ledoux-Rak, S. Deveau, J. Zyss and O. Riant, Synthesis, 2003, 3, 455 Search PubMed.
- Y.-K. Lim, S. Wallace, J. C. Bollinger, X. Chen and D. Lee, Inorg. Chem., 2007, 46, 1694 CrossRef CAS PubMed.
- A. Donoli, A. Bisello, R. Cardena, C. Prinzivalli and S. Santi, Organometallics, 2013, 32, 1029 CrossRef CAS.
- S. Santi, A. Bisello, R. Cardena and A. Donoli, Dalton Trans., 2015, 44, 5234 RSC.
- T.-Y. Dong and D. N. Hendrickson, Bull. Inst. Chem., Acad. Sin., 1987, 34, 67 CAS.
- V. Leigh, W. Ghattas, H. Mueller-Bunz and M. Albrecht, J. Organomet. Chem., 2014, 771, 33 CrossRef CAS.
- J. Kühnert, M. Dusek, J. Demel, H. Lang and P. Štěpnička, Dalton Trans., 2007, 2802 RSC.
- J. Schulz, I. Císařová and P. Štěpnička, Organometallics, 2012, 31, 729 CrossRef CAS.
- K. Škoch, I. Císařová, J. Schulz, U. Siemeling and P. Štěpnička, Dalton Trans., 2017, 46, 10339 RSC.
- U. Siemeling, T. Klemann, C. Bruhn, J. Schulz and P. Štěpnička, Dalton Trans., 2011, 40, 4722 RSC.
- M. J. Verschoor-Kirss, O. Hendricks, C. M. Verschoor, R. Conry and R. U. Kirss, Inorg. Chim. Acta, 2016, 450, 30 CrossRef CAS.
- F. Barrière, R. U. Kirss and W. E. Geiger, Organometallics, 2005, 24, 48 CrossRef.
- D. A. Durfey, R. U. Kirss, C. Frommen and W. Feighery, Inorg. Chem., 2000, 39, 3506 CrossRef CAS PubMed.
- J. Popp, S. Hanf and E. Hey-Hawkins, ACS Omega, 2019, 4, 22540 CrossRef CAS PubMed.
- P. Štěpnička and I. Císařová, Dalton Trans., 2013, 42, 3373 RSC.
- A. Hildebrandt, N. Wetzold, P. Ecorchard, B. Walfort, T. Rüffer and H. Lang, Eur. J. Inorg. Chem., 2010, 2010, 3615 CrossRef.
- U. Siemeling, T. Klemann, C. Bruhn, J. Schulz and P. Štěpnička, Z. Anorg. Allg. Chem., 2011, 637, 1824 CrossRef CAS.
- T. M. Dau, J. R. Shakirova, A. Doménech, J. Jänis, M. Haukka, E. V. Grachova, T. A. Pakkanen, S. P. Tunik and I. O. Koshevoy, Eur. J. Inorg. Chem., 2013, 28, 4976–4983 Search PubMed.
- B. Assadollahzadeh and P. Schwerdtfeger, Chem. Phys. Lett., 2008, 462, 222 CrossRef CAS.
- Q. Zheng, S. Borsley, G. S. Nichol, F. Duarte and S. L. Cockroft, Angew. Chem., Int. Ed., 2019, 58, 12617 CrossRef CAS PubMed.
- M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884 RSC.
- M. R. Ringenberg, Chem.–Eur. J., 2019, 25, 2396 CrossRef CAS PubMed.
- P. Kalck, Polyhedron, 1988, 7, 2441 CrossRef CAS.
- Homo- and Heterobimetallic Complexes in Catalysis. Cooperative Catalysis, ed. P. Kalck, Springer International Publishing, Cham, 1st edn, 2016 Search PubMed.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723 RSC.
- M. Flores-Jarillo, D. Mendoza-Espinosa, V. Salazar-Pereda and S. González-Montiel, Organometallics, 2017, 36, 4305 CrossRef CAS.
- Y. Li, E. M. W. Tsang, A. Y. C. Chan and H.-Z. Yu, Electrochem. Commun., 2006, 8, 951 CrossRef CAS.
- J. Heck and M. Dede, in Ferrocenes. Ligands, Materials and Biomolecules, ed. P. Štěpnička, J. Wiley, Chichester, England, Hoboken, NJ, 2008, pp. 319–392 Search PubMed.
- M. Iyoda, T. Kondo, T. Okabe, H. Matsuyama, S. Sasaki and Y. Kuwatani, Chem. Lett., 1997, 26, 35 CrossRef.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980 CrossRef PubMed.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395 CrossRef CAS PubMed.
- P. Zanello, A. Cinquantini, M. Fontani, M. Giardiello, G. Giorgi, C. R. Landis and B. F. M. Kimmich, J. Organomet. Chem., 2001, 637–639, 800 CrossRef CAS.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461 Search PubMed.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed.
- D. Canseco-Gonzalez, A. Petronilho, H. Mueller-Bunz, K. Ohmatsu, T. Ooi and M. Albrecht, J. Am. Chem. Soc., 2013, 135, 13193 CrossRef CAS PubMed.
- R. Pretorius, M. R. Fructos, H. Müller-Bunz, R. A. Gossage, P. J. Pérez and M. Albrecht, Dalton Trans., 2016, 45, 14591 RSC.
- L. Hettmanczyk, D. Schulze, L. Suntrup and B. Sarkar, Organometallics, 2016, 35, 3828 CrossRef CAS.
- M. Rigo, L. Hettmanczyk, F. J. L. Heutz, S. Hohloch, M. Lutz, B. Sarkar and C. Müller, Dalton Trans., 2016, 46, 86 RSC.
- A. S. K. Hashmi, M. C. Blanco Jaimes, A. M. Schuster and F. Rominger, J. Org. Chem., 2012, 77, 6394 CrossRef CAS PubMed.
- P. Veit, C. Volkert, C. Förster, V. Ksenofontov, S. Schlicher, M. Bauer and K. Heinze, Chem. Commun., 2019, 55, 4615 RSC.
- L. Hettmanczyk, S. Manck, C. Hoyer, S. Hohloch and B. Sarkar, Chem. Commun., 2015, 51, 10949 RSC.
- L. Hettmanczyk, L. Suntrup, S. Klenk, C. Hoyer and B. Sarkar, Chem.–Eur. J., 2017, 23, 576 CrossRef CAS PubMed.
- S. Klenk, S. Rupf, L. Suntrup, M. van der Meer and B. Sarkar, Organometallics, 2017, 36, 2026 CrossRef CAS.
- S. Vanicek, M. Podewitz, J. Stubbe, D. Schulze, H. Kopacka, K. Wurst, T. Müller, P. Lippmann, S. Haslinger, H. Schottenberger, K. R. Liedl, I. Ott, B. Sarkar and B. Bildstein, Chem.–Eur. J., 2018, 24, 3742 CrossRef CAS PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed.
- D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS PubMed.
- A. Homs, I. Escofet and A. M. Echavarren, Org. Lett., 2013, 15, 5782 CrossRef CAS PubMed.
- A. Zhdanko and M. E. Maier, ACS Catal., 2015, 5, 5994 CrossRef CAS.
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS PubMed.
- Y. Sota, M. Yamamoto, M. Murai, J. Uenishi and M. Uemura, Chem.–Eur. J., 2015, 21, 4398 CrossRef CAS PubMed.
- M. Chen, B. Yang and C. Chen, Angew. Chem., Int. Ed., 2015, 54, 15520 CrossRef CAS PubMed.
- E. Deck, H. E. Wagner, J. Paradies and F. Breher, Chem. Commun., 2019, 55, 5323 RSC.
- K. Arumugam, C. D. Varnado, S. Sproules, V. M. Lynch and C. W. Bielawski, Chem.–Eur. J., 2013, 19, 10866 CrossRef CAS PubMed.
- K. Rößler, T. Rüffer, B. Walfort, R. Packheiser, R. Holze, M. Zharnikov and H. Lang, J. Organomet. Chem., 2007, 692, 1530 CrossRef.
- C. García-Morales, B. Ranieri, I. Escofet, L. López-Suarez, C. Obradors, A. I. Konovalov and A. M. Echavarren, J. Am. Chem. Soc., 2017, 139, 13628 CrossRef PubMed.
- J. E. Aguado, S. Canales, M. C. Gimeno, P. G. Jones, A. Laguna and M. D. Villacampa, Dalton Trans., 2005, 3005 RSC.
- M. C. Gimeno, A. Laguna, C. Sarroca and P. G. Jones, Inorg. Chem., 1993, 32, 5926 CrossRef CAS.
- J. Wolf, F. Huber, N. Erochok, F. Heinen, V. Guerin, C. Legault, S. Kirsch and S. M. Huber, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202005214.
- I. Krossing, Chem.–Eur. J., 2001, 7, 490 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- S. Ibáñez, M. Poyatos, L. N. Dawe, D. Gusev and E. Peris, Organometallics, 2016, 35, 2747 CrossRef.
- J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 2493 CrossRef.
- L.-G. Zhuo, W. Liao and Z.-X. Yu, Asian J. Org. Chem., 2012, 1, 336 CrossRef CAS.
- The increased rate for the non-switched reaction is likely linked to the stirring (500 rpm) which, in contrast to the NMR experiment, was applied.
- L. A. Brown, J. L. Rhinehart and B. K. Long, ACS Catal., 2015, 5, 6057 CrossRef CAS.
- S. F. Hartlaub, N. K. Lauricella, C. N. Ryczek, A. G. Furneaux, J. D. Melton, N. A. Piro, W. S. Kassel and C. Nataro, Eur. J. Inorg. Chem., 2017, 2017, 424 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003 RSC.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- R. J. Webb, S. J. Geib, D. L. Staley, A. L. Rheingold and D. N. Hendrickson, J. Am. Chem. Soc., 1990, 112, 5031 CrossRef CAS.
- J. A. Kramer and D. N. Hendrickson, Inorg. Chem., 1980, 19, 3330 CrossRef CAS.
- K. Kowalski, M. Linseis, R. F. Winter, M. Zabel, S. Záliš, H. Kelm, H.-J. Krüger, B. Sarkar and W. Kaim, Organometallics, 2009, 28, 4196 CrossRef CAS.
- R. Prins, J. Chem. Soc. D, 1970, 280b RSC.
- H. B. Gray, Y. S. Sohn and N. Hendrickson, J. Am. Chem. Soc., 1971, 93, 3603 CrossRef.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- B. Artetxe, A. Iturrospe, P. Vitoria, E. Ruiz-Bilbao, J. S. Garitaonandia and J. M. Gutiérrez-Zorrilla, Molecules, 2018, 23, 3150 CrossRef PubMed.
- A. G. Furneaux, N. A. Piro, R. Hernández Sánchez, K. M. Gramigna, N. Fey, M. J. Robinson, W. S. Kassel and C. Nataro, Dalton Trans., 2016, 45, 4819 RSC.
- I. Abdul-Razak, A. Usman, H.-K. Fun, B. M. Yamin and N. A. Kasim, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m162–m164 CrossRef PubMed.
- S. Chatterjee, J. A. Krause, W. B. Connick, C. Genre, A. Rodrigue-Witchel and C. Reber, Inorg. Chem., 2010, 49, 2808 CrossRef CAS PubMed.
- A. N. Nesmeyanov, E. G. Perevalova, O. B. Afanasova and K. I. Grandberg, Izv. Akad. Nauk, Ser. Khim., 1977, 2166 CAS.
- R. Makhoul, P. Hamon, T. Roisnel, J.-R. Hamon and C. Lapinte, Chem.–Eur. J., 2020, 26, 8368 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, detailed spectral and crystallographic characterisation, DFT calculations, electrochemical data, catalysis plots. CCDC 1990272–1990279, 1990281–1990283 and 2012019. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03604h |
| ‡ Present address: Laboratory of Inorganic Chemistry, ETH Zürich, CH-8093 Zürich (Switzerland). |
| This journal is © The Royal Society of Chemistry 2020 |