
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-chain polyamide covalent adaptable networks based on renewable ethylene brassylate and disulfide exchange†
Charalampos
Pronoitis
 ,
Minna
Hakkarainen
and
Karin
Odelius
*
,
Minna
Hakkarainen
and
Karin
Odelius
*
Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, 100 44 Stockholm, Sweden. E-mail: hoem@kth.se
First published on 23rd September 2021
Abstract
Conventional thermosets cannot be recycled once they reach their end-of-life creating unavoidable waste. Covalent adaptable networks (CANs) are a promising circular solution as they can be reprocessed by conventional techniques employed for processing thermoplastics. In this work, strong and chemically resistant, long-chain polyamide (PA) CANs were developed by introducing intrinsically reactive disulfides in PA networks. Following a solvent-free strategy and utilizing ethylene brassylate, a renewable cyclic diester, our approach brings together the high strength and chemical resistance of long-chain, crosslinked PAs with the reprocessability of dynamic networks in a sustainable fashion. The structure of the PA CANs was elucidated by X-ray diffraction analysis, and the effect of the disulfides on the thermal, mechanical, viscoelastic and dynamic properties was evaluated. The PA CANs had high gel content (86–98%) and they were reprocessable over three grinding-compression molding cycles, retaining their strength (15–20 MPa), crosslink density and gel content. They exhibited rapid stress relaxation with relaxation times as low as 1.06 s and were healable within 5 min. The long-chain PA CANs are easy to prepare and feature several elements of sustainable materials design, highly valued in plastics’ circular economy.
Introduction
Unlike thermoplastics that can be recycled and/or reprocessed when they reach their end-of-life, the development of circular thermosets is more challenging. Thermosets reach their end-of-use once they are damaged, as the permanently crosslinked network does not allow reprocessing by conventional techniques, and incineration, landfill or use as lower value additives commonly follow. Associative covalent adaptable networks1 or vitrimers2 have emerged as a promising solution to the challenge of achieving recyclable thermosets. Associative CANs are thermally reprocessable and chemical recycling of the pristine synthons is possible in certain cases. Their principle is the activation of exchange reactions between dynamic bonds that overall keep the crosslink density of the network constant, but allow it to flow and rearrange its topology at high temperature. Thus, CANs meld the features of classical thermosets such as chemical resistance and dimensional stability with reprocessability, and in some cases self-healing ability and chemical recycling,2–7 all highly attractive features for many applications.8Polyamide (PA) networks have rarely been reported, let alone PA networks with exchangeable bonds. This lack of PA CAN examples could partly be due to the harsh conditions required for the synthesis of PAs and the difficulty to combine a sensitive dynamic motif in the same synthetic protocol. However, fusing the high mechanical strength, thermal and chemical resistance of PAs in a reprocessable network is an intriguing route towards high-performance, sustainable materials. Specifically, long-chain PAs, that is PAs containing aliphatic chains with more than 10 –CH2– units,9 are of particular interest not only from an application aspect but also fundamentally due to their structural similarity to polyethylene.10 Importantly, long-chain PAs are usually derived from fatty acids and/or their derivatives,11–15 and such systems have been reported to be reprocessable and to exhibit self-healing ability and shape-memory behavior. In systems that are designated as long-chain, the long aliphatic chains hold a critical role both for the design of the structure and the macroscopic properties, and for the dynamic response. To prepare such systems, elegant and efficient synthetic routes have been followed, however, they involved some complex synthesis and purification of precursors, use of transition metal catalysts and/or solvents and toxic reagents for the polymerization or crosslinking.16–19
Since the seminal work of Leibler and coworkers,20 a multitude of different bonds, namely esters,21,22 disulfides,23,24 imines,25,26 urethanes27,28 and boronic esters,29,30 have been employed for the design of reprocessable networks. Seeking for a simpler and more sustainable alternative towards long-chain PA CANs, we focused on disulfides as an intrinsically reactive bond,31,32 that can be activated under mild conditions.33 Disulfides have been incorporated into urea,34 urethane,23,35,36 ester4,37 and epoxy-based38–40 networks, and have been combined with renewable building blocks such as diacids (succinic, adipic, sebacic),4 castor oil24,41 and epoxidized soybean oil,42 leading to high-performance thermosets with impressive results regarding the ease of reprocessability and retainment of the mechanical properties after several reprocessing cycles.
Our aim was to develop biobased, long-chain PA networks that would combine the high mechanical strength and chemical resistance of PAs with the malleability of associative CANs. The long aliphatic chains should endow flexibility to the network and affect its dynamic character, enabling fast relaxation. We hypothesized that a simple and straightforward route to achieve such a system would be by leveraging a solvent-free, ring-opening aminolysis-condensation/crosslinking strategy of renewable ethylene brassylate,43,44 and the fast disulfide exchange.4,23,39 The dynamic character of the resulting long-chain PA network was verified by stress relaxation analysis and the material was thermally reprocessable over three grinding-compression molding cycles without deterioration of its properties.
Experimental
Materials
Ethylene brassylate (EB, 95%), tris(2-aminoethyl)amine (TAEA, 96%), 2,2′-(ethylenedioxy)bis(ethylamine) (EDOBA, 98%) and hexamethylenediamine (Hex, 98%) were purchased from Sigma-Aldrich. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD, 97%) was purchased from ChemScence. Cystamine (Cys) was prepared from cystamine dihydrochloride (98%, AmBeed) as previously described.23 1,1,1,3,3,3-Hexafluoropropan-2-ol (HFIP, 99%) was purchased from Apollo Scientific, UK. Chloroform-d (CDCl3, 99.8%) was purchased from VWR. All reagents were used as received without any further purification.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TAEA molar ratios were prepared, keeping the total ester:amine ratio constant to 1. The samples are named as CysXX, where XX denotes the relative molar amount of cystamine (Cys) compared to TAEA. A typical procedure is described for Cys50; Cys (0.450 g, 0.00296 mol), TAEA (0.432 g, 0.00296 mol) and TBD (0.205 g, 0.00148 mol, 5 mol% to total –NH2) were added into a 10 mL scintillation vial, which was gently heated with a heat gun until TBD was fully dissolved and the color of the solution turned yellowish-orange. The solution was cooled down to room temperature and EB (2 g, 0.0074 mol) was weighed in the vial. The mixture was thoroughly vortexed, poured in a disposable aluminium pan and placed in an oven at 100 °C for 18 h, followed by 18 h at 140 °C. All samples were stored in a desiccator when not in use.
:TAEA molar ratios were prepared, keeping the total ester:amine ratio constant to 1. The samples are named as CysXX, where XX denotes the relative molar amount of cystamine (Cys) compared to TAEA. A typical procedure is described for Cys50; Cys (0.450 g, 0.00296 mol), TAEA (0.432 g, 0.00296 mol) and TBD (0.205 g, 0.00148 mol, 5 mol% to total –NH2) were added into a 10 mL scintillation vial, which was gently heated with a heat gun until TBD was fully dissolved and the color of the solution turned yellowish-orange. The solution was cooled down to room temperature and EB (2 g, 0.0074 mol) was weighed in the vial. The mixture was thoroughly vortexed, poured in a disposable aluminium pan and placed in an oven at 100 °C for 18 h, followed by 18 h at 140 °C. All samples were stored in a desiccator when not in use.
Characterization
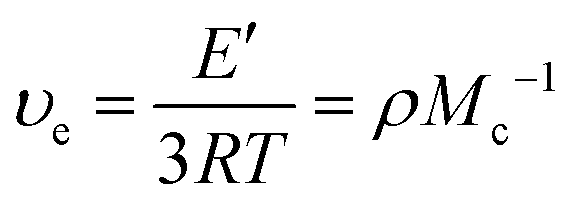 | (1) |
 | (2) |
Results and discussion
Structure, thermal and viscoelastic properties of the PA CANs
Biobased long-chain PA associative CANs were successfully prepared by incorporating exchangeable disulfide (S–S) bonds in PA networks. The molar ratio between the amine-containing reactants (cystamine and tris(2-aminoethyl)amine, TAEA) was varied to achieve CANs with different S–S content and crosslink density, and to evaluate their influence on the thermal properties, mechanical properties, and ability of the dynamic networks to rearrange their topology. Reference networks with non-dynamic bonds were also prepared for comparison reasons. The general strategy towards the long-chain PA associative CANs is presented in Fig. 1.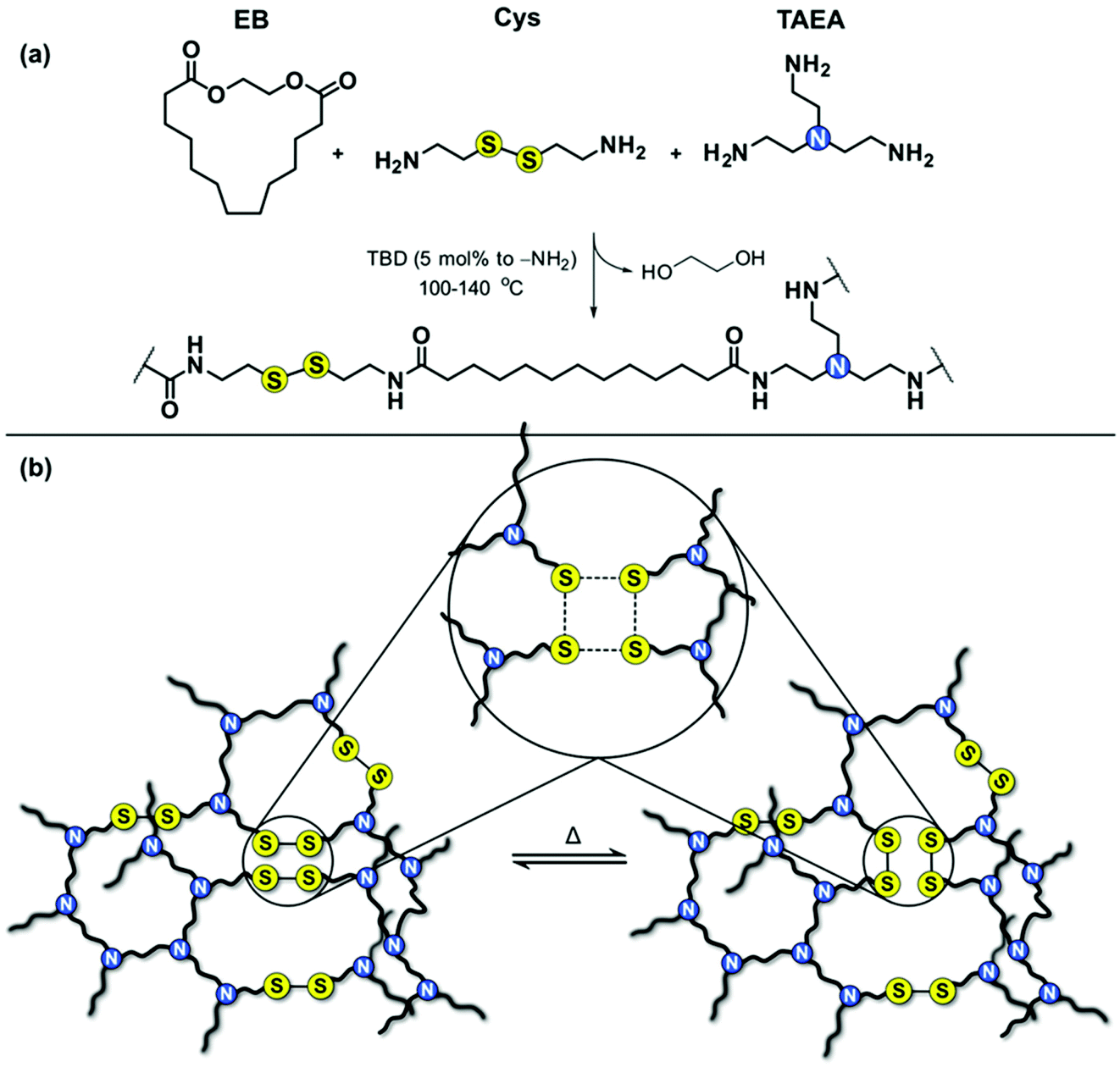 | ||
| Fig. 1 Synthesis of long-chain PA associative CANs; (a) general reaction scheme and (b) schematic representation of the disulfide exchange in the network. | ||
To achieve the synthesis of the PA CANs, we screened the reaction conditions that promoted an efficient ring-opening aminolysis-crosslinking reaction between EB, Cys and TAEA. For the neat EB/TAEA system, the reaction proceeds efficiently at 100 °C under basic catalytic conditions (4 mol% of TBD to –NH2), yielding PA networks with high gel content (∼93%).44 When the same reaction conditions were applied to the EB/Cys/TAEA mixtures with varying the Cys:TAEA molar ratio (20:80 to 80:20), networks with lower than expected gel content (89–81%) were obtained and the FTIR spectra showed some unreacted EB monomer left, Fig. S1.† Higher gel content values could not be achieved by increasing the temperature to 120 °C or by extending the reaction time (48 h) at 100 °C. Side-reactions involving Cys were excluded as the compound was stable at high temperature and in basic conditions, Fig. S2.† The lower gel content was a result of replacing part of the trifunctional TAEA with difunctional Cys, therefore reducing the crosslink density of the networks (vide infra).
Hence, to ensure the successful incorporation of Cys in the PA networks, a two-step procedure was implemented along with a slightly higher catalyst loading (5 mol% to total –NH2). The first step was performed at 100 °C for 18 h and ring-opening of the majority of EB took place. In a second step, the reaction temperature was increased to 140 °C for additional 18 h to drive the aminolysis reaction further. After the two-step procedure, the gel content was 98, 94, 90 and 86% for Cys30, Cys40, Cys50 and Cys60, respectively, and elemental mapping showed that sulfur was homogeneously distributed over the network indicating a successful network formation, Fig. S4–S7.† To further prove that most of the monomers have been incorporated into the networks, the dissolved phases, collected after swelling the networks in HFIP, were analyzed by 1H NMR spectroscopy. The 1H NMR spectrum showed that the dissolved fractions consisted mostly of TBD and traces of monomers and/or soluble oligomers, Fig. S3.† Hence, to achieve Cys-containing, dynamic PA networks with high gel content, a higher curing temperature and longer reaction time were necessary.
The appearance of the characteristic N–H stretching (3290 cm−1) and the amide I (1634 cm−1) and amide II (1541 cm−1) bands in the FTIR spectra of the resulting materials, concomitantly with the disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of EB (1736 cm−1), confirmed the formation of the amide bonds and the successful preparation of the PA networks, Fig. 2a. The presence of consecutive ester units in the network resulting from ring-opening oligomerization of EB cannot be excluded,44 but no signal in the 1740–1720 cm−1 range (ester CO stretching) was observed in the FTIR spectra. The networks were thermally stable at least up to 260 °C with Td,5% values lower than those of pure PA networks,44 Fig. S8.† The lower thermal stability is a consequence of the presence of thermally sensitive S–S bonds as confirmed by the observed decrease of the Td,5% values with increasing the Cys content, namely 293, 284, 276 and 259 °C for Cys30, Cys40, Cys50 and Cys60, respectively. These Td,5% values are similar to those reported for other aromatic or aliphatic disulfide-based urethane and epoxy associative CANs.23,40,42
O stretching of EB (1736 cm−1), confirmed the formation of the amide bonds and the successful preparation of the PA networks, Fig. 2a. The presence of consecutive ester units in the network resulting from ring-opening oligomerization of EB cannot be excluded,44 but no signal in the 1740–1720 cm−1 range (ester CO stretching) was observed in the FTIR spectra. The networks were thermally stable at least up to 260 °C with Td,5% values lower than those of pure PA networks,44 Fig. S8.† The lower thermal stability is a consequence of the presence of thermally sensitive S–S bonds as confirmed by the observed decrease of the Td,5% values with increasing the Cys content, namely 293, 284, 276 and 259 °C for Cys30, Cys40, Cys50 and Cys60, respectively. These Td,5% values are similar to those reported for other aromatic or aliphatic disulfide-based urethane and epoxy associative CANs.23,40,42
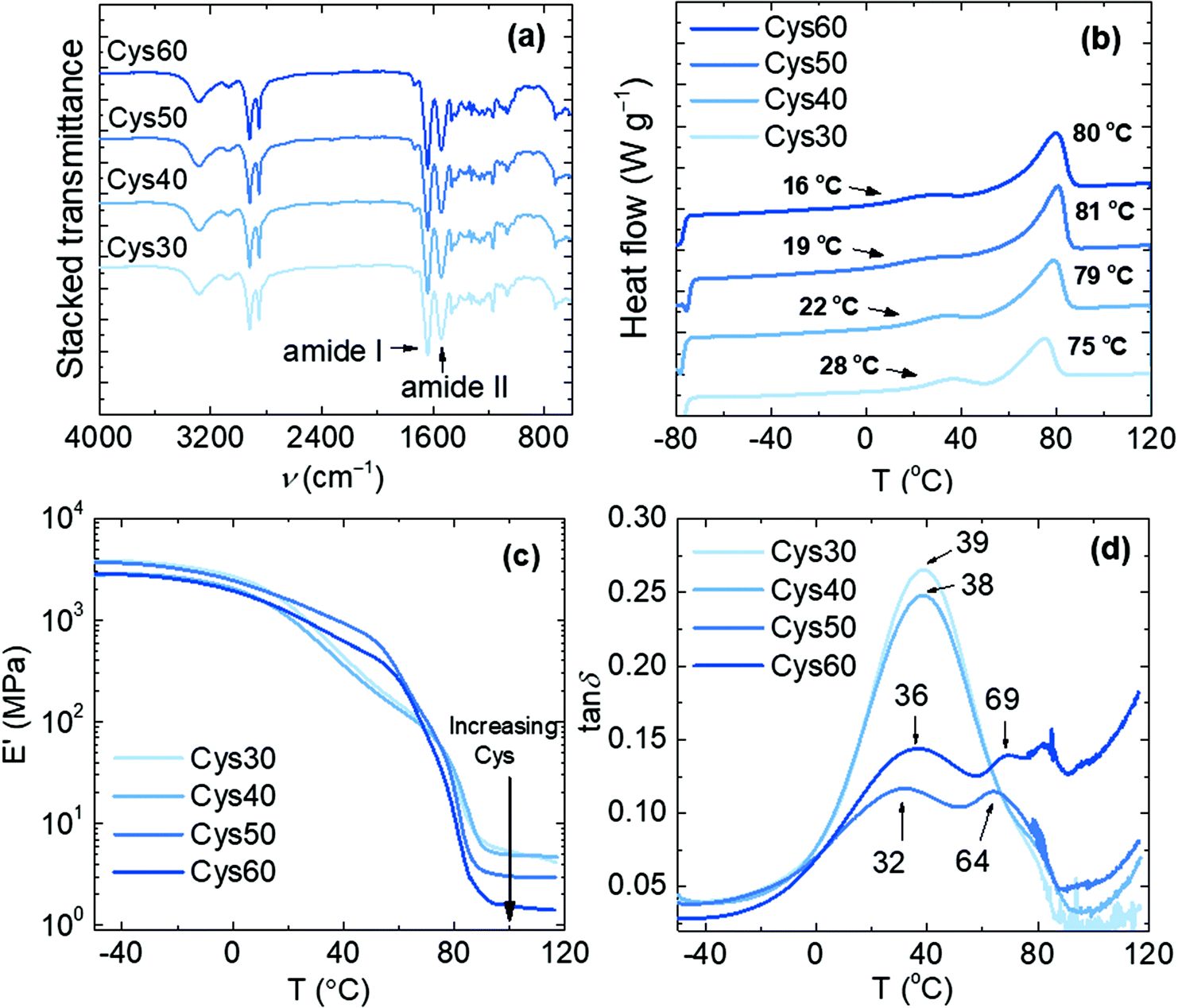 | ||
| Fig. 2 Characterization of the PA CANs; (a) FTIR spectra, (b) DSC traces of the 2nd heating scan and (c), (d) E′ and tanδ, respectively, from DMA analysis. | ||
The networks were semicrystalline and their thermal and viscoelastic properties were altered by the increasing content of Cys as proven by DSC and DMA, Fig. 2b–d, Table 1. The general trend observed by DSC was a decrease of Tg and an increase of Tm with increasing the Cys content. This trend can be explained when considering that the increasing Cys content resulted in lower crosslink density (υe) and larger molar mass between crosslinks (Mc), Table 1. Consequently, the higher chain mobility in the network was reflected on the decreasing Tg from 28 to 16 °C for Cys30–60, respectively. The crystallinity observed was attributed to the aliphatic chains of the amide strands, i.e. the linear macromolecular segments connected to the crosslinks. The increase of Tm with higher Cys content (75–81 °C) was accompanied by higher enthalpy of fusion (ΔHm) which is congruent with the behavior observed for other linear, aliphatic oligoamides of PA-10,10 exhibiting higher Tms and degree of crystallinity at higher molar mass, but also higher Tm values than the PA networks (118–188 °C).45 Four factors account for the lower Tm values of the PA networks compared to those of linear oligoamides: (i) the presence of crosslinks in the PA networks, (ii) the discrepancies in the conformation and crystal structure arising from well-known odd-even effects,46–49 (iii) the perturbing effect of the amide groups on the crystallization ability of the aliphatic chain,10 and (iv) the presence of the S heteroatoms disturbing the packing of the aliphatic chains.15 However, as previously explained, replacing TAEA with Cys entails higher degree of chain freedom as the amide strands became longer and facilitated the development of H-bonding between the amide groups. Further, due to the low electronegativity difference between S and H, S is considered a weak H-bonding partner however, it does participate in H-bonding,50–52 and/or generally in secondary interactions. Hence, when higher number of S atoms were available in the network, the possibility of H-bonding increased and as a consequence the Tm increased. For comparison, pure EB/TAEA networks and linear EB homopolymers exhibited a Tm of 67 and 69 °C, respectively.44,53 Keeping the 50:50 ratio of diamine:TAEA but replacing cystamine with 2,2′-(ethylenedioxy)bismethylamine (EDOBA) or hexamethylenediamine (Hex) resulted in networks (EDOBA50 and Hex50) with Tm = 85 and 68 °C, respectively. The Tm of Hex50 is much lower than EDOBA50 and Cys50, both of which contained additional heteroatoms participating in secondary interactions and therefore increasing the Tms of the networks. Hex50 had also a second Tm = 123 °C due to the additionally crystalline macromolecules formed with hexamethylenediamine between the crosslinks. Full characterization of EDOBA50 and Hex50 are provided in Table S2, Fig. S21–S23.†
| Network |
T
g, DSCa (°C) |
T
g, DMAb (°C) |
T m (°C) | ΔHm (J g−1) | E′c (MPa) |
υ
ed (mol m−3) |
M
cd (g mol−1) |
|---|---|---|---|---|---|---|---|
|
a Obtained from the 2nd heating scan.
b Maximum of tanδ.
c Storage modulus in the rubbery plateau.
d Calculated through eqn (1).
|
|||||||
| Cys30 | 28 ± 0.1 | 40 ± 3 | 75 ± 0.6 | 17 ± 0.3 | 5.0 ± 0.3 | 540 ± 33 | 1820 ± 110 |
| Cys40 | 22 ± 0.3 | 38 ± 0.8 | 79 ± 0.7 | 18 ± 6 | 5.0 ± 0.2 | 538 ± 21 | 1820 ± 69 |
| Cys50 | 19 ± 0.3 | 35 ± 3 | 81 ± 0.6 | 27 ± 1 | 3.8 ± 0.9 | 409 ± 92 | 2550 ± 560 |
| Cys60 | 16 ± 0.1 | 39 ± 2 | 80 ± 1 | 25 ± 1 | 1.6 ± 0.4 | 170 ± 39 | 6320 ± 1430 |
| EDOBA50 | 11 ± 0.8 | 16 ± 2 | 84 ± 0.8 | 33 ± 2 | 3.4 ± 0.4 | 364 ± 46 | 3040 ± 360 |
The viscoelastic properties of the networks were influenced by the Cys content as shown by the decreasing storage modulus in the rubbery plateau region, Fig. 2c. This drop in the elastic response is consistent with more loosely crosslinked networks exhibiting higher Mc, and corresponds with the values calculated here, Mc increased from 1820 g mol−1 for Cys30 to 6320 g mol−1 for Cys60, Table 1. The Tg values (40–35 °C) taken from the maxima of the tanδ curves, correlated with the trend in DSC, although for Cys60 (39 °C) a higher value than Cys50 (35 °C) was recorded, Table 1. It is noteworthy that if the maxima of the E′′ curves are considered as the Tg, the same decreasing trend with higher Cys content and the same phenomenon of Tg, Cys60 > Tg, Cys50 are observed, Fig. S9.† Further, some of the tested samples of Cys50 and Cys60 showed more than one maximum in the tanδ curves, Fig. 4c, Fig. S10.† Recognizing the origin of multiple peaks in tanδ curves is challenging,54 but the two maxima observed here could be explained based on subtle discrepancies in the local environment of the amide strands and in the concentration of the bulkier S atoms. Depending on their proximity to the crosslink points, different parts of the amide strands would exhibit different degrees of chain freedom, lower for those found closer to a crosslink site and higher for those farther away. Given the high sensitivity of DMA in detecting structural changes, it is possible that those different regions reflected on the tanδ curves as two maxima. The higher Tg of Cys60 than Cys50 can be rationalized on the basis of the higher number of bulkier S atoms in the former. In fact, although a lower crosslink density is usually regarded as a definitive parameter that will decrease the Tg of a network, there are cases for which this rule is not met.55,56 The expected presence of network defects (loops, dangling chains) could also contribute to above-described effects.57,58
The crystalline structure of the PA networks was elucidated by XRD analysis. All of the networks displayed a single diffraction peak at 2θ 21.4°, and the amorphous halo was observed only Cys30 and Cys40, Fig. 3. The 21.4° peak, due to the (110) crystal plane, is characteristic of the orthorhombic crystal lattice adopted by long-chain hydrocarbons,59 polyethylene-like polyesters,60 and long-chain polyamides,10 polyacetals and polycarbonates.61 Crystallization in the orthorhombic system is further supported by the characteristic –CH2– scissoring and rocking vibrational modes in the FTIR spectra at 1466 and 720 cm−1, respectively.10,62,63 The EDOBA50 control network and a network prepared only with EB and TAEA, featured exactly the same XRD patterns as the Cys-containing dynamic networks, yet the peak was slightly shifted at 21.7°. This difference in the position of the peak from 21.7 to 21.4°, indicates a slightly larger unit cell for the PA CANs specifically, a distortion of the a and b unit cell parameters, due to the addition of the larger S–S groups compared with the –CH2– groups. Similar behaviour has been reported for long-chain PAs containing sulfide bonds.15 In fact, applying Bragg's law it is seen that the distance between the crystal planes (d) slightly increased from 41 to 41.5 Å (see ESI† for calculations). The absence of peaks at 2θ > 22–23° that have been reported for other long-chain aliphatic PAs10 or EB homopolymers64 implies the absence of higher-order organization of the amide strands along the c-axis of the crystalline cells. Since the number of carbons in EB are 13 (PA-X,13), the amide strands can be considered long-chain,9,43 but with high density of amide groups (133.3 per 1000 –CH2– units). Mecking et al.10 suggested that when the density of amide groups is higher than ∼50 per 1000 –CH2– units, the orthorhombic system is not favored anymore due to the increased cohesion energy from the pronounced H-bonding. The reason why the orthorhombic system was maintained in the presented PA networks is not clear but we speculate that, despite the higher amide density compared with other long-chain PAs9 and the presence of the S heteroatoms participating in secondary interactions, the crosslinks could create an inverse “locking” effect resulting in the retainment of the orthorhombic crystal structure. Through several studies, the structure and properties of thermoplastic, long-chain PAs and their relation with purely linear polyethylene are now more fundamentally understood.9,65–68 Although the focus here was on finding a simple and sustainable pathway towards reprocessable PA networks, the above results can be the basis for future work aiming to develop crosslinked PAs with even longer aliphatic segments. The elucidation of the specific effects of the crosslinks and the presence of heteroatoms on the crystal structure, thermal, mechanical and viscoelastic properties of such networks would be valuable for a deeper understanding of the structure–property relationships.
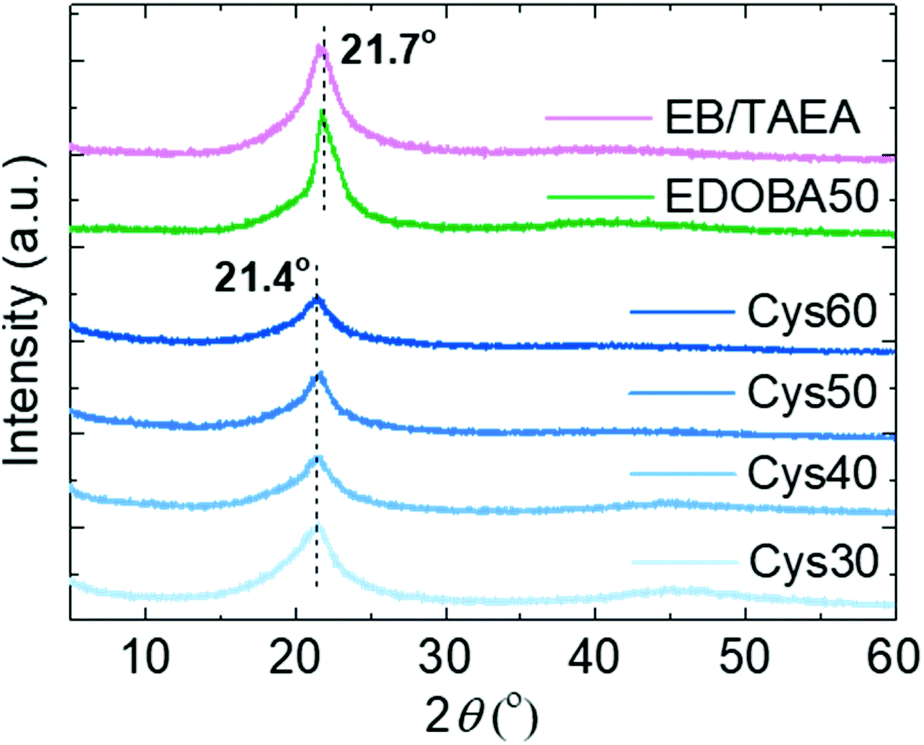 | ||
| Fig. 3 XRD patterns of the PA CANs and the control networks. | ||
Dynamic, mechanical properties and reprocessability of the PA CANs
Stress relaxation experiments proved the dynamic character of the PA networks. 1% strain was instantly applied and the decrease of the relaxation modulus (Gt) was monitored until it reached 0.37 (1/e) of its initial value (G0), according to Maxwell's model for viscoelastic fluids,69eqn (3):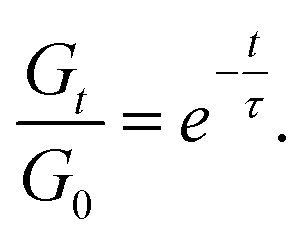 | (3) |
Disulfide exchange was proposed already in 1946 as a plausible mechanism accounting for the stress relaxation of polysulfide rubbers.70 Thanks to the fast exchange reaction, various disulfide-based CANs have displayed short relaxation times in a range of temperatures.4,23,39 In this system, the long aliphatic chains and the increasing Mc with higher Cys content should impact the relaxation times, in accordance to the impact on the thermal, viscoelastic and mechanical properties (vide infra).
To test our hypothesis, we selected Cys60 which had the highest Mc, and tested its response to stress relaxation at different temperatures, Fig. 4a and b. Cys60 fully relaxed the applied stress as a result of the disulfide exchange and the relaxation time (τ) decreased from 6.6 to 1.06 s, shorter as the testing temperature increased from 80 to 105 °C. At 105 °C τ was 1.06 s indicating an extremely rapid relaxation and a very easy activation of the disulfide exchange reaction, similar to other disulfide-based CANs containing flexible polysulfide or polyether segments.71,72 In fact, the very fast relaxation is likely ascribed to the highest Mc of Cys60 (6320 g mol−1) as the long flexible amide strands are expected to enable longer-range chain mobility compared to the other more densely crosslinked PA CANs. Hence, the exchange between disulfides is promoted as the probability of two adjacent groups meeting is increased. As the network was above its Tm (80 °C) during testing, the molten state contributed to the higher chain mobility and therefore to the low τ values. However, even at temperatures lower than the Tm (70 °C), full stress relaxation was observed in reasonably short times (87 s), while at a temperature higher than 105 °C the relaxation time was shorter than 1 s, Fig. S11.† It should be noted that the presence of TBD may have a salutary effect on the exchange reaction between disulfides,39,72–74 although it should not be the dominant factor for it. The system followed an Arrhenius-like behavior as shown by plotting τ against temperature, Fig. 4b. The activation energy (Ea) was calculated to be 76.7 kJ mol−1, a value comparable to some other disulfide-based CANs,23,36,39,71 although it can vary significantly both for the aliphatic and aromatic disulfide exchange reaction.4,24,40 The dynamic character was further proven for all the PA CANs with different content of Cys, and the EDOBA50 control network was tested under the same conditions. All of the PA CANs were capable of fully relaxing the stress, whereas the EDOBA50 control network showed minimal stress relaxation (Gt/G0 = 0.8 after 10800 s), Fig. 4c. The decrease of its relaxation modulus was largely due to the high linear content of the network. For the PA CANs, τ increased from 1.6 to 2986 s with decreasing the Cys content as a direct consequence of the decreasing number of disulfides and the increasing crosslink density. The latter hinders the diffusion of the disulfides to a higher extent in Cys30 and Cys40 compared to Cys50 and Cys60, explaining the difference of the relaxation times. Overall, the observed trend is consistent with the behavior of other associative CANs where the relaxation rate is dictated by the concentration of the exchange-controlling species (either catalyst or self-activated motif) and the crosslink density.6,22,23
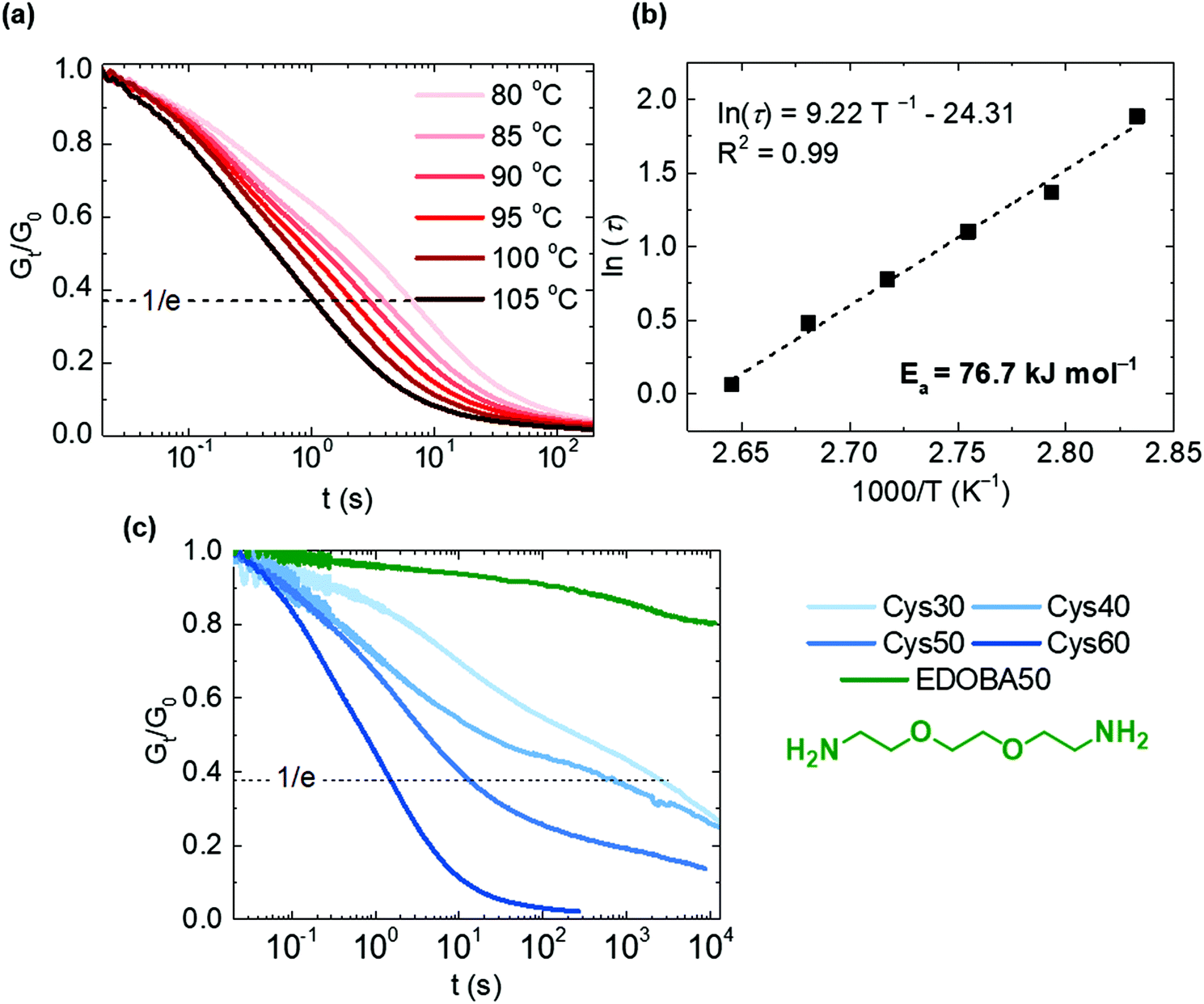 | ||
| Fig. 4 Dynamic properties of the PA CANs; (a) stress relaxation curves of Cys60 at different temperatures, (b) relaxation times (τ) as a function of the temperature and calculation of the activation energy (Ea) for Cys60 and (c) comparison of the stress relaxation of Cys30–60 at 100 °C. | ||
In an attempt to retain the crosslink density while varying the fractions of dynamic bonds, three reference networks with similar gel content and crosslink densities, yet with varying S–S content were prepared. To do so, a part of Cys was replaced by EDOBA, while the overall diamine:triamine ratio was kept constant at 50:50. Three formulations of EDOBA:Cys were created, namely, 40:10, 25:25 and 10:40, Fig. S12–S15, Table S1.† The dynamic response of these networks was vastly different. The 40:10 and 25:25 EDOBA:Cys networks did not exhibit full stress relaxation, with Gt/G0 values at the end of the testing time being 0.75 and 0.43 respectively, reflecting the increasing dynamic bond content. In contrast, the 10:40 EDOBA:Cys network having the highest number of dynamic S–S bonds fully relaxed stresses within 28.5 s, a value that is reasonably lower than Cys50 (τ = 14.7 s), Fig. S15.† Overall, the reference networks along with EDOBA50 prove that disulfide metathesis is the mechanism that enabled the stress relaxation to the Cys-containing networks.
The mechanical properties of the PA CANs were evaluated by tensile testing. The networks generally exhibited a rigid profile with high strength, typical of PAs, but without a clear yield point, Fig. 5a. Compared to PA networks made of EB and TAEA (E = 150–330 MPa, σb = 7–17 MPa, εb = 70–176%),44 the PA CANs were comparably strong (σb = 7–15 MPa) but more rigid, E varied between 350–440 MPa and increased with the Cys content, presumably due to the bulkier S atoms. If compared among them, the strength (σb) and strain at break (εb) followed the order Cys50 > Cys40 > Cys60 > Cys30 and Cys50 > Cys40 > Cys30 > Cys60, respectively, revealing an intriguing dual effect of the number of S–S bonds on the mechanical properties. As the content of Cys increased from Cys30 to Cys50, σb and εb increased as well, but that increment was discontinued for Cys60 which contained the highest number of S–S bonds. As explained previously for the thermal properties, introducing more Cys in the PA CANs increased the Mc and endowed them higher flexibility, resulting in the almost linear increase of σb and εb going from Cys30 to Cys50. However, the lower bond dissociation energy (BDE)75 of dialkyl disulfides (277 kJ mol−1) compared to other stronger bonds found in the network, e.g. C–C (BDE ≥ 350–360 kJ mol−1), could macroscopically be translated in a higher propensity of Cys60 to break upon exertion of a mechanical force. Putatively, the optimum balance between the beneficial effect of the longer, flexible amide strands on σb and εb, and the sensitivity of the S–S bonds towards breaking was reached for Cys50, which presented the highest strength and elongation among the PA CANs.
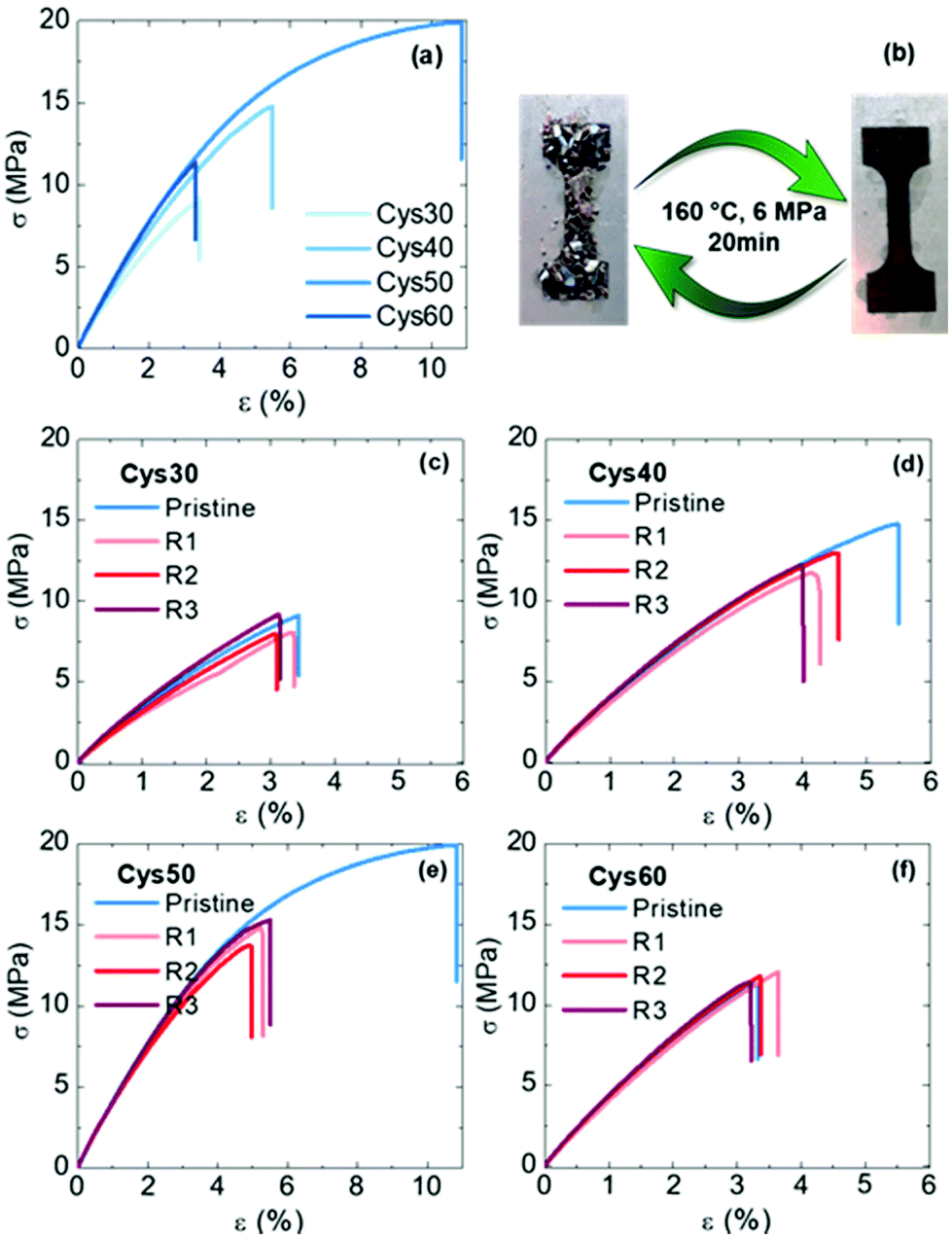 | ||
| Fig. 5 Mechanical properties of the PA CANs; (a) stress–strain curves of the pristine networks, (b) appearance of a reprocessed sample after compression molding and (c–f) stress–strain curves of the reprocessed CANs. | ||
The reprocessability of the PA CANs was demonstrated through three cycles of grinding-compression molding. The disulfide exchange promoted the topological rearrangement of the network and enabled its reformation. The grinded networks were compression-molded for 20 min at 160 °C, under 6 MPa pressure and perfectly consolidated specimens were obtained, Fig. 5b. The reprocessed specimens were subjected to tensile testing and DMA to evaluate their mechanical performance, as well as FTIR and gel content measurements to confirm that their structure remained unaffected during the reprocessing. The properties of the PA CANs for each cycle are summarized in Table 2. No significant loss of the tensile mechanical properties was observed for the reprocessed networks with the σb and εb values being very similar to the values of the pristine networks, Fig. 5c–f. An exception was Cys50 for which the reprocessed material had lower σb and εb values compared with the original material (20 MPa, 10% vs. 15 MPa, 5%) yet, with the stress–strain curves over the three reprocessing cycles being almost identical. Additionally, σb and εb of both Cys40 and Cys50 were higher than the values of Cys60, in line with the previous discussion concerning the effect of the number of S–S on the mechanical properties. The storage modulus in the rubbery plateau remained constant or slightly decreased indicating a constant crosslink density, Fig. S24,† and the gel fraction remained essentially unchanged strongly indicating that no side-reactions or permanent damage took place during reprocessing. The FTIR spectra recorded after each reprocessing cycle corroborated these results showing that the chemical structure of the networks remained unaffected, Fig. S25.† Overall, the characterization of the reprocessed networks proved the efficiency of the S–S as a dynamic motif that enabled the creation of high-performance, fully reprocessable PA CANs without deterioration of their properties.
| PA CAN | RC | E (MPa) | σ b (MPa) | ε b (%) | E′a (MPa) | Gel fraction |
|---|---|---|---|---|---|---|
| a Storage modulus in the rubbery plateau. | ||||||
| Cys30 | Pristine | 348 ± 10 | 9.1 ± 0.05 | 3.6 ± 0.3 | 5.0 ± 0.3 | 0.98 ± 0.01 |
| 1 | 321 ± 12 | 7.7 ± 1.1 | 3.1 ± 0.5 | 4.2 ± 1.4 | 0.96 ± 0.02 | |
| 2 | 348 ± 15 | 8.0 ± 0.7 | 3.0 ± 0.2 | 2.8 ± 0.6 | 0.97 ± 0.01 | |
| 3 | 380 ± 10 | 8.7 ± 0.7 | 3.0 ± 0.2 | 3.3 ± 1.0 | 0.95 ± 0.0006 | |
| Cys40 | Pristine | 373 ± 14 | 14.1 ± 0.8 | 5.4 ± 0.3 | 5.0 ± 0.2 | 0.94 ± 0.05 |
| 1 | 363 ± 4 | 11.3 ± 1.2 | 4.1 ± 0.3 | 4.7 ± 1.5 | 0.92 ± 0.04 | |
| 2 | 357 ± 61 | 11.6 ± 1.4 | 4.4 ± 0.2 | 5.4 ± 1.4 | 0.93 ± 0.03 | |
| 3 | 388 ± 23 | 11.8 ± 0.9 | 4.1 ± 0.3 | 3.9 ± 1.4 | 0.91 ± 0.01 | |
| Cys50 | Pristine | 400 ± 5 | 19.6 ± 0.3 | 10.4 ± 1.9 | 3.8 ± 0.9 | 0.91 ± 0.04 |
| 1 | 409 ± 7 | 14.7 ± 0.7 | 5.1 ± 0.3 | 2.7 ± 1.2 | 0.94 ± 0.02 | |
| 2 | 412 ± 13 | 13.5 ± 0.5 | 4.9 ± 0.2 | 2.7 ± 0.3 | 0.89 ± 0.05 | |
| 3 | 409 ± 16 | 15.5 ± 1.1 | 5.4 ± 0.07 | 3.4 ± 1.0 | 0.87 ± 0.006 | |
| Cys60 | Pristine | 435 ± 15 | 11.2 ± 1.9 | 3.3 ± 0.3 | 1.6 ± 0.4 | 0.86 ± 0.01 |
| 1 | 440 ± 23 | 12.3 ± 1.2 | 3.6 ± 0.5 | 2.2 ± 0.2 | 0.90 ± 0.01 | |
| 2 | 431 ± 32 | 12.3 ± 1.9 | 3.9 ± 1.1 | 2.0 ± 0.1 | 0.88 ± 0.01 | |
| 3 | 418 ± 9 | 15.4 ± 0.9 | 5.2 ± 0.5 | 2.2 ± 0.3 | 0.81 ± 0.06 | |
Healing and chemical resistance of the PA CANs
Disulfide metathesis has been previously exploited as a means to induce (self-)healing to associative CANs.34,36,76,77 We presumed that despite the rigidness of the PA networks due to the H-bonding, the disulfide exchange could be thermally activated to facilitate networks that are healable. As a proof of concept, a disk of Cys60 (∅ 23 mm, T = 3 mm) was cut in half with a sharp blade and the two cut surfaces were merged together while heated over a conventional heat gun operating between 150–200 °C. Already after 5 min of heating and keeping the cut surfaces pressed against each other, the as-healed disk could withstand a 2 kg load hung perpendicularly to the cut, Fig. 6. Thus, the disulfide exchange occurring at the interface of the two pieces of the disk resulted in a robust healing. Although this protocol was not optimized, it provides tangible proof that the PA CANs were healable, a highly valued property of the thermosets of the future that should ideally regain their function quickly after damage, without compromising the rest of their structure.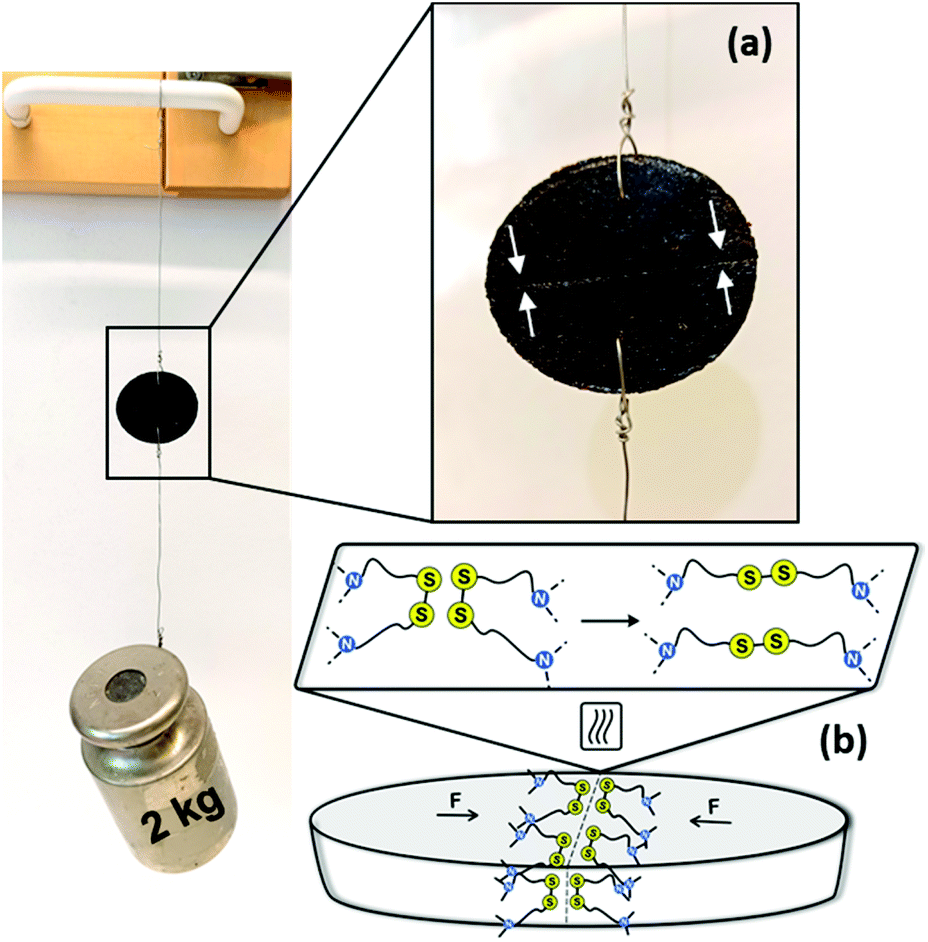 | ||
| Fig. 6 Healing of the PA CANs. (a) A disk of Cys60 can withstand a 2 kg load after being cut in half and reconnected by pressing and heating the cut surfaces, the white arrows in the enlarged image point the cut; (b) proposed healing mechanism of the sample through heat-activated disulfide exchange at the interface of the two pieces, F denotes the force that keeps the two sides in contact with each other. | ||
Finally, when associative CANs are placed in a good but non-reactive solvent they behave as conventional thermosets, they only swell and they do not dissolve. To evaluate the chemical resistance and stability of the PA CANs, samples from Cys40 that were already hot-pressed one time, were tested against an array of solvents, Fig. 7, Table S3.† Infused with the notable chemical resistance of PAs and balanced with the dynamic nature of disulfides, Cys40 showed minimal mass loss (≤5%) in most solvents. In DMF, DMSO and 1 M NaOH, partial swelling and coloration of the solutions was observed and associated to 7–8% mass loss, while in DCM, a maximum of 12% mass loss was measured. As PAs are well-known for their particular resistance against solvents due to their polar nature, the fact that they were already reprocessed one time prior to the solvent-resistance test, likely contributed to the measured mass loss. Overall, the test indicated that the PA CANs were robust materials that can perform in various chemical environments without being considerably affected.
 | ||
| Fig. 7 Chemical resistance of the PA CANs; Cys40 after being immersed in each solvent for 24 h. From left to right: hexane, toluene, diethyl ether, dichloromethane, tetrahydrofuran, ethyl acetate, acetone, methanol, acetonitrile, dimethylformamide, dimethyl sulfoxide, water, hydrochloric acid (aq. 1 M) and sodium hydroxide (aq. 1 M). | ||
Conclusions
Long-chain, PA associative CANs based on renewable ethylene brassylate were successfully developed by employing disulfide exchange as the dynamic reaction. The structure–property relation of the PA CANs was investigated and the effect of the aliphatic chains and the number of S–S bonds on their properties was elucidated. The networks were reprocessable over three cycles without deterioration of their performance, and demonstrated healing ability by applying an external stimulus. Overall, the system has several sustainable characteristics: (i) it utilizes a renewable resin, (ii) it is devoid of solvents and toxic reagents, (iii) it is easy and simple to handle, (iv) it is quickly healable and (v) it provides the opportunity to prepare fully functional materials from its own ‘waste’. These features make the developed long-chain PA CANs highly attractive for applications that require materials with the properties of crosslinked PAs combined with the ability to regain function after damage, through simple mechanical reprocessing and thus, prolonging their service life. The materials presented here are promising candidates for a closed-loop environment where maximum utilization of a material is realized.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Swedish Research Council Formas (2016-00700). Dr Karin Adolfsson is gratefully acknowledged for helping with the XRD and EDS measurements.References
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef
.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed
- Y. X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed
- W. Q. Yuan, G. L. Liu, C. Huang, Y. D. Li and J. B. Zeng, Macromolecules, 2020, 53, 9847–9858 CrossRef CAS
- H. Memon, H. Liu, M. A. Rashid, L. Chen, Q. Jiang, L. Zhang, Y. Wei, W. Liu and Y. Qiu, Macromolecules, 2020, 53, 621–630 CrossRef CAS
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed
- F. Stempfle, P. Ortmann and S. Mecking, Chem. Rev., 2016, 116, 4597–4641 CrossRef CAS PubMed
- P. Ortmann, T. A. Lemke and S. Mecking, Macromolecules, 2015, 48, 1463–1472 CrossRef CAS
- M. A. R. Meier, Macromol. Rapid Commun., 2019, 40, 1–10 CrossRef PubMed
- N. Kolb, M. Winkler, C. Syldatk and M. A. R. Meier, Eur. Polym. J., 2014, 51, 159–166 CrossRef CAS
- M. Winkler and M. A. R. Meier, Green Chem., 2014, 16, 3335–3340 RSC
- P. H. Nguyen, S. Spoljaric and J. Seppälä, Eur. Polym. J., 2018, 109, 16–25 CrossRef CAS
- P. H. Nguyen, S. Spoljaric and J. Seppälä, Polymer, 2018, 153, 183–192 CrossRef CAS
- Z. Guo, W. Wang, Y. Yang, K. Majeed, B. Zhang, F. Zhou and Q. Zhang, Polym. Chem., 2021, 12, 2009–2015 RSC
- M. Wu, L. Yuan, F. Jiang, Y. Zhang, Y. He, Y. Z. You, C. Tang and Z. Wang, Chem. Mater., 2020, 32, 8325–8332 CrossRef CAS
- L. Song, T. Zhu, L. Yuan, J. Zhou, Y. Zhang, Z. Wang and C. Tang, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed
- F. Lossada, D. Jiao, X. Yao and A. Walther, ACS Macro Lett., 2020, 9, 70–76 CrossRef CAS
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS
- J. H. Chen, W. Q. Yuan, Y. D. Li, Y. X. Weng and J. B. Zeng, ACS Sustainable Chem. Eng., 2019, 7, 15147–15153 CrossRef CAS
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2020, 8, 17272–17279 CrossRef CAS
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed
- F. Elizalde, R. H. Aguirresarobe, A. Gonzalez and H. Sardon, Polym. Chem., 2020, 11, 5386–5396 RSC
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed
- F. Cuminet, S. Caillol, E. Dantras, E. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS
- E. K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC
- A. Rekondo, R. Martin, A. Ruiz De Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC
- J. Dong, B. Liu, H. Ding, J. Shi, N. Liu, B. Dai and I. Kim, Polym. Chem., 2020, 11, 7524–7532 RSC
- C. Zhang, H. Liang, D. Liang, Z. Lin, Q. Chen, P. Feng and Q. Wang, Angew. Chem., 2020, 510642, 4289–4299 Search PubMed
- M. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2020, 53, 8367–8373 CrossRef CAS
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Polym. Chem., 2015, 6, 4271–4278 RSC
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS
- A. Ruiz De Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC
- J. H. Chen, D. D. Hu, Y. D. Li, F. Meng, J. Zhu and J. B. Zeng, Polymer, 2018, 143, 79–86 CrossRef CAS
- Y. Y. Liu, J. He, Y. D. Li, X. L. Zhao and J. B. Zeng, Ind. Crops Prod., 2020, 153, 112576 CrossRef CAS
- G. Hua and K. Odelius, Biomacromolecules, 2018, 19, 1573–1581 CrossRef CAS
- C. Pronoitis, G. Hua, M. Hakkarainen and K. Odelius, Macromolecules, 2019, 52, 6181–6191 CrossRef CAS
- X. Y. Jian, X. P. An, Y. D. Li, J. H. Chen, M. Wang and J. B. Zeng, Macromolecules, 2017, 50, 5729–5738 CrossRef CAS
- R. Hill and E. E. Walker, J. Polym. Sci., 1948, 3, 609–630 CrossRef CAS
- Y. Kinoshita, Makromol. Chem., 1959, 33, 1–20 CrossRef CAS
- W. G. Perkins and R. S. Porter, J. Mater. Sci., 1977, 12, 2355–2388 CrossRef CAS
- N. S. Murthy, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1763–1782 CrossRef CAS
- T. G. Heafield, G. Hopkins and L. Hunter, Nature, 1942, 149, 218 CrossRef CAS
- L. M. Gregoret, S. D. Rader, R. J. Fletterick and F. E. Cohen, Proteins: Struct., Funct., Bioinf., 1991, 9, 99–107 CrossRef CAS PubMed
- D. L. Howard and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2008, 10, 4113–4118 RSC
- A. Pascual, H. Sardon, A. Veloso, F. Ruipérez and D. Mecerreyes, ACS Macro Lett., 2014, 3, 849–853 CrossRef CAS
- F. Magliozzi, A. Scali, G. Chollet, H. Cramail, D. Montarnal and E. Grau, ACS Sustainable Chem. Eng., 2020, 8, 9125–9135 CrossRef CAS
- J. Meng, Y. Zeng, P. Chen, J. Zhang, C. Yao, Z. Fang, P. Ouyang and K. Guo, Macromol. Mater. Eng., 2020, 305, 1–8 Search PubMed
- G. D'escamard, C. De Rosa and F. Auriemma, AIP Conf. Proc., 2016, 1736, 020176 CrossRef
- D. Chan, Y. Ding, R. H. Dauskardt and E. A. Appel, ACS Appl. Mater. Interfaces, 2017, 9, 42217–42224 CrossRef CAS PubMed
- Y. Gu, J. Zhao and J. A. Johnson, Trends Chem., 2019, 1, 318–334 CrossRef CAS
- C. W. Bunn, Trans. Faraday Soc., 1939, 35, 482–491 RSC
- M. P. F. Pepels, M. R. Hansen, H. Goossens and R. Duchateau, Macromolecules, 2013, 46, 7668–7677 CrossRef CAS
- P. Ortmann, I. Heckler and S. Mecking, Green Chem., 2014, 16, 1816–1827 RSC
- L. Fontana, M. Santoro, R. Bini, D. Q. Vinh and S. Scandolo, J. Chem. Phys., 2010, 133, 204502 CrossRef PubMed
- K. Tashiro, S. Sasaki and M. Kobayashi, Macromolecules, 1996, 29, 7460–7469 CrossRef CAS
- J. Fernández, H. Amestoy, H. Sardon, M. Aguirre, A. Larrañaga Varga and J. R. Sarasua, J. Mech. Behav. Biomed. Mater., 2016, 64, 209–219 CrossRef PubMed
- C. W. Bunn, J. Polym. Sci., 1955, XVI, 323–343 CrossRef
- K. Saotome and H. Komoto, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 1463–1473 CrossRef CAS
- M. Ehrenstein, P. Smith and C. Weder, Macromol. Chem. Phys., 2003, 204, 1599–1606 CrossRef CAS
- M. Ehrenstein, S. Dellsperger, C. Kocher, N. Stutzmann, C. Weder and P. Smith, Polymer, 2000, 41, 3531–3539 CrossRef CAS
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS
- M. D. Stern and A. V. Tobolsky, J. Chem. Phys., 1946, 14, 93–100 CrossRef CAS
- W. Ge, B. Zhao, W. Liu, K. Nie and S. Zheng, Macromol. Rapid Commun., 2021, 42, 2000718 CrossRef CAS PubMed
- J. Deng, X. Kuang, R. Liu, W. Ding, A. C. Wang, Y. C. Lai, K. Dong, Z. Wen, Y. Wang, L. Wang, H. J. Qi, T. Zhang and Z. L. Wang, Adv. Mater., 2018, 30, 1–10 Search PubMed
- A. Takahashi, T. Ohishi, R. Goseki and H. Otsuka, Polymer, 2016, 82, 319–326 CrossRef CAS
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS
-
Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 1st edn, 2007 Search PubMed
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1–8 Search PubMed
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, ACS Macro Lett., 2019, 8, 1511–1516 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00811k |
| This journal is © The Royal Society of Chemistry 2021 |