
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NMR studies of group 8 metallodrugs: 187Os-enriched organo-osmium half-sandwich anticancer complex†
Russell J.
Needham
 ,
Ivan
Prokes
,
Abraha
Habtemariam
,
Isolda
Romero-Canelón‡
,
Guy J.
Clarkson
and
Peter J.
Sadler
*
,
Ivan
Prokes
,
Abraha
Habtemariam
,
Isolda
Romero-Canelón‡
,
Guy J.
Clarkson
and
Peter J.
Sadler
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
First published on 24th August 2021
Abstract
We report the synthesis of the organo-osmium anticancer complex [Os(η6-p-cym)(N,N-azpy-NMe2)Br]PF6 (1) containing natural abundance 187Os (1.96%), and isotopically-enriched (98%) [187Os]-1. Complex 1 and [187Os]-1 contain a π-bonded para-cymene (p-cym), a chelated 4-(2-pyridylazo)-N,N-dimethylaniline (azpy-NMe2), and a monodentate bromide as ligands. The X-ray crystal structure of 1 confirmed its half-sandwich ‘piano-stool’ configuration. Complex 1 is a member of a family of potent anticancer complexes, and exhibits sub-micromolar activity against A2780 human ovarian cancer cells (IC50 = 0.40 μM). Complex [187Os]-1 was analysed by high-resolution ESI-MS, 1D 1H and 13C NMR, and 2D 1H COSY, 13C–1H HMQC, and 1H–187Os HMBC NMR spectroscopy. Couplings of 1H and 13C nuclei from the azpy/p-cym ligands to 187Os were observed with J-couplings (1J to 4J) ranging between 0.6–8.0 Hz. The 187Os chemical shift of [187Os]-1 (−4671.3 ppm, determined by 2D 1H–187Os HMBC NMR) is discussed in relation to the range of values reported for related Os(II) arene and cyclopentadienyl complexes (−2000 to −5200 ppm).
Introduction
There is much current interest in the design of transition metal anticancer complexes.1–4 Progress depends on the elucidation of structure–activity relationships and their mechanisms of action, depends on the identification of their pharmacophores, the active species. Although some metal complexes are relatively inert (e.g. low-spin d6 complexes), many are pro-drugs which will undergo ligand exchange and redox reactions before reaching the target site. NMR is a potentially powerful method for investigating metallodrug speciation, since studies can be carried out in solution under physiologically relevant conditions, and information on the thermodynamics (equilibria) and kinetics (dynamics) of ligand exchange reactions can often be obtained. Direct observation of NMR resonances for the metals themselves (heteronuclear NMR) is potentially very informative, but often difficult to achieve.We focus here on group 8 transition metals, Fe, Ru and Os. In particular two Ru(III) complexes, NAMI-A ([trans-RuCl4(DMSO-S)(Im)]ImH, where Im = imidazole) and KP1019 ([trans-RuCl4(In)2]InH, where In = indazole) have been on clinical trials.5 Ru(III) complexes are paramagnetic, but diamagnetic low-spin 4d6 Ru(II) complexes, can in principle, be studied by 99Ru and 101Ru NMR, Table 1. Their natural abundances are reasonable (12.76% and 17.06%, respectively), and although their gyromagnetic ratios are relatively low, they should be detectable with similar sensitivities as 13C. However, both are quadrupolar with I = ![[/]](https://www.rsc.org/images/entities/char_e11f.gif) ; 101Ru has a higher quadrupole moment than 99Ru. Both isotopes give rise to broad lines unless in highly symmetrical complexes. The complexes [Ru(NH3)6]Cl2 and K4[Ru(CN)6] (notably with a temperature coefficient for the chemical shift of >1 ppm K−1) give narrow resonances and are useful references.699Ru solution studies on such symmetrical complexes are also possible.7 For tris-(polypyridyl)Ru(II) complexes in acetonitrile, a correlation between the 99Ru-NMR chemical shifts and the energy of metal-to-ligand charge transfer has been reported.8 The chemical shift range for 99Ru is >9000 ppm. The 99Ru NMR linewidth of 0.15 M [RuII(bipyridine)3]PF6 in CD3CN is 65 Hz, and for other polypyridyl complexes up to 800 Hz, depending on the counter-anion and concentration.9 There is medical interest in related complexes as photodynamic agents for the treatment of cancer. TLD-1433, [Ru(4,4′-dimethyl-2,2′-bipyridine)2(2-(2′,2′′:5′′,2′′′-terthiophene)-imidazo[4,5-f][1,10]phenanthroline)]Cl2, an octahedral tris-diimine Ru(II) complex with two methylated bipyridyl and one phenanthroline derivative as ligands, is activated by green light and currently on clinical trial for treatment of non-muscle invasive bladder cancer (NMIBC), which is refractory to Bacillus Calmette–Guérin (BCG) treatment.10,11 Complexes of lower symmetry, such as half-sandwich Ru(II) arene anticancer complexes give resonances which are usually too broad to observe easily (unpublished data).
; 101Ru has a higher quadrupole moment than 99Ru. Both isotopes give rise to broad lines unless in highly symmetrical complexes. The complexes [Ru(NH3)6]Cl2 and K4[Ru(CN)6] (notably with a temperature coefficient for the chemical shift of >1 ppm K−1) give narrow resonances and are useful references.699Ru solution studies on such symmetrical complexes are also possible.7 For tris-(polypyridyl)Ru(II) complexes in acetonitrile, a correlation between the 99Ru-NMR chemical shifts and the energy of metal-to-ligand charge transfer has been reported.8 The chemical shift range for 99Ru is >9000 ppm. The 99Ru NMR linewidth of 0.15 M [RuII(bipyridine)3]PF6 in CD3CN is 65 Hz, and for other polypyridyl complexes up to 800 Hz, depending on the counter-anion and concentration.9 There is medical interest in related complexes as photodynamic agents for the treatment of cancer. TLD-1433, [Ru(4,4′-dimethyl-2,2′-bipyridine)2(2-(2′,2′′:5′′,2′′′-terthiophene)-imidazo[4,5-f][1,10]phenanthroline)]Cl2, an octahedral tris-diimine Ru(II) complex with two methylated bipyridyl and one phenanthroline derivative as ligands, is activated by green light and currently on clinical trial for treatment of non-muscle invasive bladder cancer (NMIBC), which is refractory to Bacillus Calmette–Guérin (BCG) treatment.10,11 Complexes of lower symmetry, such as half-sandwich Ru(II) arene anticancer complexes give resonances which are usually too broad to observe easily (unpublished data).
| Metal | Isotope | Abundance (%) | Nuclear spin (I) | Quadrupole momenta | Frequencyb | Receptivityc (Rel. to 13C) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Electric quadrupole moment in units of fm2 (10−30 m2; 1 barn = 100 fm2). b 1H 400 MHz, 13C 100 MHz. c Receptivity at natural abundance relative to 13C. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Iron | 57Fe | 2.119 | 1/2 | — | 12.955 | 4.25 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ruthenium | 99Ru | 12.76 | 5/2 | 7.9 | 16.427 | 0.85 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 101Ru | 17.06 | 5/2 | 45.7 | 12.750 | 1.58 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Osmium | 187Os | 1.96 | 1/2 | — | 9.132 | 1.43 × 10−3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 189Os | 16.15 | 3/2 | 85.6 | 31.072 | 2.32 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Although both Ru(II) and Ru(III) complexes have been widely explored as anticancer agents, similar studies of osmium complexes are more recent.13–15 Os(VI) complexes, of general formula [Os{N}(N^N)Cl3] (where N^N = 2,2′-bipyridine or a phenanthroline derivative), can attack DNA and induce endoplasmic reticulum stress, probably through protein binding.16,17 Half sandwich diamine Os(II) arene anticancer complexes can also bind to DNA and inhibit DNA synthesis,18–20 whereas azopyridine complexes are more inert and have a redox mechanism of action. Particularly potent is the iodido complex [Os(η6-p-cym)(N,N-azpy-NMe2)I]PF6 (FY26; p-cym = para-cymene, azpy-NMe2 = 2-(p-((dimethylamino)phenylazo)pyridine), which is ca. 49× more active on average than the anticancer drug cisplatin towards a Sanger Institute panel of over 800 cancer cell lines, and induces formation of reactive oxygen species (ROS) in cancer cells.21,22 This complex is capable of delaying the growth of HCT-116 human colon cancer xenografts in mice,23 and is up to 64× more selective towards A2780 ovarian cancer cells over normal cells when applied synergistically with L-buthionine sulfoximine, which blocks glutathione (γ-L-Glu-L-Cys-Gly, GSH) synthesis.24 The complex FY26 is inert to hydrolysis, but undergoes activation in cancer cells by attack on the azo bond by the intracellular thiol GSH, resulting in liberation of the iodido ligand.25
Additional current interest in organo-osmium(II) half sandwich complexes arises from their activity as transfer hydrogenation catalysts. For example, chiral arene (p-toluenesulfonyl)-1,2-diphenylethylenediamine (TsDPEN) complexes, [Os(arene)(TsDPEN)], are effective catalysts for the conversion of the natural metabolite pyruvate into either natural L-lactate or unnatural D-lactate depending on the enantiomer of the catalyst used, reactions of which can be achieved even inside cancer cells.26,27
Osmium NMR studies are highly challenging, firstly, the natural abundance in the Earth's crust is very low, ca. 1 g per 200 tonnes, making it amongst the rarest stable elements in the periodic table.28 There are seven stable naturally-occurring osmium isotopes:29184Os (0.02%), 186Os (1.59%), 187Os (1.96%), 188Os (13.24%), 189Os (16.15%), 190Os (26.26%), 192Os (40.78%), one of which (186Os) is a radioisotope with a half-life of 2.011 × 1015 years (α-decay), and for practical purposes is considered ‘stable’. Of the stable isotopes, 187Os and 189Os possess a nuclear spin (I = ½ and ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) , respectively), making them both magnetically receptive.30189Os has a high quadrupole moment (Q = 0.91) and low gyromagnetic ratio (Table 1), making it relatively insensitive to detection (1.43 × 10−3 at natural abundance relative to 13C), due to quadrupolar relaxation. 189Os is therefore unsuitable for most NMR studies, with the exception of highly symmetrical molecules like 189OsO4, which yields a broad signal.
, respectively), making them both magnetically receptive.30189Os has a high quadrupole moment (Q = 0.91) and low gyromagnetic ratio (Table 1), making it relatively insensitive to detection (1.43 × 10−3 at natural abundance relative to 13C), due to quadrupolar relaxation. 189Os is therefore unsuitable for most NMR studies, with the exception of highly symmetrical molecules like 189OsO4, which yields a broad signal.
187Os is a radioactive decay product of 187Re (half-life of 4.56 × 1010 years) and has been studied extensively in the dating of terrestrial and meteoric rocks, and hydrocarbon deposits.31 It is an I = ½ nucleus capable of yielding sharp NMR peaks over a chemical shift range of >5000 ppm. Bell et al. demonstrated that the use of polarisation transfer techniques to observe J(187Os–1H/31P) couplings can greatly enhance the sensitivity of 187Os detection, with maximum gains of 1300 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for 31P and 1H-detected spectra.32 This polarisation transfer approach had been applied also to μ-hydrido and Cp complexes.32–34 Bell et al. utilised one-bond 187Os–1H (hydride) couplings of ca. 70 Hz, and 187Os–31P couplings to phosphines of ca. 270 Hz, to detect 187Os NMR peaks for 37 [Os(arene)X2(PR3)] complexes. In a previous study on the biphenyl sandwich complex [Os(η6-bip)2](OTf)2, we used 1H/187Os heteronuclear multiple bond correlation (HMBC) spectroscopy to observe the 187Os resonance for a 25 mM solution of the complex via the small 187Os–1H couplings to arene ring protons.35
000 for 31P and 1H-detected spectra.32 This polarisation transfer approach had been applied also to μ-hydrido and Cp complexes.32–34 Bell et al. utilised one-bond 187Os–1H (hydride) couplings of ca. 70 Hz, and 187Os–31P couplings to phosphines of ca. 270 Hz, to detect 187Os NMR peaks for 37 [Os(arene)X2(PR3)] complexes. In a previous study on the biphenyl sandwich complex [Os(η6-bip)2](OTf)2, we used 1H/187Os heteronuclear multiple bond correlation (HMBC) spectroscopy to observe the 187Os resonance for a 25 mM solution of the complex via the small 187Os–1H couplings to arene ring protons.35
Here we have synthesised the bromido analogue of the iodido complex FY26, [Os(η6-p-cym)(N,N-azpy-NMe2)Br]PF6 (1), and determined its X-ray crystal structure. Also we have synthesised on a milligram scale, the 187Os isotopically-enriched complex [187Os]-1, carried out multinuclear NMR and MS studies, and determined the antiproliferative activity of 1 towards human cancer cells. This appears to be the first report of NMR studies on a 187Os-enriched complex.
Results and discussion
Synthesis and characterisation
The synthesis of the 187Os isotopically-enriched complex, [187Os(η6-p-cym)(N,N-azpy-NMe2)Br]PF6 ([187Os]-1), from ca. 50 mg metallic osmium-187 (98% enriched) was achieved in five steps based on previously reported methods (Scheme 1), with an overall yield of 41%.36,37 The non-enriched complex 1 was synthesised from K2[OsO2(OH)4], also via reported methods, and is analogous to the highly active complex [Os(η6-p-cym)(N,N-azpy-NMe2)I]PF6 (FY26),21 with bromide replacing iodide as the monodentate ligand. | ||
| Scheme 1 The multi-step synthesis of [187Os(η6-p-cym)(N,N-azpy-NMe2)Br]PF6 with 187Os isotopic enrichment, starting from 187Os metal powder. | ||
The X-ray crystal structure of complex 1 shows that it adopts a similar pseudo-octahedral three-legged piano-stool geometry to its analogue, FY26, (Fig. 1, Table S1†).25 Osmium(II) is π-bonded to a p-cym ligand, and coordinated to a monodentate bromide and a bidentate azopyridine ligand, which constitute the three legs of the piano-stool. The complex crystallises as a racemate owing to the chiral Os centre, and contains a PF6− counter-anion.
 | ||
| Fig. 1 ORTEP diagram for the cation in the X-ray crystal structure of [Os(η6-p-cym)(azpy-NMe2)Br]PF6. Ellipsoids are shown at the 50% probability level and all hydrogens, counter ions and solvent molecules have been omitted for clarity. | ||
Anticancer activity
Similarly to FY26, complex 1 exhibits potent sub-micromolar antiproliferative activity against A2780 human ovarian cancer cells (IC50 = 0.40 ± 0.01 μM, 24 h drug exposure, 72 h recovery) and probably has a similar mechanism of action, inducing formation of reactive oxygen species (ROS) and perturbing the redox balance in cancer cells. The IC50 follows an expected trend of activity for change in the monodentate ligand: I > Br > Cl, whereby the iodido complex is the most active and the chlorido complex is the least, due to weakening of the Os–X bond decreasing stability, and hence greater drug deactivation before reaching the target sites (Table S2†). This trend was observed previously for a related bromido Os(II) arene azopyridine complex and its halido analogues.38 The racemic nature of the complex would be expected to have little effect on activity, since previous studies on the separated enantiomers of FY26 showed that the chiral centre does not significantly change the anticancer activity.39Mass spectrometry
The observed and calculated positive-ion high resolution mass spectra of natural abundance complex 1 ([M − PF6]+; [C23H28N4BrOs]+) and [187Os]-1 ([M − PF6]+; [C23H28N4Br187Os]+) are compared in Fig. 2A and B, respectively. The spectrum of enriched [187Os]-1 is greatly simplified due to the presence of predominantly only one osmium isotope (187Os) instead of 4 additional isotopes (188Os 13.24%, 189Os 16.15%, 190Os 26.26%, and 192Os 40.78%). The presence of 79Br (natural abundance 51%) and 81Br (49%) isotopes is apparent.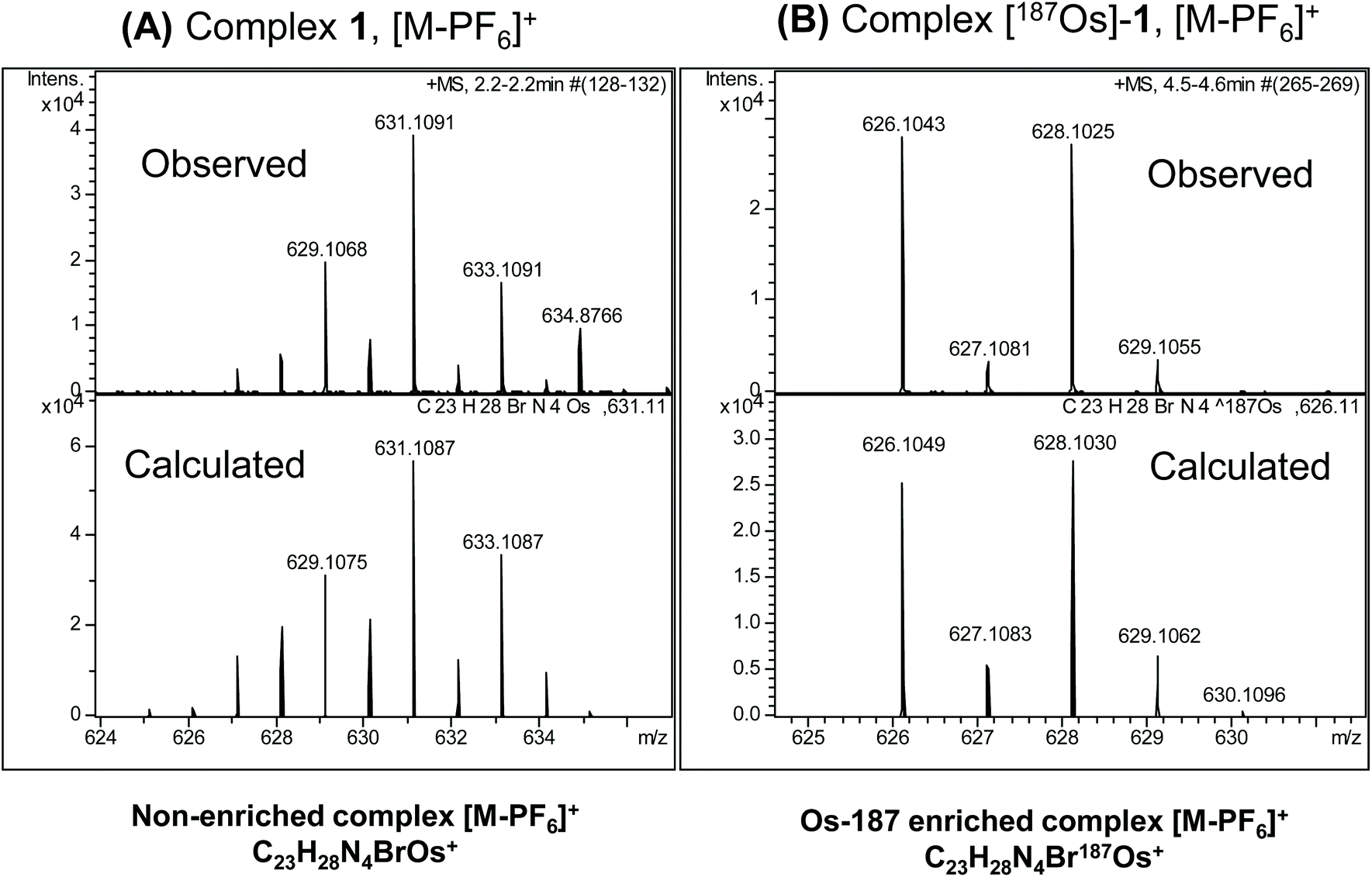 | ||
| Fig. 2 Observed and calculated positive-ion high resolution mass spectra of (A) natural abundance complex 1 ([M − PF6]+; [C23H28N4BrOs]+), and (B) 187Os-enriched [187Os]-1 ([M − PF6]+; [C23H28N4Br187Os]+). The simplification of the spectrum on enrichment due to the presence of predominantly only one osmium isotope (187Os) together with 79Br (51%) and 81Br (49%) is apparent. | ||
1H NMR
Complexes 1 and [187Os]-1 were prepared in MeCN-d3 (∼10 mg mL−1) and 1H COSY NMR spectra were recorded on 400 and 700 MHz instruments (Fig. 3), and the resonances assigned. To resolve coupling interactions between 187Os and 1H nuclei, 250 MHz 1H NMR spectra of the samples were also recorded (Fig. 4). As expected, no 187Os–1H coupling interactions were observed for 1 due to the low natural abundance of 187Os (1.96%). However, the 1H spectrum of [187Os]-1 shows the presence of 3- and 4-bond couplings between 187Os and 1Ha (3J = 1.7 Hz), and 1Hb (4J = 0.6 Hz). There was also a small unresolved coupling to 1Hd; the resolution was not sufficient to measure 4J. The resonance for proton 1Hc is overlapped with other resonances, although the 5J value might be expected to be very small compared to the linewidth.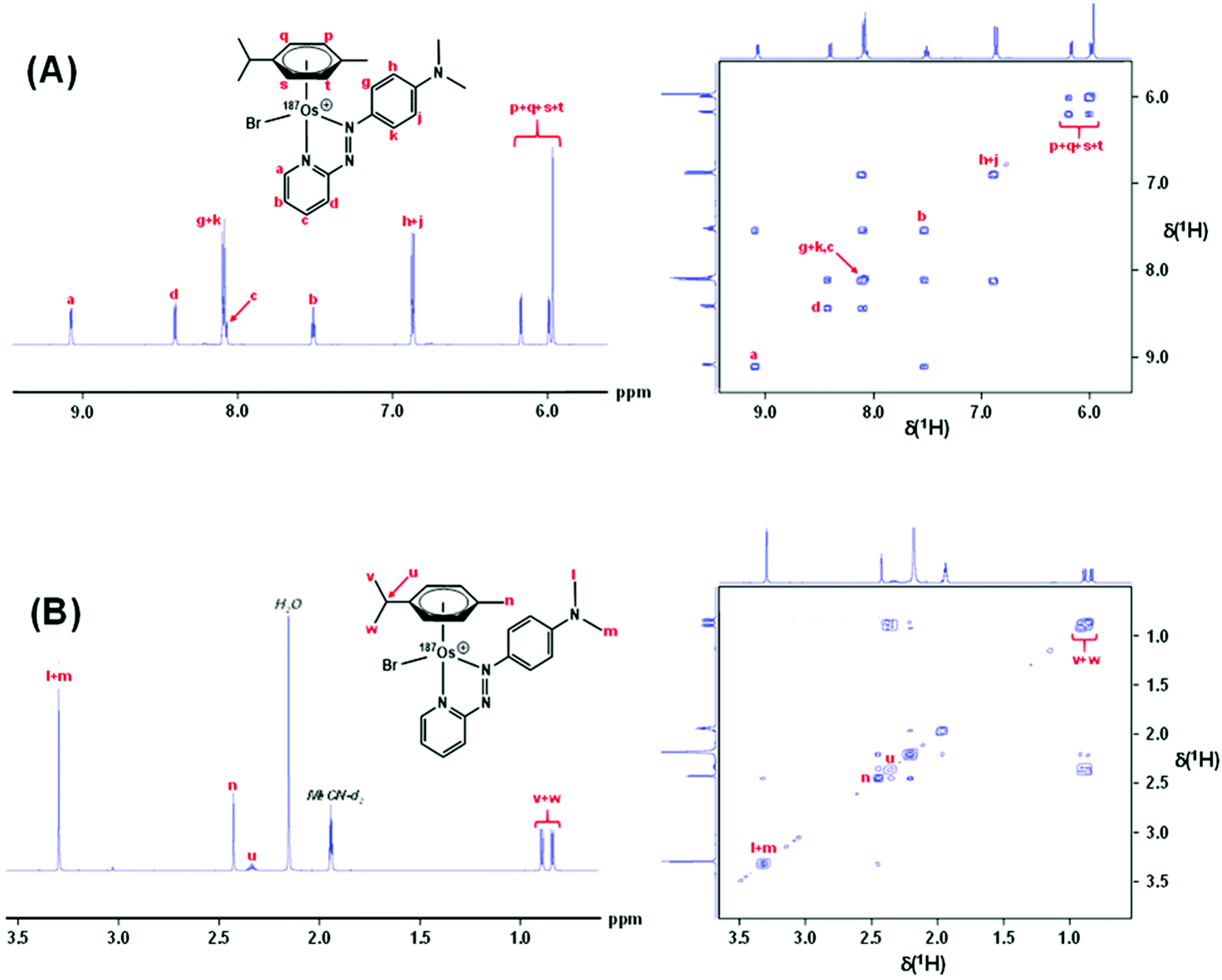 | ||
| Fig. 3 700 MHz 1H NMR spectrum of [187Os]-1 (LHS), and 400 MHz 2D 1H COSY NMR spectrum of 1 with all 1H resonances assigned. (A) The aromatic region, and (B) the aliphatic region. Samples were prepared in MeCN-d3. | ||
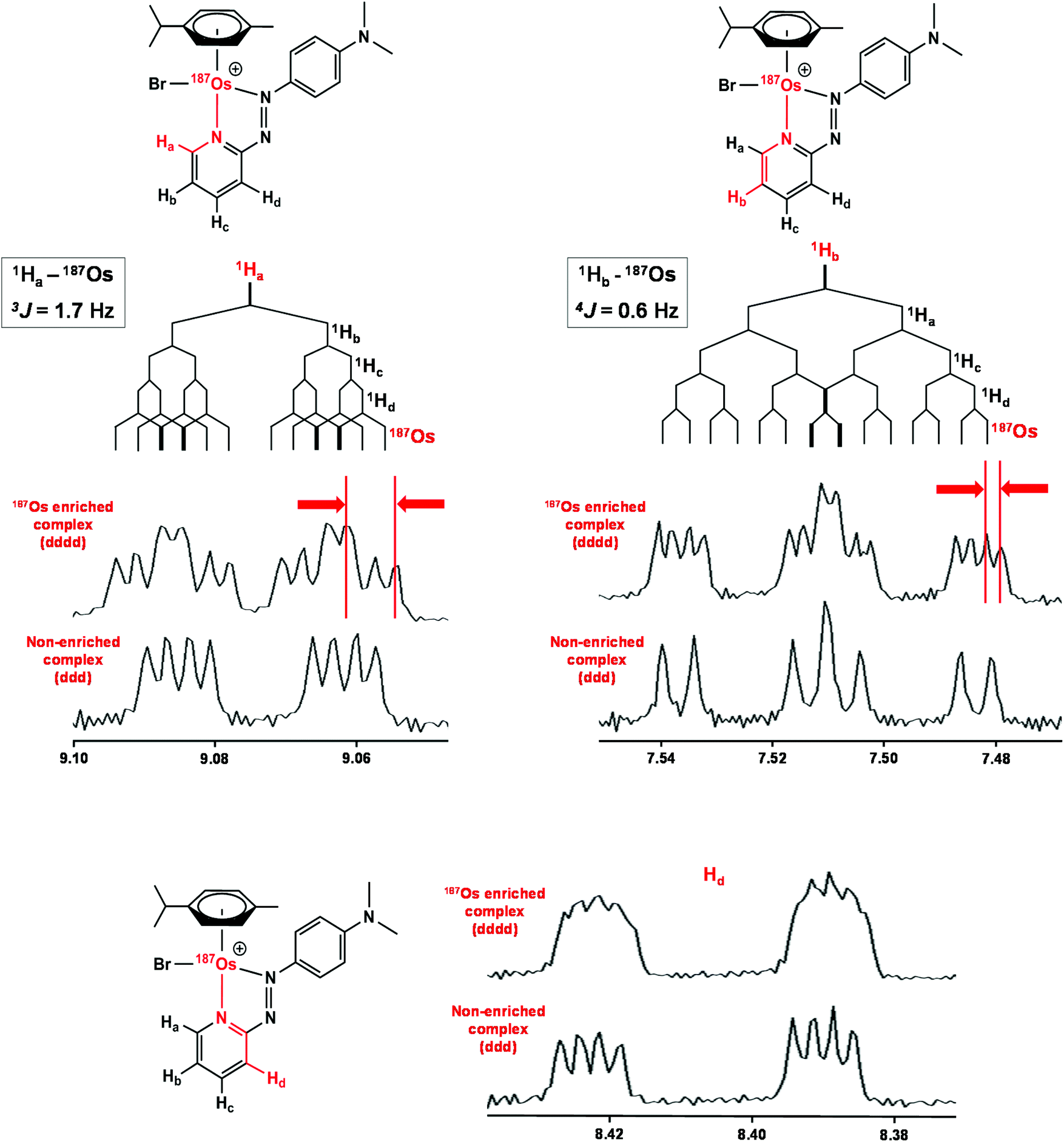 | ||
| Fig. 4 1H NMR (250 MHz) spectra of [187Os]-1 compared to natural abundance 1, with expansions of the resonances for 1Ha, 1Hb, and 1Hd illustrating their couplings to other protons and 187Os. Samples were prepared in MeCN-d3. | ||
13C NMR
The samples used for 1H NMR studies were also analysed by 13C NMR, utilising a J-MOD sequence (Fig. 5). To aid characterisation of the resonances, a 13C–1H HMQC NMR spectrum was recorded (Fig. S1†), and all 23 13C resonances were observed and characterised in the spectrum. Furthermore, expansions of some resonances reveal distinct 187Os–13C couplings for [187Os]-1 (Fig. 6). Short-range couplings are observed between 187Os and 13Cp, 13Cq, 13Cs, and 13Ct (1J = 7.2–8.0 Hz), which comprise the four CH carbons of the arene ring of the p-cym ligand, and between 187Os and the two tertiary carbons of the ring, 13Co and 13Cr (1J = 4.7 and 5.9 Hz, respectively). Furthermore, longer range couplings are observed between 187Os and 13Ce and 13Cf (2J = 2.4 and 5.0 Hz, respectively), and between 187Os and 13Cb (3J = 1.8 Hz), which comprise 13C resonances of the bidentate azpy ligand. Table 2 lists all the observed 187Os–1H and 187Os–13C couplings. | ||
| Fig. 5 176 Hz 13C NMR spectrum of [187Os]-1 using the J-MOD sequence, with 13C assignments. The sample was prepared in MeCN-d3. C and CH2 peaks are upright, CH and CH3 peaks inverted. | ||
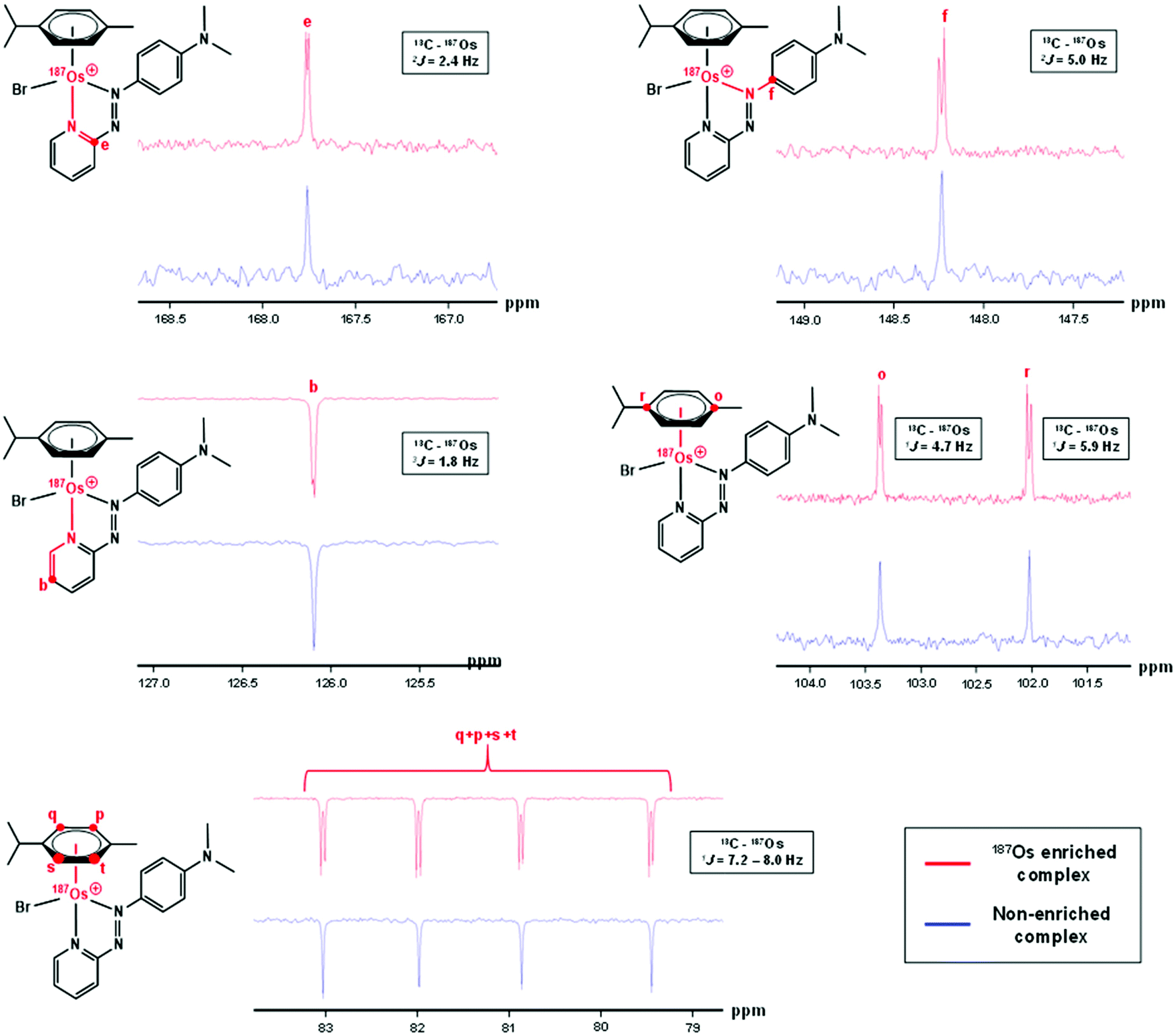 | ||
| Fig. 6 176 MHz 13C NMR spectra of [187Os]-1 compared to natural abundance 1 (150 MHz), with expansions of resonances for 13Cb, 13Ce, 13Cf, 13Co, 13Cp, 13Cq, 13Cr, 13Cs, and 13Ct, which all exhibit couplings to 187Os. Samples were prepared in MeCN-d3. | ||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coupling | J (Hz) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a The signs of the coupling constants have not been determined. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 J(1Ha – 187Os) | 1.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 J(1Hb – 187Os) | 0.6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 J(13Co – 187Os) | 4.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 J(13Cr – 187Os) | 5.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 J(13Cq,p,s,t – 187Os) | 7.2–8.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 J(13Ce – 187Os) | 2.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 J(13Cf – 187Os) | 5.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 J(13Cb – 187Os) | 1.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

1H–187Os HMBC
Utilising 1H–187Os HMBC, we were able to observe the 187Os resonance of [187Os]-1 and determine its chemical shift relative to OsO4. The resonance was observed at −4671.3 ppm through correlation with aromatic proton Ha (pyridyl ring), and aliphatic protons from the arene ligand, Hn (Fig. 7). Attempts to observe directly the 187Os resonance of [187Os]-1 were unsuccessful (Fig. S2†). The very low gyromagnetic ratio of 187Os makes direct observation difficult, despite the complex being isotopically enriched. Fig. 8 shows a comparison of 187Os chemical shifts for various organo-osmium complexes with arene and cyclopentadienyl ligands. The comparatively low chemical shift of the (high-field-shifted) 187Os resonance of [187Os]-1, similar to a bis-biphenyl arene Os(II) sandwich complex, suggests a highly shielded 187Os nucleus, perhaps surprising on account of the presence of the strong π-acceptor azopyridine ligand, although the comparator complexes mostly contain phosphines, or hydride ligands. Future work is required to elucidate the relationship between the electron densities in these complexes and the 187Os chemical shifts, e.g. DFT calculations.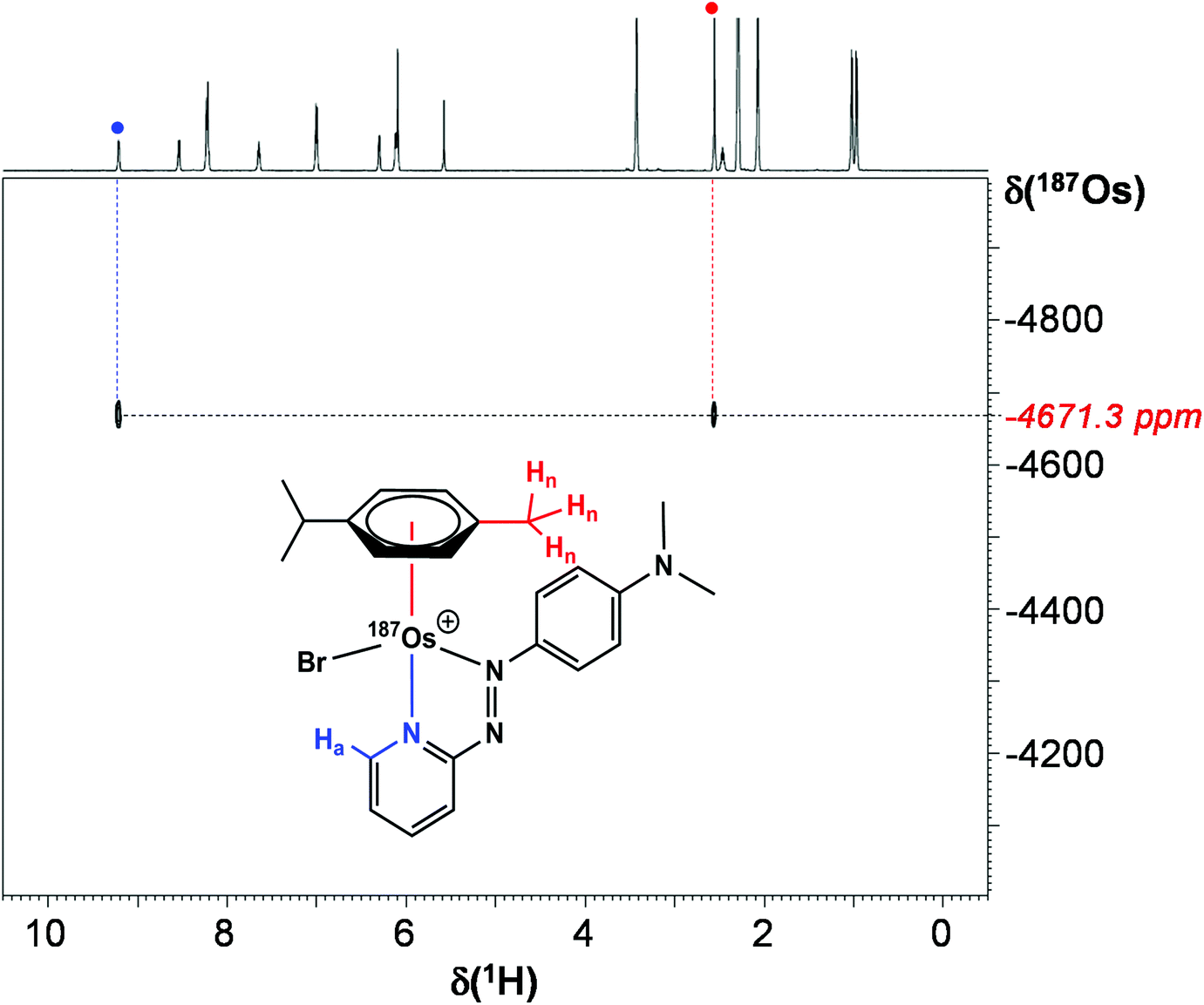 | ||
| Fig. 7 2D 1H–187Os HMBC NMR spectrum of [187Os]-1 in MeCN-d3. | ||
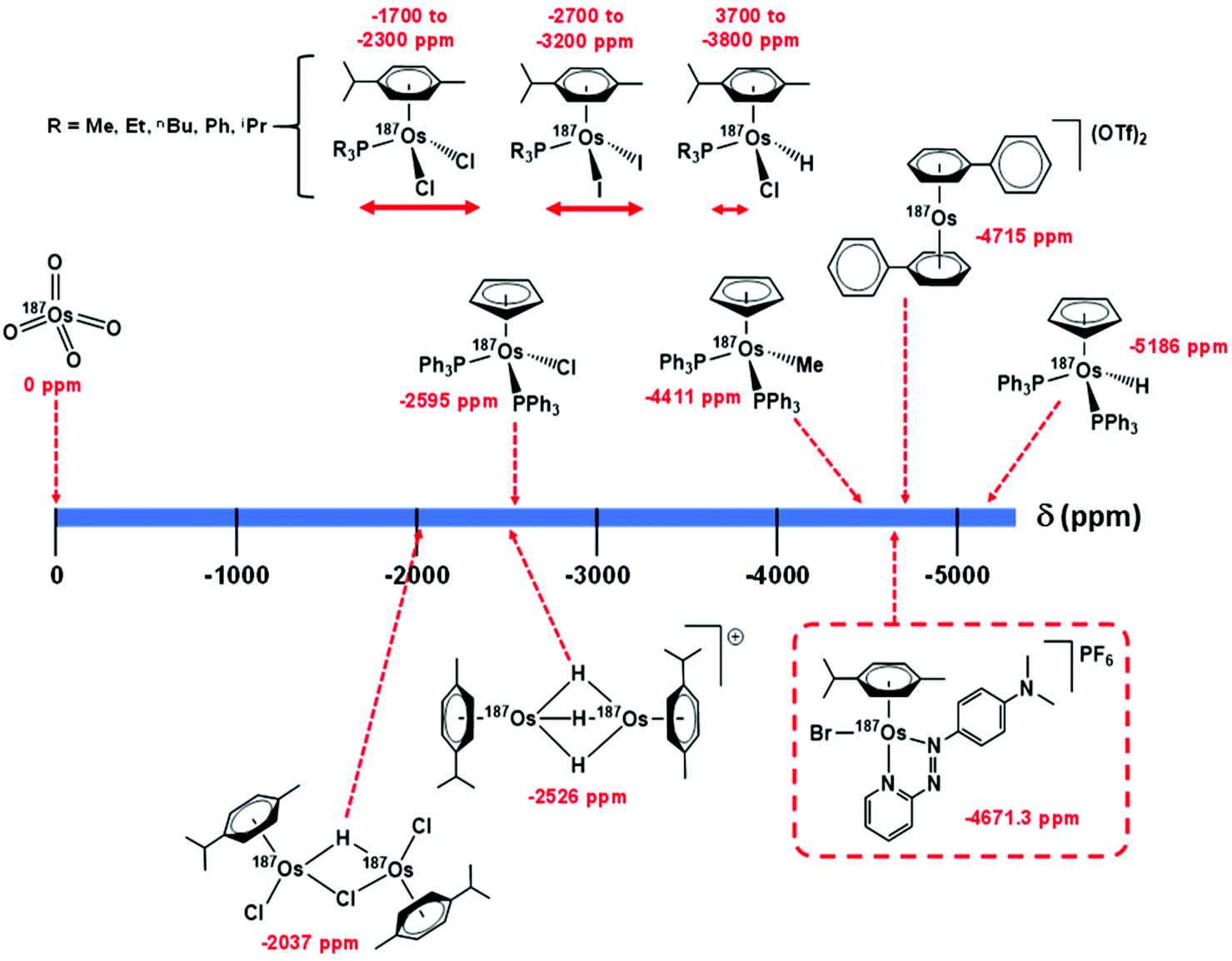 | ||
| Fig. 8 Comparison of 187Os chemical shifts for [187Os]-1 in comparison with values for arene and cyclopentadienyl Os(II) complexes in the literature.33–35,40 Shifts are either referenced directly to OsO4 or indirectly from the proton frequency of TMS. | ||
Heteronuclear NMR for metallodrugs
Most NMR spectroscopic studies of metallodrugs involve nuclei which are the most sensitive to detection. Only a few NMR nuclei offer the sensitivity for studies at pharmacological concentrations. These include 1H, 19F, 31P, and 13C and 15N with enrichment and detection by polarisation transfer methods. Studies of NMR-active isotopes of metals present significant challenges.41–43Sensitivity is a function of the size of the gyromagnetic ratio (γ, proportional to the frequency of detection of resonances), the natural abundance, and the applied magnetic field (B0). NMR signal intensity is proportional to B3/20γ5/2. In general, as nuclei become heavier, they have smaller gyromagnetic ratios and they become less sensitive to detection. The detection sensitivity of low abundance nuclei such as 13C and 15N, which are often coupled to more sensitive 1H nuclei, can be greatly enhanced by use of spin-polarisation-transfer pulse sequences such as heteronuclear single quantum coherence (HSQC), e.g. for 15N by up to a factor of 306 (|γ1H|/|γ15N|)5/2.
All elements in the periodic table from atomic number 1 to 103 have at least one isotope which possesses nuclear spin.29,44 Radioactivity can be a problem for some elements, although shielding by glass is adequate for tritium (3H), which has a higher gyromagnetic ratio than 1H and is 1.21× more sensitive to detection. A more common problem is that nuclei with spin quantum numbers >½ (quadrupolar nuclei) in environments that are not symmetrical, usually suffer line broadening via interaction with electric field gradients. For nuclei with high quadrupole moments (e.g.197Au), this can lead to extreme broadening making resonances very difficult, and sometimes almost impossible to detect. Line broadening can also be severe for paramagnetic metal complexes, especially complexes where the unpaired spins have long electron spin relaxation times (those detectable by EPR at ambient temperature), and when there are exchange processes which occur at intermediate rates on the NMR timescale. The resonances of heavier nuclei can be broadened by chemical shift anisotropy relaxation mechanisms (e.g.195Pt in square-planar complexes), which also broadens 195Pt satellites in 1H NMR spectra of Pt(II) ligands. Such broadening is proportional to B20 so can make such satellites very broad at high frequencies (e.g. at 600 MHz versus 300 MHz).45
There is also current pharmacological interest in the group 8 metal iron. Ferrocene in particular shows promise as a fragment for conjugation in drug design.46,47 Ferroquine is on clinical trial as an antimalarial drug,48 and ferrocifens (tamoxifen derivatives) have promising anticancer activity especially towards breast cancer cells possessing the estrogen receptor (ER+).4957Fe is an I = ½ nucleus, but the abundance is low (2.1%), as is the gyromagnetic ratio, Table 1. With polarisation transfer from 1H, utilising the small two-bond Cp (C–H) 1H–57Fe coupling of ca. 0.5 Hz, 57Fe resonances can readily be detected for concentrated solutions of ferrocene (ca. 0.4 M).5057Fe chemical shifts have been used to study the donor properties of amino groups on coordinated Cp rings.5157Fe isotopic enrichment (to ca. 90%) allows detection at millimolar concentrations, e.g. for haem proteins.52 Enrichment combined with polarisation-transfer methods should allow detection at pharmacological concentrations of relevance to metallodrug activity. However, oxidation to paramagnetic 3d5 Fe(III) is likely to lead to severe broadening and loss of 57Fe resonances.
Conclusions
NMR is a versatile technique for the study of metallodrugs in both the solid state and solution. Here we have focussed on solution studies, which can not only provide structural information, but also insights into both the thermodynamics and kinetics of ligand exchange reactions. Such information is vital for identifying pharmacologically active species. The common spin-½ nuclei 1H, 19F and 31P are sensitive enough for NMR studies at physiologically relevant concentrations (often sub-millimolar), and so too are 13C and 15N in isotopically-enriched compounds, especially when polarisation transfer methods such as HSQC are used. In contrast, I > ½ (quadrupolar) nuclei usually give rise to broad peaks which limits their usefulness at low concentrations.Platinum anticancer drugs are now the most widely used drugs in cancer chemotherapy, and NMR studies of the 33.4% abundant I = ½ isotope 195Pt have proved useful for studies of the activation of cisplatin and related drugs by hydrolysis and subsequent attack of DNA target sites, as well as the reduction of Pt(IV) prodrugs to Pt(II).4,41,43,53 Currently, there is much interest in the discovery of active anticancer complexes of other transition metals, and we have highlighted here the potential for group 8 of the periodic table, iron, ruthenium, and osmium. Complexes of ruthenium have already entered clinical trials,54 and one is in clinical trials as a photosensitizer for cancer treatment.10 However, ruthenium has only quadrupolar nuclei and direct studies of these drugs by Ru NMR are difficult. The heavier congener, osmium, on the other hand has a useful I = ½ isotope 187Os, although with a low natural abundance and low gyromagnetic ratio (Table 1).
We have shown here that isotopic enrichment of a potent anticancer complex [187Os(η6-p-cym)(N,N-azpy-NMe2)Br]PF6 to >98% 187Os allows facile detection of its 187Os resonance NMR using 2D 1H–187Os HMBC, and accompanying 1H and 13C NMR studies allow detection for the first time of 1J, 2J, 3J and 4J coupling constants for both the arene ring and the chelated azopyridine ligand, ranging from 0.6–8 Hz. However, despite isotopic enrichment, direct observation of the 187Os resonance (without the use of spin-polarisation-transfer techniques) was not achievable due to its very low gyromagnetic ratio. The synthesis of the enriched complex was achieved in good yield over 5 steps from metallic 187Os, the first step of which involved conversion to 187OsO4, a highly toxic volatile compound generated in a KOH/KNO3 melt at >350 °C. The mechanism of anticancer activity of this class of organo-osmium arene anticancer complexes appears to be different from that of platinum anticancer drugs, and further studies using such 187Os-enriched complexes are likely to provide new insight into their reactions in biological media and the nature of their target sites.
Experimental
Materials
Synthesis
:H2O (3:1, v/v, 3 × 15 mL). The clean precipitate was mixed with EtOH (10 mL), HBr (48%, v/v, 5 mL) and heated under reflux for 18 h. α-Terpinene (501 mg, 3.678 mmol) was added and the mixture was refluxed for a further 18 h. The product was extracted with CH2Cl2 (25 mL) and washed with H2O (3 × 20 mL). The solution was concentrated to ∼3 mL under reduced pressure and a brown precipitate formed, which was placed in a freezer overnight (253 K). The precipitate was collected via vacuum filtration and washed with EtOH (5 mL) and Et2O (5 mL). Yield (53.1 mg, 41%). No analysis was conducted at this stage to avoid loss of valuable 187Os. Note: Extreme care must be taken with the handling of the OsO4 intermediate (Scheme 1).
Natural abundance [Os(η6-p-cym)Br2]2 was synthesised from K2[OsO2(OH)4] using a previously reported method.37
Natural abundance [Os(η6-p-cym)(N,N-azpy-NMe2)Br]PF6
Synthesised using the same procedure as above with; [Os(η6-p-cym)Br2]2 (58.6 mg, 60.548 μmol), N,N-dimethyl-4-(2-pyridylazo)aniline (30.1 mg, 133.120 μmol), and NH4PF6 (98.6 mg, 0.605 mmol). Yield: (79.9 mg, 85%). 1H NMR 250 MHz (CD3CN): δ 9.07 (ddd, 1H, J = 5.8, 1.4, 0.6 Hz), 8.41 (ddd, 1H, J = 8.2, 1.3, 0.6 Hz), 8.05–8.13 (m, 3H), 7.51 (ddd, 1H, J = 7.5, 5.9, 1.4 Hz), 6.84–6.90 (m, 2H), 6.16–6.18 (m, 1H), 5.96–6.00 (m, 3H), 3.30 (s, 6H), 2.43 (s, 3H), 2.34 (sept., 1H, J = 6.9 Hz), 0.90 (d, 3H, J = 6.9 Hz), 0.85 (d, 3H, J = 6.9 Hz). 13C NMR 150 MHz (CD3CN): δ 167.75 (C), 156.56 (C), 154.20 (CH), 148.23 (C), 142.31 (CH), 130.33 (CH), 126.09 (CH), 124.41 (CH), 113.10 (CH), 103.36 (C), 102.01 (C), 83.02 (CH), 81.98 (CH), 80.86 (CH), 79.44 (CH), 40.83 (CH3), 32.08 (CH), 22.27 (CH3), 22.06 (CH3), 19.63 (CH3). ESI-MS calculated for C23H28BrN4Os+: m/z 631.1087. Found: 631.1091. CHN analysis: Found: C, 35.37%; H, 3.44%; N, 7.37%. Calculated for C23H28BrF6N4OsP: C, 35.62%; H, 3.64%; N, 7.22%.X-ray crystal structure
A single crystal of complex 1 was grown by slow evaporation of a methanolic solution (∼3 mg mL−1), and its molecular structure was determined by X-ray crystallography (Fig. 1). Diffraction data were collected on an Oxford Diffraction Gemini four-circle system with a Ruby CCD area detector. The structure was refined by full-matrix least squares against F2 using SHELXL 97 and solved by direct methods using SHELXS(TREF) with additional light atoms found by Fourier methods.55 Hydrogen atoms were added at calculated positions and refined using a riding model. Anisotropic displacement parameters were used for all non-H atoms; H-atoms were given an isotropic displacement parameter equal to 1.2 (or 1.5 for methyl and NH H-atoms) times the equivalent isotropic displacement parameter of the atom to which they are attached. The data were processed by the modelling program Mercury 1.4.1. X-ray crystallographic data for complex 1 have been deposited in the Cambridge Crystallographic Data Centre under the accession number CCDC 2067332.†Mass spectrometry
Electrospray mass spectra were obtained using a Bruker MaXis UHR-ESI-TOF instrument. Samples of 1 and [187Os]-1 were prepared in methanol and analysed in positive ion mode (500–1000 m/z).NMR spectroscopy
1H and 13C NMR spectra were acquired in 5 mm NMR tubes at 298 K on Bruker AV-250, AV-400, AV-600 or AV-700 spectrometers in MeCN-d3. Data processing was carried out using TOPSPIN version 2.1 (Bruker UK Ltd). 1H NMR chemical shifts were internally referenced to TMS via the residual solvent peak: acetonitrile (δ = 1.94 ppm).56 Likewise for 13C NMR chemical shifts: acetonitrile (δ = 1.32 ppm). 1D 1H NMR spectra were recorded using standard pulse sequences and 1D 13C NMR spectra were recorded using a J-MOD pulse sequence. Typically, 1H data were acquired with 16 transients into 32k data points over a spectral width of 14 ppm, and 13C data with 8192 transients into 64k data points over a spectral width of 220 ppm.
1H–187Os HMBC NMR spectroscopy was carried out with MeCN-d3 as solvent at 298 K on an Advance III 600 (1H = 600.13 MHz, 187Os = 13.69 MHz) spectrometer equipped with a 5 mm triple resonance broadband inverse (TBI-Lr) 1H/31P/BB probe with z-field gradients. The 90° pulse length for 187Os was 11 μs. The 187Os resonance frequency was roughly estimated from a series of 1H–187Os HMBC experiments carried out with incremented values of the carrier frequency. In order to check for the absence of folding in the F1 dimension, two gradient-enhanced HMBC spectra were recorded with different settings of the 187Os spectral width and offset (SW = 1000 ppm O1P = −3800 and −4500 respectively). The second, shown in Fig. 7, was recorded with the following experimental conditions: 128 transients collected for each FID, 32 increments, 1/2J delay set to 0.1 s, recycling delay 2.25 s; three sine-shaped gradients pulses of 1 ms duration in the intensity ratio of 60:20:41.83 for coherence (echo type) selection; sine bell multiplication in both dimensions and zero filling to 1024 data points in the F1 dimension was applied before Fourier transform followed by magnitude calculation. The reference frequency for the 187Os chemical shift was calculated from the proton frequency of internal TMS, using a conversion factor Ξ = 2.282331.
187Os NMR spectroscopy was carried out using MeCN-d3 as solventat 298 K on an Avance III 600 (1H = 600.13 MHz, 187Os = 13.69 MHz) spectrometer equipped with a 5 mm triple resonance broadband inverse (TBI-Lr) 1H/31P/BB probe with z-field gradients. The 90° pulse length for osmium was 11 μs. A series of experiments was carried out with incremented values of the carrier frequency in order to scan the full 10000 ppm scale (SW = 1000 ppm O1P = from 4500 to −4500). A standard 1D sequence with power gated decoupling was used with 256 scans, recycling delay 4 s.
Cytotoxicity assay
A stock solution of 1 was prepared in cell culture medium (5% DMSO). Approximately 5000 A2780 human ovarian cancer cells were seeded per well in 96-well plates. The cells were pre-incubated in drug-free media at 37 °C for 48 h before adding different concentrations of 1. Cells were exposed to 1 for 24 h at 37 °C and then allowed to recover for 72 h in a drug-free medium at 37 °C. Then the supernatants were removed by suction and the cells washed with PBS. The cells were The SRB assay was used to determine cell viability. Absorbance measurements of the solubilised dye (on a BioRad iMark microplate reader using a 470 nm filter) allowed the determination of viable treated cells compared to untreated controls. IC50 values (the concentration at which 50% cell death occurs) were determined as triplicates of duplicates for each complex. ICP-OES (PerkinElmer Optima 5300 DV instrument) was used to determine [Os] of the stock solution of 1, and the IC50 value was corrected for ICP factor.Conflicts of interest
There are no conflicts of interest.Acknowledgements
We thank the EPSRC (grant no. EP/F034210/1 and EP/P030572/1), ERC (grant no. 247450), and Anglo American Platinum for their support for this work, and Dr Dimitrios Bessas (ESRF) for his kind gift of osmium-187. We are grateful to Dr Lijiang Song for assistance with mass spectrometry.References
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- C. Imberti and P. J. Sadler, in Adv. Inorg. Chem, ed. P. J. Sadler and R. van Eldik, Academic Press, 2020, vol. 75, pp. 3–56 Search PubMed.
- R. J. Needham and P. J. Sadler, in The Periodic Table II: Catalytic, Materials, Biological and Medical Applications, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2019, pp. 175–201 Search PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- E. Alessio and L. Messori, in Metallo-Drugs: Development and Action of Anticancer Agents, ed. S. Astrid, S. Helmut, F. Eva and K. O. S. Roland, De Gruyter, 2018, pp. 141–170 Search PubMed.
- K. J. Ooms and R. E. Wasylishen, J. Am. Chem. Soc., 2004, 126, 10972–10980 CrossRef CAS.
- C. Brevard and P. Granger, Inorg. Chem., 1983, 22, 532–535 CrossRef CAS.
- X. Xiao, M.-I. Takeko and M. Shigeo, Chem. Lett., 1997, 26, 241–242 CrossRef.
- G. Orellana, A. Kirsch-De Mesmaeker and N. J. Turro, Inorg. Chem., 1990, 29, 882–885 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque 3rd, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- https://clinicaltrials.gov/ct2/show/NCT03945162 .
- http://kodu.ut.ee/~laurit/AK2/NMR_tables_Bruker2012.pdf .
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- M. Hanif, M. V. Babak and C. G. Hartinger, Drug Discovery Today, 2014, 19, 1640–1648 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Philos. Trans. R. Soc., A, 2015, 373, 20140185 CrossRef PubMed.
- W. X. Ni, W. L. Man, M. T. Cheung, R. W. Sun, Y. L. Shu, Y. W. Lam, C. M. Che and T. C. Lau, Chem. Commun., 2011, 47, 2140–2142 RSC.
- K. Suntharalingam, T. C. Johnstone, P. M. Bruno, W. Lin, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2013, 135, 14060–14063 CrossRef CAS PubMed.
- A. Dorcier, C. G. Hartinger, R. Scopelliti, R. H. Fish, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1066–1076 CrossRef CAS.
- E. Păunescu, P. Nowak-Sliwinska, C. M. Clavel, R. Scopelliti, A. W. Griffioen and P. J. Dyson, ChemMedChem, 2015, 10, 1539–1547 CrossRef.
- H. Kostrhunova, J. Florian, O. Novakova, A. F. A. Peacock, P. J. Sadler and V. Brabec, J. Med. Chem., 2008, 51, 3635–3643 CrossRef CAS PubMed.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- J. M. Hearn, I. Romero-Canelón, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3800–E3805 CrossRef CAS.
- S. D. Shnyder, Y. Fu, A. Habtemariam, S. H. van Rijt, P. A. Cooper, P. M. Loadman and P. J. Sadler, MedChemComm, 2011, 2, 666–668 RSC.
- I. Romero-Canelón, M. Mos and P. J. Sadler, J. Med. Chem., 2015, 58, 7874–7880 CrossRef PubMed.
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelón, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, Angew. Chem., Int. Ed., 2017, 56, 1017–1020 CrossRef CAS.
- J. P. C. Coverdale, C. Sanchez-Cano, G. J. Clarkson, R. Soni, M. Wills and P. J. Sadler, Chem. – Eur. J., 2015, 21, 8043–8046 CrossRef CAS PubMed.
- J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- G. Girolami, Nat. Chem., 2012, 4, 954–954 CrossRef CAS PubMed.
- https://www.webelements.com/isotopes.html .
- http://mriquestions.com/predict-nuclear-spin-i.html .
- D. Selby and R. A. Creaser, Science, 2005, 308, 1293–1295 CrossRef CAS PubMed.
- A. G. Bell, W. Koźmiński, A. Linden and W. von Philipsborn, Organometallics, 1996, 15, 3124–3135 CrossRef CAS.
- J. A. Cabeza, B. E. Mann, P. M. Maitlis and C. Brevard, J. Chem. Soc., Dalton Trans., 1988, 629–634 RSC.
- A. Gisler, M. Schaade, E. J. M. Meier, A. Linden and W. von Philipsborn, J. Organomet. Chem., 1997, 545–546, 315–326 CrossRef CAS.
- J. C. Gray, A. Pagelot, A. Collins, F. P. A. Fabbiani, S. Parsons and P. J. Sadler, Eur. J. Inorg. Chem., 2009, 2009, 2673–2677 CrossRef.
- H. L. Grube, in Handbook of Preparative Inorganic Chemistry, ed. G. Brauer, Academic Press Inc., 1965, vol. 2, p. 1604 Search PubMed.
- H. S. Clayton, B. C. E. Makhubela, H. Su, G. S. Smith and J. R. Moss, Polyhedron, 2009, 28, 1511–1517 CrossRef CAS.
- R. J. Needham, H. E. Bridgewater, I. Romero-Canelón, A. Habtemariam, G. J. Clarkson and P. J. Sadler, J. Inorg. Biochem., 2020, 210, 111154 CrossRef CAS.
- S. A. Kumar, R. J. Needham, K. Abraham, H. E. Bridgewater, L. A. Garbutt, H. Xandri-Monje, R. Dallmann, S. Perrier, P. J. Sadler and F. Lévi, Metallomics, 2020, 13, mfaa003 CrossRef.
- R. Benn, H. Brenneke, E. Joussen, H. Lehmkuhl and F. López Ortiz, Organometallics, 1990, 9, 756–761 CrossRef CAS.
- S. J. Berners-Price and P. J. Sadler, Coord. Chem. Rev., 1996, 151, 1–40 CrossRef CAS.
- L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2008, 252, 2239–2277 CrossRef CAS PubMed.
- T. Zou and P. J. Sadler, Drug Discovery Today: Technol., 2015, 16, 7–15 CrossRef PubMed.
- http://mriquestions.com/predict-nuclear-spin-i.html .
- I. M. Ismail, S. J. S. Kerrison and P. J. Sadler, Polyhedron, 1982, 1, 57–59 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- P. Chellan and P. J. Sadler, Chem. – Eur. J., 2020, 26, 8676–8688 CrossRef CAS PubMed.
- D. Daniel and B. Christophe, Curr. Top. Med. Chem., 2014, 14, 1684–1692 CrossRef PubMed.
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- B. Wrackmeyer, O. L. Tok and M. Herberhold, Organometallics, 2001, 20, 5774–5776 CrossRef CAS.
- B. Wrackmeyer, E. V. Klimkina, H. E. Maisel, O. L. Tok and M. Herberhold, Magn. Reson. Chem., 2008, 46, S30–S35 CrossRef PubMed.
- L. Baltzer, E. D. Becker, R. G. Tschudin and O. A. Gansow, J. Chem. Soc., Chem. Commun., 1985, 1040–1041 RSC.
- S. J. Berners-Price, L. Ronconi and P. J. Sadler, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 65–98 CrossRef CAS.
- A. Bergamo, C. Gaiddon, J. H. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2067332. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02213j |
| ‡ Current address: School of Pharmacy, Institute of Clinical Sciences, University of Birmingham, Birmingham B15 2TT, UK. |
| This journal is © The Royal Society of Chemistry 2021 |