DOI:
10.1039/D0QO01058H
(Review Article)
Org. Chem. Front., 2021,
8, 445-465
Nitriles as radical acceptors in radical cascade reactions†
Received
31st August 2020
, Accepted 24th October 2020
First published on 26th October 2020
Abstract
The cyano group is a valuable and readily available functional group for the preparation of various functional groups, such as amines, carboxylic acids, and ketones. In recent decades, the radical cascade reaction has emerged as a versatile tool to prepare a large variety of functional molecules. The application of the cyano group as a radical acceptor in cascade reactions provides diverse opportunities for the convenient construction of various important heterocycles and carbocycles. Such synthetic strategies will open new ways for the rapid buildup of molecular complexity. The focus of this review is the summary of the dynamic field of radical cascade processes using the cyano group as a radical acceptor, which has not been well documented so far.
1. Introduction
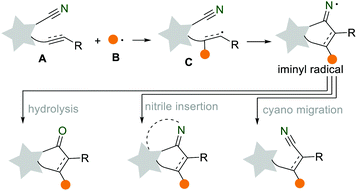
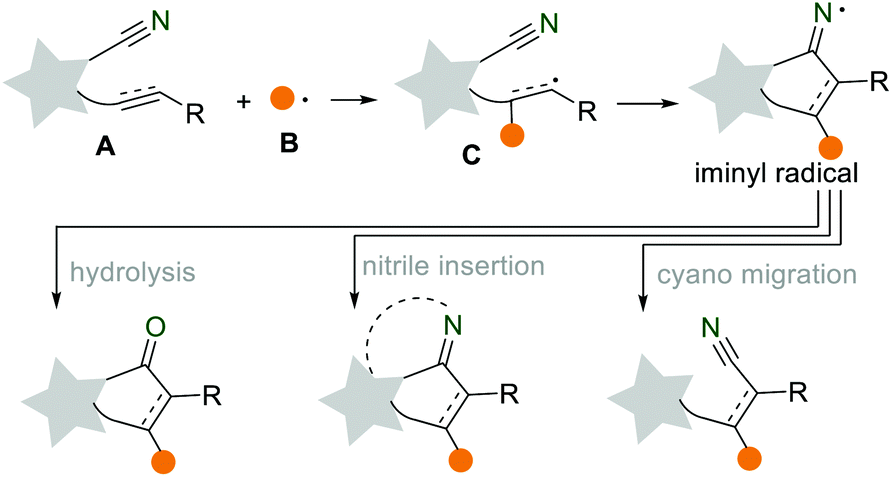
The development of simple, rapid, and efficient synthetic protocols for the preparation of complex organic molecules is always a goal, attracting the widespread interest of organic chemists. In the past few decades, the radical cascade reaction has been developed into a versatile synthetic platform to synthesize structurally diverse compounds with great atom-economy and step-economy.1 The rapid development of the radical cascade reaction largely depends on the continuous discovery of novel radical acceptors, such as alkenes, alkynes, cyano and isocyano groups.2 Among them, the cyano group is a valuable and readily available functional group for the preparation of various functional groups, such as amines, carboxylic acids, and ketones. Recently, the application of the cyano group as a radical acceptor in cascade reactions provides diverse opportunities for the convenient construction of various important heterocycles and carbocycles. As shown in Scheme 1, when the in situ generated radical B was added to the unsaturated CN-containing substrate A, an alkenyl radical C was formed. This was followed by an intramolecular cyclization to produce the key intermediate iminyl radical. Then, the iminyl radical underwent prior restoration and hydrolysis (either electron transfer or hydrogen abstraction), nitrile insertion, or cyano migration to form the corresponding ketone, N-heterocycle, and nitrile.3 We herein would like to summarize the recent progress of the radical cascade reactions using the cyano group as a radical acceptor. This review is organized into three sections based on the different transformations of iminyl radicals: (i) hydrolysis into cyclic ketones, (ii) nitrile insertion for N-heterocycles, and (iii) remote cyano migration. We hope this review provides a scientific auxiliary tool for researchers in radical chemistry, inspiring more strategic synthetic applications in this dynamic and exciting field.
 |
| | Scheme 1 The cyano group as a radical acceptor in the radical cascade reactions. | |
2. Hydrolysis into cyclic ketones
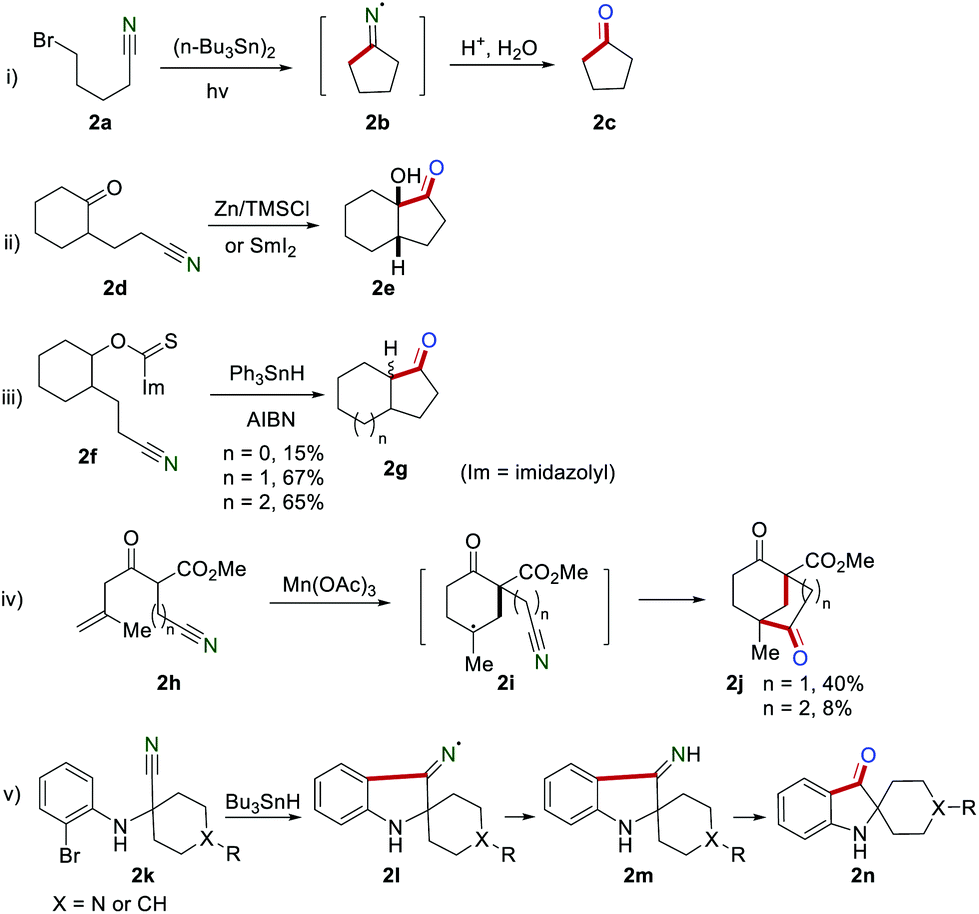
The application of the cyano group as a radical acceptor has attracted the increasing research interest of organic chemists for nearly half a century. The generated iminyl radicals are reduced before hydrolysis (either electron transfer or hydrogen abstraction) into cyclic ketones.4 For example, Ogibin's group described the preparation of cyclopentanone 2c in high yield via cyclization of 5-bromocyanopentane 2a in the presence of (n-Bu3Sn)2 as early as 1975 (Scheme 2i). Iminyl radical 2b was generated in this transformation, and the rate constant was estimated to be 4 × 104 s−1 at 80 °C reported by Grille and co-workers.5 In 1983, Corey and Pyne realized the synthesis of bicyclic ketone 2evia cyclization of the keto-nitrile 2d in the presence of zinc/trimethylchlorosiIane.6 A similar reaction could proceed in the presence of SmI2 (Scheme 2ii).7 In 1984, Clive and co-workers developed the construction of the fused ring systems 2g by employing triphenyltin hydride (Ph3SnH) and azobisisobutyronitrile (AIBN) to promote the generation of an iminyl radical via deoxygenation of alcohols and intramolecular cyclization (Scheme 2iii).8 In 1992, Snider and Buckman reported the cyclization of 2h for the generation of the bicyclic ketone 2j in the presence of Mn(OAc)3 (Scheme 2iv).9 In this study, the five-membered cyclic ketone (n = 1) could be obtained in 40% yield, while the six-membered cyclic ketone (n = 2) was only obtained in 8% yield. In 1999, Sulsky and co-workers realized the preparation of spiro[2H-indol]-3(1H)-ones 2n by cyclization of 2k in the presence of Bu3SnH via iminyl radical intermediate 2l (Scheme 2v).10
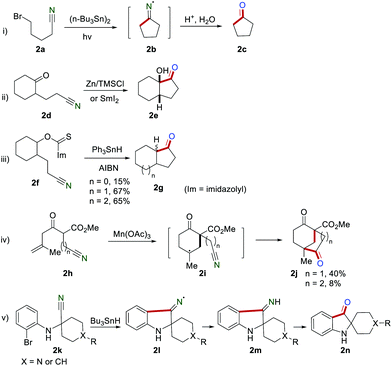 |
| | Scheme 2 Early studies on the iminyl radical for the construction of five-membered cyclic ketones. | |
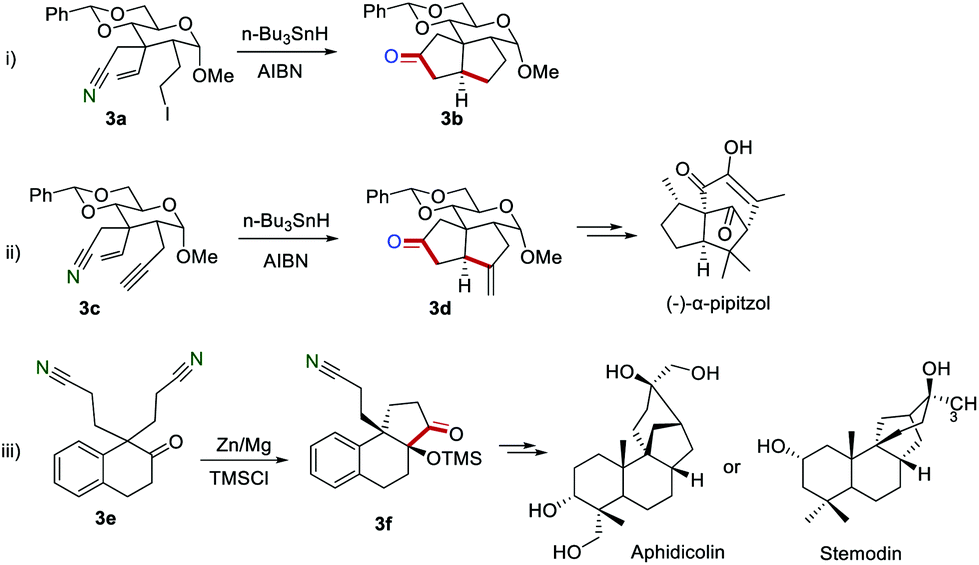
The employment of this effective strategy to synthesize the bio-related substances has also been reported. For example, Fraser-Reid and co-workers described the preparation of five-membered cyclic ketones 3b by the reaction of the carbohydrate framework 3a in the presence of n-Bu3SnH in 1986 (Scheme 3i).11 Later, the same group achieved the transformation of the carbohydrate framework 3c to 3d, which was a key intermediate for the tricyclic cedrenoid sesquiterpene (−)-α-pipitzol (Scheme 3ii).12 In 1995, Mann and Hegarty developed a new approach to access the key intermediate 3f, which could be further converted into the biologically natural products, aphidicolin or stemodin.13
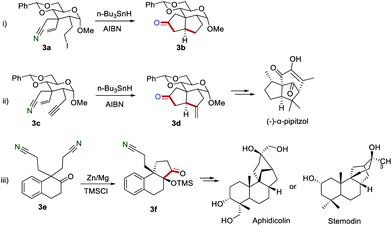 |
| | Scheme 3 The application of the cyano group as a radical acceptor to synthesize the bio-related substances. | |
The quite slow rate of the 6-exo cyclization onto the cyano group led to the challenge preparation of the six-membered cyclic ketones.14 The reaction could proceed smoothly to produce six-membered cyclic ketones only when the reactive centers were part of a more rigid system, where the unfavorable conformations were minimized. For example, Fraser-Reid and co-workers realized the synthesis of the six-membered cyclic ketones 4b by employing the constrained geometry of a 1,6-anhydrocarbohydrate 4a as the starting material in 1993 (Scheme 4i).15 In 1990, an electroreduction of the γ- and δ-cyanoketones 4c was developed by Shono and Kise to access α-hydroxyketones 4d and dehydroxylated ketones 4e (Scheme 4ii).16
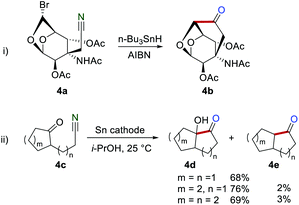 |
| | Scheme 4 Early studies on the iminyl radical for the construction of the six-membered cyclic ketones. | |
Although great achievements have been made, it is only in the last two decades that this knowledge has been widely applied in the preparation of cyclic ketones and explosive growth has been witnessed. In the following part, we will summarize the recent progress using the cyano group as a radical acceptor for the preparation of a vast array of cyclic ketones.
2.1 Synthesis of quinoline-2,4-dione derivatives
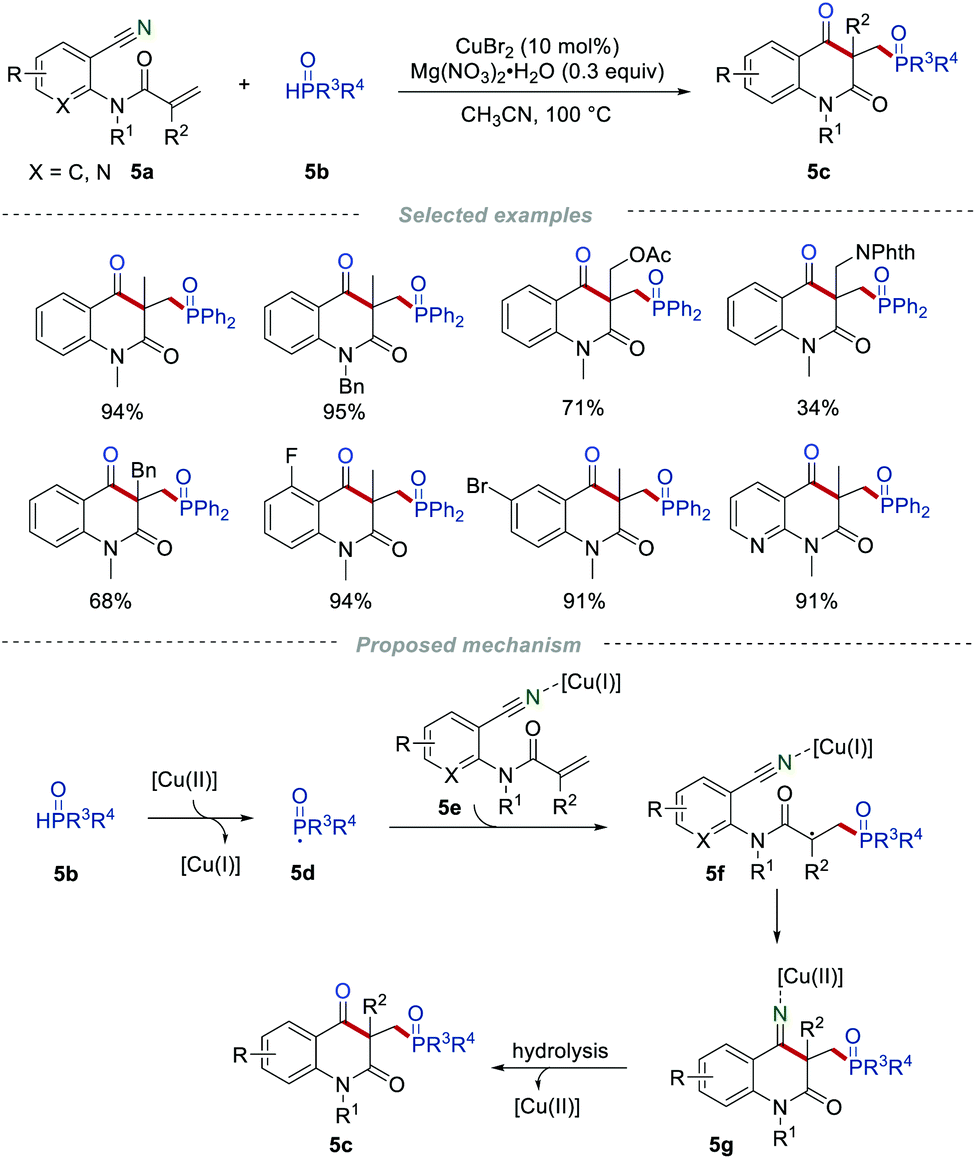
Quinoline-2,4-dione frameworks are frequently found in a large number of natural products, pharmaceuticals, and agrochemicals. In 2015, Li and co-workers reported the construction of quinoline-2,4(1H,3H)-diones via radical cascade reaction by reacting the readily accessible 2-cyanoarylacrylamides 5a with the H-phosphonates 5b (Scheme 5).17 A series of 2-cyanoarylacrylamides 5a bearing different functional groups and different kinds of H-phosphonates 5b were all examined, leading to the formation of the phosphorylated quinoline-2,4(1H,3H)-diones 5c in good yields. This efficient protocol enables the simultaneous formation of the C–C, C–O, and C–P bonds in one step, and is convenient to carry out gram-scale synthesis with similar efficiency. The radical-trapping experiments provided important support for the generation of the phosphoryl radicals in the reaction process.
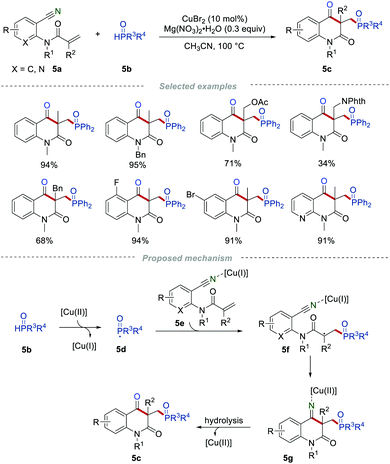 |
| | Scheme 5 The preparation of the phosphorylated quinoline-2,4-diones. | |
On the basis of a series of control experiments, a reasonable radical cyclization mechanism for this transformation was suggested. Initially, the phosphoryl radical 5d was generated from H-phosphonate 5b under the oxidation of Cu(II). Then, the selective addition of 5d to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of intermediate 5e would give an electrophilic carbon-centered radical 5f. 5f would further undergo an intramolecular addition to produce the intermediate 5g. Subsequently, the hydrolysis of 5g provided the desired product 5c with the release of the copper species.
C bond of intermediate 5e would give an electrophilic carbon-centered radical 5f. 5f would further undergo an intramolecular addition to produce the intermediate 5g. Subsequently, the hydrolysis of 5g provided the desired product 5c with the release of the copper species.
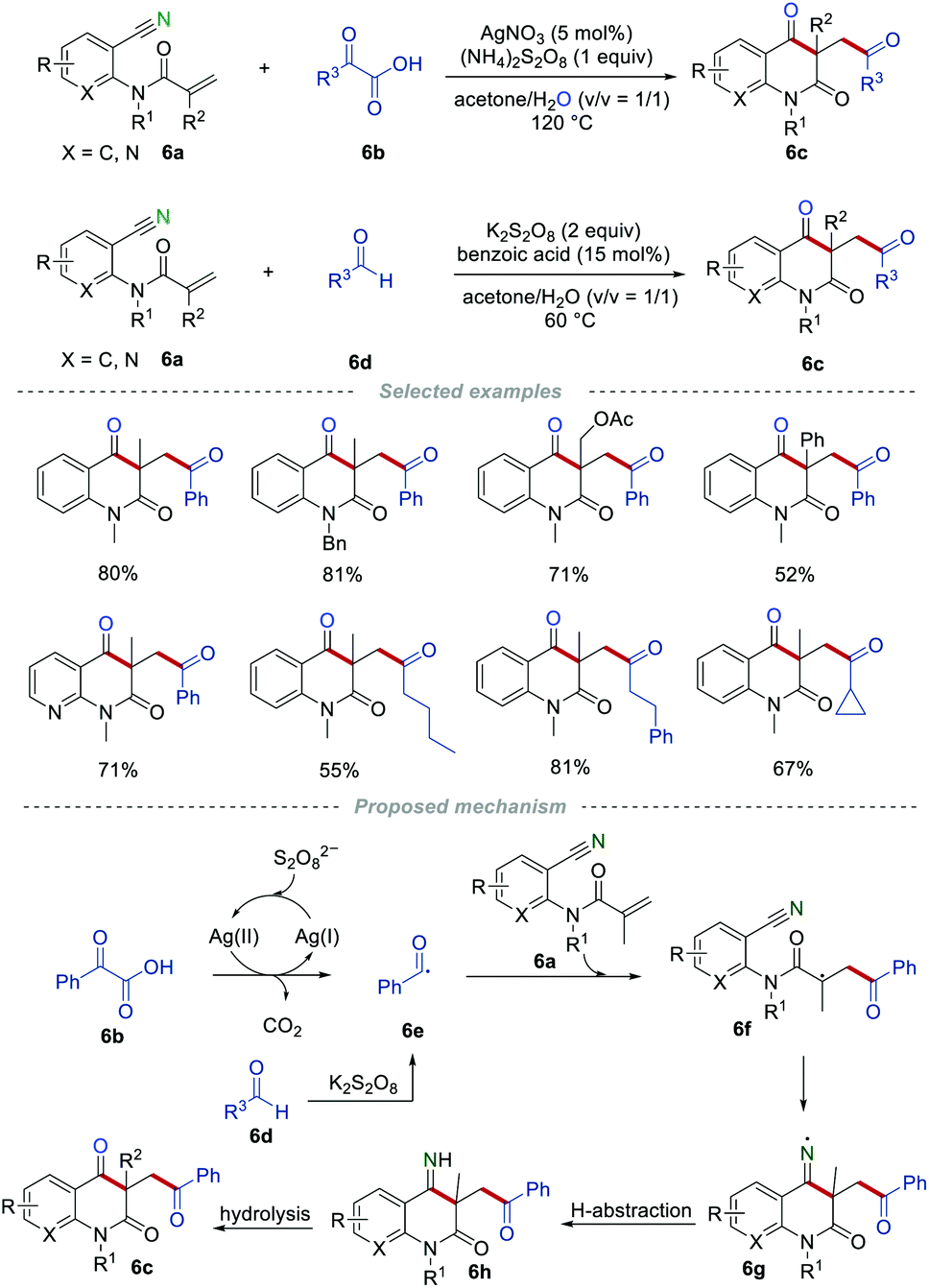
Since this pioneering study, the reactions of 2-cyanoarylacrylamides with various types of radical precursors have been studied in detail. In the past few decades, aldehydes and α-keto acids have been widely used as the acyl radical precursors in the presence of transition metals and oxidants.18 In 2016, Li and co-workers realized the construction of carbonyl-containing quinoline-2,4(1H,3H)-diones 6cvia the radical addition/cyclization of o-cyanoarylacrylamides 6a with α-keto acids 6b or aldehydes 6d (Scheme 6).19 Both aromatic and aliphatic aldehydes or α-keto acids were compatible in this transformation, and the cyclopropyl group could be tolerated during the reaction process without the ring-opening reaction. This strategy provides a simple and efficient protocol to access various carbonyl-containing quinoline-2,4(1H,3H)-diones 6c in good yields. The radical trapping experiments demonstrated that the reaction proceeds through a radical pathway. As it can be seen, acyl radical 6e was produced from α-keto acid 6b or aldehyde 6d. After undergoing a similar radical addition/cyclization process, the iminyl radical 6g was formed, which subsequently abstracted a hydrogen atom from the surrounding to give the imine 6h. Imine 6h was converted to the final product 6c through hydrolysis.
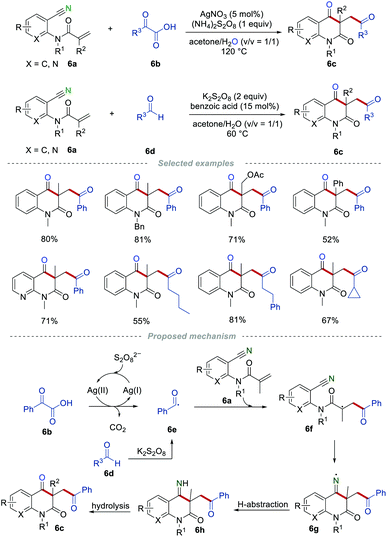 |
| | Scheme 6 The construction of the carbonyl-containing quinoline-2,4-diones. | |
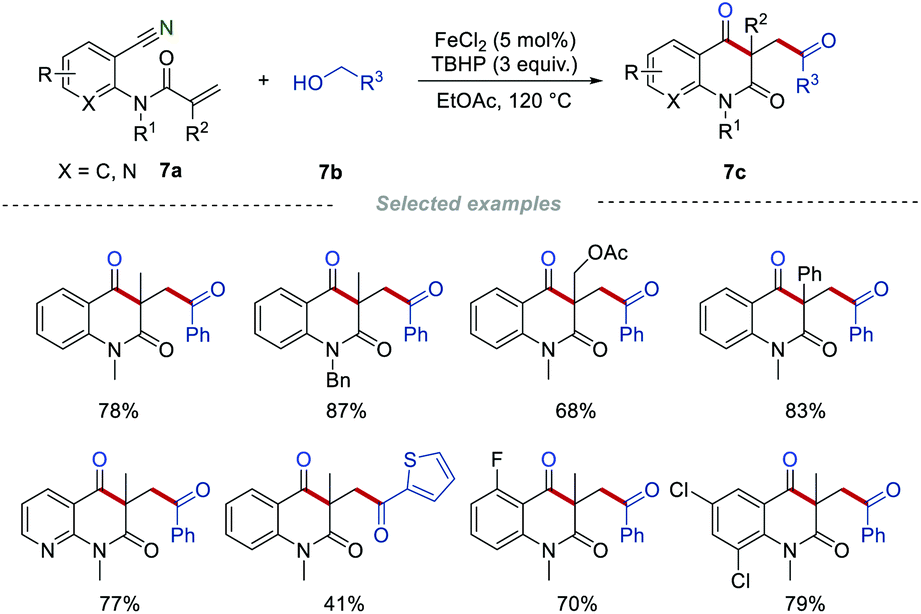
It is well known that alcohol could be oxidized into the corresponding aldehyde in the presence of suitable oxidants.20 In 2016, Li's group employed benzylic alcohols 7b instead of α-keto acids or aldehydes as acyl radical precursors to access carbonyl-containing quinoline-2,4-diones 7c (Scheme 7).21 This method provides an alternative to synthesizing the carbonyl-containing quinoline-2,4-diones 7c, showing good functional group compatibility. In this work, the oxidant of the benzylic alcohols 7b to the corresponding aldehydes in the presence of FeCl2/TBHP was critical. Mechanistic studies indicated that acyl radicals are important intermediates, which were produced from the in situ generated aldehydes.
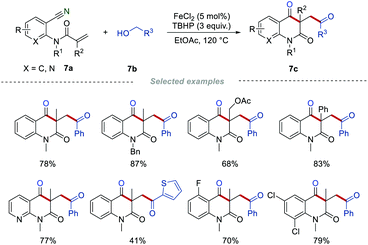 |
| | Scheme 7 The construction of carbonyl-containing quinoline-2,4-diones from alcohols. | |
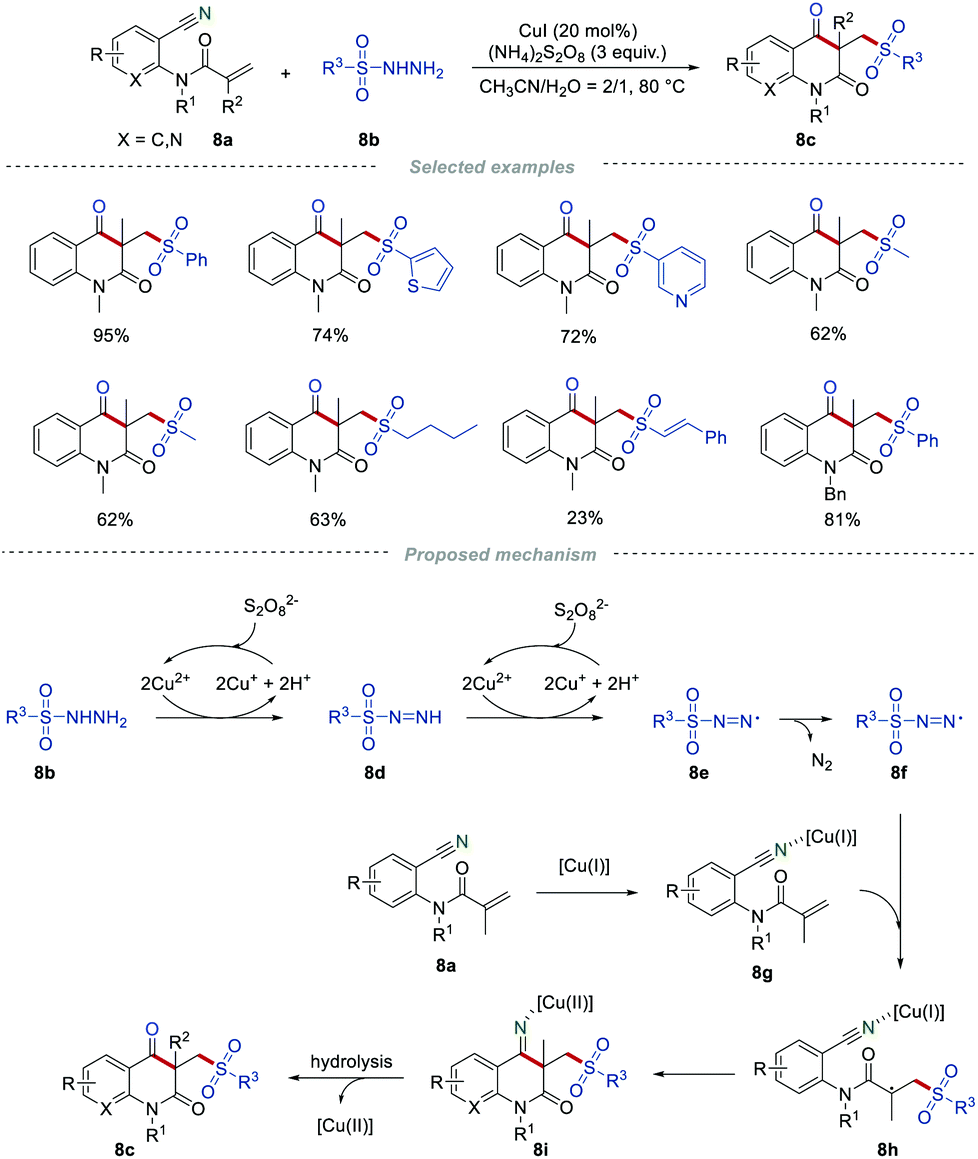
Sulfone-containing molecules are widely found in a large variety of biologically active compounds, drug molecules, and versatile synthetic intermediates.22 The addition of sulfonyl radicals to the unsaturated C–C bond has been an effective method for the synthesis of organic sulfones.23 In this context, Wang's group reported the synthesis of sulfonated quinoline-2,4-diones 8c by employing easily available sulfonyl hydrazides 8b as sulfonyl radical precursors (Scheme 8).24 There was a wide range of o-cyanoarylacrylamides 8a possessing diverse steric and electronic properties that were applicable in this transformation. Moreover, the aryl and alkyl sulfonyl hydrazides 8b, and even those sulfonyl hydrazides containing CC bonds were suitable in this reaction, producing sulfonated quinoline-2,4-diones 8c in moderate to good yields. Presumably, the sulfonyl radicals were generated from sulfonyl hydrazides via single electron transfer and the deprotonation process in the presence of CuI and (NH4)2S2O8. After the addition of the sulfonyl radical to the CC bond, intramolecular cyclization, and hydrolysis, the final product 8c was produced.
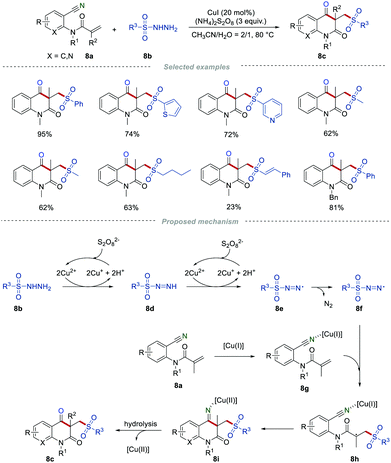 |
| | Scheme 8 The construction of sulfonated quinoline-2,4-diones. | |
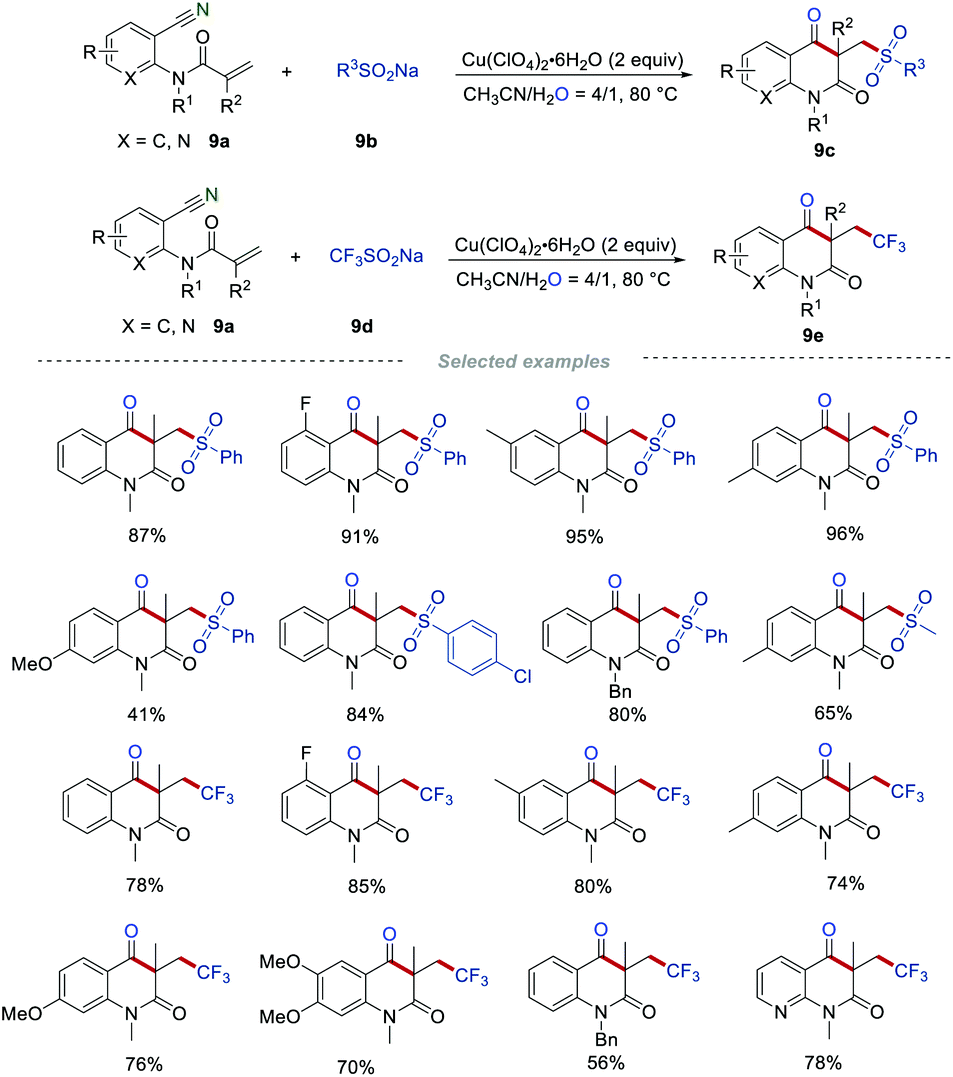
In contrast to Wang's procedure, Li's group25 employed sodium sulfinates 9b as sulfonyl radical precursors and Cu(ClO4)2 as a catalyst to access the sulfonated quinoline-2,4-diones 9c (Scheme 9). Besides the aromatic sulfonyl radical precursors, the aliphatic sulfonyl radical precursors also showed good reactivities in the preparation of the sulfonated quinoline-2,4-diones 9b, giving the corresponding heterocycles in reasonable yields. In addition, the synthesis of trifluoromethylated quinoline-2,4-diones 9cvia the radical cascade reaction was disclosed when sodium trifluoromethanesulfonate 9d (Langlois’ reagent) was applied as a reactant. Under the optimized conditions, various trifluoromethylated quinoline-2,4-diones 9e were obtained in moderate to good yields.
 |
| | Scheme 9 The construction of sulfonated and trifluoromethylated quinoline-2,4-diones. | |
2.2 Synthesis of indenones
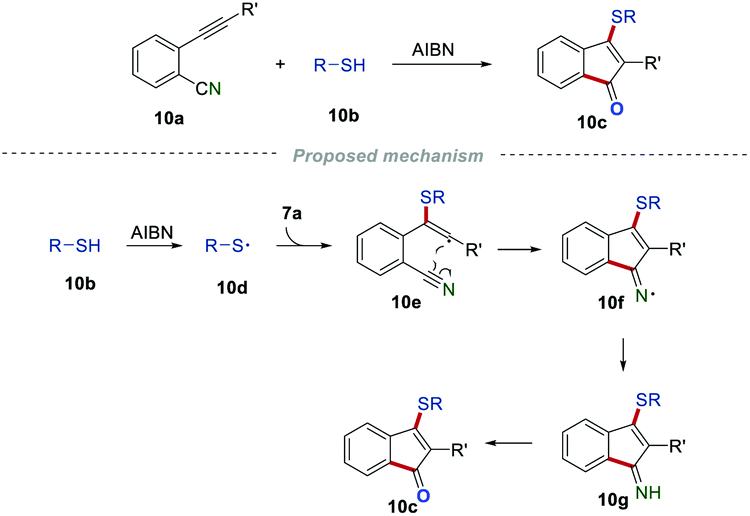
As important skeletons, indenones are extensively found in natural products and many synthetic compounds with various biological activities. Developing powerful and reliable methods for the construction of indenone frameworks has attracted a lot of attention in the past few decades. In 1998, Montevecchi and co-workers synthesized sulfanylated indenones 10c by reactions of 2-alkynylbenzonitriles 10a and thiols 10b (Scheme 10).26 When R′ was an alkyl substituent, the desired product 10c could be only obtained in low yield, which might be caused by the unstable intermediate. The regioselective addition of sulfanyl radicals 10d to the C–C triple bond generated vinyl radical 10e. The intramolecular cyclization of radical 10e led to the formation of the C–N bond to access the iminyl radical 10f, which further underwent hydrogen abstraction and hydrolysis to produce the desired products 10c.
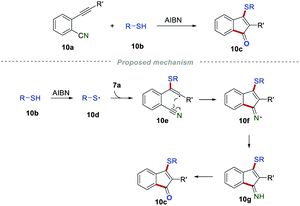 |
| | Scheme 10 The construction of sulfanylated indenones. | |
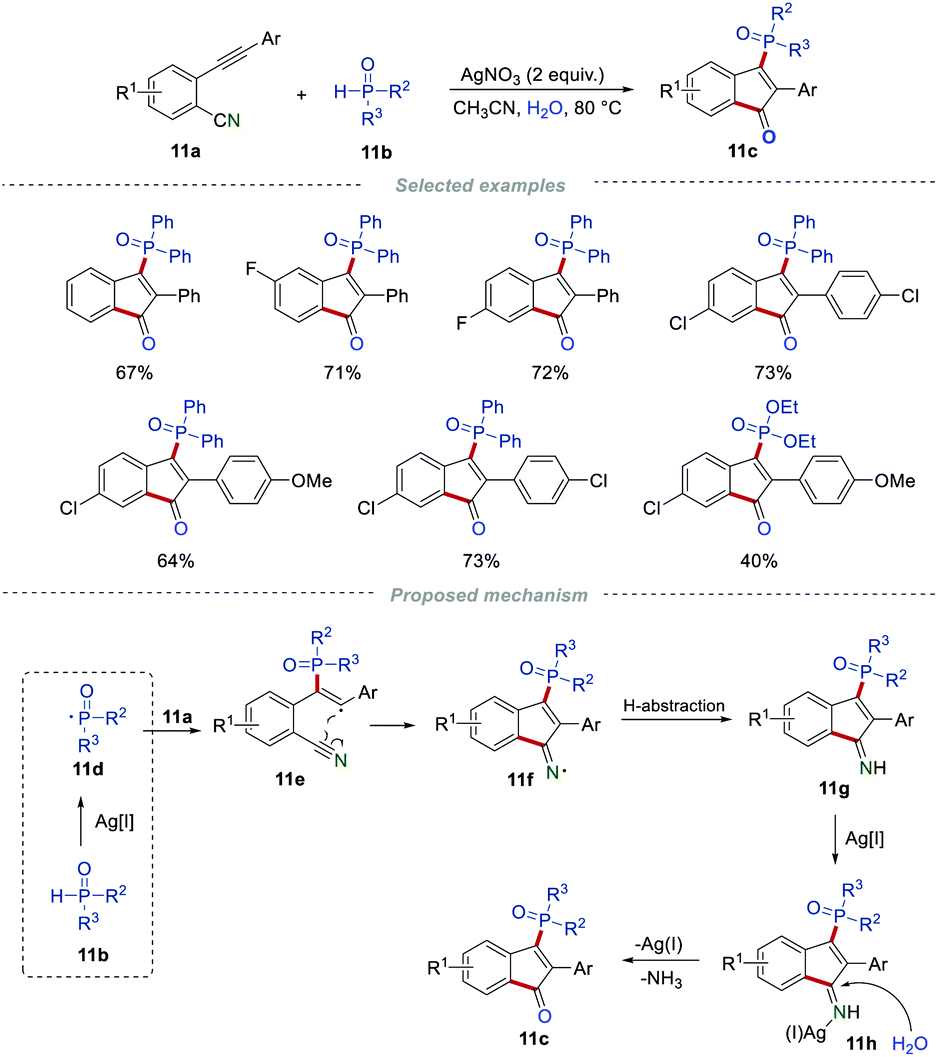
In 2016, the group of Tu and Jiang disclosed a novel and straightforward approach for the preparation of phosphinylated 1-indenones from 2-alkynylbenzonitriles and disubstituted phosphine oxides in the presence of AgNO3 (Scheme 11).27 After optimization of the reaction conditions, it was found that the highest yield was obtained in the presence of 2 equiv. of AgNO3 in CH3CN at 80 °C. The main merits of this method are that the reaction could be carried out under mild reaction conditions with a broad substrate scope and high functional group tolerance. Based on the control experiments, a reasonable mechanism was proposed, as illustrated in Scheme 11. In the presence of Ag(I), 11b could form a phosphorus-centered radical 11d, which further underwent regioselective addition to yield the alkenyl radical 11e. After an intramolecular cyclization, the radical intermediate 11f was produced. This subsequently abstracted a hydrogen atom, leading to the formation of imine 11g. Finally, the silver(I)-catalyzed hydrolysis of 11g produced the final product 11c in the presence of H2O.
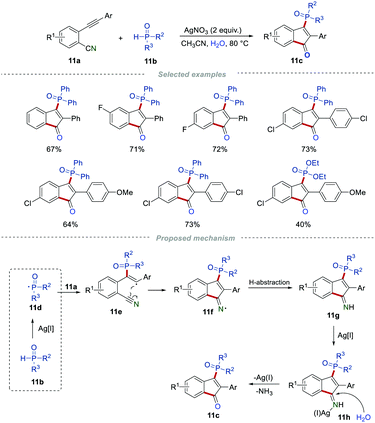 |
| | Scheme 11 The synthesis of phosphinylated indenones. | |
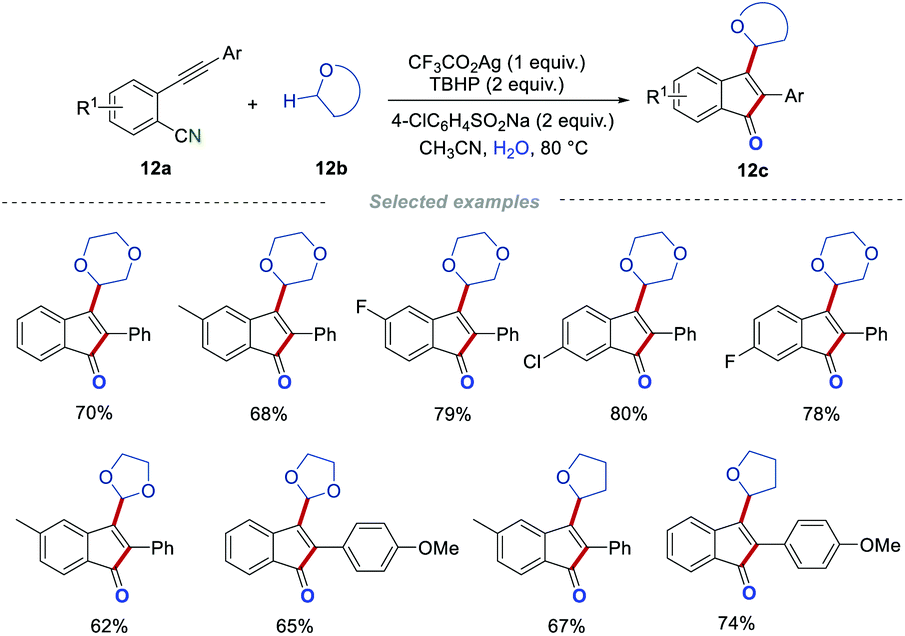
After that, the same group developed an interesting and easy-to-handle protocol, by which a large variety of 3-alkylated 1-indenones 12c were prepared from 2-alkynylbenzonitriles 12a and cyclic ethers.28 This procedure was amenable to 2-alkynylbenzonitriles with diverse functionalities, such as fluoro, chloro, bromo, methyl, ethyl, t-butyl, and methoxy groups. Moreover, 1,4-dioxane, tetrahydrofuran (THF) as well as 1,3-dioxolane were all suitable for this transformation. The control experiments showed that a radical pathway was involved in this transformation, and the oxygen atom of the carbonyl in products 12c indeed comes from water. It is particularly worth mentioning that the cyclic ether acts as both a C-centered radical precursor and a reaction medium in this reaction process. Interestingly, two different radical precursors (cyclic ethers and 4-ClC6H4SO2Na) can be well tolerated simultaneously in this transformation without disturbing the reaction process (Scheme 12).
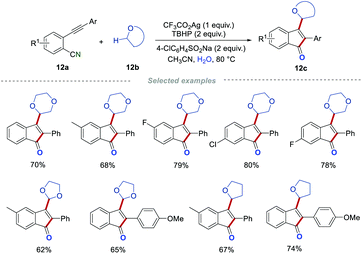 |
| | Scheme 12 The construction of 3-alkylated indenones. | |
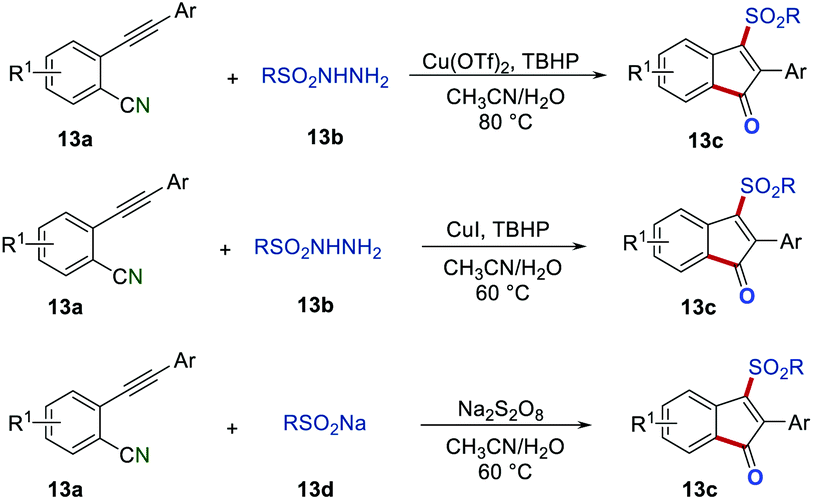
Due to the importance of sulfonyl-containing compounds, various methods were developed for the introduction of the sulfonyl group to indenones.29 For example, Jiang's group, Yu's group, and Liang's group independently reported the preparation of 3-sulfonated indenones 13cvia the sulfonyl radical-initiated cascade cyclization (Scheme 13).30 By using different sulfonyl radical precursors, there was a wide range of 3-sulfonated indenones 13c that were synthesized in satisfactory yields. Moreover, in the report of Yu's group, the synthesized compounds were proven to own typical aggregation-induced emission (AIE) properties, which showed promising application in live-cell imaging. Additionally, in Liang's report, the trifluoromethylated indenone could be obtained in 35% isolated yield by employing CF3SO2Na as a radical precursor.
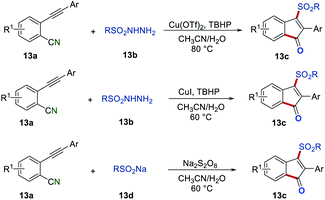 |
| | Scheme 13 The preparation of 3-sulfonated indenones. | |
3. Nitrile insertion for N-heterocycles
Nitrogen-containing heterocycles are highly privileged frameworks with unique biological activity and are widespread in various natural products, playing significant roles in biologically active molecules and pharmaceuticals. In the past decades, great efforts have been devoted to developing novel and efficient synthetic methodologies for the preparation of N-heterocycles.31 Importantly, the radical cascade reactions using the cyano group as a radical acceptor have emerged as a powerful strategy for the construction of N-heterocycles through a nitrile insertion process.
3.1 Synthesis of fused quinoline and quinoxaline derivatives
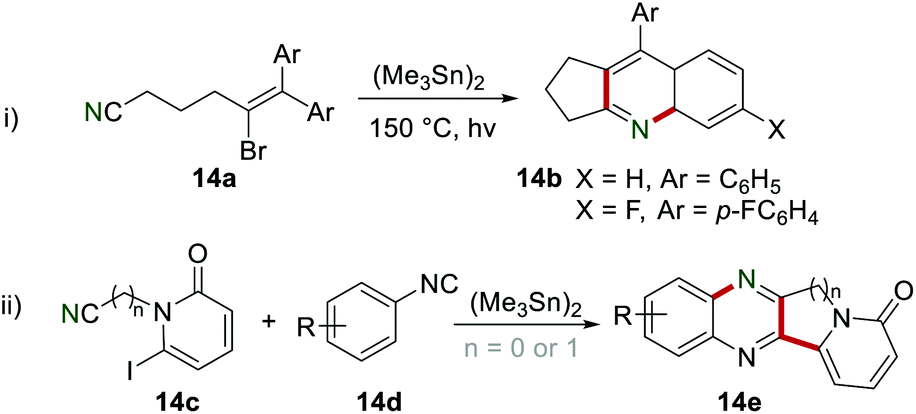
In 1991, Curran and Liu utilized the cyano group as a radical acceptor to provide a simple procedure to synthesize the cyclopenta-fused quinolines 14b by cyclization of 14a in the presence of (Me3Sn)2 (Scheme 14i).32 In 1996, the synthesis of the fused quinoxaline 14e was also achieved by the same group, leading to the formation of two C–C bonds and one C–N bond in one step (Scheme 14ii).33
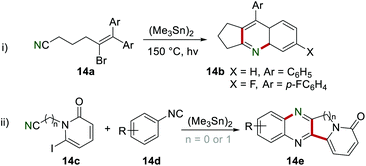 |
| | Scheme 14 The construction of fused quinoline and quinoxaline derivatives. | |
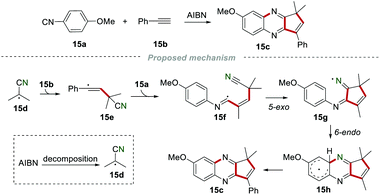
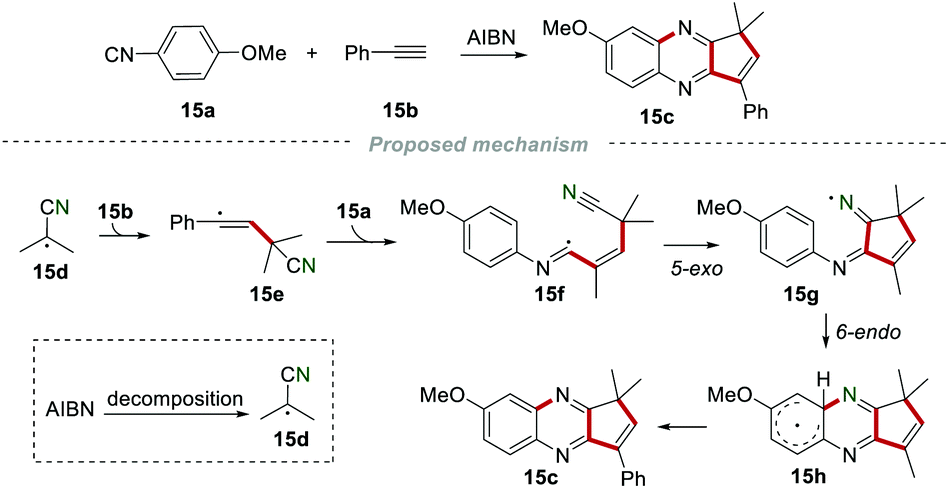
In 1995, Nanni and co-workers achieved the construction of the cyclopenta-fused quinoxaline 15c by reacting 4-methoxyphenylisonitrile 15a and phenylacetylene 15b in a refluxing benzene solution in the presence of AIBN (Scheme 15).34 The decomposition of AIBN produced the 2-cyanoprop-2-yl radical 15d, which was added to the C–C triple bond of phenylacetylene 15b to access the vinyl radical 15e. The vinyl radical 15e subsequently attacked the carbon atom of isonitrile 15a to generate imidoyl 15f. Imidoyl 15f further underwent cascade 5-exo and 6-endo cyclization to give the radical 15h, which transformed into the desired product 15cvia aromatization.
 |
| | Scheme 15 The construction of cyclopenta-fused quinoxaline. | |
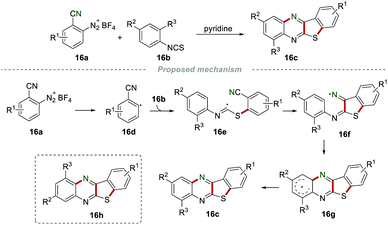
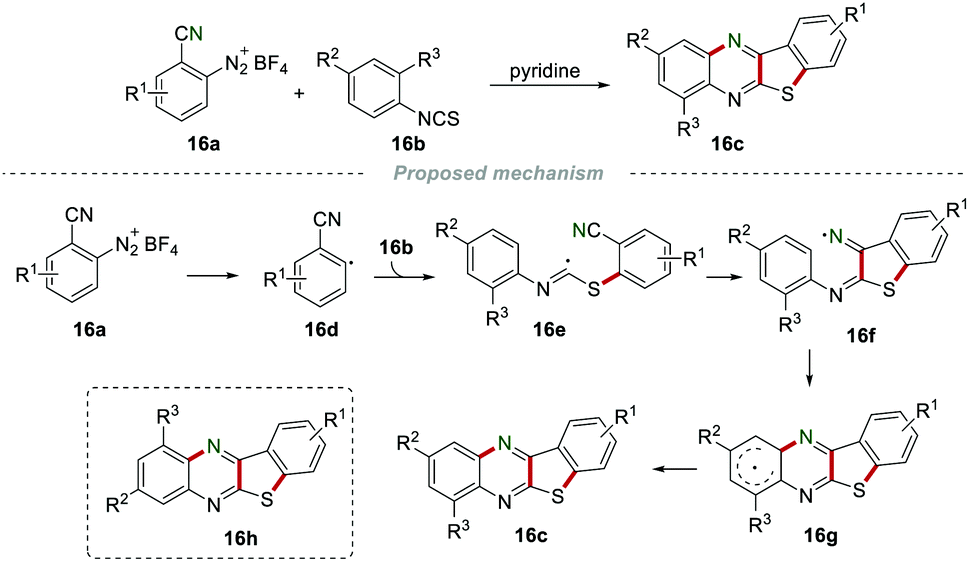
In 1997, Zanardi et al. accomplished the synthesis of benzothienoquinoxalines 16c by annulation of aryl diazonium tetrafluoroborates 16a with isothiocyanates 16b (Scheme 16).35 The aryl radical 16d attacked the sulfur atom of the isothiocyanate 16b to produce the imidoyl radical 16e, which subsequently was added to the cyano group to give iminyl 16f. Iminyl 16f further underwent an intramolecular cyclization and aromatization to access the desired product 16c. Importantly, no product 16h was observed, indicating that the intramolecular cyclization of 16fvia 5-membered ipso-cyclization followed by rearrangement was impossible.
 |
| | Scheme 16 The construction of benzothienoquinoxalines. | |
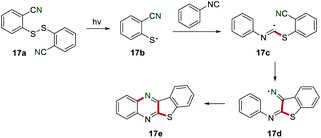
After that, Nanni and co-workers reported the photocatalytic synthesis of benzothienoquinoxalines 17e in 1998 (Scheme 17).36 The cleavage of an S–S bond generated the sulfanyl radical 17b, which was added to the isocyano group to produce an imidoyl radical 17c. Subsequently, the imidoyl radical 17c underwent an intramolecular cyclization reaction to give an iminyl radical 17d, which further suffered from cyclization to complete this transformation.
 |
| | Scheme 17 Photocatalytic synthesis of benzothienoquinoxalines. | |
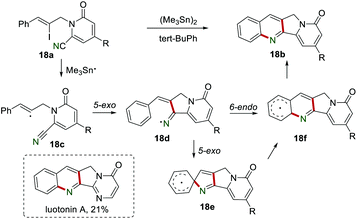
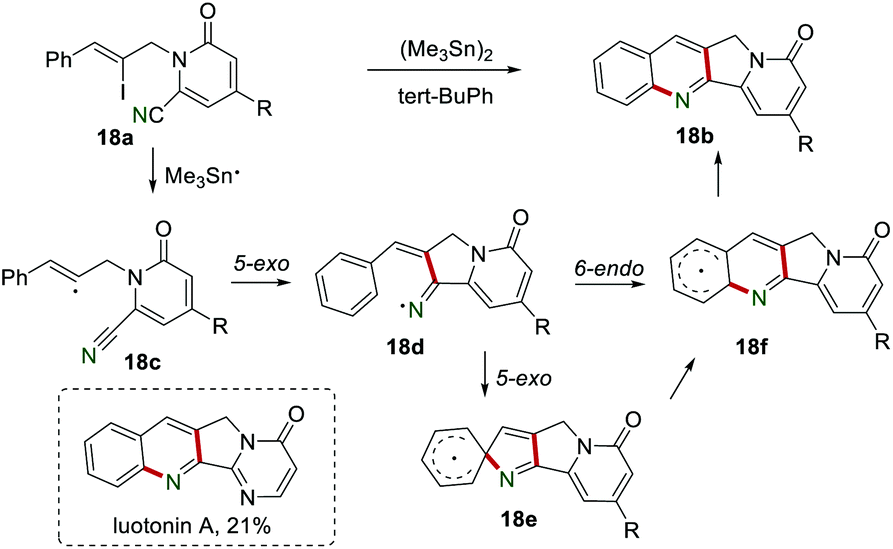
In 2005, Bowman et al. reported the synthesis of tetracycline 18b containing the 2,3-dihydro-1H-pyrrolo[3,4-b]quinoline ring system in the presence of (Me3Sn)2 and tert-BuPh (Scheme 18).37 By using this novel protocol, the biologically active luotonin A could be obtained in 21% yield. The mechanism showed that the reaction of vinyl iodide 18a and Me3Sn˙ produced vinyl radical 18c, which subsequently was added to the cyano group to afford the iminyl radical 18d. The rate of iodine abstraction by Bu3Sn˙ from the vinyl iodide 18a is theoretically faster than bromine abstraction (106–107 M−1 s−1),38 which might not be the rate-determining step. The iminyl radical 18d underwent 6-endo intramolecular cyclization (or 5-exo intramolecular cyclization followed by rearrangement) to generate the radical intermediate 18f, which lost a H˙ in a H-abstraction step to yield the desired product 18b. Both the intramolecular cyclization, including 5-exo cyclization onto the cyano group and iminyl radical onto the phenyl ring, were slow. A control experiment without (Me3Sn)2 led to the formation of the trace product 18b, suggesting that iodine homolysis or the Diels–Alder cyclization might be the possible minor mechanisms.
 |
| | Scheme 18 The synthesis of tetracycline. | |
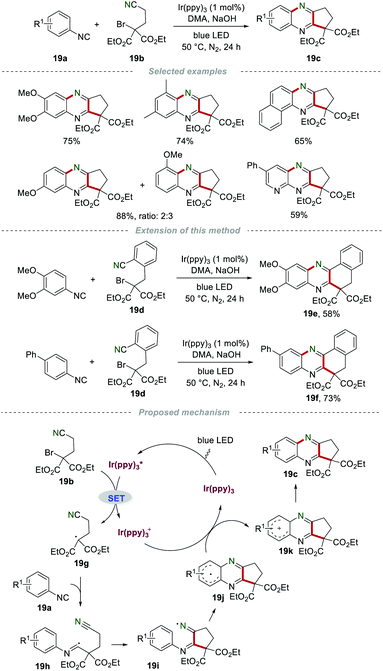
In 2014, Yu's group developed a practical and eco-friendly strategy for the construction of two C–C bonds and one C–N bond in one step to access the fused quinoxaline derivatives 19c under the irradiation of visible light (Scheme 19).39 A variety of 5-membered ring-fused quinoxalines 19c were prepared in moderate to good yields. Moreover, 6-membered ring-fused quinoxalines could be obtained by using nitrile 19d with a benzene ring as a linker. The proposed mechanism showed that the photocatalyst fac-Ir(ppy)3 was irradiated to the excited state 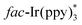 under the irradiation of visible light, which was oxidized by bromide 19b to fac-Ir(ppy)3+, along with the formation of radical 19g. Subsequently, the addition of 19g to isocyanide 19a generated the imidoyl radical 19h, which subsequently underwent intramolecular cyclization to access the iminyl radical 19i. Radical 19i underwent homolytic aromatic substitution to produce the radical 19j, which was oxidized by fac-Ir(ppy)3+ to yield the radical cation 19k, along with the regeneration of fac-Ir(ppy)3 for the next cycle. Finally, the aromatization of radical cation 19k led to the formation of the desired products 19c by deprotonation.
under the irradiation of visible light, which was oxidized by bromide 19b to fac-Ir(ppy)3+, along with the formation of radical 19g. Subsequently, the addition of 19g to isocyanide 19a generated the imidoyl radical 19h, which subsequently underwent intramolecular cyclization to access the iminyl radical 19i. Radical 19i underwent homolytic aromatic substitution to produce the radical 19j, which was oxidized by fac-Ir(ppy)3+ to yield the radical cation 19k, along with the regeneration of fac-Ir(ppy)3 for the next cycle. Finally, the aromatization of radical cation 19k led to the formation of the desired products 19c by deprotonation.
 |
| | Scheme 19 The synthesis of fused quinoxaline derivatives. | |
3.2 Synthesis of quinazolinones
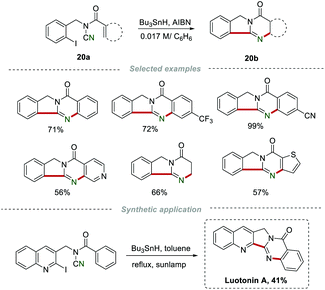
In 2007, Malacria et al. reported a radical cascade cyclization of N-acyl-N-(2-iodobenzyl)cyanamides 20a for the construction of quinazolinones 20b (Scheme 20).40 Under the optimized conditions, a large number of quinazolinones 20b with good regioselectivity were obtained in satisfactory yields. More importantly, the total synthesis of the natural product, luotonin A, with DNA topoisomerase I poison could be achieved by this method.
 |
| | Scheme 20 Radical migration for the construction of quinazolinones. | |
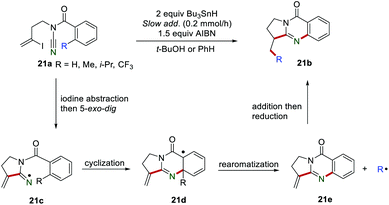
Afterward, the same group introduced a CC bond in the substrate and the reaction can proceed smoothly to access quinazolinones 21b (Scheme 21).41 The mechanism investigations indicated that the iminyl radical 21c was formed by iodine abstraction and regioselective 5-exo-dig cyclization, which immediately underwent an intramolecular cyclization onto the aromatic ring, rendering the radical intermediate 21d. Subsequently, the intermediate 21e was formed via rearomatization, releasing the radical R˙. Finally, the addition of R˙ to the CC bond of 21e gave the product 21b.
 |
| | Scheme 21 Radical migration for the construction of quinazolinones. | |
In 2010, Malacria et al. developed an efficient strategy for the preparation of bi- and tricyclic guanidines 22bvia a radical domino process involving cyanamides as relay partners (Scheme 22).42 Various functional groups, including the electron-withdrawing groups and electron-donating groups, were all well tolerated, and the corresponding guanidines 22b were obtained in moderate to good yields. Moreover, when the final acceptor aryl group was replaced by an olefin, the reaction also reacted smoothly under the standard reaction conditions.
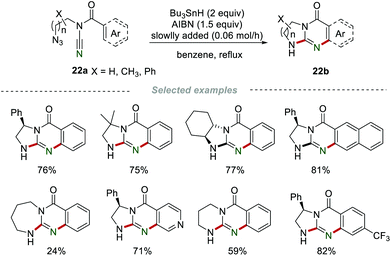 |
| | Scheme 22 The construction of guanidines. | |
The above-mentioned methods are normally performed in the presence of toxic reagents (e.g., Bu3SnH) or proceed at high reaction temperature, which might limit the application of these methods. In order to overcome these drawbacks, Yu and co-workers developed a visible-light-promoted intramolecular radical cyclization procedure for the preparation of quinazolinones 23b (Scheme 23).43 This protocol would avoid the use of toxic Bu3SnH and high reaction temperature, rendering the reaction more practical from the viewpoint of the environment and safety. In order to gain further insight into the mechanism, a series of emission quenching experiments were carried out. The result showed that the excited state  was indeed quenched by DIPEA (i-Pr2NEt), indicating that a reductive quenching was involved between DIPEA and
was indeed quenched by DIPEA (i-Pr2NEt), indicating that a reductive quenching was involved between DIPEA and 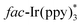 via single electron transfer. The proposed mechanism showed that
via single electron transfer. The proposed mechanism showed that 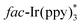 was formed under the irradiation of visible light, which further reacted with i-Pr2NEt to give fac-Ir(ppy)3−, along with the N-centered radical cation species. Subsequently, a single-electron transfer between fac-Ir(ppy)3− and reactant 23c led to the generation of the radical intermediate 23d. After a similar intramolecular radical cyclization, single-electron oxidation, and deprotonation process, the final product 23h was produced.
was formed under the irradiation of visible light, which further reacted with i-Pr2NEt to give fac-Ir(ppy)3−, along with the N-centered radical cation species. Subsequently, a single-electron transfer between fac-Ir(ppy)3− and reactant 23c led to the generation of the radical intermediate 23d. After a similar intramolecular radical cyclization, single-electron oxidation, and deprotonation process, the final product 23h was produced.
 |
| | Scheme 23 The construction of quinazolinones via visible-light-promoted radical cyclization. | |
In 2016, Cui et al. developed a silver(I)-mediated radical cascade phosphorylation/cyclization of N-cyanamide alkenes 24a for the preparation of phosphorylated quinazolinone derivatives 24c (Scheme 24).44 Different kinds of N-cyanamide alkenes were investigated in this transformation, producing the desired phosphorylated quinazolinone derivatives 24c in good yields. The radical trapping experiments were conducted to gain a deeper understanding of the mechanism. For instance, by adding radical scavengers (TEMPO or BHT), the reaction was severely inhibited. Based on the results of the control experiments, a reasonable mechanism involving a radical pathway was suggested. In the beginning, the H-phosphonates 24b was oxidized by AgNO3 to give the phosphorus-centered radical 24d. Then, the addition of the phosphoryl radical 24d to the CC bond of 24a formed the radical intermediate 24e, which subsequently underwent an intramolecular cyclization to afford the iminyl radical intermediate 24f. Afterward, an intramolecular cyclization of 24f led to the formation of the aryl radical 24g. 24g was then oxidized by Ag(I) to produce the phosphorylated quinazolinones 24c, along with the release of Ag(0). Partial Ag(0) could be reoxidized to Ag(I); therefore, 1 equiv. of AgNO3 was enough for this transformation.
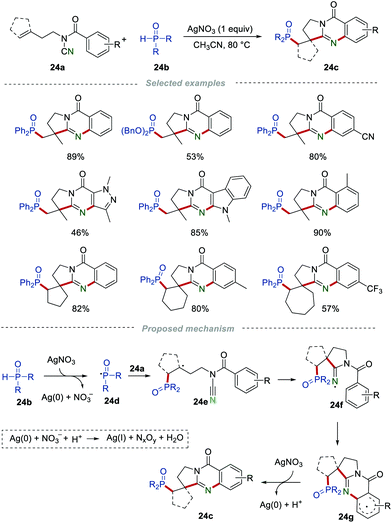 |
| | Scheme 24 The preparation of phosphorus quinazolinones. | |
Encouraged by the importance of the trifluoromethyl group in medicinal chemistry, Cui and co-workers successfully achieved the synthesis of trifluoromethylated quinazolinones 25c in 2016 (Scheme 25).45 In this transformation, the CF3 radical was generated from the Togni's reagent 25b with the assistance of the Cu(I) catalyst. A broad range of substrates bearing electron-withdrawing groups and electron-donating groups were performed smoothly, delivering the desired products in moderate to good yields. In addition, heterocyclic moieties (such as indole, pyrazole, and thiophene) were all suitable for this radical cyclization reaction.
 |
| | Scheme 25 The preparation of trifluoromethylated quinazolinones. | |
Afterward, Wang et al. reported a metal-free cascade annulation of N-cyanamide alkenes 26a by using the readily available aldehydes 26b as alkyl radical precursors for the construction of the alkyl-substituted quinazolinones 26c (Scheme 26).46 In this reaction, the alkyl radical was generated from aliphatic aldehydes via a decarbonylation process in the presence of DTBP.47 After going through a similar radical addition and cyclization process, a wide range of alkyl-substituted quinazolinones 26c could be obtained in satisfactory yields. In this transformation, a large variety of alkyl aldehydes, including secondary and tertiary alkyl aldehydes, could be successfully applied.
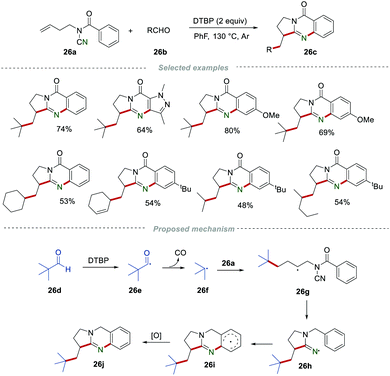 |
| | Scheme 26 The preparation of alkyl-substituted quinazolinones. | |
3.3 Synthesis of phenanthridines
As early in 1998, Zanardi et al. reported the synthesis of cyclopentaphenanthridines 27c by reacting the aryldiazonium salt 27a with phenylacetylene or trimethylsilylacetylene 27b (Scheme 27).48 Initially, the aryl radical 27d added to the C–C triple bond to give the vinyl radical 27e, which further underwent intramolecular cyclization onto the cyano group to afford the iminyl radical 27f. Subsequently, 27f underwent intramolecular cyclization and H-abstraction to access the desired product 27c.
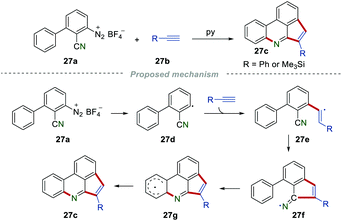 |
| | Scheme 27 The construction of cyclopentaphenanthridines. | |
In 2019, Ji and Wang's group demonstrated an efficient and regioselective construction of pyrroloporidine compounds 28c by the reaction of multifunctionalized 3-isocyano-[1,1′-biphenyl]-2-carbonitriles 28a and arylboronic acids 28b in the presence of Mn(OAc)3 (Scheme 28).49 The efficiency of this simple protocol was obviously affected by the steric hindrance effect of the substituents on arylboronic acids, and o-tolylboronic acid failed to furnish the corresponding product. Arylboronic acids bearing different substituents, including electron-withdrawing and electron-donating groups on other positions of the phenyl ring, reacted smoothly to access the desired products 28c in moderate to good yields. In addition, halo substituents were compatible in this transformation, providing the possibility for further cross-coupling reactions to afford structurally diverse pyrroloporidine compounds. On the basis of the radical trapping experiments, a possible mechanism involving a radical pathway was presented. The aryl radical 28d, generated from arylboronic acids under the assistance of Mn(III), attacked the isocyano group in 28a to produce the imdoyl radical 28e. Then, radical 28e underwent an intramolecular cyclization with the cyano group to afford the iminyl radical 28f, which underwent another intramolecular cyclization with the phenyl ring to give radical 28g. After single-electron oxidation by Mn(III) and deprotonation, the desired product 28c was generated.
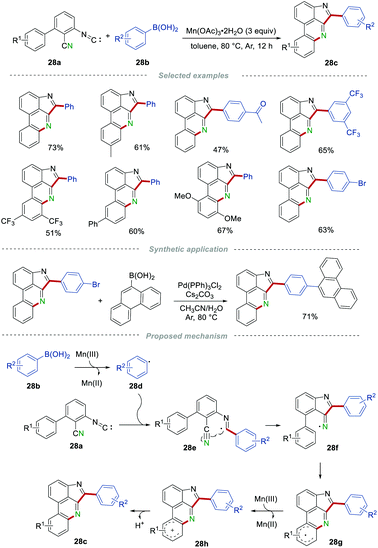 |
| | Scheme 28 The construction of pyrroloporidine compounds. | |
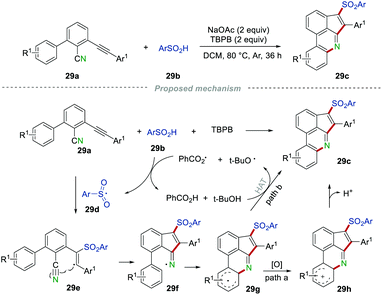
Almost at the same time, Zhou's group replaced the isocyano group with an unsaturated C–C triple bond to develop a novel and mild protocol for the synthesis of 3-sulfonated cyclopenta[gh]phenanthridines 29c (Scheme 29).50 The reaction worked well for both electron-donating and electron-withdrawing group-substituted substrates, providing the desired products 29c in moderate to good yields. Unfortunately, no desired products could be obtained when the Ar1 was replaced with the halogen or alkyl group (n-C4H9), which might be caused by the unstable alkenyl radical intermediate. A detailed assessment of the control experiments suggested that a radical process might be involved. Ultimately, the arylsulfonyl radical 29d was formed from the reaction between the sulfinic acids 29b and 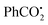 or t-BuO˙, which was generated from the thermal decomposition of TBPB. Then, the addition of radical 29d to the C–C triple bond of 29a resulted in the alkenyl radical 29e, which then underwent intramolecular cyclization with the cyano group to produce the radical intermediate 29g. The radical intermediate 29g was further oxidized to radical cation 29h, which subsequently regenerated the aromatic ring by deprotonation (path a). On the other hand, radical intermediate 29g could also convert into the desired product 29gvia hydrogen atom transfer (path b).
or t-BuO˙, which was generated from the thermal decomposition of TBPB. Then, the addition of radical 29d to the C–C triple bond of 29a resulted in the alkenyl radical 29e, which then underwent intramolecular cyclization with the cyano group to produce the radical intermediate 29g. The radical intermediate 29g was further oxidized to radical cation 29h, which subsequently regenerated the aromatic ring by deprotonation (path a). On the other hand, radical intermediate 29g could also convert into the desired product 29gvia hydrogen atom transfer (path b).
 |
| | Scheme 29 The construction of 3-sulfonated cyclopenta[gh]phenanthridines. | |
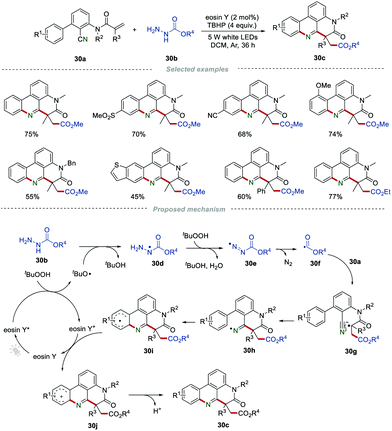
In recent years, the radical addition/cyclization of 2-cyano-3-arylaniline-derived acrylamides has been developed for the efficient preparation of phenanthridines. In 2017, Sun et al. described a novel visible-light-induced radical cascade reaction for the construction of ester-functionalized phenanthridines (Scheme 30).51 This protocol employed alkyl carbazates 30b as the alkoxycarbonyl radical precursor, providing a convenient access to 1,4-dicarbonyl compounds 30c in moderate to excellent yields. The structure of the products was interesting not only because of the generation of one C–N bond and two C–C bonds, but also the challenging all-carbon quaternary centers generated through a single step. It was proposed that the key step for the reaction was the generation of the tert-butoxyl radical via single electron transfer between eosin Y* and TBHP, which would thus lead to the formation of the nitrogen radical intermediate 30d. Then, the sequential dehydrogenation of the intermediate 30d by TBHP generated the alkoxycarbonyl diazo radical 30e, which then released a molecule of nitrogen to form the important alkoxycarbonyl radical 30f. The addition of the radical 30f to the CC bond of 30a afforded the radical intermediate 30g, which further underwent an intramolecular cyclization with a cyano group to produce the radical intermediate 30i. Subsequently, 30i was oxidized by eosin Y+ to obtain the intermediate 30j, along with the regeneration of eosin Y. Finally, 30j underwent a rearomatization process, producing the target products 30cvia deprotonation.
 |
| | Scheme 30 The construction of ester-functionalized phenanthridines. | |
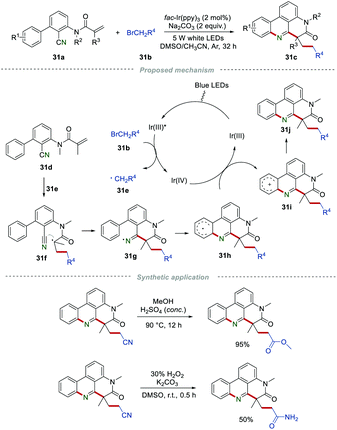
As their follow-up work, various alkylated phenanthridines 31c were synthesized by using 2-bromoacetonitrile, ethyl 2-bromoacetate, or 2-bromo-N,N-dimethylacetamide 31b as the active methylene radical precursors (Scheme 31).52 This eco-friendly procedure was conducted under visible-light, affording the desired products 31c in moderate to good yields, showing a wide substrate scope. The control experiments demonstrated that the reaction process involved a radical pathway. The reactant 31b could react with [fac-IrIII(ppy)3]* to produce active primary radicals 31e. After a similar process including radical addition and cyclization, single-electron oxidation, and deprotonation, the final product 31j was generated. Furthermore, the cyano group in the products could be further transformed into other functional groups, such as an ester group or amide group.
 |
| | Scheme 31 The construction of alkylated phenanthridines. | |
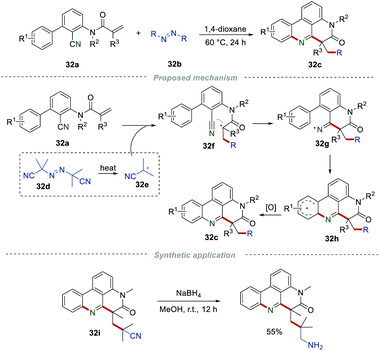
Azo compounds, such as azobisisobutyronitrile (AIBN), can generate free radicals under mild conditions. The exploration of azo compounds as radical sources to prepare different functionalized molecules is always arguably of high interest. In 2018, Sun et al. used azo compounds 32b as radical precursors to complete a radical cascade reaction for the construction of phenanthridine derivatives 32c (Scheme 32).53 Notably, most of the azo compounds attached with alkyl groups were suitable for this transformation, and a broad range of the desired products were obtained under the catalyst- and oxidant-free conditions. Under the heating reaction conditions, the thermal decomposition of AIBN (32d) led to the radical 32e, along with the release of N2. Due to their high thermal stability, diethyl azodicarboxylate and azobenzene cannot be applied to this conversion. Additionally, the cyano group in product 32i could be easily reduced by NaBH4 to generate the aminomethyl group in moderated yield.
 |
| | Scheme 32 Azo compounds as radical sources to prepare alkylated phenanthridines. | |
Using inexpensive carboxylic acids as the radical sources, Sun and co-workers reported an efficient protocol to construct alkylated phenanthridines (Scheme 33).54 The environmentally-friendly (NH4)2S2O8/eosin Y system was employed for the decarboxylation of aliphatic carboxylic acids under photoredox catalysis. It is worth noting that the primary carboxylic acids (isovaleric acid, 2-cyclohexylacetic acid) and cyclic secondary carboxylic acids, as well as the tertiary carboxylic acid, were all suitable in this transformation, leading to the formation of products 33c in good to excellent yields under mild reaction conditions. Notably, the trifluoromethyl group, which was widely found in drugs, was also compatible, affording the product 33k with good isolated yield.
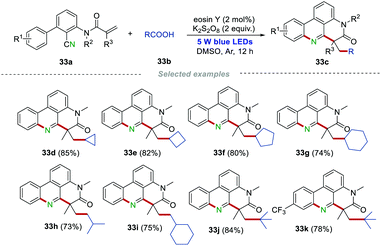 |
| | Scheme 33 Carboxylic acids as radical sources to prepare alkylated phenanthridines. | |
Moreover, α-keto acids 34b as acyl radical precursors55 were applied as substrates to react with the 2-cyano-3-arylaniline derived acrylamides 34a, proving a simple and efficient synthetic strategy to access the carbonyl-containing phenanthridines 34c (Scheme 34).56 A large variety of carbonyl-containing phenanthridines 34c were obtained in moderate to good yields under the optimized conditions without obvious electronic and steric hindrance effects. However, when the substitution of R2 was an acetyl, tosyl, or H, the reaction could not react smoothly to give the desired products. The radical trapping reaction showed that the reaction was almost completely suppressed by adding the radical inhibitor TEMPO or BHT to the standard reaction system, implying that a radical pathway should be involved in this transformation. Moreover, the results of the kinetic isotope effects (KIE) suggested that the cleavage of the C(sp2)–H bond that occurred after the formation of the C(sp2)–N bond might not be the rate-determining step.
 |
| | Scheme 34 α-Keto acids as radical sources to prepare the carbonyl-containing phenanthridines. | |
The importance of fluorine-containing compounds has prompted chemists to continuously develop new synthetic methods for the synthesis of fluorine-contained phenanthridine derivatives. In 2018, Sun et al. reported another visible-light-induced radical reaction of 2-cyano-3-arylaniline derived acrylamides 35a.57 By changing different fluorine-containing radical sources, trifluoroalkyl phenanthridine 35c or difluoroalkyl phenanthridine derivatives 35e could be obtained in moderate to good yields. This method provides a green and efficient transformation for the preparation of trifluoroalkyl or difluoroalkyl phenanthridine derivatives. Recently, the Fu group successfully employed N-trifluoromethylthiosaccharin 35f as an effective precursor of SCF3 radical to access the SCF3-containing phenanthridine derivatives 35g.58 A large variety of trifluoromethylthiolated phenanthridine derivatives with electron-donating and electron-withdrawing groups could be obtained in moderate to good yields (Scheme 35).
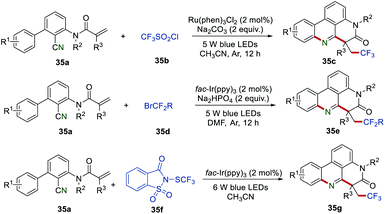 |
| | Scheme 35 The construction of trifluoroalkyl, difluoroalkyl, or trifluoromethylthiolated phenanthridine derivatives. | |
In 2018, Li et al. reported a copper-catalyzed radical cascade reaction of 2-cyano-3-arylaniline-derived acrylamides 36a and acetonitriles 36b (Scheme 36). This work provided a powerful and easy-handle approach to access cyano-substituted phenanthridines 36c using the Cu(OAc)2/TBPB system. In addition, various methyl-substituted phenanthridines 36e were obtained when acetonitriles were replaced by PhCl under metal-free conditions.59
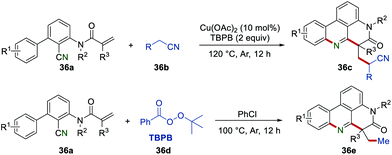 |
| | Scheme 36 The construction of cyano substituted phenanthridines and methyl-substituted phenanthridines. | |
Moreover, the Sun group successfully developed a silyl radical-initiated cyclization reaction for the synthesis of silyl-substituted phenanthridines 37c (Scheme 37).60 This protocol displayed the advantages of the wide substrate scope and good functional group tolerance, affording the silyl-substituted phenanthridines 37c in good yields. According to the proposed mechanism, the silyl radical could be generated under the oxidant of lauroyl peroxide (LPO), which then underwent a similar radical addition and intramolecular cyclization to form the radical 37g with the formation of the C–Si, C–C, and C–N bonds. Finally, radical 37g was further converted into the corresponding product 37c through hydrogen atom abstraction by the radical 37d.
 |
| | Scheme 37 The construction of silyl substituted phenanthridines. | |
The introduction of deuterium into drug molecules has attracted huge attention due to the improved pharmacokinetic properties, such as enhanced absorption, distribution, as well as metabolic stability. In 2020, the Sun group described a powerful strategy for the construction of deuterium-labeled phenanthridines 38cvia the deuterium addition cascade cyano insertion/cyclization of the 2-cyano-3-arylaniline-derived acrylamides 38a with NaBD4 (Scheme 38).61 In the presence of inexpensive Fe(NO3)3·9H2O, numerous desired products could be obtained in satisfactory yield. Moreover, the gram-scale reaction with no obvious decrease in efficiency showed the practicality of this method. The control experiments demonstrated that a radical pathway might be involved in the reaction process, and the rate-determining step was not the generation or introduction of a deuterium atom in this cyano insertion/cyclization reaction. The proposed mechanism showed that the key intermediate deuterium radical was generated via the oxidation of NaBD4 in the presence of Fe(NO3)3·9H2O. Then, the addition of the deuterium radical to the CC bond of 38a formed radical 38c, which further attacked the cyano group to yield the iminyl radical 38d. After cyclization and single-electron oxidation by Fe(III), the radical cation 38f was formed, which subsequently lost a proton to afford the product 38c.
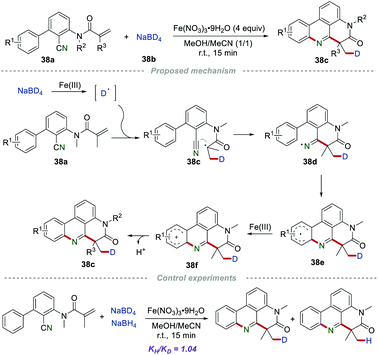 |
| | Scheme 38 The construction of deuterium-labelled phenanthridines. | |
Very recently, Li's group achieved the functionalization of the alkyl C(sp3)–H bond via oxidative cascade cyclization of 2-cyano 3-arylaniline-derived acrylamides 39a (Scheme 39).62 In this transformation, good functional group tolerance was observed and some sensitive functional groups, such as the hydroxyl group, could survive in this transformation. Moreover, toluenes, ethers, aliphatic alcohols, and simple alkanes could be applied as alkyl radical precursors for the synthesis of the alkyl-substituted phenanthridines 39cvia the formation of a C(sp3)–C(sp3)/C(sp2)–C(sp3)/C(sp2)–N bond in one step, demonstrating the wide range of the substrate applicability and step-/atom-economy.
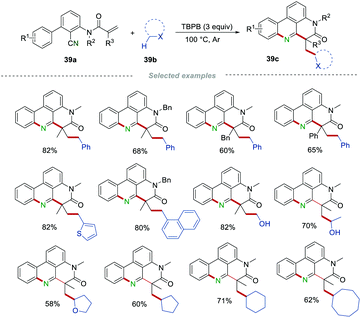 |
| | Scheme 39 The construction of alkyl-substituted phenanthridines. | |
3.4 Other heterocycles
Silicon/nitrogen heterocycles have gained particular attention due to their highly valued applications in pharmaceuticals and drug lead compounds.63 Different from the work of Sun's group in Scheme 37, He et al. developed an efficient Mn-promoted intermolecular oxidative heteroannulation of N-(2-cyanoaryl)acrylamides 40a with tertiary silanes 40b in 2018 (Scheme 40). This work provided a novel approach to the preparation of silicon/nitrogen heterocycles 40c using the MnCl2/DTBP system.64 The transformation was proposed to begin with the reaction of DTBP and Mn(II), generating a tert-butoxyl radical and the Mn(III)(tBuO) species. Then, the tert-butoxyl radical abstracted one hydrogen atom from tertiary silane 40b to form the silicon-centered radical 40d, which further underwent an addition reaction to the CC bond of 40a to afford radical intermediate 40e. Afterwards, the intramolecular cyclization of 40e with the cyano group afforded the iminyl radical 40f. Then, the 1,6-hydrogen atom transfer (1,6-HAT) of radical 40f produced the intermediate 40g, which would attack the CN bond to provide 40h. Finally, the single electron oxidation of the radical 40h by Mn(III)(tBuO), followed by subsequent deprotonation eventually provided silicon/nitrogen heterocycles 40c.
 |
| | Scheme 40 The construction of silicon/nitrogen heterocycles. | |
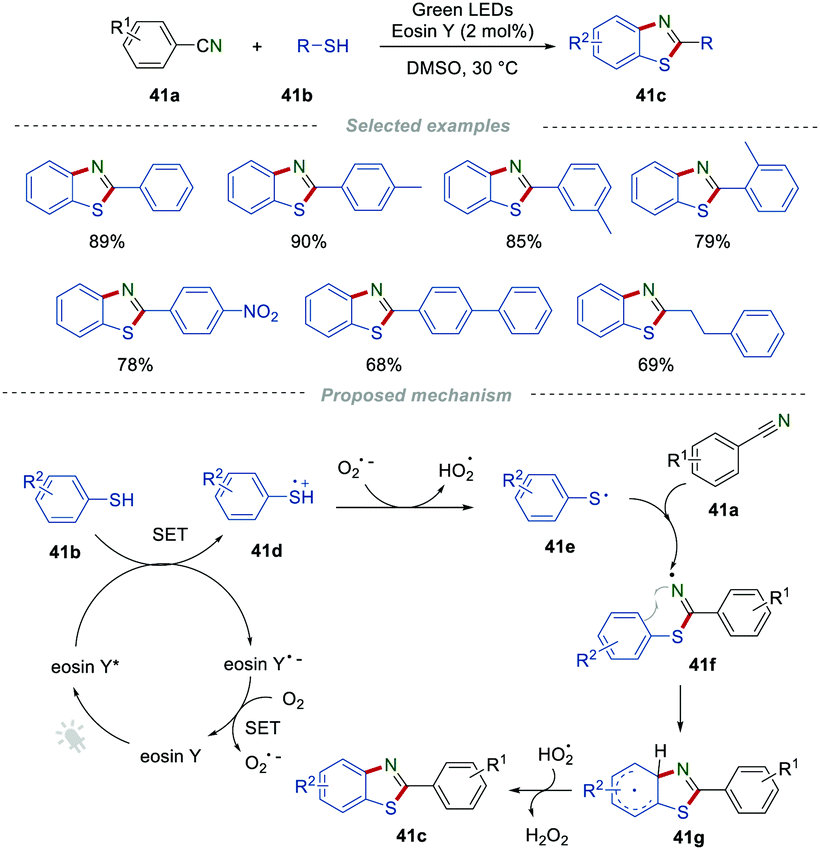
In 2018, Natarajan et al. reported a visible light-mediated radical reaction between nitriles 41a and thiophenols 41b.65 The reaction was conducted under mild reaction conditions, providing an efficient strategy for the construction of benzothiazoles 41c in good to excellent yields. A possible mechanism, as proposed by the authors, was disclosed in Scheme 41. Upon the irradiation of visible light, eosin Y was excited to eosin Y*, which then was quenched by thiophenols 41bvia single-electron transfer to form the radical cation 41d and eosin Y˙−. The eosin Y˙− was further oxidized to the ground state by oxygen, releasing a superoxide radical anion 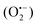 . Then, a deprotonation reaction between the radical cation 41d and
. Then, a deprotonation reaction between the radical cation 41d and 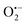 generated the thiyl radical 41e, which then attacked the cyano group in 41a to afford the iminyl radical 41f. Finally, the intramolecular cyclization of the iminyl radical 41f afforded radical 41g, which further lost a hydrogen atom to produce the desired products 41c.
generated the thiyl radical 41e, which then attacked the cyano group in 41a to afford the iminyl radical 41f. Finally, the intramolecular cyclization of the iminyl radical 41f afforded radical 41g, which further lost a hydrogen atom to produce the desired products 41c.
 |
| | Scheme 41 The construction of benzothiazoles. | |
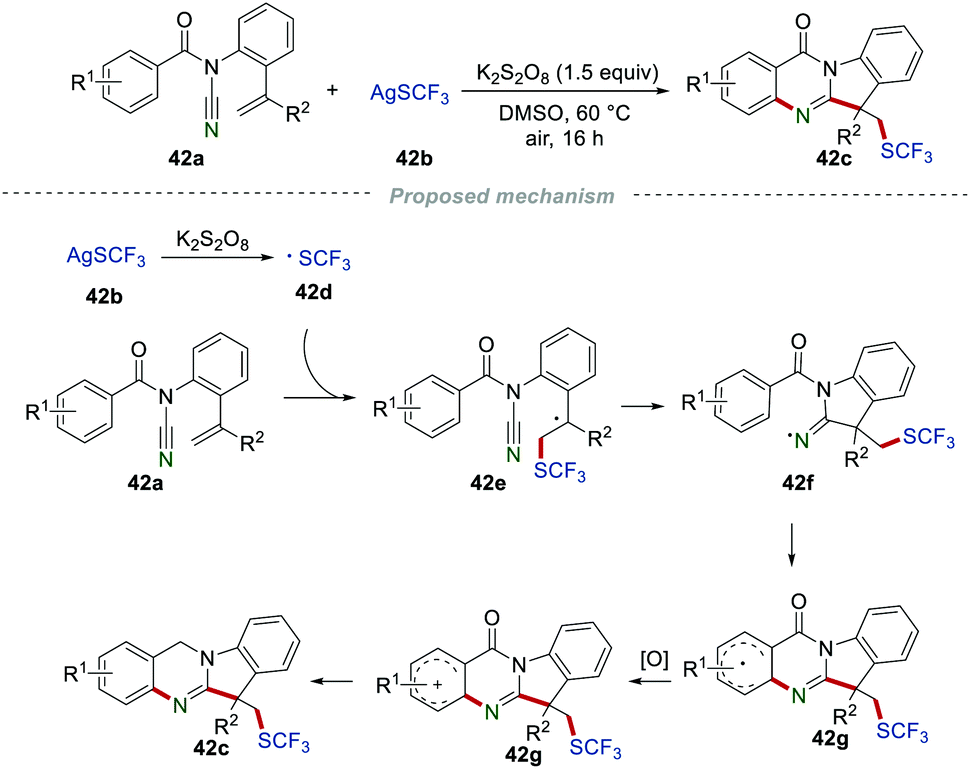
Tryptanthrin derivatives, which consist of quinazolines and indoles, were widely spread in natural products, but without simple synthetic approaches. Very recently, the synthesis of the SCF3-substituted tryptanthrin derivatives 42c was developed by the Wang group (Scheme 42).66 A large variety of desired products 42c were obtained in moderate to good yields, demonstrating the broad range of substrates. The proposed mechanism showed that ˙SCF3 was generated from AgSCF3 (42b) in the presence of K2S2O8. After the similar progress of the radical addition/cyclization, single-electron oxidation and deprotonation, the formation of the C(sp3)–SCF3 bond, C(sp2)–C bond, and C(sp2)–N bond were achieved in one step to access the desired products 42c.
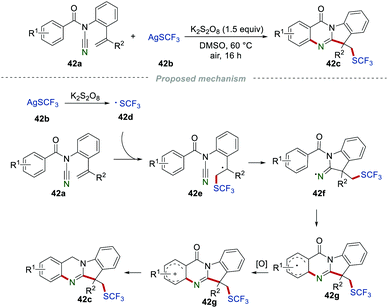 |
| | Scheme 42 Synthesis of SCF3-substituted tryptanthrin derivatives. | |
4. Remote cyano migration
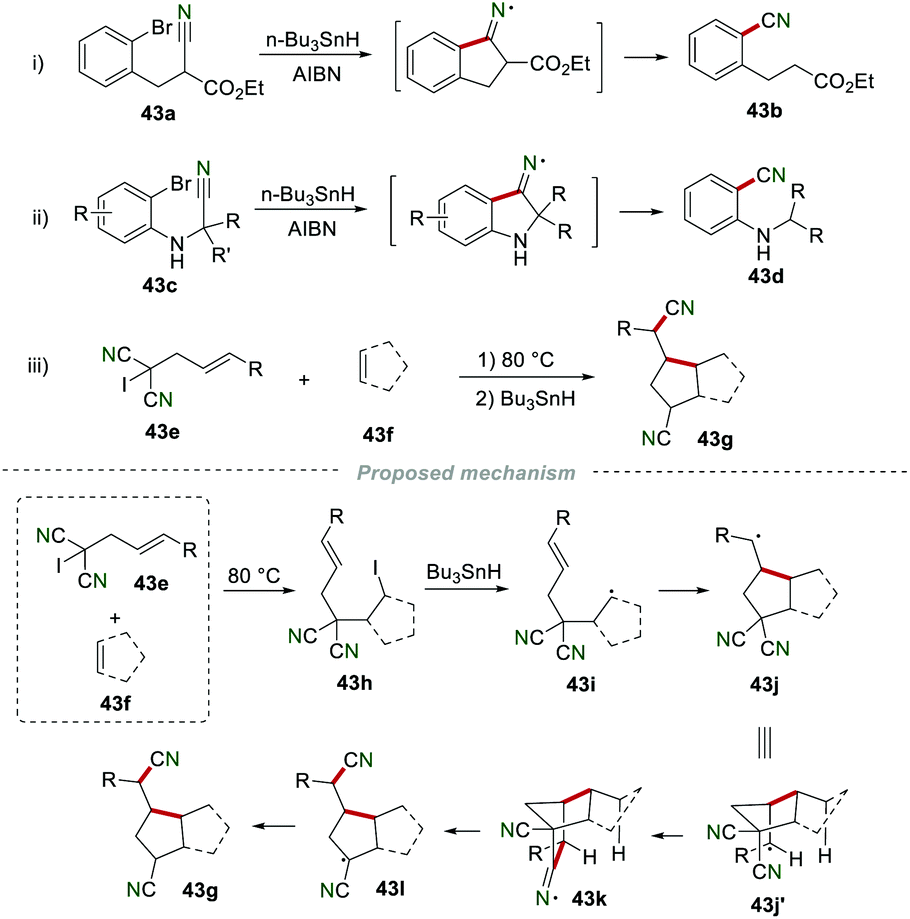
In 1987, Beckwith and co-workers reported the cyano migration to afford nitrile 43b in 59% yield by using 43a as the starting material in the presence of n-Bu3SnH and AIBN (Scheme 43i).67 Later in 1995, a similar reaction was achieved by Cossy and co-workers for the synthesis of 2-(alkylamino)benzonitriles 43d from α-(bromoarylamino)nitriles 43c (Scheme 43ii).68 In 1992, Curran and co-workers reported a novel cyano migration reaction (Scheme 43iii).69 The proposed mechanism showed that allyl iodomalononitrile 43g synthesized from the reaction of 43e and 43f could yield radical 43i in the presence of Bu3SnH. Radical 43i subsequently added to the CC bond to produce radical 43j, which further underwent intramolecular cyclization to give iminyl radical 43k. Finally, 43k underwent homolysis and H-abstraction to access the desired product 43g.
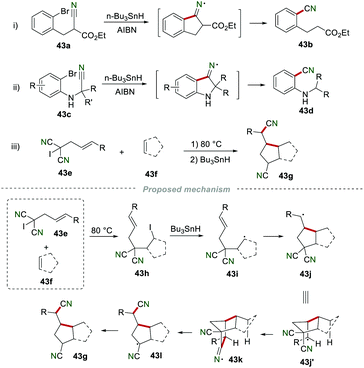 |
| | Scheme 43 Remote cyano migration to access nitriles. | |
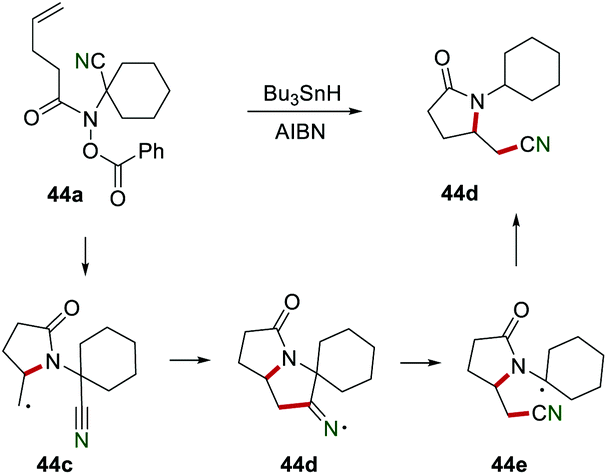
Another interesting cyano migration reaction was disclosed for the preparation of nitrile 44d in 83% yield (Scheme 44).70 In the presence of Bu3SnH and AIBN, radical 44c was produced from benzoate 44a. Radical 44c was further added to the cyano group to afford the iminyl radical 44d, which subsequently underwent homolysis and H-abstraction to yield product 44d.
 |
| | Scheme 44 Remote cyano migration to access nitrile starting from benzoate. | |
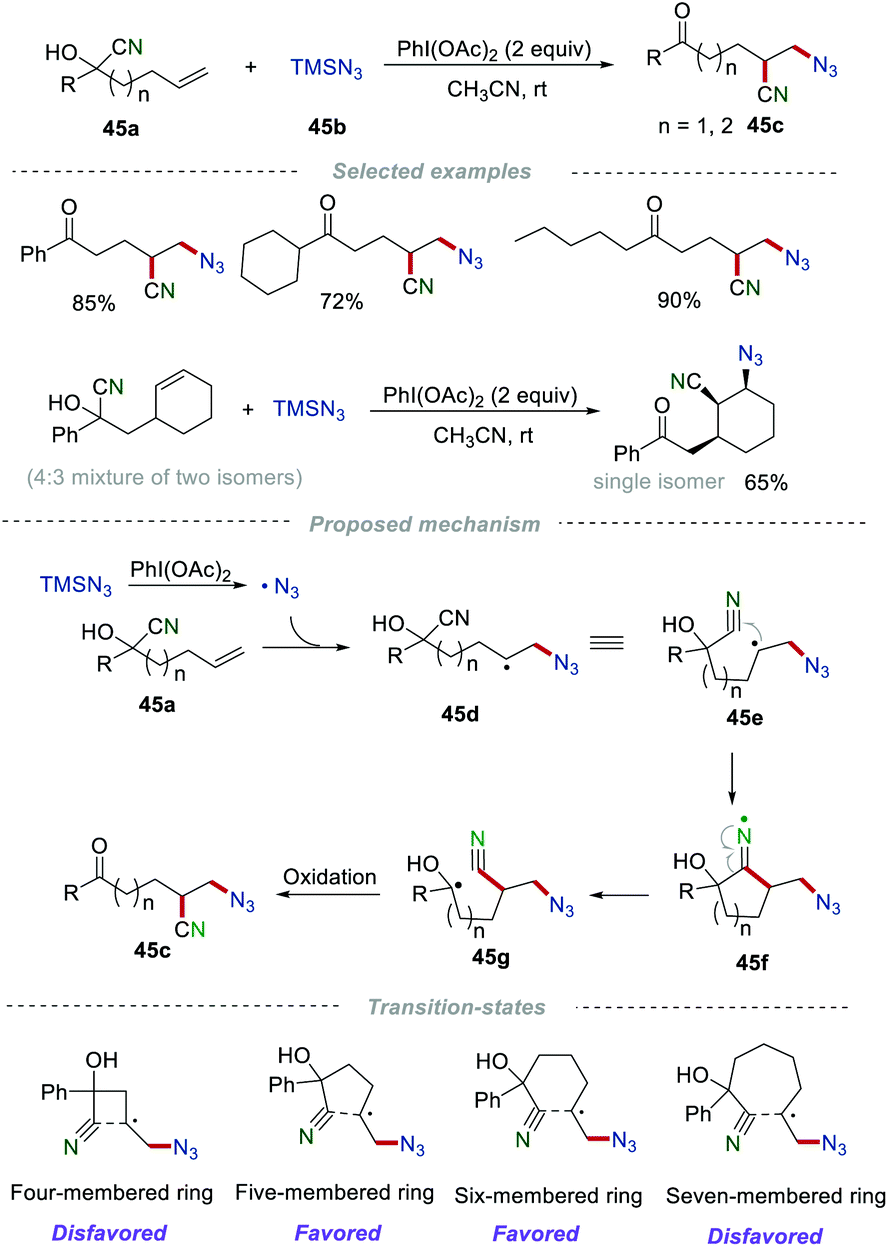
Recently, the distal migration reaction of the cyano groups has been continuously developed.71 In 2016, Zhu et al. developed a metal-free remote cyano migration triggered by the azidyl radical addition to unactivated alkenes (Scheme 45).72 In this reaction, various important azido-substituted alkyl nitriles 45c were prepared in good yields when n = 1 or 2, due to the favored five- or six-membered cyclic transition states. In sharp contrast, no satisfactory results were obtained when n = 0 or 3, which were mainly caused by the disfavored four- or seven-membered cyclic transition states. Notably, this cyano migration reaction showed excellent stereoselectivities to access corresponding single diastereomeric products. Mechanism studies showed that the azido radical (˙N3) was initially generated from TMSN3 in the presence of PhI(OAc)2. Then, the selective addition of the azido radical to alkene 45a generated the alkyl radical 45d, which then underwent intramolecular cyclization with the cyano group to afford the cyclic iminium radical 45f. Immediately, the unstable cyclic iminium radical 45f underwent homolysis to produce a more stable hydroxyalkyl radical 45g with the cyano migration. Finally, the oxidation of radical 45g produced the azido-substituted alkyl nitriles 45c.
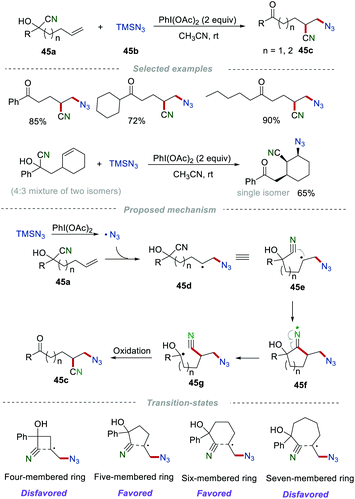 |
| | Scheme 45 Remote cyano migration for azidocyanation of unactivated alkenes. | |
Another example of long-distance cyano migration was recently reported by the Liu group. The diverse radical-mediated 1,2-cyanofunctionalization of unactivated alkenes 46a was successfully developed (Scheme 46).73 This simple strategy provided an efficient procedure for the synthesis of various important β-functionalized alkyl nitriles 46c bearing carbonyl, cyano, and other various functional groups. This synthetic approach showed a broad substrate scope and excellent selectivity. The addition of a radical to the unsaturated CC bond and the cyclization with the cyano group led to the formation of the alkyliminyl radical 46e. Through selective β-fission, the TMS-protected radical 46f was generated. Afterward, it underwent single-electron oxidation and TMS deprotection to access the final product 46c. The control experiment indicated that the TMS-protection of the hydroxyl group was a necessary factor for high yields.
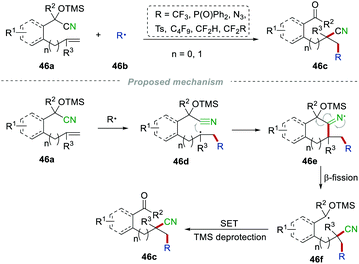 |
| | Scheme 46 Remote cyano migration of TMS-protected tertiary alcohols. | |
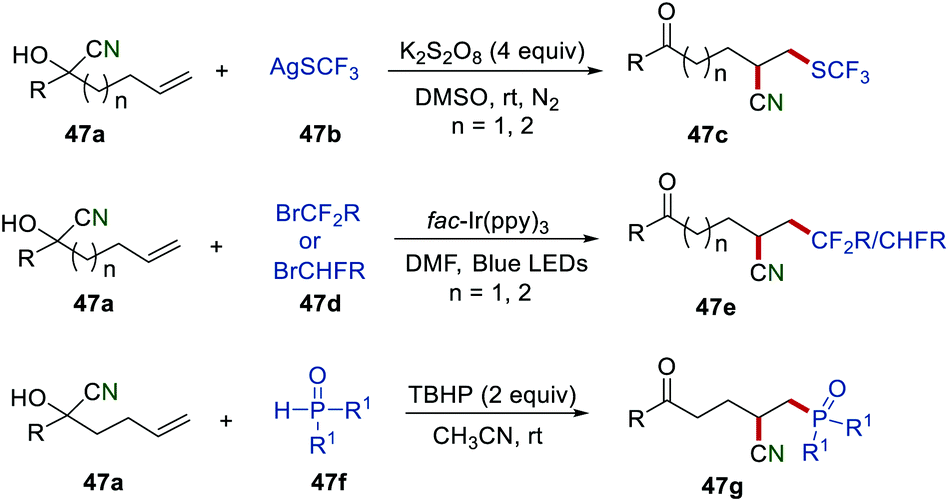
After these disclosures, Zhu and co-workers further applied this powerful and efficient strategy to achieve more structurally diverse alkyl nitriles by using a different radical source (Scheme 47). In the presence of K2S2O8, various CF3S-substituted alkyl nitriles 47c were obtained in good yields by employing AgSCF3 as a radical precursor.74 Afterwards, a visible light-driven photoredox catalysis for the cyanofluoroalkylation of unactivated alkenes 47avia remote cyano migration was developed. Under optimized conditions, a large variety of di- and monofluorinated alkyl nitriles 47e could be readily obtained in good yields under mild reaction conditions. Recently, a phosphinoyl radical-triggered cyanophosphinoylation of unactivated alkenes via the distal migration of cyano groups has been described.75 During the optimization of the reaction conditions, it was found that the phosphinoyl radical could be generated from 47f at room temperature under metal-free conditions.
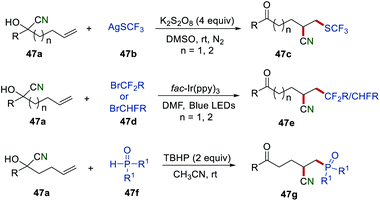 |
| | Scheme 47 Remote cyano migration for structurally diverse alkyl nitriles. | |
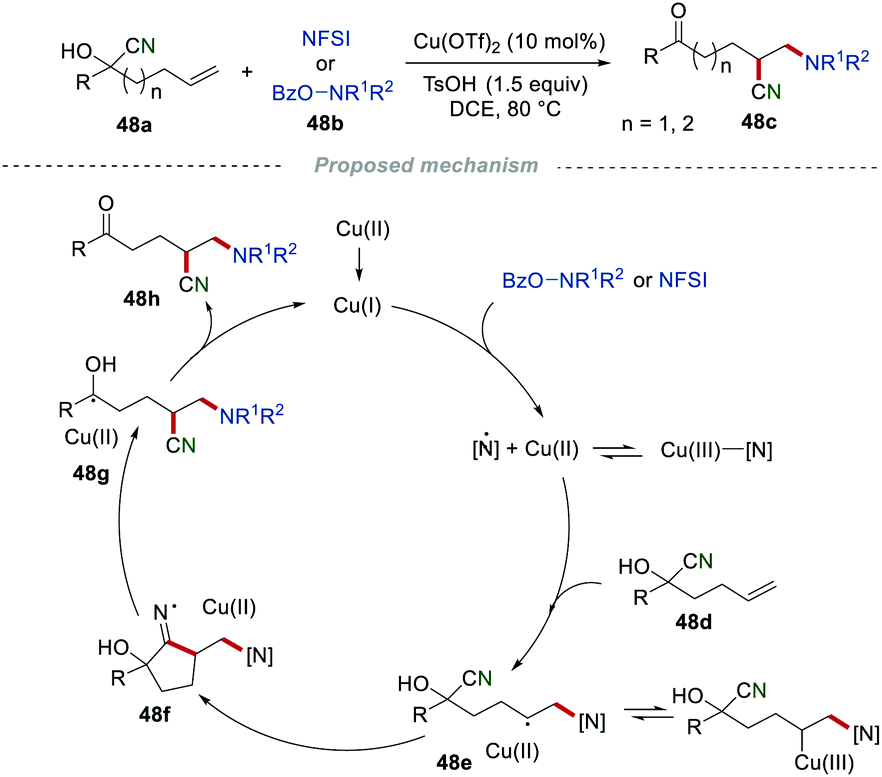
Stimulated by the importance of nitrogen-containing molecules and their wide applications in organic synthesis, pharmaceuticals, and materials, the Wang group discovered a highly regioselective approach for the aminocyanation of unactivated alkenes via distal cyano migration by reacting o-benzoylhydroxylamines 48a with N-fluorobenzenesulfonimides (NFSI) 48b (Scheme 48).76 In the presence of copper, the aminocyanation of alkenes via similar cyano migration protocol readily proceeded in moderate to good yields with a broad substrate scope. The proposed mechanism showed that o-benzyolhydroxylamine or NFSI 48b was oxidized by Cu(I) to generate an amino–Cu(III) complex, which was selectively added to the CC bond of alkene 48d, delivering the intermediate 48e. Subsequently, the intramolecular cyclization of intermediate 48e afforded the cyclic iminium radical intermediate 48f, followed by β-cleavage, leading to the formation of the hydroxyalkyl radical 48g. Finally, radical 48g was further oxidized to access the desired product 48h, along with the regeneration of Cu(I).
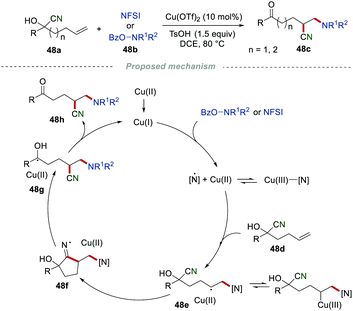 |
| | Scheme 48 Aminocyanation of alkenes. | |
5. Summary and outlook
By using cyano groups as radical acceptors, radical cascade cyclization reactions afford diverse opportunities for the convenient construction of various important heterocycles, carbocycles, and structurally diverse alkyl nitriles. In this review, the recent advances in radical cascade reactions using cyano groups as radical acceptors have been comprehensively discussed. It is shown that quinoline-2,4-diones, indenones, quinazolinones, phenanthridines, and benzothiazoles are readily accessible by using this approach. Moreover, various important functionalized alkyl nitriles can also be realized via this straightforward modular way. Although great progress has been achieved in this area in recent years, there are still many challenges remaining to be solved. For example, all of these reactions discussed in this review were carried out in conventional volatile organic solvents, which would make a great impact on the environment. Therefore, the development of novel catalytic systems in green solvents for these transformations is highly desired. Moreover, technologies including photocatalysis and electrocatalysis have emerged as novel tools for radical reactions. Although some photocatalytic systems have been reported in the above-mentioned reactions, the application of photo/electrochemical approaches to initiate various radicals for the radical cascade cyclization reaction should be attractive. We hope this review will stimulate the advancement of studies in this field.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 21501010, 21971224), 111 Project (D20003), and the Hunan Provincial Natural Science Foundation of China (No. 2019JJ20008).
Notes and references
-
(a) H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Catalytic cascade reactions by radical relay, Chem. Soc. Rev., 2019, 48, 4626 RSC;
(b) M. P. Plesniak, H.-M. Huang and D. J. Procter, Radical cascade reactions triggered by single electron transfer, Nat. Rev. Chem., 2017, 1, 0077 CrossRef;
(c) A. Studer and D. P. Curran, The electron is a catalyst, Nat. Chem., 2014, 6, 765 CrossRef CAS;
(d) Y. Zhao, Y. Lv and W. Xia, Synthesis of Cyclic Compounds via Photoinduced Radical Cyclization Cascade of C=C bonds, Chem. Rec., 2019, 19, 424 CrossRef CAS;
(e) M.-H. Huang, W.-J. Hao and B. Jiang, Recent Advances in Radical-Enabled Bicyclization and Annulation/1,n-Bifunctionalization Reactions, Chem. – Asian J., 2018, 13, 2958 CrossRef CAS;
(f) M.-H. Huang, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Recent advances in radical transformations of internal alkynes, Chem. Commun., 2018, 54, 10791 RSC;
(g) Z. Cao, Q. Zhu, Y.-W. Lin and W.-M. He, The concept of dual roles design in clean organic preparation, Chin. Chem. Lett., 2019, 30, 2132 CrossRef CAS;
(h) P. Mao, J. Zhu, J. Yuan, L. Yang, Y. Xiao and C. Zhang, Recent Advances on the Catalytic Functionalization of Quinoxalin-2(1H)-ones via C-H Bond Activation, Chin. J. Org. Chem., 2019, 39, 1529 CrossRef CAS;
(i) X.-Y. Yuan, F.-L. Zeng, H.-L. Zhu, Y. Liu, Q.-Y. Lv, X.-L. Chen, L. Peng and B. Yu, A Metal-Free Visible-Light-Promoted Phosphorylation/Cyclization Reaction in Water towards 3-Phosphorylated Benzothiophenes, Org. Chem. Front., 2020, 7, 1884 RSC.
-
(a) H. Jiang and A. Studer, Chemistry With N-Centered Radicals Generated by Single-Electron Transfer-Oxidation Using Photoredox Catalysis, CCS Chem., 2019, 1, 38 CAS;
(b) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Recent Advances of 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzI.PN) in Photocatalytic Transformations, Chem. Commun., 2019, 55, 5408 RSC.
-
(a) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts, Acc. Chem. Res., 2020, 53, 1066 CrossRef CAS;
(b) W. Yin and X. Wang, Recent advances in iminyl radical-mediated catalytic cyclizations and ring-opening reactions, New J. Chem., 2019, 43, 3254 RSC;
(c) P. Xiong and H.-C. Xu, Chemistry with Electrochemically Generated N-Centered Radicals, Acc. Chem. Res., 2019, 52, 3339 CrossRef CAS;
(d) S. Z. Zard, Iminyl Radicals: A Fresh Look at a Forgotten Species (and Some of its Relatives), Synlett, 1996, 1148 CrossRef CAS;
(e) W. R. Bowman, C. F. Bridge and P. Brookes, Radical cyclisation onto nitriles, Tetrahedron Lett., 2000, 41, 8989 CrossRef CAS.
- J. W. Grissom, D. Klingberg, S. Meyenburg and B. L. Stallman, Enediyne- and Tributyltin Hydride-Mediated Aryl Radical Additions onto Various Radical Acceptors, J. Org. Chem., 1994, 59, 7876 CrossRef CAS.
- D. Griller, P. Schmid and K. U. Ingold, Cyclization of the 4-cyanobutyl radical, Can. J. Chem., 1979, 57, 831 CrossRef CAS.
- E. J. Corey and S. G. Pyne, Conversion of ketones having δ, ε-π-functions to cyclopentanols by zinc-trimethylchlorosilane, Tetrahedron Lett., 1983, 24, 2821 CrossRef CAS.
- G. A. Molander and C. N. Wolfe, Intramolecular Ketone–Nitrile Reductive Coupling Reactions Promoted by Samarium(II) Iodide, J. Org. Chem., 1998, 63, 9031 CrossRef CAS.
- D. L. J. Clive and P. L. Beaulieu, Synthesis of ketones by cyclization of cyano and acetylenic radicals: use of .delta.-hydroxy nitriles and .delta.- or .epsilon.-hydroxy acetylenes, J. Org. Chem., 1984, 49, 1313 CrossRef CAS.
- B. B. Snider and B. O. Buckman, Termination of manganese(III)-based oxidative free-radical cyclizations by addition to nitriles. Formation of cyclopentanones and cyclohexanones, J. Org. Chem., 1992, 57, 322 CrossRef CAS.
- R. Sulsky, J. Z. Gougoutas, J. DiMarco and S. A. Biller, Conformational Switching and the Synthesis of Spiro[2H-indol]-3(1H)-ones by Radical Cyclization, J. Org. Chem., 1999, 64, 5504 CrossRef CAS.
- R. Tsang and B. Fraser-Reid, Serial radical cyclization via a vinyl group immobilized by a pyranoside. A route to bis-annulated pyranosides, J. Am. Chem. Soc., 1986, 108, 2116 CrossRef CAS.
- H. Pak, I. I. Canalda and B. Fraser-Reid, Carbohydrates to carbocycles: a synthesis of (-)-.alpha.-pipitzol, J. Org. Chem., 1990, 55, 3009 CrossRef CAS.
- P. Hegarty and J. Mann, A new approach to the skeletons of both the aphidicolin and stemodin diterpenoids, Tetrahedron, 1995, 51, 9079 CrossRef CAS.
-
(a) P. M. Esch, H. Hiemstra and W. N. Speckamp, Synthesis of cyclic α-amino acids through ring closure of glycine derived free radicals, Tetrahedron Lett., 1990, 31, 759–762 CrossRef CAS;
(b) S. Knapp, F. S. Gibson and Y. H. Choe, Radical based annulations of iodo lactams, Tetrahedron Lett., 1990, 31, 5397 CrossRef CAS;
(c) B. Chenera, C. P. Chuang, D. J. Hart and L. Y. Hsu, Observations regarding .delta.- and .epsilon.-cyano radical cyclizations, J. Org. Chem., 1985, 50, 5409 CrossRef CAS.
- R. A. Alonso, C. S. Burgey, B. V. Rao, G. D. Vite, R. Vollerthun, M. A. Zottola and B. Fraser-Reid, Carbohydrates to carbocycles: synthesis of the densely functionalized carbocyclic core of tetrodotoxin by radical cyclization of an anhydro sugar precursor, J. Am. Chem. Soc., 1993, 115, 6666 CrossRef CAS.
- T. Shono and N. Kise, Electroreductive intramolecular coupling of γ- and δ-cyanoketones, Tetrahedron Lett., 1990, 31, 1303 CrossRef CAS.
- Y.-M. Li, S.-S. Wang, F. Yu, Y. Shen and K.-J. Chang, Copper-catalyzed cascade addition/cyclization: highly efficient access to phosphonylated quinoline-2,4(1H,3H)-diones, Org. Biomol. Chem., 2015, 13, 5376 RSC.
- L. Xiong, H. Hu, C.-W. Wei and B. Yu, Radical reactions for the synthesis of 3-substituted chroman-4-ones, Eur. J. Org. Chem., 2020, 1588 CrossRef CAS.
- S.-S. Wang, H. Fu, Y. Shen, M. Sun and Y.-M. Li, Oxidative Radical Addition/Cyclization Cascade for the Construction of Carbonyl-Containing Quinoline-2,4(1H,3H)-Diones, J. Org. Chem., 2016, 81, 2920 CrossRef CAS.
-
(a) K.-J. Liu, S. Jiang, L.-H. Lu, L.-L. Tang, S.-S. Tang, H.-S. Tang, Z. Tang, W.-M. He and X. Xu, Bis(methoxypropyl) ether-promoted oxidation of aromatic alcohols into aromatic carboxylic acids and aromatic ketones with O2 under metal- and base-free conditions, Green Chem., 2018, 20, 3038 RSC;
(b) P. Li, Y. Wang, X. Wang, Y. Wang, Y. Liu, K. Huang, J. Hu, L. Duan, C. Hu and J. Liu, Selective Oxidation of Benzylic C–H Bonds Catalyzed by Cu(II)/{PMo12}, J. Org. Chem., 2020, 85, 3101 CrossRef CAS;
(c) D. Zhai and S. Ma, Copper catalysis for highly selective aerobic oxidation of alcohols to aldehydes/ketones, Org. Chem. Front., 2019, 6, 3101 RSC.
- S.-S. Wang, H. Fu, G. Wang, M. Sun and Y.-M. Li, , Iron-catalyzed addition/cyclization cascade of activated alkenes with alcohols: access to carbonyl substituted quinoline-2,4-diones, RSC Adv., 2016, 6, 52391 RSC.
-
(a) K. Sun, X.-L. Chen, X. Li, L.-B. Qu, W.-Z. Bi, X. Chen, H.-L. Ma, S.-T. Zhang, B.-W. Han, Y.-F. Zhao and C.-J. Li, H-phosphonate-mediated sulfonylation of heteroaromatic N-oxides: a mild and metal-free one-pot synthesis of 2-sulfonyl quinolines/pyridines, Chem. Commun., 2015, 51, 12111 RSC;
(b) J.-N. Desrosiers and A. B. Charette, Catalytic Enantioselective Reduction of β,β-Disubstituted Vinyl Phenyl Sulfones by Using Bisphosphine Monoxide Ligands, Angew. Chem., Int. Ed., 2007, 46, 5955 CrossRef CAS;
(c) L. Tang, K. Du, B. Yu and L. He, Oxidation of aromatic sulfides with molecular oxygen: Controllable synthesis of sulfoxides or sulfones, Chin. Chem. Lett., 2020, 31(12), 2991 Search PubMed;
(d) S. Peng, Y.-X. Song, J.-Y. He, S.-S. Tang, J.-X. Tan, Z. Cao, Y.-W. Lin and W.-M. He, TsCl-promoted sulfonylation of quinoline N-oxides with sodium sulfinates in water, Chin. Chem. Lett., 2019, 30, 2287 CrossRef CAS.
-
(a) F.-L. Yang and S.-K. Tian, Sulfonyl hydrazides as sulfonyl sources in organic synthesis, Tetrahedron Lett., 2017, 58, 487 CrossRef CAS;
(b) Y.-Q. Jiang, J. Li, Z.-W. Feng, G.-Q. Xu, X. Shi, Q.-J. Ding, W. Li, C.-H. Ma and B. Yu, Ethylene Glycol: A Green Solvent for Visible Light-Promoted Aerobic Transition Metal-Free Cascade Sulfonation/Cyclization Reaction, Adv. Synth. Catal., 2020, 362, 2609 CrossRef CAS;
(c) Z. Wang and W.-M. He, Radical Cyclization Strategy towards Indolo[1,2-a]quinolines, Chin. J. Org. Chem., 2019, 39, 3594 CrossRef CAS.
- S. Wang, X. Huang, Q. Wang, Z. Ge, X. Wang and R. Li, An efficient synthesis of sulfonated quinoline diones by copper catalyzed sulfonylation of activated alkenes with sulfonylhydrazides, RSC Adv., 2016, 6, 11754 RSC.
- H. Fu, S.-S. Wang and Y.-M. Li, Copper-Mediated Oxidative Radical Addition/Cyclization Cascade: Synthesis of Trifluoromethylated and Sulfonated Quinoline-2,4(1H,3H)-diones, Adv. Synth. Catal., 2016, 358, 3616 CrossRef CAS.
- P. C. Montevecchi, M. L. Navacchia and P. Spagnolo, Attempts at vinyl radical carbonylation through cyclization onto carbonyl and cyano groups, Tetrahedron, 1998, 54, 8207 CrossRef CAS.
- X.-T. Zhu, Q. Zhao, F. Liu, A.-F. Wang, P.-J. Cai, W.-J. Hao, S.-J. Tu and B. Jiang, Silver-mediated radical 5-exo-dig cyclization of 2-alkynylbenzonitriles: synthesis of phosphinylated 1-indenones, Chem. Commun., 2017, 53, 6828 RSC.
- Z. Xiao-Tong, Z. Tian-Shu, Z. Qi, C. Pei-Jun, H. Wen-Juan, T. Shu-Jiang and J. Bo, Sulfinate-Salt-Mediated Radical Relay Cyclization of Cyclic Ethers with 2-Alkynylbenzonitriles toward 3-Alkylated 1-Indenones, Chem. – Asian J., 2018, 13, 1157 CrossRef.
- Y. Zhang, K. Sun, Q. Lv, X. Chen, L. Qu and B. Yu, Recent applications of radical cascade reaction in the synthesis of functionalized 1-indenones, Chin. Chem. Lett., 2019, 30, 1361 CrossRef CAS.
-
(a) X.-T. Zhu, Q.-L. Lu, X. Wang, T.-S. Zhang, W.-J. Hao, S.-J. Tu and B. Jiang, Substrate-Controlled Generation of 3-Sulfonylated 1-Indenones and 3-Arylated (Z)-Indenes via Cu-Catalyzed Radical Cyclization Cascades of o-Alkynylbenzonitriles, J. Org. Chem., 2018, 83, 9890 CrossRef CAS;
(b) B. Zhou, W. Chen, Y. Yang, Y. Yang, G. Deng and Y. Liang, A radical cyclization cascade of 2-alkynylbenzonitriles with sodium arylsulfinates, Org. Biomol. Chem., 2018, 16, 7959 RSC;
(c) K. Sun, X.-L. Chen, S.-J. Li, D.-H. Wei, X.-C. Liu, Y.-L. Zhang, Y. Liu, L.-L. Fan, L.-B. Qu, B. Yu, K. Li, Y.-Q. Sun and Y.-F. Zhao, Copper-Catalyzed Radical Cascade Cyclization To Access 3-Sulfonated Indenones with the AIE Phenomenon, J. Org. Chem., 2018, 83, 14419 CrossRef CAS.
- B. Zhang and A. Studer, Recent advances in the synthesis of nitrogen heterocycles via radical cascade reactions using isonitriles as radical acceptors, Chem. Soc. Rev., 2015, 44, 3505 RSC.
- D. P. Curran and H. Liu, 4+1 Radical annulations with isonitriles: a simple route to cyclopenta-fused quinolines, J. Am. Chem. Soc., 1991, 113, 2127 CrossRef CAS.
- D. P. Curran, H. Liu, H. Josien and S.-B. Ko, Tandem radical reactions of isonitriles with 2-pyridonyl and other aryl radicals: Scope and limitations, and a first generation synthesis of (±)-camptothecin, Tetrahedron, 1996, 52, 11385 CrossRef CAS.
- D. Nanni, P. Pareschi, C. Rizzoli, P. Sgarabotto and A. Tundo, Radical annulations and cyclisations with isonitriles: the fate of the intermediate imidoyl and cyclohexadienyl radicals, Tetrahedron, 1995, 51, 9045 CrossRef CAS.
- R. Leardini, D. Nanni, P. Pareschi, A. Tundo and G. Zanardi, α-(Arylthio)imidoyl Radicals: [3 + 2] Radical Annulation of Aryl Isothiocyanates with 2-Cyano-Substituted Aryl Radicals, J. Org. Chem., 1997, 62, 8394 CrossRef CAS.
- C. M. Camaggi, R. Leardini, D. Nanni and G. Zanardi, Radical annulations with nitriles: Novel cascade reactions of cyano-substituted alkyl and sulfanyl radicals with isonitriles, Tetrahedron, 1998, 54, 5587 CrossRef CAS.
- W. R. Bowman, M. O. Cloonan, A. J. Fletcher and T. Stein, Synthesis of heteroarenes using cascade radical cyclisation via iminyl radicals, Org. Biomol. Chem., 2005, 3, 1460 RSC.
- D. P. Curran, C. P. Jasperse and M. J. Totleben, Approximate absolute rate constants for the reactions of tributyltin radicals with aryl and vinyl halides, J. Org. Chem., 1991, 56, 7169 CrossRef CAS.
- X. Sun, J. Li, Y. Ni, D. Ren, Z. Hu and S. Yu, Synthesis of Fused Quinoline and Quinoxaline Derivatives Enabled by Domino Radical Triple Bond Insertions, Asian J. Org. Chem., 2014, 3, 1317 CrossRef CAS.
- A. Servais, M. Azzouz, D. Lopes, C. Courillon and M. Malacria, Radical Cyclization of N-Acylcyanamides: Total Synthesis of Luotonin A, Angew. Chem., Int. Ed., 2007, 46, 576 CrossRef CAS.
- M.-H. Larraufie, C. Courillon, C. Ollivier, E. Lacôte, M. Malacria and L. Fensterbank, Radical Migration of Substituents of Aryl Groups on Quinazolinones Derived from N-Acyl Cyanamides, J. Am. Chem. Soc., 2010, 132, 4381 CrossRef CAS.
- M.-H. Larraufie, C. Ollivier, L. Fensterbank, M. Malacria and E. Lacôte, Radical Synthesis of Guanidines from N-Acyl Cyanamides, Angew. Chem., Int. Ed., 2010, 49, 2178 CrossRef CAS.
- Y.-Y. Han, H. Jiang, R. Wang and S. Yu, Synthesis of Tetracyclic Quinazolinones Using a Visible-Light-Promoted Radical Cascade Approach, J. Org. Chem., 2016, 81, 7276 CrossRef CAS.
- J. Zheng, Y. Zhang, D. Wang and S. Cui, Silver(I)-Mediated Phosphorylation/Cyclization Cascade of N-Cyanamide Alkenes for Divergent Access to Quinazolinones and Dihydroisoquinolinones, Org. Lett., 2016, 18, 1768 CrossRef CAS.
- J. Zheng, Z. Deng, Y. Zhang and S. Cui, Copper-Catalyzed Divergent Trifluoromethylation/Cyclization of Unactivated Alkenes, Adv. Synth. Catal., 2016, 358, 746 CrossRef CAS.
- X. Liu, P. Qian, Y. Wang and Y. Pan, Metal-free sequential decarbonylative annulation of N-cyanamides for the construction of 2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-ones, Org. Chem. Front., 2017, 4, 2370 RSC.
-
(a) W.-C. Yang, J.-G. Feng, L. Wu and Y.-Q. Zhang, Aliphatic Aldehydes: Novel Radical Alkylating Reagents, Adv. Synth. Catal., 2019, 361, 1700 CrossRef CAS;
(b) L. Kong, Y. Zhou, F. Luo and G. Zhu, Recent Advances on Oxidative Radical Addition to Aldehydes, Chin. J. Org. Chem., 2018, 38, 2858 CrossRef CAS.
- R. Leardini, D. Nanni, A. Tundo and G. Zanardi, Novel [3 + 2] radical
annulations of cyano-substituted aryl radicals with alkynes, Tetrahedron Lett., 1998, 39, 2441 CrossRef CAS.
- P. Xu, Y.-M. Zhu, F. Wang, S.-Y. Wang and S.-J. Ji, Mn(III)-Mediated Cascade Cyclization of 3-Isocyano-[1,1′-biphenyl]-2-carbonitrile with Arylboronic Acid: Construction of Pyrrolopyridine Derivatives, Org. Lett., 2019, 21, 683 CrossRef CAS.
- N. Zhou, M. Wu, M. Zhang, X. Zhou and W. Zhou, TBPB-initiated cascade cyclization of 3-arylethynyl-[1,1′-biphenyl]-2-carbonitriles with sulfinic acids: access to sulfone-containing cyclopenta[gh]phenanthridines, Org. Biomol. Chem., 2020, 18, 1733 RSC.
- X. Li, X. Fang, S. Zhuang, P. Liu and P. Sun, Photoredox Catalysis: Construction of Polyheterocycles via Alkoxycarbonylation/Addition/Cyclization Sequence, Org. Lett., 2017, 19, 3580 CrossRef CAS.
- Y. Yu, Z. Cai, W. Yuan, P. Liu and P. Sun, Radical Addition/Insertion/Cyclization Cascade Reaction To Assemble Phenanthridines from N-Arylacrylamide Using Cyano as a Bridge under Photoredox Catalysis, J. Org. Chem., 2017, 82, 8148 CrossRef CAS.
- C. Zhang, J. Pi, S. Chen, P. Liu and P. Sun, Construction of a 4H-pyrido[4,3,2-gh]phenanthridin-5(6H)-one skeleton via a catalyst-free radical cascade addition/cyclization using azo compounds as radical sources, Org. Chem. Front., 2018, 5, 793 RSC.
- Y. Yu, W. Yuan, H. Huang, Z. Cai, P. Liu and P. Sun, Visible-Light-Mediated Decarboxylative Alkylation Cascade Cyano Insertion/Cyclization of N-Arylacrylamides under Transition-Metal-Free Conditions, J. Org. Chem., 2018, 83, 1654 CrossRef CAS.
- L. Ruan, C. Chen, X. Zhang and J. Sun, Recent Advances on the Photo-Induced Reactions of Acyl Radical, Chin. J. Org. Chem., 2018, 38, 3155 CrossRef CAS.
- J.-Q. Shang, X.-X. Wang, Y. Xin, Y. Li, B. Zhou and Y.-M. Li, Decarboxylative cascade cyclization of α-keto acids with 2-cyano-3-arylaniline-derived acrylamides, Org. Biomol. Chem., 2019, 17, 9447 RSC.
- X. Liu, Z. Wu, Z. Zhang, P. Liu and P. Sun, Synthesis of trifluoroalkyl or difluoroalkyl phenanthridine derivatives via cascade reaction using an intramolecular cyano group as a radical acceptor under photoredox catalysis, Org. Biomol. Chem., 2018, 16, 414 RSC.
- M. Zhu, W. Fu, W. Guo, Y. Tian, Z. Wang and B. Ji, Visible-light-induced radical trifluoromethylthiolation of N-(o-cyanobiaryl)acrylamides, Org. Biomol. Chem., 2019, 17, 3374 RSC.
- J.-Q. Shang, S.-S. Wang, H. Fu, Y. Li, T. Yang and Y.-M. Li, Oxidative radical cascade cyclization involving C(sp3)–C(sp3), C(sp3)–C(sp2) and C(sp2)–N bonds formation: direct construction of cyano and methyl substituted polyheterocycles, Org. Chem. Front., 2018, 5, 1945 RSC.
- C. Zhang, J. Pi, L. Wang, P. Liu and P. Sun, Silyl radical initiated radical cascade addition/cyclization: synthesis of silyl functionalized 4H-pyrido[4,3,2-gh]phenanthridin-5(6H)-ones, Org. Biomol. Chem., 2018, 16, 9223 RSC.
- L. Ji, W. Gu, P. Liu and P. Sun, Iron-mediated deuterium addition cascade cyano insertion/cyclization of N-arylacrylamides to access deuterium-labelled phenanthridines, Org. Biomol. Chem., 2020, 18, 6126 RSC.
- W.-J. Xia, Y. Xin, Z.-W. Zhao, X. Chen, X.-X. Wang, Y. Li, G. Wang and Y.-M. Li, Oxidative cascade cyclization of 2-cyano-3-arylaniline derived acrylamides with toluenes, ethers, aliphatic alcohols or simple alkanes, Org. Chem. Front., 2020, 7, 1997 RSC.
- D. Hernández, L. Nielsen, K. B. Lindsay, M. López-García, K. Bjerglund and T. Skrydstrup, Stereocontrolled Synthesis of 2-Substituted-1,3-Azasilaheterocycles, Org. Lett., 2010, 12, 3528 CrossRef.
- L.-J. Wu, Y. Yang, R.-J. Song, J.-X. Yu, J.-H. Li and D.-L. He, An access to 1,3-azasiline-fused quinolinones via oxidative heteroannulation involving silyl C(sp3)–H functionalization, Chem. Commun., 2018, 54, 1367 RSC.
- P. Natarajan, Manjeet, Muskan, N. K. Brar and J. J. Kaur, Visible light photoredox catalysis: conversion of a mixture of thiophenols and nitriles into 2-substituted benzothiazoles via consecutive C–S and C–N bond formation reactions, Org. Chem. Front., 2018, 5, 1527 RSC.
- J. Guo, Y. Hao, G. Li, Z. Wang, Y. Liu, Y. Li and Q. Wang, Efficient synthesis of SCF3-substituted tryptanthrins by a radical tandem cyclization, Org. Biomol. Chem., 2020, 18, 1994 RSC.
-
(a) A. L. J. Beckwith, D. M. O'Shea and S. W. Westwood, Rearrangement of suitably constituted aryl, alkyl, or vinyl radicals by acyl or cyano group migration, J. Am. Chem. Soc., 1988, 110, 2565 CrossRef CAS;
(b) A. L. J. Beckwith, D. M. O'Shea, S. Gerba and S. W. Westwood, Cyano or acyl group migration by consecutive homolytic addition and β-fission, J. Chem. Soc., Chem. Commun., 1987, 666 RSC.
- J. Cossy, C. Poitevin, D. G. Pardo and J. L. Peglion, Synthesis of 2-(Alkylamino)benzonitriles from α-(Bromoarylamino)nitriles, Synthesis, 1995, 1368 CrossRef CAS.
- D. P. Curran and C. M. Seong, Radical annulation reactions of allyl iodomalononitriles, Tetrahedron, 1992, 48, 2175 CrossRef CAS.
-
(a) A.-C. Callier, B. Quiclet-Sire and S. Z. Zard, Amidyl and carbamyl radicals by stannane mediated cleavage of O-benzoyl hydroxamic acid derivatives, Tetrahedron Lett., 1994, 35, 6109 CrossRef CAS;
(b) J. Boivin, A.-C. Callier-Dublanchet, B. Quiclet-Sire, A.-M. Schiano and S. Z. Zard, Iminyl, amidyl, and carbamyl radicals from O-benzoyl oximes and O-benzoyl hydroxamic acid derivatives, Tetrahedron, 1995, 51, 6517 CrossRef CAS.
- X. Wu and C. Zhu, Radical-Mediated Remote Functional Group Migration, Acc. Chem. Res., 2020, 53, 1620 CrossRef CAS.
- Z. Wu, R. Ren and C. Zhu, Combination of a Cyano Migration Strategy and Alkene Difunctionalization: The Elusive Selective Azidocyanation of Unactivated Olefins, Angew. Chem., Int. Ed., 2016, 55, 10821 CrossRef CAS.
- N. Wang, L. Li, Z.-L. Li, N.-Y. Yang, Z. Guo, H.-X. Zhang and X.-Y. Liu, Catalytic Diverse Radical-Mediated 1,2-Cyanofunctionalization of Unactivated Alkenes via Synergistic Remote Cyano Migration and Protected Strategies, Org. Lett., 2016, 18, 6026 CrossRef CAS.
- M. Ji, Z. Wu, J. Yu, X. Wan and C. Zhu, Cyanotrifluoromethylthiolation of Unactivated Olefins through Intramolecular Cyano Migration, Adv. Synth. Catal., 2017, 359, 1959 CrossRef CAS.
- D. Chen, Z. Wu, Y. Yao and C. Zhu, Phosphinoyl-functionalization of unactivated alkenes through phosphinoyl radical-triggered distal functional group migration, Org. Chem. Front., 2018, 5, 2370 RSC.
- Y. Kwon and Q. Wang, Copper-Catalyzed 1,2-Aminocyanation of Unactivated Alkenes via Cyano Migration, Org. Lett., 2020, 22, 4141 CrossRef CAS.
Footnote |
| † Dedicated to the 100th anniversary of Chemistry at Nankai University. |
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Qi-Yan
Lv
ab,
Ying-Wu
Lin
a,
Qi-Yan
Lv
ab,
Ying-Wu
Lin


 under the irradiation of visible light, which was oxidized by bromide 19b to fac-Ir(ppy)3+, along with the formation of radical 19g. Subsequently, the addition of 19g to isocyanide 19a generated the imidoyl radical 19h, which subsequently underwent intramolecular cyclization to access the iminyl radical 19i. Radical 19i underwent homolytic aromatic substitution to produce the radical 19j, which was oxidized by fac-Ir(ppy)3+ to yield the radical cation 19k, along with the regeneration of fac-Ir(ppy)3 for the next cycle. Finally, the aromatization of radical cation 19k led to the formation of the desired products 19c by deprotonation.
under the irradiation of visible light, which was oxidized by bromide 19b to fac-Ir(ppy)3+, along with the formation of radical 19g. Subsequently, the addition of 19g to isocyanide 19a generated the imidoyl radical 19h, which subsequently underwent intramolecular cyclization to access the iminyl radical 19i. Radical 19i underwent homolytic aromatic substitution to produce the radical 19j, which was oxidized by fac-Ir(ppy)3+ to yield the radical cation 19k, along with the regeneration of fac-Ir(ppy)3 for the next cycle. Finally, the aromatization of radical cation 19k led to the formation of the desired products 19c by deprotonation.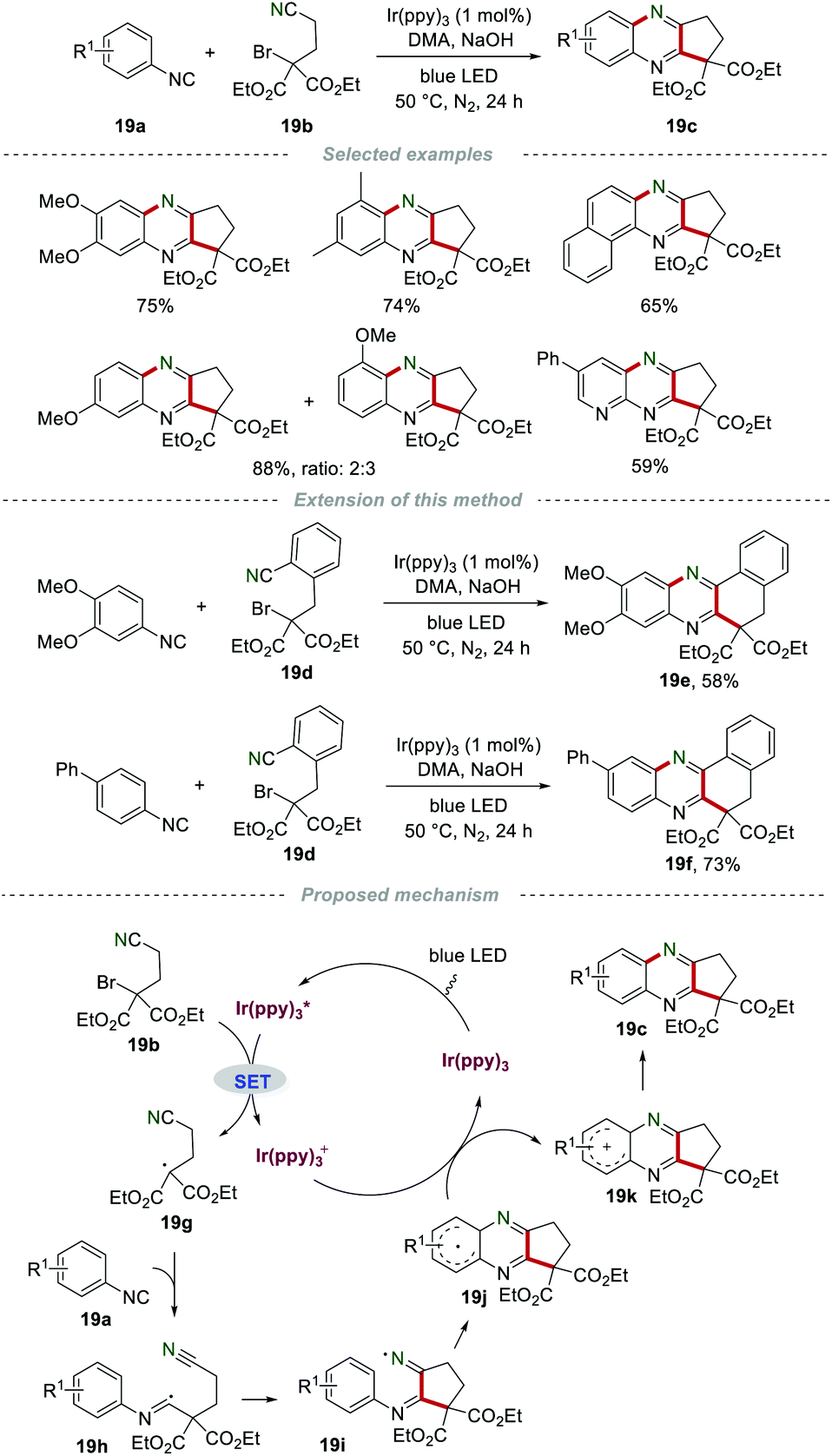
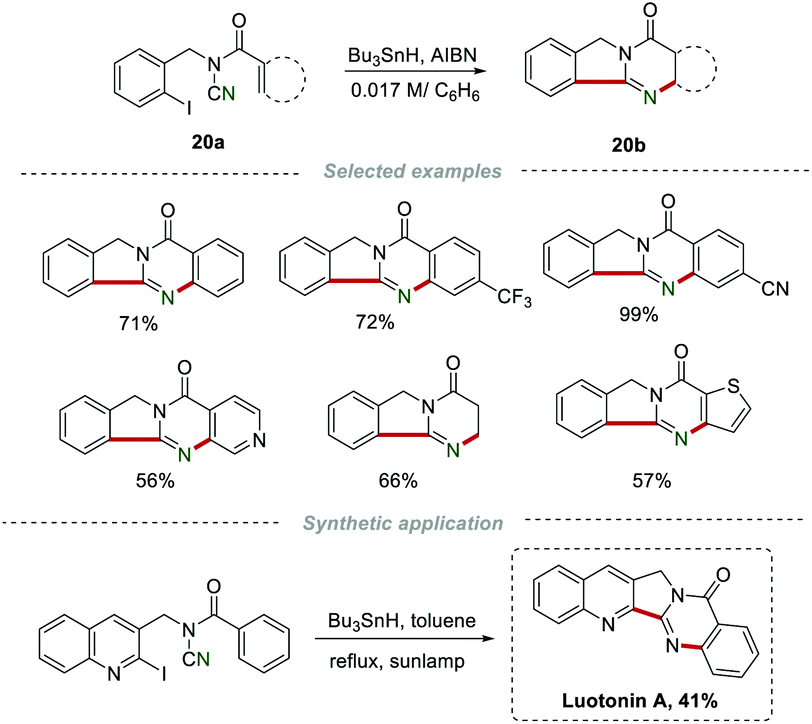
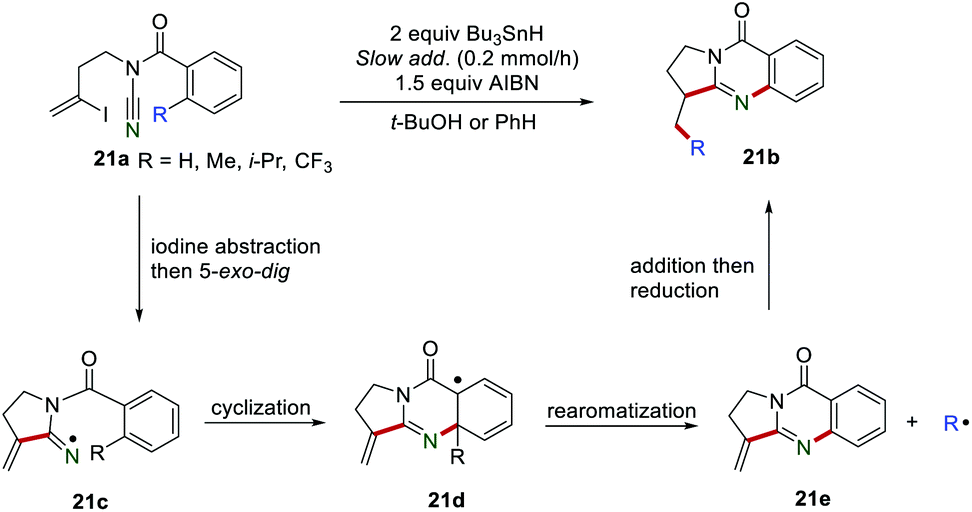

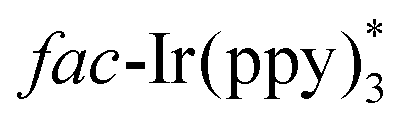 was indeed quenched by DIPEA (i-Pr2NEt), indicating that a reductive quenching was involved between DIPEA and
was indeed quenched by DIPEA (i-Pr2NEt), indicating that a reductive quenching was involved between DIPEA and 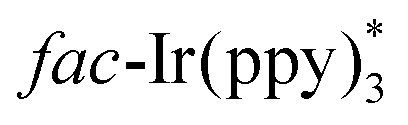 via single electron transfer. The proposed mechanism showed that
via single electron transfer. The proposed mechanism showed that 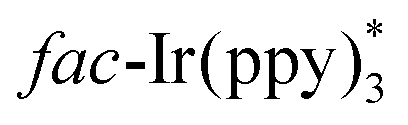 was formed under the irradiation of visible light, which further reacted with i-Pr2NEt to give fac-Ir(ppy)3−, along with the N-centered radical cation species. Subsequently, a single-electron transfer between fac-Ir(ppy)3− and reactant 23c led to the generation of the radical intermediate 23d. After a similar intramolecular radical cyclization, single-electron oxidation, and deprotonation process, the final product 23h was produced.
was formed under the irradiation of visible light, which further reacted with i-Pr2NEt to give fac-Ir(ppy)3−, along with the N-centered radical cation species. Subsequently, a single-electron transfer between fac-Ir(ppy)3− and reactant 23c led to the generation of the radical intermediate 23d. After a similar intramolecular radical cyclization, single-electron oxidation, and deprotonation process, the final product 23h was produced.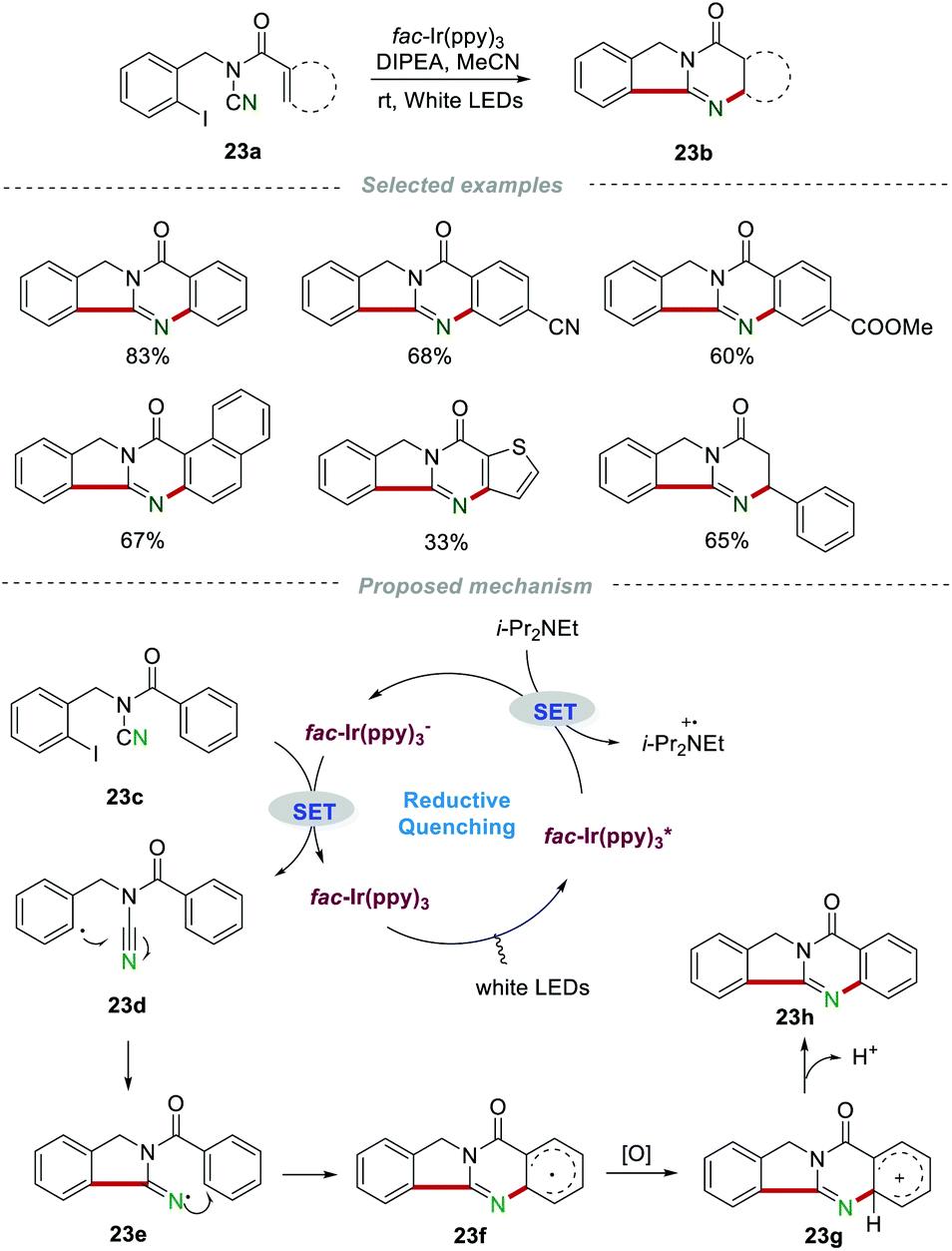
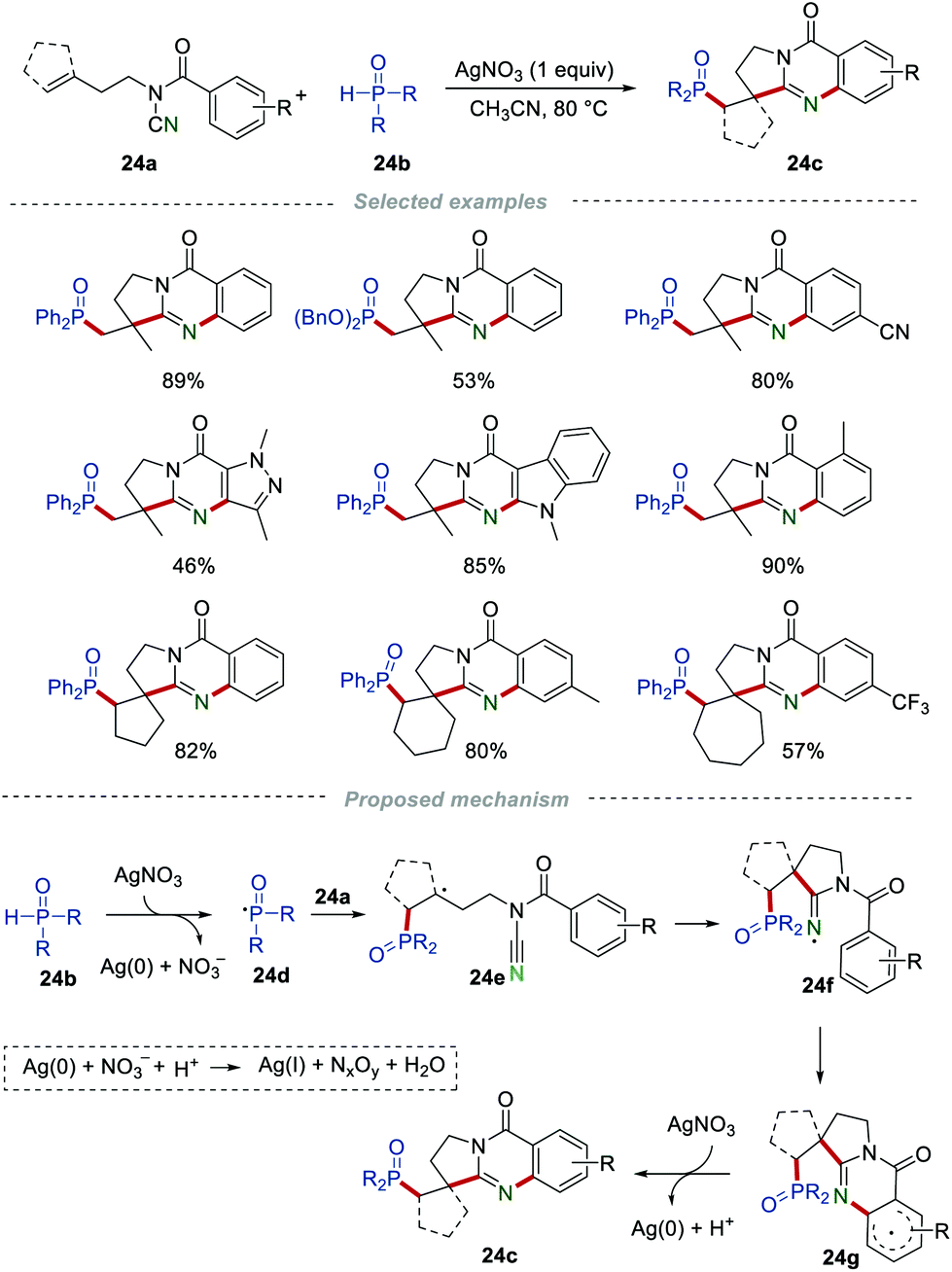

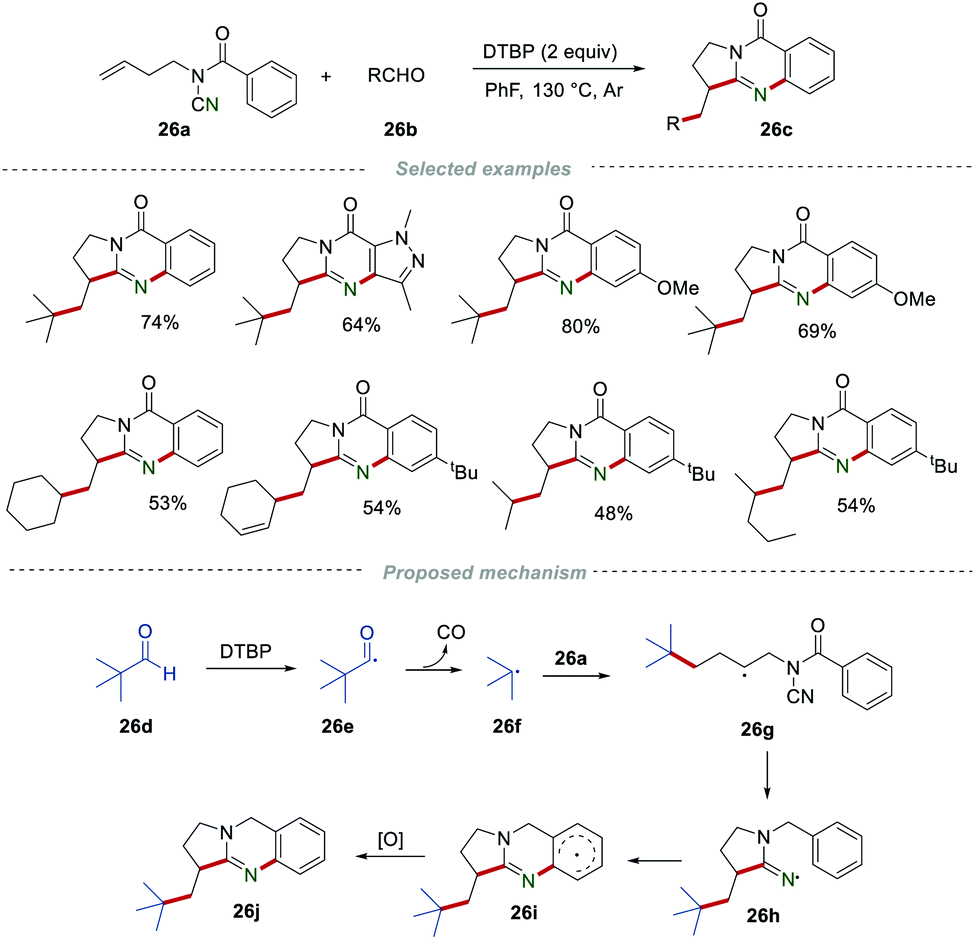
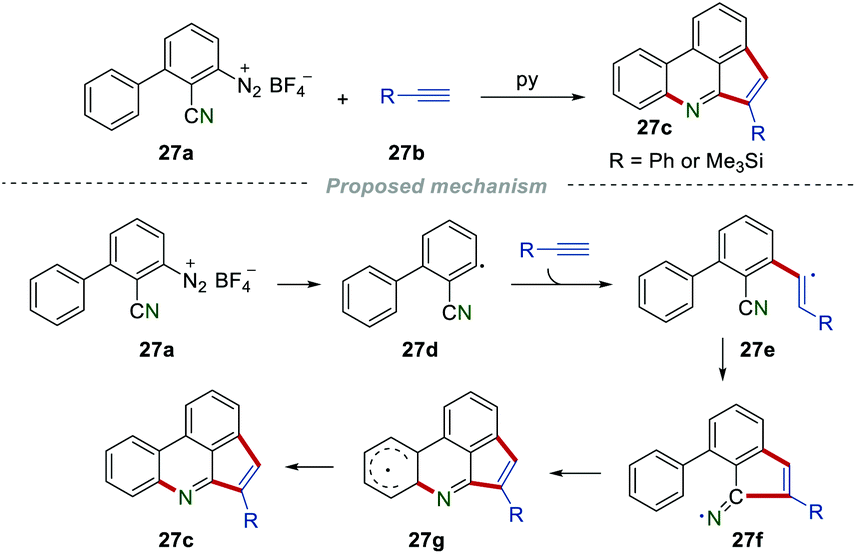
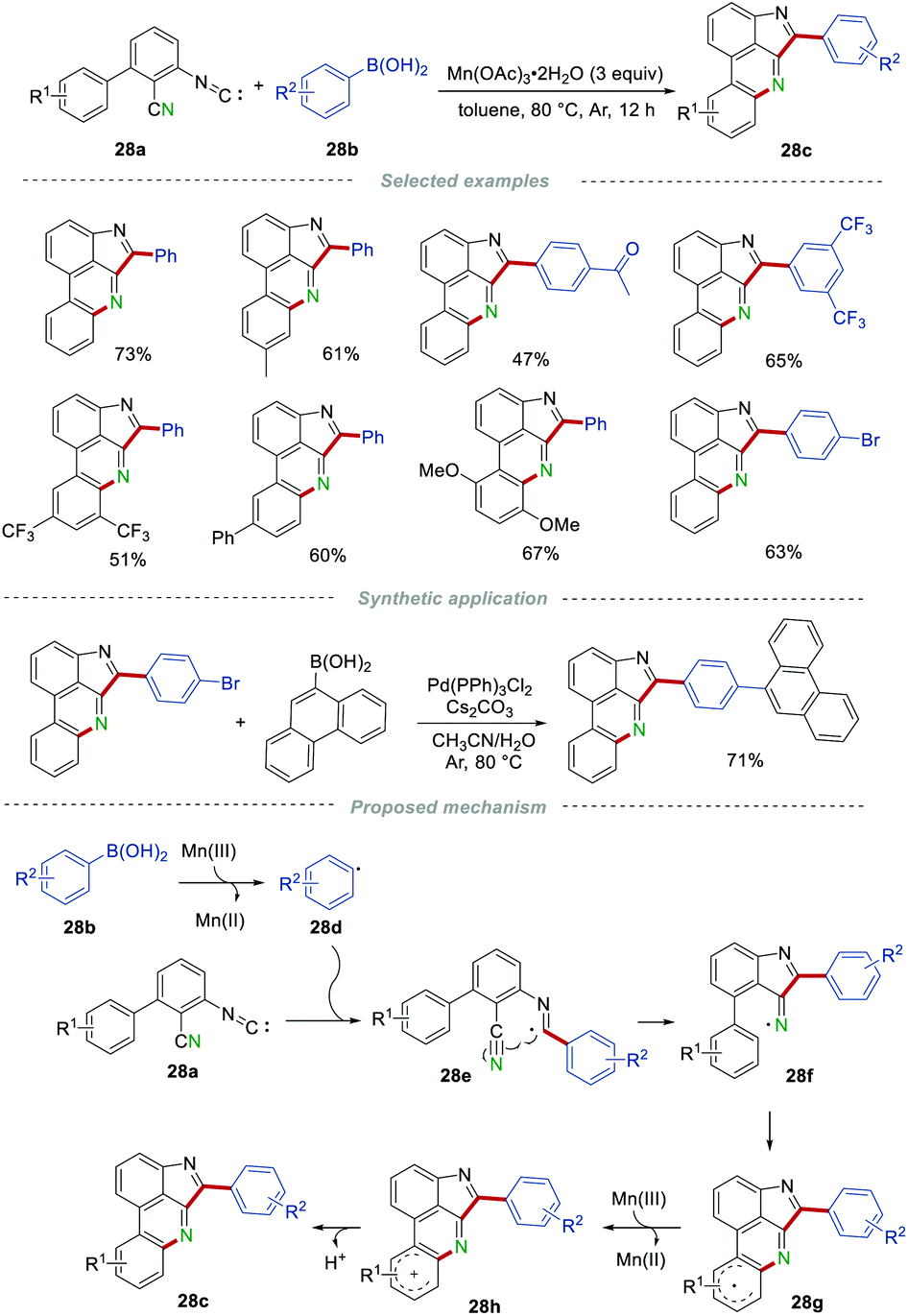
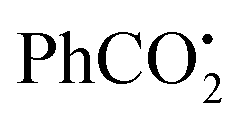 or t-BuO˙, which was generated from the thermal decomposition of TBPB. Then, the addition of radical 29d to the C–C triple bond of 29a resulted in the alkenyl radical 29e, which then underwent intramolecular cyclization with the cyano group to produce the radical intermediate 29g. The radical intermediate 29g was further oxidized to radical cation 29h, which subsequently regenerated the aromatic ring by deprotonation (path a). On the other hand, radical intermediate 29g could also convert into the desired product 29gvia hydrogen atom transfer (path b).
or t-BuO˙, which was generated from the thermal decomposition of TBPB. Then, the addition of radical 29d to the C–C triple bond of 29a resulted in the alkenyl radical 29e, which then underwent intramolecular cyclization with the cyano group to produce the radical intermediate 29g. The radical intermediate 29g was further oxidized to radical cation 29h, which subsequently regenerated the aromatic ring by deprotonation (path a). On the other hand, radical intermediate 29g could also convert into the desired product 29gvia hydrogen atom transfer (path b).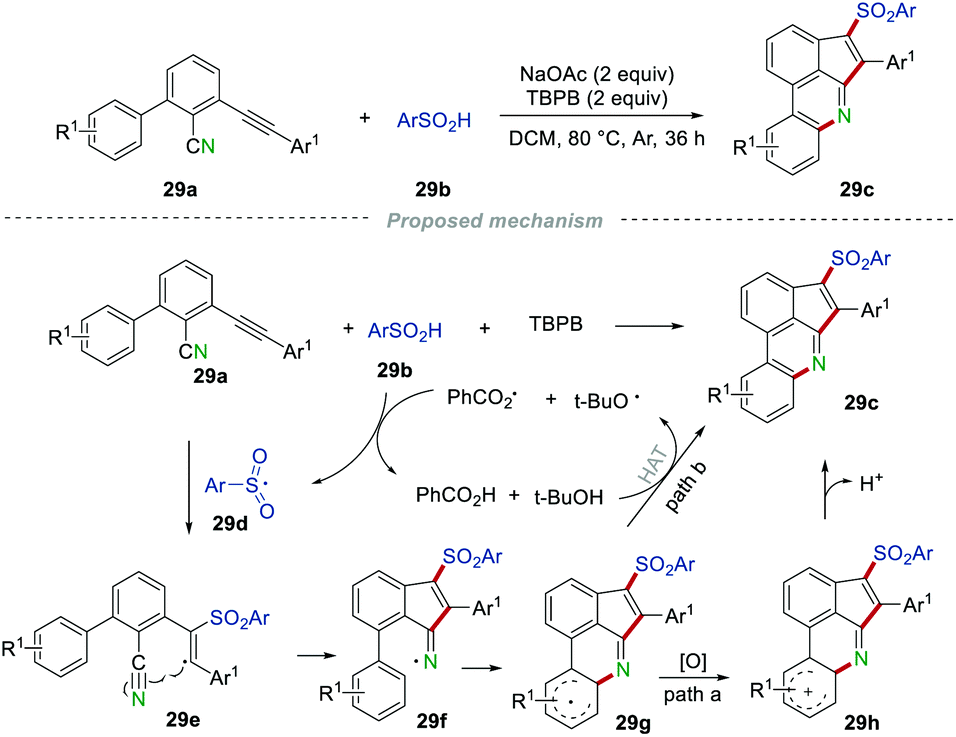
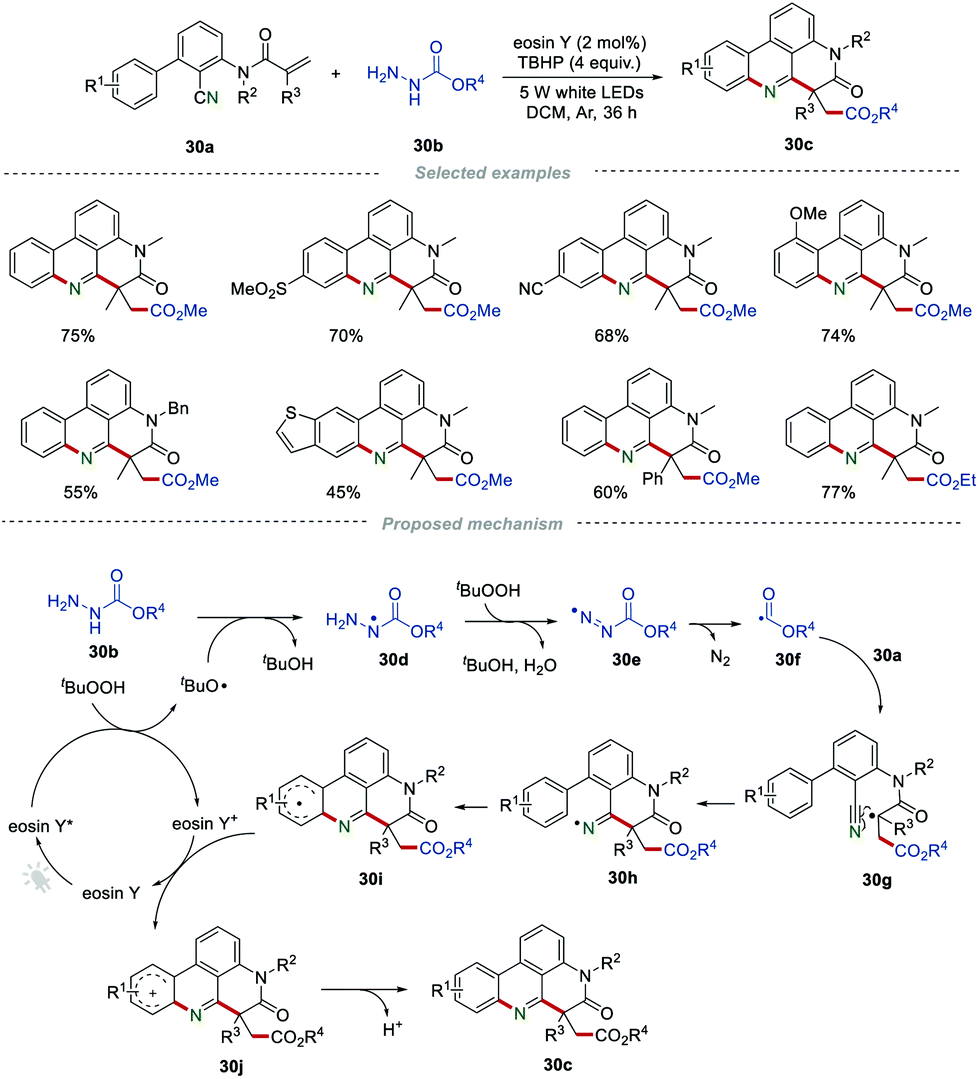
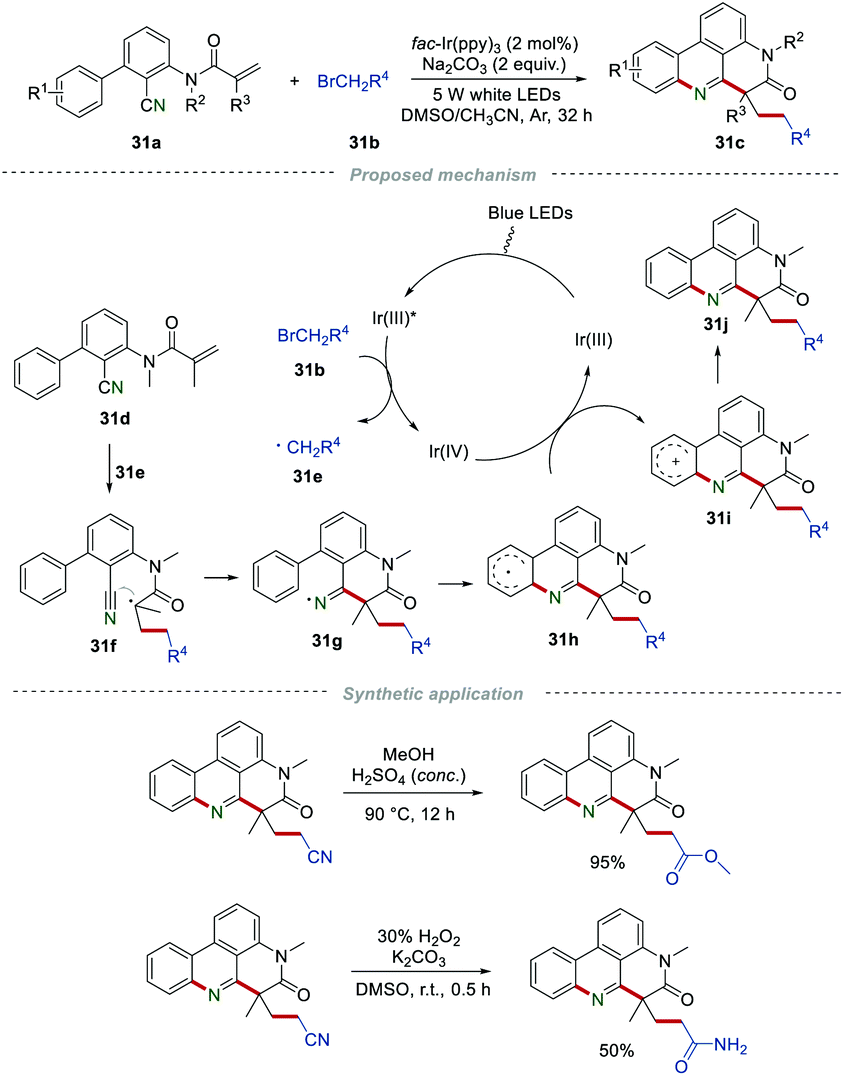
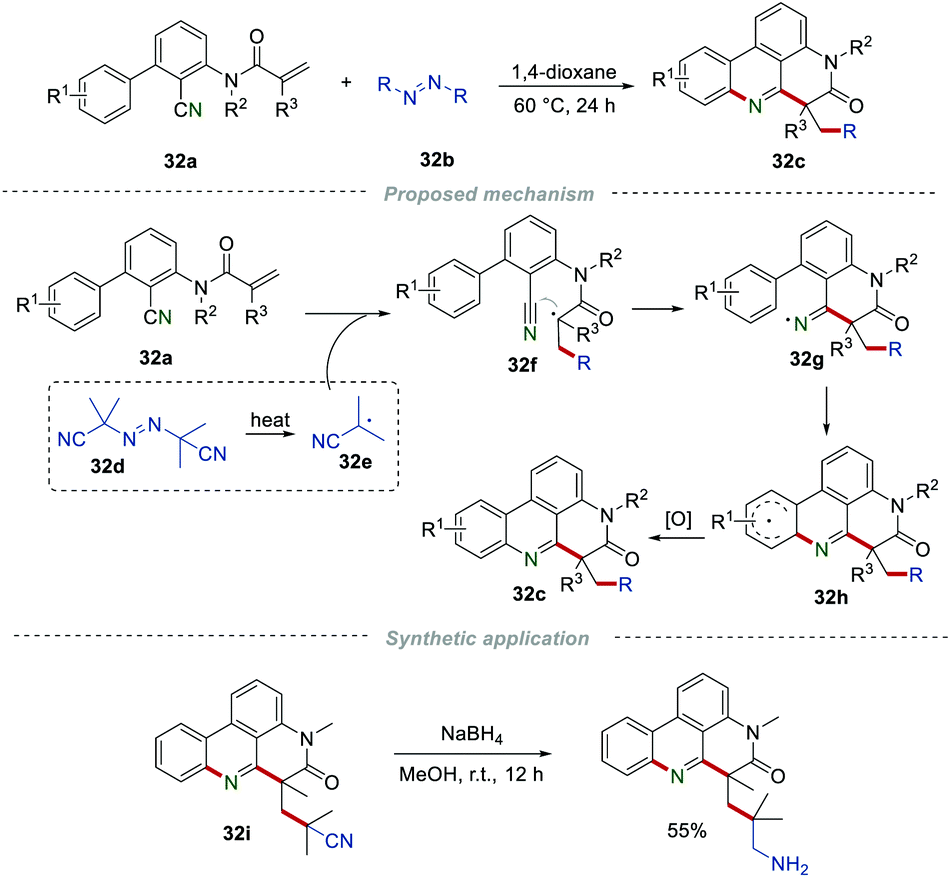
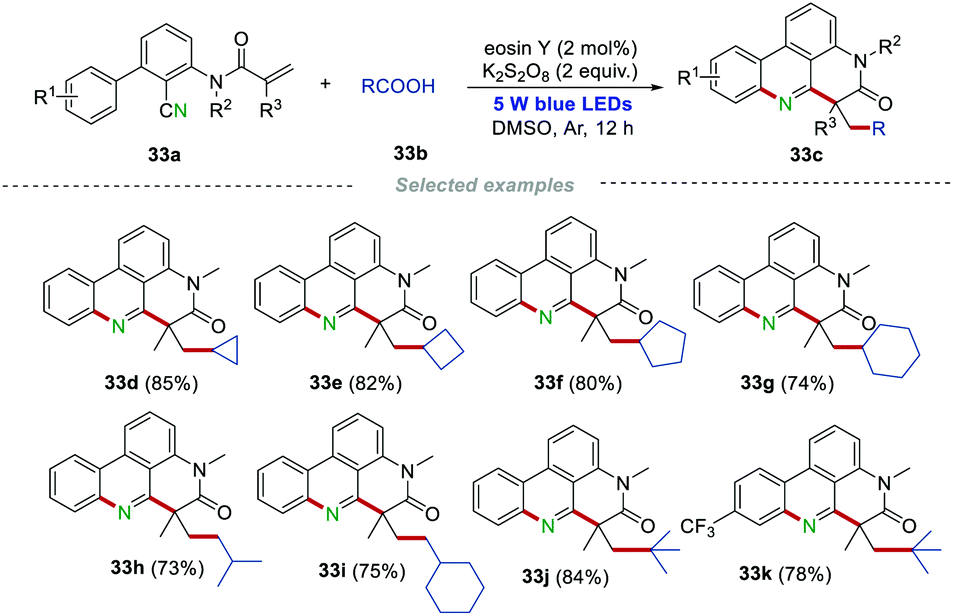
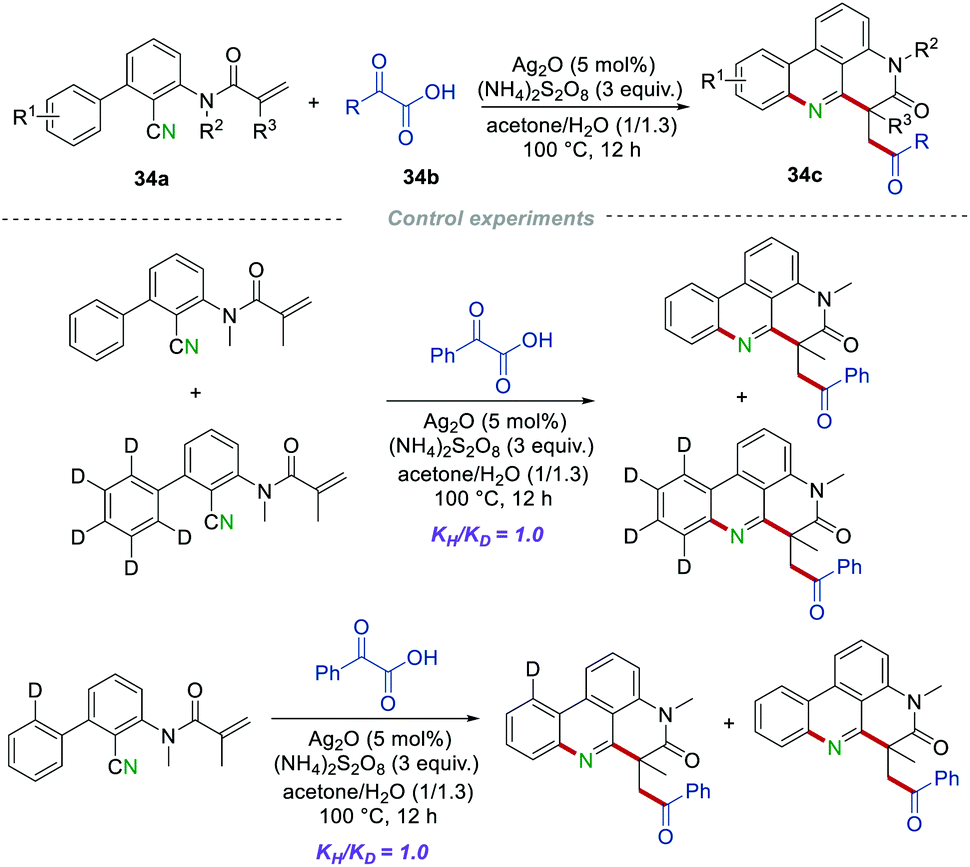
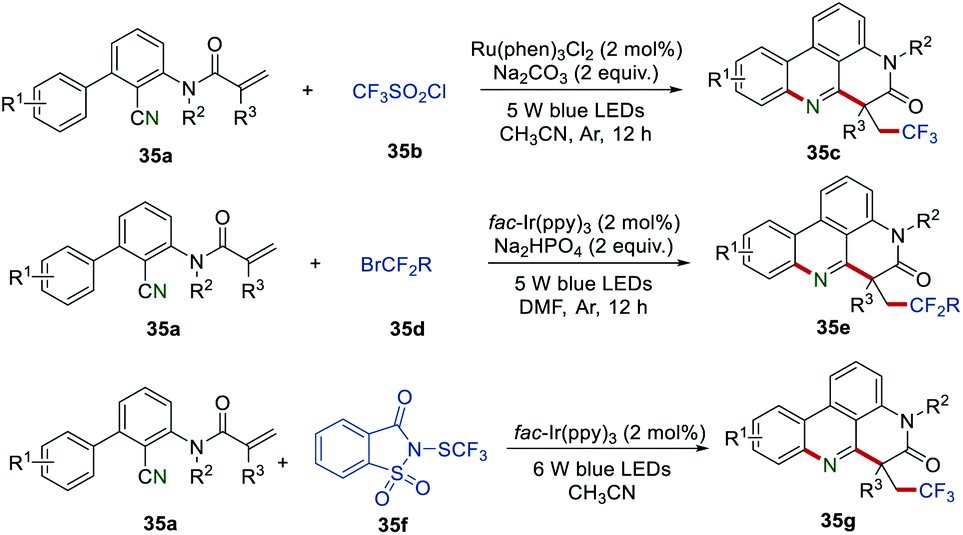
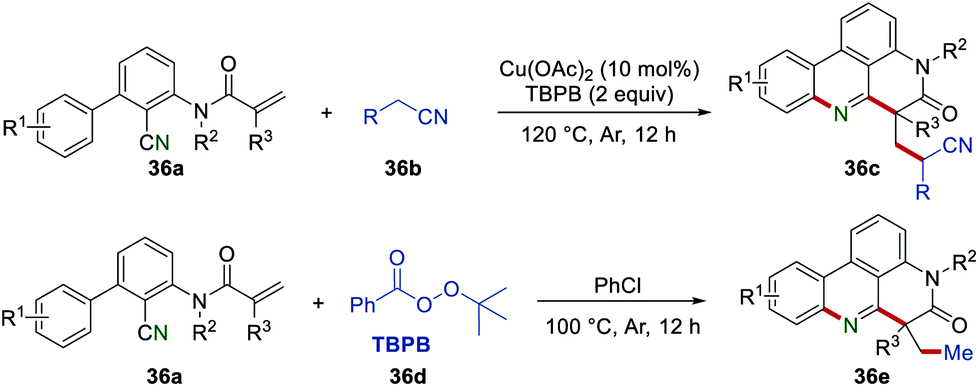
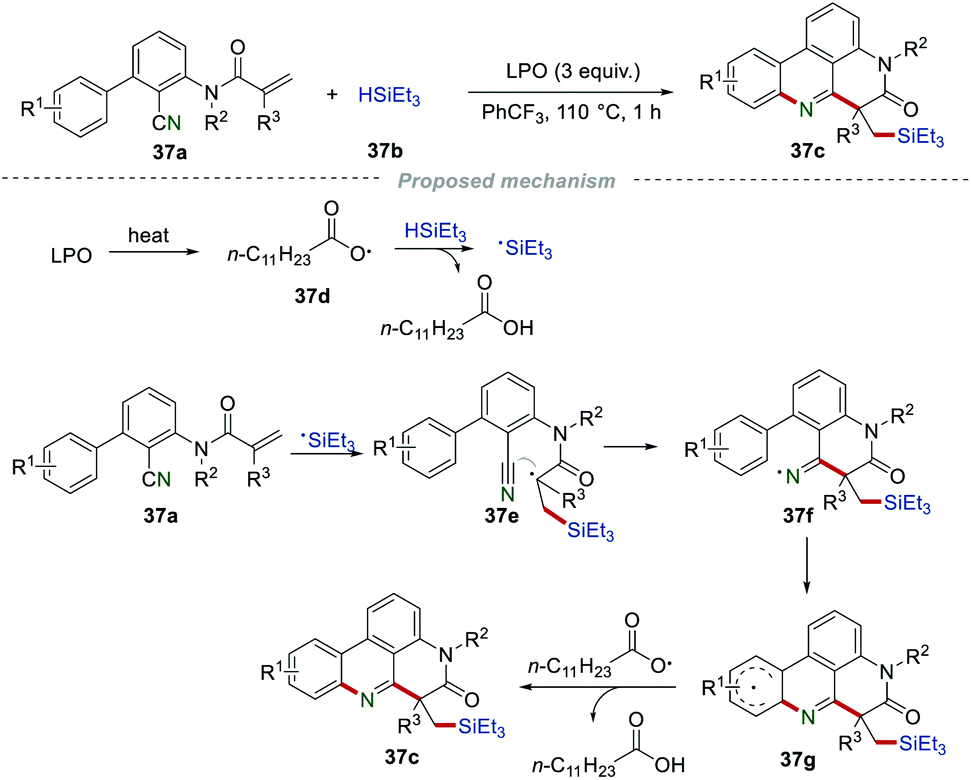
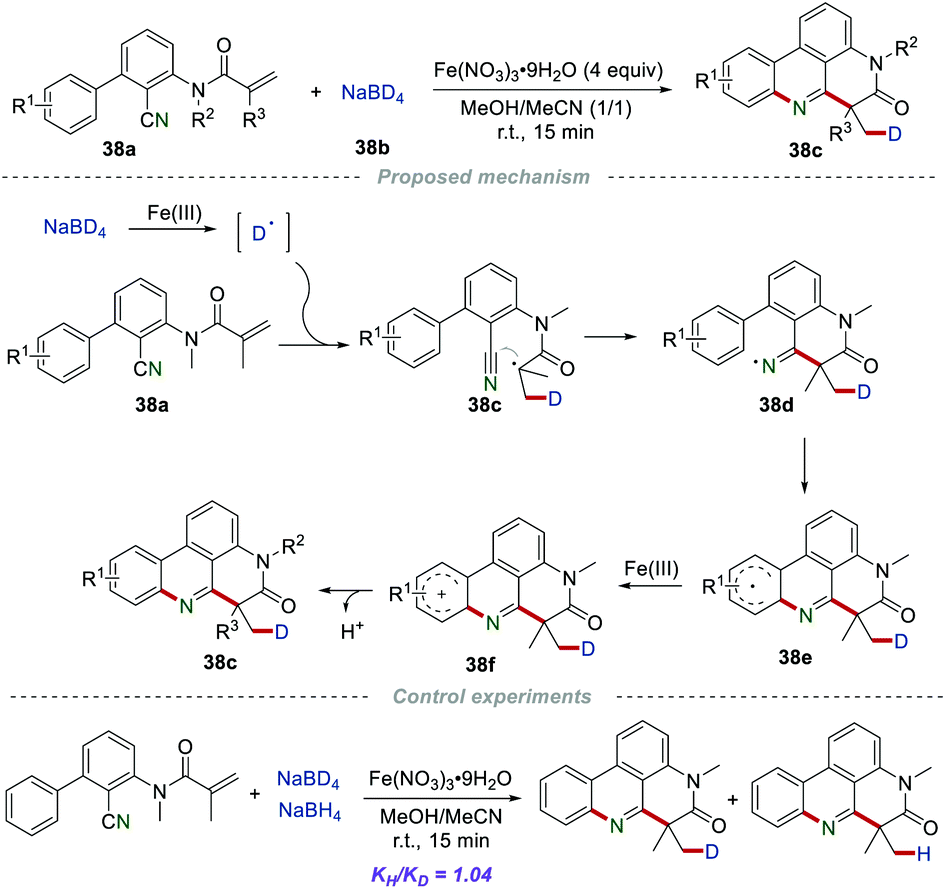
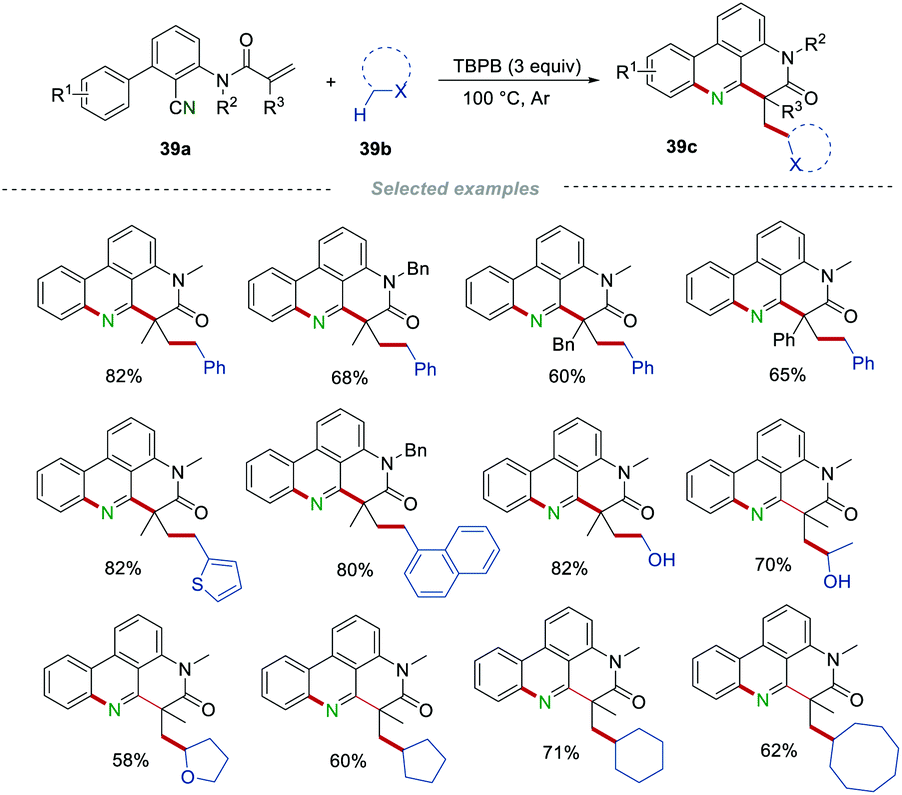
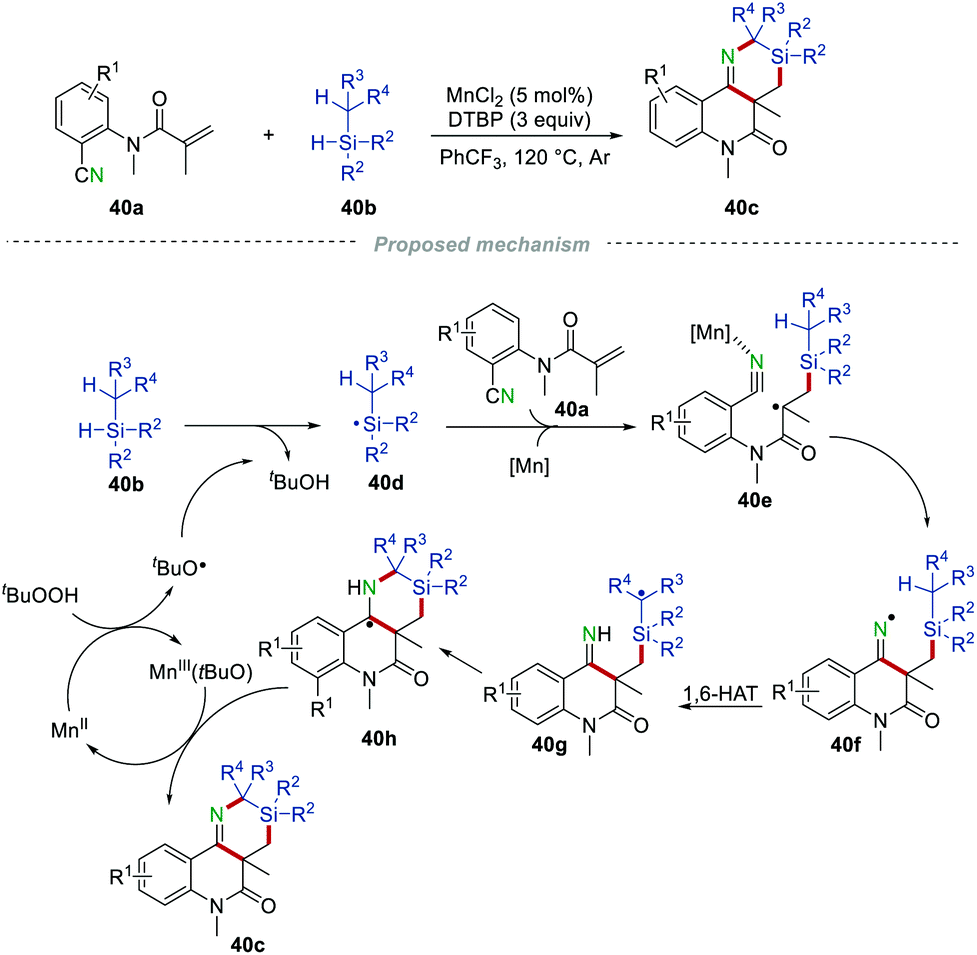
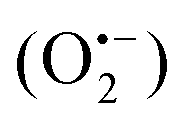 . Then, a deprotonation reaction between the radical cation 41d and
. Then, a deprotonation reaction between the radical cation 41d and 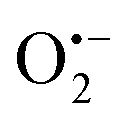 generated the thiyl radical 41e, which then attacked the cyano group in 41a to afford the iminyl radical 41f. Finally, the intramolecular cyclization of the iminyl radical 41f afforded radical 41g, which further lost a hydrogen atom to produce the desired products 41c.
generated the thiyl radical 41e, which then attacked the cyano group in 41a to afford the iminyl radical 41f. Finally, the intramolecular cyclization of the iminyl radical 41f afforded radical 41g, which further lost a hydrogen atom to produce the desired products 41c.