
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of isostructural hydrogen-bonded organic frameworks: limitations and possibilities of pore expansion†
Yuto
Suzuki
a,
Mario
Gutiérrez
 b,
Senri
Tanaka
c,
Eduardo
Gomez
b,
Norimitsu
Tohnai
d,
Nobuhiro
Yasuda
e,
Nobuyuki
Matubayasi
*c,
Abderrazzak
Douhal
*b and
Ichiro
Hisaki
*a
b,
Senri
Tanaka
c,
Eduardo
Gomez
b,
Norimitsu
Tohnai
d,
Nobuhiro
Yasuda
e,
Nobuyuki
Matubayasi
*c,
Abderrazzak
Douhal
*b and
Ichiro
Hisaki
*a
aDivision of Chemistry, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: hisaki@chem.es.osaka-u.ac.jp
bDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, INAMOL, Universidad de Castilla-La Mancha, Avenida Carlos III, S/N, 45071 Toledo, Spain. E-mail: Abderrazzak.Douhal@uclm.es
cDivision of Chemical Engineering, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: nobuyuki@cheng.es.osaka-u.ac.jp
dDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-7891, Japan
eJASRI, 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
First published on 23rd June 2021
Abstract
The library of isostructural porous frameworks enables a systematic survey to optimize the structure and functionality of porous materials. In contrary to metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), a handful of isostructural frameworks have been reported for hydrogen-bonded organic frameworks (HOFs) due to the weakness of the bonds. Herein, we provide a rule-of-thumb to develop isostructural HOFs, where we demonstrate the construction of the third and fourth generation of isostructural HAT-based HOFs (TolHAT-1 and ThiaHAT-1) by considering three important structural factors, that are (1) directional H-bonding, (2) shape-fitted docking of the HAT core, and (3) modulation of peripheral moieties. Their structural and photo-physical properties including HCl vapor detection are presented. Moreover, TolHAT-1, ThiaHAT-1, and other isostructural HOFs (CPHAT-1 and CBPHAT-1) were thoroughly compared from the viewpoints of structures and properties. Importantly, molecular dynamics (MD) simulation proves to be rationally capable of evaluating the stability of isostructural HOFs. These results can accelerate the development of various isostructural molecular porous materials.
Introduction
Isostructural porous frameworks, whose molecular arrangements and network topology are the same, but with different pore sizes and chemical and physical properties, can provide an important library for a systematic design and optimization of the structure and properties of functional porous materials.1–6 Therefore, it is a touchstone for reticular chemistry to construct such frameworks.7 The expansion of the pores may be readily accomplished by elongating the length of ligand molecules or spacer moieties. Indeed, isostructural MOFs and COFs were successfully manufactured with this methodology as follows.8–10 Yaghi and coworkers reported isostructural MOFs with pore size ranging from 1.4 to 9.8 nm constructed from a series of oligophenylene ligands.8 Lin and coworkers reported a family of isostructural MOFs and their catalytic activities.9 Dichtel and coworkers reported the lattice expansion of 2D phthalocyanine COFs by using linear diboronic acid linkers.10 However, it still remains difficult to prepare isostructural frameworks in the case of porous molecular crystals formed through weak intermolecular interactions,11,12 such as hydrogen-bonded organic frameworks (HOFs)13–18 connected through reversible H bonds.HOFs are available as highly crystalline materials with a large domain via a facile recrystallization process. Additionally, if the frameworks are damaged, they are capable of recovering and regenerating via a wet process, such as solvent vapor annealing, owing to the non-covalent nature of the H bonds.19 On the other hand, the design principle to construct stable HOFs with permanent porosity still remains unsatisfactory when compared with MOFs and COFs, although reports of fascinating HOFs with permanent porosity have been increasing over the last two decades.20–26
Homologous building block molecules of HOFs with different lengths of arms often gave non-isostructural, but diverse frameworks with different molecular arrangements and H-bonded network topologies.21–27 Only a handful of isostructural HOFs are known so far. Cooper, Day, and coworkers employed triptycenetrisbenzimidazolone and its expanded analogue to yield isostructural honeycomb frameworks.28,29 Farha's, Chao's, and Chen's groups independently reported pyrene-based isostructural HOFs based on pyrene derivatives with different substituents (carboxy, 4-carboxyphenyl, and 6-carboxynaphthalen-2-yl).19,30,31 Miljanić and coworkers also reported a series of quasi-isotropic fluorinated honeycomb HOFs.32,33 The difficulty to obtain a set of isostructural HOFs is clearly originated from weakness of H-bonds. Therefore, a further design principle is necessary to construct isostructural HOFs.
In connection with this, we have recently achieved the fabrication of various isostructural hexagonal networks (HexNets) through predictable H-bonding of C3-symmetric carboxylic acids with π-conjugated cores, such as triphenylene (Tp) derivatives,34,35 where the most important structural factors are the rigidity of the molecular skeletons and the highly directional and predictable H bonds (key 1). However, the resultant HexNets stack in different ways to form non-isostructural HOFs. Moreover, we show that two derivatives with even the same Tp core (Tp and BPTp) with carboxy-phenyl and -biphenyl arms form non-isostructural frameworks (Fig. 1a). This is caused by the lack of preferable unique stacking geometry between the molecules. Meanwhile, we have demonstrated that two hexaazatriphenylene (HAT) derivatives (CPHAT and CBPHAT) with carboxy-phenyl and -biphenyl arms, respectively, yielded isostructural HOFs (Fig. 1b), where the crucial structural factor is a twisted conformation of the HAT core that enables the molecule to stack uniformly through shape-fitted docking (key 2).36,37
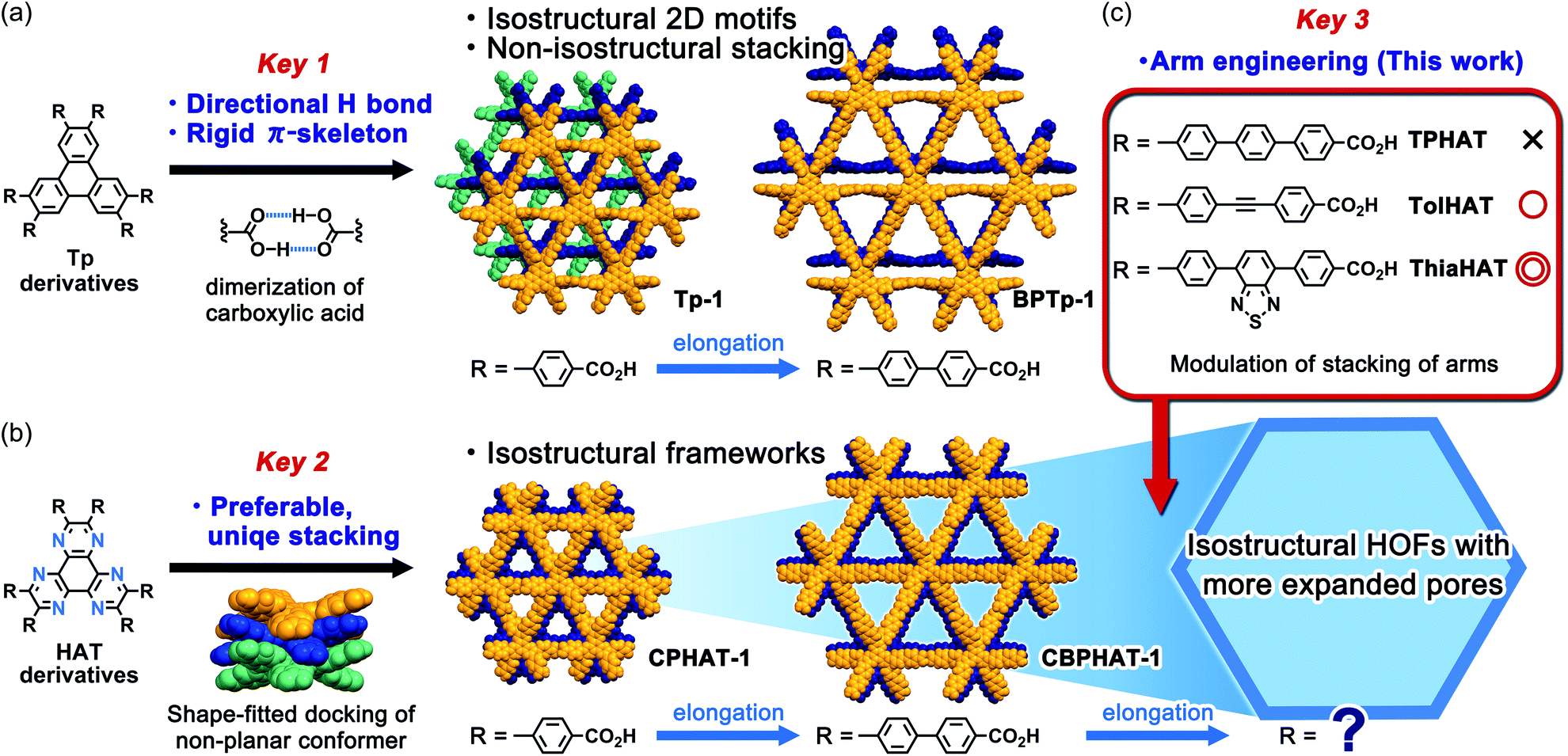 | ||
| Fig. 1 Illustration of keys 1, 2 and 3 to construct isostructural HOFs with different pore sizes. (a) Frameworks formed from triphenylene derivatives. The molecules form isostructural hexagonal network motifs through directional H-bonding of the carboxy group (key 1), while the stacking configurations of the derivatives differ from each other to result in non-isostructural HOFs (Tp-1 and BPTp-1). (b) Frameworks formed from HAT-based molecules. Both molecules with carboxy-phenyl and -biphenyl groups form rigid and stable isostructural HOFs (CPHAT-1 and CBPHAT-1) due to the uniform shape-fitted docking of the twisted HAT core (key 2). (c) Construction of isostructural frameworks with larger pores by a systematic engineering of the arm moieties (key 3). | ||
Can HAT derivatives with further elongated arms provide another isostructural HOF with larger pores? Herein, we provide an answer to this question by constructing the third and fourth generation of isostructural HAT-based HOFs with larger pores. The simple elongation of the arms from biphenylene to terphenylene (TPHAT) quickly reached the limit, while the modification of the arms by 1,2-diphenylethyne (tolane) and 4,7-diphenyl-benzo-2,1,3-thiadiazole allowed the formation of expanded HOFs (TolHAT-1 and ThiaHAT-1), evidencing the importance of suitable arm engineering (key 3 in Fig. 1c). To our knowledge, the present system has so far the largest number of isostructural HOFs. In addition to their structures and properties, including solid-state and solution fluorescence behaviours, structural comparison between the four isostructural HOFs is also described. Interestingly, ThiaHAT-1 exhibits a large sensitivity to HCl vapours which can be recorded by UV-Vis absorption or emission experiments. Moreover, it is noteworthy that the stability of HOFs can be precisely evaluated by molecular dynamics (MD) simulation, reflecting the importance of considering not only the interaction energy, but also the fluctuation of the molecules. These results give an insight to develop a systematic series of porous molecular crystalline materials.
Results and discussion
Synthesis and crystallization
HAT derivatives, TPHAT, TolHAT, and ThiaHAT, were synthesized as shown in Scheme 1. 1,2-Dione derivative 3a with a tolane moiety was prepared by the Sonogashira coupling reaction of 4,4′-dibromobenzil (1) and hexyl 4-ethynyl-benzoate (2). Dione derivatives 3b with terphenyl moieties and 3c with 4,7-diphenyl-benzo-2,1,3-thiadiazole moieties were synthesized by the Suzuki–Miyaura cross-coupling reaction of 4,4′-(pinacolboryl)benzil derivative 4 with hexyl 4′-bromobiphenylcarboxylate derivatives 5 and 6, respectively. Then hexaaminobenzene, which was prepared by the reduction of 1,3,5-triamino-2,4,6-trinitrobenzene (7),38 was triply condensed with the corresponding dione derivatives 3a–c under acidic conditions to afford HAT derivatives with ester groups 9a–c, respectively. The hydrolysis of 9a–c gave the building block molecules TolHAT, TPHAT, and ThiaHAT, respectively. As a reference compound, BPTp with carboxybiphenyl groups was also synthesized and crystallized (for details, see ESI Sections 2 and 3†).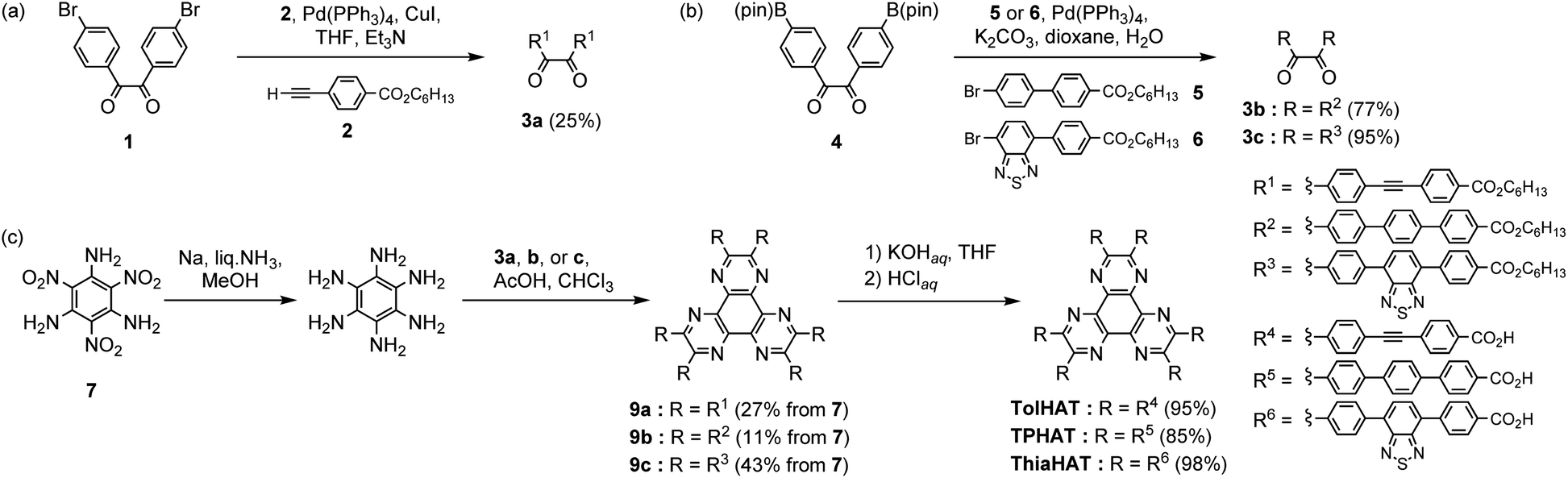 | ||
| Scheme 1 Synthesis of (a) 3a, (b) 3b and 3c, and (c) TPHAT, TolHAT, and ThiaHAT. | ||
To get the HOFs, the synthesized HAT derivatives were recrystallized by slow evaporation from a mixed solution of N,N-dimethylacetamide (DMA) and an aromatic solvent [1,2,4-trichlorobenzene (TCB) or 1,2,4-trimethylbenzene (TMB)] at 60 °C for TolHAT and TPHAT and at 120 °C for ThiaHAT. TPHAT gave pale green precipitates (TPHAT-P) (Fig. 2a), which only showed weak and broad peaks in the powder X-ray diffraction (PXRD) pattern (Fig. S1†). TolHAT, on the other hand, gave yellow green needle-like single crystals (TolHAT-1) with a width of 5–20 μm and a length of over 100 μm (Fig. 2b). Similarly, ThiaHAT yielded yellow needle-like microcrystals (ThiaHAT-1) with a width of approximately 10 μm and a length of over 100 μm (Fig. 2c). TolHAT-1 and ThiaHAT-1 were subject to single-crystalline X-ray diffraction (SCXRD) measurements with synchrotron X-ray radiation to pin-point their crystalline structure.
 | ||
| Fig. 2 Polarized optical microscopy (POM) images of (a) TPHAT-P, (b) TolHAT-1, and (c) ThiaHAT-1 under daylight. Scale bar: 100 μm. | ||
Crystallography
TolHAT and ThiaHAT crystallized into the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c to yield porous frameworks (TolHAT-1 and ThiaHAT-1) (Fig. 3). TolHAT-1 has the cell parameters of a = b = 59.529(1) Å, c = 6.9637(2) Å, and V = 21
c to yield porous frameworks (TolHAT-1 and ThiaHAT-1) (Fig. 3). TolHAT-1 has the cell parameters of a = b = 59.529(1) Å, c = 6.9637(2) Å, and V = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 371.4(8) Å3 and ThiaHAT-1 has a = b = 65.827(5) Å, c = 6.9879(7) Å, and V = 26223(4) Å3 (Table S1†). The latter has a slightly elongated framework due to the longer arms of the building block molecule. Both molecules have D3h-symmetry and are stacked via shape-fitted docking of the HAT cores with intermolecular distances of 3.49 Å to form one-dimensional (1D) columns. The root means square deviations (RMSDs) of the HAT cores of TolHAT and ThiaHAT in the crystals are 0.215 Å and 0.229 Å, respectively. Their torsion angles between adjacent aryl arms in the ortho-positions are 23.5° and 24.5°, respectively. The optimized structures obtained at the B3LYP/6-31G(d) level show more planar conformations (RMSD on the HAT core: 0.111 Å for TolHAT and 0.108 Å for ThiaHAT; and torsion angles between arms: 17.8° for TolHAT and 16.7° for ThiaHAT (see ESI Section 5†)), indicating that packing forces significantly induced the molecular distortion of the HAT moieties in the crystalline states.
371.4(8) Å3 and ThiaHAT-1 has a = b = 65.827(5) Å, c = 6.9879(7) Å, and V = 26223(4) Å3 (Table S1†). The latter has a slightly elongated framework due to the longer arms of the building block molecule. Both molecules have D3h-symmetry and are stacked via shape-fitted docking of the HAT cores with intermolecular distances of 3.49 Å to form one-dimensional (1D) columns. The root means square deviations (RMSDs) of the HAT cores of TolHAT and ThiaHAT in the crystals are 0.215 Å and 0.229 Å, respectively. Their torsion angles between adjacent aryl arms in the ortho-positions are 23.5° and 24.5°, respectively. The optimized structures obtained at the B3LYP/6-31G(d) level show more planar conformations (RMSD on the HAT core: 0.111 Å for TolHAT and 0.108 Å for ThiaHAT; and torsion angles between arms: 17.8° for TolHAT and 16.7° for ThiaHAT (see ESI Section 5†)), indicating that packing forces significantly induced the molecular distortion of the HAT moieties in the crystalline states.
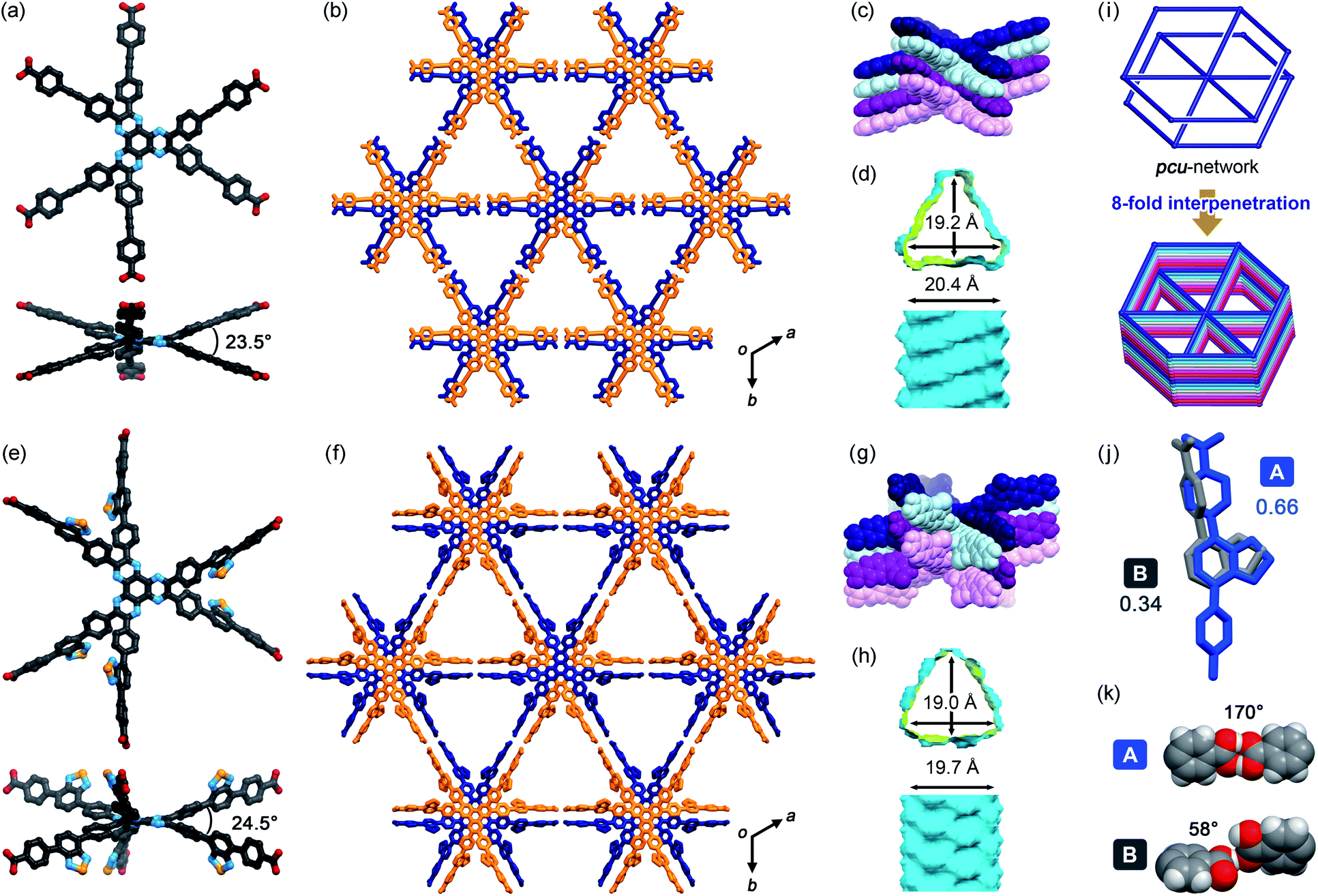 | ||
| Fig. 3 Crystal descriptions of (a–d) TolHAT-1 and (e–h) ThiaHAT-1. (a and e) Twisted nonplanar conformations viewed down the c axis (top) and the ab planes (bottom). (b and f) Packing diagrams. (c and g) 1D columns formed by shape-fitted docking of the HAT derivatives. (d and h) Visualized void surfaces viewed down the c axis (top) and the ab planes (bottom) (yellow: inside and cyan: outside). (i) A H-bonded pcu-topological network, which interpenetrates by 8-fold to form the frameworks of TolHAT-1 and ThiaHAT-1. (j) The disordered arms of ThiaHAT-1 into two positions A and B with an occupancy of 0.662 and 0.338, respectively. (k) H-bonded dimers formed from the arms A and B. A structure in site B is not shown in (e–h). | ||
In TolHAT-1 and ThiaHAT-1, the HAT derivatives form dimers of carboxyl groups through H bonds to provide 3D networks with a pcu-topology (Fig. 3i). The networks are 8-fold interpenetrated to give the whole frameworks possessing large C3-symmetric 1D pores with a triangular aperture. The height and base of the aperture in TolHAT-1 are 19.2 Å and 20.4 Å, respectively, and those of ThiaHAT-1 are 18.0 Å and 19.4 Å, respectively (Fig. 3d and h). Although ThiaHAT has a longer arm than TolHAT, the pores of ThiaHAT-1 are smaller than those of TolHAT-1 because of the bulky thiadiazol groups of ThiaHAT. The void ratios of ThiaHAT-1 and TolHAT-1 are 48% and 55%, respectively, which were calculated by using PLATON software with a probe radius of 1.2 Å.39
It should be noted that the arms of the ThiaHAT molecule are disordered in two positions A and B with a site occupancy of 0.662 and 0.338, respectively (Fig. 3j). The carboxy group in site A forms a typical complementary H-bonded dimer with an O–H⋯O distance and an angle of 2.6 Å and 170°, respectively, while the group in site B forms a truncated dimer with an O–H⋯O distance and a dihedral angle between carboxylate planes of 2.7 Å and 59°, respectively (Fig. 3i). In both conformations, the benzothiadiazole groups are inclined into the pore and aligned vertically along the c axis (Fig. S4 and S5†).
Further structural comparison is conducted focusing on the assembling position of the arm moieties in TolHAT-1 and ThiaHAT-1, as well as in previously reported HOFs, CPHAT-1 and CBPHAT-1 (Fig. 4). The benzene rings directly bonded to the HAT core are labeled Ar-I. Similarly, the benzene rings adjacent to the terminal carboxyl group are labeled Ar-III in CBPHAT, TolHAT, and ThiaHAT. The middle moieties between Ar-I and Ar-III rings are labeled Ar-II in ThiaHAT. The dihedral angles of the neighbouring aryl groups (i.e. the HAT core, Ar-I, Ar-II, and Ar-III) are listed in Table 1. The torsion angles between Ar-I and the core are 23.5° for CPHAT, 25.5° for CBPHAT, 25.6° for TolHAT, and 28.2° for ThiaHAT. The distortions are similar to each other. Ar-I rings are aligned via edge-to-face CH/π interaction with a contact angle of 33.6° (CPHAT), 38.1° (CBPHAT), 37.1° (TolHAT), and 46.5° (ThiaHAT). The contact angles of all these HOFs follow the same tendency as the distortion described above with the exception of ThiaHAT due to the bulky group.
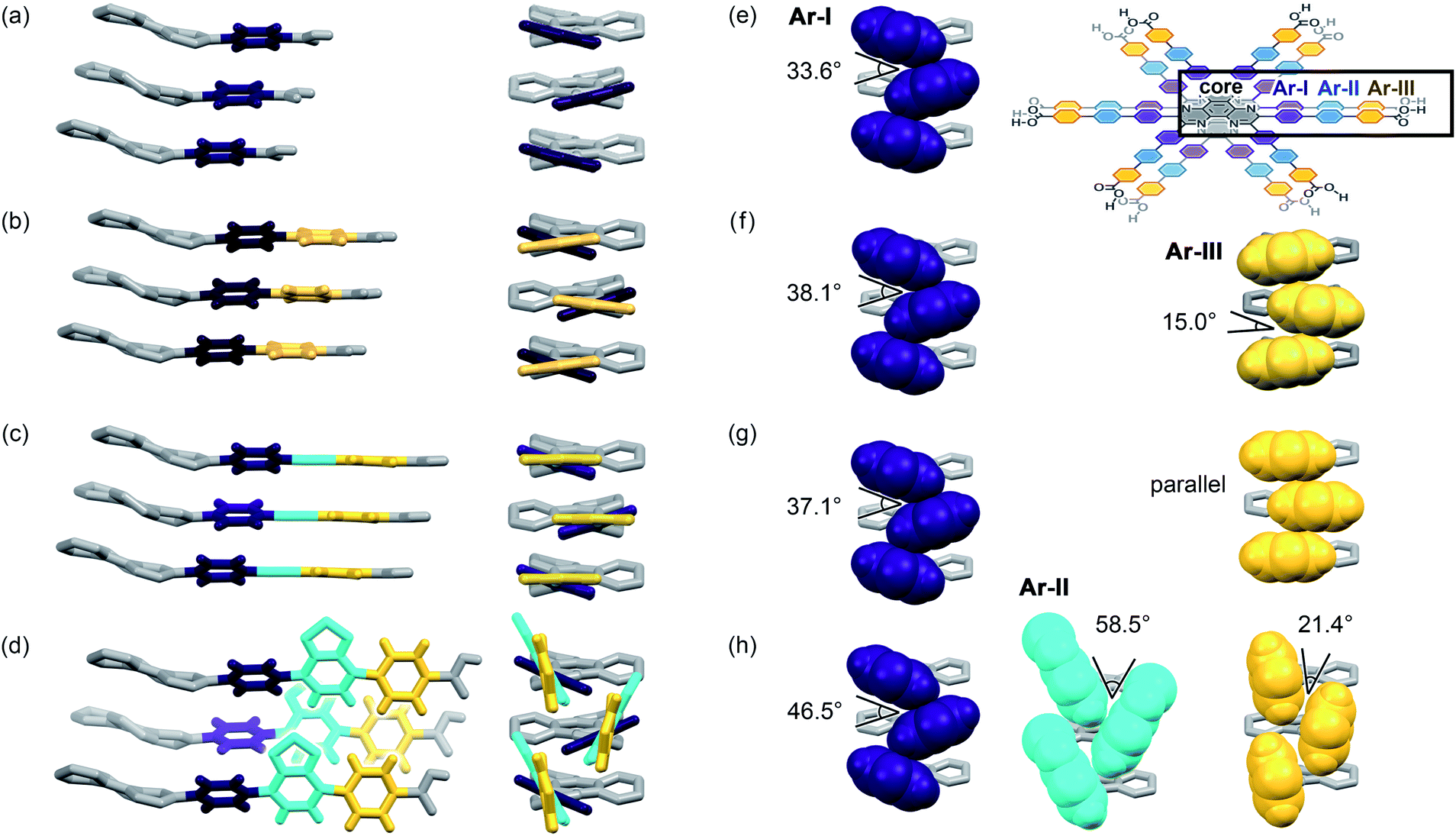 | ||
| Fig. 4 Arm assembling behaviour of (a) CPHAT-1, (b) CBPHAT-1, (c) TolHAT-1, and (d) ThiaHAT-1 from the orthogonal two different views (left and right). (e–h) Contact structures of aryl rings (left: Ar-I, center: Ar-II, and right: Ar-III) in CPHAT-1, CBPHAT-1, TolHAT-1, and ThiaHAT-1, respectively. (Inset) Chemical structures of stacking HAT derivatives. The aromatic rings in the order adjacent to the HAT core are defined as I (purple), II (cyan), and III (yellow), respectively. The HAT core is coloured in gray. | ||
| CPHAT/° | CBPHAT/° | TolHAT/° | ThiaHAT (A site)/° | ThiaHAT (B site)/° | |
|---|---|---|---|---|---|
| a Details are shown in Fig. S6. | |||||
| Core-I | 23.5 | 25.5 | 25.6 | 28.2 | 28.2 |
| Core-II | — | — | — | 61.6 | 62.6 |
| Core-III | — | −16.0 | −19.1 | 80.4 | 41.7 |
| I and II | — | — | — | 37.4 | 38.5 |
| I–III | — | −26.7 | −15.1 | 55.5 | 65.6 |
| II and III | — | — | — | 18.8 | 76.6 |
In previously reported CBPHAT-1, Ar-I and Ar-III were twisted moderately (−26.7°), and their rotation direction was opposite to the rotation direction between the core and Ar-I. Therefore, the torsion angle between the core and Ar-III was only −16.0°. Adjacent Ar-III moieties were assembled nearly parallel (15.0°) via face-to-face π stacking. Similarly, TolHAT-1 has a small torsion angle (−15.1°) between Ar-I and Ar-III rings because of the less sterically hindered ethynyl moiety, resulting in parallel face-to-face π stacking of the adjacent Ar-III rings. On the other hand, it is difficult for ThiaHAT-1 to form the same parallel assembly of the arms due to bulky Ar-II rings, i.e. a benzothiadiazole (Tz) group. The Tz group is twisted by 37.4° and 18.8° against Ar-I and Ar-III, respectively. Adjacent Tz groups interact through face-to-edge CH/π interactions with a contact angle of 58.5°. Every other Tz group is oriented in the same direction and assembled through multiple dipole–dipole interactions.40,41 There is also an intermolecular H-bonding interaction between Ar-II nitrogen and Ar-I hydrogen atoms of two molecules away (Fig. S5†). Consequently, the introduction of the Tz group forces the arm to have a limited conformation, which, combined with shape-fitted docking of the HAT core, induces the ThiaHAT molecules to be organized into a crystalline framework.
On the basis of the aforementioned results, we suggest that, in the case of TPHAT, the conformations of the terphenyl arm may not be suitable either for attractive face-to-face or for face-to-edge interaction, preventing the formation of crystalline porous frameworks. Additionally, considering the above interpretation, it could be possible to construct isostructural HOFs with further expanded pores by varying the bulkiness of elongated arms. Namely, molecules with no steric hindrance between arm components, such as TolHAT, could form HOFs through simply face-to-face π stacking, while those with sterically hindered arm components, such as ThiaHAT, could also produce HOFs through edge-to-face interactions among conformationally restricted arm components.
Thermal analysis and activation
To investigate their thermal behavior, thermal gravimetric (TG) and variable temperature (VT) PXRD analyses were conducted on the as-formed crystals of HOFs. The crystalline bulk of TolHAT-1 was prepared from a mixed solution of DMA and 1,2,4-trimethylbenzene (TMB) instead of DMA and TCB because of its more prominent PXRD pattern, although both of them have the same framework structure (Fig. S7†). TG analysis of TolHAT-1 shows a stepwise curve with no clear plateau, indicating that solvent molecules were released from the pore in two steps: up to approximately 80 °C and up to 300 °C (Fig. S8†).37 PXRD patterns were recorded with increasing temperatures to follow the structural changes in the framework induced by guest removal (Fig. 5a). The intensity of diffraction peaks such as those at 2θ = 2.96° and 5.94°, ascribable to the (2![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and (4
0) and (4 ![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) planes, respectively, slightly increases upon removal of the disordered solvent molecules up to 85.5 °C (red line). Then, the intensity of the peaks at 2θ = 2.96° and 5.94° decreases at higher temperatures, and instead, weak peaks at 2θ = 3.60° and 5.06° were recorded, indicating a structural transition. This temperature corresponds to the second stage of guest removal in the TG curve, indicating that the structural transition coincides with the removal of guest molecules from the framework.
0) planes, respectively, slightly increases upon removal of the disordered solvent molecules up to 85.5 °C (red line). Then, the intensity of the peaks at 2θ = 2.96° and 5.94° decreases at higher temperatures, and instead, weak peaks at 2θ = 3.60° and 5.06° were recorded, indicating a structural transition. This temperature corresponds to the second stage of guest removal in the TG curve, indicating that the structural transition coincides with the removal of guest molecules from the framework.
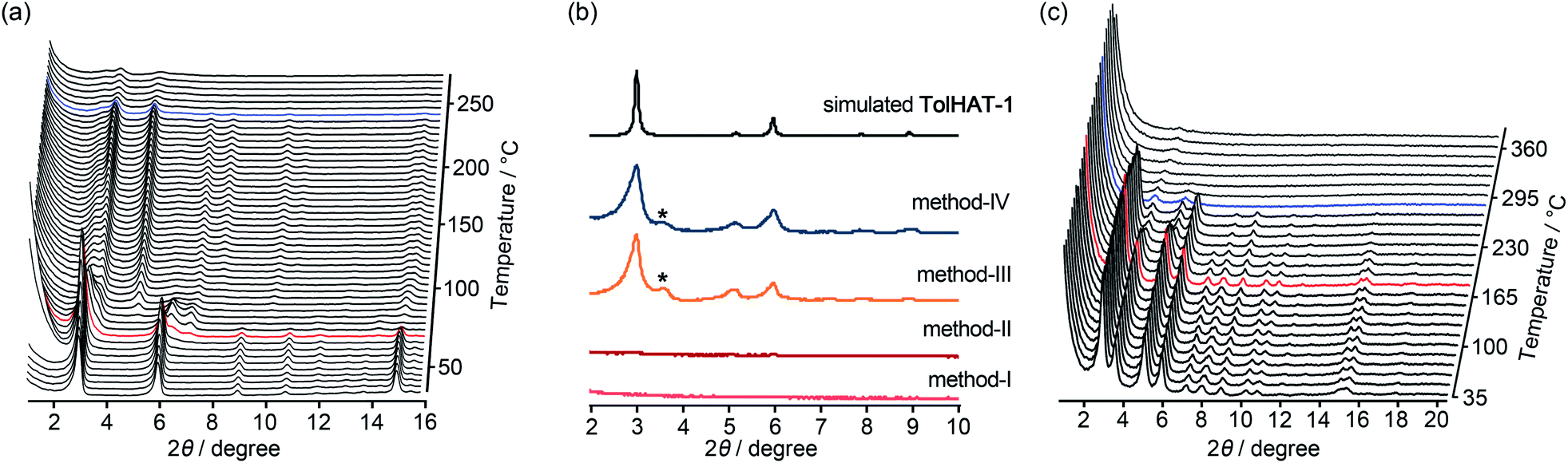 | ||
| Fig. 5 (a) Variable temperature (VT)-PXRD patterns of the as-formed HOF TolHAT-1. (b) PXRD patterns of activated HOF TolHAT-1aN (N = I–IV) with the simulation pattern of TolHAT-1. (c) VT-PXRD patterns of TolHAT-1aIV from rt to 360 °C. Red line indicates the temperature where the network begins to collapse and blue line corresponds to the temperature where the framework is fully collapsed. The asterisk (*) denotes a non-identifiable peak appeared for the activated TolHAT samples. | ||
Keeping this in mind, the activation (i.e. removal of solvent molecules filling the pores) of TolHAT-1 was performed by the following four methods: (I) heating at high temperature (120 °C) under vacuum; (II) immersing in benzene to exchange the trapped solvent with benzene molecules followed by heating at relatively low temperature (80 °C) under vacuum; (III) immersing in benzene followed by freeze-drying; (IV) immersing sequentially in dichlorobenzene, chlorobenzene, toluene, and benzene, followed by freeze-drying. The complete removal of the solvent from the activated materials was confirmed by 1H-NMR spectroscopy (Fig. S9†). PXRD patterns of the materials activated by methods I–IV, termed TolHAT-1aN (N = I–IV), were recorded (Fig. 5b). The crystallinity of TolHAT-1aI and TolHAT-1aII was completely lost. However, TolHAT-1aIII retained its crystallinity, while one non-identifiable peak appeared at 3.60°, which may be ascribable to another phase partially formed by the transformation of the original framework. By a more controlled exchange of the solvent molecules, the intensity of the peak became weaker (TolHAT-1aIV), but still present, revealing that the framework of TolHAT-1a is quite fragile and prone to undergoing phase transition.
The obtained TolHAT-1aIV was then subject to VT-PXRD measurements (Fig. 5c). The intensity of the peak at 2θ = 2.96° starts to decrease at 190 °C (the pattern coloured in red) and vanishes at 298 °C (blue line) (Fig. 5c and S10†). This behaviour is in contrast with that of a HOF based on hexakis[(4-carboxyphenyl)ethynyl]benzene, which is stable at high temperatures up to 300 °C, as reported by Chen and coworkers.27
Thermal analysis of ThiaHAT-1 was conducted (Fig. 6). The TG curve shows two-step release of solvent (TCB) molecules, suggesting that TCB molecules are in two different locations in the voids corresponding to the middle and the corner ones.37 The curve reaches a plateau at 180 °C, indicating a complete removal of solvent molecules. The HOF was also subject to VT-PXRD experiments (Fig. 6c). The PXRD peaks of the as-formed ThiaHAT-1 show no shift at different temperatures, while their intensities increase over 100 °C until reaching a plateau at 170 °C due to the removal of severely disordered TCB molecules, which is frequently observed for HOFs.35–37 The original diffraction peaks are retained up to 305 °C; however, higher temperatures produce a rapid decrease of the intensity of the (100) peak, evidencing the collapse of the framework.
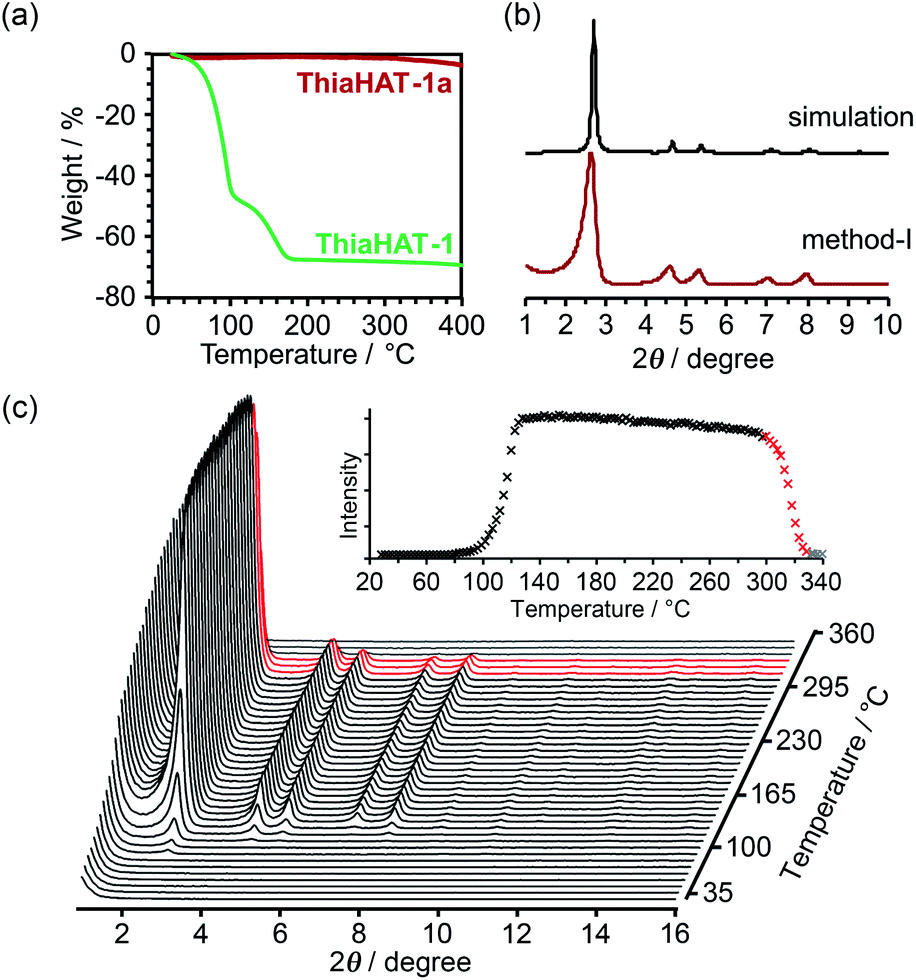 | ||
| Fig. 6 (a) TG profiles of ThiaHAT-1 (green line) and ThiaHAT-1a (red line). (b) PXRD pattern of ThiaHAT-1a activated via method I with the simulation pattern of ThiaHAT-1. (c) VT-PXRD patterns of ThiaHAT-1 heated from RT to 360 °C. Inset: changes of the (100) peak intensity of ThiaHAT-1 with temperature. The intensity rapidly decreased from 305 °C (red line), which indicated the collapse of the framework and no peak was detected at over 330 °C. | ||
The activation of ThiaHAT-1 was, therefore, conducted by method I described above. Heating under vacuum allowed the formation of an activated framework ThiaHAT-1a (Fig. 6b), which maintained its structure and crystallinity. Other activation methods also yielded the same activated HOFs (Fig. S11†). The activation completion was confirmed by the 1H-NMR spectrum of the material dissolved in DMSO-d6 (Fig. S12†). This ThiaHAT-1a was also subject to VT-PXRD experiments, showing that the crystalline network is stable up to 305° (Fig. S13†).
Evaluation of the stability and porosity of HOFs
To evaluate the chemical stability, the activated HOFs were immersed in various solvents and solutions (water, conc. HClaq, NaOHaq, ethanol, CHCl3, and diethyl ether) and tested through PXRD measurements (Fig. S14†). For the case of TolHAT-1aIV, its crystallinity is lower than that of the original, indicating that the introduction and removal of guest molecules caused a partial collapse of the framework. The PXRD patterns of ThiaHAT-1a, on the other hand, retain the original diffraction profile, indicating its good stability in various solvents. However, the porosity after being immersed in conc. HClaq and NaOHaq became lower than that of pristine ThiaHAT-1a (Fig. S15, S20, and Table S2†).The permanent porosity of the activated frameworks of TolHAT-1 and ThiaHAT-1, as well as TPHAT-1 (Fig. S18–S20†), was evaluated by gas adsorption experiments at −196 °C for N2, O2, and H2 and at −78 °C for CO2. For the case of TolHAT-1aN (N = I–IV), the uptake of N2 gas varied with the activation method (Fig. S19, S20, and Table S3†) and TolHAT-1aIV showed the highest porosity among them (SA: 330 m2 g−1 and pore width: 1.66 nm) (Fig. 7a). This demonstrates the importance of a controlled activation. The gas uptakes and BET surface area of TolHAT-1aIV are yet lower than those of CBPHAT-1a (SA: 1288 m2 g−1, Table S4†), suggesting that a large part of the original structure was collapsed or transformed into a less- or non-porous structure. The gas sorption isotherms of ThiaHAT-1a, on the other hand, show a significant uptake of gasses [N2: 18.3, O2: 21.9, CO2: 13.8, and H2: 3.28 mmol(STP) g−1] (Fig. 7b). The BET surface area and pore width were calculated to be 1394 m2 g−1, and 1.58 nm, respectively, which are larger than those of CBPHAT-1a.
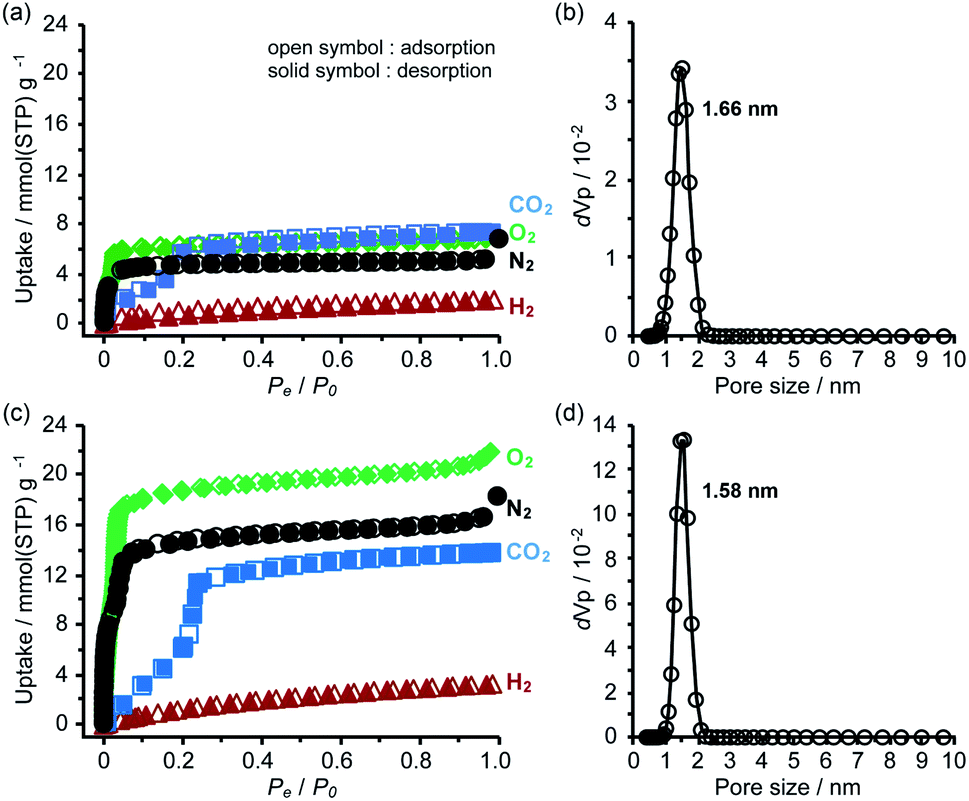 | ||
| Fig. 7 (a and c) Gas sorption isotherms and (b and d) pore distribution of (a and b) TolHAT-1aIV and (c and d) ThiaHAT-1a. The isotherms were recorded at −196 °C for N2, O2, and H2 and at −78 °C for CO2. Solid and open symbols denote absorption and desorption processes, respectively. | ||
Computational evaluation of stability
To investigate the stability of the HOFs, the intermolecular interactions and extents of structural disorder were calculated. Firstly, complexation energies between two stacked molecules in HOFs were calculated (Fig. 8). The stability increases proportionally to the arm length in the cases of CPHAT-1 (−38.4 to −58.4 kcal mol−1), CBPHAT-1, (−63.9 to −90.9 kcal mol−1) and TolHAT-1 (−82.1 to −108.6 kcal mol−1). This result suggests that TolHAT should have been more stable than CPHAT-1 and CBPHAT-1. In the case of ThiaHAT, the increase of stabilization energy (−74.0 to −112.7 kcal mol−1) becomes slow; particularly the energy calculated at the M062X level is smaller than that of TolHAT, which is caused by the face-to-edge contact of the arm moieties. These results indicate that the stability of the HOFs cannot be evaluated from complexation energies.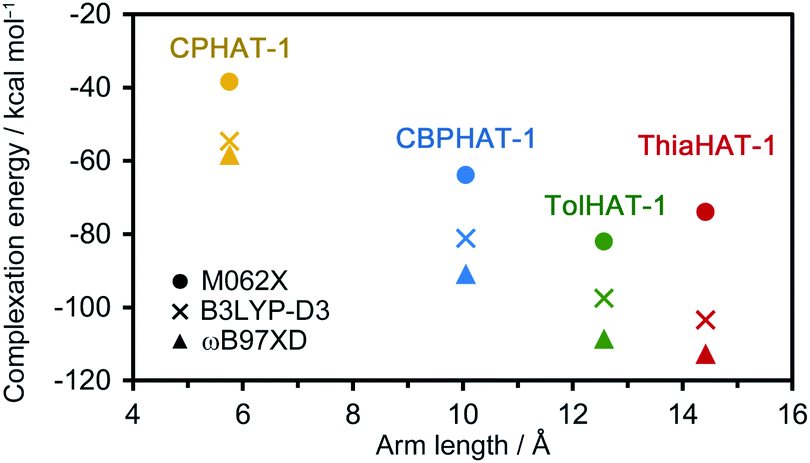 | ||
| Fig. 8 Complexation energy–linker length plot of CPHAT-1, CBPHAT-1, TolHAT-1, and ThiaHAT-1, calculated at the B3LYP-D3/6-311G(d,p), M062X/6-311G(d,p) and wB97XD/6-311(d,p) levels with the counterpoise method for BSSE correction. | ||
Secondly, to assess the structural disorders of the HOFs, all-atom MD (molecular dynamics) simulations were conducted at 300 K and 1 bar on the frameworks of CPHAT-1, CBPHAT-1, TolHAT-1, and ThiaHAT-1. The procedures of MD are described in the ESI,† and snapshot configurations of CPHAT-1 and TolHAT-1 seen from the c-axis are depicted in Fig. 9a as representative examples. The structural disorder is more evident in the arm parts than in the HAT cores, and the disorder of the arm is larger for TolHAT-1 than for CPHAT-1. The extent of disorder can be quantified in terms of the root-mean-square displacement (RMSD) averaged over all the heavy (non-hydrogen) atoms in the system. In the present work, RMSD was computed for the fluctuations in the a- and b-directions. A RMSD plot over the time course of MD for 10 ns (Fig. 9b) shows that the frameworks formed by the H bonds are stable at 300 K. The disorder of the framework represented by RMSD is larger in the order of TolHAT-1 > CBPHAT-1 > ThiaHAT-1 > CPHAT-1, and this is in fair correspondence with the experimentally observed decomposition temperatures of the frameworks, which reflects a higher stability of the framework in the order of CPHAT-1 > CBPHAT-1 ∼ ThiaHAT-1 > TolHAT-1. The above correspondence is actually reminiscent of the Lindemann criterion which states that the crystal with a larger RMSD will be “closer” to melting. Our RMSD (computed along the lateral directions) can thus describe the crystal stability. RMSD is a quantity readily obtained in MD simulations, and it will be of use for predicting the stability of a HOF.
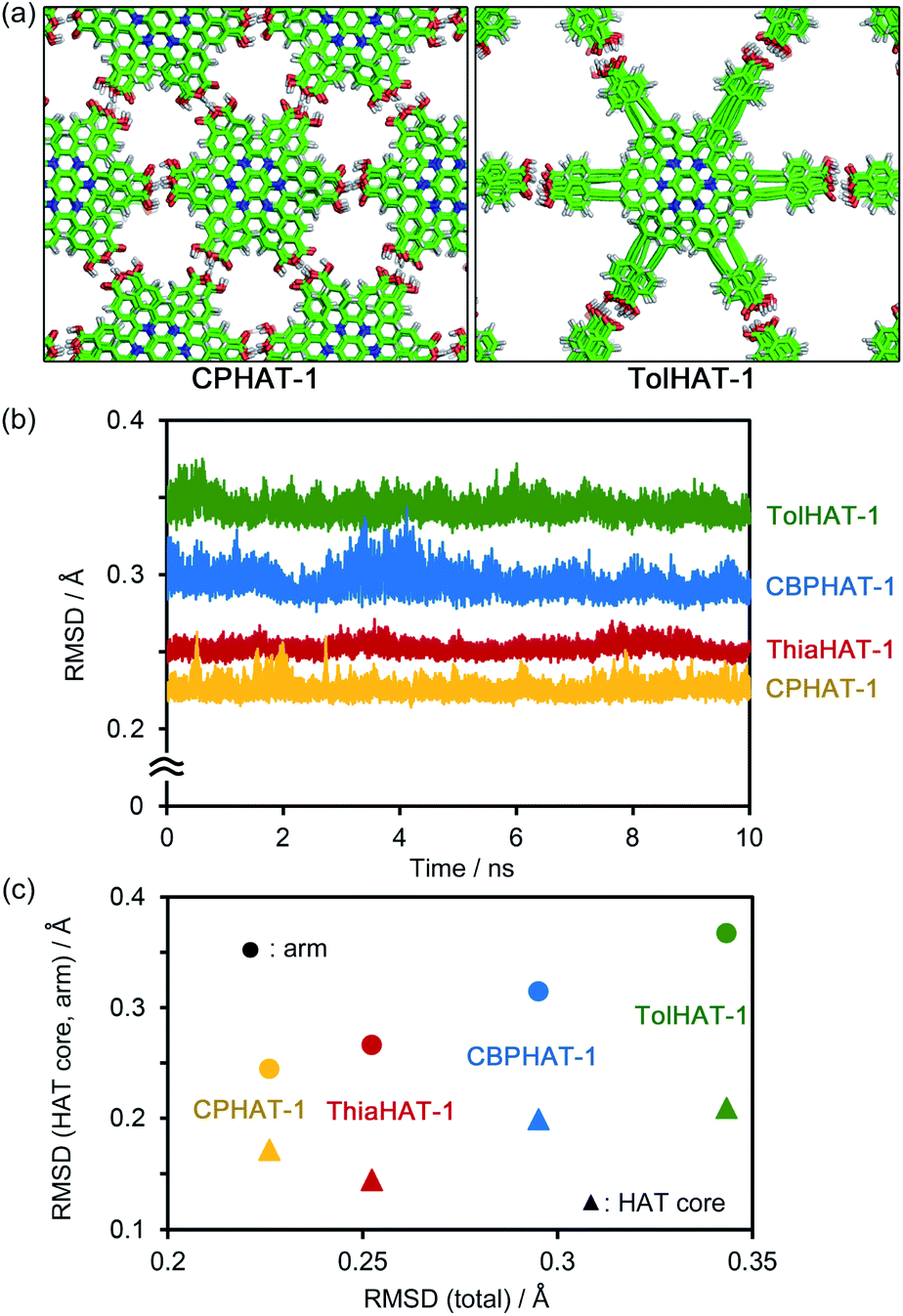 | ||
| Fig. 9 (a) Snapshot configurations of CPHAT-1 and TolHAT-1 seen from the c-axis. (b) RMSD during the time course of MD. (c) Correlation plots of the total RMSD against the contributions from the HAT core and arm parts. RMSD was computed for the fluctuations of the heavy (non-hydrogen) atoms in the system along the lateral (a and b) directions. The (total) RMSD values that are plotted in (b) and refer to the abscissa in (c) were calculated over all the heavy atoms. In (c), the RMSDs averaged over 10 ns are shown. | ||
Fig. 9c further shows the RMSDs computed separately over the HAT core and arm parts. The RMSD is smaller for the HAT core than for the arm. As Fig. 9a shows, the structural disorder is more pronounced in the arm part. The correlation with the total RMSD is seen for the arm RMSD. The crystal stability is thus governed by the extent of disorder of the arm part, and therefore, a rule-of-thumb for producing more stable HOFs would be the design of arms with suppressed fluctuations.
Photophysical properties
Since porous molecular crystals are promising materials for photophysical materials,42 the photophysical properties of TolHAT-1 and ThiaHAT-1 were evaluated by a combination of UV-Vis steady-state absorption and emission spectroscopy and time-resolved emission and single crystal fluorescence microscopy. Unfortunately, during the experiments, TolHAT-1 was revealed to be unstable under UV light excitation, probably due to the presence of the ethynyl moieties of TolHAT-1, which can be a breaking point of the structure upon its excitation with high energies (Fig. S22†). In contrast, ThiaHAT-1 exhibits an excellent stability under light irradiation, even under fs-laser excitation conditions. ThiaHAT-1 crystalline powder displayed a bright yellow emission upon illumination with UV light (365 nm), with an emission quantum yield of 8%. A foreground of the photoproperties of ThiaHAT-1 is shown in Fig. 10.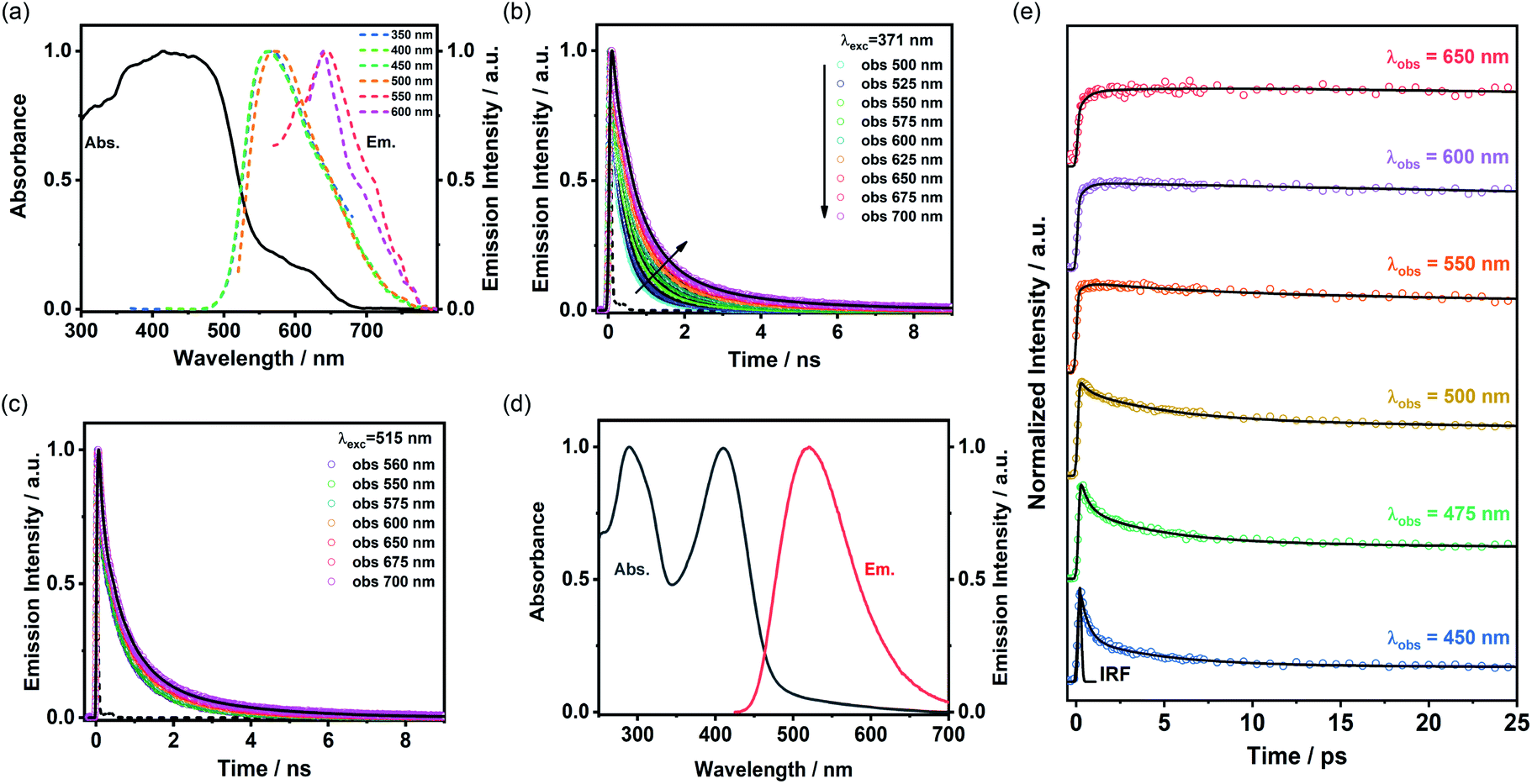 | ||
| Fig. 10 (a) Absorption and emission spectra of ThiaHAT-1 in the powder form. The excitation wavelengths are indicated in the inset. (b and c) Emission decays of ThiaHAT-1 in the powder form upon excitation at (b) 371 nm and (c) 515 nm. The solid lines are from the best multiexponential global fit and the recorded wavelengths are indicated in the figure. (d) Absorption and emission spectra of ThiaHAT-1 in DMF suspension. For emission, the excitation wavelength was 410 nm. (e) fs-Emission decays of ThiaHAT-1 in DMF suspension. The solid lines are from the best multiexponential global fit and the IRF is the instrumental response function. | ||
UV-Vis steady state properties
The absorption spectrum of ThiaHAT-1 crystalline powder consists of two broad bands with intensity maxima located at ∼435 and 600 nm (Fig. 10a). Consequently, the emission spectrum strongly depends on the excitation wavelength. For excitation wavelengths lower than 500 nm, the emission spectrum of ThiaHAT-1 is a broad band with the maximum intensity centered at 565 nm, while its excitation with wavelengths longer than 500 nm produces a red-shifted emission spectrum, with the intensity maximum showing up at 644 nm (Fig. 10a). Both, the absorption and emission spectra of ThiaHAT-1 are red-shifted when compared to its isostructural HOF, CBPHAT-1 (abs. 435 nm; em. 500 nm),37,43 reflecting a stronger charge transfer (CT) character. Interestingly, as commented above, ThiaHAT-1 presents additional absorption (600 nm) and emission (644 nm) bands that are not observed for CBPHAT-1.37,43 There exists two plausible explanations: (i) the building blocks of ThiaHAT-1 are strongly interacting through H bonds, preforming anionic species in the ground state. Similar red-shifted absorption and emission bands owing to anions have been described for other HOFs in the presence of NaOH.43 (ii) The intrinsic structure of ThiaHAT-1 induces a higher interaction between crystals, leading to the formation of aggregates, which will absorb and emit less energetic electromagnetic radiation. To shed more light on this and other aspects of the photodynamical properties of ThiaHAT-1, its time-resolved photobehavior was investigated by means of a picosecond (ps) time-correlated single photon counting (TCSPC) system.TCSPC observations
The photodynamic properties of ThiaHAT-1 were explored by exciting (ps laser pulses) the sample at 371 and 515 nm and collecting the emission decays at several wavelengths, which were then analysed using a multiexponential global fit method. The obtained results show a comparable multiexponential photobehavior independently of the excitation wavelength, with time constants of: τ1 = 160–180 ps, τ2 = 710–720 ps, and τ3 = 2.3–2.5 ns (Fig. 10b, c and Table S8†). The contribution of the shortest component (τ1) to the signal is higher in the bluest region of the spectrum, while the longest component (τ3) contributes more to the emission decays recorded at longer wavelengths. Based on that, and following the steady state discussion, the τ1 component can be attributed to the species initially excited, which deactivate from the first singlet excited state (S1), while the longest τ3 component corresponds to the emission of the anionic species. This is further supported by the increase in the total contribution of this component when excited at 515 nm, which may indicate that the red-shifted absorption and emission bands could have originated from the anionic species. On the other hand, as described previously, ThiaHAT-1 also exhibits a strong CT character, and therefore, the intermediate component (τ2) can be ascribed to the emission of these CT species. The isostructural CBPHAT-1 had also exhibited a multiexponential behavior with time components assigned to species initially excited, others undergoing a CT and anions.37,43Ultrafast dynamics
To unveil the ultrafast dynamics of the proton transfer (PT) and CT reactions, ThiaHAT-1 crystals dispersed in a DMF solution were investigated by means of an ultrafast femtosecond (fs) up-conversion (emission) technique. Prior to the fs-experiments, we evaluated the steady state properties of ThiaHAT-1 in DMF (Fig. 10d). The absorption spectrum is now better defined with narrow bands having their intensity maxima at 208 and 410 nm, while the emission spectrum has its maximum at 510 nm. Subsequently, a suspension of ThiaHAT-1 in DMF was pumped with a 410-nm fs-laser and its emission was probed at different wavelengths ranging from 450 to 650 nm (Fig. 10e). The fs-emission decays are well fitted to a double exponential function with time constants of τ1 = 450 fs and τ2 = 4.4 ps and an offset of ∼320 ps (Table S9†). Both time components decay in the highest energetic regions and rise in the lowest ones, reflecting the occurrence of different photoreactions. However, while the shortest τ1 component rises from wavelengths longer than 500 nm, the longest τ2 one only rises in the reddest part (from 650 nm). Based on our previous explanations, where the anion species fluoresce at the longest wavelengths, it is reasonable to attribute the 4.4 ps component to an excited-state PT reaction. The shortest 450-fs component, which rises from 500 nm, can be therefore associated with an ultrafast CT event, leading to the formation of the species having a CT character, which relax to the ground state within a time of 720 ps as described in the TCSPC part.Single crystal microscopy
The emission properties of ThiaHAT-1 were also appraised at a single crystal level using a ps time-resolved confocal fluorescence microscope. The emission spectra obtained for different isolated crystals are reminiscent of that obtained for the bulk material (Fig. S23†). Similarly, the ps-photodynamical behaviour provides time constants of 300 ps, 800 ps and 2.7 ns (Fig. S24 and Table S10†), which are comparable to those observed in the bulk, and therefore, can be attributed to the initially excited species (300 ps), species having CT character (800 ps) and anions (2.7 ns). Interestingly, the emission recorded for the ThiaHAT-1 single crystals is highly anisotropic, showing a histogram with a value of around 0.45 when the crystals are oriented perpendicularly to the plane of observation, and a value of −0.1 after their rotation by 90° (Fig. S25†). This fact clearly evidences that ThiaHAT-1 has a highly ordered structure, where the fundamental units are arranged through π–π stacking interactions along the length axis of the crystal while the dipole moments are oriented perpendicularly.Luminescence vapochromic sensing
ThiaHAT-1 has exhibited an outstanding response to the presence of HCl vapors, which can be easily inspected by the naked eye under daylight or UV irradiation. Short time exposure (5 min to 1 h) to HCl vapors induced a strong colour change of ThiaHAT-1 powder from vibrant yellow to dark brown, and disappearance of strong yellow emission was observed upon UV light illumination (Fig. 11a). Indeed, the absorption spectrum of ThiaHAT-1 crystals after being dosed with HCl displays an increase in the intensity of the band at 600 nm (Fig. 11b). The emission spectrum of ThiaHAT-1 suffers from a strong quenching (up to 98% of the initial intensity) upon interaction with HCl vapors (Fig. 11c). Additionally, a new band with its intensity maximum located at 700 nm is recorded (Fig. 11d). The increase in the intensity of the red-shifted bands reflects a protonation of ThiaHAT-1. Apart from the thiadiazole group, this HOF contains several N atoms susceptible to being protonated, so the interaction with a strong acid like HCl will induce the protonation, producing the observed changes.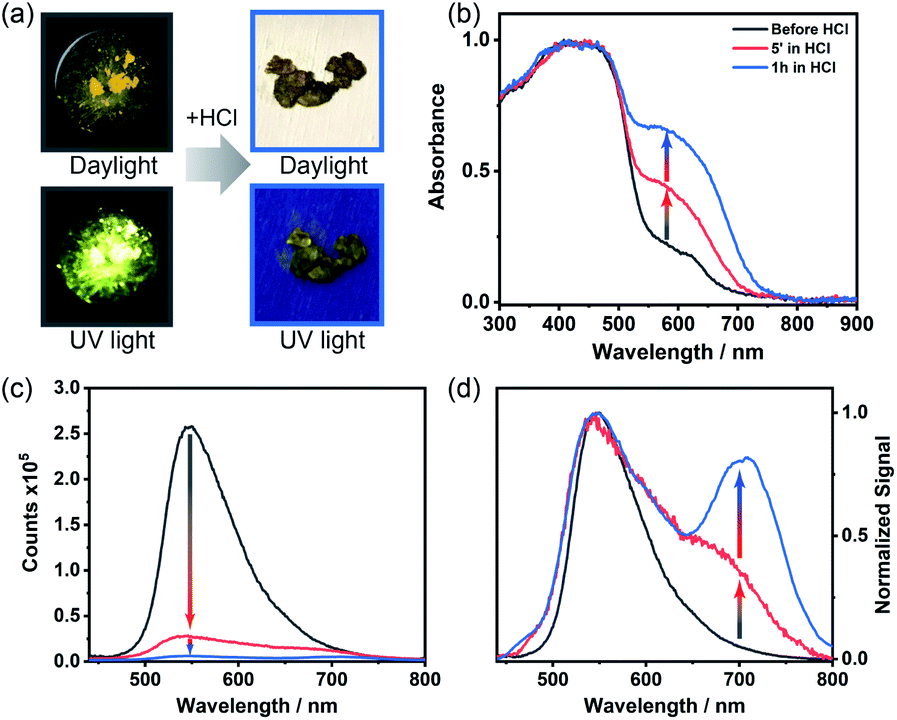 | ||
| Fig. 11 Acid responsiveness of ThiaHAT-1. (a) Photographs of the crystalline powder before (left) and after (right) being exposed to HCl under daylight (top) and UV light (365 nm, bottom). (b) Absorption, (c) emission and (d) normalized emission spectra of ThiaHAT-1 before and after being exposed to vapors of HCl. | ||
Remarkably, ThiaHAT-1 retains the dark brown colour and quenched emission after its dosing with HCl, making this HOF a promising candidate to fabricate an HCl vapochromic smart sensor. The idea behind this is that if at any time the HOF interacts with HCl, it will retain the information until checked. Additionally, the yellow colour and the emission of ThiaHAT-1 can be partially recovered (up to 55% of the initial emission intensity) by heating the sample at 130 °C for 18 hours (Fig. S26†), so there is a possibility to reuse this material. It is noteworthy that acetic acid (AcOH) did not alter the absorption spectrum of ThiaHAT-1 and just slightly quenched its emission (Fig. S27†). This can be explained in terms of the weak acidity of acetic acid, which cannot efficiently protonate the HOF.
Conclusion
In this work, we synthesized three HAT derivatives (TPHAT, TolHAT, and ThiaHAT) and explored the construction of isostructural HOFs with larger pores through arm engineering. The simple elongation of arms from biphenylene to terphenylene quickly reached the limit, while the modification of the arms with 1,2-diphenylethyne and 4,7-diphenylbenzo-2,1,3-thiadiazole allowed the formation of expanded HOFs (TolHAT-1 and ThiaHAT-1). Especially, ThiaHAT-1 showed a great stability even at high temperatures (up to 305 °C), a high BET surface area of 1394 m2 g−1, and irreversible HCl responsivity in spectroscopy. Moreover, it is noteworthy that the stability of HOFs can be precisely evaluated by MD simulation, reflecting the importance of considering not only the interaction energy, but also the fluctuation of the molecules. We gave details on the photobehavior of ThiaHAT-1 as crystals and in solutions providing information on the dynamics from the fs to the ns regime. Remarkably, ThiaHAT-1 shows a large sensitivity to HCl vapors which can be followed by either absorption or emission measurements. These results give an insight to develop a systematic series of porous molecular crystalline materials. To summarize, a comparison of the structural, chemical and robustness properties of this series of isostructural HAT HOFs is shown in Table 2.|
|
|
|
|
|
|---|---|---|---|---|
| CPHAT-1 | CBPHAT-1 | TolHAT-1 | ThiaHAT-1 | |
| a Not determined. | ||||
| Periodicity of the framework/Å | 18.66 | 19.75 | 29.77 | 32.91 |
| RMSD of the HAT core plane/Å | 0.267 | 0.205 | 0.215 | 0.229 |
| Stacking distance/Å | 3.59 | 3.57 | 3.49 | 3.49 |
| Torsion angle of arms/° | 22.5 | 22.1 | 23.5 | 24.5 |
| Number of interpenetrations | 4 | 6 | 8 | 8 |
| Height of the channel aperture/Å | 6.4 | 14.5 | 19.2 | 18.0 |
| Void ratio | 0.31 | 0.45 | 0.55 | 0.48 |
| Pore width based on NLDFT/Å | —a | 12.4 | 16.6 | 15.5 |
| BET surface area/m2 g−1 | 649 | 1288 | 440 | 1394 |
| N2 uptake/mL(STP) g−1 | 21.39 | 361.7 | 155.2 | 415.7 |
| CO2 uptake/mL(STP) g−1 | 137.4 | 304.5 | 168.6 | 313.9 |
| Decomposition temp./°C | 339 | 307 | 190 | 305 |
| Ref. | Ref. 36 | Ref. 37 | This work | This work |

Author contributions
Y. S. designed, synthesized and characterized all materials, evaluated the properties of the materials and co-wrote the paper. M. G. designed, performed and analyzed the femtosecond experiments, and co-wrote the paper. E. G. performed and analyzed the absorption and fluorescence experiments, and evaluated acid responsiveness. S. T. conducted the MD calculation. N. T. contributed to the gas sorption and VTPXRD measurements. N. Y. contributed to the synchrotron X-ray radiation experiments. N. M., A. D. and I. H. planned and supervised the research, analyzed the data and co-wrote the paper. All authors discussed the results and commented on the manuscript. I. H. led the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by KAKENHI (JP18H01966, JP19H04206, and JP21H01919) from MEXT and by Bilateral Programs (JPJSBP120207401) from JSPS, Japan. I. H. thanks the Izumi Science and Technology Foundation. N. M. is supported by the Elements Strategy Initiative for Catalysts and Batteries (No. JPMXP0112101003) and the Fugaku Supercomputer Project (No. JPMXP1020200308) from the Ministry of Education, Culture, Sports, Science, and Technology. A. D. acknowledges the financial support from MINECO through project MAT2017-86532-R, JCCM through project SBPLY/19/180501/000212, and UCLM (FEDER, proposal number: 2020-GRIN-28929). Y. S. acknowledges a Grant-in-Aid for JSPS Research Fellow (20J20301). X-ray diffraction data, including preliminary datasets, were collected at BL40XU in SPring-8 with the approval of JASRI (proposal no. 2018B1244, 2019A1161, and 2019B1134).Notes and references
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, aat9180 CrossRef PubMed.
- C. Gropp, S. Canossa, S. Wuttke, F. Gándara, Q. Li, L. Gagliardi and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1255–1273 CrossRef CAS PubMed.
- J. Li, P. M. Bhatt, J. Li, M. Eddaoudi and Y. Liu, Adv. Mater., 2020, 32, 2002563 CrossRef CAS PubMed.
- W. Xu, B. Tu, Q. Liu, Y. Shu, C. C. Liang, C. S. Diercks, O. M. Yaghi, Y. B. Zhang, H. Deng and Q. Li, Nat. Rev. Mater., 2020, 5, 764–779 CrossRef CAS.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks, Wiley, 2019, ISBN 978-3-527-3450-1 Search PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS.
- T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed.
- E. L. Spitler, J. W. Colson, E. J. Uribe-Romo, A. R. Woll, M. R. Giovino, A. Saldivar and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 2623–2627 CrossRef CAS PubMed.
- I. Bassanetti, S. Bracco, A. Comotti, M. Negroni, C. Bezuidenhout, S. Canossa, P. P. Mazzeo, L. Marchió and P. Sozzani, J. Mater. Chem. A, 2018, 6, 14231–14239 RSC.
- G. Xing, I. Bassanetti, S. Bracco, M. Negroni, C. Bezuidenhout, T. Ben, P. Sozzani and A. Comotti, Chem. Sci., 2019, 10, 730–736 RSC.
- Y. F. Han, Y. X. Yuan and H. B. Wang, Molecules, 2017, 22, 266 CrossRef.
- J. Luo, J. W. Wang, J. H. Zhang, S. Lai and D. C. Zhong, CrystEngComm, 2018, 20, 5884–5898 RSC.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS.
- I. Hisaki, J. Inclusion Phenom. Macrocyclic Chem., 2020, 96, 215–231 CrossRef CAS.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS.
- Q. Yin, P. Zhao, R. J. Sa, G. C. Chen, L. Jian, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691–7696 CrossRef CAS PubMed.
- F. Hu, C. Liu, M. Wu, J. Pang, F. Jiang, D. Yuan and M. Hong, Angew. Chem., Int. Ed., 2017, 56, 2101–2104 CrossRef CAS PubMed.
- B. Han, H. Wang, C. Wang, H. Wu, W. Zhou, B. Chen and J. Jiang, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed.
- P. Li, Y. He, H. D. Arman, R. Krishna, H. Wang, L. Weng and B. Chen, Chem. Commun., 2014, 50, 13081–13084 RSC.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- P. Li, Z. Chen, M. R. Ryder, C. L. Stern, Q. H. Guo, X. Wang, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 12998–13002 CrossRef CAS PubMed.
- K. Kobayashi, T. Shirasaka, E. Horn and N. Furukawa, Tetrahedron Lett., 2000, 41, 89–93 CrossRef CAS.
- P. Li, P. Li, M. R. Ryder, Z. Liu, C. L. Stern, O. K. Farha and J. F. Stoddart, Angew. Chem., Int. Ed., 2019, 58, 1664–1669 CrossRef CAS PubMed.
- X. Zhang, L. Li, J. X. Wang, H. M. Wen, R. Krishna, H. Wu, W. Zhou, Z. N. Chen, B. Li, G. Qian and B. Chen, J. Am. Chem. Soc., 2020, 142, 633–640 CrossRef CAS PubMed.
- A. Pulido, L. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Nature, 2017, 543, 657–664 CrossRef CAS PubMed.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- K. Ma, P. Li, J. H. Xin, Y. Chen, Z. Chen, S. Goswami, X. Liu, S. Kato, H. Chen, X. Zhang, J. Bai, M. C. Wasson, R. R. Maldonado, R. Q. Snurr and O. K. Farha, Cell Rep. Phys. Sci., 2020, 1, 100024 CrossRef.
- B. Wang, X. L. Lv, J. Lv, L. Ma, R. B. Lin, H. Cui, J. Zhang, Z. Zhang, S. Xiang and B. Chen, Chem. Commun., 2019, 56, 66–69 RSC.
- T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. Š. Miljanić, Nat. Commun., 2014, 5, 5131 CrossRef CAS PubMed.
- M. I. Hashim, H. T. M. Le, T. H. Chen, Y. S. Chen, O. Daugulis, C. W. Hsu, A. J. Jacobson, W. Kaveevivitchai, X. Liang, T. Makarenko, O. Miljanić, I. Popovs, H. V. Tran, X. Wang, C. H. Wu and J. I. Wu, J. Am. Chem. Soc., 2018, 140, 6014–6026 CrossRef CAS PubMed.
- I. Hisaki, S. Nakagawa, N. Ikenaka, Y. Imamura, M. Katouda, M. Tashiro, H. Tsuchida, T. Ogoshi, H. Sato, N. Tohnai and M. Miyata, J. Am. Chem. Soc., 2016, 138, 6617–6628 CrossRef CAS PubMed.
- I. Hisaki, Y. Suzuki, E. Gomez, Q. Ji, N. Tohnai, T. Nakamura and A. Douhal, J. Am. Chem. Soc., 2019, 141, 2111–2121 CrossRef CAS PubMed.
- I. Hisaki, N. Ikenaka, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Chem.–Eur. J., 2017, 23, 11611–11619 CrossRef CAS PubMed.
- I. Hisaki, Y. Suzuki, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Angew. Chem., Int. Ed., 2018, 57, 12650–12655 CrossRef CAS PubMed.
- D. Z. Rogers, J. Org. Chem., 1986, 51, 3904–3905 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. Song, Y. He, B. Li, Y. Ling, H. Wang, Y. Feng, R. Krishna and B. Chen, Chem. Commun., 2014, 50, 12105–12108 RSC.
- T. Ikeda, T. Iijima, R. Sekiya, O. Takahashi and T. Haino, J. Org. Chem., 2016, 81, 6832–6837 CrossRef CAS PubMed.
- M. R. Nunzio, I. Hisaki and A. Douhal, J. Photochem. Photobiol. C: Photochem. Rev., 2021, 47, 100418 CrossRef.
- E. Gomez, Y. Suzuki, I. Hisaki, M. Moreno and A. Douhal, J. Mater. Chem. C, 2019, 7, 10818–10832 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, characterization, thermal analysis, spectroscopic data, theoretical calculation, and crystallographic data. CCDC 2081131, 2081132, and 2081133. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02690a |
| This journal is © The Royal Society of Chemistry 2021 |