
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel chemical biology and computational approach to expedite the discovery of new-generation polymyxins against life-threatening Acinetobacter baumannii†
Xukai
Jiang
ab,
Nitin A.
Patil
 b,
Mohammad A. K.
Azad
b,
Hasini
Wickremasinghe
b,
Heidi
Yu
b,
Jinxin
Zhao
b,
Xinru
Zhang
b,
Mengyao
Li
b,
Bin
Gong
c,
Lin
Wan
c,
Wendong
Ma
d,
Philip E.
Thompson
e,
Kai
Yang
d,
Bing
Yuan
d,
Falk
Schreiber
fg,
Lushan
Wang
h,
Tony
Velkov
i,
Kade D.
Roberts
b and
Jian
Li
*b
b,
Mohammad A. K.
Azad
b,
Hasini
Wickremasinghe
b,
Heidi
Yu
b,
Jinxin
Zhao
b,
Xinru
Zhang
b,
Mengyao
Li
b,
Bin
Gong
c,
Lin
Wan
c,
Wendong
Ma
d,
Philip E.
Thompson
e,
Kai
Yang
d,
Bing
Yuan
d,
Falk
Schreiber
fg,
Lushan
Wang
h,
Tony
Velkov
i,
Kade D.
Roberts
b and
Jian
Li
*b
aNational Glycoengineering Research Center, Shandong University, Qingdao, China
bBiomedicine Discovery Institute, Infection & Immunity Program, Monash University, Melbourne, Australia. E-mail: Jian.Li@monash.edu; Fax: +61 3 9905 6450; Tel: +61 3 9903 9702
cSchool of Software, Shandong University, Jinan, China
dCentre for Soft Condensed Matter Physics and Interdisciplinary Research, School of Physical Science and Technology, Soochow University, Suzhou, China
eMedicinal Chemistry, Monash Institute of Pharmaceutical Science, Faculty of Pharmacy and Pharmaceutical Sciences, Monash University, Melbourne, Australia
fDepartment of Computer and Information Science, University of Konstanz, Konstanz, Germany
gFaculty of Information Technology, Monash University, Melbourne, Australia
hState Key Laboratory of Microbial Technology, Shandong University, Qingdao, China
iDepartment of Pharmacology & Therapeutics, University of Melbourne, Melbourne, Australia
First published on 19th August 2021
Abstract
Multidrug-resistant Gram-negative bacteria represent a major medical challenge worldwide. New antibiotics are desperately required with ‘old’ polymyxins often being the only available therapeutic option. Here, we systematically investigated the structure–activity relationship (SAR) of polymyxins using a quantitative lipidomics-informed outer membrane (OM) model of Acinetobacter baumannii and a series of chemically synthesized polymyxin analogs. By integrating chemical biology and all-atom molecular dynamics simulations, we deciphered how each residue of the polymyxin molecule modulated its conformational folding and specific interactions with the bacterial OM. Importantly, a novel designed polymyxin analog FADDI-287 with predicted stronger OM penetration showed improved in vitro antibacterial activity. Collectively, our study provides a novel chemical biology and computational strategy to expedite the discovery of new-generation polymyxins against life-threatening Gram-negative ‘superbugs’.
1. Introduction
Antibiotic resistance is an urgent threat to global health.1 Due to high morbidity and mortality, multidrug-resistant (MDR) Gram-negative Acinetobacter baumannii, Klebsiella pneumoniae and Pseudomonas aeruginosa have been identified by the World Health Organization (WHO) as the top three priority pathogens desperately requiring new therapeutic options.2 The lack of new antibiotics to treat infections caused by such pathogens has revived the use of ‘old’ polymyxins (polymyxin B and colistin) as a last-line therapy to combat infections caused by these MDR ‘superbugs’.3,4 Worryingly, the emergence and spread of polymyxin resistance threatens the utility of these agents and would render life-threatening MDR infections virtually untreatable.5 Therefore, new-generation polymyxins are urgently needed. To date, >2000 polymyxin analogs have been generated in various medicinal chemistry programs using empirical-based approaches.6–8 Unfortunately, most analogs are less active than native polymyxins, although a very small number of analogs (e.g. CA824, CA900 and CA1049) show slightly improved activity against Gram-negative pathogens.7 The knowledge gap in our understanding of the structure–activity relationship (SAR) of polymyxins has significantly hindered the discovery of new-generation polymyxins.Polymyxin antibacterial activity occurs primarily via binding to lipopolysaccharides (LPS) and disrupting the bacterial outer membrane (OM).6,9 Therefore, understanding the interaction between polymyxins and the OM is essential for elucidation of the mechanism and rational drug design. However, examining polymyxin–membrane interactions using current experimental approaches is challenging. Previous attempts to characterize polymyxin SAR focused on polymyxin interactions with a single LPS molecule;6,8,10,11 this approach identified the hydrophobic segments (e.g. fatty acyl group, D-Phe6 and Leu7 of polymyxin B) and positively-charged L-α-γ-diaminobutyric (Dab) acid residues as essential for LPS binding. However, these simplified LPS-based SAR models fail to mimic polymyxin interactions with the bacterial OM, thereby hampering our understanding of their exact mode of action.12 Here, we employed an experimentally-based bacterial OM model rather than a single LPS molecule to develop a novel polymyxin SAR model through integrating all-atom molecular dynamics (MD) simulations and chemical biology approaches. Our in vitro results supported the utility of this SAR model to predict the antimicrobial activity of polymyxin analogs, which will significantly accelerate the discovery of next-generation polymyxins against problematic Gram-negative pathogens.
2. Results and discussion
2.1. Polymyxin B formed a unique folded conformation upon penetration into the OM
A very small number of MD simulation studies have previously been conducted with polymyxins.13–16 In these studies only polymyxin B and colistin A were used to examine the interactions with the bacterial OM and limited SAR information of polymyxins could be inferred. Therefore, we firstly developed a precision bacterial OM model containing lipid A, phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and cardiolipin; the composition and chemotype of each component were determined based upon our quantitative lipidomics data from A. baumannii (Fig. S1, ESI†).17 The asymmetricity and heterogeneity of our lipidomics-informed OM model well represents the complexity of the bacterial OM18,19 and allowed us to precisely determine membrane–polymyxin interactions at the membrane level.12 Furthermore, we synthesized a series of polymyxin B3 (PMB3) analogs with alanine-substitution at each amino acid position except the bridging Dab4 (Fig. S2–S12, ESI†). PMB3 was selected as a representative of polymyxins because it has similar antibacterial activity to polymyxin B1 and colistin A11,17 and lacks the chiral methyl group in the N-terminal fatty acyl chain. The combination of experimental and computational approaches enabled us to better understand the OM-based SAR of polymyxins and will assist in the discovery of novel polymyxin lead compounds.First, we employed all-atom MD simulations to comprehensively examine polymyxin penetration through the OM outer leaflet, the major permeability barrier of bacterial cells.18 PMB3 transitioned from an extended to a folded conformation upon penetration into the OM hydrophobic center (Fig. 1A, Band S13, ESI†). The distance between its fatty acyl chain (using its first carbon atom) and Leu7 (using its Cα atom) was 1.89 ± 0.24 nm in the OM headgroup but significantly decreased to 1.14 ± 0.19 nm in the hydrophobic center (Fig. 1C), showing a folding process during penetration. In the folded state, hydrophobic D-Phe6, Leu7 and the fatty acyl chain oriented toward the OM hydrophobic center, while the polar (Thr2 and Thr10) and positively-charged Dab residues bound to the headgroups of four lipid A molecules (Fig. 1D). Specifically, Dab1 interacted with the 1-phosphate group of #1 lipid A; Dab3 and Dab8 bound to the 4′-phosphate group of #2 lipid A; Dab5 interacted with the 4′-phosphate group of #3 lipid A; Dab9 interacted with the 4′-phosphate group of #3 lipid A and the 1-phosphate group of #4 lipid A. Our findings reveal that modifications of the lipid A phosphate groups substantially minimize their interaction with polymyxins.12,13 Additionally, the fatty acyl chain, D-Phe6 and Leu7 made hydrophobic contacts with the tails of lipid A. Energy analysis revealed electrostatic interactions from Dab1 (−51.6 ± 2.9 kcal mol−1) and Dab9 (−96.5 ± 8.1 kcal mol−1) made prominent contributions to OM binding, which is ∼5–10 fold greater than the hydrophobic interactions from D-Phe6 (−12.6 ± 0.4 kcal mol−1), the fatty acyl group (−10.9 ± 0.5 kcal mol−1), and Leu7 (−10.4 ± 0.2 kcal mol−1) (Fig. 1E). These results reveal that the unique conformational folding is critical for the interaction of polymyxins with the bacterial OM with both polar and hydrophobic interactions involved.
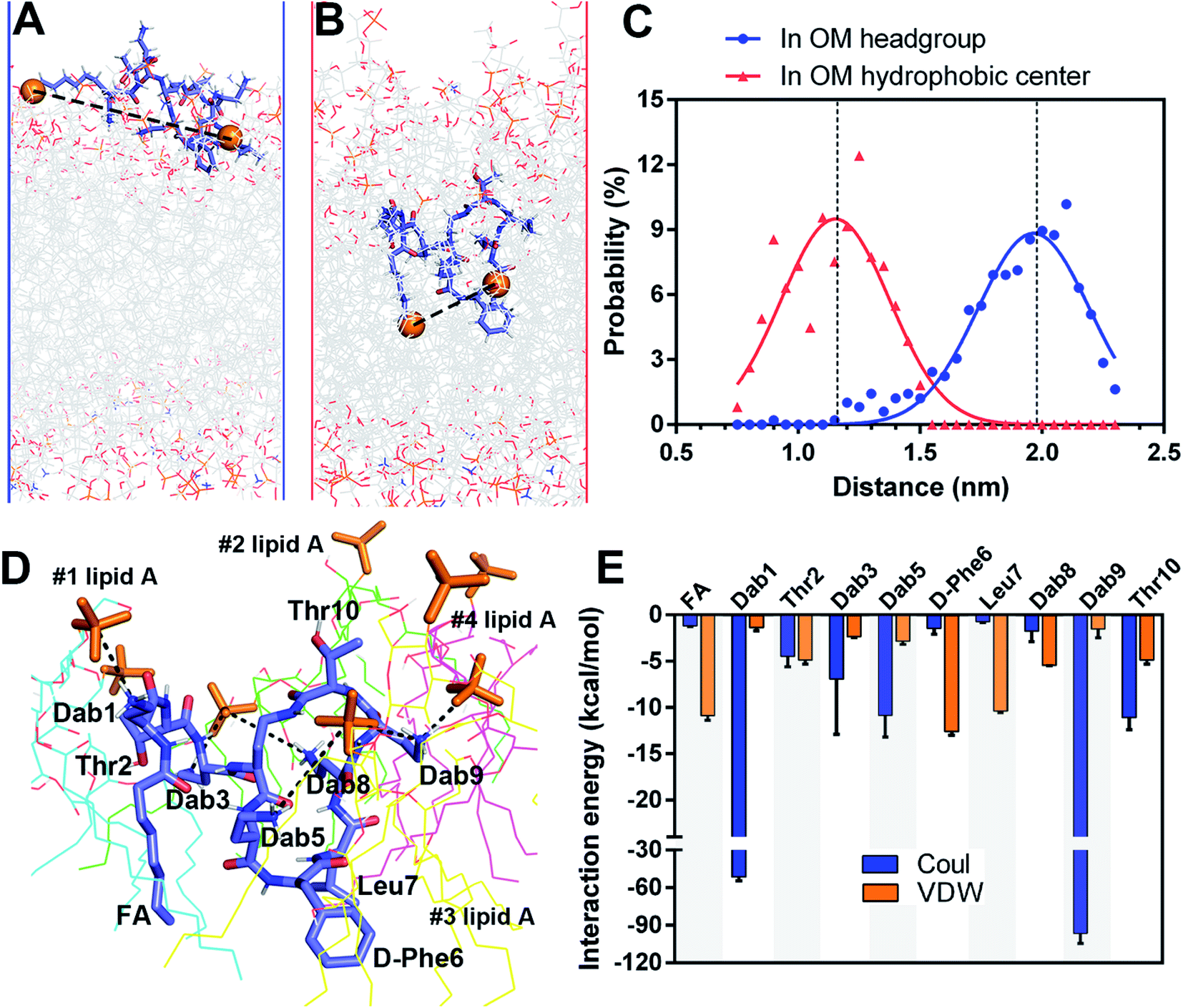 | ||
| Fig. 1 Simulations of polymyxin B3 with the A. baumannii OM. (A and B) Polymyxin B3 conformations in the headgroup region and hydrophobic center of the OM. (C) Distance distribution of polymyxin geometry in the OM. (D) Interaction of polymyxin B3 with lipid A molecules in the OM. (E) Interaction energy between polymyxin B3 and the OM. | ||
2.2. Alanine-substitutions differentially impacted polymyxin folding and penetration in the OM
Following the alanine-substitutions at different PMB3 positions, the conformational transitions in the OM were impacted differentially (Fig. 2 and S14–S22, ESI†). [Ala10]-PMB3, [Ala8]-PMB3 and [Ala1]-PMB3 retained a similar conformational transition to that of PMB3 (Fig. 1B). In contrast, [Ala2]-PMB3, [Ala3]-PMB3, [Ala6]-PMB3 and [Ala7]-PMB3 adopted extended conformations throughout penetration. Thr2 and Dab3 sit on the turn of the folded conformation of PMB3 and interacted with lipid A headgroups, whereas D-Phe6 and Leu7 were deeply inserted into the OM making hydrophobic interactions with the lipid A tails (Fig. 1D). The failure of the conformational folding with these analogs indicates that Thr2 and Dab3 might serve as pivot points for rotation of the polymyxin fatty acyl chain and cyclic ring, while D-Phe6 and Leu7 drive conformational changes necessary for OM binding.20 Interestingly, [Ala5]-PMB3 and [Ala9]-PMB3 adopted folded conformations in both the OM headgroup and hydrophobic center, demonstrating a dramatic loss of conformational flexibility. Dab5 and Dab9 were located in the middle of the folded PMB3 (Fig. 1D) and their positively-charged side chains inevitably generated repulsion with the adjacent hydrophobic fatty acyl chain, D-Phe6 and Leu7. Such hinderance was removed following alanine-substitutions at positions 5 and 9, favouring the folded conformation in the absence of LPS. While previous NMR experiments examined the structural dynamics of polymyxin B in aqueous trifluoroethanol,21 our novel atomic-scale, OM-level findings revealed the diverse roles of the side chains of residues within the polymyxin cyclic ring in regulating the conformational folding in the bacterial OM, albeit via different mechanisms. | ||
| Fig. 2 Conformational distribution of alanine-substituted polymyxin B3 analogs upon interacting with the A. baumannii OM. | ||
To quantitatively compare OM interactions of these analogs, the free energy of OM penetration was measured through umbrella sampling22–24 (Fig. 3, S23 and S24, ESI†). Compared to PMB3, alanine-substitutions of Dab1, Thr2, Dab5, D-Phe6 and Dab9 significantly increased free energy (19.2–31.2 kcal mol−1), whereas alanine-substitutions of Dab3 and Leu7 caused moderate increases (12.3–13.6 kcal mol−1); free energy was only slightly increased (3.1 kcal mol−1) by Dab8 substitution, but decreased (−6.2 kcal mol−1) with Thr10 substitution. Given only one polymyxin molecule was included in the calculations due to technical settings of steered MD,25,26 potential cooperation of multiple polymyxin molecules was not considered. Overall, alanine-substitutions at most positions attenuated the penetration of polymyxins into the OM; furthermore, the residues critical to OM penetration were identified, namely Dab1, Thr2, Dab5, D-Phe6 and Dab9.
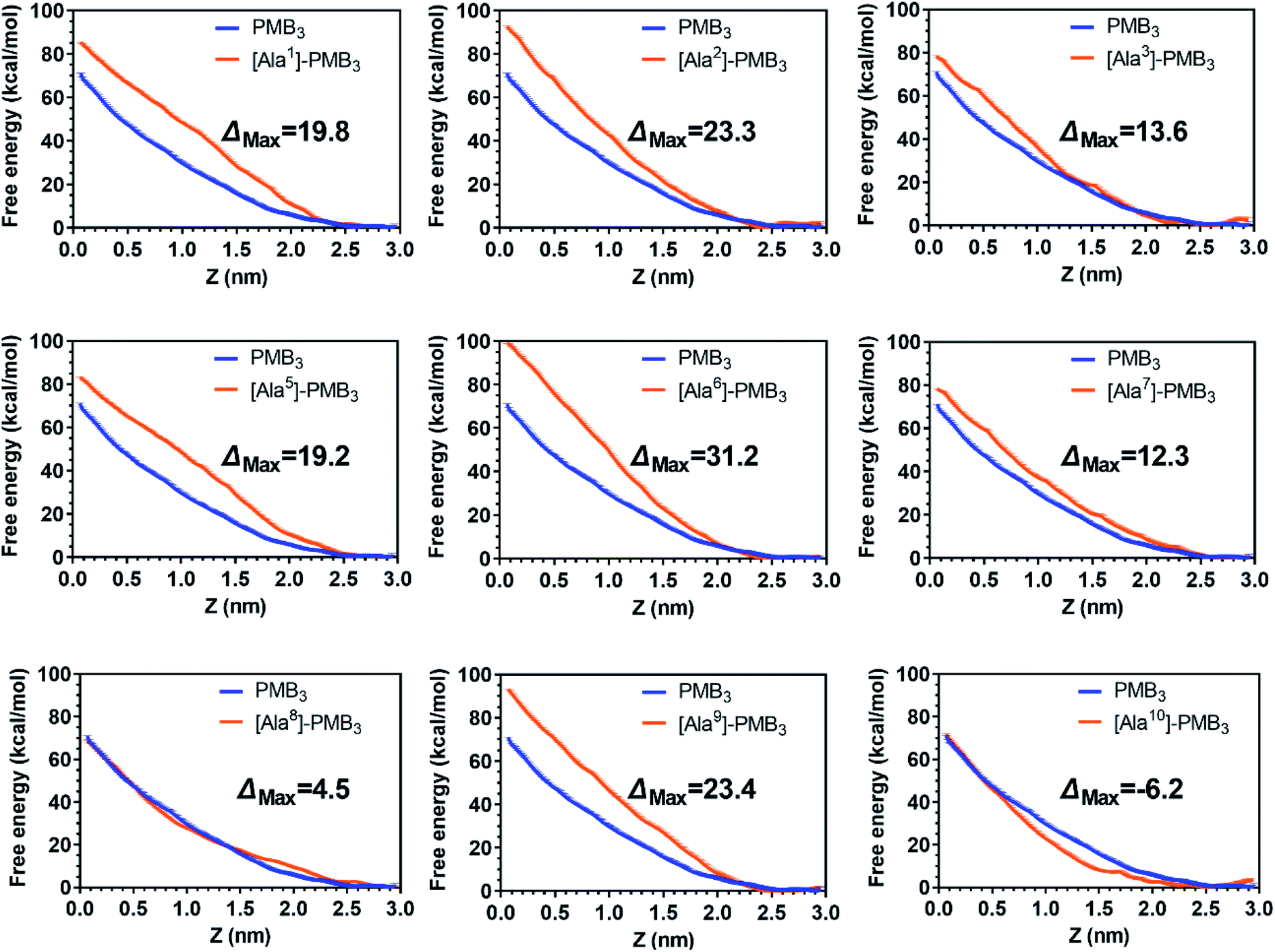 | ||
| Fig. 3 Free energy profiles of polymyxin B3 and analogs upon penetration into the A. baumannii OM. Z = 0 and 2.5 nm indicate the OM hydrophobic center and headgroup region, respectively. Error bars are determined using boot strapping. | ||
2.3. Alanine-substitutions differentially impacted OM disorganization by polymyxins
Polymyxins caused membrane deformation near the penetration site and increased water permeability therein (Fig. S25, ESI†). Flow cytometry using bis-(1,3-dibutylbarbituric acid) trimethineoxonol (DiBAC4(3))27,28 was employed to quantitatively measure the OM disorganization by different analogs. Compared to vehicle-treated controls, DiBAC4(3) cellular fluorescence intensity increased ∼36.8-fold after PMB3 treatment (Fig. 4A), indicating substantially impaired OM integrity. In contrast, fluorescence was not much increased by [Ala1]-PMB3, [Ala2]-PMB3, [Ala5]-PMB3, [Ala6]-PMB3, [Ala8]-PMB3 and [Ala9]-PMB3, indicating these alanine-substitutions dramatically attenuated OM disorganization. [Ala7]-PMB3 decreased fluorescence by ∼30% compared to PMB3, indicating moderately weakened OM disorganization. [Ala3]-PMB3 and [Thr10]-PMB3 showed comparable fluorescence to PMB3, revealing these alanine-substitutions minimally impacted OM disorganization by PMB3. Interestingly, the in silico free energies for OM penetration correlated well with the in vitro measurements of OM disorganization (indicated by fluorescence intensity [Pearson coefficient = 0.68]) (Fig. 4B). These findings reveal that polymyxin OM disruption is mechanistically related to their OM penetration abilities.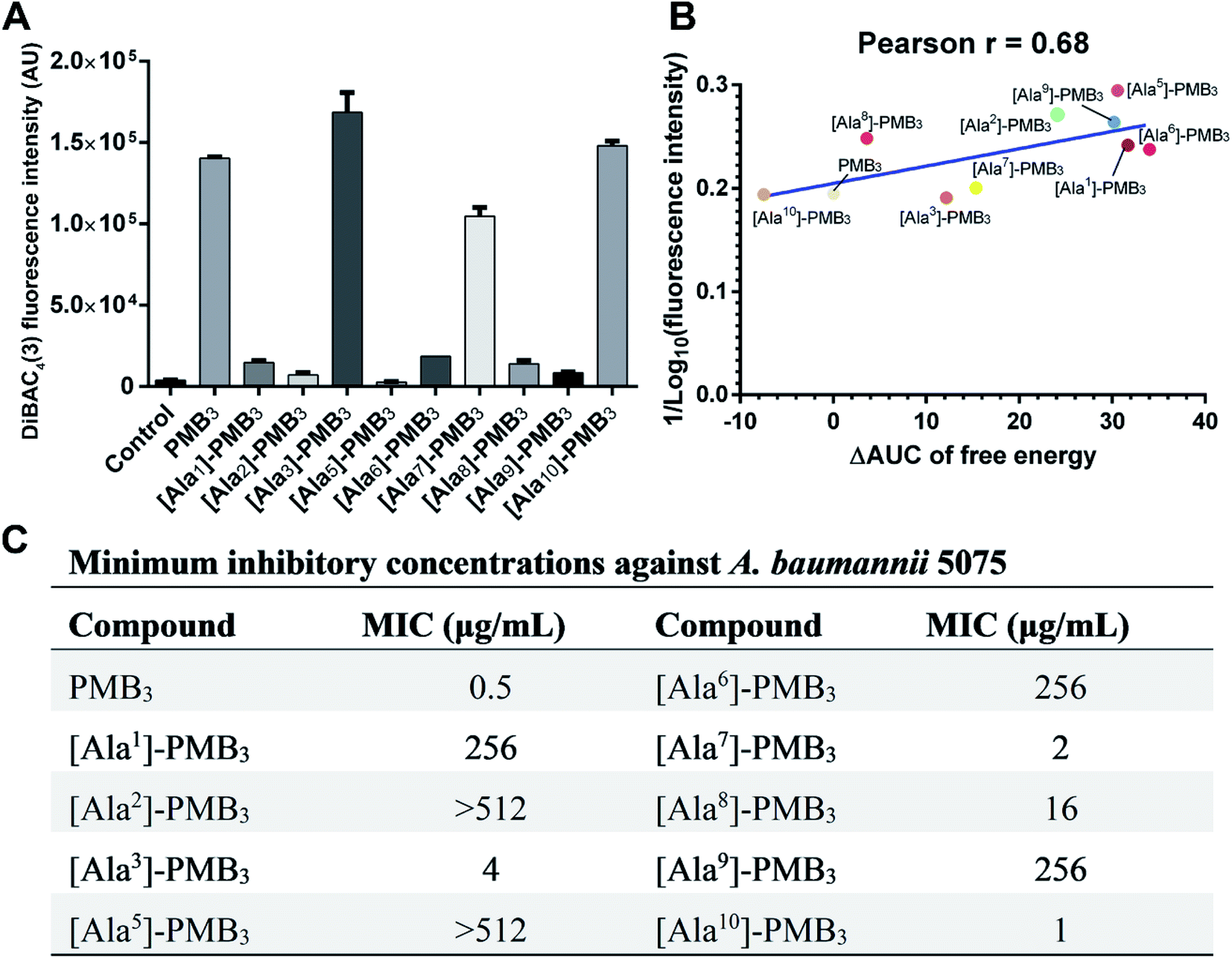 | ||
| Fig. 4 In vitro characterization of polymyxin B3 (PMB3) and analogs. (A) Fluorescence intensity in A. baumannii 5075 following 1 h polymyxin treatment (n = 3). (B) Correlation between free energy and fluorescence intensity. ΔAUC is the difference in free energy between polymyxin B3 and the analog. (C) Minimum inhibitory concentrations (MICs) of polymyxin B3 and analogs. | ||
2.4. Effect of alanine-substitutions on the antimicrobial activity of polymyxins
The minimum inhibitory concentrations (MICs) of all analogs were measured against MDR A. baumannii 5075 (Fig. 4C) and four additional A. baumannii strains (Table S1†). Alanine-substitutions at Dab1, Thr2, Dab5, D-Phe6 and Dab9 led to loss of antibacterial activity compared to unsubstituted PMB3 (MICs ≥256 μg mL−1) which perfectly matched our OM penetration free energy results (Fig. 3). As Dab1 and Dab9 in unsubstituted PMB3 formed intensive electrostatic interactions with lipid A phosphate groups (Fig. 1E), their alanine-substitutions attenuated OM binding and substantially decreased OM disorganization and antibacterial activity (Fig. 4A). Alanine-substitutions of Thr2 and D-Phe6 inhibited the conformational transition and OM penetration (Fig. 2 and 3), which explains their decreased activity. The significantly decreased activity and OM disorganization observed for [Ala5]-PMB3 (Fig. 4A and C) supports the hypothesis that loss of conformational flexibility is a major disadvantage for polymyxin antimicrobial activity.21 Alanine-substitution of Dab8 increased the MIC to 16 μg mL−1, whereas alanine-substitutions of Dab3 and Leu7 only slightly increased MICs (to 4 and 2 μg mL−1, respectively) despite the negative effect on their conformational transition. Alanine-substitution of Thr10 minimally increased the MIC (from 0.5 to 1 μg mL−1), consistent with its minor impact on conformational transition and OM penetration (Fig. 2 and 3).For an out-of-sample validation, we performed simulations with a novel polymyxin analog (FADDI-287) to examine the thermodynamics of penetration into the OM (Fig. 5A).29 FADDI-287 folded similarly to PMB3 during OM penetration and displayed a lower free energy barrier (−7.2 kcal mol−1) (Fig. 5B and C), which perfectly matched its superior activity (MICs = 0.125–0.5 μg mL−1) against A. baumannii compared to polymyxin B.29 These consistent computational and experimental results support the use of our novel all-atom MD system for predicting polymyxin antibacterial activity through examining their interactions with bacterial OMs.
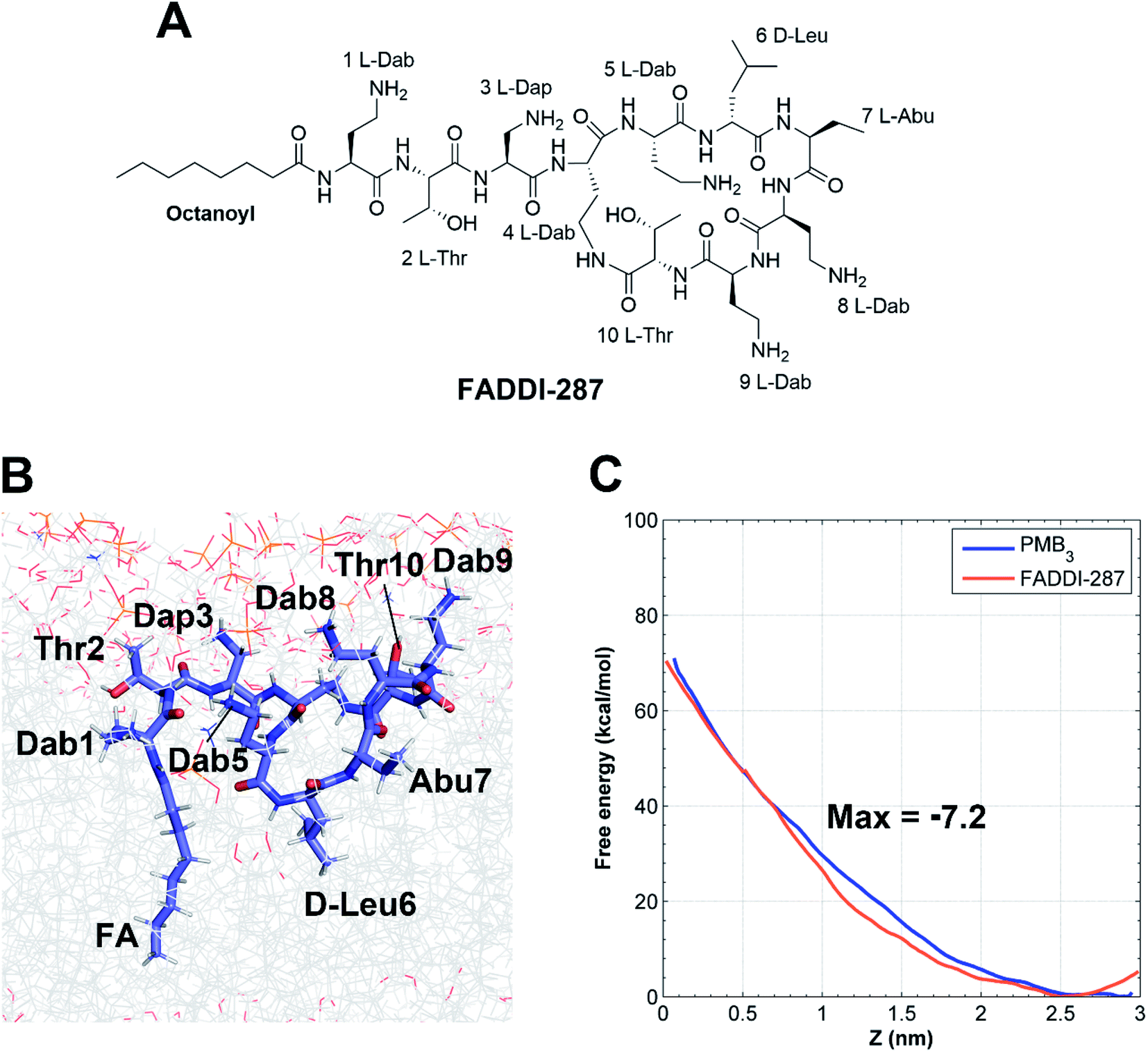 | ||
| Fig. 5 Penetration of FADDI-287 into the A. baumannii OM. (A) Chemical structure of FADDI-287. (B) The folded conformation of FADDI-287 in the hydrophobic center of the OM. (C) Free energy profiles of the penetration of PMB3 and FADDI-287 through the outer leaflet of the OM. | ||
2.5. A novel SAR model of polymyxins based on computational and experimental results
Finally, we developed a new SAR model for the interaction of polymyxins with the OM of A. baumannii (Fig. 6) that is representative of Gram-negative pathogens. Our OM-based SAR model represents significant improvements on previous SAR models utilizing a single LPS molecule.6,8,10,11 Our OM-based SAR model revealed for the first time the specific role of each residue in facilitating polymyxin folding and interactions with LPS and the various bacterial phospholipids in the OM. This is a significant advancement from our previous SAR model,6 which only focused on the interaction of one polymyxin molecule with a single LPS. The importance of the hydrophobic groups at the N-terminus and positions 6 and 7 of the polymyxin ring was highlighted by their interaction with the lipid A hydrocarbon tails. Alanine-substitutions of Thr2 and Dab3 significantly perturbed conformational folding because both residues modulate rotation of the polymyxin fatty acyl chain and cyclic ring. Alanine-substitution at Dab3 decreased activity 8-fold (Fig. 4C). Given the residue at position 3 is variable in natural polymyxins (e.g.D-Ser in polymyxin D), it seems that the positively-charged residue at position 3 is not essential for antibacterial activity against A. baumannii. In contrast, the positive charges from Dab1, Dab5 and Dab9 are necessary due to their key roles in regulating the structural flexibility of polymyxins. Alanine-substitution of Dab8 minimally impacted the conformational transition and OM penetration but dramatically attenuated OM disorganization and activity, likely due to Dab8 forming stronger hydrophobic interactions with the OM via side chain carbon atoms than electrostatic interactions (Fig. 1E). This discrepancy suggests that mechanism(s) other than OM penetration contribute to OM disorganization and polymyxin antimicrobial activity.30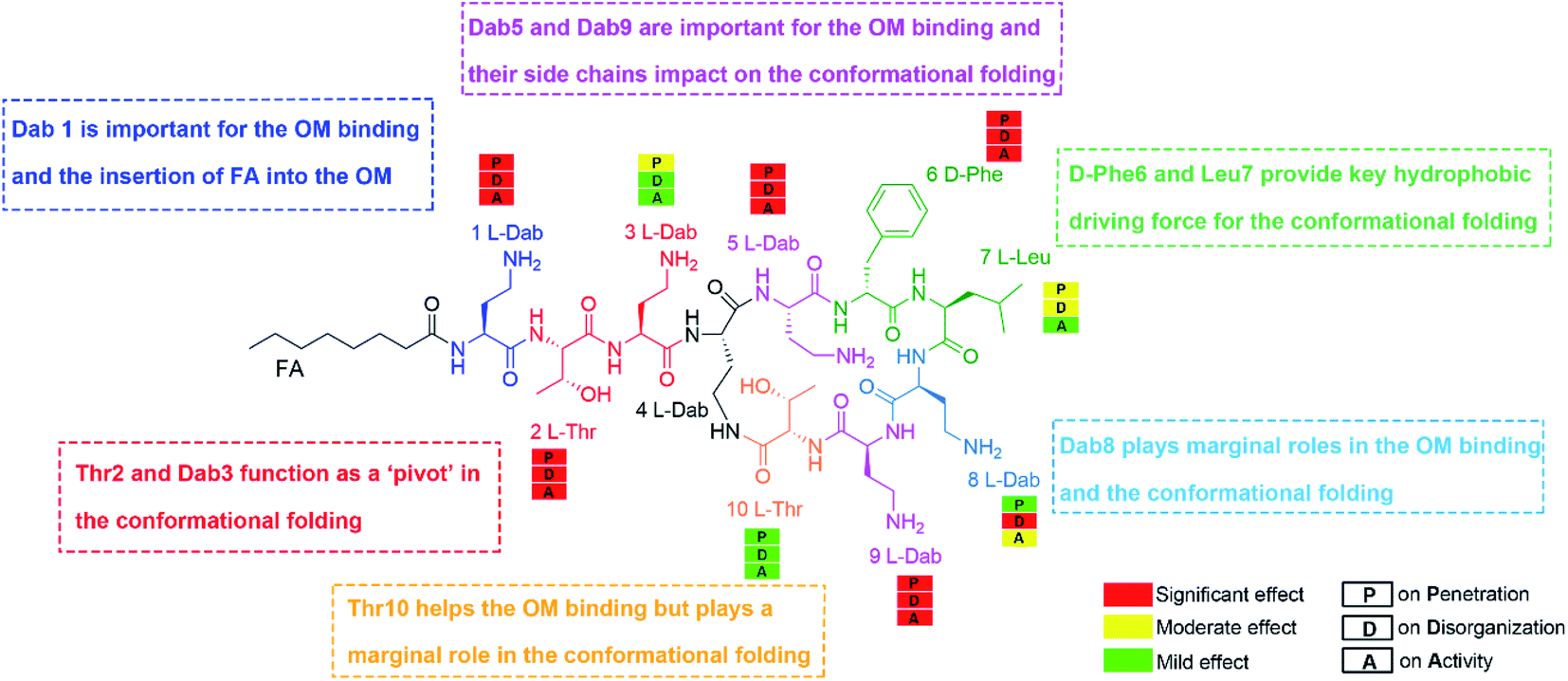 | ||
| Fig. 6 An outer membrane based structure–activity relationship model of polymyxins. | ||
Our multiscale results provide an integrative picture of how polymyxins act on the Gram-negative OM. In solution, Dab5 and Dab9 enable polymyxins to maintain extended conformations, maximizing the OM binding surface. Subsequently, the hydrophobic segments (i.e., fatty acyl chain, D-Phe6 and Leu7) reorientate towards the OM hydrophobic center with Thr2 and Dab3 acting as pivot points, forming a unique folded conformation. Importantly, this folded conformation divides the polymyxin molecule into hydrophilic and hydrophobic domains, enabling optimal interactions with the headgroup and hydrocarbon tails of lipid A, respectively. These interactions disorganize the OM and increase its permeability, which may lead to the leakage of intracellular contents and eventually bacterial death.9
3. Conclusion
Collectively, we systematically investigated the interactions of alanine-substituted polymyxin analogs with the OM of A. baumannii at the atomic level, and a novel OM-based SAR model was developed for polymyxins. For the first time, the role of each amino acid residue in polymyxin antimicrobial activity was elucidated. Importantly, the well-matched in silico and experimental results demonstrated the capability of our OM-based SAR model for predicting the activity of novel polymyxin analogs. Our integrated computational and chemical biology approaches will significantly accelerate the discovery of new-generation polymyxins against Gram-negative ‘superbugs’.4. Materials and methods
4.1. Molecular dynamics simulations
Chem3D and SwissParam server were employed to construct the structural models and topology parameters of all polymyxin analogs, respectively.31 An asymmetric OM model was constructed using the CHARMM-GUI membrane builder.32 The size of the OM model was approximately 7.6 × 7.6 nm2 (x–y plane). Importantly, previous quantitative lipidomics results attained from A. baumannii 5075 were employed to determine the exact lipid composition of the bacterial OM used in the simulations.17 Initially, the polymyxin molecules were placed above the outer leaflet of the OM by replacing the overlapping water molecules (Fig. S1, ESI†).33To track the penetration of polymyxins into the A. baumannii OM, steered simulations with three independent replicates were performed for each polymyxin analog.34–36 An adaptive force was applied on the centres of mass (COMs) of both the polymyxin molecules and the OM along the Z-axis. The force constant of the harmonic potential was initially set to 500 kJ mol−1 nm−2 and changed adaptively according to the interaction between the polymyxin molecule and the OM. The pulling force profiles from different steered simulations of PMB3 showed a similar changing trend (Fig. S26†); therefore, we selected the first steered simulation to initiate the umbrella sampling simulations.
Umbrella sampling simulations were performed to characterize the energetics during penetration of polymyxins into the OM.34–36 The system configurations from the steered simulation trajectory were used to build the umbrella sampling windows where the polymyxin molecule was harmonically restrained at various fixed positions at the Z-axis with an interval of 0.2 nm, resulting in approximately 18 simulation windows in each polymyxin simulation system; additionally, several extra windows were supplemented at the positions where sampling was poor (Fig. S23†). Each window was simulated for 50 ns; hence, a total of approximately 1100 ns all-atom simulations were performed to obtain a single free energy profile that quantitatively describes the penetration of the polymyxin from bulk water to the hydrophobic centre of the OM. The WHAM integration method was used to calculate the free energy profile.37 For the umbrella sampling simulations with PMB3, we performed 200 ns simulations for each window. In these simulations, the free 5 and the free energy profiles calculated from different parts of the simulation trajectories of analogs showed almost identical free energy profiles (Fig. S28†). The statistical errors of the free energy profiles were estimated using the bootstrap analysis method as previously described.37 In the reaction coordinates, Z >0 indicated the bulk water shell and the outer leaflet of the OM; Z = 0 corresponded to the hydrophobic centre of the OM.
All MD simulations were performed using GROMACS 5.1.2 with the CHARMM36 all-atom force field.33,38 Energy minimization was performed using the steepest descent method with the maximum force tolerance of 1000 kJ mol−1 nm−1. To fully equilibrate the simulation system, six equilibration cycles were carried out by gradually turning off the position restraints on the lipids. Subsequently, 400 ns nonrestraint simulations were performed to further equilibrate the OM. The area per lipid A molecule fluctuated around 1.76 ± 0.02 nm2 (Fig. S29†), which is consistent with the literature.13 Periodic boundary conditions were considered. All production simulations were conducted at constant temperature and pressure (NPT ensemble). The temperature and pressure were maintained at 313 K using the Nose–Hoover algorithm and 1 bar using the semi-isotropic pressure coupling method with Parrinello–Rhaman barostat, respectively.39–41 The electrostatic and Van der Waals interactions were calculated using the Particle Mesh Ewald method and Lennard–Jones potential algorithm, respectively.42 The time step was 2 femtoseconds and the trajectory was recorded every 10 picoseconds in production simulations.
4.2. Materials for peptide synthesis
The general synthetic methodology used for the synthesis of the peptides has been previously described in detail elsewhere by our group.43 The materials and synthesis protocol used are also presented below.Piperidine, diisopropylethylamine (DIPEA) and trifluoroacetic acid (TFA) were purchased from Auspep (Melbourne, Australia). Fmoc-Dab(Boc)-OH, was purchased from Try-lead Chem (Hangzhou, China). Fmoc-D-Phe-OH, Fmoc-Dab(ivDde)-OH, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and 1H-benzotriazolium-1-[bis(dimethylamino)methylene]-5-chloro hexafluoro phosphate-(1-),3-oxide (HCTU) were purchased from Chem-Impex International (Wood Dale, IL, USA). Fmoc-Ala-OH, Fmoc-Leu-OH and Fmoc-Thr(tBu)-OH were purchased from Mimotopes (Melbourne, Australia). Dimethylformamide (DMF), methanol (MeOH), diethyl ether, dichloromethane (DCM), hydrochloric acid (HCl) and acetonitrile were purchased from Merck (Melbourne, Australia). Fmoc-Thr(tBu)-TCP-Resin was purchased from Intavis Bioanayltical Instruments (Germany). Triisopropylsilane (TIPS), 1,2-ethanedithiol (EDT), diphenylphosphorylazide (DPPA) and hydrazine were obtained from Sigma-Aldrich (Castle Hill, Australia).
4.3. Protocol of peptide synthesis
Synthesis of the protected linear peptide was conducted on a Protein Technologies Prelude automated peptide synthesizer using standard Fmoc solid-phase peptide chemistry. Synthesis was carried out using TCP-Resin pre-loaded with Fmoc-Thr(tBu)-OH (0.1 mmol scale). Coupling of all Fmoc-amino acids was performed using the same standard protocol 3 molar equivalents (relative to resin loading) of the Fmoc amino acid and the coupling reagent HCTU in DMF, with activation in situ using 6 molar equivalents of DIPEA. The reaction was left to proceed for 50 min at room temperature which was followed by washing of the resin with DMF. Fmoc deprotection was conducted using the protocol 20% piperidine in DMF (1 × 5 min, 1 × 10 min), followed by washing of the resin with DMF at room temperature. Removal of the ivDde protecting group was achieved with 3% hydrazine in DMF (4 × 15 min), followed by washing of the resin with DMF. The protected linear peptide was then cleaved from the resin by treating the resin with 10% hexafluoroisopropanol (HFIP) in DCM (1 × 30 min, 1 × 5 min). This solution was concentrated in vacuo to give the crude protected linear peptide. The protected linear peptide was dissolved in DMF (5 mL) to which DIPEA 0.6 mmol, 104 μL and DPPA, 0.3 mmol, 0.65 μL were added, and the resulting solution stirred for 6 h at room temperature. The cyclisation solution was concentrated under vacuum overnight and the resulting residue taken up in a solution of 2.5% EDT and 5% TIPS in TFA and stirred for 2 h at room temperature. To this solution, 40 mL of diethyl ether was added to precipitate the crude peptide. The resulting precipitate was collected by centrifugation, washed twice with diethyl ether (40 mL), then dried to give the crude cyclic peptide product as a white solid. The crude peptide was dissolved in Milli-Q water (5 mL), de-salted using a Vari-Pure IPE SAX column, then purified by RP-HPLC on a Waters Prep LC system with a Waters 486 tuneable absorbance detector (214 nm) and a Phenomenex Axia Luna C8(2), 250 × 21.2 mm i.d., 100 Å, 10 μm column. Peptides were eluted from the column with a gradient of 0–60% buffer B over 60 min at a flow rate of 15 mL min−1; buffer A was 0.1% TFA/water, and buffer B was 0.1% TFA/acetonitrile.4.4. LC-MS analysis of peptides
The purity of the fractions collected was determined by LC-MS analysis on a Shimadzu 2020 LC-MS system incorporating a photodiode array detector (214 nm) coupled to an electrospray ionization source and a single quadrupole mass analyser. A Phenomenex column (Luna C8(2), 100 × 2.0 mm ID) was used, eluting with a gradient of 0–60% solvent B over 10 min at a flow rate of 0.2 mL min−1. Solvent A was 0.05% TFA in water and Solvent B 0.05% TFA in acetonitrile. Mass spectra were acquired in the positive ion mode with a scan range of 200–2000 m/z. Fractions with the desired purity were combined and lyophilised for two days to give the peptide product as its corresponding TFA salt. The yields obtained for each peptide and their corresponding analytical data are shown in ESI Appendix and Fig. S3–S12.†4.5. Flow cytometric analysis of the OM integrity
The cell membrane potential in depolarised cells was examined using an oxonol dye: bis-(1,3-dibutylbarbituric acid) trimethine oxonol (DiBAC4(3)).44–46 For flow cytometric analysis, bacterial samples (600 μL of bacterial suspension after appropriate dilution to keep the event per s <1000) were stained with the aforementioned stain. The final concentrations of DiBAC4(3) in the bacterial suspension was 1.67 μg mL−1 and stained samples were placed on ice in the dark for 1–2 min after a brief vortex before flow cytometric analysis.Flow cytometric analysis was performed using an ACEA NovoCyte® high-performance benchtop flow cytometer (ACEA Biosciences, San Diego, CA, USA).47 The forward scatter (FSC), side scatter (SSC), violet (VL2) and blue (BL1 and BL4) fluorescence of each cell were measured. DiBAC4(3) fluorescence emission (Ex/Em, 488/660–690 nm) was detected in the BL4 channel.45,48 All registered signals were logarithmically amplified. A gate was created in the dot plot of FSC vs. SSC and pre-set to distinguish bacterial cells from medium components. A linear plot was made of the FSC-H vs. FSC-A to avoid all the events from doublets. First unstained samples were examined through the flow cytometer to identify the cells labelled with dye combinations following staining.47 Data acquisition was set to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events at a low flow rate of <1000 events per s and collected data were analyzed (NovoExpress® software V2.1, ACEA Biosciences, San Diego, CA, USA). The mean fluorescence response from each treatment was determined and presented as mean ± standard deviation (SD).
000 events at a low flow rate of <1000 events per s and collected data were analyzed (NovoExpress® software V2.1, ACEA Biosciences, San Diego, CA, USA). The mean fluorescence response from each treatment was determined and presented as mean ± standard deviation (SD).
4.6. Determination of MICs
MICs of polymyxin B3 and its analogs were determined against five A. baumannii strains: A. baumannii 5075, A. baumannii ATCC 17978, A. baumannii ATCC 19606, A. baumannii 246-01-C.246, and A. baumannii 248-01-C.248 using the Clinical and Laboratory Standards Institute (CLSI) recommended microbroth dilution method. Two-fold dilutions of each antimicrobial agent ranging from 0–512 μg mL−1 were prepared in cation-adjusted Mueller-Hinton Broth (CAMHB; Oxoid, Hampshire, UK). Bacterial suspensions were prepared by suspending colonies grown on nutrient agar (School of Biomedical Sciences Media Prep Services, Clayton, Australia) in 0.9% NaCl to match a 0.5 McFarland standard (Densichek, BioMerieux). Bacterial suspensions were further diluted in CAMHB to yield ∼106 CFU mL−1, with 100 μL of this diluted suspension then added to 100 μL of antimicrobial solution in 96-well micro-titre plates (Techno Plas, St Marys, Australia). Quality control was assessed using Pseudomonas aeruginosa ATCC 27853. Plates were incubated at 37 °C and visually examined after 20 h incubation to determine the MICs.Data availability
Additional information can be found in the ESI.†Author contributions
J. L. conceived and supervised the project. X. J. conducted molecular dynamics simulations and wrote the manuscript. N. A. P. and K. D. R. synthesized the polymyxin analogs. M. A. K. A. and H. W. conducted FACS experiments. H. Y. conducted in vivo experiments. J. Z., X. Z., and M. L. analyzed simulation data. B. G., L. W., W. M., K. Y., B. Y., F. S., and L. W. contributed to molecular dynamics simulations. P. E. T. and T. V. contributed to data analysis. All authors contributed to manuscript revision.Conflicts of interest
J. L., T. V., K. D. R., P. E. T. are listed as inventors on the patent WO2015149131 ‘Polymyxin Derivatives as Antimicrobial Compounds’ which covers FADDI-287 and has been licenced to Qpex Biopharma.Acknowledgements
This work was supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI132154). XKJ is a recipient of the Faculty Bridging Fellowship, Monash University. NAP is an Australia National Health and Medical Research Council (NHMRC APP1158171) Peter Doherty Fellow. JL is an Australia NHMRC Principal Research Fellow. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. The MD simulations in this study were performed on the HPC Cloud Platform (National Key Research Program, 2016YFB0201702) at Shandong University and the supercomputer at eResearch, Monash University.References
- R. C. MacLean and A. San Millan, Science, 2019, 365, 1082 CrossRef CAS PubMed.
- World Health Organization, https://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/, 2017.
- T. Velkov, K. D. Roberts, P. E. Thompson and J. Li, Future Med. Chem., 2016, 8, 1017 CrossRef CAS PubMed.
- D. Landman, C. Georgescu, D. A. Martin and J. Quale, Clin. Microbiol. Rev., 2008, 21, 449 CrossRef CAS PubMed.
- Y.-Y. Liu, Y. Wang, T. R. Walsh, L.-X. Yi, R. Zhang, J. Spencer, Y. Doi, G. Tian, B. Dong and X. Huang, Lancet Infect. Dis., 2016, 16, 161 CrossRef CAS PubMed.
- T. Velkov, P. E. Thompson, R. L. Nation and J. Li, J. Med. Chem., 2010, 53, 1898 CrossRef CAS PubMed.
- F. Rabanal and Y. Cajal, Nat. Prod. Rep., 2017, 34, 886 RSC.
- T. Velkov, K. D. Roberts, R. L. Nation, J. Wang, P. E. Thompson and J. Li, ACS Chem. Biol., 2014, 9, 1172 CrossRef CAS PubMed.
- R. E. Hancock, Annu. Rev. Microbiol., 1984, 38, 237 CrossRef CAS PubMed.
- J. Mares, S. Kumaran, M. Gobbo and O. Zerbe, J. Biol. Chem., 2009, 284, 11498 CrossRef CAS PubMed.
- K. Kanazawa, Y. Sato, K. Ohki, K. Okimura, Y. Uchida, M. Shindo and N. Sakura, Chem. Pharm. Bull., 2009, 57, 240 CrossRef CAS PubMed.
- A. Khondker and M. C. Rheinstädter, Chem. Pharm. Bull., 2020, 3, 1 Search PubMed.
- A. Rice and J. Wereszczynski, Biophys. J., 2018, 114, 1389 CrossRef CAS PubMed.
- L. Fu, M. Wan, S. Zhang, L. Gao and W. Fang, Biophys. J., 2019, 118, 138 CrossRef PubMed.
- N. A. Berglund, T. J. Piggot, D. Jefferies, R. B. Sessions, P. J. Bond and S. Khalid, PLoS Comput. Biol., 2015, 11, e1004180 CrossRef PubMed.
- D. E. S. Santos, L. Pol-Fachin, R. D. Lins and T. A. Soares, J. Chem. Inf. Model., 2017, 57, 2181 CrossRef CAS PubMed.
- Y. Zhu, J. Lu, M.-L. Han, X. Jiang, M. A. K. Azad, N. A. Patil, Y.-W. Lin, J. Zhao, Y. Hu, H. H. Yu, K. Chen, J. D. Boyce, R. A. Dunstan, T. Lithgow, C. K. Barlow, W. Li, E. K. Schneider-Futschik, J. Wang, B. Gong, B. Sommer, D. J. Creek, J. Fu, L. Wang, F. Schreiber, T. Velkov and J. Li, Adv. Sci., 2020, 7, 2000704 CrossRef CAS PubMed.
- E. R. Rojas, G. Billings, P. D. Odermatt, G. K. Auer, L. Zhu, A. Miguel, F. Chang, D. B. Weibel, J. A. Theriot and K. C. Huang, Nature, 2018, 559, 617 CrossRef CAS PubMed.
- A. Hossein and M. Deserno, Biophys. J., 2020, 118, 624 CrossRef CAS PubMed.
- Y. Kinoshita, F. Yakushiji, H. Matsui, H. Hanaki and S. Ichikawa, Bioorg. Med. Chem. Lett., 2018, 28, 2713 CrossRef CAS PubMed.
- J. J. Meredith, A. Dufour and M. D. Bruch, J. Phys. Chem. B, 2009, 113, 544 CrossRef CAS PubMed.
- X. Jiang, K. Yang, M.-L. Han, B. Yuan, J. Li, B. Gong, T. Velkov, F. Schreiber, L. Wang and J. Li, ACS Infect. Dis., 2020, 6, 2698 CrossRef CAS PubMed.
- X. Jiang, K. Yang, B. Yuan, B. Gong, L. Wan, N. A. Patil, J. D. Swarbrick, K. D. Roberts, F. Schreiber, L. Wang, T. Velkov and J. Li, J. Biol. Chem., 2020, 295, 15902 CrossRef CAS PubMed.
- X. Jiang, K. Yang, B. Yuan, M. Han, Y. Zhu, K. D. Roberts, N. A. Patil, J. Li, B. Gong, R. E. Hancock, T. Velkov, F. Schreiber, L. Wang and J. Li, J. Antimicrob. Chemother., 2020, 75, 3534 CrossRef CAS PubMed.
- K. Huang and A. E. Garcia, Biophys. J., 2013, 104, 412 CrossRef CAS PubMed.
- I. C. Yeh, M. A. Olson, M. S. Lee and A. Wallqvist, Biophys. J., 2008, 95, 5021 CrossRef CAS PubMed.
- E. Zahavy, S. Rotem, D. Gur, R. Aloni-Grinstein, M. Aftalion and R. Ber, J. Fluoresc., 2018, 28, 1151 CrossRef CAS PubMed.
- D. Mason, R. Allman, J. Stark and D. Lloyd, J. Microsc., 1994, 176, 8 CrossRef CAS PubMed.
- K. D. Roberts, J. Wang, H. Yu, L. Wang, O. Lomovskaya, D. Griffith, S. Hecker, M. Dudley, P. E. Thompson, R. L. Nation, T. Velkov and J. Li, Poster-434, ASM Microbe, Boston, MA, 2016 Search PubMed.
- M. J. Trimble, P. Mlynárčik, M. Kolář and R. E. Hancock, Cold Spring Harbor Perspect. Med., 2016, 6, a025288 CrossRef PubMed.
- V. Zoete, M. A. Cuendet, A. Grosdidier and O. Michielin, J. Comput. Chem., 2011, 32, 2359 CrossRef CAS PubMed.
- J. Lee, D. S. Patel, J. Ståhle, S.-J. Park, N. R. Kern, S. Kim, J. Lee, X. Cheng, M. A. Valvano and O. Holst, J. Chem. Theory Comput., 2018, 15, 775 CrossRef PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701 CrossRef CAS PubMed.
- S. Yesylevskyy, S. J. Marrink and A. E. Mark, Biophys. J., 2009, 97, 40 CrossRef CAS PubMed.
- J. Zhao, C. Zhao, G. Liang, M. Zhang and J. Zheng, J. Chem. Inf. Model., 2013, 53, 3280 CrossRef CAS PubMed.
- D. Sun, J. Forsman, M. Lund and C. E. Woodward, Phys. Chem. Chem. Phys., 2014, 16, 20785 RSC.
- J. S. Hub, B. L. De Groot and D. Van Der Spoel, J. Chem. Theory Comput., 2010, 6, 3713 CrossRef CAS.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- C. A. Galea, M. Han, Y. Zhu, K. Roberts, J. Wang, P. E. Thompson and T. Velkov, J. Nat. Prod., 2017, 80, 1264 CrossRef CAS PubMed.
- R. LopezAmoros, S. Castel, J. ComasRiu and J. VivesRego, Cytometry , 1997, 29, 298 CrossRef CAS.
- D. Deere, J. Porter, C. Edwards and R. Pickup, FEMS Microbiol. Lett., 1995, 130, 165 CAS.
- H. Strauber and S. Muller, Cytometry, Part A, 2010, 77, 623 CrossRef PubMed.
- X. Z. Gong, E. Garcia-Robledo, A. Schramm and N. P. Revsbech, Appl. Environ. Microbiol., 2016, 82, 1412 CrossRef CAS PubMed.
- F. Joux and P. Lebaron, Microbes Infect., 2000, 2, 1523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03460j |
| This journal is © The Royal Society of Chemistry 2021 |