
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of heterogeneous and homogeneous polymerisation on the structure of pNIPAm nanogels†
Alena
Vdovchenko
a,
Amanda K.
Pearce
 b,
Mark
Freeley
a,
Rachel K.
O'Reilly
b and
Marina
Resmini
*a
b,
Mark
Freeley
a,
Rachel K.
O'Reilly
b and
Marina
Resmini
*a
aSchool of Physical and Chemical Science, Queen Mary University of London, London E1 4NS, UK. E-mail: m.resmini@qmul.ac.uk
bSchool of Chemistry, University of Birmingham, Birmingham B15 2TT, UK
First published on 12th November 2021
Abstract
The thermoresponsive behaviour of cross-linked poly(N-isopropylacrylamide) (pNIPAm) nanogels makes these materials particularly attractive for a variety of applications. Literature data report the use of different methodologies for preparing nanogels, which can be divided into heterogeneous and homogeneous polymerisation approaches. Heterogeneous polymerisation occurs above the volume phase transition temperature (VPTT) of pNIPAm due to water expulsion from the network of the forming polymer. On the contrary, homogeneous polymerisation is conducted below the VPTT, so that the nanogel is in the swollen state during the polymerisation process. Here, we study the effect of phase separation during polymerisation, which reveals a significant influence on the particle size and internal structure, as well as on the thermoresponsive and interfacial behaviour of pNIPAm nanomaterials. We propose that heterogeneous polymerisation leads to preferential localisation of hydrophilic initiator residues on the particle surface, while during homogeneous polymerisation, the initiator groups are distributed within the nanogel network. These results highlight the importance of the choice of polymerisation temperature as well as initiator for the synthesis of pNIPAm gels, as this significantly affects their characteristics and application.
Introduction
The attraction of using poly(N-isopropylacrylamide)-based (pNIPAm) materials as drug delivery systems1–3 stems from their thermoresponsive character, with volume phase transition temperatures (VPTT) occurring at physiological temperature.4–6 The isopropyl side groups of pNIPAm contribute to the hydrophobic character and surface activity of the polymer,7 while the overall pNIPAm remains highly hydrophilic.7,8 These features have led to pNIPAm gels being developed for multiple applications, such as temperature-sensitive emulsifiers,9 smart sensors,10–12 reversible switches,13,14 artificial muscles,15,16 and tissue engineering.17,18Different synthetic methodologies have been reported for the preparation of pNIPAm microgels/hydrogels, based on heterogeneous or homogeneous polymerisations.19 In the former approach the starting monomers are dissolved in the feed solution, but the resulting polymer is in the form of a dispersion in an immiscible liquid,20 often referred to as dispersion or precipitation polymerisation,21,22 or emulsion polymerisation if surfactant is used.23 In homogeneous polymerisation, both monomers and the resulting polymer are dissolved in the media, or swollen in the case of cross-linked gels.21 In the case of homogeneous polymerisation, the synthesis is performed either in aprotic solvents, such as DMSO24 or in water (at room temperature).25,26
Literature data frequently report information on polymer compositions; however, the effect of synthetic protocols on particle structures is not often discussed. The use of different synthetic methodologies raises the question of whether the same formulation can result in different structures and properties. The thermoresponsive nature of pNIPAm makes it an ideal system for studying the role of phase separation during polymerisation on the morphology of nanoparticles. When pNIPAm gels are dispersed in water, their behaviour is influenced by temperature. Below the VPTT, pNIPAm chains are in a highly hydrated state, while above the VPTT, the loss of the hydration shell leads to phase separation.27 By varying the temperature of the polymerisation solution, but keeping all other experimental conditions constant, the effect of heterogeneous and homogeneous polymerisation can be evaluated.
The impact of polymerisation temperature on the structure of cross-linked pNIPAm has previously been studied for macroscopic hydrogels.28 The main conclusion was that polymerisation at higher temperature, close to the VPTT, leads to inhomogeneities in the polymer network,29–31 which results in lower swelling of the hydrogels.30 Another approach was conducted for RAFT polymerisation of NIPAm, where the hetero-/homogeneous nature of polymerisation was controlled by the choice of the solvent, not the temperature.32 In this case, the phase separation that occurred during polymerisation in water as opposed to a water/alcohol mixture was shown to influence the reaction rate and molecular weight of polymers.
Even though literature data offer an insight into the changes in structure of pNIPAm as a result of phase separation during the synthesis, to the best of our knowledge, a thorough evaluation of the impact of homogeneous/heterogeneous polymerisation on the structure of pNIPAm nanogels has yet to be reported. In this study, we evaluate the effect of phase separation during free radical polymerisation on the morphology and properties of pNIPAm-based nanogels. By changing the temperature of polymerisation in aqueous solution, it was possible to switch between heterogeneous and homogeneous polymerisation (Fig. 1), while the use of different initiators allowed us to evaluate their impact on nanogel properties. The thermoresponsive behaviour of the different nanogels was investigated using dynamic and electrophoretic light scattering, as well as UV-Vis spectroscopy. The interfacial behaviour and surface properties of the nanogels were characterised using surface tensiometry and liquid-phase atomic force microscopy (AFM). These data, together with transmission electron microscopy (TEM) studies, provide evidence of the role that synthetic methodologies have in influencing the structure and properties of nanogels.
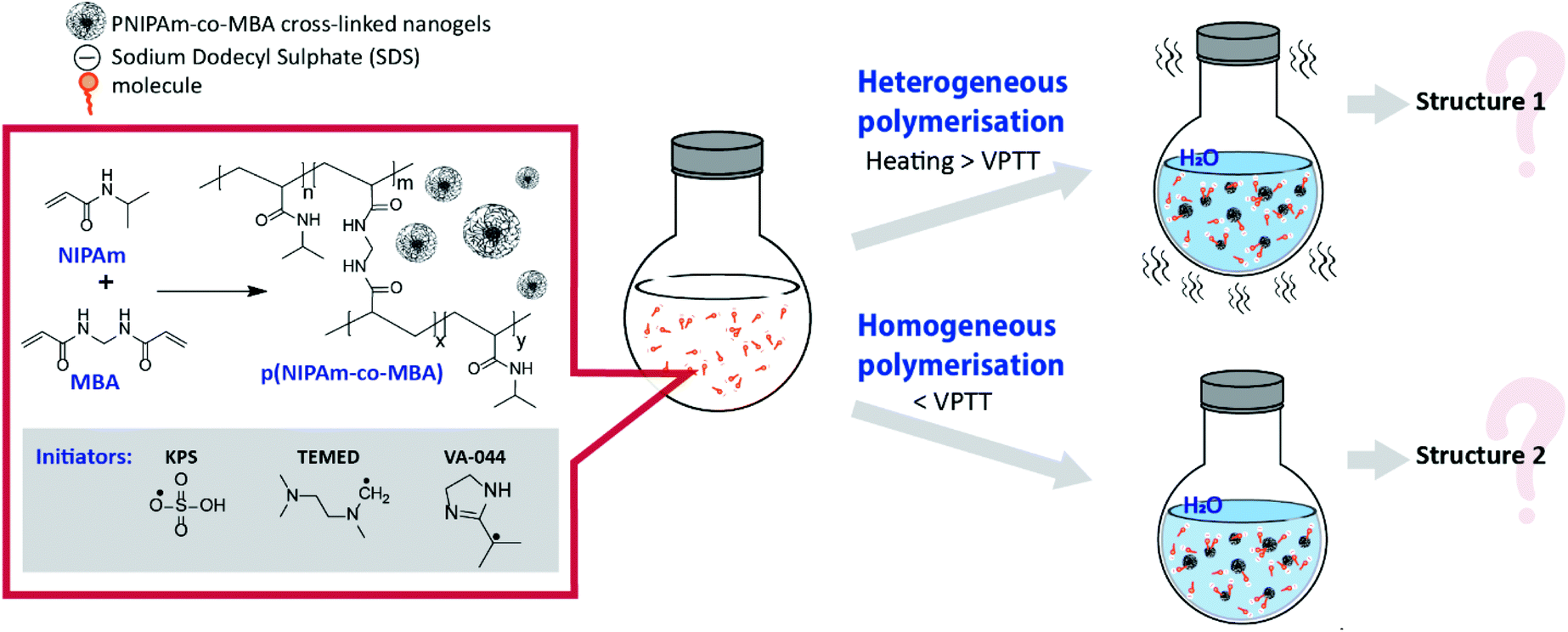 | ||
| Fig. 1 Scheme of synthesis of pNIPAm-based crosslinked gels by heterogeneous and homogeneous polymerisation. | ||
Experimental
Materials
All chemicals were used as received unless otherwise stated. N,N′-Methylenebisacrylamide (MBA), tetramethylethylenediamine (TEMED), sodium dodecyl sulphate (SDS), 1,2,4,5-tetramethylbenzene, deuterated DMSO ((CD3)2SO) were purchased from Sigma Aldrich (Gillingham, UK). N-Isopropylacrylamide (NIPAm) and azobisisobutyronitrile (AIBN) were purchased from Sigma Aldrich and used after recrystallisation from n-hexane and methanol, respectively. Potassium peroxydisulphate (KPS) was purchased from Alfa Aesar (Heysham, UK). 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) was purchased from FUJIFILM Wako (Hampshire, UK). Regenerated cellulose dialysis membranes Spectrum Spectra/Por (MWCO 6800 Da) were purchased from Fisher Scientific UK Ltd (Loughborough, UK). Silicon Blocks (N-type, Orientation 〈111〉, Dia 100 × 10 mm, Alineason Materials Technology GmbH, Frankfurt Germany) with one face optically polished (Lambda/10) were used for AFM studies, preliminarily coupled with trichloro(octyl)silane (Sigma Aldrich).Synthesis of nanogels
NIPAm and MBA in the molar ratio of 80/20% were dissolved in deionised water to give a total monomer concentration (Cm) of 1 w/v%. The solution was sealed in a round bottom flask (RBF), purged with N2 for 1 hour and heated (oil bath) until the polymerisation temperature (30, 50 or 80 °C) was reached. Solutions of the appropriate initiator (KPS, TEMED, VA-044) and SDS (where required) were prepared and sealed in separate RBFs, purged with N2 for 1 hour and added to the polymerisation mixture to start the reaction. The final amount of initiator (KPS or VA-044) used was 1% of the total moles of double bonds, while TEMED was used with a final concentration of 5%. For those nanogels requiring SDS, the final concentration of the surfactant in the polymerisation solution was either 0.5 mg mL−1 or 0.08 mg mL−1. The polymerisation reaction was maintained at the chosen temperature with stirring until full monomer conversion was achieved, as confirmed by 1H NMR spectroscopy. For polymerisations performed below the VPTT the temperature was monitored with a thermometer immersed into the feeding solution, to ensure that no temperature increase due to polymerisation would occur.33 This was not observed as a result of the low total monomer concentration (1 w/v%) used. Nanogel solutions were dialysed for 3 days (MWCO 3500 Da) against deionised water, then lyophilised (LTE scientific Lyotrap). 1H NMR spectroscopy was used to confirm the removal of SDS and any unreacted monomers (Fig. S1, ESI†). Reported yields refer to the isolated nanogels following freeze drying. The isolated nanogels, as powders, were stored in glass vials at room temperature.Quantification of monomer conversions by 1H NMR spectroscopy
Aliquots of the polymerisation solution were taken before the addition of the initiator and 0.5 h and 24 h after the start of the reaction to evaluate the conversion of NIPAm and MBA. 50 μL of the polymerisation solution was transferred into an NMR tube and added to 1,2,4,5-tetramethylbenzene dissolved in 450 μL of deuterated DMSO. The final concentration of the 1,2,4,5-tetramethylbenzene was 0.6 mg mL−1. 1H NMR spectra were recorded at 298 K using a Bruker DPX-400 spectrometer at 400 MHz or a Bruker DPX-300 spectrometer at 300 MHz. Spectra were processed with Bruker Topspin 4.0.6 software.The amount of monomer in the initial and final polymerisation solutions were determined by comparing the intensities of peaks at 5.61–5.67 ppm for NIPAM and 5.68–5.73 ppm for MBA, against the intensity of peak of the internal standard 1,2,4,5-tetramethylbenzene at 6.3 ppm.
Light scattering measurements
Assessment of thermoresponsive behaviour by UV-Vis
Optical transmittance at 500 nm was determined using Evolution 350 UV-Vis spectrophotometer equipped with Xenon Flash Lamp light source, Dual Matched Silicon Photodiodes detector and precise temperature controller between 0 °C and 90 °C. Analysis was performed in the temperature range between 20 and 65 °C. All measurements were carried out in deionised water. The nanogel solutions were transferred to quartz cells (Hellma) prior to measurements. The samples were heated at a rate of 0.5 °C min−1. The VPTT was determined as the temperature at which 50% loss of transmittance was observed. The calculations were made using OriginPro 9.0 software though the Gaussian fitting of the differential of transmittance depending on the temperature change.Transmission electron microscopy (TEM)
The evaluation of the nanogel morphology was performed using dry-state stained TEM imaging on JEOL JEM1230 microscope operating at an acceleration voltage of 100 kV. A suspension of nanoparticles prepared in deionised water at a concentration of 0.1 mg mL−1 were stained with aqueous 1 wt% uranyl acetate solution and dried on a copper grid covered with carbon film (S160, Mesh 200, Agar Scientific).Liquid-phase atomic force microscopy (AFM)
Liquid-phase AFM was carried out on a Bruker Dimension Icon system (Bruker, Billerica, MA) in PeakForce Quantitative Nanomechanical Mapping (QNM) mode. Samples were imaged at a resolution of 512 samples per line across 512 lines at a rate of 0.5 Hz with Bruker ScanAsyst Fluid + tips. AFM samples were prepared by submerging hydrophobic SiC8 substrates in glass Petri dishes filled with 0.1 mg mL−1 nanogel solution for 20 minutes prior to AFM imaging. The samples were imaged directly in the nanogel solution submerging the substrates by lowering the AFM probe into the Petri dishes. Images were analysed using Bruker Nanoscope Analysis 1.7 software.Surface tensiometry
Kinetics of nanogels’ adsorption at the air/water interface were analysed using Kruss K100 Force Tensiometer. Nanogels were dispersed in deionised water and poured into the PTFE trough prior to measurement. Analysis was conducted at 25 °C at nanogel concentrations in the range of 0.01–0.5 mg mL−1. Each measurement was repeated at least twice. When the plateau was reached steady-state surface tension was calculated as an average value over 1000 s of the analysis.Results and discussion
Effect of phase separation on nanogel morphology
In order to evaluate the effect of phase separation during polymerisation on the morphology of the resultant nanoparticles, two pNIPAm-based nanogels, NG1 and NG2, were prepared in water, both with 20% N,N′-methylenebisacrylamide (MBA) crosslinker, 1% total monomer concentration (Cm), SDS as surfactant and the same redox initiator KPS/TEMED (Table 1). The latter was chosen because it allows polymerisations to be carried out both below and above VPTT, while limiting the influence of temperature on the initiation process and polymer chain length.34| Nanogel | Initiator | C m, % | T, °C | [SDS], mg mL−1 | Conversion, % | Yield, % | Size, nm | PD | |
|---|---|---|---|---|---|---|---|---|---|
| NIPAm | MBA | ||||||||
| Data on NG1–5 represent the average value of three preparations with identical formulation, standard errors are included. All preparations were synthesised in water, with 20% MBA as crosslinker. Cm (%), T (°C), [SDS] (mg mL−1) – total monomer concentration, temperature and SDS concentration used during polymerisation. | |||||||||
| NG1 | 1% KPS, 5% TEMED | 1 | 80 | 0.5 | 85 ± 1 | 99 | 82 ± 1 | 154 ± 23 | 0.03 |
| NG2 | 30 | 99 | 99 | 78 ± 8 | 55 ± 13 | 0.27 | |||
| NG3 | 1% KPS | 80 | 99 | 99 | 91 ± 2 | 139 ± 8 | 0.03 | ||
| NG4 | 1% VA-044 | 50 | 99 | 99 | 94 ± 1 | 148 ± 13 | 0.12 | ||
| NG5 | 30 | 99 | 99 | 96 ± 4 | 55 ± 8 | 0.35 | |||
| NG6_KPS_45 | 1% KPS | 0.5 | 80 | 0.5 | ∼99 | ∼99 | 95 | 45 | 0.24 |
| NG7_KPS_130 | 1 | ∼99 | ∼99 | 95 | 130 | 0.03 | |||
| NG8_KPS_200 | 1.2% KPS | 1.2 | 0.08 | ∼99 | ∼99 | 85 | 200 | 0.07 | |
| NG9_TEMED_40 | 1% KPS, 5% TEMED | 0.5 | 30 | — | 90 | ∼99 | 86 | 40 | 0.26 |
| NG10_TEMED_80 | 1 | ∼99 | ∼99 | 76 | 80 | 0.24 | |||
SDS was used as a stabilizer for pNIPAm chains. The interactions between SDS and pNIPAm are known to be temperature dependent, with stronger binding occurring at lower temperatures, when considered in the range of 22–57 °C.35 This may have a significant influence on the polymerisation process studied at different temperatures since the association of SDS with pNIPAm leads to the formation of a polyelectrolyte complex.36,37 However, the use of SDS was deemed necessary to obtain nanosized particles by heterogeneous polymerisation without significantly decreasing the monomer concentration. The concentration of SDS was kept constant at 0.5 mg mL−1 for both preparations; this was above the critical aggregation concentration for pNIPAm-SDS complex when preparing nanogels.38,39 In addition, this concentration ensured that the polydispersity remained low26,27 and uniform nano-sized particles could be obtained25 (Table 1).
The pNIPAm nanogel preparations were synthesised in water by heterogeneous polymerisation at 80 °C (NG1) or homogeneous polymerisation at 30 °C (NG2), with monomer conversions >85% and yields of isolated polymers >78% (Table 1). High monomer conversions coupled with good chemical yields ensure a good correlation between feed formulations and polymer structure, allowing us to evaluate with confidence any changes in properties and morphologies. Particle size for NG1 and NG2, determined by DLS (Table 1), showed significant differences. High temperature polymerisation (NG1) led to the formation of larger particles with hydrodynamic diameter around 150 nm (measured at room temperature), while low temperature polymerisation led to nanogels (NG2) with particle size around 55 nm. It was previously reported that polymerisation above the VPTT of pNIPAm is characterised by approximately 20–40% lower water content in the forming polymer network.7,40 Since the nucleation process is driven by hydrophobic polymer-to-polymer interactions,41 relatively higher hydrophobicity during heterogeneous polymerisation may result in increased intermolecular attraction between polymer globules, leading to a lower particle concentration and, subsequently, larger size.
Interestingly particle size was not the only property that was affected by the choice of polymerisation temperature. The thermoresponsive behaviour of NG1 and NG2 was found to be different. NG2 exhibits aggregation above the VPTT due to increased attractive interactions between particles in the collapsed state,42,43 while NG1 is colloidally stable, but shrinks in size due to expulsion of water molecules from the polymer matrix (Fig. 2a). Besides DLS measurements, aggregation of NG2 is also observed by UV-Vis as a significant drop in transmittance of 0.5 mg mL−1 aqueous solution from 99% at 20 °C to 32% at 50 °C (Fig. 2b). For NG1, the drop in transmittance for 0.5 mg mL−1 solution is much less prominent, being about just 3%, where transmittance decreases from 88% at 20 °C to 85% at 50 °C (Fig. 2b). Higher concentration of particles in the colloidal solution may lead to increased aggregation and, therefore, higher decrease in transmittance of aqueous solution. Aggregation with more than 90% drop in transmittance is observed at higher concentrations of NG2 as well, however, for NG1 the drop in transmittance remains low (below 12%) with increasing concentration up to 4 mg mL−1 (Fig. S4 and S5, ESI†). This decrease in transmittance occurring for NG1 can be attributed to increased density of the polymer matrix due to particle shrinkage,44,45 but not to particle aggregation, as confirmed by DLS measurements in the concentration range evaluated (Fig. S6, ESI†). In literature, stabilisation of pNIPAm microgels/nanogels against coagulation above the VPTT is usually attributed to the presence of electrostatic repulsion originating from charged initiator residues incorporated into the particle structure.27 Indeed, this was previously reported for pNIPAm-based microgels below 100 nm in size, bearing anionic sulphate groups due to the KPS initiator.46,47 Therefore, the stability of NG1 to aggregation contrary to NG2 may be a result of electrostatic repulsion between particles, which also reflects the potential differences in particle structures. To study this further, we evaluated the presence of charges at the particle interface by measuring the electrophoretic mobility of their aqueous solutions (Fig. 2c). Electrophoretic mobility can be correlated with ζ-potential at the surface of solid particles; however, this conversion is less meaningful for soft gels with draining structure.48–50 Positive electrophoretic mobility of NG1 is retained over the entire investigated temperature range (20–50 °C), and it increases linearly with temperature. Positive mobility of NG1 reflects the presence of positive charged groups on the particle surface, which is probably due to the amine groups of TEMED. Therefore, this result is in agreement with the assumption made on initiator location, based on the stability of this nanogel above VPTT. The increase in electrophoretic mobility of NG1 with temperature can be associated with an increase in charge density due to the expulsion of water from the polymer matrix and rearrangement of the charged initiator residues towards the surface.51–55 On the contrary, for NG2, a sharp increase in electrophoretic mobility at VPTT corresponds to the formation of stabilised positively charged aggregated particles, since aggregation of NG2 above VPTT is observed by DLS and UV-Vis at this concentration (Fig. 2a and b). However, at a lower temperature below the VPTT, NG2 has near zero electrophoretic mobility. Since only the charges present in the draining shell of the particle contribute to the electrophoretic mobility,56,57 it can be speculated that NG2 does not bear a significant amount of charged initiator groups on the surface. At the same time, lower values of electrophoretic mobility can indicate a more soft and free-draining structure of gels, as was shown previously.57 Overall, the analysis of electrophoretic mobility of the two types of nanogels synthesised by heterogeneous and homogeneous polymerisation provides evidence of significant variations in their structure and therefore properties. Both nanogels have units of TEMED incorporated in their chemical structure, however the different synthetic methodology has led to the TEMED groups being positioned differently within the nanogel structure, therefore having an impact on morphology and behaviour.
 | ||
| Fig. 2 Comparison of physicochemical properties of NGs, synthesised by heterogeneous (NG1) and homogeneous (NG2) polymerisation; (a) changes in particle size upon volume phase transition measured by DLS for diluted solutions of NGs (0.5 mg mL−1), data presented as an average value over three measurements for one nanogel, error bars are below 1%; (b) transmittance of 0.5 mg mL−1 aqueous solutions of NGs at temperatures below (20 °C) and above (50 °C) the VPTT; (c) electrophoretic mobility of NG dispersions (0.5 mg mL−1) measured during heating of their solutions in 0.1 mM NaCl from 20 to 50 °C. | ||
To obtain additional information about the morphology and, in particular, the surface properties of nanogels, their interfacial behaviour was studied by surface tensiometry. Adsorption of pNIPAm at the air/water interface leads to a multilayer structure where the particles are deformed to various degrees depending on crosslinker-content.58–62 The adsorption time depends on the rate of particle diffusion to the interface as well as the adsorption energy.62 It was shown that pNIPAm with higher molecular weight reaches a surface equilibrium more slowly than the corresponding low molecular weight polymers,63 and similar behaviour can be expected for low crosslinked gels of different sizes. Our results indeed confirm this observation; the adsorption at the air/water interface of NG1, with larger average particle size, was slower than the smaller nanogel NG2 (Fig. 3a). However, adsorption kinetics can also be affected by structural features such as rigidity, determined by the cross-linker density, and surface chemistry, e.g. due to charged functional groups.22,63,64 The latter can create an electrostatic adsorption barrier due to repulsion between adjacent particles.62,64–66
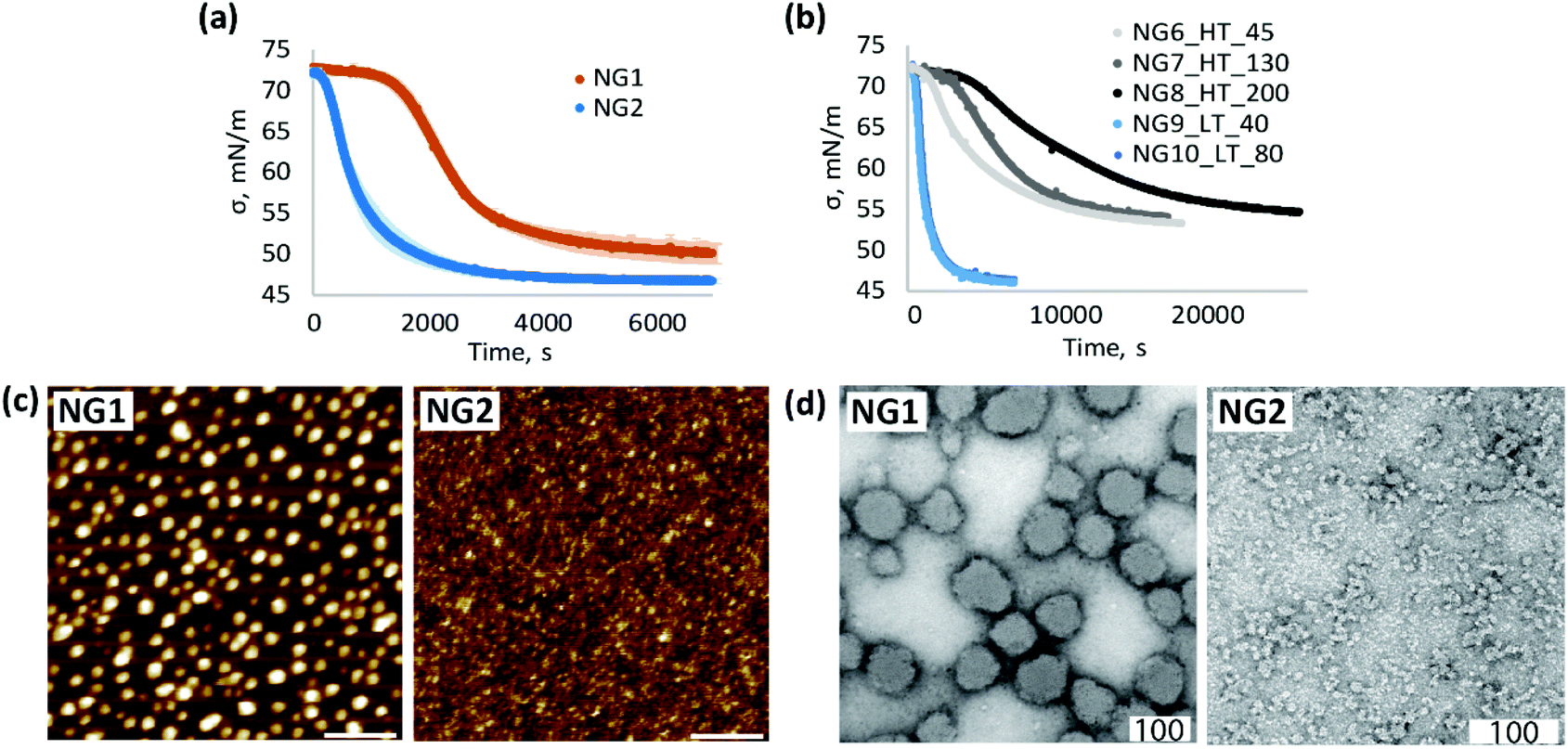 | ||
| Fig. 3 Comparison of physicochemical properties of NGs, synthesised by heterogeneous (NG1, NG6–8) and homogeneous (NG2, NG9–10) polymerisation (a) changes in surface tension of air/water interface over time due to adsorption of NGs (0.01 mg mL−1 bulk concentration), measured at room temperature; (b) adsorption kinetic at the air/water interface measured for nanogels NG6–NG8 synthesised at high temperature (HT) with Z-average sizes determined by DLS at RT of 45, 60 and 200 nm respectively and nanogels NG9–10 synthesised at low temperature (LT) with Z-average sizes of 40 and 80 nm; (c) liquid AFM images of 0.1 mg mL−1 aqueous solution of NGs adsorbed at hydrophobic interface, scale bar is 400 nm; (d) TEM images of NGs stained with 1 wt% uranyl acetate, scale bar is 100 nm. | ||
In order to study the impact of size, a new set of nanogels were prepared by varying the total monomer concentration in the formulation. Cm of 1% resulted in a Z-average particle size of 130 nm for the heterogeneous polymerisation (NG7_HT_130) and 80 nm for the homogeneous process (NG10_LT_80). A decrease in Cm to 0.5% led to nanogels with particle size of 45 nm (NG6_HT_45) and 40 nm (NG9_LT_40) at high and low temperature respectively. A Cm of 1.2% and a lower SDS concentration of 0.08 mg mL−1 accounted for the larger particle size of NG8_HT_200, synthesised by heterogeneous polymerisation. Surface tensiometry data of these nanogels are shown in Fig. 3b (polymerisation conditions are summarised in Table 1).
The results provide evidence that the polymerisation temperature plays a role in determining the adsorption kinetic profiles. In the case of heterogeneous polymerisation, at high temperature, changes in particle size result in different absorption profiles (NG6–NG8). Instead, at low temperature, variation in particle size has no influence. These results suggest that changes in size are not the only contributing factors influencing interfacial behaviour and adsorption kinetics, and in fact, the synthetic methodology is also important; comparison of the data for N6 and N9 shows a remarkable difference in the adsorption profiles despite the two nanogels having very similar particle sizes.
Based on the data presented in Fig. 3b we can therefore conclude that the slower adsorption of NG1 is not only due to the larger size but also to a change in particle morphology. The higher amount of positively charged initiator groups on the surface of NG1 compared to NG2, as evidenced by the electrophoretic mobility measurements, governs the increased hydrophilicity of the particle surface, subsequently slowing down the adsorption kinetics and reducing the surface activity.
At the same time, higher steady-state surface tension values of NG1 may be indicative of less material adsorbed at the air/water interface (Fig. 3a and Fig. S7, ESI†). One possible explanation of this difference can be the presence of charged TEMED residues on the particle surface of NG1 preventing full occupation of the hydrophobic surface and reducing the surface activity of the material. This trend is confirmed by liquid AFM measurements of a hydrophobic template immersed into aqueous solutions of nanogels (Fig. 3c). By using the same concentration of NG1 and NG2 solutions, it can be visually recognised that NG2 appears more densely adsorbed on the hydrophobic substrate, while NG1 particles stay isolated with no visible clustering.
A significant impact of the polymerisation temperature on the internal structure of nanogels can be observed not only through indirect characterisation of their structure, but also visually by TEM imaging (Fig. 3d and Fig. S8, S9, ESI†). According to TEM micrographs, NG1 particles in a dry state have a more uniform spherically shaped structure, while NG2 is more amorphous and non-spherically shaped. This is consistent with the data on polydispersity obtained by DLS (Table 1), where it is clear that phase separation during polymerisation led to the formation of particles with a very low PD of 0.03, compared to nanogels prepared in the homogeneous phase (PD = 0.27). The possible explanation of the observed differences in the particle morphology could be that polymerisation of NG1 above VPTT leads to increased hydrophobic interactions between isopropyl groups and minimisation of interaction of formed polymer chains with poor solvent, which results in the formation of a more spherical structure. At the same time, polymerisation below VPTT allows nanogels to stay in a swollen state, which leads to the formation of less spherical particles. However, it is worth keeping in mind that TEM images show structures of particles in the dry state, which can be affected by the deformability of a nanomaterial, and the non-spherical structure of NG2 may also reflect the formation of softer and more deformable nanogels than NG1.
Overall, the data indicate that the choice of the polymerisation temperature plays a key role in determining the particle internal structure. Nanogels synthesised by heterogeneous polymerisation have shown stability to aggregation above VPTT, high electrophoretic mobility and lower surface activity in comparison to nanogels synthesised at low temperature in the homogeneous solution. These differences indicate that the localisation of initiator residues in the polymer matrix is affected by the polymerisation method. The higher hydrophobicity of pNIPAm during polymerisation at temperatures above the VPTT leads to orientation of more hydrophilic initiator groups towards the particle surface, providing stability for the growing nanogels.67 A similar behaviour was observed in the synthesis of highly hydrophobic persulphate-initiated polystyrene particles, where covalently bonded sulphate groups were located at the particle interface.68,69 Although a pNIPAm latex is more hydrophilic than polystyrene, the same effect can occur due to hydrophobic interactions between isopropyl groups of pNIPAm, and it becomes prominent when comparing nanogels synthesised by heterogeneous and homogeneous polymerisation.
These differences in particle structure lead us to suggest that in the case of heterogeneous polymerisation conducted at high temperatures, the surface properties of nanomaterial are highly affected by the initiator choice, while this is less prominent in the case of homogeneous polymerisation conducted at low temperatures. To study this further, we evaluated the influence of the initiator choice on the physicochemical properties of nanogels synthesised by heterogeneous and homogeneous polymerisation.
Effect of the initiator choice during heterogeneous and homogeneous polymerisation
The incorporation of ionisable groups into the structure of pNIPAm-based gels can lead to changes in their thermoresponsive behaviour and swelling properties.70 Additionally, the hydrophobicity and bulkiness of chemical groups integrated into the network of pNIPAm particles can affect their thermoresponsive behaviour,71 cellular uptake and interactions with blood proteins.72 Therefore, the choice of initiator may play a role in influencing morphology, size, thermal response,73,74 biological performance75 and ultimately the application of these nanomaterials. Our data suggests that this effect can be further influenced by whether the particles are synthesised by homogeneous or heterogeneous polymerisation. To study this further, we compared properties of nanogels synthesised with KPS/TEMED and two radical thermal initiators KPS and VA-044 using heterogeneous and homogeneous polymerisation processes.Thus far, our results suggest that the hydrophilic initiator residues are predominantly located on the particle surface during heterogeneous polymerisation, and so the choice of initiator is expected to affect the surface properties of nanogels synthesised at high temperature. Indeed, a comparison of nanogels synthesised at high temperatures with KPS/TEMED (NG1), KPS (NG3) and VA-044 (NG4) reveals significant variations in their properties (Fig. 4a–e).
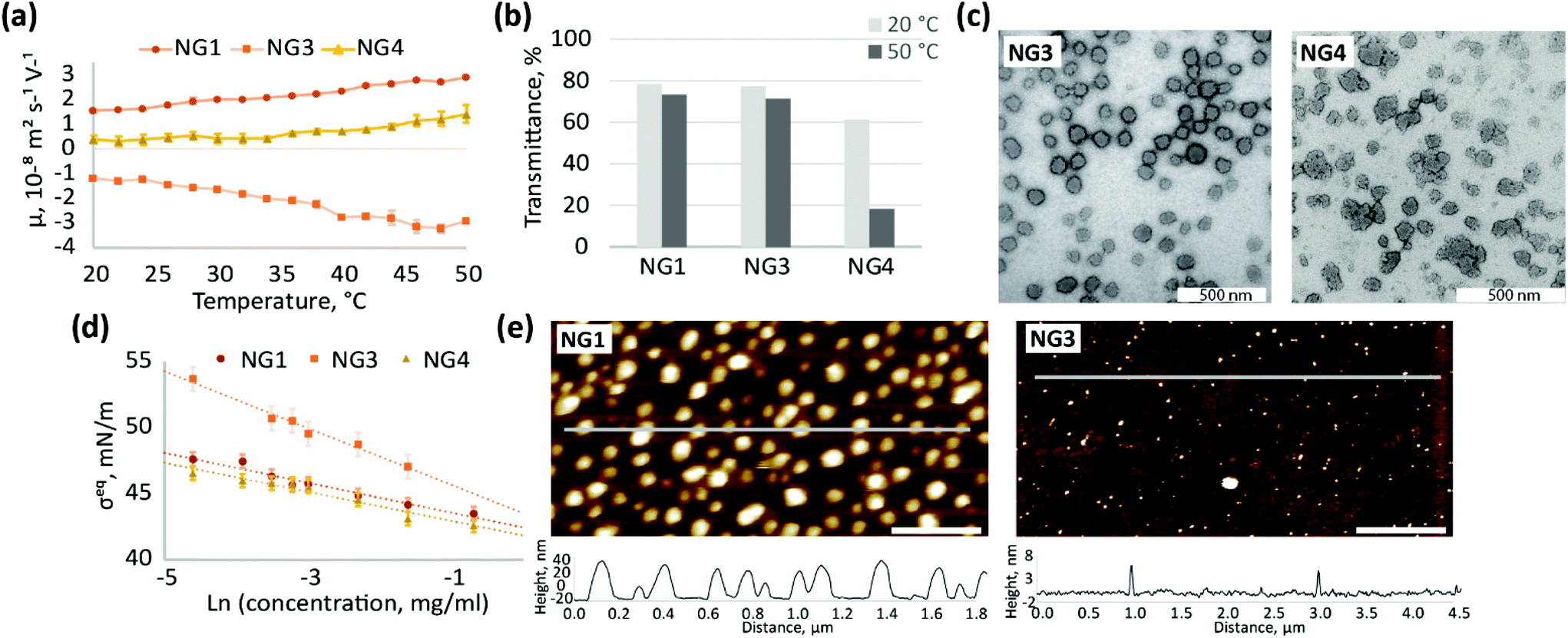 | ||
| Fig. 4 Comparison of physicochemical properties of NGs, synthesised by heterogeneous polymerisation with different initiators – NG1 (KPS/TEMED), NG3 (KPS), NG4 (VA-044); (a) electrophoretic mobility of NG dispersions (0.5 mg mL−1) at different temperatures; (b) transmittance of 1 mg mL−1 aqueous solutions of NGs recorded below (20 °C) and above (50 °C) the VPTT; (c) TEM images of NGs stained with 1 wt% uranyl acetate, scale bar is 500 nm; (d) changes in steady-state surface tension of air/water interface due to adsorption of aqueous solutions of NGs, measured at room temperature; (e) liquid AFM images of 0.1 mg ml−1 aqueous solution of NGs adsorbed at hydrophobic interface, scale bar is 400 nm. | ||
Different initiators (Fig. 1) result in different charge on the particle surface, and as a consequence, different electrophoretic mobility (Fig. 4a). The observed high positive charge of NG1 is due to amino groups originating from TEMED, while the negative charge observed for NG3 can be attributed to the incorporation of sulphate groups from KPS, as well as carboxyl groups from the hydrolysis of alkyl-sulphates as previously reported.76 The weak ionisation of imidazoline residues (pKa = 10.2) coming from azo-initiator VA-044 leads to a decrease in the absolute values of electrophoretic mobility of NG4. Utashiro et al. observed similar results, where the absolute value of the ζ-potential increased with temperature, while the sign depended on the choice of the initiator for NIPAm synthesised in heterogeneous polymerisation.48 They also reported the influence of an initiator on the size and swelling behaviour of microgels, although this is not evident in the preparation of nanosized particles in our study (Fig. S10, ESI†). However, we observe that the ionisation state of the particle surface affects their macroscopic behaviour and colloidal stability at temperatures above VPTT. Thus, NG4 shows a larger drop in transmittance of about 40% as temperature increases from 20 to 50 °C in comparison to a very small drop observed for NG1 and NG3 (Fig. 4b). The more significant loss in transmittance observed for NG4 is hypothesised to result from aggregation due to insufficient electrostatic repulsion between particles, which is supported by the lower electrophoretic mobility of this nanogel (Fig. 4a). At 50 °C the absolute value of mobility of NG4 in the electric field is 1.4 ± 0.4 × 10−8 m2 s−1 V−1, in comparison to 2.9 ± 0.1 × 10−8 m2 s−1 V−1 for both NG1 and NG3. Therefore, since NG1 and NG3 possess higher absolute values of charge on their surface, they remain colloidally stable at elevated temperatures.
The use of an initiator with lower ionisation results not only in higher propensity for agglomeration, but also in the formation of particles with a more amorphous and less uniform shape, which can be seen in TEM images for NG4 in comparison to NG1 and NG3 (Fig. 4c, further TEM images can be found on Fig. S8, S11 and S12, ESI†). Similarly to TEM, the analysis of the size distribution by DLS revealed a higher degree of polydispersity for NG4 (PD = 0.12) in comparison to NG1 and NG3 (both with PD = 0.03, Table 1), which indicates the formation of less uniform particles when an initiator with lower charge is used.
The localisation of initiator residues on the surface of nanogels should result in changes in surface activity and hydrophilicity between particles depending on the chemical structure of the initiator used. Indeed, the incorporation of highly hydrophilic sulphate groups from KPS into the structure of NG3 leads to higher steady-state surface tension values at the air/water interface in comparison to NG1 and NG4 over the studied concentration range between 0.01 and 0.5 mg mL−1 (Fig. 4d). The lower surface activity of NG3 also results in lower adsorption of nanogels at the hydrophobic interface, which is observed by AFM (Fig. 4e, more AFM images on Fig. S13, ESI†). The difference in the interfacial behaviour of NG3, observed by surface tensiometry and liquid AFM, can be attributed not only to the high hydrophilicity of the sulphate groups originating from KPS, but also to additional crosslinking on the particle surface that has been shown in the literature for these types of nanogels. NIPAm is able to form a network via self-crosslinking,77,78 which was confirmed for the KPS-initiator in contrast to the KPS/TEMED system, where the absence of self-crosslinking was shown.34,79 Therefore, lower surface activity of NG3 in comparison to NG1 can also be explained by higher crosslinking density, since a similar effect was shown for pNIPAm nanogels with different MBA content.59
As we suggested, using the example of NG2 synthesised with KPS/TEMED, the initiator residues are distributed within the polymer network of nanogels obtained by homogeneous polymerisation. Therefore, it can be expected that the effect of the initiator on surface properties of nanogels synthesised by homogeneous polymerisation be less pronounced than on those synthesised in heterogeneous conditions. Indeed, nanogels synthesised at low temperature with KPS/TEMED (NG2) and VA-044 (NG5) initiators do not show electrophoretic mobility below the VPTT (Fig. 5a), which reflects a neutral charge on the particle surface accessible to ions from the medium. When the temperature is above the VPTT of pNIPAm, the electrophoretic mobility of both nanogels NG2 and NG5 increases. However, the higher mobility above the VPTT reflects the behaviour of aggregated particles, since both NG2 and NG5 are prone to aggregation as observed by UV-Vis measurements (Fig. 2b and Fig. S4, S14, ESI†). Therefore, the electrostatic repulsion on the particles’ interface is insufficient to prevent their aggregation. The increase in electrophoretic mobility observed for aggregated particles can be attributed to the reorientation of the initiator residues in the polymer network. At low temperature, these are hidden inside the polymer matrix, while when the temperature increases water molecules are expelled and the more hydrophilic groups tend to localise at the interface.
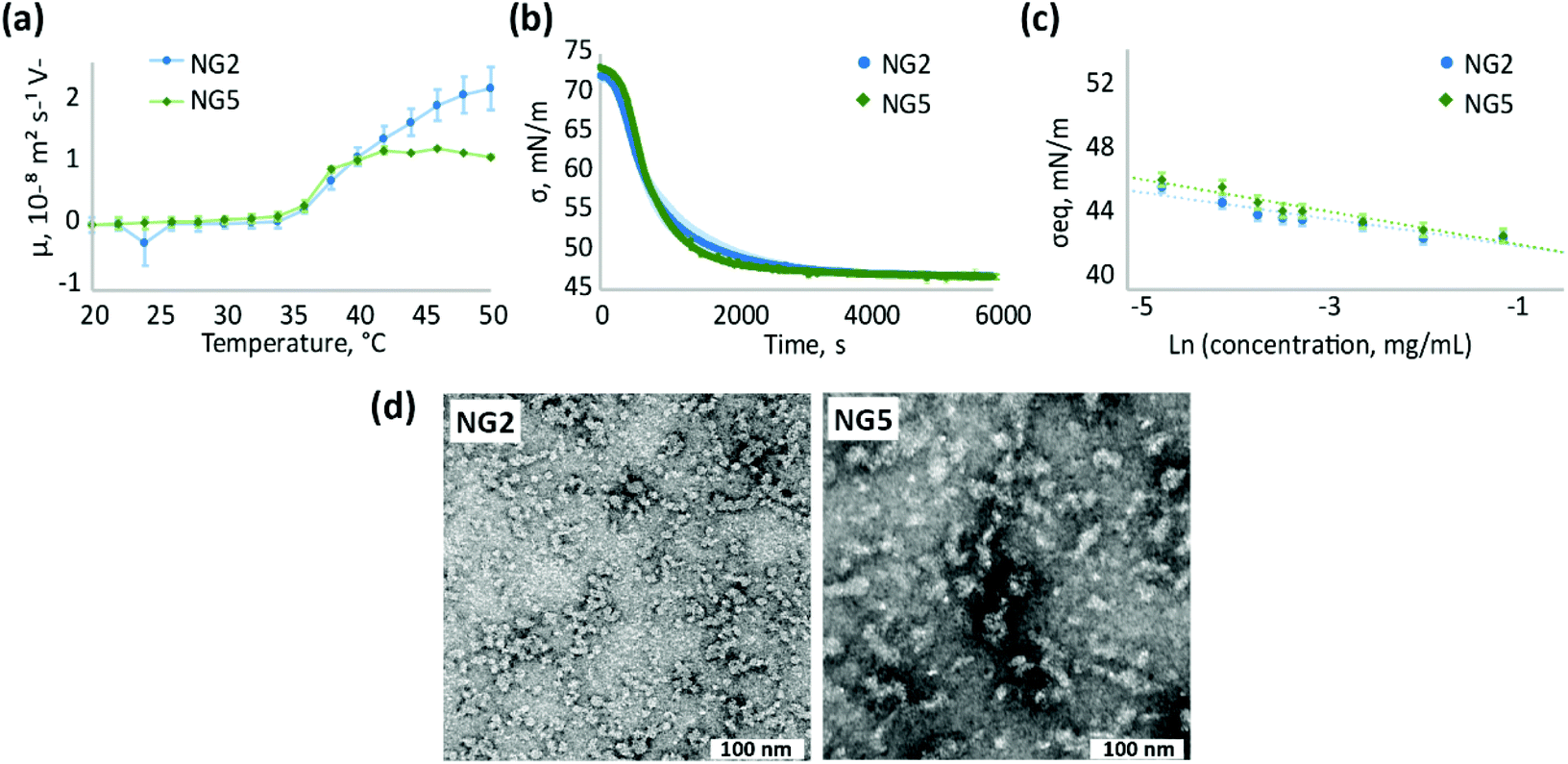 | ||
| Fig. 5 Comparison of physicochemical properties of NGs, synthesised by homogeneous polymerisation with different initiators – NG2 (KPS/TEMED), NG5 (VA-044); (a) electrophoretic mobility of NG dispersions (0.5 mg ml−1) at different temperatures; (b) changes in surface tension of air/water interface over time due to adsorption of NGs (0.01 mg ml−1 bulk concentration), measured at room temperature; (c) steady-state surface tension of air/water interface measured for aqueous solutions of NGs at different concentrations; (d) TEM images of NGs stained with 1 wt% uranyl acetate, scale bar is 100 nm. | ||
Similar to electrophoretic mobility measurements, NG2 and NG5 exhibit alike surface activity and adsorption kinetics at the air/water interface (Fig. 5b and c), confirming the suggestion that the choice of the initiator does not significantly affect the surface properties of nanogels synthesised at low temperature. At the same time, the change in initiator used does not affect particle morphology observed by TEM (Fig. 5c and Fig. S9, S15, ESI†) and particle size, analysed by DLS, since both NG2 and NG5 are characterised with Z-average hydrodynamic diameter around 55 nm (Table 1). Both nanogels show similar polydespersity (PD ∼ 0.3) that is considerably larger in comparison to particles formed during heterogeneous polymerisation (PD < 0.12).
Even though the surface properties are not significantly affected by the initiator choice in the case of homogeneous polymerisation, the effect can still be substantial for the application of nanomaterials. For example the distribution of charged groups within the polymer matrix can be of great importance in drug uploading. It has previously been shown that particles with core-localised carboxylic acid groups bind more cationic drug than microgels with surface-localised functionalisation.44 For particles synthesised with heterogeneous polymerisation, the surface functionalisation with charged residues from the initiator can define the behaviour of nanogels in vivo. Thus, a positive charge on the particle surface could lead to toxicity of the nanomaterials or promote their interaction with blood proteins.80
Conclusions
This study provides evidence that phase separation during free radical polymerisation of NIPAm has an impact on size, internal structure, thermoresponsive behaviour, and surface properties of nanoparticles. These parameters are important when developing nanomaterials for a variety of applications. Compared to a heterogeneous process, homogeneous polymerisation conducted below the VPTT results in the formation of smaller particles with initiator residues buried inside the nanogel network. The choice of initiator therefore has little influence on the surface properties of the pNIPAm nanogels synthesized at low temperature; however, it may have an impact on drug uploading or other characteristics of nanomaterial. The heterogeneous polymerisation conducted above the VPTT of pNIPAm, on the contrary, leads to the formation of uniform spherically shaped particles with initiator residues localised predominantly on the particle surface. This results in the significant influence of initiator chemical structure on the surface properties of the nanomaterials, and, subsequently, makes the choice of initiator an essential parameter to be considered, depending on the intended application.Author contributions
A. V.: writing – original draft, investigation, visualisation; A. P.: project administration, review & editing; M. F.: investigation, review & editing; R. OR.: conceptualisation, supervision, review & editing; M. R.: conceptualisation, funding acquisition, supervision, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 764958. The authors wish to thank Dr Zehua Li and Ms. Giulia Mastroianni for their help with TEM imaging.References
- R. Rajput, J. Narkhede and J. Naik, ADMET DMPK, 2020, 8, 1–15 CrossRef.
- A. Singh, K. Vaishagya, R. K. Verma and R. Shukla, AAPS PharmSciTech, 2019, 20, 1–14 CrossRef CAS PubMed.
- Y. Xu, M. Yang, Q. Ma, X. Di and G. Wu, New J. Chem., 2021, 45, 3079–3087 RSC.
- M. J. Taylor, P. Tomlins and T. S. Sahota, Gels, 2017, 3, 4 CrossRef PubMed.
- S. A. Papadimitriou, M. P. Robin, D. Ceric, R. K. O’Reilly, S. Marino and M. Resmini, Nanoscale, 2016, 8, 17340–17349 RSC.
- M. Cao, Y. Wang, X. Hu, H. Gong, R. Li, H. Cox, J. Zhang, T. A. Waigh, H. Xu and J. R. Lu, Biomacromolecules, 2019, 20, 3601–3610 CrossRef CAS PubMed.
- R. Pelton, J. Colloid Interface Sci., 2010, 348, 673–674 CrossRef CAS PubMed.
- M. Kwok and T. Ngai, J. Taiwan Inst. Chem. Eng., 2018, 92, 97–105 CrossRef CAS.
- R. Chen, N. Ren, X. Jin and X. Zhu, Colloids Surf., A, 2018, 538, 789–794 CrossRef CAS.
- D. G. Seo, Y. M. Kim, H. Ahn and H. C. Moon, Nanoscale, 2019, 11, 16733–16742 RSC.
- H. Zhang, T. Gao, S. Zhang, P. Zhang, R. Li, N. Ma, H. Wei and X. Zhang, Macromol. Chem. Phys., 2021, 222, 1–9 Search PubMed.
- Y. Zhang, K. Chen, Y. Li, J. Lan, B. Yan, L. Shi and R. Ran, ACS Appl. Mater. Interfaces, 2019, 11, 47350–47357 CrossRef CAS PubMed.
- C. Wang, W. Dong, P. Li, Y. Wang, H. Tu, S. Tan, Y. Wu and M. Watanabe, ACS Appl. Mater. Interfaces, 2020, 12, 42202–42209 CrossRef CAS PubMed.
- C. Wang, K. Hashimoto, R. Tamate, H. Kokubo and M. Watanabe, Angew. Chem., Int. Ed., 2018, 57, 227–230 CrossRef CAS PubMed.
- N. Park and J. Kim, Adv. Intell. Syst., 2020, 2, 1900135 CrossRef.
- Y. Tian, X. Wei, Z. J. Wang, P. Pan, F. Li, D. Ling, Z. L. Wu and Q. Zheng, ACS Appl. Mater. Interfaces, 2017, 9, 34349–34355 CrossRef CAS PubMed.
- C. A. Ribeiro, M. V. S. Martins, A. H. Bressiani, J. C. Bressiani, M. E. Leyva and A. A. A. de Queiroz, Mater. Sci. Eng., C, 2017, 81, 156–166 CrossRef CAS PubMed.
- Z. Atoufi, S. K. Kamrava, S. M. Davachi, M. Hassanabadi, S. S. Garakani, R. Alizadeh, M. Farhadi, S. Tavakol, Z. Bagher and G. H. Motlagh, Int. J. Biol. Macromol., 2019, 139, 1168–1181 CrossRef CAS PubMed.
- J. Ramos, A. Imazb and J. Forcada, Polym. Chem., 2012, 3, 852–856 RSC.
- R. Arshady, Colloid Polym. Sci., 1992, 270, 717–723 CrossRef CAS.
- N. Sanson and J. Rieger, Polym. Chem., 2010, 1, 965–977 RSC.
- M. Rey, X. Hou, S. Julia and N. Vogel, Soft Matter, 2017, 13, 8717–8727 RSC.
- Z. Li, N. J. Van Zee, F. S. Bates and T. P. Lodge, ACS Nano, 2019, 13, 1232–1243 CAS.
- P. Liu, C. M. Pearce, R. Anastasiadi, M. Resmini and A. M. Castilla, Polymers, 2019, 11, 353 CrossRef PubMed.
- J. Wei, H. Yu and H. Liu, J. Mater. Sci., 2018, 53, 12056–12064 CrossRef CAS.
- A. R. Kim, S. L. Lee and S. N. Park, Int. J. Biol. Macromol., 2018, 118, 731–740 CrossRef CAS PubMed.
- L. Pérez-Fuentes, D. Bastos-Gonzalez, J. Faraudo and C. Drummond, Soft Matter, 2018, 14, 7818–7828 RSC.
- S. Seiffert, Polym. Chem., 2017, 8, 4472–4487 RSC.
- J. Nie, B. Du and W. Oppermann, Macromolecules, 2004, 37, 6558–6564 CrossRef CAS.
- Y. Hirokawa, T. Okamoto, K. Kimishima, H. Jinnai, S. Koizumi, K. Aizawa and T. Hashimoto, Macromolecules, 2008, 41, 8210–8219 CrossRef CAS.
- Y. Hirokawa, H. Jinnai, Y. Nishikawa, T. Okamoto and T. Hashimoto, Macromolecules, 1999, 32, 7093–7099 CrossRef CAS.
- X. Wang, S. Li, Y. Su, F. Huo and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2188–2198 CrossRef CAS.
- S. Kara, O. Okay and O. Pekcan, J. Appl. Polym. Sci., 2002, 86, 3589–3595 CrossRef CAS.
- O. L. J. Virtanen, H. M. Ala-mutka and W. Richtering, Macromol. Chem. Phys., 2015, 216, 1431–1440 CrossRef CAS.
- N. P. Truong, C. Zhang, T. A. H. Nguyen, A. Anastasaki, M. W. Schulze, J. F. Quinn, A. K. Whittaker, C. J. Hawker, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2018, 7, 159–165 CrossRef CAS.
- J. Chen, X. Gong, H. Yang, Y. Yao, M. Xu, Q. Chen and R. Cheng, Macromolecules, 2011, 44, 6227–6231 CrossRef CAS.
- P. W. Zhu, J. Phys. Chem. B, 2015, 119, 359–371 CrossRef CAS PubMed.
- C. L. Usma, B. Lindman, V. Alfredsson, P. Taboada, C. S. Renamayor and I. E. Pacios, J. Mol. Liq., 2018, 265, 46–52 CrossRef CAS.
- M. Peng, S. Yuan, X. Shi and X. Lu, J. Appl. Polym. Sci., 2019, 47933, 6–11 Search PubMed.
- T. E. Oliveira, D. Mukherji, K. Kremer and P. A. Netz, J. Chem. Phys., 2017, 146, 034904 CrossRef PubMed.
- L. E. Theune, R. Charbaji, M. Kar, S. Wedepohl, S. Hedtrich and M. Calderón, Mater. Sci. Eng., C, 2019, 100, 141–151 CrossRef CAS PubMed.
- D. M. Heyes and A. C. Brańka, Soft Matter, 2009, 5, 2681–2685 RSC.
- L. Arleth, X. Xia, R. P. Hjelm, J. Wu and Z. Hu, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 849–860 CrossRef CAS.
- T. Hoare and R. Pelton, Curr. Opin. Colloid Interface Sci., 2008, 13, 413–428 CrossRef CAS.
- T. Hoare and R. Pelton, J. Colloid Interface Sci., 2006, 303, 109–116 CrossRef CAS PubMed.
- M. Andersson and S. L. Maunu, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3305–3314 CrossRef CAS.
- M. Rasmusson, A. Routh and B. Vincent, Langmuir, 2004, 20, 3536–3542 CrossRef CAS PubMed.
- Y. Utashiro, M. Takiguchi and M. Satoh, Colloid Polym. Sci., 2017, 295, 45–52 CrossRef CAS.
- H. Ohshima, J. Colloid Interface Sci., 1994, 163, 474–483 CrossRef CAS.
- M. Rasmusson, B. Vincent and N. Marston, Colloid Polym. Sci., 2000, 278, 253–258 CrossRef CAS.
- M. U. Witt, S. Hinrichs, N. Mo, S. Backes, B. Fischer and R. Von Klitzing, J. Phys. Chem. B, 2019, 123, 2405–2413 CrossRef CAS PubMed.
- W. Su, M. Yang, K. Zhao and T. Ngai, Macromolecules, 2016, 49, 7997–8008 CrossRef CAS.
- M. R. Islam, M. Tumbarello and L. A. Lyon, Colloid Polym. Sci., 2019, 297, 667–676 CrossRef CAS PubMed.
- S. Bochenek, A. Scotti, W. Ogieglo, A. Miguel, M. F. Schulte, R. A. Gumerov, N. V. Bushuev, I. I. Potemkin, M. Wessling, L. Isa and W. Richtering, Langmuir, 2019, 35, 16780–16792 CrossRef CAS PubMed.
- T. Lopez-Leon, J. L. Ortega-Vinuesa, D. Bastos-Gonzalez and A. Elaıssari, J. Phys. Chem. B, 2006, 110, 4629–4636 CrossRef CAS PubMed.
- I. Varga, A. Kardos, A. Borsos and T. Gilányi, J. Mol. Liq., 2020, 302, 111979 CrossRef CAS.
- T. Hoare and D. Mclean, J. Phys. Chem. B, 2006, 110, 20327–20336 CrossRef CAS PubMed.
- K. Zielińska, H. Sun, R. A. Campbell, A. Zarbakhsh and M. Resmini, Nanoscale, 2016, 8, 4951–4960 RSC.
- K. Zielińska, R. A. Campbell, A. Zarbakhsh and M. Resmini, Phys. Chem. Chem. Phys., 2017, 19, 17173–17179 RSC.
- Y. Cohin, M. Fisson, G. G. Fuller, N. Sanson and L. T. C, Rheol. Acta, 2013, 52, 445–454 CrossRef CAS.
- Z. Li, K. Geisel, W. Richtering and T. Ngai, Soft Matter, 2013, 9, 9939–9946 RSC.
- V. R. Dugyala, J. S. Muthukuru, E. Mani and M. G. Basavaraj, Phys. Chem. Chem. Phys., 2016, 18, 5499–5508 RSC.
- J. Zhang and R. Pelton, Colloids Surf., A, 1999, 156, 111–122 CrossRef CAS.
- M. Charlotte, E. Laurichesse, A. Perro, V. Ravaine and V. Schmitt, J. Colloid Interface Sci., 2019, 548, 1–11 CrossRef PubMed.
- S. A. Onaizi, Eur. Biophys. J., 2018, 47, 631–640 CrossRef CAS PubMed.
- O. Y. Milyaeva, G. Gochev, G. Loglio, R. Miller and B. A. Noskov, Colloids Surf., A, 2017, 532, 108–115 CrossRef CAS.
- E. Daly and B. R. Saunders, Langmuir, 2000, 16, 5546–5552 CrossRef CAS.
- R. H. Pelton, M. Pelton, A. Morphesis and R. L. Rowell, Langmuir, 1989, 5, 816–818 CrossRef CAS.
- J. A. Bonham, F. Waggett, M. A. Faers and J. S. van Duijneveldt, Colloid Polym. Sci., 2017, 295, 479–486 CrossRef CAS PubMed.
- R. Rivero, F. Alustiza, V. Capella, C. Liaudat, N. Rodriguez, P. Bosch, C. Barbero and C. Rivarola, Colloids Surf., B, 2017, 158, 488–497 CrossRef CAS PubMed.
- M. L. Ohnsorg, S. D. Jones, F. S. Bates and T. M. Reineke, Polym. Chem., 2019, 10, 3469–3479 RSC.
- D. Burnand, A. Milosevic, S. Balog, M. Spuch-Calvar, B. Rothen-Rutishauser, J. Dengjel, C. Kinnear and T. L. Moore, Small, 2018, 14, 1–9 Search PubMed.
- S. Chen, X. Jiang, L. Sun, S. Chen, X. Jiang and L. Sun, Part A: Pure Appl. Chem., 2014, 51, 447–455 CAS.
- S. Chen, L. Jiang, Y. Dan, S. Chen, L. Jiang and Y. I. Dan, J. Macromol. Sci., Part B: Phys., 2012, 51, 1057–1068 CrossRef CAS.
- S. Uchiyama, T. Tsuji, K. Kawamoto, K. Okano, E. Fukatsu, M. Kato, H. Koizumi and H. Tokuyama, Angew. Chem., 2018, 130, 1–6 CrossRef.
- A. Kardos, T. Gilányi and I. Varga, J. Colloid Interface Sci., 2019, 557, 793–806 CrossRef CAS PubMed.
- L. C. Kro, W. A. Kopp and K. Leonhard, J. Phys. Chem. B, 2017, 121, 2887–2895 CrossRef PubMed.
- M. Brugnoni, A. C. Nickel, L. C. Kröger and A. Scotti, Polym. Chem., 2019, 10, 2397–2405 RSC.
- O. Virtanen, Insight into Precipitation Polymerization of N-isopropylacrylamide: Reaction Mechanism, Particle Formation and Particle Structure, RWTH Aachen University, 2016 Search PubMed.
- J. Lee, D. Jeong, S. Seo and K. Na, J. Pharm. Invest., 2014, 43, 63–69 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01333e |
| This journal is © The Royal Society of Chemistry 2021 |