
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of (2R,3R)-2-(3,4-dihydroxyphenyl)chroman-3-yl-3,4,5-trihydroxy benzoate as multiple inhibitors of SARS-CoV-2 targets; a systematic molecular modelling approach†
Jubie Selvaraja,
Shyam Sundar Pa,
Logesh Rajanb,
Divakar Selvarajc,
Dhanabal Palanisamyb,
Krishnan Namboori PKd and
Suresh Kumar Mohankumar *be
*be
aDepartment of Pharmaceutical Chemistry, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Rockland's, Ooty, Nilgiris, 643001, Tamil Nadu, India
bTIFAC CORE in Herbal Drugs, Department of Pharmacognosy, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Rockland's, Ooty, Nilgiris, 643001, Tamil Nadu, India
cDepartment of Pharmacology, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Rockland's, Ooty, Nilgiris, 643001, Tamil Nadu, India
dAmrita Molecular Modelling and Synthesis (AMMAS) Research Lab, Amrita Vishwavidyapeetham, Amrita Nagar, Ettimadai, Coimbatore, Tamil Nadu, India
eSwansea University Medical School, Swansea University, Singleton Park, Swansea, Wales SA2 8PP, UK. E-mail: s.k.mohankumar@swansea.ac.uk
First published on 7th April 2021
Abstract
Coronavirus disease of 2019 (COVID-19) is a zoonotic disease caused by a new severe acute respiratory syndrome (SARS-CoV-2) which has quickly resulted in a pandemic. Recent anti-COVID-19 drug discoveries are leaning towards repurposing phytochemicals which have been previously reported for SARS and MERS-CoV outbreaks. However, they have been either virtually screened or tested so far against mono targets and the potent derivatives of virtually sorted lead molecules remain elusive. We aimed to identify the phytochemicals having potentials to inhibit SARS CoV-2 infection via multiple targets. The selected 132 phytochemicals were virtually screened using a structure based in silico technique against main protease (Mpro) which is a potential target of SARS CoV-2. Six compounds were selected based on the LibDock scores and further subjected to induced fit docking using the CDOCKER module of DS. Two compounds namely cinnamtannin-B and gallocatechin gallate were identified as top HITS against main protease (Mpro). Based on the Lipinski rule of five (L-ROF) and synthetic feasibility, gallocatechin gallate was taken for our further studies. Six analogues of gallocatechin gallate were screened against the next important targets such as RNA-dependent RNA polymerase (RdRp), angiotensin converting enzyme-2 (ACE2), transmembrane protease serine -2 (TMPRSS2) and interleukin-6 (IL-6) along with main protease (Mpro). Our molecular docking results reveal that a gallocatechin analogue (GC-2) namely (2R,3R)-2-(3,4-dihydroxyphenyl)chroman-3-yl-3,4,5-trihydroxy benzoate has shown potential to inhibit multiple targets of SARS CoV-2. Further, the molecular dynamics study was carried out to ascertain the stability of the GC-2 and RdRp complex.
The novel coronavirus disease of 2019 (COVID-19) is an infectious disease and responsible for the 2019–2020 viral pneumonia outbreaks. The World Health Organization (WHO) declared that the outbreak of COVID-19 is pandemic on the 22nd of April 2020. Sadly, this pandemic is still enduring and hence there is an urge to find novel prophylactic or therapeutic measures to combat the COVID-19 pandemic.1–4 However, to date there have been no specific therapeutics proven to eradicate COVID-19, which warrants multi-focused and multi-targeted strategies like, drug candidates having potentials to act on multiple antiviral targets and concurrently boost the host immune system.5,6 The US FDA has launched the Coronavirus Treatment Acceleration Program (CTAP) and specific guidance on COVID-19 clinical trials. Additionally, the FDA has issued emergency use authorizations for some drugs, for example, chloroquine, hydroxychloroquine, and Remdesivir. However, recent clinical trials investigating this drug concluded that the treatment was not significantly correlated to intubation risk or mortality by multiple analyses. Remdesivir, a nucleotide prodrug, inhibits viral RNA-dependent RNA polymerase and has been reported to have in vitro activity against SARS-CoV-2.7 There are now seven known coronaviruses that cause disease in humans to include, HCoV-229E, HCoV-OC43, HCoV-NL63, HCoV-HKU1, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory virus coronavirus (MERS-CoV), and now SARS-CoV-2.8 The first four CoVs cause mild and self-limiting disease. However, the last three coronaviruses are highly pathogenic, leading to communicable outbreaks causing fatal respiratory diseases. Coronaviruses have the largest genomes among all known RNA viruses, ranging from 26 to 32 kb in length, which encode structural and nonstructural proteins. Several structural components of the coronavirus family viruses have been described as bio targets for drug discovery. These targets are related to viral nucleic acids, enzymes, spike glycoprotein, and envelope (membrane, nucleocapsid, and accessory proteins).7 A massive effort has been undertaken by the international scientific community to obtain structural information about complexes between SARS-CoV-2 targets and different types of inhibitors, providing information to use ligand-based (LB) and structure-based (SB) approaches to drug repurposing. The drugs such as amodiaquine and darunavir have also been repurposed for Covid therapy through in silico tools.9 There are also reports for the organic compounds such as alpha ketoamides and 3-alkynyl substituted 2-chloroquinoxaline frame work as lead compounds for Covid therapy.10,11 The present work evaluated the druggability of the SARS-CoV-2 main protease (SARS-CoV-2 Mpro) as a potential target for the approved drug for the treatment of COVID-19 as it plays a crucial role in the cleavage of viral polyproteins involved in transcription and replication. Many research teams have exploited the major protease (Mpro) of SARS-CoV-2, also named chymotrypsin-like protease (3CLpr),12 as a potential drug target to fight COVID-19. Sequence alignment revealed that the SARS-CoV-2Mpro shares a 96% similarity to that of SARS-CoV-1. This enzyme is reported to be inhibited by several classes of compounds, some of which exhibit anti-CoV activities in vitro, in vivo, and even in nonrandomized trials (such as lopinavir).7 RNA-dependent RNA polymerase (RdRp) is a key enzyme essential for the viral replication of SARS-CoV-2.13 Due to their crucial roles, these viral proteins are considered imperative targets for developing antiviral compounds against COVID-19. The SARS-CoV-2 genome encodes more than 20 proteins, which include the main protease (Mpro), a 3C-like protease (3CLP) that shares 96.1% similarity with 3CLP of SARS-CoV.7 Mpro, a homodimeric cysteine protease, plays an important role in SARS virus replication and transcription. When the mRNA of the virus is translated polyproteins, Mpro is first auto-cleaved to become a mature enzyme, which in turn cleaves all of the 11 remaining downstream nonstructural proteins of the polyproteins to polypeptides, which are required for the replication process of the virus. The prime anti-COVID-19 target proposed is the main protease (Mpro; also known as 3 chymotrypsin like protease or 3CLpro), which is regarded as responsible for the cleavage of viral peptides into functional units for virus replication and packaging within the host cells. Likewise, the enzyme RNA dependent RNA polymerase (RdRp) responsible for viral RNA synthesis is also considered a druggable target to inhibit SARS-CoV-2. Conceivably, the interaction of viral spike proteins with its receptor, angiotensin-converting enzyme-2 (ACE2) on host cells, and subsequent viral endocytosis into the cells, may also are regarded as a viable drug target. Similarly, the enzyme transmembrane protease serine-2 (TMPRSS2) produced by the host cells plays an important role in proteolytic processing of S protein priming to the receptor ACE2 binding in humans and has gained attention.14–18 Whilst, these targets have been currently investigated extensively with numerous drugs and biologicals, the poly targeting approaches such as the leads having the potential to act on synergistic pathways concurrently by which SARS-CoV-2 infection is inhibited in the host cells. The cytokines, including interleukins, play a vital role in regulating host cell immune responses. Upon the SARS-CoV infection, a rapid and severe immune reaction occurs, and this triggers the synthesis and release of relatively higher levels of interleukins into the bloodstream. Consequently, the raise of pro-inflammatory cytokines leads to severe respiratory organ failure, high fever, and other associated diseases.19 Owing to these reasons, we hypothesize that the drug candidates having the potential to elicit anti-viral effects on multiple targets along with beneficial immunomodulatory effects in host cells, organs or systems would enable novel therapeutic leads to combat the COVID-19 pandemic. Thus, our study intended to search for the leads having the potential to act on multi druggable targets of COVID-19 along with beneficial immunomodulatory effects.
Whilst, the drug discovery and developmental research has undergone significant modernization in recent decades, the three fourth of humankind still depend largely on the bioactive from natural sources.20 The plants and their parts have been serving human mankind as sources of herbal medicine since ancient times. They possess a variety of constituents, such as flavonoids, alkaloids, glucosides, and polyphenolic compounds. These phytochemicals offer a wide range of therapeutic properties and novel scaffolds to design new drugs.21,22 Of note, several phytochemicals have been reported beneficial in preventing or treating zoonotic viral diseases including SARS and MERS-CoV. The plant constituents having bi, mono &heterocyclic rings specially bioflavonoids adapt to the geometry of the 3CLpro substrate site and have prominent interactions with the surrounding amino acid residues.23 This is further substantiated by the findings of cannabisin A and isoacetoside as potential 3CLpro inhibitors by Free Energy Perturbation studies.24 In regards, we have collated the potent phytochemicals which have been previously reported to inhibit SARS and MERS-CoV. This study aims to narrow and sort the selective 132 phytomolecules (ESI Table 1†) based on its binding affinities and interactions on major druggable target of COVID-19 using in silico computational tools. Besides, the analogues of the sorted lead molecule were prepared and examined for its molecular docking, ADMET, and molecular dynamics.
Materials and methods
Docking software and ligand library
LIBDOCK and CDOCKER modules of Discovery Studio 3.5 software (DS3.5, Accelrys, Inc., San Diego, CA, USA) were utilized for virtual screening and ligand binding analysis of inhibitors. A library consisting of 132 plants secondary metabolites already reported for SARS-CoV (2003) and MERS-CoV (2012), and other viruses after sessional outbreaks caused by dengue and influenza virus was generated (ESI Table 1†). All phytochemicals were screened for Mpro and the top hit and its analogs were further docked with the proteins Mpros, RdRp, ACE-2, TMPRSS2, and IL-6. The binding energies and the interactions were compared with the known inhibitors of the selected proteins Lopinavir, Favipiravir, Ramipril, camostat, and tartaric acid respectively. A detailed workflow is depicted in Fig. 1a.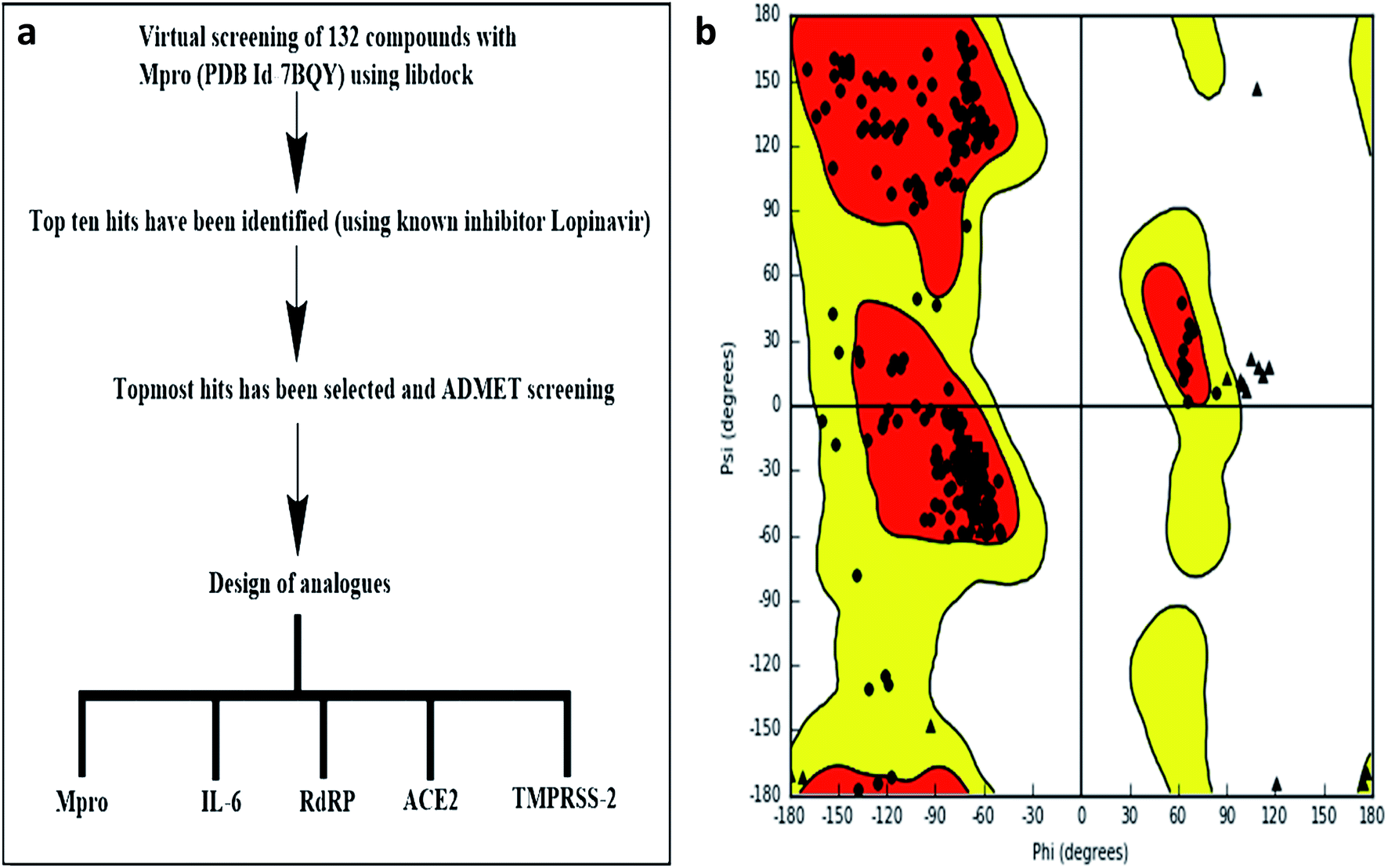 | ||
| Fig. 1 (a) Experimental workflow (b) Ramachandran plot of Mpro. | ||
Structure-based virtual screening using LIBDOCK
LIBDOCK is a rigid-based docking program. It calculates hotspots for the protein using a grid placed into the binding site and using polar and polar probes (San Diego, CA, USA). Then the hotspots are further used to align the ligands to form favorable interaction. After minimized, all the ligand poses are ranked based on the ligands score. The 1.7 Å crystal structure of CoV main proteases (Mpros) in complex with N3 a mechanism-based inhibitor (PDB ID: 7BQY) were downloaded from the protein data bank (PDB) and imported to the working environment of LIBDOCK. The protein was prepared by removing crystal water and other heteroatoms (except N3), followed by the addition of hydrogen, protonation, ionization, and energy minimization. The CHARMm (Cambridge, MA, USA)25 force field and the Smart minimizer algorithm were applied for energy minimization. The minimization performed 2000 steps with an RMS gradient tolerance of 0.1, and the final RMS gradient was 0.09463. The prepared protein was used to define the binding site from the “Edit binding site” option on the receptor–ligand interaction toolbar. Using the bound ligands (N3) binding positions, the active sites for docking were generated. Virtual screening was carried out by docking all the prepared ligands at the defined active site using LIBDOCK. Based on the LIBDOCK score, all the docked poses were ranked and grouped by name. All compounds were ranked according to their LIBDOCK score.The molecular docking (CDOCKER) module of Discovery Studio was used for the molecular docking study. CDOCKER is an implementation of a CHARMm based docking tool. The receptor is held rigid while the ligands were allowed to flex during the docking process. For each complex pose, the CHARMm energy (interaction energy plus ligand strain) and the interaction energy, which indicate ligand binding affinity was calculated. The crystal structures of Mpros (PDB ID: 7BQY, 1.7 Å), RdRp (PDB ID: 6WTC, 1.85 Å), ACE-2 (PDB ID: 6ACK, 4.5 Å), TMPRSS-2 (PDB ID: 2E7V, 1.92 Å) and IL-6 (PDB ID: 1ALU, 1.90 Å)26 were obtained from the protein data bank. The crystal water molecules were generally removed in rigid and semi-flexible docking process27,28 since the fixed water molecules might affect the formation of the receptor–ligand complex. The water molecules were removed, and hydrogen atoms were added to the protein. The structure was completed by modelling the missing loops. The missing atoms were inserted by standardizing the names of the atom. The titratable residues were protonated by pKa prediction. The potential energy van der Waals energy, Electrostatic energy, and RMS gradient of the complexes were checked before and after protein minimization. Finally, the hydrogen receptors were merged to the target receptor molecule using discovery studio 4.1 clients.29 The prepared proteins were validated through a Ramachandran plot (Fig. 1b).25 The selected plant constituents were generated and optimized using ChemSketch software. All the tautomer's and isomers of the ligand were regarded and of the ligands possessing the lowest energy had been approved for docking.
The CHARMm force field was used for energy minimisation. The binding site spheres of all the proteins were defined as the regions that come within a radius of 15 Å from the geometric centroid of the native ligands. The binding site spheres of all the proteins were defined by structural alignment. During the docking process, the ligands were allowed to bind to the residues within the binding site spheres. After being extracted from the binding site, the native ligands were re-docked into the crystal structure of all proteins. The RMSD between the docked pose and the crystal structure of the complex was less than 1 Å, indicating the CDOCKER module was highly reliable for reproducing the experimentally observed binding mode of all proteins. The structures of identified hits were prepared and docked into respective proteins. Different poses for each test molecule were generated and analyzed based on CDOCKER interaction energy. The designed compounds were minimized and used as input ligands in the protocol explorer of CDOCKER. The molecular dynamic protocol was used to generate various conformations for ligand and the initially generated structures were refined using a simulated annealing protocol. CDOCKER energies were calculated for each compound. The various binding interactions between the ligand and protein were predicted. The protocol uses a CHARMm-based molecular dynamics (MD) scheme to dock ligands into a receptor binding site. Random ligand conformations are generated using high-temperature MD. The conformations are then translated into the binding site. Candidate poses are then created using random rigid-body rotations followed by simulated annealing. A final minimization is then used to refine the ligand poses. The CDOCKER energy as an estimation of molecular complex binding affinity was used in this study.
Molecular dynamics
The molecular dynamic (MD) simulation was conducted to study the stability of the ligand–target complex.30 The simulation is set to constant number (N), volume (V), and temperature (T) (NVT) ensemble with temperature varying from 300 K, run: 80 ns, time step: 0.002, and quality threshold (QT) (the temperature response set enforce constant temperature in a molecular dynamics simulation) is set to 0.2 (Fig. 1c). It took 12.55 h to complete the simulation. The ligand–target complexes were then subjected to molecular dynamics simulation using NAMD with the following parameters; minimization-2000 steps; annealing-144![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps; equilibration & MD-500000 steps; NVT temperature 27 °C/300 K; NPT pressure-1 atm and simulation time-80 ns.
000 steps; equilibration & MD-500000 steps; NVT temperature 27 °C/300 K; NPT pressure-1 atm and simulation time-80 ns.
The highly constructive conformation, pose and favorable bonded complexes form receptor ligand interaction was used an input to study molecular dynamics simulation (MD) for the compound GC-6. The CHARMm (Chemistry at Harvard Macromolecular Mechanics) force field were applied to each complex system followed by two step minimization of 500 cycles with steep descent (SD) and conjugate gradient (CG) to eliminate possible steric strain. The relaxed and energy minimized complex were then subjected to three steps of dynamics and simulation. Initially, receptor–ligand complex was gradually simulated and heated from a temperature of 50 K followed by its ramping to 300 K and then equilibrated by 100 steps at 310 K for even distribution of atoms in a system (protein–ligand) to be constrained. Finally, production of CHARMm simulation was performed for 80 ns and conformations were allowed to save for every 2 ps. Furthermore, the Spherical Cutoff method of electrostatics was implemented to study the all-non-bonded energy. In order to fix hydrogen bonding during simulation leap frog verlet a dynamics integrator was introduced with SHAKE constraint. Equilibration of the complex system was monitored by convergence in terms of the H-bond, root-mean-square fluctuation (RMSF) and the RMSD (root-mean-squared deviations).
Results
Virtual screening of phytochemicals database against CoV main proteases (Mpros)
The prime target, coronavirus main protease (Mpros), which is a chief culprit enzyme, guides in mediating the transcription and replication process of the virus31 has been assigned for our study. There are ∼30000 nucleotides have compressed together in the virus genome of novel COVID-19, its gene may perhaps encode with two polyproteins namely, pp1a and pp1ab, which are desirable for viral replication and transcription.32,33 The main Proteolytic enzyme, Mpros, through the Proteolytic process could release the polypeptides from these polyproteins. Besides, this Mpro enzyme is the main protein in the viral cell life cycle together with the absence of closely related homolog's of human protein, recognizes the Mpros as a promising target for the anti-viral drug discovery. Therefore, the inhibition of Mpro may perhaps block the biosynthesis of the viral polyprotein, resulting in viral death. To identify new compounds that could potentially inhibit Mpros through binding to the ligand binding pocket, virtual screening was carried out using the LibDock module of DS.
The top 10 phytochemicals which have higher binding affinities with Mpro relative to all docked molecules are listed in Table 1. The binding affinities obtained for phytochemicals were compared with a conventional Mpro inhibitor, Remdesivir which is also extensively used for the treatment of COVID-19. The results reveal that the phytochemicals namely isotheaflavin 3′-gallate, dieckol, gallocatechin gallate, broussoflavan A, and hygromycin B have shown LibDock scores similar to Remdesivir (LibDock score: 177.63). Of note, the LibDock score of cinnamtannin B1 was higher than the score obtained for Remdesivir.
| S. no. | Molecule | LIBDOCK Score |
|---|---|---|
| 1 | Isotheaflavin 3′-gallate | 179.72 |
| 2 | Cinnamtannin B1 | 184.88 |
| 3 | Dieckol | 175.51 |
| 4 | Gallocatechin gallate | 173.33 |
| 5 | Broussoflavan A | 172.76 |
| 6 | Hygromycin B | 172.23 |
| 7 | Epi-gallocatechin gallate | 167.03 |
| 8 | Ginkgetin | 165.38 |
| 9 | Amentoflavone | 165.32 |
| 10 | Epi-catechin gallate | 164.68 |
| 11 | Remdesivir | 177.63 |
Ligand binding analysis
To study ligand binding mechanisms, the top six ranked compounds isotheaflavin 3′-gallate, dieckol, gallocatechin gallate, broussoflavan A, hygromycin B, and cinnamtannin B1 were docked into the 3D structure of (Mpros) by using the CDOCKER module of DS. The CDOCKER energy as an estimation of molecular complex binding affinity was used in this study. As shown in Table 2, the CDOCKER energy of gallocatechin gallate and cinnamtannin B1 were lower than the reference Remdesivir, indicating that these two compounds elicit a higher binding affinity with Mpros. The energy of the other compound dieckol is close to Remdesivir. However, the interaction energy of compounds broussoflavan, isotheaflavin 3′-gallate, and hygromycin were not significant when compared to the reference Remdesivir. The increased binding energies of gallocatechin gallate and cinnamtannin B1 were further supported by the comparative analysis of receptor–ligand interactions with the active amino acid residues of Mpros. The major non-bonded interactions such as hydrophilic, hydrophobic, and pi–pi interactions of these two compounds were investigated and compared with the standard Remdesivir (Fig. 2a–c ESI Tables 2 and 3†).| S. no. | Molecule | -CDOCKER interaction energy (kcal mol−1) | Pose |
|---|---|---|---|
| 1 | Gallocatechin gallate | 43.69 | 1 |
| 2 | Cinnamtannin B1 | 34.54 | 1 |
| 3 | Dieckol | 29.90 | 1 |
| 4 | Broussoflavan A | 4.85 | 1 |
| 5 | Isotheaflavin 3′-gallate | 22.85 | 1 |
| 6 | Hygromycin B | −9.57 | 1 |
| 7 | Remdesivir | 33.84 | 1 |
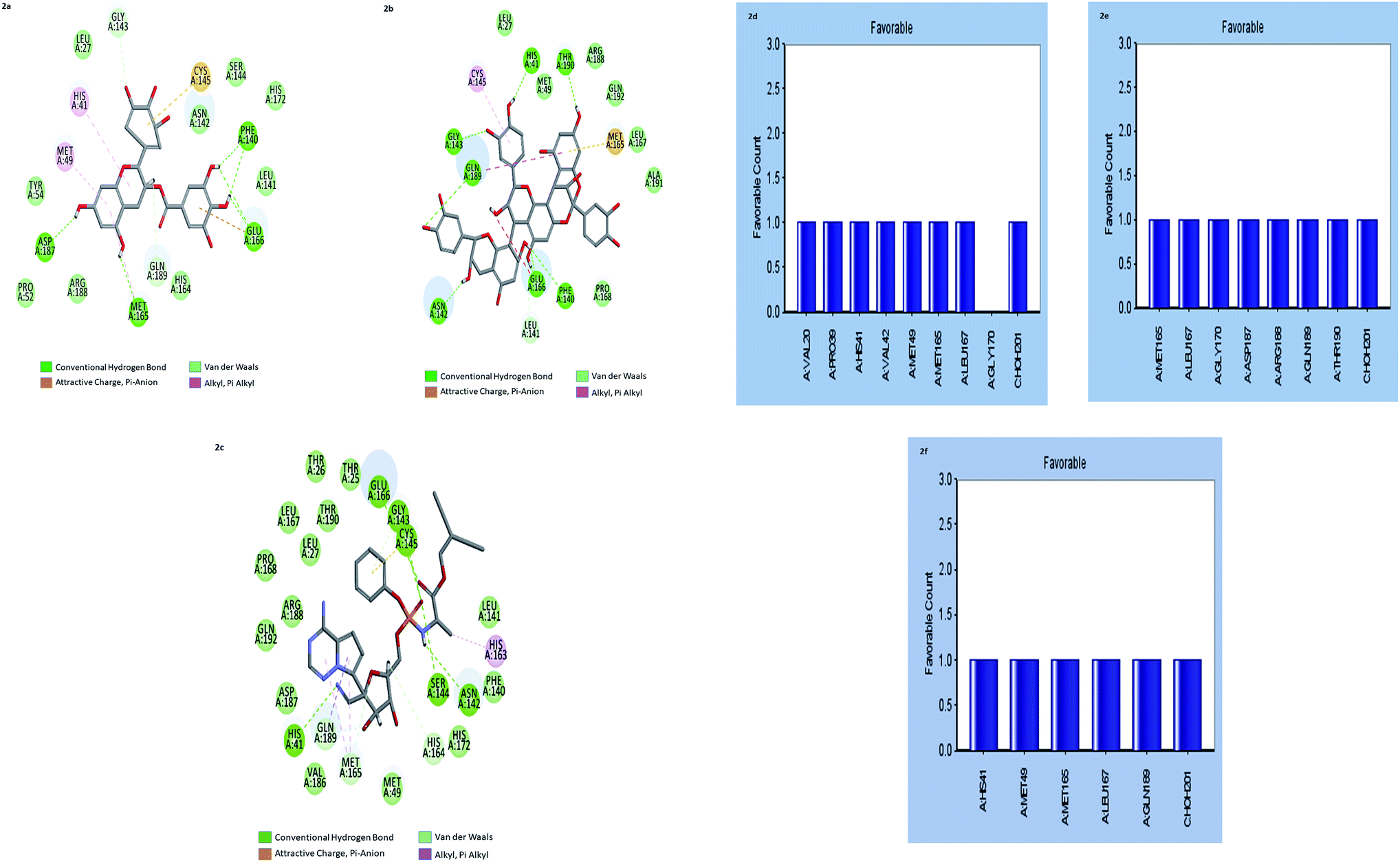 | ||
| Fig. 2 (a–c) the inter-molecular interaction of the predicted binding modes of (a) gallocatechin gallate; (b) cinnamtannin B1 and (c) Remdesivir to Mpros. (d–f) Bar diagrams depicting the favourable interactions of the residues of Mpros towards (d) gallocatechin gallate (e) cinnamtannin B1 and (f) Remdesivir. | ||
The results showed that both gallocatechin gallate and cinnamtannin B1 formed eight and seven pairs of hydrogen bonds with Mpros, respectively whereas the drug Remdesivir has shown five pairs of hydrogen bonds only. The additional hydrogen bonds formed may be the reason for the increased binding affinity of these two compounds than Remdesivir. Both of the compounds formed three pair Pi–Pi interactions with Mpros, by the centroid of the benzene ring of methionine and the centroid of the benzene ring of gallocatechin gallate and cinnamtannin respectively. The interactions of the amino acid residues are depicted in (Fig. 2d–f). These results indicated that gallocatechin gallate and cinnamtannin B1 probably bind to Mpros with similar or even better affinity than Remdesivir.
Design of gallocatechin analogs Lipinski rule of five (L-ROF) is an important parameter for oral drug candidates. When we analyzed the L-ROF of two compounds gallocatechin gallate and cinnamtannin, violations of the gallocatechin were 3 and 2 respectively (Table 3).
| Molecule | logP |
Mol. wt. (g) | H bond donor | H bond acceptor | No. of violations |
|---|---|---|---|---|---|
| GA | 0.25 | 460.4 | 8 | 11 | 2 |
| CT | 3.3 | 864.8 | 14 | 18 | 3 |
It is a well-known fact that beyond the violation of 2 is questionable. Based on these results, we decided to proceed with gallocatechin further. Chemical modification of naturally derived compounds may be required to increase the potency.
For the drug discovery process, beginning with the understanding of the structural conformity (i.e. structure including potential isomers) of the naturally derived compound(s) can timelines and greatly increase the chance. Working on this principle we designed six analogs of gallocatechin (Fig. 3a and b). To alter the hydrogen bond donor/acceptor and to increase the hydrophobicity, the OH groups of ring A and C of gallocatechin are removed in a sequential manner to derive the analogs (Table 4).
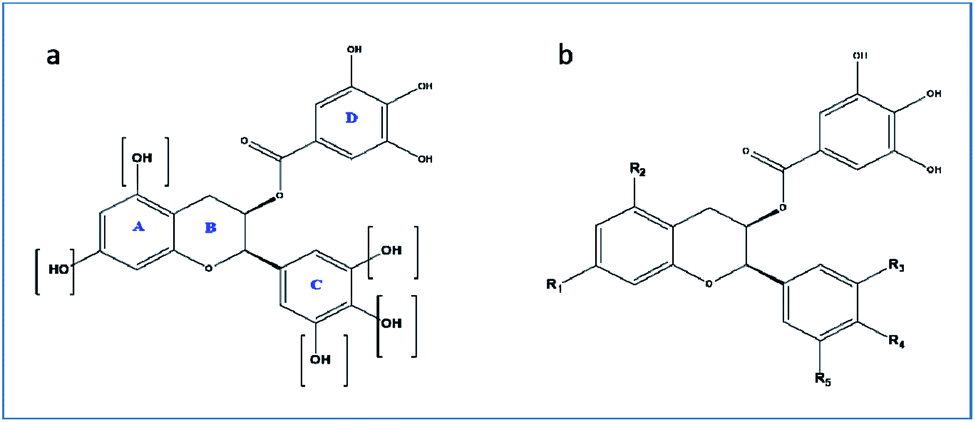 | ||
| Fig. 3 (a and b) Gallocatechin gallate scaffold. | ||
| Compound | R1 | R2 | R3 | R4 | R5 |
|---|---|---|---|---|---|
| GC-1 | OH | OH | OH | OH | OH |
| GC-2 | OH | OH | H | H | H |
| GC-3 | OH | H | OH | OH | OH |
| GC-4 | OH | H | H | H | H |
| GC-5 | OH | H | OH | H | H |
| GC-6 | H | H | OH | OH | OH |
| GC-7 | H | H | H | H | H |
The designed analogs were further docked with the proteins Mpros, RdRp, ACE-2, TMPRSS2, and IL-6. The binding energies and the interactions were compared with the known inhibitors of the selected proteins lopinavir, favipiravir, Ramipril, camostat, and tartaric acid respectively. As shown in Table 5 the CDOCKER energies of designed analogs were compared with known inhibitors. The selected compounds have shown the least CDOCKER energy towards ACE-2, IL-6, and RdRp among the five targets compared to the known standards (>2.11781, 19.1244, and 3263 kcal mol−1) respectively. Against COVID main protease, and transmembrane protease serine-2 proteins, the CDOCKER energy of tested compounds were not significant. The energies were bigger than the standard lopinavir (<51.2735 kcal mol−1), (<48.1854). However, the compounds GC-3 and GC-6 have shown the CDOCKER energies which are nearing the standard. Interestingly, the CDOCKER energies of tested compounds were lower towards the proteins ACE-2, IL-6, and RdRp than the reference compounds Ramipril, tartaric acid, and favipiravir, respectively. These results indicating the compounds may possess COVID-19 combating activity through the inhibition of these proteins. Among all, compound GC-3 and compound GC-6 shows significant activity towards all the enzymes. However, a further detailed in vitro study is needed to prove this concept.
| Compound | -CDOCKER interaction energy (kcal mol−1) | ||||
|---|---|---|---|---|---|
| MPRO | TMPRSS2 | ACE-2 | IL-6 | RdRP | |
| GC-1 | 41.61 | 36.07 | 30.40 | 31.69 | 42.11 |
| GC-2 | 34.12 | 31.73 | 22.23 | 24.78 | 30.58 |
| C-3 | 44.07 | 37.68 | 28.49 | 31.64 | 37.37 |
| GC-4 | 35.24 | 32.41 | 22.25 | 28.53 | 33.80 |
| GC-5 | 37.82 | 33.60 | 21.98 | 26.43 | 33.28 |
| GC-6 | 40.60 | 37.83 | 25.43 | 31.54 | 43.09 |
| GC-7 | 33.53 | 27.87 | 17.13 | 23.34 | 27.96 |
| Lopinavir | 49.42 | — | — | — | — |
| Ramipril | — | — | 25.12 | — | — |
| Camostat | — | 54.24 | — | — | — |
| Favipravir | — | — | — | — | 22.57 |
| Tartaric acid (co-crystal) | — | — | — | 17.61 | — |
Molecular dynamics study
In computational drug discovery, MD is a technique that sheds light on the allosteric binding site of the protein, and conformation of the ligand–protein complex. Also, it can simulate the conditions that are hard to perform in wet experiments. Herein, compound GC-6 was subjected to MD simulations bound to the SARS-CoV-2 RdRp. The initial conformations were acquired from the molecular docking experiments by CDOCKER. It can be seen from Fig. 4a that compound GC-6 retained its binding affinity and was firmly bound to its respective binding site. The RMSD curves of the receptor structures from each complex and the potential energy profiles of each complex are shown in (Fig. 4b and c). | ||
| Fig. 4 (a) Hydrogen bond interactions (b) RMSD graph (c) RMSF graph of GC-6 and 6WTC complex. | ||
The trajectories of complexes reached equilibrium after 10 ns, RMSD, and the potential energy of the complexes gets stabilized with time. The H-bond distances formed between GC-6 and RdRp were within a range of around 3.0 and 2.3 Å. Besides, GC-6 formed three pairs of hydrogen bonds with the water molecules after molecular dynamics simulation (Table 6). These hydrogen bonds might contribute to the stability of the complexes. The RMSD values of each receptor-ligand complex were plotted to elucidate the effect of the compounds on the structural stability and integrity. From this plot, 6WTC_GC-6 showed sharp increase in RMSD up to 2 nano seconds. Later 6WTC_GC-6 deviations are optimized up to 80 nano seconds, then it increase slightly to 0.75 Å further the complex attained equilibrium till the end of dynamics.
| Ligand | Interaction score (kcal mol−1) | Ligand atoms (number) | Receptor atoms (number) | Residue (number) | Chain (number) | Type | Score (%) | Distance (Å) |
|---|---|---|---|---|---|---|---|---|
| GC-6 | −9.2003 | H 5948 | OE 3340 | GLU 23 | 6WTC 3 | H-donor | 21.60 | 1.42 |
| H 5962 | O 1702 | ASP 112 | 6WTC 2 | H-donor | 25.00 | 3.64 | ||
| H 5964 | O 1702 | ASP 112 | 6WTC 2 | H-donor | 38.20 | 1.77 | ||
| H 5962 | OD 2043 | ASP 134 | 6WTC 2 | H-donor | 42.40 | 1.23 | ||
| H 5966 | OD 2043 | ASP 134 | 6WTC 2 | H-donor | 34.00 | 1.53 |
Considering all the evaluation indexes, the compound GC-6 might interact with RdRp steadily and have potential negative modulator effects on RdRp.
Discussion
One family of compounds that demonstrate antiviral activity across several studies is the polyphenols. It was well known that hydrophobicity plays a major role in predicting the binding affinity towards SARS-CoV-1. Several natural sources from flora and fauna have been tested in terms of their anti-SARS-CoV-1 activity and used as a scaffold in drug development since its outbreak in 2003 (ESI Table 1). These natural metabolites include flavones, flavonols, fatty acids, tannins, terpenes, and alkaloids.The diversity of these chemical classes is related to the different mechanisms used by each phytochemicals class capable to inhibit coronaviruses. Jo et al. (2019) stated that SARS-CoV-1 inhibition requires chemical structures containing a hydrophobic aromatic ring, hydroxyl groups, and carbohydrates moieties. Although not all of the anti-SARS-CoV-1 compounds (ESI Table 1†) present the aromatic ring, their molecular structures have lipophilic and hydrophilic regions and the ability to form multiple hydrogen bonds through hydroxyl groups. It was well evidenced by our results that the identified compound has hydrophobic and multiple hydroxyl groups. Several investigations revealed the anti-SARS-CoV-2 activity of catechins or polyphenols through the inhibitory potential of MProS alone, which have been highlighted by R. Ghosh et al. recently. Although the importance of MProS has been established as a prime target for COVID-19, there are targets like RdRp and ACE-2. TMPRSS-2 and IL-6 have not been explored yet. There were also limited studies of the polyphenols and catechins against the mentioned targets. Poly targeting is also a reasonable approach to avoid issues related to resistance and toxicity.
Keeping all these views, we wanted to study the GC and its analogs against these multiple targets. Interestingly, we observed the best binding affinities of GC against the targets RdRp, ACE-2, TMPRSS2, and IL. Though it is a potential HIT against these multiple targets, it fails to satisfy L-ROF. L-ROF is a very basic filter for oral drug candidates. In view of these, we designed analogs of GC where the OH groups have been modified (to satisfy HBD = 5; HBA = 10) and all were within the range (0–1 violation) (Table 5). All the designed analogs (GC-2 to GC-7) have shown a comparable binding affinity with GC itself; however, the compound GC-6 was identified as the topmost HIT. By considering binding energies, amino acid residues interactions nod molecular dynamics studies, we propose the following pharmacophoric query for having multitarget inhibitory potential of SARS-CoV-2. Ring aromatics >3; hydrogen bond acceptors >8; hydrogen bond donors >5.
Conclusion
The rapidly spreading outbreak of COVID-19 has challenged the healthcare sector of the world in the last few months. Several investigations have demonstrated that a variety of plant secondary metabolites especially polyphenolic have been reported for anti-SARS-CoV-1& 2 activities since 2002. However, there is very little evidence as yet that these constituents work to prevent or treat SARS-CoV-1& 2 in humans. Moreover, these plant secondary metabolites inhibit anyone's target and not much of their analogs have been reported so far against this SARS CoV-2. To overcome the less efficacious/resistance problems of plant secondary metabolites against SARS CoV-2, we aimed to identify any single molecule that can target all important targets involved in SARS CoV-2. A systematic structure based in silico study was carried out to meet our objective. A gallocatechin analog namely (2R,3R)-2-(3,4-dihydroxyphenyl)chroman-3-yl-3,4,5-trihydroxy benzoate has been identified as a multiple targeting inhibitor. Gallocatechin gallate, a catechin and a constituent of green tea, is an epimer of epigallocatechin gallate. There were strong evidences at in silico level for its effect SARS CoV-2 therapy. An in silico molecular docking study with SARS CoV-2 3CL protease showed that EGCG strongly interacted with Mpro indicating that EGCG potentially inhibit SARS CoV-2. 3CL protease-cleavage of viral proteins by protease is a vital step for viral replication.34–36Further, it is supported by Minsu Jang et al., that EGCG inhibits 3CL protease activity of SARS-CoV-2 in dose dependent manner and the half inhibitory concentration (IC50) was 7.58 μg ml−1 examine the effect of EGCG on coronavirus. EGCG treatment decreases 3CL-protease activity of HCoV-OC43 and HCoV-229E. Moreover, EGCG treatment decreased HCoV-OC43-induced cytotoxicity. Finally, they also found that EGCG treatment decreased the levels of coronavirus RNA and protein in infected cell media. These results indicate that EGCG inhibits coronavirus replication.37,38 In another study, the polyphenol EGCG exhibited significant effect on various druggable targets of SARS CoV-2. These results suggest the potential activity of tea polyphenols in the treatment of COVID-19. The use of tea phyto constituents over synthetic drugs is an exciting treatment option since they are safer, and a higher dose is feasible. However, the bulkiness of these polyphenols and violations of Lipinski's rules could be hindrances in their development. Oral bioavailability is a concern for these polyphenols for which to overcome their derivatives are being developed. EGCG derivatives are being tested for enhancement in physicochemical properties and better efficacy. In line of these, we wanted to explore the efficacy of gallocatechin gallate against various druggable targets of SARS-CoV-2.39 However, further studies are warranted for the validation of these compounds using in vitro and in vivo models to pave a way for these compounds in anti-COVID-19 drug discovery.
Conflicts of interest
No potential conflicts of interest are reported by the authors. There are no conflicts to declare.Acknowledgements
The authors are grateful to ALTEM Technologies private Ltd for providing an evaluation license for COVID-19 research and Ms. Divya Shanmugarajan, Senior Application Scientist for her assistance throughout the study. We acknowledge the generous infrastructure and research supports from JSS College of Pharmacy, JSS Academy of Higher Education & Research, Ooty.References
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu, P. Niu, F. Zhan, X. Ma, D. Wang, W. Xu, G. Wu, G. F. Gao and W. Tan, N. Engl. J. Med., 2020, 382, 727–733 CrossRef CAS PubMed.
- Q. Li, X. Guan, P. Wu, X. Wang, L. Zhou, Y. Tong, R. Ren, K. S. M. Leung, E. H. Y. Lau, J. Y. Wong, X. Xing, N. Xiang, Y. Wu, C. Li, Q. Chen, D. Li, T. Liu, J. Zhao, M. Liu, W. Tu, C. Chen, L. Jin, R. Yang, Q. Wang, S. Zhou, R. Wang, H. Liu, Y. Luo, Y. Liu, G. Shao, H. Li, Z. Tao, Y. Yang, Z. Deng, B. Liu, Z. Ma, Y. Zhang, G. Shi, T. T. Y. Lam, J. T. Wu, G. F. Gao, B. J. Cowling, B. Yang, G. M. Leung and Z. Feng, N. Engl. J. Med., 2020, 382, 1199–1207 CrossRef CAS PubMed.
- S.-C. Cheng, Y.-C. Chang, Y.-L. Fan Chiang, Y.-C. Chien, M. Cheng, C.-H. Yang, C.-H. Huang and Y.-N. Hsu, J. Formosan Med. Assoc., 2020, 119, 747–751 CrossRef CAS PubMed.
- P. Zhou, X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li, C.-L. Huang, H.-D. Chen, J. Chen, Y. Luo, H. Guo, R.-D. Jiang, M.-Q. Liu, Y. Chen, X.-R. Shen, X. Wang, X.-S. Zheng, K. Zhao, Q.-J. Chen, F. Deng, L.-L. Liu, B. Yan, F.-X. Zhan, Y.-Y. Wang, G.-F. Xiao and Z.-L. Shi, Nature, 2020, 579, 270–273 CrossRef CAS PubMed.
- P. S. Masters and S. Perlman, Fields virology, 2013, vol. 1, pp. 825–858 Search PubMed.
- J. Cui, F. Li and Z.-L. Shi, Nat. Rev. Microbiol., 2019, 17, 181–192 CrossRef CAS PubMed.
- W. R. Ferraz, R. A. Gomes and A. L. S. Novaes, Future Med. Chem., 2020, 12, 1815–1828 CrossRef CAS PubMed.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020, 15, 907–932 CrossRef CAS PubMed.
- M. Hagar, H. A. Ahmed, G. Aljohani and O. A. Alhaddad, Int. J. Mol. Sci., 2020, 21, 3922 CrossRef CAS PubMed.
- S. Shahinshavali, K. A. Hossain, A. V. D. N. Kumar, A. G. Reddy, D. Kolli, A. Nakhi, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2020, 61, 152336 CrossRef PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368(6489), 409–412 CrossRef CAS PubMed.
- D. Gentile, V. Patamia, A. Scala, M. T. Sciortino, A. Piperno and A. Rescifina, Mar. Drugs, 2020, 19 Search PubMed.
- A. V. Ravi, Front. Microbiol., 2020, 11, 15 CrossRef PubMed.
- T. P. Sheahan, A. C. Sims, S. R. Leist, A. Schäfer, J. Won, A. J. Brown, S. A. Montgomery, A. Hogg, D. Babusis, M. O. Clarke, J. E. Spahn, L. Bauer, S. Sellers, D. Porter, J. Y. Feng, T. Cihlar, R. Jordan, M. R. Denison and R. S. Baric, Nat. Commun., 2020, 11, 222 CrossRef CAS PubMed.
- R. U. Kadam and I. A. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 206–214 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280 CrossRef CAS PubMed .e8.
- G. Li and E. De Clercq, Nat. Rev. Drug Discovery, 2020, 19, 149–150 CrossRef CAS PubMed.
- Y. Li, Z. Xie, W. Lin, W. Cai, C. Wen, Y. Guan, X. Mo, J. Wang, Y. Wang, P. Peng, X. Chen, W. Hong, G. Xiao, J. Liu, L. Zhang, F. Hu, F. Li, F. Zhang, X. Deng and L. Li, Med, 2020, S2666634020300015 Search PubMed.
- M. A. Mesaik, A. Jabeen, A. Halim, A. Begum, A. Shukralla, M. Asif, Z. Ul-Haq and M. Iqbal, Chem. Biol. Drug Des., 2012, 79(3), 290–299 CrossRef CAS PubMed.
- G. M. Cragg and J. D. J. Newman, Biochim. Biophys. Acta, 2013, 1830(6), 3670–3695 CrossRef CAS PubMed.
- I. Aanouz, A. Belhassan, K. El-Khatabi, T. Lakhlifi, M. El-ldrissi and M. Bouachrine, J. Biomol. Struct. Dyn., 2020, 1–9 Search PubMed.
- M. K. Gupta, S. Vemula, R. Donde, G. Gouda, L. Behera and R. Vadde, J. Biomol. Struct. Dyn., 2020, 1–11 CAS.
- O. O. Olubiyi, M. Olagunju, M. Keutmann, J. Loschwitz and B. Strodel, Molecules, 2020, 25, 3193 CrossRef CAS PubMed.
- S. T. Ngo, N. Quynh Anh Pham, L. Thi Le, D.-H. Pham and V. V. Vu, J. Chem. Inf. Model., 2020, 60, 5771–5780 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- H. Beard, A. Cholleti, D. Pearlman, W. Sherman and K. A. Loving, PLoS One, 2013, 8, e82849 CrossRef PubMed.
- S. Sarvagalla, V. K. Singh, Y.-Y. Ke, H.-Y. Shiao, W.-H. Lin, H.-P. Hsieh, J. T. A. Hsu and M. S. Coumar, J. Comput.-Aided Mol. Des., 2015, 89–100 CrossRef CAS PubMed.
- B. K. Nganou, P. Tane, A. Nchiozem, S. A. Jubie Selvaraj and C. Nanjan, J. Pharm. Sci. Res., 2017, 9, 95–99 CAS.
- H. Yang, M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G. F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld and Z. Rao, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13190–13195 CrossRef CAS PubMed.
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes and Y.-Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- R. Ghosh, A. Chakraborty, A. Biswas and S. Chowdhuri, J. Biomol. Struct. Dyn., 2020, 1–16 Search PubMed.
- R. Ghosh, A. Chakraborty, A. Biswas and S. Chowdhuri, J. Biomol. Struct. Dyn., 2020, 1–13 Search PubMed.
- K. A. Peele, C. Potla Durthi, T. Srihansa, S. Krupanidhi, V. S. Ayyagari, D. J. Babu, M. Indira, A. R. Reddy and T. C. Venkateswarulu, Informatics in Medicine Unlocked, 2020, 19, 100345 CrossRef PubMed.
- V. K. Bhardwaj, R. Singh, J. Sharma, V. Rajendran, R. Purohit and S. Kumar, J. Biomol. Struct. Dyn., 2020, 1–10 CrossRef PubMed.
- M. Jang, R. Park, Y.-I. Park, Y.-E. Cha, A. Yamamoto, J. I. Lee and J. Park, Biochem. Biophys. Res. Commun., 2021, 547, 23–28 CrossRef CAS PubMed.
- M. Jang, Y.-I. Park, Y.-E. Cha, R. Park, S. Namkoong, J. I. Lee and J. Park, J. Evidence-Based Complementary Altern. Med., 2020, 2020, 1–7 Search PubMed.
- S. Mhatre, S. Naik and V. Patravale, Comput. Biol. Med., 2021, 129, 104137 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01603b |
| This journal is © The Royal Society of Chemistry 2021 |