
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of BODIPY dyes as triplet photosensitizers: electronic properties tailored for solar energy conversion, photoredox catalysis and photodynamic therapy
Elena
Bassan
 ,
Andrea
Gualandi
,
Pier Giorgio
Cozzi
* and
Paola
Ceroni
*
,
Andrea
Gualandi
,
Pier Giorgio
Cozzi
* and
Paola
Ceroni
*
Department of Chemistry "Giacomo Ciamician", University of Bologna, Italy. E-mail: paola.ceroni@unibo.it
First published on 14th April 2021
Abstract
BODIPYs are renowned fluorescent dyes with strong and tunable absorption in the visible region, high thermal and photo-stability and exceptional fluorescence quantum yields. Transition metal complexes are the most commonly used triplet photosensitisers, but, recently, the use of organic dyes has emerged as a viable and more sustainable alternative. By proper design, BODIPY dyes have been turned from highly fluorescent labels into efficient triplet photosensitizers with strong absorption in the visible region (from green to orange). In this perspective, we report three design strategies: (i) halogenation of the dye skeleton, (ii) donor–acceptor dyads and (iii) BODIPY dimers. We compare pros and cons of these approaches in terms of optical and electrochemical properties and synthetic viability. The potential applications of these systems span from energy conversion to medicine and key examples are presented.
 Prof. Paola Ceroni, second row on the left; Ms Elena Bassan first row on the left; Prof. Pier Giorgio Cozzi, second row on the right; Dr Andrea Gualandi, second row on the right. | The group picture displays the PhotoChemistry group (Prof. Paola Ceroni, second row on the left; Ms Elena Bassan first row on the left) and Organic Chemistry group (Prof. Pier Giorgio Cozzi, second row on the right; Dr Andrea Gualandi, second row on the right) in front of the Ciamician's Department of Bologna University. The face masks are a reminder of this difficult period. Currently, the two groups are actively collaborating in developing new photoredox reactions, new dyes for photocatalysis and synergistic metal photoredox catalytic reactions. Prof. Pier Giorgio Cozzi is full professor at the Bologna University and Degree Programme Director of the Master Course in Chemistry. Dr Andrea Gualandi is Senior Assistant Professor at the Bologna University. Prof. Paola Ceroni is full professor at the Bologna University and Degree Programme Director of the Master Course in Photochemistry and Molecular Materials. Ms Elena Bassan is a PhD student working in the group of Paola Ceroni. |
1. Introduction
BODIPYs (abbreviation for boron-dipyrromethenes, 4,4-difluoro-4-borata-3a-azonia-4a-aza-s-indacene is reported in Fig. 1) are a class of organoboron compounds characterized by high molar absorption coefficients (up to 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1), excellent thermal and photo-stability and a high fluorescence quantum yield. Thanks to these outstanding optical properties BODIPY dyes are traditionally used as light-harvesting antennas in artificial photosynthesis,1 fluorescent sensors,2 fluorescent labels for bioimaging,3 photodynamic therapy,4 electrochemiluminescence5 and optoelectronic devices,6 comprising solar cells, organic light emitting diodes (OLED) and non-linear optical devices.
000 M−1 cm−1), excellent thermal and photo-stability and a high fluorescence quantum yield. Thanks to these outstanding optical properties BODIPY dyes are traditionally used as light-harvesting antennas in artificial photosynthesis,1 fluorescent sensors,2 fluorescent labels for bioimaging,3 photodynamic therapy,4 electrochemiluminescence5 and optoelectronic devices,6 comprising solar cells, organic light emitting diodes (OLED) and non-linear optical devices.
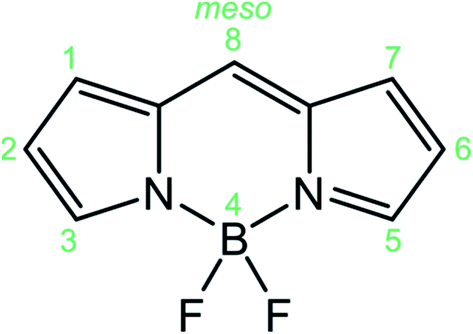 | ||
| Fig. 1 Structure of the BODIPY core and IUPAC numeration. | ||
The ease of synthetic functionalization7 at the meso position, as well as at the pyrrolic positions of the BODIPY skeleton (Fig. 1), has led to the exploration of a huge family of derivatives, demonstrating that substitution at the latter positions strongly perturbs the photophysical and electrochemical properties, enabling a fine tuning of their electronic properties, while meso substitution has a lower impact on the electronic properties and it can be used to append other chromophores in a supramolecular system or to append stimuli responsive units for sensing applications.
Recently, chemical modifications of the BODIPY skeleton have been devised to make BODIPYs efficient triplet photosensitizers: in these chromophores, the lowest singlet excited state, populated by visible-light excitation, deactivates mainly by a non-radiative decay to the triplet state instead of by the typical fluorescence decay of the most commonly investigated BODIPYs. The long-lived triplet excited state may then be involved in bimolecular sensitization processes, e.g. production of singlet oxygen used in photocatalysis and photodynamic therapy.4
Among the triplet photosensitizers, transition metal complexes are the most commonly investigated,8 since the presence of heavy metal atoms induces a strong mixing between singlet and triplet characters of their excited states (determined by SOC, or spin orbital coupling). Although the efficiency of population of the lowest triplet state can reach unity, as is the case for [Ru(bpy)3]2+,9 considerations on the toxicity and scarcity of heavy metals is shifting interest on the development of organic chromophores. Most of the organic molecules possess very weak spin orbit coupling and the rate of population of the lowest triplet state is extremely low. Efficient population of triplet excited states of organic chromophores strongly absorbing in the visible spectral region remains a very challenging task and BODIPY dyes are ideal candidates.
In this perspective, we will present, at first, the most important approaches employed to induce efficient inter system crossing (ISC) in organic dyes, followed by a selection of BODIPY examples in which the photophysical as well as electrochemical properties are discussed and compared to correlate structure and properties. Three main classes of BODIPY dyes will be discussed: (i) halogenated chromophores; (ii) BODIPY coupled to electron donating chromophores in donor–acceptor dyads; (iii) BODIPY dimers. The choice of examples was based not only on the intrinsic interest of the topic itself or its possible applications, but also on its educational value. It is worth mentioning that thiophene-fused BODIPYs,10–12 BODIPY coupled to transition metal complexes13 and, more recently, twisted BODIPYs14,15 have been reported as efficient triplet sensitizers, but they will not be covered in this review and interested readers can refer to the above-mentioned literature. The fourth section of the review discusses the synthetic procedures used to synthesize the BODIPY dyes described in the previous section. The last part is devoted to their applications with a special focus on energy conversion and photocatalysis, which have been gaining increasing attention over the last five years.
2. Inter system crossing in organic dyes
Most of the organic molecules are characterized by closed-shell ground electronic configuration, so that the ground state is a singlet state (S0), and the excited states are either singlets (S1, S2, etc.) or triplets (T1, T2, etc.). In principle, transitions between states having the same spin value are allowed, whereas those between states of different spin are forbidden. Fluorescence (FLUO), the spin-allowed radiative process, deactivates S1 to S0 and competes with non-radiative deactivations to S0 (internal conversion, IC) and to T1 (inter system crossing, ISC) (Fig. 2). Therefore, high fluorescence quantum yield implies inefficient population of T1, as it is usually the case for highly fluorescent BODIPY dyes.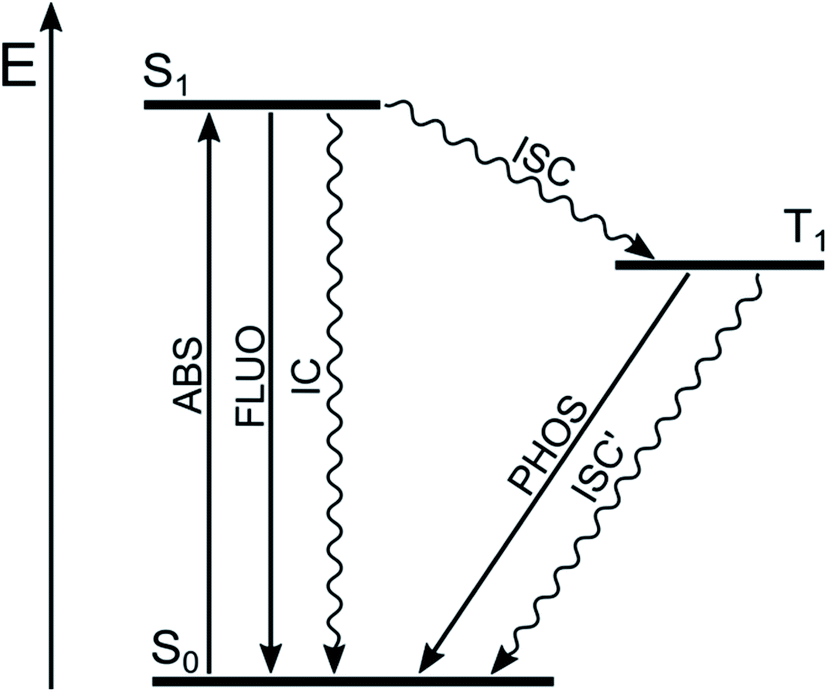 | ||
| Fig. 2 Jablonski diagram for organic molecules with radiative (straight lines) and non-radiative processes (wavy lines). | ||
The spin selection rule is valid to the extent to which spin and orbital functions can be separated rigorously. The selection rule is relaxed and the rate of ISC is strongly increased by spin–orbit coupling (SOC), namely the interaction of electron spin with the orbital magnetic moment. The SOC and thus the rate of ISC increases when (i) heavy atoms are present (spin–orbit coupling is directly proportional to Z,4 where Z is the atomic number); (ii) the involved singlet and triplet states belong to different electronic configurations, according to El-Sayed rules; (iii) the energy difference between the involved electronic states is low.16–18 The energy separation between the singlet and triplet states of a given electronic configuration arises from the repulsion experienced by neighboring electrons. When the electronic transition is (n → π*), the two orbitals barely overlap since they lie in orthogonal planes, so that the singlet-triplet splitting is small. The same holds true for electronic transitions with charge-transfer character, i.e. electronic transition in which an electron is transferred from an orbital mainly localized on the electron donating moiety to another orbital mainly localized on the electron acceptor moiety, since the spatial orbital overlap is limited. On the other hand, the splitting of singlet-triplet states deriving from (π → π*) transitions is relatively large since the orbital overlap is considerable.
SOC is not the only mechanism to promote ISC: hyperfine interaction, i.e. the magnetic interactions between electronic and nuclear spins, is an alternative mechanism. The name derives from the hyperfine splitting of the peaks observed in EPR (electron paramagnetic resonance) spectra. The main nuclear spin present in organic molecules is related to protons, so that it is also named proton hyperfine interaction. ISC promoted by hyperfine interaction is particularly important in the case of radical centers well separated in space or biradicals where SOC is not operative (see below the discussion on radical pair inter system crossing).
The choice of a proper triplet photosensitizer is dictated not only by the efficiency of population of the lowest triplet excited state, but also by: (i) the energy of the lowest triplet excited state; (ii) the redox properties of the dye; (iii) the absorption range and photostability of the dye. It is worth noting that the energy level of the lowest triplet state is usually lower by 25–30% compared to the lowest singlet excited state, e.g. in the case of BODIPY dyes, so that there is an unavoidable energy loss between excitation energy and resulting position of the sensitizing excited state. This parameter should be considered when choosing the proper triplet sensitizer.
In the following, we will discuss two main approaches for efficient ISC, namely halogenation and charge transfer, showing that they are general approaches already implemented for other organic dyes. Other paths, such as the presence of twisted π-systems as in the cases of C60 (ref. 19) and helicenes,20,21 will not be discussed in this review; interested readers can refer for instance to recently published reviews.22,23
Halogenation
The presence of halogen atoms, particularly iodine or bromine in an organic molecule strongly increases the spin–orbit coupling by heavy atom effect, particularly when the heavy halogen atom is part of the chromophoric unit, meaning that the HOMO and/or LUMO orbitals extend to the halogen atom.One representative example is that of xanthene derivatives (Scheme 1). While fluorescein is a recognized fluorescence standard (ΦFLUO = 97%),24 its halogenated counterparts are well-established triplet photosensitizers. The incorporation of progressively heavier halogen atoms into the fluorogenic core increases the rate constant of S1 → T1 ISC and, as a consequence, their fluorescence quantum yield decreases. Eosin Y exhibits a fluorescence quantum yield ΦFLUO of 69% and for erythrosin B ΦFLUO further decreases to 8%.25 Accordingly, the triplet quantum yield of eosin Y is 33% and that of erythrosin B is 83%.24
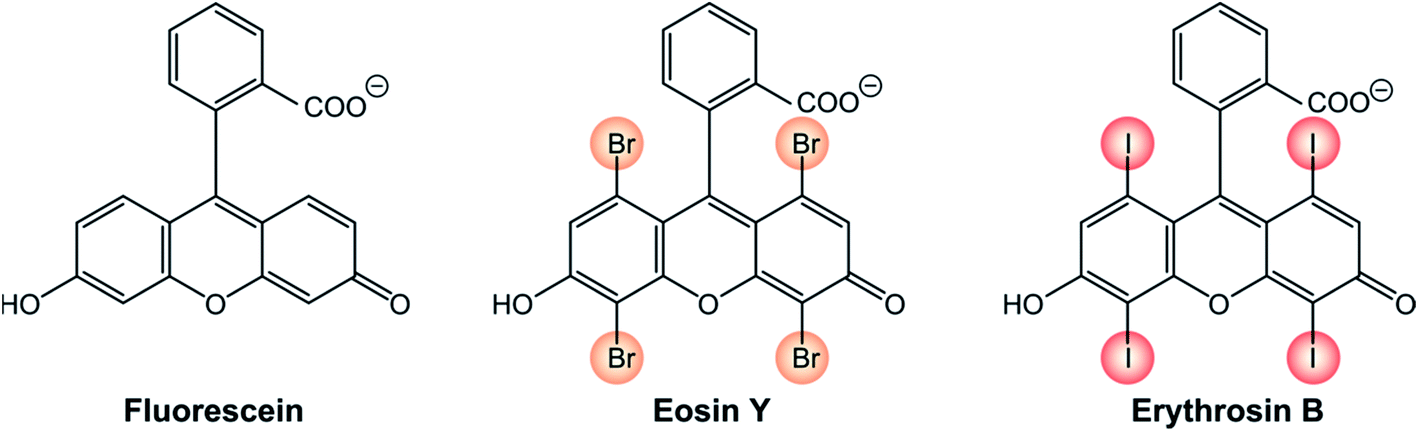 | ||
| Scheme 1 | ||
Charge-transfer mediated ISC
In a dyad constituted by an electron-donating unit (D) linked to an electron-accepting one (A), upon absorption of a suitable photon that populates the locally-excited, lowest singlet excited state of A or D (Fig. 3) a photoinduced electron transfer process (PeT) from D to A can occur, resulting in the formation of a charge-transfer singlet excited state (1CT). The following charge recombination process may populate the long-lived triplet excited states of D or A (T1). Two mechanisms are possible (Fig. 3): radical-pair intersystem crossing (RP-ISC) which populates 3CT at first and then T1, or direct population of T1 by spin–orbit charge transfer intersystem crossing (SOCT-ISC).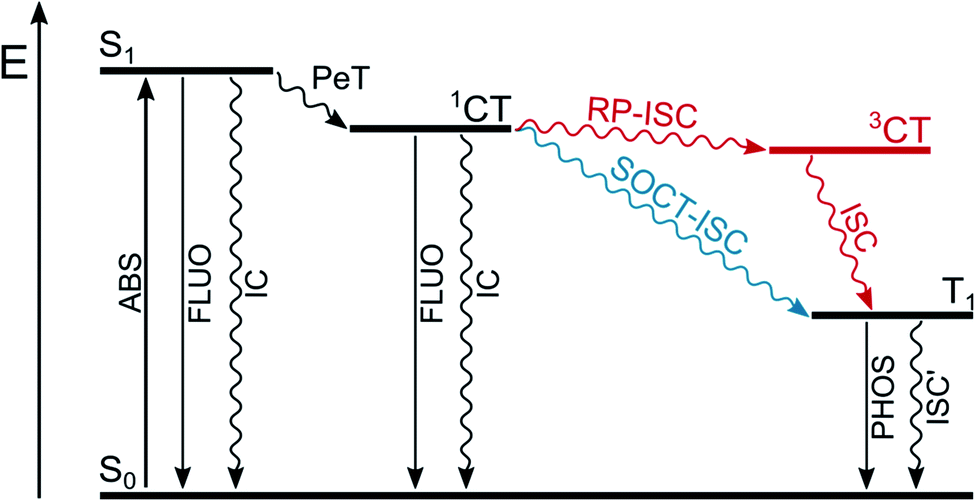 | ||
| Fig. 3 Population of triplet excited state T1 by radical-pair intersystem crossing (RP-ISC) or by spin–orbit charge transfer intersystem crossing (SOCT-ISC). | ||
The radical-pair intersystem crossing (RP-ISC) is effective when the two radical centers are well separated in space by the presence of a linker, so that the electronic exchange interaction between the donor and the acceptor is minimized and the energy gap between 3CT and 1CT becomes very small. The hyperfine interaction between the electronic and nuclear spins favors intersystem crossing between these two charge-transfer states and, once 3CT is populated, spin-allowed charge recombination can selectively produce the lowest triplet excited state of A or D, depending on which one lies lower in energy.26 This mechanism has been widely used in the design of artificial charge separation systems that mimic photosynthetic reaction centres.27,28
The spin–orbit charge transfer intersystem crossing (SOCT-ISC) is favored by high degree of charge separation, perpendicular orientation of the orbitals involved in the charge transfer, and large magnitude of the electronic coupling between the donor and acceptor. This situation is facilitated by direct linkage of the donor and acceptor moieties in an orthogonal arrangement. Thanks to their perpendicular orientation, the change in spin multiplicity is accompanied by a change in the orbital angular momentum which facilitates ISC to form directly T1 of the electron donor or acceptor. The charge transfer process usually involves the non-bonding n orbital located on the donor and the π* orbital of the acceptor and it is, in some way, similar to that which occurs in an (n,π*) electronic transition within a single chromophore.
Some examples of electron donor–acceptor dyads have recently proved to undergo SOCT-ISC.29–31 One illustrative example is the case of the dyad composed of a perylene accepting unit linked at the N-position of the phenothiazine donating one (Scheme 2).32 The steric hindrance between the perylene and phenothiazine units secures the orthogonal geometry between the two moieties, and maximizes the overlap of the nitrogen lone pair with the π system of the perylene ring. Femtosecond transient absorption spectroscopy demonstrates the formation of the charge-transfer excited state, which decays to the perylene triplet state.
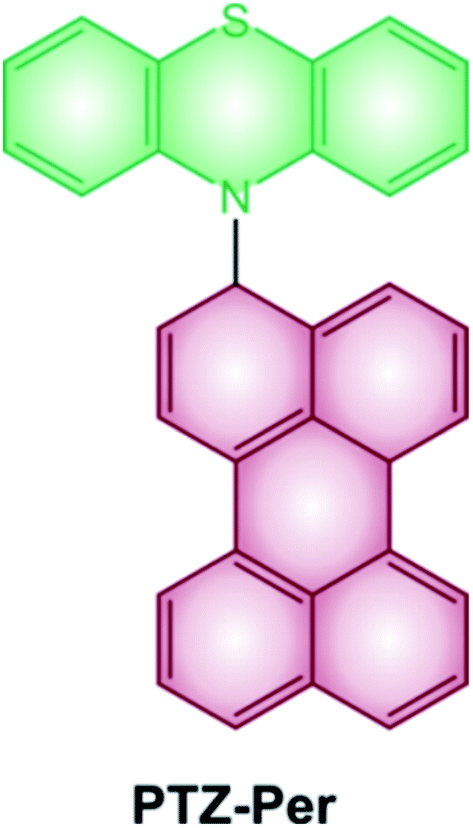 | ||
| Scheme 2 | ||
The difference between the two mechanisms reported in Fig. 3 is very subtle and difficult to ascertain. SOC-ISC shows a strong distance and orientation dependence on the contrary of RP-ISC, which is not distance dependent being a localized interaction between electron spin and nearby nuclear spins. RP-ISC is usually slower (kISCca. 108 s−1) than SOC-ISC.33 The RP-ISC is generally recognized by the fact that the yield of T1 population by RP-ISC is sensitive to the application of an external magnetic field, which splits the triplet RP manifold into its Zeeman sublevels, modulating the efficiency of singlet-triplet mixing. However, the absence of a magnetic field effect cannot be taken as a definitive proof of the SOCT-ISC mechanism since these effects can be so small that they cannot be experimentally detected. It is possible to distinguish the two mechanisms by analysis of the time-resolved EPR measurements, as reported by Wasielewski.29 The examples of BODIPY dyads presented in the following consists of very closely spaced chromophores, so that the SOCT-ISC mechanism is expected to be operative, although detailed studies on magnetic field effects are rare.
3. Photophysical and electrochemical properties of BODIPY photosensitizers
In this section, BODIPY triplet photosensitizers are classified according to their functionalization, and a special focus is dedicated to the impact of the functionalization on their photophysical and electrochemical properties. Redox potentials are reported in V vs. SCE.† We decided to name BODIPYs as follows: substituents linked to the BODIPY core (shortened as BDP) are abbreviated before BDP except for those linked at the meso position, which follow BDP. Methyl groups in position 1, 3, 5, 7 are omitted when they are not necessary in the discussion.Halogenated BODIPYs
The introduction of heavy halogen atoms directly onto the chromophoric core was proposed in 2005 by Nagano's group.34 For example, I2-BDP (Scheme 3) has a molar absorption coefficient (110000 M−1 cm−1 at λ = 534 nm) similar to that of BDP (120000 M−1 cm−1 at λ = 502 nm), but displays a much lower fluorescence quantum yield (ΦFLUO = 2%) compared to BDP (ΦFLUO = 70%) and is able to generate singlet oxygen with a quantum yield (ΦΔ) 1.34 times higher than the benchmark photosensitizer Rose Bengal.
 | ||
| Scheme 3 | ||
It is worth analyzing how the photophysical and electrochemical properties of BODIPY dyes are affected by the presence of different halogen substituents (Scheme 4 and Table 1).5,7 Absorption and emission maxima red-shift as the halogen's atomic number is increased, whereas molar absorption coefficients are only slightly decreased by halogenation. Fluorescence quantum yields strongly decrease from 67.5% to 1.8% on going from chlorine to iodine, and the quantum yields of singlet oxygen formation concomitantly increase from 22% to 84%. As expected, the heavy-atom effect makes ISC more efficient if halogen atoms with a higher atomic number are present on the BODIPY core. In addition, the electron-withdrawing nature of halogen atoms makes the reduction process easier.
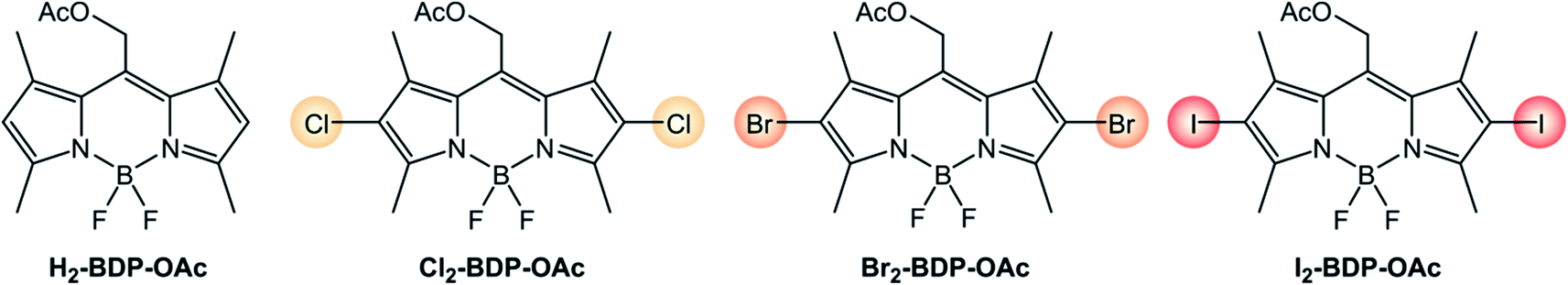 | ||
| Scheme 4 | ||
| Molecule | ε (M−1 cm−1)a | λ maxABS (nm)a | λ maxFLUO (nm)a | Φ FLUO | Φ Δ | E(A/A˙−)bd | E(A˙+/A)bd |
|---|---|---|---|---|---|---|---|
| a In MeOH. b In MeCN. c Chemically irreversible electron transfer process. d In V vs. SCE. | |||||||
| H2-BDP-OAc (ref. 35 and 36) | 71000 |
517 | 529 | 73.4% | <0.05% | −1.05c | +1.14c |
| Cl2-BDP-OAc (ref. 35 and 37) | 48000 |
544 | 562 | 67.5% | 22% | −0.85c | +1.46c |
| Br2-BDP-OAc (ref. 35 and 36) | 56300 |
545 | 565 | 15.5% | 54% | −0.74c | +1.33 |
| I2-BDP-OAc (ref. 35 and 36) | 49000 |
553 | 575 | 1.8% | 84% | −0.75c | +1.30 |
The effect of the number and position of halogen substituents is evident from the comparison (Table 2) of the compounds reported in Scheme 5. The presence of iodine atoms on the BODIPY skeleton red-shifts absorption and fluorescence maxima and increases the quantum yield of singlet oxygen formation. Interestingly, the fluorescence quantum yield of BODIPY dyes functionalized with iodine atom in the chromophore skeleton is generally decreased compared to the pristine BDP-tol, apart in the case of I3-BDP-tol and I4-BDP-tol.
| Molecule | ε (M−1 cm−1) | λ maxABS (nm) | λ maxFLUO (nm) | Φ FLUO | Φ Δ |
|---|---|---|---|---|---|
| a In cyclohexane. b In MeCN. c In MeOH. d In DCM. | |||||
| BDP-tol (ref. 38) | 69000a |
500a | 516a | 3.6%a | n.r. |
| I-BDP-tol (ref. 38) | 22000a |
523a | 540a | 3.4%a | n.r. |
| I2-BDP-tol (ref. 38) | 43000a |
548a | 567a | 1.2%a | 83%b |
| I3-BDP-tol (ref. 38) | 48000a |
563a | 577a | 6.0%a | 86%b |
| I4-BDP-tol (ref. 38) | 116000a |
581a | 593a | 9.9%a | 87%b |
| Me4-BDP-Ph (ref. 39) | 44000c |
498c | 509c | 65%c | 1%d |
| Me4-BDP-Ph-I (ref. 39) | 81000c |
499c | 511c | 49%c | 1%d |
 | ||
| Scheme 5 | ||
It is worth noting that if the halogen is present in the para position of the phenyl ring (Me4-BDP-Ph-I), its photophysical properties remain similar to those of the parent compound (Me4-BDP-Ph) without any significant heavy-atom effect. The site of iodination is therefore critical in the design of a BODIPY-based photosensitizer.
The photophysical properties of 2,6-diiodinated BODIPYs (Table 3 and Scheme 6) are almost unaffected by the nature of the meso substituent. Computational studies have in fact revealed that HOMO and LUMO orbitals in meso-aryl BODIPYs are located on the chromophoric core and do not involve the aryl substituent.39 A small influence by the nature of the meso substituent on the redox properties of I2-BDP-mes, I2-BDP-Py and I2-BDP-PhOH can be observed, but the main role is exerted by groups appended directly on the BODIPY core.
 | ||
| Scheme 6 | ||
BODIPY dyads
An entirely different approach to the design of BODIPY-based photosensitizers is that of utilizing a dyad composed of an electron-donating moiety and an electron-accepting BODIPY unit.43–46 As explained in Section 2, when an electron donor and an electron acceptor are directly linked and orthogonally oriented, spin orbit charge transfer intersystem crossing can take place and the triplet excited state of the donor or the acceptor can be populated.Nagano's group reported a very simple BODIPY derivative, namely BDP-Ph(OMe)3.47 Its fluorescence quantum yield is significantly reduced by a photoinduced electron transfer (PeT) from the electron-donating trimethoxybenzene moiety to the electron accepting BODIPY core47 and BDP-Ph(OMe)3 is able to generate singlet oxygen,45 suggesting that the recombination of the charge-transfer (CT) state populates the long-lived BODIPY triplet excited state.
If we compare dyads comprising different electron-rich moieties combined with the same BODIPY core (Scheme 7 and Table 4), we can notice that the lowest energy absorption band and the fluorescence band are unaffected by the nature of the donating unit and no charge-transfer band is observed in UV-Vis spectra, implying that no significant electronic coupling exists between the two moieties in the ground state. Quantum yields of singlet oxygen formation are, conversely, dependent on the electron-donating ability of the meso substituent and the solvent polarity. Indeed, an increase of the solvent polarity or electron-donating ability of the substituent is expected to stabilize the CT state. Whereas the ΦΔ of BDP-PTZ is 67% in apolar toluene, that of BDP-ANPh is just 10% and its ΦFLUO is 81%: photoinduced electron transfer is rather inefficient in this solvent for BDP-ANPh, which features a less electron donating unit (ANPh) compared to PTZ. On the other hand, ΦΔ of BDP-ANPh increases to 95% in more polar DCM solution.
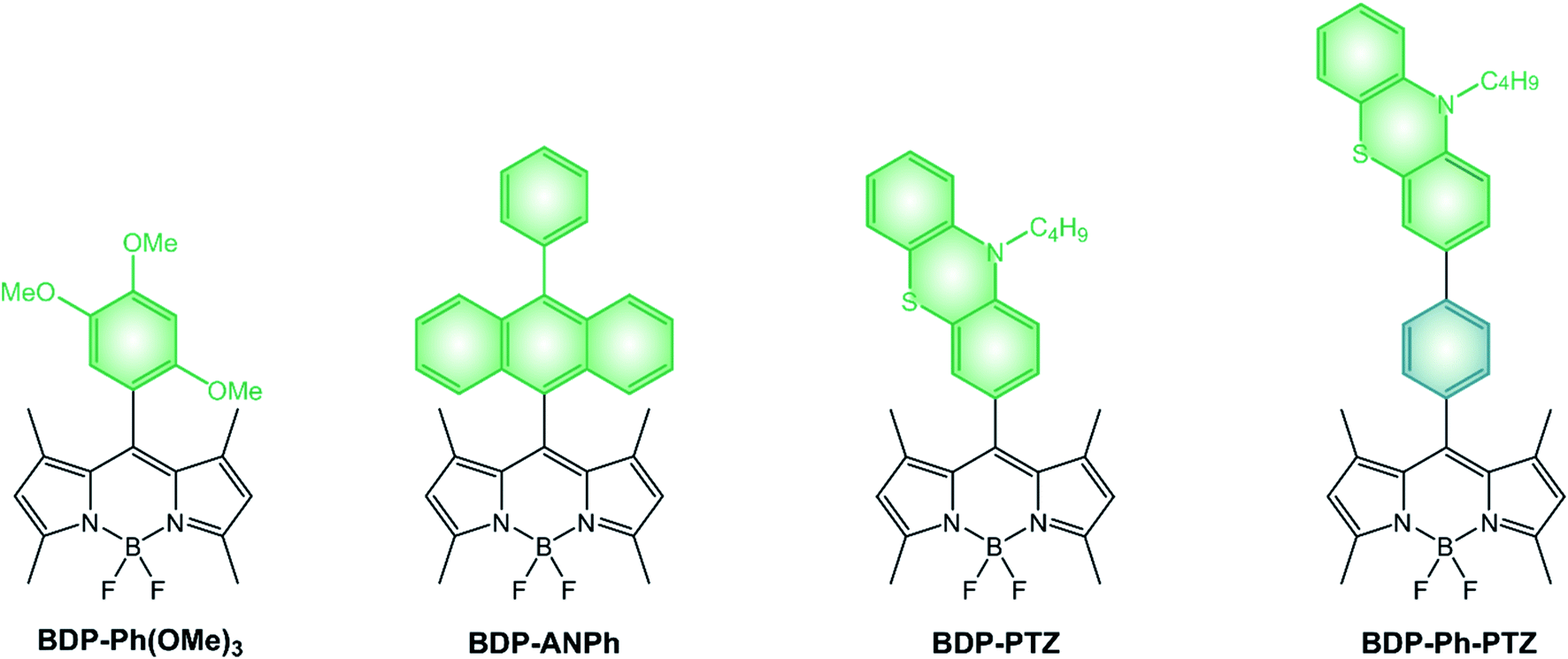 | ||
| Scheme 7 | ||
| Molecule | ε (M−1 cm−1)a | λ maxABS (nm) | λ maxFLUO (nm) | Φ FLUO | Φ Δ | E(A/A˙−)b | E(A˙+/A)b |
|---|---|---|---|---|---|---|---|
| a In DCM. b V vs. SCE. c In THF. d In MeCN. e In toluene. | |||||||
| BDP-Ph(OMe)3 (ref. 45 and 47) | n.r. | 503c | 514c | 0.5%c | 36%c | −1.21d | +1.11; +1.29d |
| BDP-ANPh (ref. 48) | 86000 |
506a | 518a | 1%a | 95%a | −1.20a | +1.24a |
| 81%e | 10%e | ||||||
| BDP-PTZ (ref. 49) | 49000 |
502a | 510a | 2.7%a | 67%e | −1.23a | +0.82; +1.30a |
| BDP-Ph-PTZ (ref. 49) | 48700 |
502a | 510a | 7.2%a | 25%e | −1.23a | +0.78; +1.24a |
Since the electron donor and the electron acceptor units are electronically decoupled, redox potentials of BODIPY dyads correspond to those of the constituent units and can be assigned by comparison with appropriate model compounds. Reduction potentials of dyads BDP-Ph(OMe)3, BDP-ANPh and BDP-PTZ are similar (Table 4) and have been ascribed to the one-electron reduction of the BOIDPY core. BDP-ANPh shows one oxidation at +1.24 V vs. SCE, attributed to the oxidation of phenylanthracene,‡ and BDP-PTZ at +0.82 V vs. SCE (imputed to phenothiazine) and one at +1.30 V vs. SCE (attributed to BODIPY).
When the electron donor and acceptor are separated by a spacer, such is the case for BDP-Ph-PTZ, PeT is less efficient and, consequently, its fluorescence quantum yield slightly increases at the expenses of the ΦΔ quantum yield. Besides distancing the two units, the presence of the phenyl spacer additionally induces their co-planarity which contributes to an even lower value of ΦΔ. Redox potentials are almost unaffected by the spacer, confirming the absence of electronic coupling between the two units for both BDP-Ph-PTZ and BDP-PTZ.
It is interesting to analyze the photophysical properties of a series of pyrene-BODIPY dyads in which the number of alkyl substituents on the BODIPY core ranges from zero to six (Table 5 and Scheme 8). Stokes shift is small for all compounds, except for BDP-Pyr, which is characterized by a considerably lower fluorescence quantum yield. The larger Stokes shift observed for BDP-Pyr is consistent with the formation of an emissive charge-transfer state. ΦΔ quantum yields decline with the number of alkyl groups present on the BODIPY core. Alkylation, in fact, increases the electronic density of the BODIPY core and moves the CT state to higher energies and this brings about a reduction of the PeT efficiency.
 | ||
| Scheme 8 | ||
The energy of the CT state, in relation to the energy of S1 and T1 states of BODIPY dyes, is a crucial parameter to maximize the efficiency of T1 population, as clearly described in a recent paper on BODIPY dyads (Fig. 4).51 In order to maximize the efficiency of T1 population, the spin-forbidden charge recombination from 1CT to T1 (CRT1) should be as fast as possible, i.e. an activationless process at the top of the Marcus parabola (blue lines in Fig. 4A and B), while the free energy associated to the recombination from 1CT to S0 (ΔG(CRS0)) should be so negative that the process lies in the Marcus inverted region, and its kinetics is slow (blue lines in Fig. 4B). Contrarily, if the initially-formed charge transfer state 1CT were located at lower energy values (yellow lines in Fig. 4A), the free energies associated to both charge recombination processes (ΔG(CRS0) and ΔG(CRT1)) would decrease. The kinetic constant for charge recombination to the ground state could then increase and compete with the recombination to the triplet excited state (yellow lines in Fig. 4B): this would be detrimental for the efficient population of the triplet state.
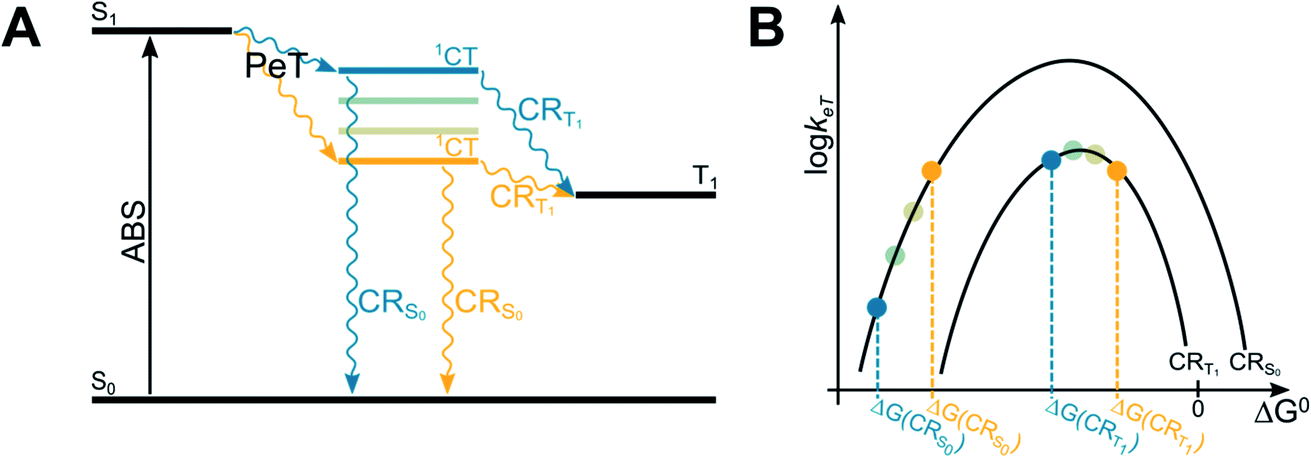 | ||
| Fig. 4 Jablonski diagram illustrating the energies of the excited states involved in the SOCT-ISC mechanism (A) and related Marcus curve (B). | ||
In order to adjust the energy of 1CT in donor–acceptor dyads it is therefore necessary to choose electron-donating and accepting units with appropriate redox properties. Alternatively, one can tune the energy of the charge-transfer state by changing the solvent polarity.
BODIPY dimers
The linkage of two BODIPY units to form a dimer is an alternative route to the efficient population of T1 excited state. For example, BDP-3-BDP (Scheme 9 and Table 6), in which two chromophoric units are linked at the 3-position displays novel optical properties: the absorption band centered at 534 nm for the parent monomer compound BDP-Mon is split into two intense bands peaked at 492 nm and 565 nm in the case of BDP-3-BDP, due to an exciton coupling interaction18 between the chromophoric units which leads to a loss of degeneracy in the S1 state. Additionally, BDP-3-BDP's emission is red-shifted compared to BDP-Mon, its fluorescence quantum yield is lower, and it is able to generate singlet oxygen with a quantum yield of 40%. A strong solvent dependence is observed for dimers: for example, ΦΔ of BDP-3-BDP is much lower than 10% in acetonitrile. This suggests that, on top of the smaller S1–T1 energy gap due to exciton coupling, symmetry-breaking charge-transfer states52 are involved in the triplet state population, in accordance with computational studies and with a detailed study reported by Wasielewski on perylenediimide dimers.53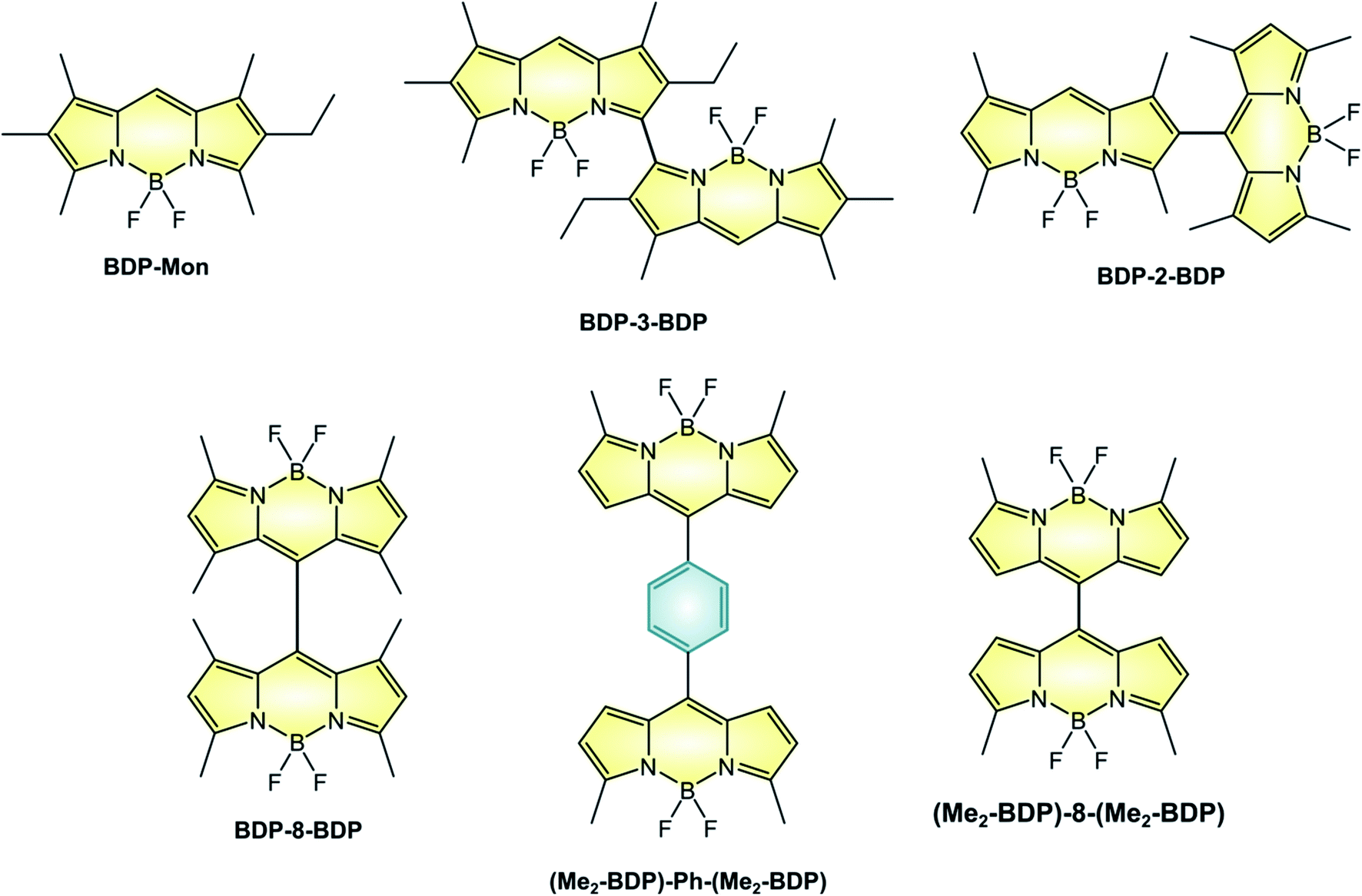 | ||
| Scheme 9 | ||
| Molecule | ε (M−1 cm−1) | λ maxABS (nm) | λ maxFLUO (nm) | Φ FLUO | Φ Δ | E(A/A˙−)f | E(A˙+/A)f |
|---|---|---|---|---|---|---|---|
| a In toluene. b In CHCl3. c In DCM. d In MeCN. e Chemically irreversible electron transfer process. f In V vs. SCE. | |||||||
| BDP-Mon 54 | 82000 |
534 | 540 | 100% | ≤ 0.1 | — | — |
| BDP-3-BDP 54 | 64400 |
492 | 648 | 71% | 40% | — | — |
| 73600 |
565 | ≪10%d | |||||
| BDP-2-BDP 55 | n.r. | 515 | 588 | 3% | 46% | — | — |
| BDP-8-BDP 55 | n.r. | 514 | 527 | 31% | 51% | — | — |
| (Me2-BDP)-Ph-(Me2-BDP) 56 | n.r. | 515 | 538 | 9.7% | n.r. | −0.91c | +1.40ce |
| (Me2-BDP)-8-(Me2-BDP) 56 | n.r. | 526 | 585 | 35% | n.r. | −0.73 | +1.42ce |
| −0.92c | +1.60ce | ||||||
Not all dimers are characterized by exciton coupling. For example, BDP-8-BDP (in which the two units are linked in the meso-position) and BDP-2-BDP (linked in position 2) possess absorption and emission spectra that closely relate to those of monomeric BODIPYs, but are nonetheless able to sensitize singlet oxygen with quantum yields of 46% and 51%, respectively. Therefore, the position at which the two moieties are linked does not play a major role in the efficiency of population of the triplet state, as long as orthogonality is secured. Compared to BDP-8-BDP, (Me2-BDP)-Ph-(Me2-BDP) possess more rotational freedom of the constituent units and therefore lower values of ΦFLUO, with that of the former being lower than that of the latter because of the smaller steric hindrance of the meso substituent. For these two dimers, population of a CT state was proven by femtosecond transient absorption spectroscopy, but the efficiency of triplet population was not studied. Interestingly, only one reversible reduction is observed for (Me2-BDP)-Ph-(Me2-BDP) whereas two separate reductions are seen for (Me2-BDP)-8-(Me2-BDP), as reported in Table 6. In fact, the two BODIPY units in (Me2-BDP)-8-(Me2-BDP) are much closer to each other and strongly interacting.
4. Rational design and synthesis of BODIPYs dyes with tailored properties
The strong interest in the properties and applications of BODIPY dyes has stimulated many successful synthetic strategies§ for their effective preparation. The chemistry of BODIPYs has been carefully discussed by Burgess et al.,7 by Ziessel et al.57 and a recent review has been published by Boens, Dehaen et al.58In a quite simplified discussion, the standard synthetic methodology is an acid-catalyzed condensation of pyrrole derivatives with carbonyl compounds, followed by a boron chelation step of the nitrogen atoms. One of the most efficient approaches for the acid promoted condensation was reported by Lindsey.59 The condensation of an aldehyde with two equivalents of a 2-substituted pyrrole derivative first forms a symmetric dipyrromethane. To avoid polycondensation, when unsubstituted pyrroles are used, a large excess of pyrrole is employed. The dipyrromethane intermediate is then oxidized by quinones (p-chloranil or DDQ) to dipyrromethene, that is transformed into the desired BODIPY by an amine mediated condensation with BF3.
An alternative strategy that avoids the oxidation step is the condensation between substituted pyrrole and an acyl chloride, that produces directly the dipyrromethene.60 An orthoester, in place of the acid chloride, can also be employed.61 These three strategies are well suitable for precursors to BODIPY dyes described in this review. Modular approaches and post functionalization are widely used for the decoration of BODIPY dyes obtained with the presented standard methodologies.58,62
In this paragraph, we will report the approaches relevant for the synthesis of the three classes of BODIPY dyes discussed in this review, namely halogenated BODIPY, BODIPY dyads and dimers. Briefly, the simple post-functionalization by halogenation of BODIPY precursors is used for halogenated BODIPYs. The approach towards BODIPY dyads involves the construction of the BODIPY core incorporating the appropriate meso substituents. In the case of BODIPY dimers, either post-functionalization of the BODIPY monomer or the construction of the linked framework is considered for the synthetic approach (Scheme 10).
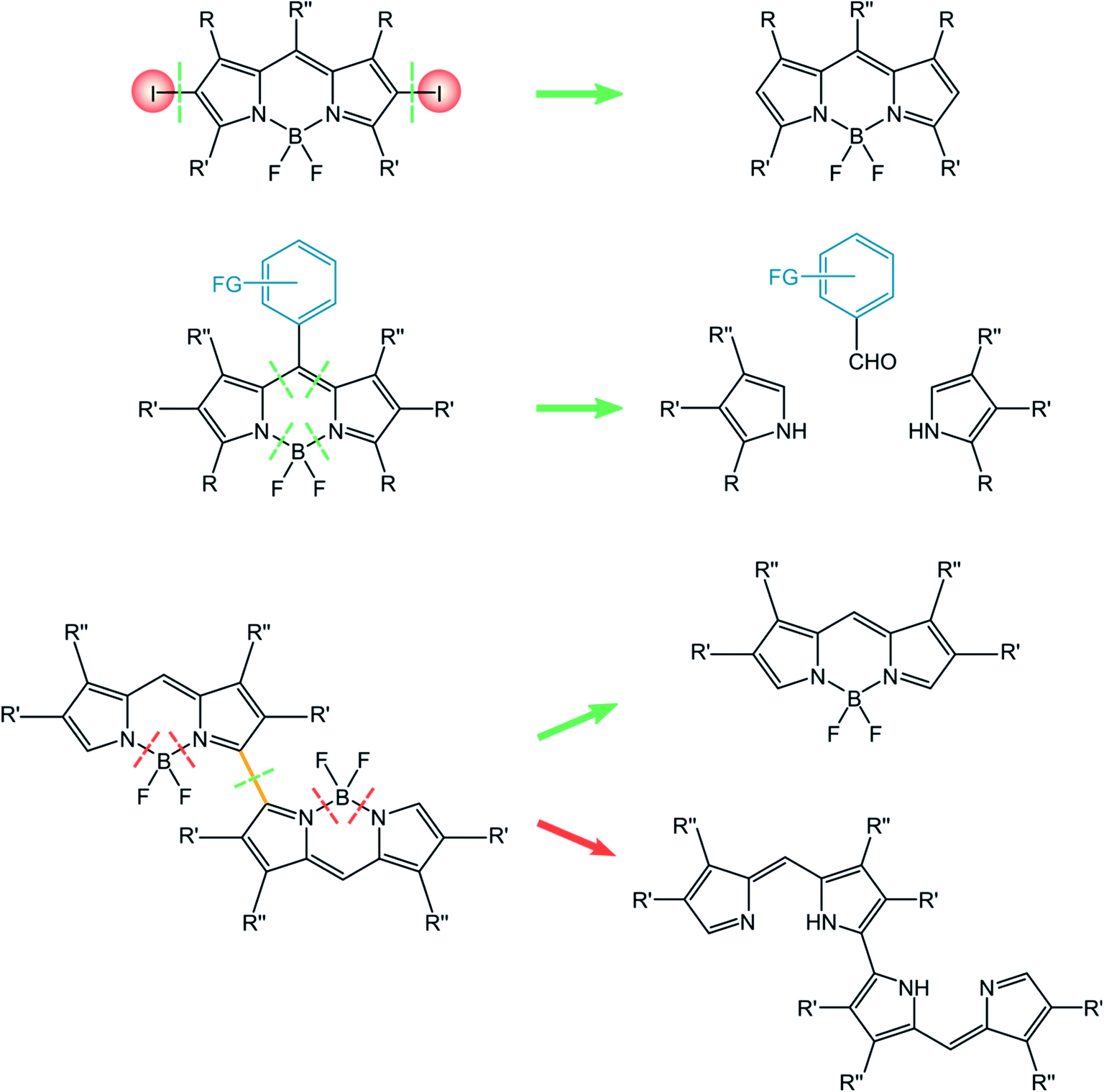 | ||
| Scheme 10 | ||
The approach towards halogenated BODIPYs is simple and direct, and consists in the halogenation of the BODIPY parent compound.63 The possibility of introducing halogens directly onto BODIPY dyes is due to their electron-rich core and to electrophilic aromatic substitutions. Halogenations can be performed by using Br2,64 or HI,34 but milder conditions use N-halosuccinimide (NXS, NCS, NBS, and NIS).65 Moreover, Zhang described halogenated BODIPYs for which the reaction is performed in fluorinated alcohols (1,1,1,3,3,3-hexafluoro-2-propanol (HFIP)) as the solvent in the presence of NXS66 or for which the reaction involves the use of CuBr2 in the presence of oxygen.67I2-BDP-Mes, I2-BDP-Py, and I2-BDP-PhOH were obtained using these approaches. Derivatives I-BDP-tol, I2-BDP-tol, I3-BDP-tol, and I4-BDP-tol can be prepared by using iodine monochloride in methanol or iodine/iodic acid starting from the corresponding BODIPY derivative.38 Derivative I2-BDP-Py was prepared by using iodine/iodic acid,34 while derivative I2-BDP-PhOH was prepared using N-iodosuccinimide.42
BODIPY dyads described in the review are generally obtained by the above-mentioned condensation reactions. All the pyrene derivatives BDP-Pyr, R2-BDP-Pyr, R4-BDP-Pyr, and R6-BDP-Pyr were obtained via the general condensation methodology of the corresponding pyrrole derivatives with pyrene carboxaldehyde.50 The other dyads BDP-Ph(OMe)3 (ref. 45) and BDP-ANPh (ref. 48) can be simply obtained via acid condensation with the appropriate aldehyde. BDP-PTZ (ref. 49) is synthesized thorough the BF3 catalyzed condensation of 10-butyl-10H-phenothiazine-2-carbaldehyde with 2,4-dimethyl-pyrrole, followed by standard reactions used to prepare BODIPYs. BDP-Ph-PTZ,49 was prepared starting from 2-bromo-10-butyl-10H-phenothiazine (1) by Suzuki-type coupling reaction with BDP-Ph-dioxaborolane (2) (Scheme 11).
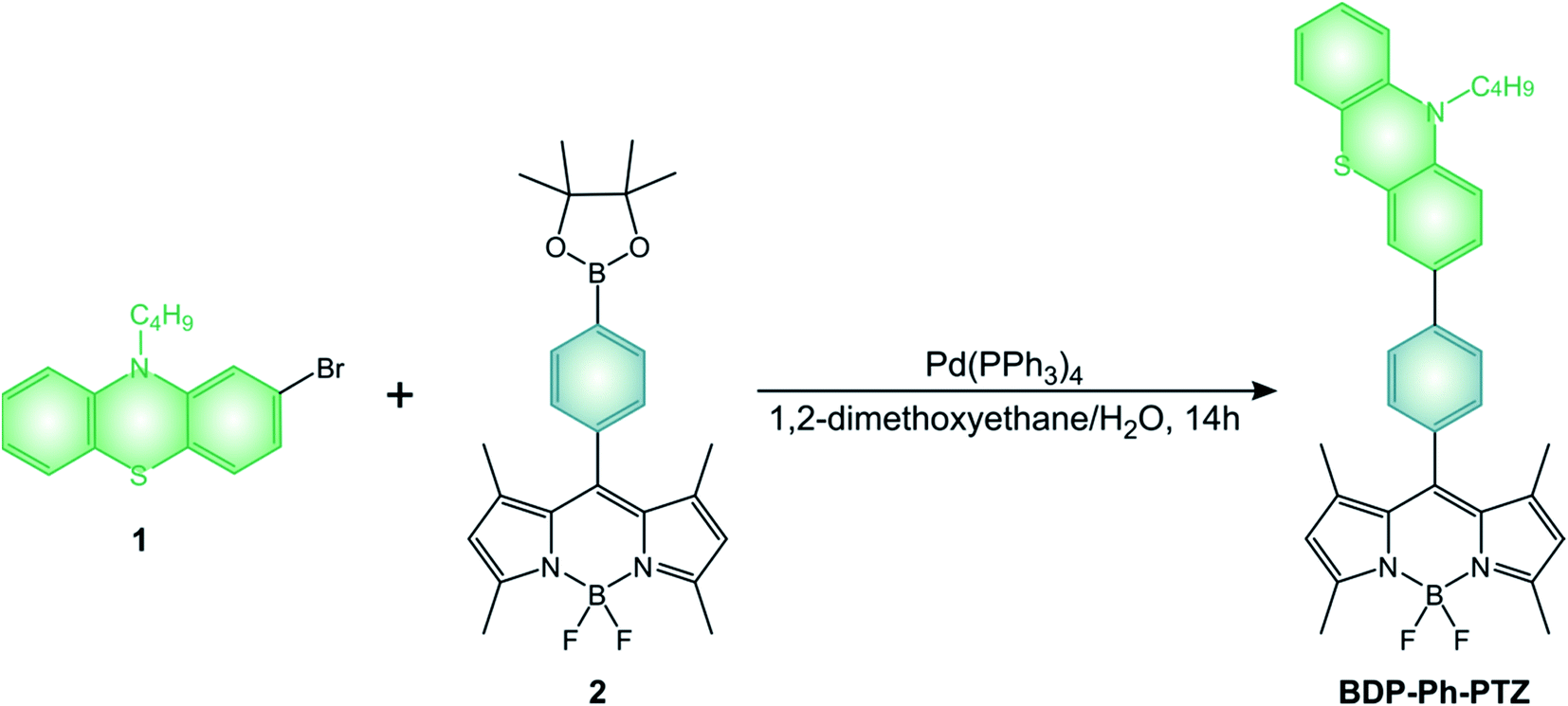 | ||
| Scheme 11 | ||
The dyads described in the review were obtained by the following strategies: BDP-3-BDP was synthesized68 by addition of BF3 to the corresponding tetrapyrrole derivative; BDP-2-BDP was obtained via a Vilsmeier formylation of the related BODIPY,55 followed by the standard synthetic strategy to access BODIPYs (namely, the condensation of the pyrrole derivative in the presence of TFA, oxidation with chloranil, and complexation with BF3). Compound BDP-8-BDP was prepared with an analogue strategy,55 starting from the meso formyl BODIPY derivative. Molecule (Me2-BDP)-Ph-(Me2-BDP) was synthesized56 by acid-catalysed condensation of terephthalaldehyde with four equivalents of 2-methylpyrrole, followed by the standard complexation procedures. Finally, (Me2-BDP)-8-(Me2-BDP) was prepared56 by reaction of 2-methylpyrrole with meso-formyl BODIPY, followed by oxidation with DDQ and complexation with BF3.
The preparation of BODIPY dimers requires quite elaborated synthetic procedures. One interesting strategy for the preparation of dimeric compounds relies on the oxidative dimerization with oxidants like [bis(trifluoroacetoxy)iodo]benzene (PIFA).69 Alternative approaches use electrochemistry,70 or stoichiometric Fe(III) salts.71 A quite interesting and innovative approach was recently reported72 and involves the photoinduced, Ag(I)-promoted oxidative dimerization of BODIPY dyes. This methodology does not require a strong oxidant, and is promoted by a PeT involving the excited BODIPY dye that forms a persistent radical species able to dimerize in the reaction conditions.
5. Applications in energy conversion, photocatalysis and medicine
We will discuss the applications of BODIPY triplet photosensitizers in energy conversion processes, including energy upconversion and photocatalysis for the production of chemicals and solar fuels. Finally, we will briefly discuss a medical application, namely photodynamic therapy. Halogenated BODIPYs have been extensively studied for this purpose, and readers interested in their application can refer to previously published reviews.4,73,74 Here we will report only the most recent examples which employ different strategies to populate the sensitiser's triplet state.Energy upconversion by sensitized triplet–triplet annihilation (TTA)
Energy upconversion is the process by which incident photons of a given energy are converted into photons of higher energy. It can be realized by a variety of mechanism, among which sensitized triplet–triplet annihilation is gaining increasing attention because it does not require coherent excitation sources and it can be conveniently performed by low power excitation.75–77 For that reason, it is now an emerging technique with possible applications in different fields: wavelength shifting for spectroscopy, sensitized photoreaction by low-energy photons, photovoltaic devices, and luminescent probes for bioimaging.The process can be exemplified by considering two chromophores displaying singlet and triplet excited states, where A molecules absorb lower energy photons and B molecules emit higher energy photons according to the following processes:
| A(S0) + hν → *A(S1) | (1) |
| *A(S1) → *A(T1) | (2) |
| *A(T1) + B(S0) → A(S0) + *B(T1) | (3) |
| *B(T1) + *B(T1) → *B(S1) + B(S0) | (4) |
| *B(S1) → B(S0) + hν′ | (5) |
The energy requirements for A and B are that: (i) the S1 excited state of B is at higher energy than that of A, while the corresponding T1 excited states with the reverse order, (ii) combined triplet energy of *B(T1) greater or equal to the energy of *B(S1). Process (3) and (4) are bimolecular processes, namely the sensitized population of T1 excited state of B by energy transfer from A and triplet–triplet annihilation of B molecules.
BODIPY dyes have been reported in both roles, either absorbing (A) or emitting (B) molecules,78,79 but, in the present case, we focus on BODIPY dyes acting as A molecules and sensitizing the population of the T1 excited state of B molecules. In this context, BODIPY dyes have the undoubted advantage of a large molar absorption coefficient, maximizing the probability of step (1) and high photostability, reducing side photoreactions. They have been reported to play the role of the sensitizer when perylene is used as the triplet energy annihilator.48,80 For example, BDP-PTZ's ability to populate its triplet excited state has been exploited to convert green laser excitation radiation to perylene's typical blue emission (Fig. 5).49 As expected, the fluorescence of perylene is delayed and the upconversion quantum yield is 3.2%. In comparison, the use of BDP-Ph-PTZ leads to a reduction of the quantum yield (ΦUP = 0.6%).49 In fact, in this case the efficiency of step (2) is reduced, since the phenyl spacer decreases the efficiencies of both PeT and SOCT-ISC, as described in Section 3.
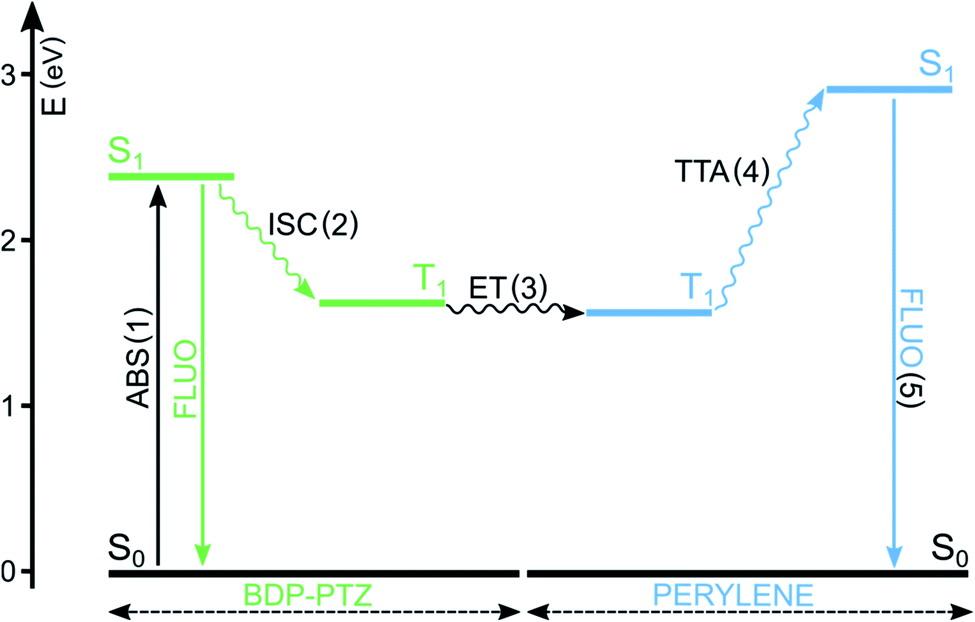 | ||
| Fig. 5 Simplified Jablonski diagram illustrating the main excited states' energy levels and processes involved in TTA upconversion. In this example, A = BDP-PTZ and B = perylene. | ||
An interesting example is that of derivative (I2-BDP)-BDP (Scheme 12), that comprises a fluorescent BODIPY chromophore and an iodinated BODIPY one linked to form a dyad.81 Thanks to this peculiarity, a broader range of the visible spectrum can be absorbed compared to the 2,6-diiodinated reference monochromophoric compound (I2-BDP-Ph), since iodination on the BODIPY core is known to red-shift the absorption band. Moreover, both units' triplet states can be populated due to the feasibility of a triplet–triplet energy transfer from the iodinated moiety to the adjacent fluorescent one. While the broader absorption in the visible spectrum correlates to step (1) of TTA upconversion, this second feature increases the probability of step (3), given that it increases the sensitiser's triplet excited state lifetime. Consequently, the upconversion quantum yield is enhanced as well (ΦUP = 8.1% for (I2-BDP)-BDP; ΦUP = 7.5% for I2-BDP-Ph). Additionally, by multiplying the molar absorption coefficient of the photosensitiser at the irradiation wavelength and the upconversion quantum yield, it is possible to compare the performances of different sensitisers. For example, the value obtained for a platinum complex82 (1140, with ε = 11400 M−1 cm−1 and ΦUP = 10%) is about five times smaller than that obtained for (I2-BDP)-BDP (5500, with ε = 67900 M−1 cm−1 and ΦUP = 8.1%).81
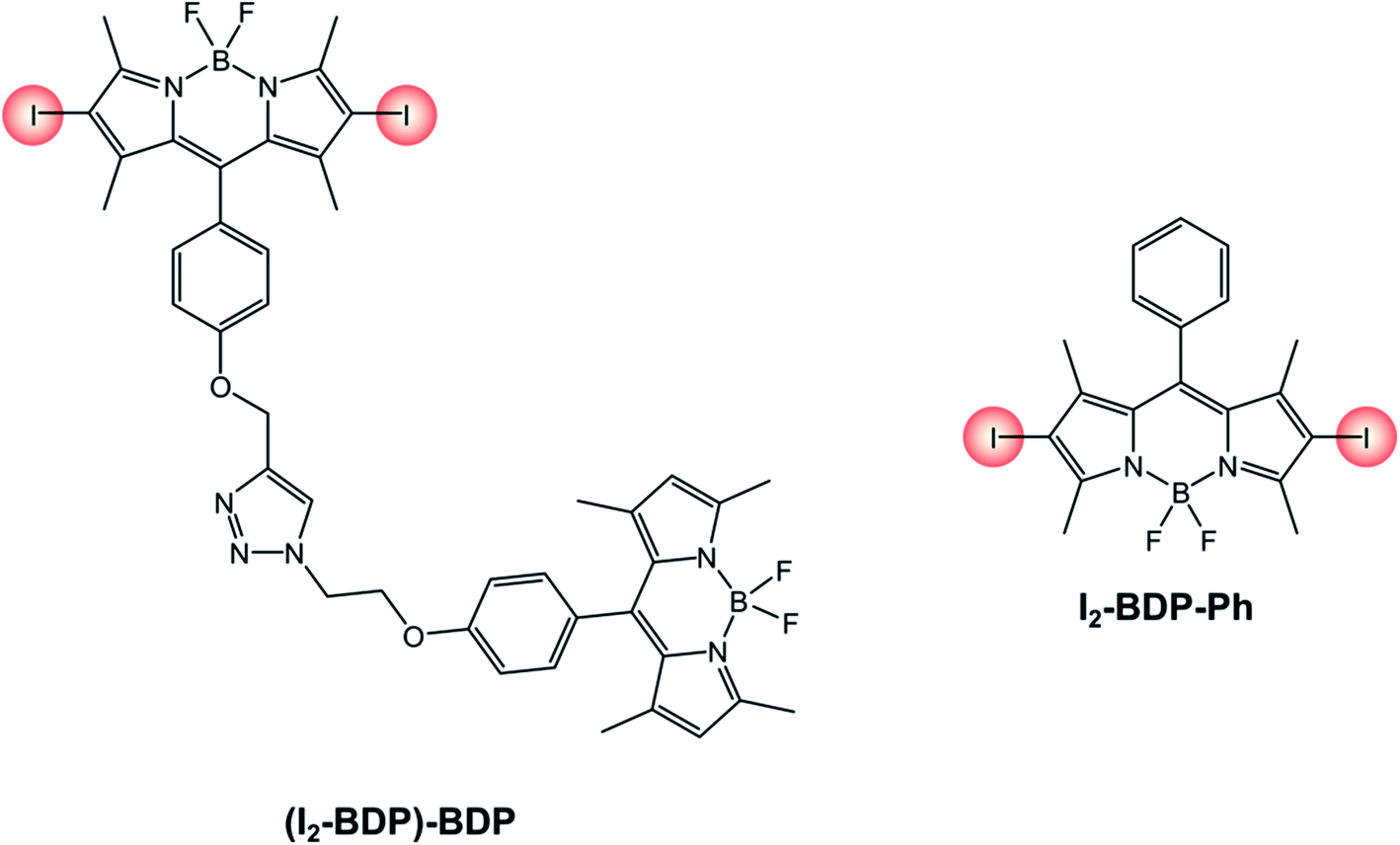 | ||
| Scheme 12 | ||
Photocatalysis
Photocatalysis exploits light absorption to increase reaction rates, efficiencies or to pave the way for reaction pathways that would not otherwise be accessible.In the field of photocatalysis, 2,6-halogenated BODIPYs have been employed to carry out a variety of reactions.83 The most common photochemical mechanisms of BODIPY triplet sensitizers are the following: (i) photo-oxidation due to sensitized singlet oxygen population by BODIPY excitation; (ii) photoredox reaction by oxidative or reductive quenching of BODIPY lowest-energy triplet state T1. The former is the most widely investigated approach and, besides an efficient population of T1 excited state, it requires a high resistance to photobleaching by 1O2. Conversely, in the second case this is not a major concern. In fact, the presence of oxygen must be avoided since it would quench the BODIPY's long-living triplet excited state and would re-oxidize the reduced form of the photosensitizer in the event of a reductive quenching. However, it's important to keep in mind that when photoredox events need to take place, the photosensitizer is not only required to efficiently undergo ISC but also to satisfy electrochemical constraints imposed by the system. This implies that, for this type of reactions, electrochemical properties need to be taken into consideration more carefully.
I2-BDP-Ph has been employed for the singlet oxygen-mediated oxidation of thioanisole39 and the brominated analogue has found application for aerobic oxidative amidations.84I2-BDP-Ph has also been utilized for the oxidative coupling of amines (Scheme 13A) and in the one-pot synthesis of N-aryl-2-amino-1,4-naphthoquinones, which are anticancer and antibiotic reagents (Scheme 13B).85 In the latter case, naphthalene derivatives are oxidized by 1O2 to 1,4-naphthoquinones and subsequently react with an amine to yield the final products. The yields of this reaction are generally higher with BODIPY as the sensitiser than with tetraphenylporphyrin, probably due to the better absorption in the visible region of the first compared with the latter. However, a comparison of photoreaction quantum yields would be desirable, but these data are not available in the literature. A similar BODIPY comprising styryl groups, I2-St-BDP-Ph has been used for the same reactions as well: red-shifted absorption allows for the use of longer-wavelength irradiation.85
 | ||
| Scheme 13 | ||
A bifunctional photosensitizer composed of an iodinated BODIPY and a quinine (I2-BDP-quinine, Scheme 14A) has also been employed for the asymmetric photooxygenation of β-dicarbonyl compounds,86 while a functionalized porous organic polymer comprising a 2,6-halogenated BODIPY monomeric unit (I2-BDP-POP, Scheme 14B) has been employed for the photodegradation of chemical warfare agent 2-chloroethyl ethyl sulphide to its respective sulfoxide.87
 | ||
| Scheme 14 | ||
As for the second mechanism involving oxidative or reductive quenching of the T1 excited state of BODIPY dyes, 2,6-halogenated BODIPYs have been extensively employed. I2-BDP-Ph has been used as a sensitizer for the aza-Henry reaction of tetrahydroisoquinoline (Scheme 15A), the oxidation/[3 + 2] cycloaddition/oxidative aromatization tandem reaction between tetrahydroisoquinolines and maleimides (Scheme 15B), and the C–H arylation of heteroarenes with diazonium salts (Scheme 15C).88
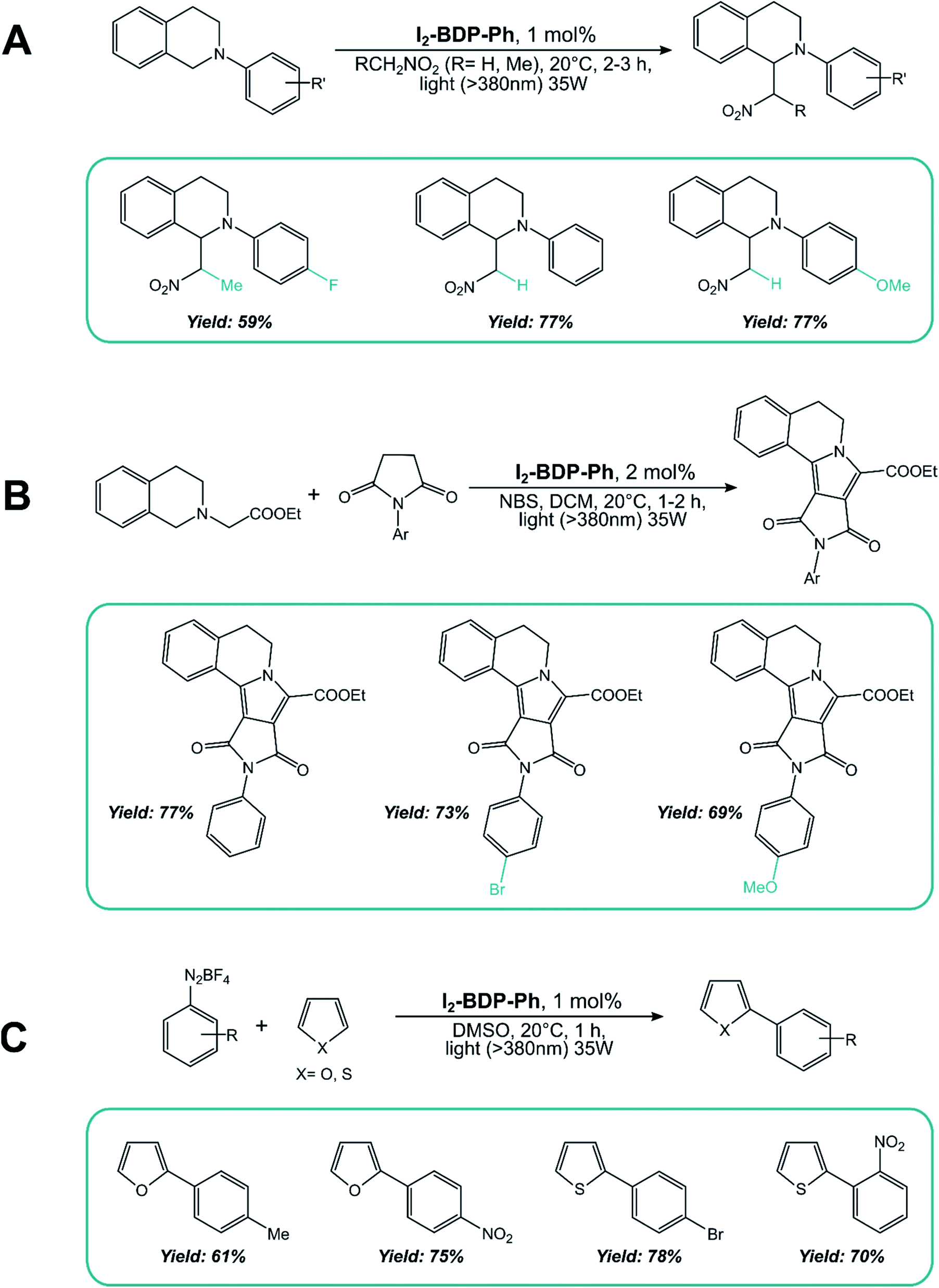 | ||
| Scheme 15 | ||
I2-BDP-Ph has additionally been utilized as a photocatalyst to promote ATRA (Atom-Transfer Radical Addition) reactions between various bromoderivatives and different alkenes (Scheme 16).89 The interaction between the triplet excited state of the BODIPY derivative with possible reaction partners was studied by Stern–Volmer experiments, which revealed a reductive quenching by sodium ascorbate. The so-obtained reduced form of the photocatalyst is a sufficiently strong reductant to reduce the organic halide, a step which triggers the sequence presented by Stephenson90 that eventually leads to the ATRA product.
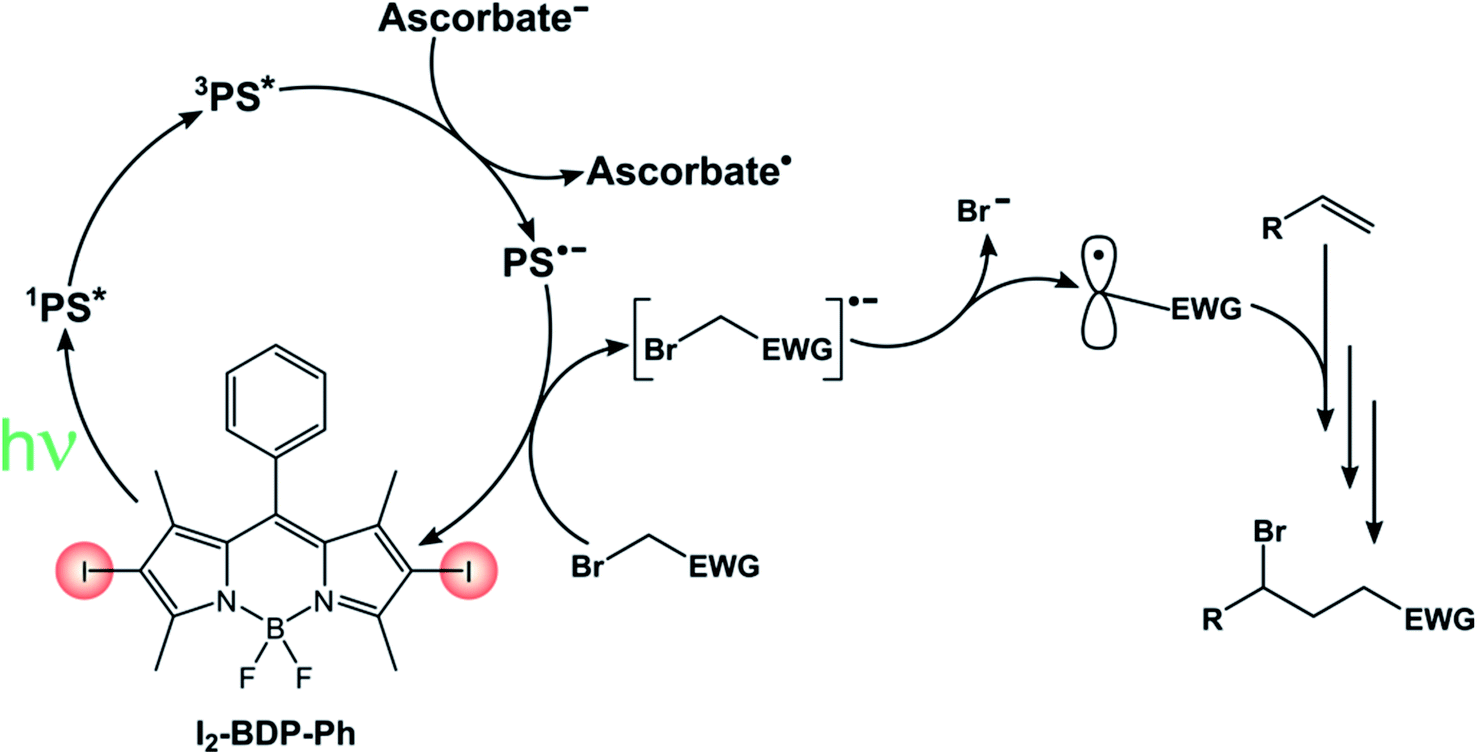 | ||
| Scheme 16 | ||
Moreover, halogenated BODIPYs (Scheme 17) have very recently been employed to promote light-driven polymerizations, since they allow for the utilization of a lower catalyst loading and lower light intensity compared to current UV photocuring methods.91 For example, a photocurable resin containing Br2-BDP-Mes (the photosensitizer), N,N-dimethyl acrylamide (the monomer) and trimethylolpropane triacrylate (the crosslinker) was employed to demonstrate the feasibility of 3D-printing a complex lattice using a green LED.
 | ||
| Scheme 17 | ||
Solar fuels
The sustainable production of energy is one of the biggest challenges mankind is currently facing. A very appealing, but extremely challenging option is the conversion of sunlight into chemical energy, mimicking natural photosynthesis to obtain the so-called solar fuels. Within this field, many researchers focused their attention to photosensitized water splitting into molecular oxygen and hydrogen, which can be used as an energy carrier.In this sense, much effort is being devoted towards the study of green systems for photocatalytic hydrogen production and BODIPY dyes have been used to replace heavy metal-based photosensitizers.92–94 For instance, I2-BDP-Py has been employed to sensitize a cobaloxime catalyst in a single-component system whose turnover number is 82.41 Following reductive quenching of *PS3 by TEOA (the sacrificial electron donor), a rapid electron transfer from PS˙− to cobaloxime can take place thanks to the complexation of the BODIPY sensitiser to the catalyst. Upon absorption of a second photon, this process is repeated again and the catalyst eventually reaches the oxidation state that enables it to evolve hydrogen. On the contrary, if BDP-Py is used no hydrogen is produced since its inability to populate its triplet state does not allow quenching by TEOA to take place. Binding of I2-BDP-Py with a hydrogen evolution catalyst offers the advantage of suppressing undesired side-reactions involving PS˙−, since it can be quickly restored to PS. However, in the case of cobaloxime catalysts the lability of the N–Co(II) bond that forms during the catalytic cycle needs to be considered as well.95,96 For example, by increasing the basicity of the pyridyl linker it is possible to improve the TON by a factor of three.92 These complications considered, untethering the BODIPY sensitiser from the cobaloxime catalyst has been demonstrated to considerably increase the TONs of these systems (TON up to 670 for I2-BDP-mes, Scheme 18).96 Given its bimolecular nature, the components' ratio needs to be carefully tuned so that (i) the catalyst concentration is not too low, to avoid photosensitiser's bleaching due to degradation of PS˙− and (ii) the catalyst concentration is not too high, to minimize the induction period needed to convert Co(III) to Co(II) before starting hydrogen evolution.
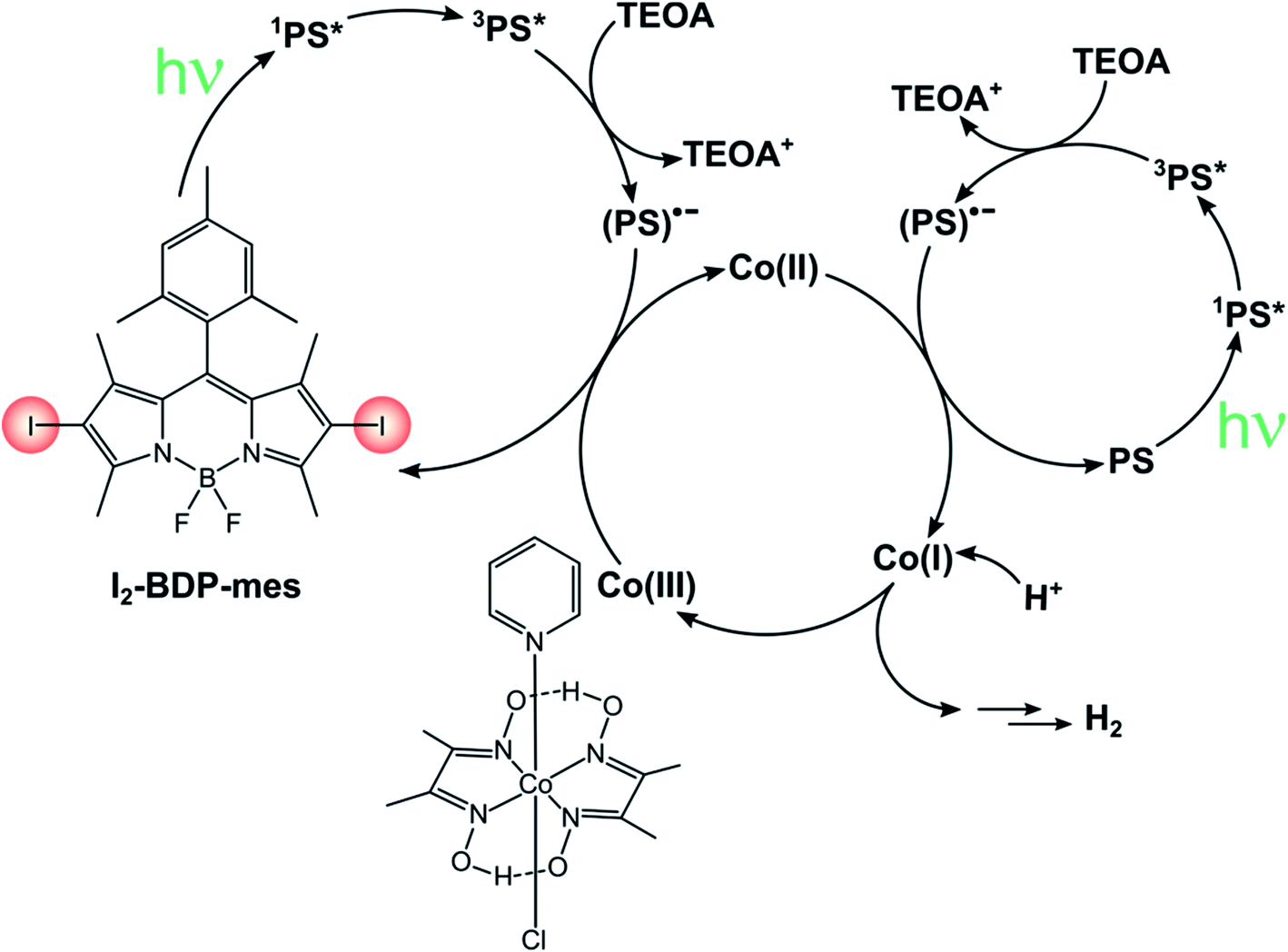 | ||
| Scheme 18 | ||
A different approach in the production of solar fuels is the use of a BODIPY derivative (albeit unable to undergo ISC alone) as ligand in a multichromophoric iridium complex.97 This strategy exploits the strong absorption of BODIPY dyes in the visible region, allowing for a lower amount of scarce iridium to be used. When coupled with a similar cobaloxime catalyst as that shown in Scheme 18 and with N,N-dimethyl-p-toluidine as the sacrificial electron donor, the turnover number reaches the exceptionally high value of 1.16 × 105.
Furthermore, their efficient absorption of green light and tunability of their absorption maxima make BODIPY dyes ideal candidates to construct light-harvesting antennae. For example, BODIPY-based artificial antennae have been demonstrated to sensitize photocurrent in amorphous silicon98 and NIR-absorbing derivatives have been employed in dye-sensitised solar cells (DSSC).99
Photodynamic therapy
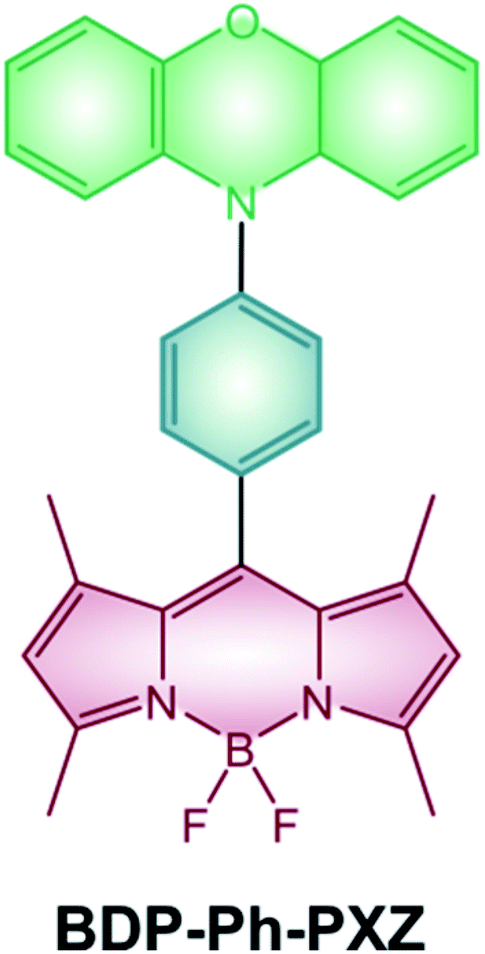
Photodynamic therapy (PDT) is a strategy developed to treat tumors and other proliferative pathologies by the combined use of light, oxygen and a photosensitizer: cytotoxic singlet oxygen is locally produced thanks to the initial absorption of light by the sensitizer and subsequent interaction of its triplet excited state with ground-state molecular oxygen.Halogenated BODIPYs have been widely studied and reviewed4,73,74 as emerging agents for PDT given that they use the most established strategy to efficiently populate their triplet excited state. Moreover, the possibility of red-shifting BODIPYs' absorption maxima to the NIR region (e.g. by addition of styryl groups or extension of the π-system) makes them promising candidates for this application.100,101 Unfortunately, the presence of heavy atoms, although beneficial for an efficient ISC, leads to a quite high dark toxicity which can undesirably damage healthy tissues. Conversely, BODIPY dyads and dimers have only recently started to be investigated in the field of photodynamic therapy and compared to halogenated BODIPYs benefit from higher light-to-dark cytotoxicity ratios. For example, dimer BDP-2-BDP (Scheme 9) is able to populate its triplet states with a good quantum yield and shows cytotoxicity towards K562 human erythroleukemia cells upon irradiation with a green LED.55 On the other hand, cells kept in the dark in the presence of the dimer did not shown any significant change in their viability. Dyad BDP-Ph-PXZ (Scheme 19) has also demonstrated cytotoxic activity towards HeLa cells and has been used to reduce tumor growth in a mice.102
 | ||
| Scheme 19 | ||
6. Future perspectives
If we look at the development of BODIPY chemistry in a historical perspective (Fig. 6), the first BODIPY dye, namely 4,4-difluoro-4-bora-3a, 4a-diaza-s-indacene, was synthesized by Treibs and Kreuzer in 1968 × 103 In 1989, BODIPY was used as a fluorescent probe for bioactive ligands for the first time.104 Since then, a variety of applications have been devised for fluorescent BODIPYs, spanning from bioimaging to sensing and optoelectronic devices. | ||
| Fig. 6 Historical development of BODIPY dyes. | ||
More recently, BODIPYs have emerged as viable alternatives to transition metal complexes as triplet photosensitizers, being characterized by strong and easily tunable absorption in the visible spectral region and high photostability. The first examples were halogenated BODIPY dyes reported by Nagano in 2005. Since then, many halogenated derivatives have been explored and employed in diverse applications such as photodynamic therapy, photocatalysis and solar fuel production. More recently, BODIPY dimers and dyads, where electron-accepting BODIPY dyes are linked to electron donating units, have emerged. Most of these studies are mainly focused on the photophysical properties and their relationship to the molecular structure. Some applications are emerging, e.g. in the photodynamic therapy. We expect an increasing utilization of these systems for a variety of applications in the next future.
A comparison of the three approaches reported in the present review enable us to derive important conclusions for future developments.
• Halogenation of the BODIPY core leads to the highest efficiency of T1 population, but strongly affects the electrochemical properties, shifting the reduction potential to less negative values. On the other hand, meso-linked substituents play a minor role on the electrochemical properties, but they are much less effective in inducing spin–orbit coupling by heavy-atom effect because HOMO and LUMO orbitals are not significantly extended to these substituents.
• Dyad photosensitizers couple quite efficient triplet population with strongly negative reduction potential of the BODIPY core. This is made viable by a proper choice of the substituents on the pyrrolic rings of the BODIPY skeleton and of proper electron-donating moiety attached to the meso position. By this approach, the T1 excited state can be engineered to be a strong reductant, an outstanding property for photocatalytic applications in organic reactions and solar fuel production.
• Dimers are the least explored class of triplet photosensitizers; the optical properties are modified compared to the monomer species when exciton coupling occurs, but most of the reported examples commonly preserve the monomer's optical properties and can populate the T1 state quite efficiently via symmetry-breaking charge transfer state. Although their synthesis is more demanding, it is worth noting that a recently published photodimerization procedure can pave the way to more easily accessible dimers. Besides the above mentioned symmetry-breaking charge transfer states, some authors105 invoke singlet fission, a process in which a chromophore in an excited singlet state shares its excitation energy with a neighboring ground-state chromophore and both are converted into triplet excited states, as the main mechanism to populate the T1 excited state in these dimers. However, the topic is still controversial106 and detailed studies are needed to clarify this aspect.
Photostability is, of course, of the utmost importance for a photosensitizer. In this regard, BODIPY dyes are generally reported to have high photostability.3,4,34 Iodinated BODIPY dyes are the most susceptible to photodegradation, but literature reports show that they are, in any case, better performing than gold-standard triplet photosensitisers as rose Bengal107 and, because of that, they are currently attracting increasing interest for photodynamic therapy.101 When BODIPY dyes are involved in electron transfer processes, the chemical stability of the so-formed radical cation or anion is a key requisite: the presence of alkyl substituents in the positions 2,3,5 and 6 improves their chemical stability, preventing dimerization/polymerization processes.5
Rather unexplored aspects that may influence the triplet excited state population are: (i) the effect of aggregation in solid state or confined environments compared to solution studies,108 reminiscent of the co-facial π-stacked arrangements of chromophores found in nature (e.g., photosynthetic reaction center); (ii) the radical pair induced ISC that is expected to have potential applications in the field of solar energy conversion; (iii) the switching behavior of BODIPY dyads and dimers by external stimuli that can modify the energy of the charge transfer state; (iv) the shift of BODIPY absorption to the red and near-infrared spectral region coupled to efficient triplet population with potential applications in biomedicine.100
In conclusion, the present challenge in the development of light energy conversion and photocatalysis is to design organic dyes which couple (i) strong visible absorption, (ii) efficient population of a long-lived triplet excited state T1 and (iii) strong reducing and/or oxidizing power. BODIPY dyes are ideal and unrivaled candidates for such purposes. To better evaluate their performance in comparison to other organic or metal-based triplet photosensitizers, it is worth estimating the product of the molar absorption coefficient at the excitation wavelength and the intersystem crossing quantum yield (or an associated value), in analogy with the brightness defined for luminescent molecules as the product of the molar absorption coefficient at the excitation wavelength times the luminescence quantum yield. For instance, if quantum yields of singlet oxygen formation are used, the value for I2-BDP-tol (at 548 nm) is five times greater than that of the most famous metal-based triplet photosensitizer [Ru(bpy)]32+ (at 452 nm). The higher the estimated value, the higher the efficiency of the process at the same photosensitizer concentration.
To further improve BODIPY optical properties, there is a clear need of a deeper understanding of the relationship between the molecular structure and the efficiency of triplet excited state population, above all for the last two classes of compounds. This will enable the design of optimized systems with tailored optical and electrochemical properties and the emergence of BODIPY applications in diverse fields. Furthermore, these studies may have important implications for the design of other classes of organic chromophores as triplet photosensitizers: as briefly outlined in the first section, the same approaches can be extended to other classes of organic chromophores.
Author contributions
A. G. and P. G. C. contributed to Section 4 and part of Section 5, and provided input and feedback to the whole article. E. B. and P. C. conceived and wrote the rest of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Bologna and national PRIN 2017 project (ID: 20174SYJAF, SURSUMCAT) are acknowledged for financial support of this research.References
- M. E. El-Khouly, S. Fukuzumi and F. D'Souza, ChemPhysChem, 2014, 15, 30–47 CrossRef CAS PubMed.
- L. Y. Niu, H. Li, L. Feng, Y. S. Guan, Y. Z. Chen, C. F. Duan, L. Z. Wu, Y. F. Guan, C. H. Tung and Q. Z. Yang, Anal. Chim. Acta, 2013, 775, 93–99 CrossRef CAS PubMed.
- T. Kowada and K. Kikuchi, Chem. Soc. Rev., 2015, 4953–4972 RSC.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- A. B. Nepomnyashchii and A. J. Bard, Acc. Chem. Res., 2012, 45, 1844–1853 CrossRef CAS PubMed.
- M. Poddar and R. Misra, Coord. Chem. Rev., 2020, 421, 213462 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- S. Ji, J. Ge, D. Escudero, Z. Wang, J. Zhao and D. Jacquemin, J. Org. Chem., 2015, 80, 5958–5963 CrossRef CAS PubMed.
- R. L. Watley, S. G. Awuah, M. Bio, R. Cantu, H. B. Gobeze, V. N. Nesterov, S. K. Das, F. D'Souza and Y. You, Chem. – Asian J., 2015, 10, 1335–1343 CrossRef CAS PubMed.
- S. Qi, N. Kwon, Y. Yim, V. N. Nguyen and J. Yoon, Chem. Sci., 2020, 11, 6479–6484 RSC.
- J. Sun, F. Zhong, X. Yi and J. Zhao, Inorg. Chem., 2013, 52, 6299–6310 CrossRef CAS PubMed.
- Y. Dong, B. Dick and J. Zhao, Org. Lett., 2020, 22, 5535–5539 CrossRef CAS PubMed.
- Z. Wang, L. Huang, Y. Yan, A. M. El-Zohry, A. Toffoletti, J. Zhao, A. Barbon, B. Dick, O. F. Mohammed and G. Han, Angew. Chem., Int. Ed., 2020, 59, 16114–16121 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, California, 2010 Search PubMed.
- V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics. Concepts, Research, Applications, Wiley, Weinheim, Germany, 2014 Search PubMed.
- P. Klán and J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice, Wiley, Chichester, United Kingdom, 2009 Search PubMed.
- J. W. Arbogast, A. P. Darmanyan, C. S. Foote, F. N. Diederich, R. L. Whetten, Y. Rubin, M. M. Alvarez and S. J. Anz, J. Phys. Chem., 1991, 95, 11–12 CrossRef CAS.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J. L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS PubMed.
- M. Sapir and E. Vander Donckt, Chem. Phys. Lett., 1975, 36, 108–110 CrossRef CAS.
- R. Kumar, H. Aggarwal and A. Srivastava, Chem. – Eur. J., 2020, 10653–10675 CrossRef CAS PubMed.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis Group, Boca Raton, United States, III., 2006 Search PubMed.
- X. F. Zhang, I. Zhang and L. Liu, Photochem. Photobiol., 2010, 86, 492–498 CrossRef CAS PubMed.
- E. A. Weiss, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2003, 107, 3639–3647 CrossRef CAS.
- K. Hasharoni, H. Levanon, S. R. Greenfield, D. J. Gosztola, W. A. Svec and M. R. Wasielewski, J. Am. Chem. Soc., 1995, 117, 8055–8056 CrossRef CAS.
- E. A. Weiss, M. J. Ahrens, L. E. Sinks, A. V. Gusev, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 5577–5584 CrossRef CAS PubMed.
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed.
- N. Rehmat, A. Toffoletti, Z. Mahmood, X. Zhang, J. Zhao and A. Barbon, J. Mater. Chem. C, 2020, 8, 4701–4712 RSC.
- Y. Zhao, R. Duan, J. Zhao and C. Li, Chem. Commun., 2018, 54, 12329–12332 RSC.
- M. Imran, A. A. Sukhanov, Z. Wang, A. Karatay, J. Zhao, Z. Mahmood, A. Elmali, V. K. Voronkova, M. Hayvali, Y. H. Xing and S. Weber, J. Phys. Chem. C, 2019, 123, 7010–7024 CrossRef CAS.
- D. J. Gibbons, A. Farawar, P. Mazzella, S. Leroy-Lhez and R. M. Williams, Photochem. Photobiol. Sci., 2020, 19, 136–158 CrossRef CAS PubMed.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- A. M. Durantini, L. E. Greene, R. Lincoln, S. R. Martínez and G. Cosa, J. Am. Chem. Soc., 2016, 138, 1215–1225 CrossRef CAS PubMed.
- K. Krumova and G. Cosa, J. Am. Chem. Soc., 2010, 132, 17560–17569 CrossRef CAS PubMed.
- M. J. Ortiz, A. R. Agarrabeitia, G. Duran-Sampedro, J. Bañuelos Prieto, T. A. Lopez, W. A. Massad, H. A. Montejano, N. A. García and I. Lopez Arbeloa, Tetrahedron, 2012, 68, 1153–1162 CrossRef CAS.
- W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417–13421 RSC.
- L. Xu, Y. Li, R. Jiang, Z. Qin and Y. Li, Dyes Pigm., 2014, 107, 90–96 CrossRef CAS.
- G. G. Luo, K. Fang, J. H. Wu, J. C. Dai and Q. H. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 23884–23894 RSC.
- C. Zhang, J. Zhao, X. Cui and X. Wu, J. Org. Chem., 2015, 80, 5674–5686 CrossRef CAS PubMed.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Phys. Chem. Chem. Phys., 2018, 20, 8016–8031 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- W. Hu, X. F. Zhang, X. Lu, S. Lan, D. Tian, T. Li, L. Wang, S. Zhao, M. Feng and J. Zhang, J. Lumin., 2018, 194, 185–192 CrossRef CAS.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay, V. K. Voronkova, Z. Wang, M. Ivanov, J. Zhao and D. Donato, J. Phys. Chem. C, 2019, 123, 1–51 Search PubMed.
- S. Hattori, K. Ohkubo, Y. Urano, H. Sunahara, T. Nagano, Y. Wada, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, J. Phys. Chem. B, 2005, 109, 15368–15375 CrossRef CAS PubMed.
- Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492–4495 CrossRef CAS PubMed.
- K. Chen, W. Yang, Z. Wang, A. Iagatti, L. Bussotti, P. Foggi, W. Ji, J. Zhao and M. Di Donato, J. Phys. Chem. A, 2017, 121, 7550–7564 CrossRef CAS PubMed.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606–615 CrossRef CAS.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2019, 5, 138–155 CAS.
- M. Kellogg, A. Akil, D. S. Muthiah Ravinson, L. Estergreen, S. E. Bradforth and M. E. Thompson, Faraday Discuss., 2019, 216, 379–394 RSC.
- K. M. Lefler, K. E. Brown, W. A. Salamant, S. M. Dyar, K. E. Knowles and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 10333–10345 CrossRef CAS PubMed.
- B. Ventura, G. Marconi, M. Bröring, R. Krüger and L. Flamigni, New J. Chem., 2009, 33, 428–438 RSC.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS PubMed.
- M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Chem. Commun., 2012, 48, 284–286 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577–6595 CrossRef CAS.
- R. W. Wagner and J. S. Lindsey, Pure Appl. Chem., 1996, 68, 1373–1380 CAS.
- J. Godoy, G. Vives and J. M. Tour, Org. Lett., 2010, 12, 1464–1467 CrossRef CAS PubMed.
- C. B. Reese and H. Yan, Tetrahedron Lett., 2001, 42, 5545–5547 CrossRef CAS.
- L. Jean-Gérard, W. Vasseur, F. Scherninski and B. Andrioletti, Chem. Commun., 2018, 54, 12914–12929 RSC.
- V. Lakshmi, M. Rajeswara Rao and M. Ravikanth, Org. Biomol. Chem., 2015, 13, 2501–2517 RSC.
- M. Shah, K. Thangaraj, M. -L. Soong, L. T. Wolford, J. H. Boyer, I. R. Politzer and T. G. Pavlopoulos, Heteroat. Chem., 1990, 1, 389–399 CrossRef CAS.
- G. Duran-Sampedro, A. R. Agarrabeitia, I. Garcia-Moreno, A. Costela, J. Bañuelos, T. Arbeloa, I. López Arbeloa, J. L. Chiara and M. J. Ortiz, Eur. J. Org. Chem., 2012, 6335–6350 CrossRef CAS.
- L. Wang, J. W. Wang, A. J. Cui, X. X. Cai, Y. Wan, Q. Chen, M. Y. He and W. Zhang, RSC Adv., 2013, 3, 9219–9222 RSC.
- J.-H. Ye, G. Wang, C. Huang, Z. Hu, W. Zhang and Y. Zhang, Synthesis, 2012, 44, 104–110 CrossRef CAS.
- M. Bröring, R. Krüger, S. Link, C. Kleeberg, S. Köhler, X. Xie, B. Ventura and L. Flamigni, Chem. – Eur. J., 2008, 14, 2976–2983 CrossRef PubMed.
- S. Rihn, M. Erdem, A. De Nicola, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 1916–1919 CrossRef CAS PubMed.
- A. B. Nepomnyashchii, M. Bröring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 19498–19504 CrossRef CAS PubMed.
- L. Gai, H. Lu, B. Zou, G. Lai, Z. Shen and Z. Li, RSC Adv., 2012, 2, 8840–8846 RSC.
- D. Wang, Q. Wu, X. Zhang, W. Wang, E. Hao and L. Jiao, Org. Lett., 2020, 22, 7694–7698 CrossRef PubMed.
- S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC.
- M. L. Agazzi, M. B. Ballatore, A. M. Durantini, E. N. Durantini and A. C. Tomé, J. Photochem. Photobiol., C, 2019, 40, 21–48 CrossRef CAS.
- P. Ceroni, Chem. – Eur. J., 2011, 17, 9560–9564 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 1, 3776–3778 RSC.
- N. Kiseleva, M. A. Filatov, M. Oldenburg, D. Busko, M. Jakoby, I. A. Howard, B. S. Richards, M. O. Senge, S. M. Borisov and A. Turshatov, Chem. Commun., 2018, 54, 1607–1610 RSC.
- T. N. Singh-Rachford, A. Haefele, R. Ziessel and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164–16165 CrossRef CAS PubMed.
- W. Wu, X. Cui and J. Zhao, Chem. Commun., 2013, 49, 9009–9011 RSC.
- C. Zhang, J. Zhao, S. Wu, Z. Wang, W. Wu, J. Ma, S. Guo and L. Huang, J. Am. Chem. Soc., 2013, 135, 10566–10578 CrossRef CAS PubMed.
- K. Chen, M. Hussain, S. S. Razi, Y. Hou, E. A. Yildiz, J. Zhao, H. G. Yaglioglu and M. Di Donato, Inorg. Chem., 2020, 59, 14731–14745 CrossRef CAS PubMed.
- P. De Bonfils, L. Péault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2021, 1809–1824 CrossRef CAS.
- X. F. Wang, S. S. Yu, C. Wang, D. Xue and J. Xiao, Org. Biomol. Chem., 2016, 14, 7028–7037 RSC.
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed.
- J. Fischer, L. Mele, H. Serier-Brault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2019, 2019, 6352–6358 CrossRef CAS.
- A. Atilgan, M. M. Cetin, J. Yu, Y. Beldjoudi, J. Liu, C. L. Stern, F. M. Cetin, T. Islamoglu, O. K. Farha, P. Deria, J. F. Stoddart and J. T. Hupp, J. Am. Chem. Soc., 2020, 142, 18554–18564 CrossRef CAS PubMed.
- L. Huang and J. Zhao, RSC Adv., 2013, 3, 23377–23388 RSC.
- G. Magagnano, A. Gualandi, M. Marchini, L. Mengozzi, P. Ceroni and P. G. Cozzi, Chem. Commun., 2017, 53, 1591–1594 RSC.
- C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- A. Stafford, D. Ahn, E. K. Raulerson, K.-Y. Chung, K. Sun, D. M. Cadena, E. M. Forrister, S. R. Yost, S. T. Roberts and Z. A. Page, J. Am. Chem. Soc., 2020, 142, 14733–14742 CrossRef CAS PubMed.
- J. Bartelmess, A. J. Francis, K. A. El Roz, F. N. Castellano, W. W. Weare and R. D. Sommer, Inorg. Chem., 2014, 53, 4527–4534 CrossRef CAS PubMed.
- L. Dura, M. Wächtler, S. Kupfer, J. Kübel, J. Ahrens, S. Höfler, M. Bröring, B. Dietzek and T. Beweries, Inorganics, 2017, 5, 1–17 CrossRef.
- G. G. Luo, K. Fang, J. H. Wu and J. Mo, Chem. Commun., 2015, 51, 12361–12364 RSC.
- J. C. Manton, C. Long, J. G. Vos and M. T. Pryce, Phys. Chem. Chem. Phys., 2014, 16, 5229–5236 RSC.
- R. P. Sabatini, B. Lindley, T. M. McCormick, T. Lazarides, W. W. Brennessel, D. W. McCamant and R. Eisenberg, J. Phys. Chem. B, 2016, 120, 527–534 CrossRef CAS PubMed.
- P. Wang, S. Guo, H. J. Wang, K. K. Chen, N. Zhang, Z. M. Zhang and T. B. Lu, Nat. Commun., 2019, 10, 3155 CrossRef PubMed.
- R. Ziessel, G. Ulrich, A. Haefele and A. Harriman, J. Am. Chem. Soc., 2013, 135, 11330–11344 CrossRef CAS PubMed.
- S. P. Singh and T. Gayathri, Eur. J. Org. Chem., 2014, 4689–4707 CrossRef CAS.
- P. Chinna Ayya Swamy, G. Sivaraman, R. N. Priyanka, S. O. Raja, K. Ponnuvel, J. Shanmugpriya and A. Gulyani, Coord. Chem. Rev., 2020, 411, 213233 CrossRef CAS.
- C. S. Kue, S. Y. Ng, S. H. Voon, A. Kamkaew, L. Y. Chung, L. V. Kiew and H. B. Lee, Photochem. Photobiol. Sci., 2018, 17, 1691–1708 CrossRef CAS PubMed.
- V. N. Nguyen, Y. Yim, S. Kim, B. Ryu, K. M. K. Swamy, G. Kim, N. Kwon, C. Y. Kim, S. Park and J. Yoon, Angew. Chem., Int. Ed., 2020, 59, 8957–8962 CrossRef CAS PubMed.
- A. Treibs and F. -H. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- F. J. Monsma, A. C. Barton, H. Chol Kang, D. L. Brassard, R. P. Haugland and D. R. Sibley, J. Neurochem., 1989, 52, 1641–1644 CrossRef CAS PubMed.
- R. Montero, V. Martínez-Martínez, A. Longarte, N. Epelde-Elezcano, E. Palao, I. Lamas, H. Manzano, A. R. Agarrabeitia, I. López Arbeloa, M. J. Ortiz and I. Garcia-Moreno, J. Phys. Chem. Lett., 2018, 9, 641–646 CrossRef CAS PubMed.
- Y. E. Kandrashkin, Z. Wang, A. A. Sukhanov, Y. Hou, X. Zhang, Y. Liu, V. K. Voronkova and J. Zhao, J. Phys. Chem. Lett., 2019, 10, 4157–4163 CrossRef CAS PubMed.
- R. Prieto-Montero, R. Sola-Llano, R. Montero, A. Longarte, T. Arbeloa, I. López-Arbeloa, V. Martínez-Martínez and S. Lacombe, Phys. Chem. Chem. Phys., 2019, 21, 20403–20414 RSC.
- J. Gemen, J. Ahrens, L. J. W. Shimon and R. Klajn, J. Am. Chem. Soc., 2020, 142, 17721–17729 CrossRef CAS PubMed.
Footnotes |
| † The following conversions were used: E(Fc+/Fc) = +0.46 V vs. SCE in DCM; E(Fc+/Fc) = +0.39 V vs. SCE in MeCN; E(AgNO3/Ag) = +0.30 V vs. SCE in MeCN. |
| ‡ Although the oxidation that occurs at +1.24 V vs. SCE has been attributed by authors to phenylanthracene, that of the BODIPY unit is located in close proximity and a safe attribution is difficult. |
| § A search in Web of Science for the words “BODIPY synthesis” as topic has generated more than 1700 items. |
| This journal is © The Royal Society of Chemistry 2021 |