
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining free energy calculations with tailored enzyme activity assays to elucidate substrate binding of a phospho-lysine phosphatase†
Anett
Hauser
ab,
Songhwan
Hwang
c,
Han
Sun
 *c and
Christian P. R.
Hackenberger
*ab
*c and
Christian P. R.
Hackenberger
*ab
aDepartment of Chemical Biology II, Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Berlin, Germany. E-mail: hackenbe@fmp-berlin.de
bInstitute for Chemistry, Humboldt-Universität zu Berlin, Berlin, Germany
cGroup of Structural Chemistry and Computational Biophysics, Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Berlin, Germany. E-mail: hsun@fmp-berlin.de
First published on 8th September 2020
Abstract
Studying enzymes that are involved in the regulation of dynamic post-translational modifications (PTMs) is of key importance in proteomics research. Such investigations can be particularly challenging when the modification itself is intrinsically labile. In this article, we elucidate the enzymatic activity of Phospholysine Phosphohistidine Inorganic Pyrophosphate Phosphatase (LHPP) towards different O- and N-phosphorylated peptides by a combined experimental and computational approach. LHPP has been previously described to hydrolyze the phosphoramidate bonds in different small molecule substrates, including phosphorylated lysine (pLys). Taking the instability of the phosphoramidate bond into account, we conducted a carefully adjusted enzymatic assay with various pLys pentapeptides to confirm enzymatic phosphatase activity with LHPP. Molecular docking was employed to explore possible binding poses of the substrates in complex with the enzyme. Molecular dynamics based free energy calculations, which are unique in their accuracy and solid theoretical basis, were further applied to predict relative binding affinity of different substrates. Comparison of simulations with experiments clearly suggested a distinct binding motif of pLys peptides as well as a very narrow promiscuity of LHPP. We believe this integrated approach can be widely adopted to study the structure and interaction of poorly characterized enzyme–substrate complexes, in particular with synthetically challenging or labile substrates.
Introduction
A balanced interplay of protein phosphorylation and dephosphorylation is a key factor to functioning cellular processes.1–3 So far, many detailed studies on structure–activity relationships, substrates and mechanisms have been conducted to understand the various processes taking place during the introduction of phosphate groups into proteins by kinases.4–6 Meanwhile, enzymes responsible for the cleavage of phosphates or other phosphate ester derivatives like phosphoramidates or phosphorothioates are less often subjects of studies.7 Challenging aspects include short-term interactions with substrates, high complexity regarding evolutionary development, regulatory pathways and possible substrates as well as experimental demands, especially if the phosphorylated peptide substrates have a low stability profile.7A prime example in this regard is the Phospholysine Phosphohistidine Inorganic Pyrophosphate Phosphatase (LHPP). First reported in 1997 by Hiraishi et al., LHPP was isolated from bovine liver. Subsequently the enzymatic hydrolase activity was shown, first for pyrophosphate (PPi) and imidodiphosphate (PNP), later for the single amino acids ε-phospho-lysine (pLys) and 3-phospho-histidine (3-pHis).8,9 While researchers could observe slightly higher activity for PNP compared to PPi (0.44 μmol min−1 mg−1 and 0.11 μmol min−1 mg−1 released phosphate at pH 8.2 in the presence of 1 mM MgCl2) as well as for pLys compared to pHis (1.22 μmol min−1 mg−1 and 1.09 μmol min−1 mg−1 released phosphate at pH 8.2 in the presence of 1 mM MgCl2), no structural information about the binding event of pLys or pHis residues to LHPP is currently known. Furthermore, besides small molecules, no endogenous pLys-containing peptide or protein substrates for LHPP were reported to date and thus, no standardized value that describes catalytic potency has been established. Nevertheless, LHPP is known to occur in several species10 and to have an influence in different diseases.11–13 Very recently, its role as a tumor suppressor was highlighted by Hindupur et al., Zheng et al. and Li et al. shortly afterwards.12–14 In all cases, findings were supported by knockout/overexpression experiments, but no specific substrate was investigated. Moreover, LHPP is assigned to the haloacid dehydrogenase (HAD) super-family of hydrolases,15 but its catalytic mechanism remains unknown and therefore, information about the scope or sequence dependency is missing.16 Although the enzymatic activity was shown to be higher for pLys, all the later findings described LHPP in the role of a phosphoramidate hydrolase selective for pHis.17,18
Although the phosphorylation of lysine residues was described for the first time more than 45 years ago, its biological role has been, to date, only rudimentarily elucidated. Initially described to occur as an acid-labile group in histone H1,19,20 pLys could be detected in rat liver in vivo later.21,22 The reported chemical and thermal instability23 has hampered further identification and characterization with conventional proteomics techniques. Despite recent reports on incident pLys detection by empirico-statistical methods,24,25 an in-depth study on endogenous pLys sites has yet to be published. Along those lines, also no enzyme being selectively active on phosphorylated lysine peptides or proteins is known so far. It should be noted that the phosphoramidate bond in pLys is intrinsically labile at physiological pH, which means that high background hydrolysis during enzymatic studies is to be expected.23 Our group has recently developed suitable synthetic and analytical methods to advance research questions for this most intriguing post-translational modification.23,26–29 Here, we present a systematic study to elucidate the structure–activity relationship as well as the interaction of different N- and O-phosphorylated substrates with LHPP including phosphorylated lysine. In particular, we probed the enzymatic activity of LHPP towards phosphate- or phosphoramidate-containing peptide substrates with different steric and electronical demand. All peptidic substrates were obtained via elaborated organic synthetic protocols specifically optimized for each compound class. This was particularly important for studying the hydrolase activity of pLys-containing peptides due to their intrinsic lability. It has been shown that the phosphorous–nitrogen bond was hydrolyzed very fast under acidic conditions and at elevated temperatures.23 For example, the half-life time of pLys is approx. 2.5 h at room temperature and pH 3.5 or below and even less than 1 h at 60 °C and neutral pH.23 Therefore, a fine-tuned assay with less delay between each step was required.
Since no structural information about the binding mode of phosphoramidate substrates was available, we used molecular docking originating from a known crystal structure with a bound pyrophosphate substrate to explore possible binding poses. To further evaluate binding of different substrates quantitatively, we used molecular dynamics (MD) based alchemical free energy calculations, which has emerged in recent years as a promising tool for in silico drug optimizations,30,31 quantifying protein–protein interactions32 and understanding drug resistance.33 Taking advantage of high accuracy of this first-principle statistical mechanics-based method, we predicted free energy changes (ΔΔG) upon mutation in the pLys-peptides for a number of different docking poses. Comparison of the predicted free energy changes with the experimental rates verified enzymatic activity for pLys dephosphorylation and pointed to low substrate promiscuity.
Results and discussion
Phosphoramidate hydrolase and phosphatase assay to evaluate the LHPP substrate scope
To minimize phosphoramidate bond hydrolysis even without enzyme being present, fast sample preparation, distinct pH adjustment and storage at low temperature were crucial. Whereas previous studies to monitor LHPP activity relied on detection of released inorganic phosphate (Pi) using Rosenberg's reagent9 or malachite green,8,10 we considered both methods as too time consuming to meet the stability profile of pLys peptides. State of the art real-time detection of released Pi can be conducted conveniently in cuvette- or microplate-based experiments and we desired a similar set up to conduct kinetic experiments. The EnzChek® Phosphate Assay kit enables continuous measurement of released Pi by the phosphate dependent conversion of 2-amino-6-mercapto-7-methylpurine riboside to a UV-active thione.34 In order to identify optimal assay conditions, we first conducted LHPP activity measurements with the previously reported substrate pLys (1a).9,23 Varying the concentration of 1a and LHPP at pH 7.4, we identified 100 μM substrate 1a in a 200 μL reaction volume with 0.25 μg enzyme (7.4 × 10−3 nmol, 0.037 μM, 3.7 × 10−4 eq.) as suitable for conducting enzyme kinetics within 90 minutes incubation time. It should be noted that pLys background hydrolysis gave a significant absorbance already at the starting point of each experiment (Fig. S1†). Any possible synthesis route of phospho-lysine or pLys-peptides would fail to deliver pure phosphorylated substrates, since either the conversion would not be quantitative or the phosphoramidate would partially hydrolyze during purification.23,28,35 Hence, we conducted the kinetic measurements with mixtures of pLys and Lys + Pi.Additionally, the intrinsic lability led to more Pi release over time even without enzyme being present (Fig. 1A, grey line). Therefore, we used the difference between the detected value for released phosphate from incubation with LHPP (Fig. 1A, black line) and the value without addition of enzyme (Fig. 1A, grey line) to evaluate the enzymatic hydrolysis. As shown for 100 μM 1a, more than 40% of substrate were hydrolyzed after the experiment without LHPP being present. This means, roughly 50 μM pLys were degraded by LHPP, according to 50% enzymatically hydrolyzed substrate, after 60 minutes, when the conversion was complete. With the standardized conditions, we were able to determine kinetic parameters, e.g. initial rates v0 and first order rate constant k, from the measured data. Fig. 1B shows the data for the sample experiment of 100 μM pLys 1a without enzyme and with 0.25 μg LHPP. It can be seen that the hydrolysis occurred one magnitude faster upon LHPP addition and thus, the enzyme could accelerate the reaction rate despite the substrate's intrinsic lability.
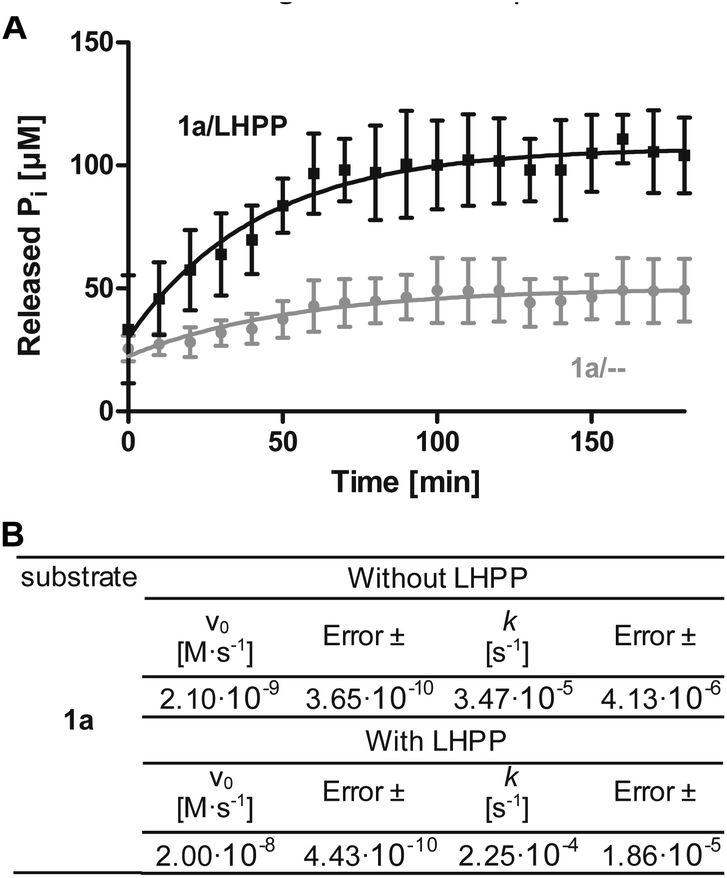 | ||
Fig. 1 (A) Example for kinetics of 100 μM 1a with 0.25 μg LHPP over 180 min. Amount of released inorganic phosphate without ( ) or with (—) LHPP being present. (B) Initial rates and first order rate constants of the kinetic studies of 1a. ) or with (—) LHPP being present. (B) Initial rates and first order rate constants of the kinetic studies of 1a. | ||
Next, we investigated the enzymatic activity of LHPP towards different types of phosphorylated amino acids (Fig. 2A), which were synthesized following published protocols.36,37 LHPP activity with various substrates was determined on a microplate reader by photometric detection of released inorganic phosphate (ESI, Section 1†). All substrates were incubated at a concentration of 100 μM in 200 μL of 50 mM Tris–HCl buffer containing 1 mM MgCl2 at pH 7.8 in the presence or absence of 0.25 μg LHPP (3.7 × 10−4 eq., 7.4 × 10−3 nmol, 0.037 μM). Absorbance was measured every ten minutes for 90 min. In accordance with the results published by Hiraishi et al. (see above),9 phosphorylated lysine monomer 1a was hydrolyzed at a rate of 0.88 μmol min−1·mg−1, which corresponded to 56.26% enzymatically hydrolyzed pLys after 90 min (background hydrolysis subtracted). For the other N-phospho-amino acids (pHis 1f, pArg 1e) and for the phosphates (pSer 1b, pThr 1c, pTyr 1d) drastically lower values for the hydrolyzed substrates were observed after 90 min (17.57%, 6.64%, 5.75%, 3.67%, 4.81%, respectively, see Fig. 3A).
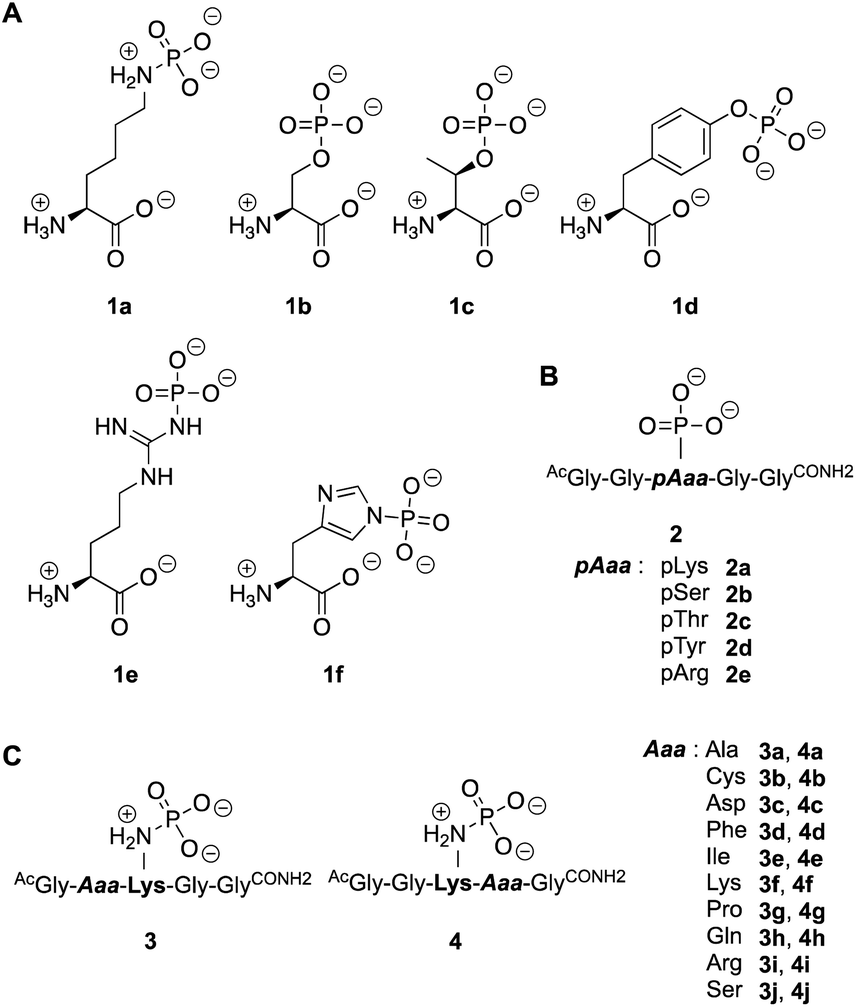 | ||
| Fig. 2 Substrates employed in this study. Different phospho-amino acids (A) and phospho-peptides (B) to examine the activity towards phosphates and phosphoramidates. (C) Pentapeptides to study the neighboring effect of amino acid side chains. | ||
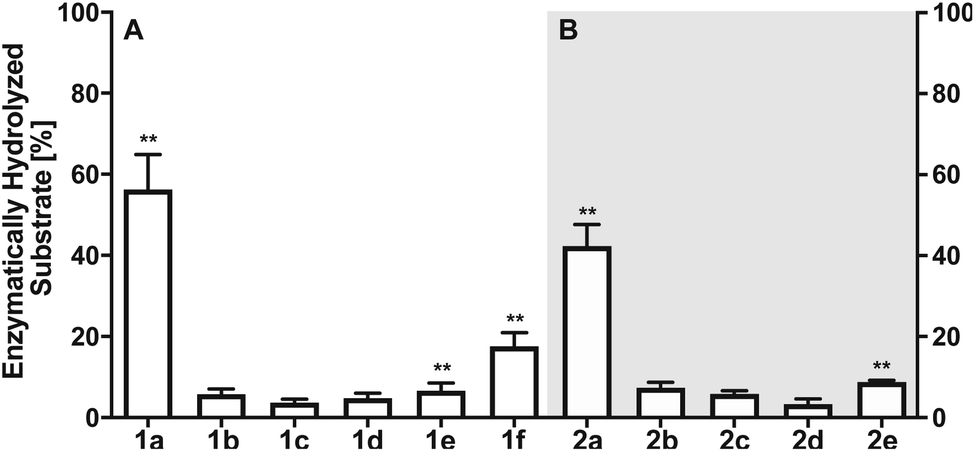 | ||
| Fig. 3 (A) Percentage of enzymatically hydrolyzed substrate after 90 minutes of incubation of monomeric phospho-amino acids 1a–1f with LHPP. (B) Percentage of enzymatically hydrolyzed substrate after 90 minutes of incubation of differently phosphorylated pentapeptides 2a–2e with LHPP. Experiments have been conducted as biological and technical triplicates. ** percentage of hydrolyzed substrate without enzyme being present has been subtracted. | ||
To evaluate different N- and O-phosphorylated peptide substrates of LHPP we synthesized a set of simple pentapeptides with the sequence AcGlyGlypAaaGlyGlyCONH2 (pAaa: pLys 2a, pSer 2b, pThr 2c, pTyr 2d or pArg 2e) (Fig. 2B). Even though we intended to analyze the pHis substrate as well, the reaction to form the corresponding pHis peptide could neither be driven to sufficient conversion nor be purified from the phosphorylation reagent or the unphosphorylated peptide and was thus excluded from this study. For the synthesis of pLys peptides, we used our previously reported protocols to obtain pLys peptides.23 This allowed us to take advantage of the chemoselectivity during a reaction between an azide and a phosphite for the incorporation of a photocaged pLys precursor at a given site in the peptide starting from commercially available Fmoc-protected azido-lysine. Subsequently, the Staudinger-phosphite reaction with tris(1-(2-nitrophenyl)ethyl) phosphite and subsequent photodeprotection yielded desired, free pLys (ESI, Section 2.1†).
The enzymatic activity observed for the pentapeptides corroborated with the amino acid findings from before, for instance 37.90% enzymatically hydrolyzed AcGlyGlypLysGlyGlyCONH22a after 90 min, while all other substrates showed significantly lower hydrolysis yield (2b: 7.37%, 2c: 5.86%, 2d: 3.34%, 2e: 8.78%, Fig. 3B). To our knowledge, this is the first time that N- and O-phosphorylated peptides have been assayed with LHPP, which serves as corroboration that LHPP is a rather a selective phosphoramidate hydrolase but not a phosphate ester hydrolase. In addition, the decreased hydrolysis yield obtained for peptide 2a when compared to 1a points towards a narrower substrate scope.
To investigate this further, we systematically evaluated two additional sets of peptides, in which the amino acids N- or C-terminal to the pLys residue were exchanged (AcGlyAaapLysGlyGlyCONH23, AcGlyGlypLysAaaGlyCONH24, Aaa: Ala, Cys, Asp, Phe, Ile, Lys, Pro, Gln, Arg, Ser, Fig. 2C). The amino acid substitutions were chosen to address different electronic and steric scenarios. All substrates were assayed as described above. Indeed, we observed a distinct impact on the phosphoramidates hydrolase activity of LHPP by modifications adjacent to the phosphorylation site (Fig. 4). While most of the modifications led to a drastic decrease of enzymatic hydrolysis, only peptides 3a and 3i with N-terminal Ala and Arg or peptide 4f with C-terminal Lys could retain or surpass the hydrolysis yields after 90 minutes (3a: 37.33%, 3i: 40.70%, 4f: 29.81%, respectively) (Fig. 4). These findings were particularly interesting since modifications with switched positions (C-terminal Ala 4a and Arg 4j or N-terminal Lys 3f) displayed reduced conversions. Interestingly, peptides 3c and 4c containing a negatively charged Asp residue showed hydrolysis values as compared to substrates with positively charged residues (Lys: 3f and 4f, Arg: 3i and 4i), in which only up to one third of Pi could be released. These observations hint towards electrostatic repulsion of 3c and 4c in the binding pocket of LHPP as it exhibits a high negative electron density as discussed later, thus further suggesting that LHPP is sensitive to sequence alterations and accepts only a limited scope of pLys substrates. Similar findings have been reported recently for LHPP activity on histidine phosphorylations by Choi et al. as mentioned above.18
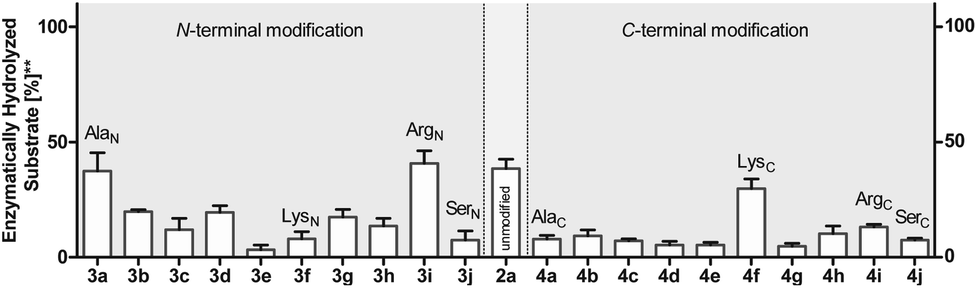 | ||
| Fig. 4 Phosphoramidate hydrolysis yields for N- and C-terminally modified pentapeptides after 90 min of incubation. Experiments have been conducted as biological and technical triplicates. ** percentage of hydrolyzed substrate without enzyme being present has been subtracted. | ||
Next, we evaluated the kinetics for the hydrolysis of pLys peptides with and without LHPP addition for peptides 2a, 3a and 3j, which included the substrates with the highest and lowest hydrolysis yields after 90 min (Fig. 5 and Table 1). We observed the highest first order rate constants with LHPP for peptide 3a, which was one order of magnitude higher than for 3j. In addition, the experiments conducted without LHPP pointed towards a distinct intrinsic lability for each substrate depending on the amino acid sequence and revealed a higher lability for peptide 3a as compared to the other peptides. As already observed with pLys 1a, the background hydrolysis could have a significant impact on the obtained amount of enzymatically hydrolyzed substrate. This could be observed with 2a but was particularly clear in the case of Ala-modified substrate 3a, which yielded more than 40% hydrolysis without LHPP being present and thus, only approx. 38% enzymatically hydrolyzed substrate was eventually observed.
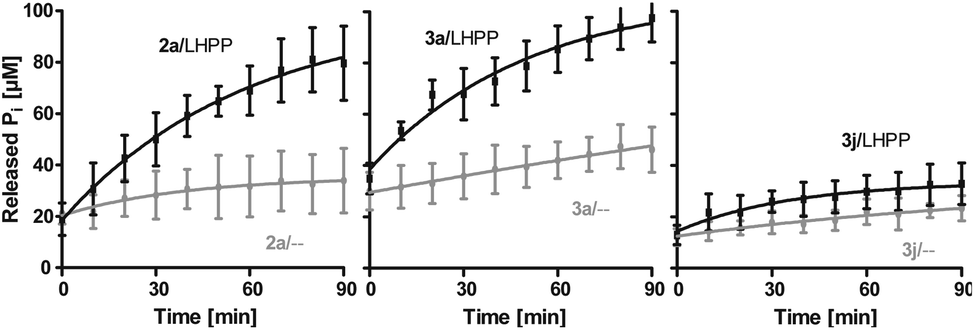 | ||
Fig. 5 Kinetics for 2a, 3a and 3j. The rate of background hydrolysis was strongly impacted by the peptide sequence. — with LHPP,  without addition of LHPP. without addition of LHPP. | ||
| Substrate | v 0 [M s−1] | Error ± | k [s−1] | Error ± |
|---|---|---|---|---|
| Without LHPP | ||||
| 2a | 4.95 × 10−9 | 2.16 × 10−9 | 3.59 × 10−5 | 4.35 × 10−6 |
| 3a | 2.48 × 10−9 | 1.53 × 10−10 | 5.60 × 10−5 | 2.95 × 10−6 |
| 3j | 3.09 × 10−9 | 2.58 × 10−10 | 2.47 × 10−5 | 2.00 × 10−6 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| With LHPP | ||||
| 2a | 1.97 × 10−8 | 5.80 × 10−11 | 3.44 × 10−4 | 2.07 × 10−5 |
| 3a | 2.72 × 10−8 | 2.26 × 10−9 | 4.29 × 10−4 | 3.93 × 10−5 |
| 3j | 6.46 × 10−9 | 1.94 × 10−9 | 4.24 × 10−5 | 4.90 × 10−6 |
Also, both, the higher initial rate v0 and the increased first order rate constant k indicated higher substrate lability in reactions without enzyme being present (Table 1, upper part). We hypothesize that the increased stability of Ser-modified peptide 3j could be based on hydrogen bonds formed between the protons on ε-nitrogen in pLys and the free electron pairs on the Ser oxygen, which resulted in a decreased effective positive charge on the Lys side chain.
Molecular modeling and MD-based free energy calculation of LHPP-substrate complexes
The precise binding mode of the peptide substrate GGpKGG (2a, Fig. 6A) in complex with LHPP was unknown. Instead, a crystal structure of human LHPP has been available since 2010 in the protein database (RCSB PDB, ID: 2X4D), but has not been published anywhere else yet. This structure revealed the catalytic center of the LHPP comprising a phosphate group and a magnesium ion. In order to explore possible binding poses of 2a within the catalytic center of LHPP, we performed a molecular docking study by defining Mg2+ in the catalytic center as the center of the docking pocket. Docking of 2a into the pocket was carried out using the Glide module38–40 implemented in the software Schrödinger.41 Out of ten best docking poses with the highest scores, seven poses exhibited considerable overlap of the phosphate position of pLys with the one from the crystal structure. From these seven poses, we chose four poses (Fig. 6B & ESI, Section 4.6†), that showed significant variation in their binding conformations as the starting structures in the MD simulations. As no force field parameter for pLys can be found in the literature, we generated here the AMBER-based force field parameters for pLys using the previously proposed approach for parameterizing phosphorylated serine, threonine, tyrosine, and histidine.42 Detailed procedure of the parameterization as well as newly introduced parameters of phosphorylated lysine are given in the ESI in Section 4.2, Tables S4 and S5.† During a 100 ns simulation of 2a solvated in water, the energy values remained to be stable. Furthermore, no obvious molecular distortion was observed during the simulations, additionally verifying these new force field parameters of phosphorylated lysine to be suitable for the MD simulations.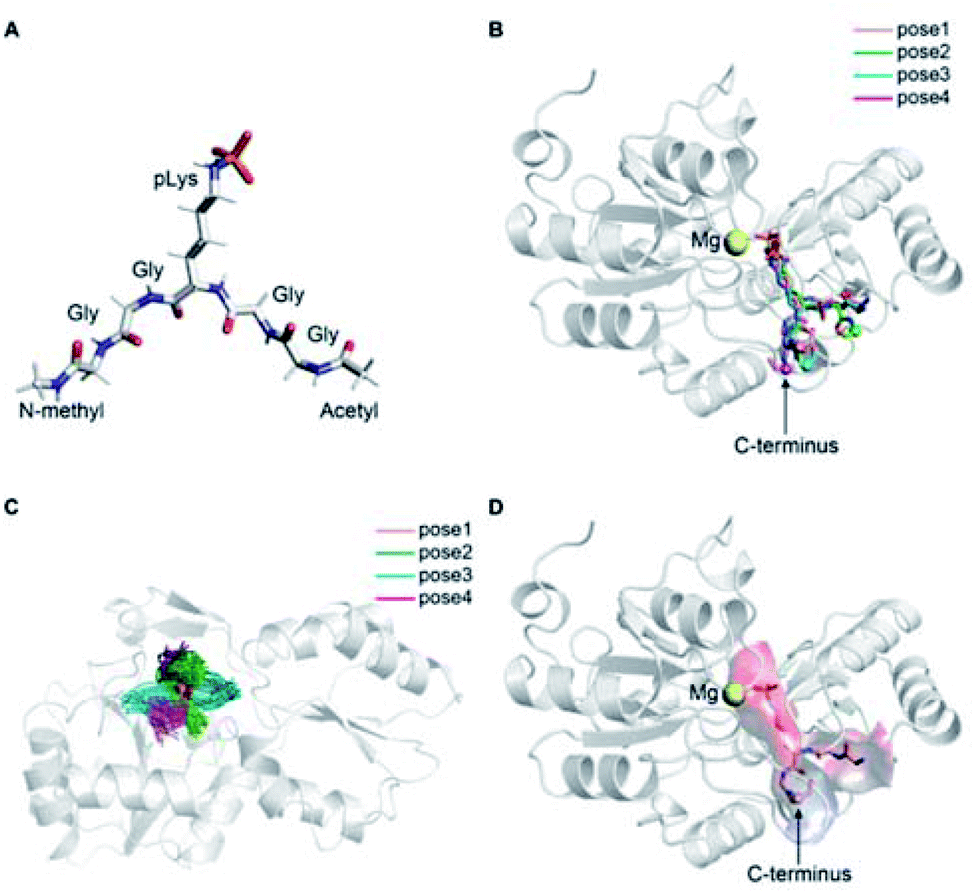 | ||
| Fig. 6 (A) Molecular structure of model substrate 2a. In the simulations N-methyl and acetyl were capped at C- and N-termini, respectively. (B) Overlaid docking poses of 2a in the binding pocket of LHPP, showing pLys interacts with Mg2+. Peptide 2a in four different docking poses is shown as sticks with the C-terminal highlighted as sphere, and the LHPP as cartoon in grey. (C) Superposition of simulation snapshots of 2a starting from 4 different docking poses. Peptides are shown as ribbon and LHPP as cartoon in grey. (D) Determined binding mode of 2a in complex with LHPP (pose 1). Peptides are shown as ribbon with the C-terminal highlighted in sphere. LHPP is shown as cartoon and the electrostatic potential (red: negative, blue: positive) of the binding pocket has been mapped onto the molecular surface. | ||
In order to theoretically evaluate the free energy changes of different pLys-based pentapeptides, we employed alchemical free energy calculations for 3a, 3f, 3i, 3j, 4a, 4f, 4i and 4j using the non-modified peptide 2a as the reference. 3a/4a (Ala), 3f/4f (Lys) and 3i/4i (Arg) were selected in the simulations, as experimentally introducing these three amino acids in either C- or N-terminus showed large differences in their enzymatic activities (Fig. 4). As a comparison, we calculated free energy changes of 3j/4j (Ser) relatively to 2a, which revealed a remarkable decrease in the phosphoramidate hydrolase activity of LHPP when introducing the serine at both C- and N-termini.
The general idea of MD-based free energy calculation is illustrated in Fig. 7A. The method requires a combination of the equilibrium MD simulations to sample the conformational space of the non-modified and modified peptides, as well as fast non-equilibrium transitions from each other. In the equilibrium MD simulations of 2a starting from four different docking poses, the binding conformations of the peptide remained relatively stable and did not interconvert to each other (Fig. 6C). In order to evaluate which docking pose represents the best binding conformation of the peptide, we performed the equilibrium simulations for 3a, 3f, 3i, 3j, 4a, 4f, 4i and 4j, respectively, starting from each four poses. The subsequent non-equilibrium alchemical transition simulations were carried out according to previously established protocol43,44 using a combination of the GROMACS45,46 and pmx47,48 software packages. In the free energy calculations upon amino acid switching that cause a net charge change, we adopted the double-system/single-box setup,49 while the charge-conserving simulations were performed using the double-system/double-box setup (Fig. 7B). Both Crooks Gaussian Intersection (CGI) and Bennet's Acceptance Ratio (BAR) estimators were employed in the analysis of free energy changes ΔΔG.44 Details of the simulations are given in the ESI in Section 4.4 & 4.5.†
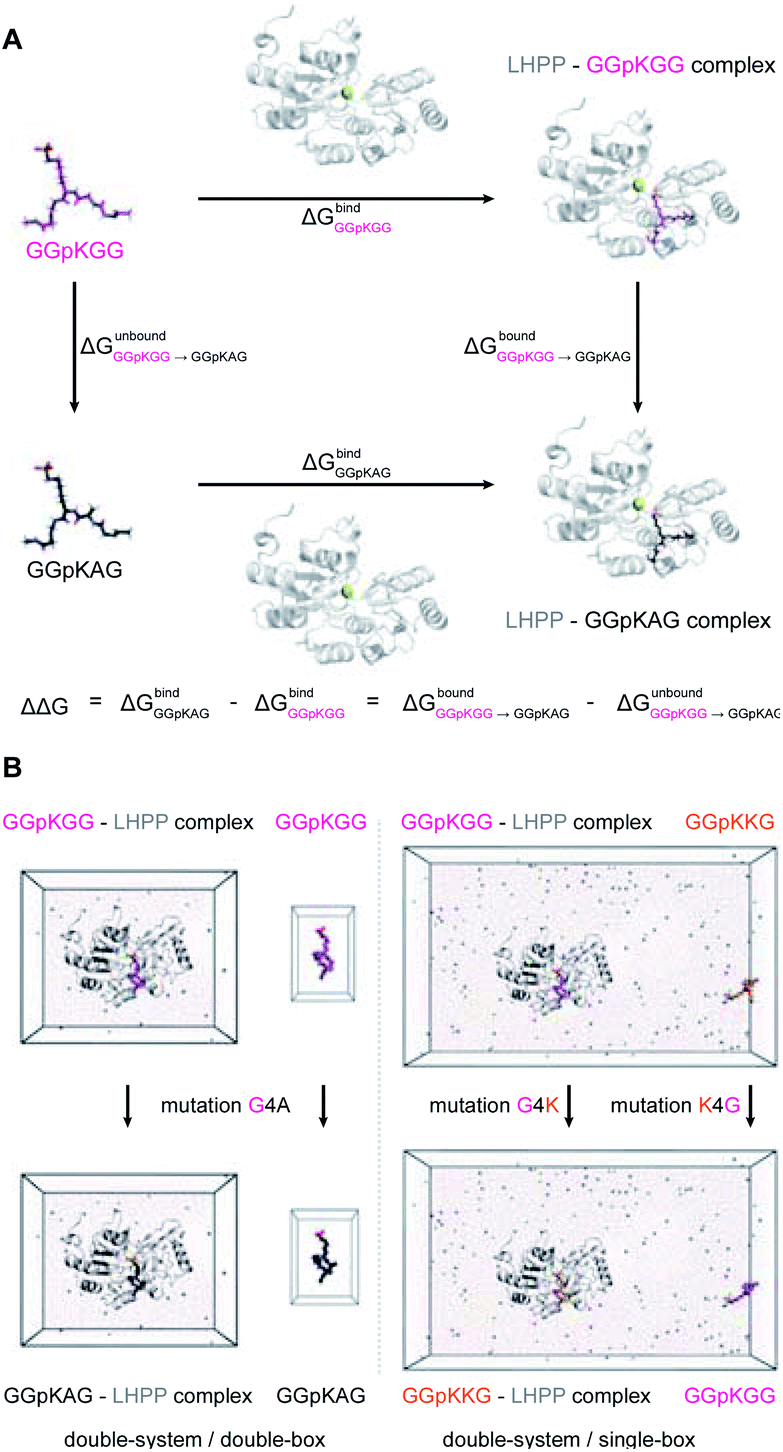 | ||
| Fig. 7 (A) Thermodynamic cycle used in this study to calculate the free energy difference between the GGpKGG 2a and other mutated peptides, e.g. GGpKAG 4a or GGpKKG 4f. (B) Double-system/double-box and double-system/single-box setups employed in the ΔΔG calculation of charge-conserving and charge-changing mutations, respectively. | ||
Results summarized in Table S7† suggested that the predicted free energy changes upon amino acid changes in the peptide were considerably different for the simulations starting from different docking poses. Qualitatively, free energy changes derived from the simulations of the docking pose 1 revealed unambiguously the best agreement with the experimental data (Fig. 4 and 8). The large difference in free energy changes of 3f/4f and 3i/4i as well as a significant decrease of free energy in both 3j/4j compared to the WT 2a were all well reproduced in the simulations. Nevertheless, experimentally introducing the Ala at the N-terminal (3a) retained the hydrolysis yields, while the counter modification at the C-terminus (4a) diminished conversion. This result did not fit to the simulation data, as slight increases in relative binding free energy were predicted for both 3a/4a compared to 2a. We hypothesize that this was, because introducing small steric perturbation in amino acid sequence may require much longer simulation time than that was performed in the current study (a few hundred of nanoseconds) to capture the differences in binding.
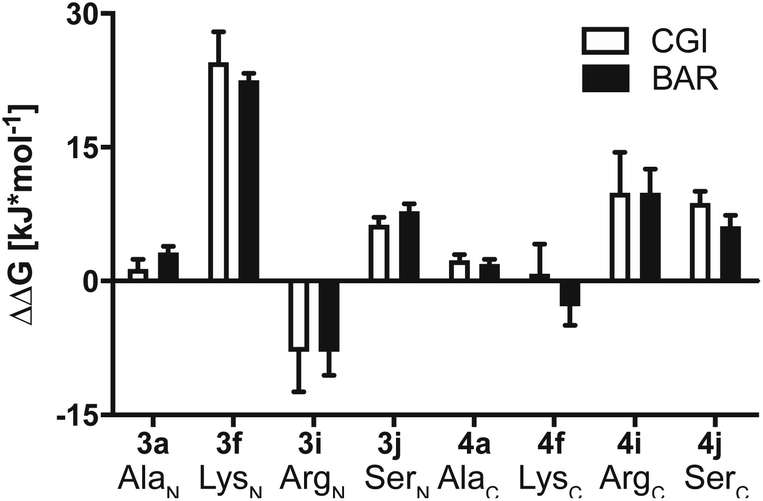 | ||
| Fig. 8 Calculated ΔΔG together with errors of mutated substrates relative to the WT model substrate 2a estimated from the MD simulations starting from docking pose 1. Crooks Gaussian Intersection (CGI) and Bennet's Acceptance Ratio (BAR) estimators were employed for the analysis of ΔΔG. | ||
In the best binding mode revealed by the alchemical free energy calculations (pose 1, Fig. 6D), the side chain of the pLys is nicely fitted in a small cavity that is negatively charged and completely buried from solvent, with the doubly charged magnesium ion compensating the negative charges both from the substrate and the protein environment. In contrast, electronic potential of the binding site for accommodating the substrate main chain was mostly negative. This was in good agreement with the enzymatic assay revealing peptides containing a negatively charged Asp residue (3c/4c) and showed considerably decreasing hydrolysis rate compared to the WT 2a (Fig. 4). Nevertheless, we also observed significant difference in the electrostatic potential of the binding pocket for C- and N-termini regions (Fig. 6D), explaining the experimental finding that substrates with positively charged residues (3f/4f and 3i/4i) exhibited very different enzymatic activity in their C- or N-terminus modifications (Fig. 4).
Conclusions
In conclusion, we present an integrated experimental and computational approach to evaluate the activity and binding affinity of an enzyme towards intrinsically labile substrates, for which no information about the substrate scope was available prior to our studies. Specifically, we elucidate the enzymatic reactivity of LHPP towards different N-and O-phosphorylated amino acids and peptides. Building upon reliable synthetic protocols to obtain a variety of phosphorylated substrates, we expand the previous reports by Hiraishi et al.8,9 that LHPP acts as primarily as phosphoramidate hydrolase, for example on pLys or pHis. Furthermore, we studied in detail the sequence dependency in synthetic pLys substrates and observed a high sequence dependency rather than promiscuity on peptidic substrates. By applying molecular docking, we explored the possible binding poses of the non-modified peptide 2a in the catalytic center of LHPP, which were further evaluated using MD-based alchemical free energy calculations. The latter calculations delivered precious insight into the sequence dependency of LHPP's enzymatic activity. The combination of biochemical assays and computational simulations suggested a defined binding mode of the labile substrates in complex with LHPP, which is difficult to establish using conventional approaches such as X-ray crystallography. Overall, in light of the recent emergence of labile phosphorylations, we show in a case study a novel binding event of LHPP specific for pLys substrates. Moreover, we strongly envision that this integrated approach can deliver valuable information about interactions of other enzymes with labile substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the DFG (SFB 765), the Evonik Stiftung (Doctoral fellowship to A. H.) and DFG under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat. We thank Dr Jens von Kries for equipment support. The authors thank Prof. Maja Köhn for advice on the phosphatase assay. Furthermore, we also acknowledge the North-German Supercomputing Alliance (HLRN) for providing High Performance Computing (HPC) resources that have contributed to the research results reported in this paper.Notes and references
- J. A. Ubersax and J. E. Ferrell Jr, Nat. Rev. Mol. Cell Biol., 2007, 8, 530–541 CrossRef CAS.
- M. K. Tarrant and P. A. Cole, Annu. Rev. Biochem., 2009, 78, 797–825 CrossRef CAS.
- S. J. Humphrey, D. E. James and M. Mann, Trends Endocrinol. Metab., 2015, 26, 676–687 CrossRef CAS.
- G. Manning, G. D. Plowman, T. Hunter and S. Sudarsanam, Trends Biochem. Sci., 2002, 27, 514–520 CrossRef CAS.
- B. A. Haubrich and D. C. Swinney, Curr. Drug Discovery Technol., 2016, 13, 2–15 CrossRef CAS.
- M. J. Suskiewicz, B. Hajdusits, R. Beveridge, A. Heuck, L. D. Vu, R. Kurzbauer, K. Hauer, V. Thoeny, K. Rumpel, K. Mechtler, A. Meinhart and T. Clausen, Nat. Chem. Biol., 2019, 15, 510–518 CrossRef CAS.
- S. Fahs, P. Lujan and M. Köhn, ACS Chem. Biol., 2016, 11, 2944–2961 CrossRef CAS.
- H. Hiraishi, T. Ohmagari, Y. Otsuka, F. Yokoi and A. Kumon, Arch. Biochem. Biophys., 1997, 341, 153–159 CrossRef CAS.
- H. Hiraishi, F. Yokoi and A. Kumon, Arch. Biochem. Biophys., 1998, 349, 381–387 CrossRef CAS.
- F. Yokoi, H. Hiraishi and K. Izuhara, J. Biochem., 2003, 133, 607–614 CrossRef CAS.
- E. Koike, S. Toda, F. Yokoi, K. Izuhara, N. Koike, K. Itoh, K. Miyazaki and H. Sugihara, Biochem. Biophys. Res. Commun., 2006, 341, 691–696 CrossRef CAS.
- J. Zheng, X. Dai, H. Chen, C. Fang, J. Chen and L. Sun, Biochem. Biophys. Res. Commun., 2018, 503, 1108–1114 CrossRef CAS.
- S. K. Hindupur, M. Colombi, S. R. Fuhs, M. S. Matter, Y. Guri, K. Adam, M. Cornu, S. Piscuoglio, C. K. Y. Ng, C. Betz, D. Liko, L. Quagliata, S. Moes, P. Jenoe, L. M. Terracciano, M. H. Heim, T. Hunter and M. N. Hall, Nature, 2018, 555, 678 CrossRef.
- Y. Li, X. Zhang, X. Zhou and X. Zhang, Biosci. Rep., 2019, 39(7), BSR20182270 CrossRef.
- A. M. Burroughs, K. N. Allen, D. Dunaway-Mariano and L. Aravind, J. Mol. Biol., 2006, 361, 1003–1034 CrossRef.
- H. Jung, S. H. Shin and J.-M. Kee, ChemBioChem, 2019, 20, 623–633 CrossRef.
- A. Gohla, Biochim. Biophys. Acta, Mol. Cell Res., 2019, 1866, 153–166 CrossRef.
- Y. Choi, S. H. Shin, H. Jung, O. Kwon, J. K. Seo and J.-M. Kee, ACS Sens., 2019, 4, 1055–1062 CrossRef.
- D. L. Smith, B. B. Bruegger, R. M. Halpern and R. A. Smith, Nature, 1973, 246, 103–104 CrossRef.
- D. L. Smith, C.-C. Chen, B. B. Bruegger, S. L. Holtz, R. M. Halpern and R. A. Smith, Biochemistry, 1974, 13, 3780–3785 CrossRef CAS.
- C. C. Chen, B. B. Bruegger, C. W. Kern, Y. C. Lin, R. M. Halpern and R. A. Smith, Biochemistry, 1977, 16, 4852–4855 CrossRef CAS.
- C.-C. Chen, D. L. Smith, B. B. Bruegger, R. M. Halpern and R. A. Smith, Biochemistry, 1974, 13, 3785–3789 CrossRef CAS.
- J. Bertran-Vicente, R. A. Serwa, M. Schümann, P. Schmieder, E. Krause and C. P. R. Hackenberger, J. Am. Chem. Soc., 2014, 136, 13622–13628 CrossRef.
- K. Adam, S. Fuhs, J. Meisenhelder, A. Aslanian, J. Diedrich, J. Moresco, J. La Clair, J. R. Yates and T. Hunter, bioRxiv, 2019, 691352, DOI:10.1101/691352.
- G. Hardman, S. Perkins, P. J. Brownridge, C. J. Clarke, D. P. Byrne, A. E. Campbell, A. Kalyuzhnyy, A. Myall, P. A. Eyers, A. R. Jones and C. E. Eyers, EMBO J., 2019, 38, e100847 CrossRef.
- J. Bertran-Vicente, M. Penkert, O. Nieto-Garcia, J.-M. Jeckelmann, P. Schmieder, E. Krause and C. P. R. Hackenberger, Nat. Commun., 2016, 7, 12703 CrossRef.
- J. Bertran-Vicente, M. Schümann, C. P. R. Hackenberger and E. Krause, Anal. Chem., 2015, 87, 6990–6994 CrossRef.
- J. Bertran-Vicente, M. Schümann, P. Schmieder, E. Krause and C. P. R. Hackenberger, Org. Biomol. Chem., 2015, 13, 6839–6843 RSC.
- M. Penkert, A. Hauser, R. Harmel, D. Fiedler, C. P. R. Hackenberger and E. Krause, J. Am. Soc. Mass Spectrom., 2019, 30, 1578–1585 CrossRef CAS.
- J. D. Chodera, D. L. Mobley, M. R. Shirts, R. W. Dixon, K. Branson and V. S. Pande, Curr. Opin. Struct. Biol., 2011, 21, 150–160 CrossRef CAS.
- B. J. Williams-Noonan, E. Yuriev and D. K. Chalmers, J. Med. Chem., 2018, 61, 638–649 CrossRef CAS.
- S. Michielssens, J. H. Peters, D. Ban, S. Pratihar, D. Seeliger, M. Sharma, K. Giller, T. M. Sabo, S. Becker, D. Lee, C. Griesinger and B. L. de Groot, Angew. Chem., Int. Ed., 2014, 53, 10367–10371 CrossRef CAS.
- K. Hauser, C. Negron, S. K. Albanese, S. Ray, T. Steinbrecher, R. Abel, J. D. Chodera and L. Wang, Commun. Biol., 2018, 1, 70 CrossRef.
- M. R. Webb, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4884–4887 CrossRef CAS.
- H. R. Matthews, Pharmacol. Ther., 1995, 67, 323–350 CrossRef CAS.
- Y.-F. Wei and H. R. Matthews, in Methods Enzymol., ed. B. M. S. Tony Hunter, Academic Press, 1991, vol. 200, pp. 388–414 Search PubMed.
- F. T. Hofmann, C. Lindemann, H. Salia, P. Adamitzki, J. Karanicolas and F. P. Seebeck, Chem. Commun., 2011, 47, 10335–10337 RSC.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS.
- L. Schrödinger, in Schrödinger Release 2018-1, New York, 2018 Search PubMed.
- N. Homeyer, A. H. C. Horn, H. Lanig and H. Sticht, J. Mol. Model., 2006, 12, 281–289 CrossRef CAS.
- M. Aldeghi, V. Gapsys and B. L. de Groot, ACS Cent. Sci., 2018, 4, 1708–1718 CrossRef CAS.
- M. Aldeghi, B. L. de Groot and V. Gapsys, in Computational Methods in Protein Evolution, ed. T. Sikosek, Springer New York, New York, NY, 2019, pp. 19–47, DOI:10.1007/978-1-4939-8736-8_2.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- D. Seeliger and B. L. de Groot, Biophys. J., 2010, 98, 2309–2316 CrossRef CAS.
- V. Gapsys, S. Michielssens, D. Seeliger and B. L. de Groot, J. Comput. Chem., 2015, 36, 348–354 CrossRef CAS.
- V. Gapsys, S. Michielssens, J. H. Peters, B. L. de Groot and H. Leonov, Calculation of Binding Free Energies, in: Molecular Modeling of Proteins, Methods in Molecular Biology (Methods and Protocols), ed. A. Kukol, Humana Press, New York, NY, 2015, vol. 1215, DOI:10.1007/978-1-4939-1465-4_9.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03930f |
| This journal is © The Royal Society of Chemistry 2020 |