
Contributing to energy sustainability: a review of mesoporous material supported catalysts for Fischer–Tropsch synthesis
Leonard Uchejim
Okonye
 *a,
Yali
Yao
a,
Diane
Hildebrandt
*a and
Reinout
Meijboom
b
*a,
Yali
Yao
a,
Diane
Hildebrandt
*a and
Reinout
Meijboom
b
aInstitute for the Development of Energy for African Sustainability (IDEAS), University of South Africa (UNISA), Private Bag X6, Florida, 1710, South Africa. E-mail: yaoy@unisa.ac.za
bDepartment of Chemistry, University of the Johannesburg, PO Box 524, Auckland Park, Johannesburg, 2006, South Africa
First published on 19th November 2020
Abstract
Fischer–Tropsch synthesis (FTS) provides an alternative route for the conversion of carbon-containing feedstocks, such as biomass and municipal waste, to valuable end products. The concern, however, is how to make the process more efficient in terms of the activity of the catalyst and selectivity to desired products and in this way provide an economically competitive and sustainable technology to supply fuels and chemicals to global energy suppliers. This review centres on the optimisation of metals and the use of mesoporous materials to enhance the structure–performance relationship of catalysts used in FTS. The enhancement takes advantage of active site accessibility, increased surface-to-volume ratios and improved chemical potential, to facilitate optimum catalyst performance, therefore improving the entire process and minimising CO2 emissions. The unique properties of nanoparticles and mesoporous materials, higher volumetric activity, the controllability afforded by morphology modification of mesoporous materials and the various oxidation states of mesoporous metal oxides can be exploited for catalyst development.
 Leonard Uchejim Okonye | Leonard Okonye is a research fellow at the Institute for the Development of Energy for African Sustainability (IDEAS), University of South Africa. He obtained his M.Sc. (Eng) degree from The University of the Witwatersrand in 2011, and a PhD degree from the University of Johannesburg in 2019. His research interests are in energy sustainability, and the design of catalysts and efficient processes with reduced CO2 emissions, for energy conversion technologies. |
 Yali Yao | Yali Yao is an associate professor at the Institute for the Development of Energy for African Sustainability (IDEAS) at the University of South Africa, South Africa. She obtained her PhD degree from the University of the Witwatersrand, South Africa, and master's degree from East China University of Science and Technology, China. Her research areas focus on the Coal/Biomass/NaturalGas/Waste-to-Liquid Fuels via Fischer–Tropsch Syntheses (FTS) and Deep Desulfurization of Diesel Fuels. |
 Diane Hildebrandt | Diane Hildebrandt is the Director of the Institute for the Development of Energy for African Sustainability (IDEAS) and a Professor of Chemical Engineering at the University of South Africa. She is also a Member of the Council of the University of Johannesburg. She is a B1 rated researcher by the NRF. Diane has been appointed the Director of the newly formed Comprehensive Biomass Utilization Laboratory at the Hubei University of Science and Technology (HBUST), China. Diane obtained her B.Sc., M.Sc. and Ph.D. from the University of the Witwatersrand. She has authored or co-authored over 170 scientific papers, including an invited paper in Science, and has supervised over 100 postgraduate students. She has been a plenary speaker or invited speaker at numerous local and international conferences. She was awarded the President's Award by the Foundation for Research and Development and the Distinguished Researcher Award by the University of the Witwatersrand in 1996. In 1997 she became the first engineer to be awarded the Royal Society of South Africa's Meiring Naude Medal. In 2000 she and a colleague were the first academics to be awarded the Bill Neale-May Gold Medal by the South African Institute of Chemical Engineers. In 2002 she was made a Fellow of the Royal Society of South Africa and also received the Vice Chancellor's Research Award of the University of the Witwatersrand. In 2003 she was elected as a member of the Academy of Sciences of South Africa. In 2005 she was recognized as a world leader in her area of research when she was awarded an A rating by the National Research Foundation. In 2006 she was elected a Fellow of the Academy of Engineering of South Africa. In 2009 she won the Distinguished Woman Scientist Award from the Department of Science and Technology and the African Union Continental Scientific Award in the category Basic Science, Technology and Innovation. In 2010 she was awarded the ASSAf ‘Science-for-Society’ Gold Medal Award. African Union Continental Scientific Award in the category Basic Science, Technology and Innovation. In 2017, Diane was conferred the NSTF Research and Capacity Development award. In addition she was chosen as one of the 100 Foreign Experts to advise Government of Hubei on the development of new, green and sustainable technologies for the province. |
 Reinout Meijboom | Reinout Meijboom was born and raised in the Netherlands. He studied chemistry at the University of Groningen where he completed his MSc research under Prof Jan Teuben. Subsequently, he moved to Cape Town to pursue his PhD in organometallic chemistry under the supervision of Prof John R. Moss. Following his PhD, he pursued a post-doctoral fellowship with Prof Andre Roodt at the then Rand Afrikaans University (currently University of Johannesburg) and the University of the Free State. Reinout took up the position of Senior Lecturer at the University of Johannesburg in 2008. There he changed his research direction towards heterogeneous catalysis, while continuing to pursue the crystallography of silver coordination compounds. These compounds were found to show apoptotic behavior in cancer cells, and this project is ongoing. He raised through the ranks at UJ to full professor and was the former head of department. Reinout has published over 175 research papers. |
1. Introduction
The Fischer–Tropsch Synthesis (FTS) process is an important industrial-scale heterogeneously catalysed reaction, and a promising alternative to convert syngas derived from coal/natural gas/biomass/waste into environmentally responsive fuels and chemicals, which has received much attention both from industry and academia.1–22 Iron (Fe), cobalt (Co), nickel (Ni), and occasionally ruthenium (Ru), that are generally supported on an inert oxide (for example silica, titania, or alumina), have been used for the conversion. The reaction is sometimes promoted with an alkali metal oxide to improve the catalytic performance. The industrial catalysts of choice have been iron and cobalt-based.5,23An iron-based catalyst is most commonly used because it is: less expensive; effective under different process conditions (temperature, pressure, and H2/CO ratio); and more suitable for H2 deficient syngas sources.24 However, it does have flaws, including: low alkane selectivity, even at high conversion; the tendency to form more alkenes and oxygenates; it is easily deactivated, due to carbon deposition, coking and carbide formation.1,23,25,26 A cobalt-based catalyst is preferred, particularly for low temperature applications, because of high activity and selectivity for linear and long-chain alkanes. The cobalt-based catalyst is resistant to deactivation and has low selectivity to alkenes and oxygenates.1,27
Technically the performance of catalysts11–17,28–34 is managed by several parameters, such as the method of preparation; catalyst pre-treatment; dispersion; metal loading; nature of the support; and effect of the promoter if used. Amongst other things, support type and pore structure (pore size, distribution, pore volume and surface area) are believed to influence the performance of many catalysts significantly, in a number of reactions.25,35–44
Structural promotion using the mesoporous metal oxides as supports, combined with chemical promotion could enhance activity per unit weight of the metal, when the metals are adequately dispersed on the support material, which would, in turn, would increase the surface-to-volume ratio. The various molecular-scale interactions between the bimetallic metal oxide support and the metal particle of choice (such as changes in electronic state, metal–support interaction, surface structure and oxidation states), could improve the performance of the catalyst. This could also increase the catalyst lifespan and could aid in the adjustment of product yield to specifically targeted products. Thus, the efficient control of metal dispersion and the reduction of the metal could lead to a new design for metal-supported catalysts with improved catalytic activity and selectivity.35–37
It is proposed that the use of mesoporous materials could improve catalytic performance, due to easy control of the catalyst structure (morphology), which could affect catalyst activity, selectivity and stability.45–50
The assumption is that mesoporous material will have a positive effect on mass and heat transfer, but in particular mass transfer due to enhanced diffusion.50–55 The ease with which the catalyst morphology can be controlled may help to understand the mechanism and kinetics of reactions. Another advantage that the material offers is the porosity, which would offer stability (by reducing agglomeration) and aid the uniform dispersion of the active metal, thereby increasing the dispersion and specific surface area. It would also: improve reactant and product diffusion in the mesoporous channel; interception of intermediates and secondary reactions; and limit chain growth by means of shape selectivity.8 This would improve the distribution of the hydrocarbon product yield from the FTS.
The effect of porosity of the support on activity and selectivity remain unclear, because of the difficulties involved in doing such a study. An adequate understanding of the catalyst active metal particle size and the relationship between the pore structure and support type could lead to the development of improved, efficient and selective catalysts.35–37
This review discusses the potential role that mesoporous material could play to improve the possible, desirable and necessary products. The studies done on nanostructured and mesoporous materials will help to build an understanding of the complex systems at the atomic and molecular level, and the phenomena could, therefore, be exploited, in order to design new heterogeneous catalytic systems that would provide a more efficient and sustainable process.56–63
2. Mesoporous materials in catalysis
Adequate coordination of metal particles within the pores of mesoporous materials is important, as it helps with elucidating the effect of pore size and structure. The strength of the interaction between a metal complex and the support affects the reducibility of the metal oxide loaded onto the support. Although the role of TiO2, Al2O3, ZrO2 and SiO2 as a support for active metals is well-known, there is a great deal of interest in the need to develop new supports that are better at influencing catalytic activity.64–74 An promising material that can play this role is ordered mesoporous materials (OMMs). The reasons for the growing interest in OMMs are large surface area, narrow pore size distribution and the tuneable pore size. Depending on the porous material, the surface area can sometimes attain a value of several thousand square meters per gram.49–51,73–83According to IUPAC classification, mesoporous materials84–86 are materials with pores within the scope of between 2–50 nm.87–90 The material is of great significance to the materials community, as their synthetic pore organisation, adsorption, and conductive and catalytic properties can be readily modified.45,48,50,73–75,91–101
Mesoporous materials are favoured for several applications, because of their highly ordered mesostructure and high surface area range, this allows for the adsorption and dispersion of molecular particles for a wide range of applications.87,90 The high surface area and highly ordered support texture of mesoporous materials, makes it possible to control the size as well as the dispersion of supported metal particles, which allows maximisation of the available reactive metal usage.25,35–37,46,50,102
After the discovery of mesoporous material, the demand for better reaction and separation generated much interest in metal-substituted and silica-based mesoporous materials. This was due to their potential applications in catalysis, separation, adsorption, biomaterials and nano-scaled devices.79,80,103–106 A huge assortment of nano-phase isolated composite materials can, therefore, be imagined, whereby a selection of the block copolymer species composition and chain architecture is employed in order to tune and self-assemble the material. This is achieved with the aid of process variables (for example, pH, temperature and environments), which are controlled, to direct the complexation of the desired resultant structures.45,46,54,73,93,103,104,107
Mesoporous silicas with important unique properties were first reported by Mobil Corporation scientists in 1992, with the discovery of the M41S family of mesoporous materials.73,79,80,91,92,108–115 Over the years, highly ordered mesoporous silica have received much attention, because of their controllable surface chemistry, very large surface area, tunable pore size (which can be modified over a wide range), excellent stability (chemical and thermal) and a large pore volume.116,117 This makes it a very promising catalyst support (or carrier), in general for a wide range of valuable prospective applications in the field of adsorption, separation and catalysis.79,80,91,92,112,113 Following the discovery of mesoporous silica, noteworthy advancements were made in controlling their morphology, composition variation, pore size alteration, synthesis methods and application developments.46,48–50,101,118–122
In 1998, interesting research was done by Zhao and colleagues,73,74 who produced clustered pores and a parallel hexagonal cylindrical array of pores of meso-structured silica, which came to be known as “Santa Barbara Amorphous no. 15 (SBA-15)”. The pore size range was 4.6–30 nm.74 This proved to be a research gamble that paved the way for further development and improvement of the mesoporous material.73–75 Since its discovery, SBA-15 has shown potential for use as a support material for the synthesis of novel catalytic materials, due to its uniform hexagonal array of channels, the tuneable pore size and narrow pore size distribution (see Fig. 1).115,123
 | ||
| Fig. 1 HRTEM (high resolution TEM) image of SBA-15: (a) long 1 D channels; (b) P6mm plane group and hexagonal structure. Reproduced from ref. 123 with permission from John Wiley and Sons, Copyright 2019. | ||
The tuneable pore diameter (2–50 nm) and the wall thickness of the framework give it a higher mechanical and hydrothermal stability than is seen with other materials.25,74,91,117,124 It became the preferred choice as catalytic support, because of these interesting properties. Nevertheless, sometimes there is a need to modify, functionalise or even immobilise other active elements into the porous structure, to enhance and optimise the catalytic activity of a synthesised catalyst for specific catalytic reactions and reusability.87,125–134
Recently, mesoporous transition metal oxides have been considered in some reactions, because of their multiple oxidation states, and the effect of the ensuing metal-oxide interface on catalytic activity and selectivity.43,122,135,136 Among them, iron and cobalt mesoporous materials are found/expected to generate higher CO conversion as well as C5+ hydrocarbon selectivity when compared to other conventional supports such as TiO2, SiO2 and Al2O3 for the FTS reaction.23Fig. 2 plots the TEM images of mesoporous cobalt oxide.
 | ||
| Fig. 2 TEM images of mesoporous Co3O4 heat-treated at 250 °C for 3 h: (a) 50 nm; (b) 5 nm scale bar. Insets: (a) 10 nm scale bar; (b) SAED (selected area electron diffraction image). The Co3O4 nanoparticles are 8–16 nm in diameter. Reproduced from ref. 122 with permission from AIP Publishing, Copyright 2014. | ||
2.1. Mesoporous materials synthesis
Generally, mesoporous silica materials are framed within conditions suitable for the self-associating molecules. The silica-surfactant self-assembly occurs simultaneously with the condensation of the inorganic species and yields mesoscopically-ordered composites (see Fig. 3).115,118,142,143 Numerous engineered pathways have accounted for the synthesis of these porous materials (with either a disordered or ordered pore framework with different structures), with the assistance of supportive surfactant templating, which can meet the demand of the intended application – both from an academic and industrial perspective.46,48–51,53,79–81,106,107,118,119,135,142–158
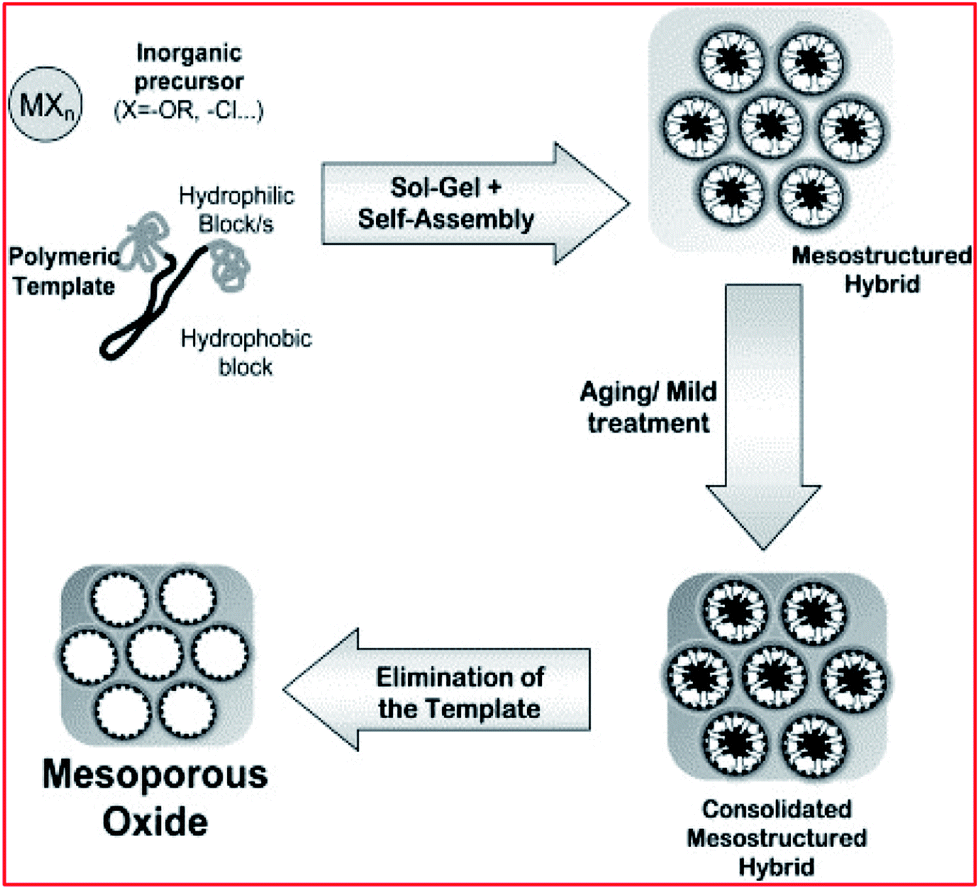 | ||
| Fig. 3 Schematic illustration of mesoporous material synthesis. Reproduced from ref. 143 with permission from Elsevier, Copyright 2003. | ||
The pathways were developed to take advantage of the structure-directing functionality of hydrogen-bonding, electrostatic, and van der Waals interactions linked utilizing the amphiphilic molecules for the self-assembly. The surfactant self-assembly then takes place concurrently with the condensation of the inorganic species and produces mesoscopically-ordered composites. This is achieved with the help of cosolvent organic molecules and extended hydrothermal treatment (ageing step) during synthesis, which enlarges the pore size up to 10 nm (100 Å)73,91,92,98,105,159–163 (see Fig. 4) With the synthesis of ordered mesoporous silica, template removal is critical, as the process could alter the final properties of the preferred permeable porous material. A discretionary washing step before calcination may assist with the removal of the organic polymer specie.87–90,137 The standard technique for removing the template is calcination, usually at 500 °C ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 73,164–166 (see Fig. 4).
73,164–166 (see Fig. 4).
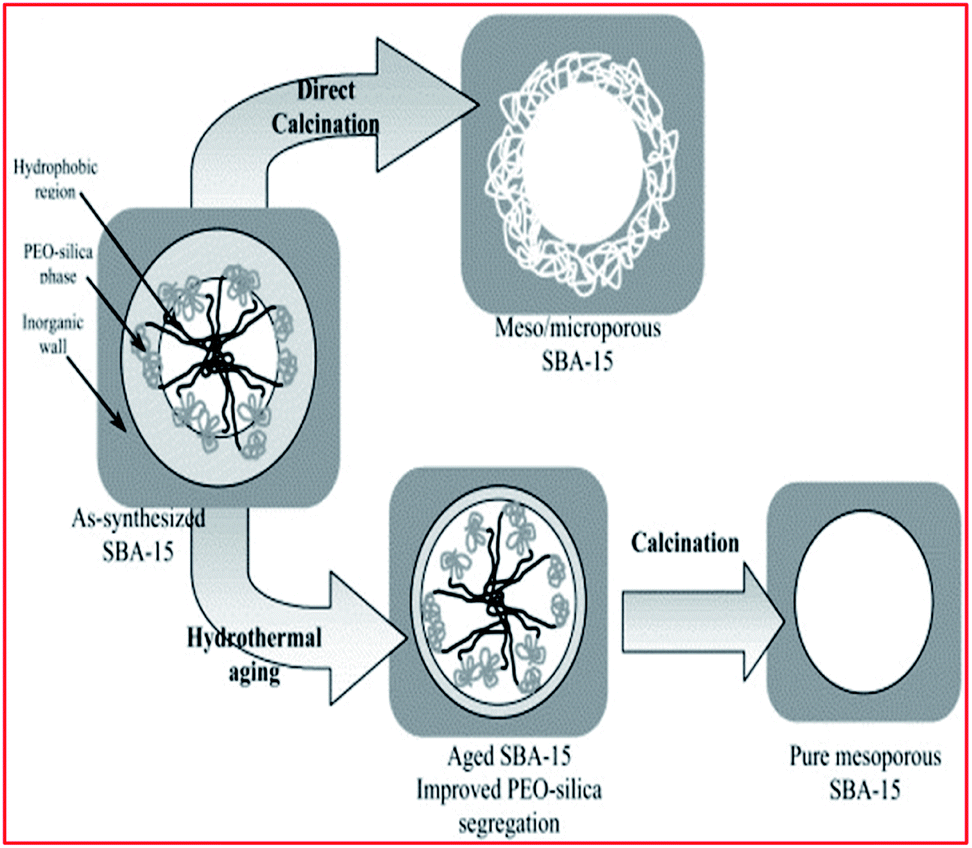 | ||
| Fig. 4 Schematic illustration of aging and calcination steps. Reproduced from ref. 143 with permission from Elsevier, Copyright 2003. | ||
The successful synthesis of ordered mesoporous silica (SBA-15) entails a summarised two-step approach that comprises: an initial step at 35–40 °C; and a vital extended hydrothermal crystallisation step (usually after 24 h of ageing) between 80–120 °C.73–75,117,142,143,167,168 Variation of pore size and the silica wall thickness is related to the temperature heating rate (35–140 °C) and the reaction time (between, 11–72 hours) of the synthesised SBA-15 in the reaction mixture, used to generate systematically different structures with uniform pores. Larger pore size and thinner silica walls in the material are achieved by means of higher synthesis temperature and/or longer reaction time. It is a direct result of protonation and temperature-dependent hydrophilicity of the poly (ethylene oxide) specie of the block copolymer, under the acidic conditions.73–75,88,167,168
The use of co-solvent organic molecules and extended thermal treatment during synthesis can be employed to control the morphology of the composite mesoporous material by expanding the pore size and increasing the dimension of the pore structures during synthesis.167,170 This was accounted for by Mesa and colleagues,169 who argued that a co-surfactant (CTAB) can be used to modify the shape and size of particles when determining the morphology of SBA-15 mesoporous silica. Besides, the ethanol co-solvent used plays a key role in the arrangement and formation of flawless morphology. However, this ethanol addition might diminish the polarity of the solvent and so decrease the rate of nucleation and development of the mesostructured material that is produced.73,164–166
Unlike silica-based mesoporous materials used with the well-established synthesis method, the synthesis method for transition metal oxides – by either soft templating, hard templating or other templating pathways – is somewhat difficult and challenging.47,81,103,104,120,142,143,149,153,157,170 However, the successful synthesis of such materials has been reported when using soft templating inverse micelle for the creation of the mesoporosity. Further thermal treatment of the gel at different heating cycles, then results in the nano-crystallite material that contains mesopores, as reported.120,122,136,170
3. Mesoporous materials in FT
The dependence of FTS on the number of surface atoms available for catalysis makes mesoporous materials useful as supports when studying the structural effects on FTS catalytic activity and selectivity.36,171–173 This is due to the possibility of modifying the pore length, volume and diameter of the material, which, in turn, has an impact on the structural effect of the supports on catalytic performance. These highly ordered mesoporous materials (sometimes modified) have been employed as supports in FTS reaction studies,174–183 in order to determine the structural effect of the catalyst, supports on catalytic performance.36 The high surface area of the ordered mesoporous materials (>800 m2 g−1) and its regular porous structure, with narrow pore diameter distribution, allows for higher metal dispersion, even at high metal loadings. The mesoporous material is also able to exert control on metal particle size distribution, which influences the catalytic properties.Previous studies that were done on different silica-based mesostructured materials focused on the effect of the porosity, the reducibility and dispersion of the active metal, and the overall catalytic activity of the catalysts in the FTS reaction.184–192 The results of the studies showed a noticeable influence of porosity on metal size, metal dispersion, reducibility (degree of reduction (DOR)) and selectivity toward C5+ hydrocarbons. This was ascribed to diffusional limitation and a much less sterical hindrance within the support, due to the pore size and pore volume of the catalysts.
Investigations have been done on modifications to the catalyst texture and porosity, to improve catalyst reducibility. These modifications were achieved by the introduction of oxides (such as MnO and ZrO2),193–199 trimethylbenzene (TMB),200 aluminium and/or titanium;201–204 or by silylation205–207 of the mesoporous silica (SBA-15); or, in some cases, by metal promotion.188,191,208–210 The modified catalysts had higher reducibility, which led to better catalytic performance, due to high dispersion and readily available surface atoms (active sites).
The research was recently done on the use of penetrable pores211 and pore-expanded (3D mesocellular pore structured)212 mesoporous materials (MCM-41, SBA-15, SiO2) as supports for FT catalysts.213 Dispersion and degree of reduction of the catalyst was affected by pore size and the structure of the mesoporous silica support, which led to higher catalytic properties. An interesting recent development involves the use of other mesoporous materials (carbon, zeolite, alumina), and a combination of macroporous/microporous–mesoporous pored materials (to enhance mass transport in the mesoporous material) as supports for the Fischer–Tropsch synthesis reaction.125–134
In all the cases mentioned above, various features of the catalyst structure were exploited, to improve the performance of the catalysts, which resulted in better performance of the catalysts. The wider-pore materials facilitated better reducibility of the active metals and also allowed for controllability of the metal particle size, which enhanced the catalytic activity of the catalysts. It also led to rapid re-adsorption of 1-alkenes and subsequent chain growth and selectivity towards the C5+ hydrocarbons. This is accredited to an increase in mass transport restrictions; a lowered CO diffusion barrier to the active site due to a decrease in pore length (compared to the narrow-pored catalysts) and fewer diffusion restrictions.
3.1 Mesoporous materials supported cobalt-based catalysts
Li et al.214 studied the effect of meso–macroporous structured support on the performance of silica-based cobalt catalysts in the FTS reaction. The authors find the catalyst with a mesoporous size of 8.5 nm (Co/S3 catalyst) to have exhibited a higher catalytic activity with the maximum conversion of CO, which was ascribed to the balanced blend (synergy) of Co dispersion and reduction degree. The product distribution is shown in Fig. 5, more or less had a very strong dependence on the mesoporous diameter and Co particle size.
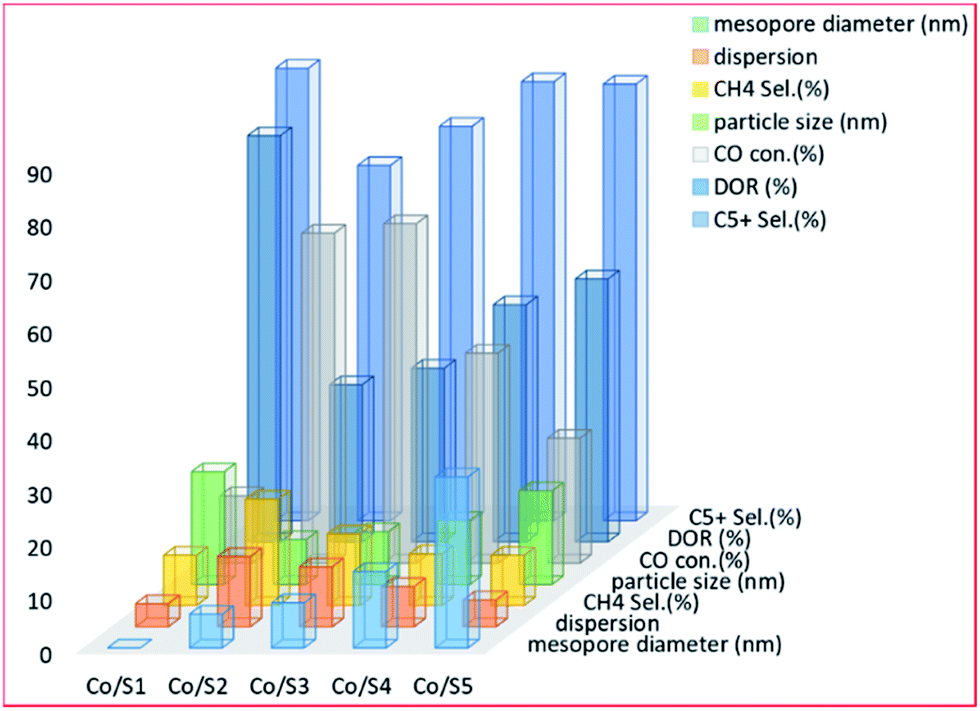 | ||
| Fig. 5 The effect of mesopore diameter size on catalyst performance for the catalysts [Co/S1, mesopore diameter 0 (nm); Co/S2, mesopore diameter 6.3 (nm); Co/S3, mesopore diameter 8.46 (nm); Co/S4, mesopore diameter 14.3 (nm) and Co/S5, with mesopore diameter of 32 (nm)], adapted from ref. 214. | ||
The macropores, however, played a role in reducing internal diffusion limitation through enhanced mass transfer, while the mesopores provided the required high surface area (greater availability of the surface-active site) and suitable dispersion of the active component, thereby improving the CO conversion, FT reaction rate, and the general catalyst performance.
Prieto and colleagues186 studied the effect of pore length while using a cobalt-based catalyst prepared by wet impregnation on the SBA-15 support material. They found that the wider-pored catalysts have increased activity and selectivity towards C5+ hydrocarbons. The catalysts had a pore diameter of approximately 11 nm, and different pore lengths (0.33, 1.15 and 5.70 μm). The increase in selectivity was associated with a decrease in the pore length of the support, which resulted in a lowered CO diffusion barrier to the active site. This is coupled with an overall decrease in methane selectivity since wider pores facilitate the formation of longer hydrocarbon chains. The increase in these long-chain hydrocarbons is ascribed to the lower H2/CO ratio near the active sites of the short-pored catalysts, which experience fewer diffusion restrictions. This increase in CO monomer concentration near active sites allows chain propagation during FTS reactions. The authors inferred that for non-interconnected 1D-mesopores, such as SBA-15, the distance required for CO diffusion to become kinetically relevant is relatively shorter than that of other mesoporous materials. Fig. 6 shows the effect on the catalytic activity of a Co-based SBA-15 catalyst at 55 ± 2 CO conversion, of the pore size; wide (11 nm) and narrow (7 nm) pore supports; and nature (S, M or L, which refers to short, medium or long). Although the wide-long pored catalyst (Co/11SBA_L) had a better particle size (17.8 nm) and better reducibility (∼100%), it had low dispersion (6%) and displayed poor selectivity towards C5+ hydrocarbons, compared to the wide-short pored catalyst, i.e. Co/11SBA_S (10.2 nm). The latter had the least particle size, better dispersion and higher selectivity to C5+ hydrocarbons.
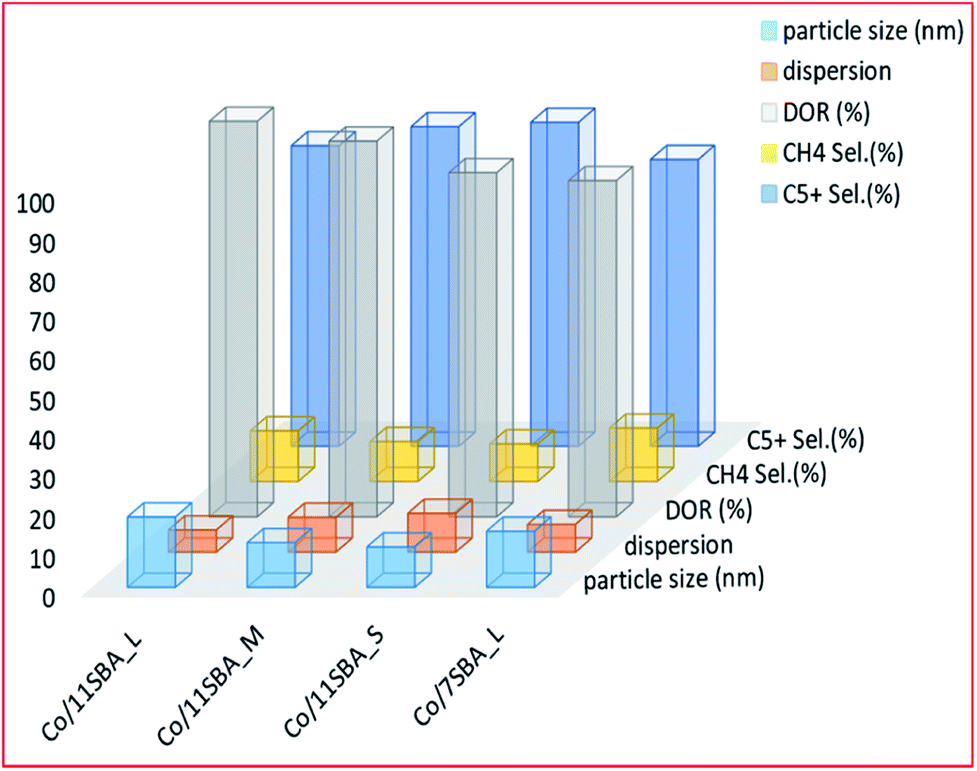 | ||
| Fig. 6 The effect of pore length and particle size on the catalyst performance for Co/SBA-15 catalysts [Co/11SBA_L, with a pore size of 11 (nm) long pores; Co/11SBA_M, with a pore size of 11 (nm) medium pores; Co/11SBA_S, with a pore size of 11 (nm) short pores and Co/7SBA_L, with a pore size of 7 (nm) long pores], adapted from ref. 186. | ||
While studying the role of pore size (using a 30% Co/SBA-15 catalyst, prepared by incipient wetness impregnation, IWI), Xiong et al.188,215 concluded that catalysts with larger pores gave rise to a larger cobalt cluster size, lower dispersion and higher reducibility and more bridge-type CO adsorbed on FTS. The researchers188 found the selectivity to be largely affected by the pore size: larger pores allow for the formation of larger C5+ alkanes. The reported results show solid and clear evidence that has been supported by other36,216 studies. However, the authors of this paper believe that an additional factor also played a role, i.e. shape selectivity.
Bartolini et al.187 investigated the effect of porosity on hydrocarbon distribution in FTS reaction, using 30% Co-SBA-15 catalysts with different pore sizes (5.0, 10.7 and 14.6 nm) prepared by impregnation. From the results, they concluded that pore size affects the size and reducibility of Co particles, and has a strong influence on liquid product distribution, with larger pores producing a higher molecular weight hydrocarbon fraction (see Fig. 7). This was credited to a shape selectivity effect induced by the pore size and pore volume of the catalysts.
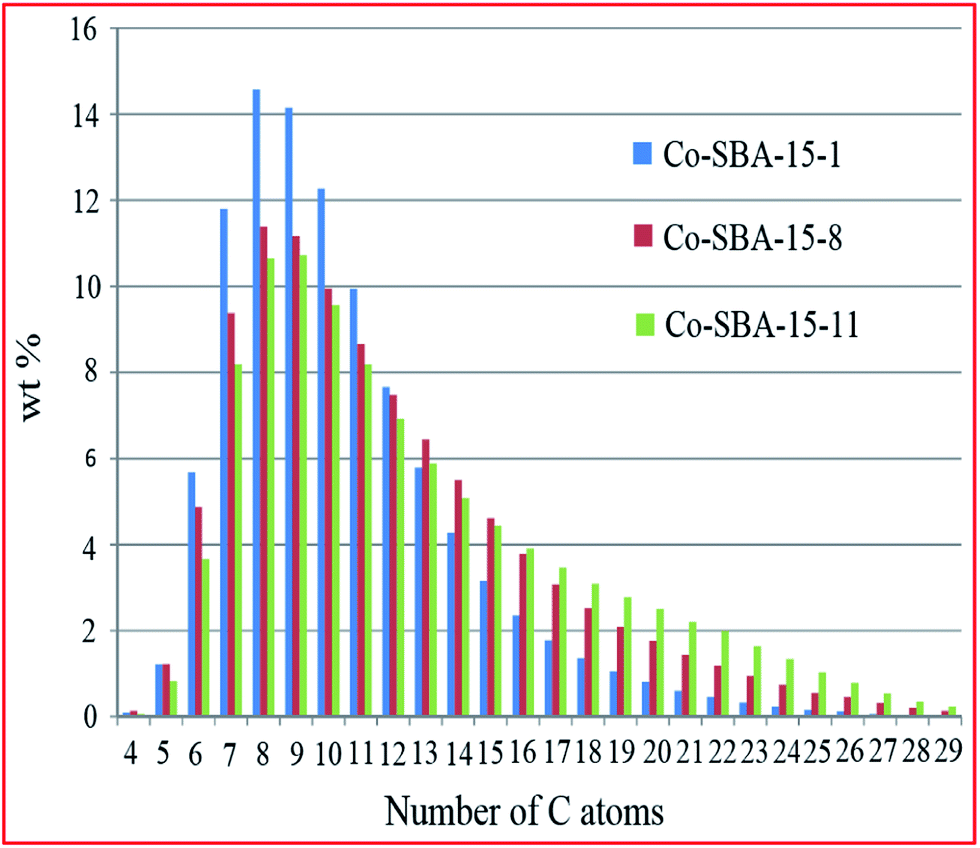 | ||
| Fig. 7 Comparison of the liquid hydrocarbons product distribution for the studied catalysts (Co-SBA-15-1 with a pore size of 5 nm; Co-SBA-15-8 with a pore size of 11 and Co-SBA-15-11 with a pore size of 15). Reproduced from ref. 187 with permission from Elsevier, Copyright 2015. | ||
Wang et al.189 used 20% Co-based SBA-15 catalysts with various pore diameters (3.6–12.0 nm), prepared by impregnation for their investigation. They used trimethyl-benzene (TMB) as a swelling agent to increase pore diameter and observed fewer CO conversions (6.7–23 mol%) compared to those (30–50 mol%) reported with Co-based catalysts loaded on conventional supports (SiO2, Al2O3, TiO2). They found that catalysts with larger pores and larger Co particles have a slightly higher conversion, at 250 °C. However, the authors did not provide a conclusive reason for the observed low activity and poor selectivity, but this could be due to difficulty in reducibility of the Co metal complexes, as a result of the strong Co interaction with the SBA-15 framework.
Li et al.190 investigated the effect of pore size on the FTS reaction for a series of silica-supported (MCM 48 and SBA-15) cobalt catalysts and metal loading effect, using MCM 48.217 The initial and steady-state CO conversions obtained for the SBA-15 based catalyst was intermediate, compared to the larger-pored MCM-48 catalyst. The catalytic behaviour of silica-supported cobalt catalysts in FT synthesis was found to be dependent on the nature of the Co species, the Co particle size and the mesoporous structure. The authors suggested that the mesoporous structure and pore size have a significant effect on the properties of the cobalt particles, and a positive effect on the selectivity.
Khodakov et al.218 also investigated the porosity and loading (5–55.9 wt%) effects on the reducibility and dispersion of a mesoporous material-supported cobalt-based catalyst for the FTS reaction. To account for the variable cobalt loadings, the cobalt time yield was used as a measure for catalytic activity. The results of the study show that the cobalt time yield increased with an increase in Co surface density when SBA-15 was used as a support. The selectivity was not as strongly affected and there was no significant difference between different metal loadings. The authors further investigated the relationship between pore size and performance of Co-based FTS catalysts supported on periodic-mesoporous silica.36 They found the reducibility of the Co species to be higher with larger-pored silica, because of the larger Co3O4 crystallites that are easier to reduce. The narrower-pored catalysts have smaller Co particles and showed lower activity in CO conversion, with selectivity tending towards methane formation.
Kim and colleagues219 also investigated cobalt-based SBA-15 catalysts for the FTS reaction. They investigated the impact of the impregnation method (incipient wetness (IW), post-synthesis (PS) impregnation and a supercritical solvent (SS)) on the reducibility of cobalt species. The supercritical technique resulted in a larger crystallite size and gave better reducibility.
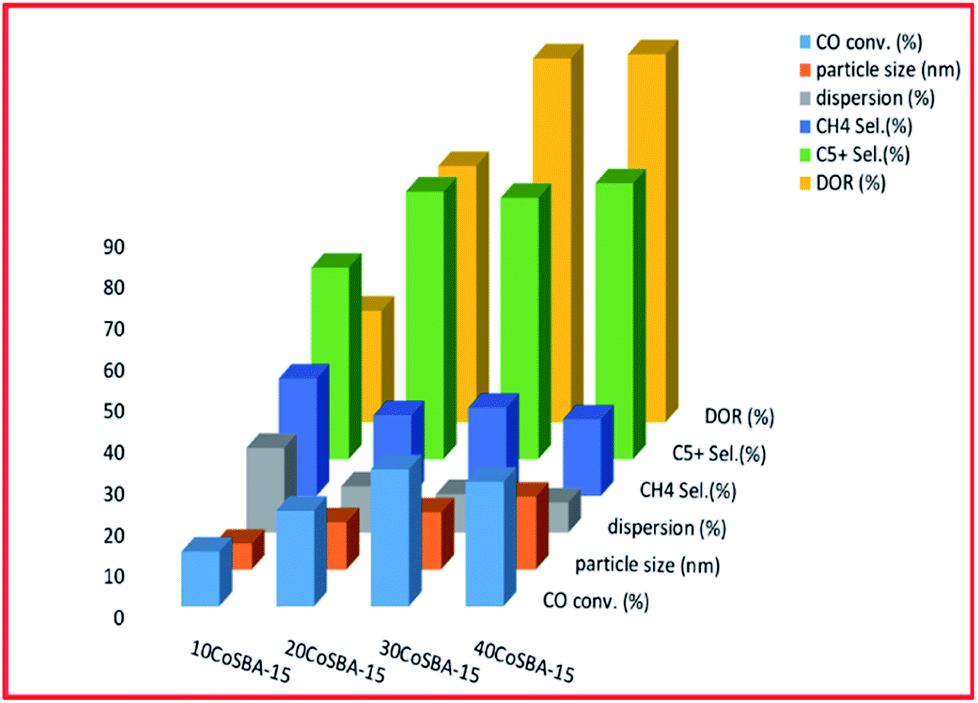 | ||
| Fig. 8 The effect of metal loading on FTS (for 10% CoSBA-15, 20% CoSBA-15, 30% CoSBA-15, and 40% CoSBA-15 catalysts), adapted from ref. 220. | ||
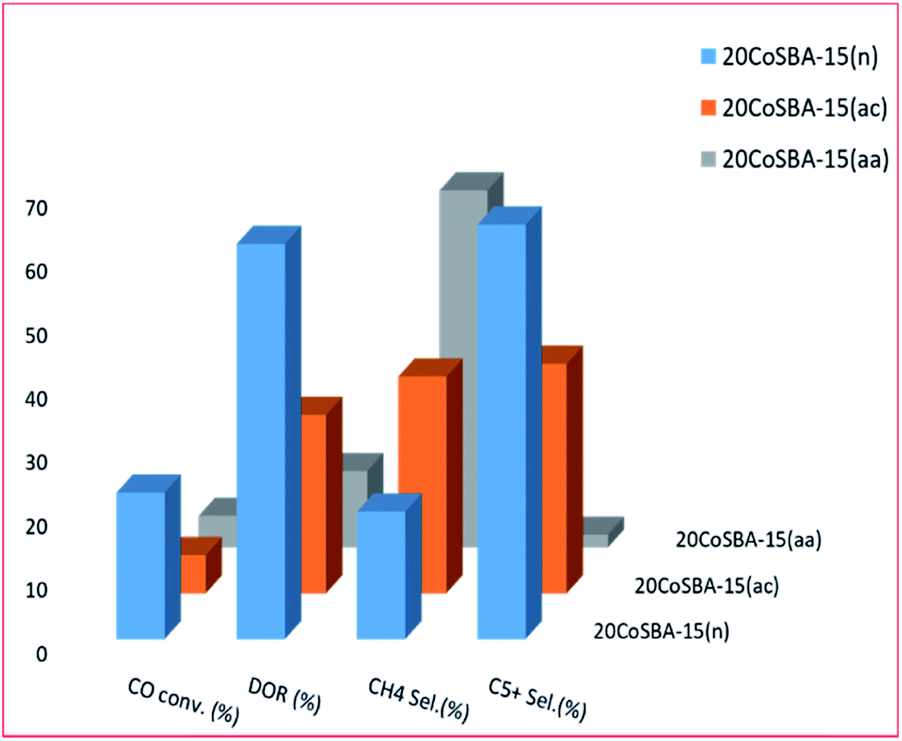 | ||
| Fig. 9 The effect of precursor on FTS (aa: cobalt acetylacetonate; ac: cobalt acetate, and n: cobalt nitrate), adapted from ref. 220. | ||
Fig. 8 shows that an increase in the metal content of the catalyst resulted in larger particle size, and improved the reducibility, as well as the selectivity to C5+ hydrocarbons; but the least dispersity. Additionally, the 30 wt% loaded catalyst exhibited better conversion (which was attributed to better dispersion, high concentration of surface Co0 sites and higher reducibility). Fig. 9 plots the effect of the metal precursors, which shows that cobalt nitrate prefers to higher catalyst activity and C5+ product selectivity. Other researchers also reported that cobalt nitrate exhibit high CO conversion and good selectivity to long-chain hydrocarbons.221–223
Lu et al.192 also investigated the influence of cobalt loading and pore size of SBA-15 on the catalytic performance of Co/SBA-15 catalysts for FTS reaction. The SBA-15 supported catalysts (10–30 wt% Co) were prepared by wet impregnation with cobalt(II) nitrate dissolved in ethanol. With an increase in either the pore size of SBA-15 support or Co loading, there was an increase in the extent of cobalt specie reduction and a decrease in the dispersion of the Co specie on the catalyst. The 20 wt% Co/SBA-15 catalyst with larger pores (which led to a larger Co crystallite size), higher reducibility and lower dispersion, exhibited the maximum CO conversion. They observed an increase in CO conversion with the increase in pore size with the larger catalyst having higher selectivity for C5+ hydrocarbons.
Rodrigues and colleagues177 reported the activity of a 15 wt% and 20% Co-based SBA-15-supported catalyst in a slurry reactor. The catalysts were synthesised by impregnation. The conversion rate for CO was observed to be 40%, with a selectivity of 53.9% for C5+ hydrocarbon after 8 hours. The group also investigated two 20% (w/w) cobalt-based SBA-15 catalysts that were synthesised by microwave (MW) and conventional heating (CH) methods.168 They found that the catalyst synthesised using the microwave method to have slightly higher CO conversion and selectivity towards C5+ hydrocarbons, compared to the catalyst that was synthesised using the conventional heating method. This was attributed to the higher reducibility of the Co/SBA-15 catalyst, which made more Co0 sites available.
Ohtsuka and co-workers175 reported a study conducted on the use of Co-based mesoporous material supported catalysts for the production of the diesel fuel fraction. The catalysts were prepared using cobalt acetate and cobalt nitrate precursors. The catalysts prepared from cobalt acetate proved almost inactive, due to the formation of a less reducible cobalt species. The active catalysts all achieved a high space-time yield for C10–C20 diesel fraction hydrocarbons. The catalysts showed good stability since the pore structure and dispersion states of the catalyst remained relatively unchanged after the FTS reaction.
The SBA-15 based catalyst showed an increase of almost 1.5 times the catalytic activity of amorphous silica-based catalysts with similar Co loading.220 The study192 also showed that the Co/SBA-15 catalysts have almost twice the CO conversion compared to Co/SiO2 samples, with only a minor difference in product selectivity, see Fig. 10.
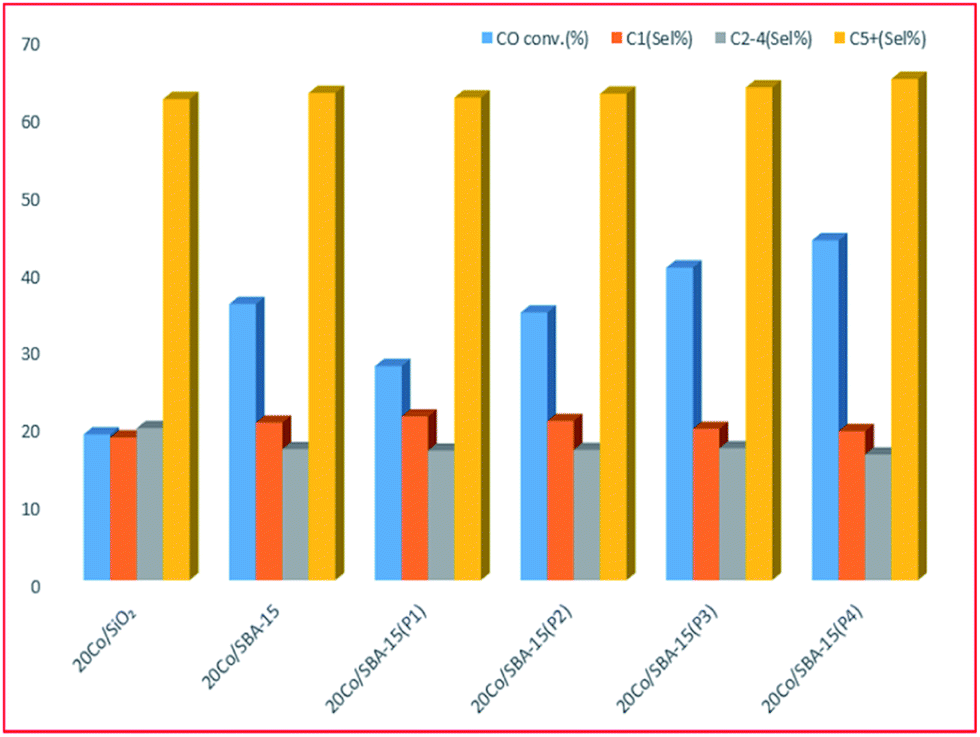 | ||
| Fig. 10 Comparison of the effect of SBA-15 (with different pore sizes, P1, P2, P3, P4) with the conventional support SiO2, adapted from ref. 192. | ||
The effect of porosity on the properties and hydrocarbon selectivity for some Co-based mesoporous material used in FTS are shown in Table 1. It was observed that an increase in either the pore size or metal loading content generally led to increased catalytic activity and selectivity towards C5+ hydrocarbons. In most cases, this is due to the larger metal particle size and higher reducibility of the catalysts.
| Catalyst | CO conversion | Textural properties of the support and catalystb | CH4 selectivity | C5+ selectivity | Comments | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| D p (nm) | V p (cm3 g−1) | BET (m2 g−1) | ||||||
| a CH – conventional heating, HDMS – hexamethyldisilazane, IW – incipient wetness, IWI – incipient wetness impregnation, MW – microwave, PS – post-synthesis, SMI – support–metal interaction, SS – supercritical solvent. b Textural properties of the catalyst. | ||||||||
| 18.1% CoSBA-15 | 63.2 (39.6, @ iso conversion condition) | 4.9 [b4.8 fresh, 5.1 used] | 0.87 [b0.62 fresh, 0.2 used] | 781 [b538 fresh, 205 used] | 22.3 (15.6) | 71.1 (79.6) | The catalyst was prepared by impregnation. The test was carried out using micro-activity reference equipment, at: T = 250 °C; P = 1 MPa; H2/CO = 2; GSHV = 0.15 mol kg−1 s−1; catalyst mass = 450 mg; and 70 h TOS. Co3O4 crystallite diameter = 14.6 nm; dispersion = 3%; reduced fraction = 81%; alpha value 0.70 (0.76); CO2 selectivity 0.9 (0.3) | 185 |
| 10% Co-SBA-15 | 15.47 | [b9.8] | [b1.12] | [b563] | 18.07 | 71.28 | Prepared by IWI. The reaction was carried out: in a fixed bed, at: T = 230 °C; P = 1 MPa; H2/CO = 2; SV = 8 SL g−1 h−1; catalyst mass = 500 mg. Corrected metal cluster diameter = 4.2 nm; corrected dispersion = 32.61%; DOR = 22.05% | 193 |
| 10–30% CoSBA-15 | 18.3–43.8 | 8 [b4.9–9.7] | 1.85 [b0.2–1.72] | 460 [b354–457] | 19.2–21.1 | 61.4–64.6 | Catalyst prepared by wet impregnation and reaction carried out in a fixed bed reactor, at: T = 245 °C; P = 20 bar; H2/CO = 2; GSHV = 2000 h−1; mass = 500 mg. Crystallite size (Co0) = 7.9–15.5; dispersion = 6.2–12.1%; reducibility = 37–86%; alpha value = ∼0.8, for C8–C14 range hydrocarbons; CO2 selectivity = 0.4–1.8 | 192 |
| 20% CoSBA-15 | 6.7–23 (mol%) | 3.6–12 [b5.5] | 0.48–2.8 [b0.35–1.8] | 819–1000 | NR | NR | Prepared by impregnation, using TMB/TCP to modify pore properties. FTS test conducted in a fixed bed, at: T = 230–250 °C; P = 2 MPa; H2/CO = 2; mass = 500 mg. Estimated catalyst size = 0.51–0.96 nm | 189 |
| 15% Co-MCM-48, (SBA-15) | Initial: 27.2 (61.8) | 2.6 (5.3) [b2.4 (b5.2)] | 1.1 (0.88) [b0.58 (b0.6) ] | 1127 (662) [b626 (b453)] | 17.79 (10.97) | 74.78 (85.18) | Prepared by IWI. FTS was done in a fixed bed, at: T = 230 °C; P = 1 MPa; H2/CO = 2; SV = 2 SL g−1 h−1 (@273 K, 0.1 MPa); mass = 6 gcat with 36 g carborundum. Estimated Co3O4 crystallite size = 14 (14.8) nm, by XRD; 9.6 (11.7) nm, TPD; 6.8 (8) nm, TEM. Dispersion = 14.2 (11.7)%, from TPD and O2 titration. Extent of reduction = 21.3 (28)%; CO2 selectivity = 1.44 (1.32)% | 190 |
| Steady: 25.8 (59.4) | ||||||||
| 5–15% Co-MCM-48 | 1.56–27.1 | 2.6 [b2.3–2.4] | 1.1 [b0.58–0.72] | 1127 [b626–776] | 17.79–23.84 | 59.88–74.78 | Prepared by IWI. FTS was done in a fixed bed, at: T = 230 °C; P = 1 MPa; H2/CO = 2; SV = 2 SL g−1 h−1 (@273 K, 0.1 MPa); mass = 6 gcat with 36 g carborundum. Estimated Co3O4 crystallite size = 5–14 nm, by XRD; 3–6.5 nm, TEM. Dispersion = 9.1–25.6% (14.2–59.2%, from TPD and O2 titration); extent of reduction = 1–21.3%; CO2 selectivity = 1.44–3.96% | 217 |
| 15% Co SBA-15 | 40 | 7.1 [b6.8] | 0.79 [b0.78] | 641 [b420] | 7 | 53.9 | Synthesized by WI. FTS carried out in a slurry (stirred batch 500 mL autoclave) reactor, at: T = 240 °C; P = 2 MPa; H2/CO = 1; mass = 3 g; mechanical agitation of 800 rpm. Average Co3O4 crystallite size 18.2 nm | 177 |
| 20% Co SBA-15 | 35.5–37 (*39–40) | 7.1 (*6.5) [b6.8 (*6.4)] | 0.79 (*0.69) [b0.73 (*0.62)] | 641 (*655) [b418 (427)] | 5.9–12.1 (6.2–12.8) | 47–71.9 (*65.6–78.5) | Support was synthesised by MW, as well as CH, while the synthesis of the catalyst was achieved by wet impregnation. FTS was carried out in a slurry (stirred batch 500 mL autoclave) reactor, at: T = 240 °C; P = 2 MPa; H2/CO = 1; mass = 3 g; mechanical agitation of 800 rpm. Average Co3O4 crystallite = 18.5 nm. O/P ratio 1.6–4.1 (*1.0–1.5). The crystallite size for a MW catalyst was not shown, but was said to not be significantly different from that of CH. The Co/SBA-15MW catalyst showed higher selectivity to C5+ compared to the Co/SBA-15CH catalyst. Co/SBA-15MW also showed better reducibility and dispersion of Co3O4 compared to the catalyst Co/SBA-15CH | 178 and 209 |
| 30% Co-SBA-15 | 28–29 | 5–15 [b5–11] | 0.77–2 [b0.32–0.74] | 716–813 [b285–357] | 25/32 | 49–58 | Prepared by WI. FTS was carried out in a fixed bed, at: T = 230 °C; P = ∼2 MPa; H2/CO = 2; GSHV = 7000 mL (NTP) gcat−1 h−1; mass = 300 mg. Particle size 12.4–18.7 (XRD), 6.3–20.7 (TEM). Reducibility = 67–84%; H2 consumption = 4553–5681 μmolH2 gcat−1. Alpha value = 0.72–0.83 (alpha increased with increasing pore size); CO2 selectivity = 2–6 | 187 |
| 10% Co SBA 15 | 12.3–20.4 | 6.5 [b6.2–6.5] | 1.29 [b0.89–1.06] | 844.1 [b499.3–661.8] | 7.56–11.39 | 76.78–84.48 | Prepared by IW. Reaction carried out in a fixed bed, at: T = 215 °C; P = 2 MPa, H2/CO = 2; flow = 8 SL (h g); mass = 400 mg diluted with 5 g carborundum. Metal cluster diameter = 3.51–8.46 nm; corrected dispersion = 12.1–26.27%; reducibility = 16.98–34.8%. Silylation with TMCS was employed in the synthesis | 206 |
| 20% Co/SBA-15 | 52.82–77.15 | 6.8–7.1 [b6.3–7.3] | 0.73–0.94 [b0.5–0.6] | 407–552 [b276–381] | 9.53–19.18 | 65.93–83.92 | Prepared by IW. FTS was carried out in fixed bed, at: T = 240 °C; P = 2 MP; H2/CO = 2; GHSV = 1000 h−1. Co3O4 particle size = 14.3–16.4 nm; reducibility = 35.8–68.3%. Silylation with TMCS | 207 |
| 15% Co/SBA-15 | 39.6 | 5.6 [b5.6] | 0.94 [b0.72] | 771 [b587.5] | 12.8 | 72.8 | Prepared by IWI. FTS was carried out in fixed bed, at: T = 240 °C; P = 1 MPa; H2/CO = 2; GHSV = 8000 mL (gcat h)−1; mass = 100 mg mixed with 300 mg carborundum. Particle size = 11.6 nm (Co3O4), 8.7 nm (Co); dispersion 11%; alpha value = 0.78 | 211 |
| 12% Co/MCM-41 | 4.4–27/*10–28 | 2.6/*7.5 [b3.1/*7.9] | 0.9/*1.1 [b0.8] | *940/1200 [b*721/990] | 8.6–12.2/*13.6–18.1 | 76.3–83.5/*62.3–74.4 | Prepared by IWI. FTS reaction conditions were: T = 210 °C; P = 2 MPa; H2/CO = 2; GHSV = 1500–6000 (SBA-15: 2550–6000) mL (gcat h)−1 NTP; mass = 1 g with 5 g SiC. Particle size (after reduction): Co3O4 = 7/*15.1 nm, Co0, 5.3/*11.3 nm (XRD), 4.7/*7.2 nm (chemisorption); corrected dispersion = 7.1/*8%; DOR = 35/*60% CO2 selectivity = 1–1.4/*1.8–2.2 | 212 |
| 12% Co/SBA-15 (*) | ||||||||
| 5/*30% Co/SBA-15 | 5.3/*29.1 | 6.46 [b*6.17] | 1.37 [b*0.69] | 897 [b*473] | 44.8/*9.61 | 47.6/*79 | Catalyst was prepared by IWI. FTS reaction condition was in a tubular fixed bed, at: T = 210 °C; P = 2 MPa; H2/CO = 2; GHSV = 8000 h−1 (8 SL h−1 g−1); 0.5 g with 5 g carborundum. Corrected metal cluster diameter = *26.3 nm, cobalt particle metal diameter = *12.3. Dispersion = *3.9%; reducibility = *49.7%; CO2 selectivity 1.6/*0.1. H2 spill-over to Co via strong electronic interaction and not support–metal interaction (SMI) was responsible for the observed activity | 215 |
| 6% Co SBA-15 | 4.6–21.1 | 8.09 [b8.08] | 1.24 [b0.81–0.86] | 724 [b461–472] | 7.3–15.8 | 27.4–60.3 | The supports were synthesized by direct synthesis, the catalyst was prepared with IW, PS, and using SS. FTS reaction conditions in a fixed bed reactor: T = 265 °C; P = 1 MPa; H2/CO = 2; SV = 2100 h−1; 12 h TOS after stabilization of 4 h. Co3O4 diameter 11.1–11.6 nm; DOR 18–63% alpha value 0.82–0.88. SS sample gave better activity (@21.1% CO conversion), due to the higher crystallite size and better reducibility | 219 |
| 6% Co SBA-15 | 5.2–10.7 | 8.09 [b8.07–8.09] | 1.24 [b0.76–0.9] | 724 [b421–463] | 3.9–12.5 | 38.7–81.7 | The supports were synthesized by silylation with HMDS. FTS reaction conditions in a fixed bed reactor: T = 265 °C; P = 1 MPa; H2/CO = 2; SV = 2100 h−1; 12 h TOS after stabilization of 4 h. Co3O4 diameter = 10.4–14.7 nm; DOR = 14–49% alpha value 0.83–0.9 | 205 |
| 12% Co SBA-15 | 13.7–40.3 (*42.3/34.6 added 20 & 33% water respectively) | 9.7 [b5] | 0.95 [b0.77] | 713 [b655] | 11.6–16 (*6.4/5.1) | 68.6–76 (*84.4/86.4) | The catalyst was synthesized with IW. Fixed bed FT synthesis reaction conditions: T = 200 °C; P = 2 MPa; H2/CO = 2; GHSV = 6686 N cm3 g−1 h−1; mass = 700 mg diluted with 5 g SiC. Co0 diameter 9 nm (XRD), 15 nm (chemisorption); dispersion 6%, 6.6% (corrected); DOR 91%; TOF = 0.08 S−1(10 hours on stream); O/P ratio 1.9–2.4 (*2.8/3.9). The conversion and C5+ selectivity increased significantly with H2O addition | 203 |
| 10% Co/SBA-15 | Rate CO × 10−2 (mol CO per gcat per h) = 1.4/*1.8 | 10.6/*11.2 [b7.7/*10.4] | 1.45/*1.48 [b0.97/*0.94] | 633/*487 [b488/*358 ] | 20.3/*24.2 | 67.1/*63.2 | Supports were synthesized conventionally and with the addition of decane (as the swelling agent to control the pore size, *M). The catalyst was then synthesised impregnation. FTS (fixed bed) reaction conditions: T = 230 °C; P = 0.5 MPa; H2/CO = 2; mass = 1 g. Co3O4 metal crystal size 10.5/*9.7 nm. O/P ratio = 0.31/*0.2 (C2–C4); alpha value = 0.8/*0.76; CO2 selectivity = 2.3/*3.4. The SBA-15(M) catalyst displayed enhanced activity due to shorter cavity length and the presence of macropores, which enhanced the diffusion of reactants, intermediates and products | 191 |
| 10–30% Co SBA-15 | 18.3–43.8 | 8 [b4.9–9.7] | 1.85 [b0.72–1.72] | 460 [b354–457] | 19.2–21.1 | 61.4–64.6 | The catalyst was prepared by WI, and FTS was conducted in a fixed bed. Reaction conditions: T = 245 °C, P = 2 MPa; H2/CO = 2; GHSV = 2000 h−1; mass 500 mg diluted with quartz. Average crystallite size = 10.5–20.6 nm (Co3O4), 7.9–15.5 nm (Co0); dispersion = 6.2–12.1%; reducibility = 37–86%; alpha value = 0.81–0.83; CO2 selectivity = 0.4–1.8 | 192 |
| 10–30% Co SBA-15 | 18.3–43.8 | 8 [b4.9–9.7] | 1.85 [b0.72–1.72] | 460 [b354–457] | 19.2–21.1 | 61.4–64.6 | The catalyst was prepared by WI, and FTS was conducted in a fixed bed. Reaction conditions: T = 245 °C, P = 2 MPa; H2/CO = 2; GHSV = 2000 h−1; mass 500 mg diluted with quartz. Average crystallite size = 10.5–20.6 nm (Co3O4), 7.9–15.5 nm (Co0); dispersion = 6.2–12.1%; reducibility = 37–86%; alpha value = 0.81–0.83; CO2 selectivity = 0.4–1.8 | 192 |
| 10% Co/MCM-41 (10% Co/SBA-15) | 1/(3.3–*9.3) | 3.4/(4.4–*5.7) [b(4.5–*6.1)] | 1.05/(0.92–*1.13) [b0.3/(0.56–*0.67)] | 1180/(*935–1029) [b349/(471–*534)] | 21.9/(9.8–*9.9) | 45.7/(*75.1–76) | The catalyst was prepared by IWI and FTS was conducted in a fixed bed, at: T = 190 °C; P = 0.1 MPa; H2/CO = 2; GHSV = 1800 mL (g−1 h−1); mass = 500 mg. Co3O4 crystallite diameter = 8.7/(9.2–*9.8) nm, (XRD), *7.2 nm (XPS); dispersion = *17.8% (XPS); CTY 0.4/(1.4–*3.9) (10−4 s−1); alpha value 0.58/(0.8). The MCM-41 catalyst is bimodal (with pore diameter 2 nm mesopore, and 30 nm micropore). One SBA-15 catalyst was synthesized with 5 times higher concentration of HCl (*) | 208 |
| 10% Co/SBA-15 (10% Co/MCM-41) | 17.7 (4.8) | 7.7 (3.4) [b7.2 (2.8)] | 1.13 (1.05) [b0.96 (0.65)] | 935 (1189) [b647 (548)] | 21.1 (33) | 55 (31.4) | FT reaction conditions (in a fixed bed tubular micro reactor): T = 200 °C; P = 2 MPa; H2/CO = 2; GHSV = 18000 mL (g h)−1; mass = 500 mg. CO3O4 crystallite size 9.3 (7.6) nm, (from XRD); dispersion 10.3 (12.7)% (from propene chemisorption); TOF = 4.6 (2.1) × 10−2 s−1; FT reaction rate = 6.9 (1.9) × 10−4 s−1. The TOF was calculated from CO conversions, GSV and normalized by propene chemisorption instead of overall cobalt contents |
197 |
| 5% Co (0–0.5% Ru) SBA-15 | 6.51–20.51 | 7.7 | 1.41 | 860 | 19.17–49.39 | 42.43–77.14 | The catalyst was prepared by IWI. Fixed bed reaction conditions: T = 230 °C; P = 1 MPa; H2/CO = 2; GHSV = 3000 h−1; mass = 3 g mixed with 36 g carborundum and 80 g glass beads. Average Co3O4 diameter = 11.5–13.4 nm (XRD); DOR = 17.8–28.7%; TOF = 27.1–30.8 (10−3) s−1. Alpha value = 0.79/0.81 | 179 |
| 15% Co/SBA-15 | 90 | 9.1 [b4.5] | 1.18 [b0.25] | 831 [b376] | 18 | 49 | The catalyst was prepared by IWI. Fixed bed reaction conditions: T = 240 °C; P = 2 MPa; H2/CO = 2; GHSV = 1000 h−1. Co3O4 crystallite size = 14 nm; dispersion = 6.8%; DOR = 80%; TOF 0.02 (×10−3) s−1 @ CO conversions of 5–10% & GSVH of 10000–18000 h−1. Alpha value = 0.81; CO2 selectivity = 24 |
198 |
| Co/Al-SBA-15 (Al: 0–10) | 32.06–49.59 | 4.7 [b6.2–10.4] | 1.03 [b1.03–1.14] | 867 [b415–732] | 12.32–19.6 | 69.98–82.44 | The catalyst was prepared by dry impregnation (DI). FTS reaction was carried out in a fixed bed reactor, at: T = 230 °C; P = 2 MPa; H2/CO = 2; WHSV = 10000 mL gcat−1 h−1; mass = 300 mg. Co3O4 crystallite size = 9.4–12.5 nm; Co0 13–14.4 nm (after reduction); dispersion = 10.08–13.62%; O/P ratio = 0.43–1.21. Si/Al molar ratio (0–10) |
204 |
3.2 Mesoporous materials supported iron-based catalysts
Compared to iron-based catalysts, cobalt-based catalysts are more active in the FTS reaction, which meant that the bulk of the research has been done on mesoporous material supported Co-catalyst, with only a few studies being done using an iron-based catalyst. Cano and colleagues studied the effect of active iron species crystallite size on catalytic performance using an iron-based mesoporous-supported (SBA-15 and MCM-41) catalyst and evaluated them in the FTS reaction.181 The catalysts were prepared by impregnating the support with aqueous and ethanolic solutions of Fe(NO3)3·9H2O, to give a Fe loading of 8.9 wt% and 14.0 wt%, respectively. After impregnation, there was no significant change in the structural properties of the support; however, larger iron crystallites were formed with SBA-15 (with larger pores) than with MCM-41 (with smaller pores). The catalysts with larger pores exhibited higher conversion, higher chain growth, and lower selectivity towards CH4. The researchers also studied the effect of the activation environment (H2 or H2/CO) on the iron species and resulting catalytic activity and selectivity, using SBA-15 supported iron catalysts.181 Pre-treatment with H2 resulted in higher CO conversion of total hydrocarbons, and more CH4 and light gases were also observed over 6 days. Additionally, higher selectivity toward light olefins was obtained with H2 activation, during the first 24 hours, but this decreased with time on stream. The researchers concluded that H2 pre-treatment delivered the most active iron carbine carbide species.Cano et al.125 investigated the effect of a promoter (Cu and K) and support type (SiO2 and SBA-15) on FTS for Fe based catalysts. The Fe/SBA-15 showed higher activity compared to SiO2 catalysts. The authors observed Cu addition resulted in increased reducibility and stability of the catalyst, while K addition enhanced the activity of the catalysts and favoured selectivity towards olefins. The positive effect of the promoted SBA-15 supported catalysts was ascribed to the porosity afforded by the SBA-15 and metal promotion. The use of SBA-15 resulted in the stability of the nanoparticles and increasing the dispersion of the active metal.
Toncón-Leal et al.126 studied the behaviour of iron nanoparticles confined in an ordered mesoporous material (SBA-15, with pore diameter ∼ 8 nm) for the FTS reaction. The characterization of the materials showed that in the Fe@SBA-15 material, iron nanoparticles were confined inside the mesopores of the SBA-15 support (pore diameter ∼ 8 nm). The authors observed a high selectivity to methane and light hydrocarbons with Fe@SBA-15 and Fe@Aerosil®-200 compared to the bulk catalyst. The bulk iron, however, showed a higher selectivity to C5–C9 hydrocarbons and oxygenates. The Fe@SBA-15 displayed the highest activity per total iron loading and the highest selectivity to ethylene. This was attributed to the confinement of the iron nanoparticles inside the pores of the ordered mesoporous material (SBA-15).126
Oschatz and co-workers182 investigated ordered mesoporous materials (SBA-15, CMK-3 and OM-SiC) with comparable textural properties to supports for stable iron (promoted with Na and S) catalysts in the FTS of lower alkenes (see Fig. 11). They showed that the properties of the Fe-based catalysts were influenced by the supports in the presence of promoters. The lower activity displayed by the SBA-15 supported catalysts (compared to the non-oxidic supported catalysts) was attributed to the formation of iron silicate species during synthesis, and not the effect of the support on promoter dispersion. They further studied the influence of S promotion (using 0–3 wt% S for Fe) and showed that selectivity towards methane decreased, while selectivity to lower alkene products increased, with an increase in the sulphur content. Promoter addition led to 3–5 times higher catalytic activity.
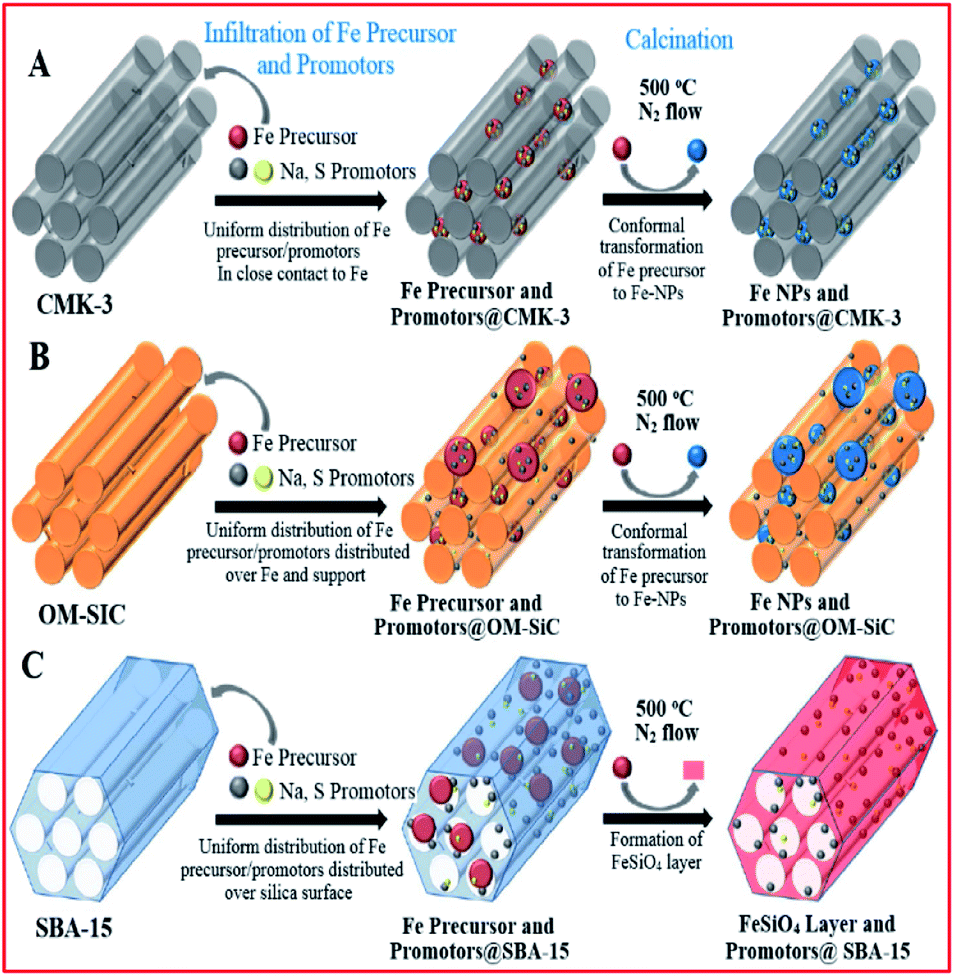 | ||
| Fig. 11 Ordered mesoporous materials as model supports for Fe-based FTO catalysts and their effects on the properties of the catalysts. Reproduced from ref. 182 with permission from John Wiley and Sons, Copyright 2016. | ||
Kim and colleagues201 also investigated the performance of iron-based SBA-15 catalysts, as well as the effect on the morphology and catalyst activity for higher hydrocarbon synthesis when incorporating aluminium (Al:Si = 0.010–0.033) into the support structure. The catalyst without Al had an induction period of just over 10 hours – a typical result for a Fe-based FTS catalyst, and which is ascribed to the gradual formation of the iron-carbide phase, which is responsible for the catalytic activity. With an Al:Si ratio of 0.010, the induction period was reduced: a 30% conversion was reached in the first two hours. Further increases in the Al:Si ratio led to a decrease in CO conversion, although the shorter induction period was maintained (see Fig. 12A). The selectivity profile (shown in Fig. 12B) indicated an increase in C10+ fractions from 50% to 80% upon increasing the Al:Si ratio to 0.010; but a gradual decrease was observed with an increase in Al content. The researchers concluded that the addition of trace amounts of Al may, therefore, increase the rate at which active iron-carbide is formed; thus, it has a positive effect on catalyst activity and selectivity to higher hydrocarbons products.
 | ||
| Fig. 12 Activity and selectivity of SBA-15-supported iron catalysts as a function of aluminium loading: (A) CO conversion and (B) product selectivity. Reproduced from ref. 201 with permission from American Chemical Society, Copyright 2006. | ||
Table 2 shows the results obtained from studies on Fe-based catalysts supported on different mesoporous materials used in FTS. A larger crystallite size resulted in better conversion and lower methane selectivity. The catalytic performance (activity ad selectivity) was also found to be dependent on the ratio of Al:Si, for the Al promoted catalyst.
| Catalyst | CO conversion | Textural properties of the support and catalystb | CH4 selectivity | C5+ selectivity | Comments | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| D p (nm) | V p (cm3 g−1) | BET (m2 g−1) | ||||||
| a CH – conventional heating, HDMS – hexamethyldisilazane, IW – incipient wetness, IWI – incipient wetness impregnation, MW – microwave, PS – post-synthesis, SMI – support–metal interaction, SS – supercritical solvent. b Textural properties of the catalyst. | ||||||||
| 8.9% Fe/MCM-41, 14% Fe/SBA-15 | 3/(12.2) | 2.7/(2.9) [b8.1/(7.1)] | 0.88/(0.64) [b1.09/(0.57)] | 912/(691) [b893/(519)] | 34.1/(7.0) | ∼78 (20) | The catalyst was prepared by WI, catalytic activity was carried out in a fixed bed reactor, at reaction conditions: T = 430 °C; P = 0.1 MPa; H2/CO = 2; SV = 1176 h−1; mass 400 mg. O/P ratio = 3.5/4.2; CO2 selectivity = 5.8 (5.4). Total HC production 5.8/(5.7) × 1017 [HC molecules per s per gFe]. Support wall thickness: MCM-41 = 2 nm, SBA-15 = 7 nm. Crystal size of iron in MCM 41 = 20 nm (7%) and 3 nm, while SBA-15 = 10 nm | 180 |
| 13% Fe/SBA-15 | 18.7–20.1/*8.6–12.1 | 8.1 [b7.1] | 1.09 [b0.57] | 893 [b519] | ∼18/*7 | ∼64/*83.5 | Catalyst prepared by IWI, then activated with H2 and syngas (*) prior to FT evaluation in a fixed bed, at conditions: T = 430 °C; P = 0.1 MPa; H2/CO = 2; SV = 1176 h−1; mass = 450 mg; O/P ratio = 0.28–2.06 (*0.97–1.13); CO2 selectivity = 8.3 (*5.4). Total HC production (13.2–23.5) × 1017/*(4.3–7) × 1017 [HC molecules per s per gFe]. Crystal size = 8 nm | 181 |
| 7.1–8.1% Fe/0.2–2.3Na/0–0.08 S/CMK | ∼20 | 3.8 [b3.5–3.7] | 1.55 [b1.45–1.62] | 1543 [b1530–1555] | ∼40–43 (*∼15–18) | ∼5–6 (*∼21–28) | Catalyst prepared by impregnation. Result for sulphur addition (*). FTS conditions: T = 340 °C; P = 0.1 MPa; H2/CO = 2; GHSV = 10000–30000 h−1. Iron particle size 5.58–27.54 nm |
182 |
| 20% Fe/Al-SBA-15 | ∼4–37 | [b5.4–5.5] | [b0.74–0.76] | [b490–580] | ∼5–8 | ∼(79–90) | Prepared by IWI, and tested in fixed bed reactor for the production of diesel fraction, at reaction conditions: T = 230 °C; P = 2 MPa; H2/CO = 2; WHSV = 10000 mL gcat−1 h−1; mass = 400 mg. Diesel fraction = ∼45–80%; CO2 selectivity = ∼3–8. Al:Si ratio 0–0.033. The catalysts retained the textural properties of the support |
201 |
| 10% Fe@SBA-15 | 6–8 | 8 [b7] | 1.05 [b0.65] | 960 [b630] | ∼33 | ∼−60 | Catalyst prepared by IW and calcined at 300 °C. FT evaluation in a fixed bed, at conditions: T = 275 °C; P = 2 MPa; H2/CO = 2; mass = 200 mg | 126 |
| 15% Fe@Aerosil®-200 | 0.53 [b0.83] | 200 [b200] | 45 | 27 | ||||
3.3 Other mesoporous materials (promoted/modified) catalyst for FT
3.3.1.1 Ruthenium. Several metals (Ru, Pd, Re, etc.), as well as the silanol group of materials (CH3)3SiSi(CH3)3, have been used to promote the reducibility of cobalt metal complexes supported on mesoporous materials. Ruthenium is most commonly used to promote cobalt-based catalysts in the FT reaction, because of the weak electronic interaction between ruthenium and cobalt. The addition of ruthenium: reduces the amount of Co2+ and Co3+ species, and the formation of cobalt active sites (Co0); and lowers the temperature required for reduction (i.e. it helps with the reducibility of cobalt oxide specie). Xiong and co-workers215 investigated the use of ruthenium (0.05–0.5 wt%) as a promoter for an SBA-15 supported cobalt (30 wt%) catalyst, as seen in Fig. 13. The ruthenium-promoted catalysts showed slightly higher selectivity for C5+ hydrocarbons, compared to the unpromoted catalyst. An increase in the ruthenium content gave rise to an increase in CO adsorption of the catalysts, thereby improving the catalytic activity on FTS. The larger cobalt particles were proposed by the group, to facilitate the formation of bridged CO species, which have weaker C–O bonds than the linear-type, and which can be more easily dissociated into carbon and oxygen. This process is essential to FT synthesis.
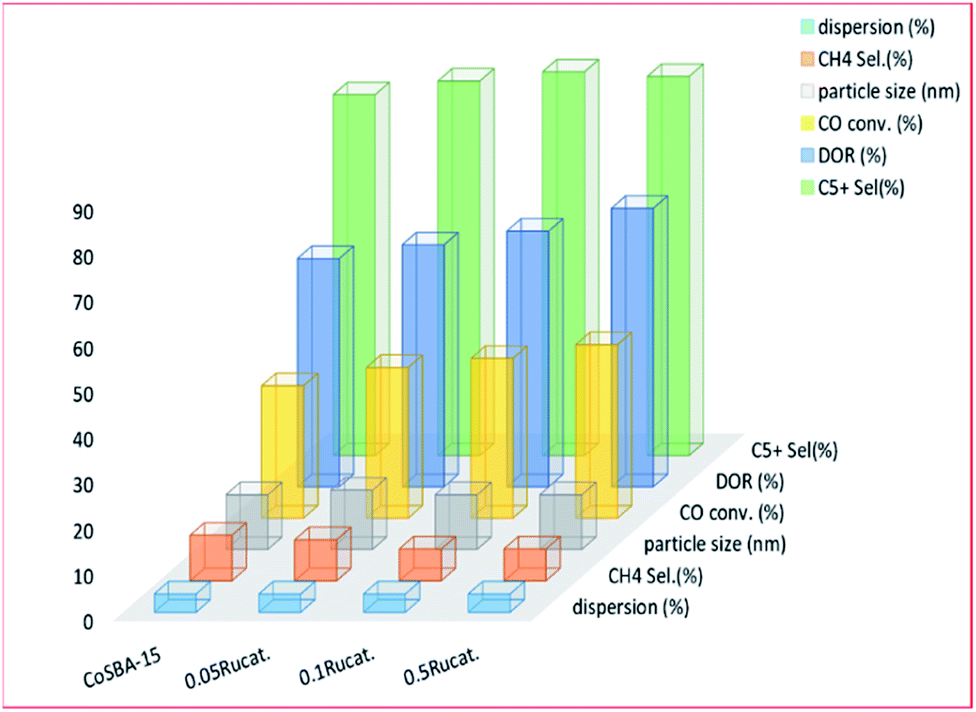 | ||
| Fig. 13 The effect of ruthenium promotion on the activity of a 30% CoSBA-15 catalyst, adapted from ref. 215. | ||
Rodrigues and co-workers also reported Ru promotion of a Co-based SBA-15 catalyst for FTS in a slurry reactor.210 The resulting increase in the catalytic activity was attributed to the increase in the number of active sites, which is due to the higher reducibility and the synergy between Ru and Co. The study was done by Li and Cai on the activity of SBA-15-supported cobalt (5 wt%) catalysts,179 when using various amounts of ruthenium (0–0.5 wt%), also showed an increase in CO conversion and selectivity towards C5+ hydrocarbons. Hong et al.197 also studied the effect of ruthenium (0.3 wt%) promotion on the reducibility and performance of a 10% Co-based FT catalyst supported on mesoporous silica with different pore sizes. The activity and selectivity of the catalysts were reported during a period of quasi-steady-state behaviour. There was a significant increase in the dispersion of Co and the activity for the small pored catalyst compared to the large pored catalyst, which was lower.
Xiong and co-workers researched pore-confined, supported Ru catalysts with a metal loading of 3.72–3.97%.188,215 They found that larger pores facilitated reactant diffusion and increased catalytic rates to an extent. They also found that the more confined catalysts exhibited higher selectivity for C5+ hydrocarbons, which was attributed to restriction of product diffusion out of the pores; rapid re-adsorption of alkenes; and subsequent chain growth to waxes that eventually limit CO diffusion.
3.3.1.2 Palladium. Osakoo et al.191 investigated the effect of support morphology and Pd promotion on a Co/SBA-15 catalyst for FT synthesis. The SBA-15 material was synthesised, both with and without the addition of decane (for pore size control), and further impregnated with Co and Co–Pd, to give 10 wt% Co-based catalysts. The catalyst prepared with the addition of decane, displayed a higher and steadier conversion and selectivity to C5–C9 products, while palladium (Pd) promotion improved the reducibility of cobalt oxides and led to the production of more methane and light alkanes.
3.3.1.3 Cerium. Albuquerque and colleagues,127 evaluated the FT products formed according to the ASF distribution using cerium promoted and unpromoted cobalt mesoporous SBA-15 molecular sieve catalysts. At steady state, the Co/Ce/SBA-15 catalyst had better activity than the unpromoted catalyst, however, the authors didn't explain the observation, it appears to be a promotional effect. The product distribution reflected two chain growth probabilities (α1 and α2) with the light olefins range hydrocarbons having maximum production followed by the gasoline range (see Fig. 14).
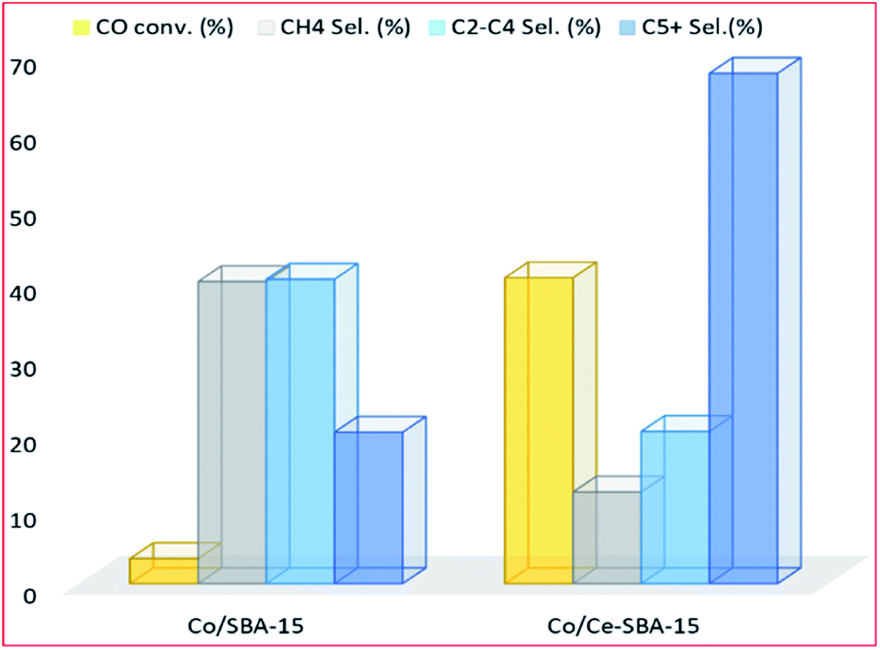 | ||
| Fig. 14 Catalytic activity of cerium promoted and unpromoted Co catalyst, adapted from ref. 127. | ||
Besides, Vosoughi et al.128a investigated the promotional effect of noble and transition metals (Ru, Ir, Re, Pt, Mn, Y) on a 15 wt% cobalt catalyst supported on mesoporous alumina for FT synthesis. The promotion increased cobalt dispersion and improved reducibility, which in turn, lead to higher active site density and an improved activity, with Ru and Re having a better promotional impact on activity and selectivity of the catalysts (see Fig. 15).
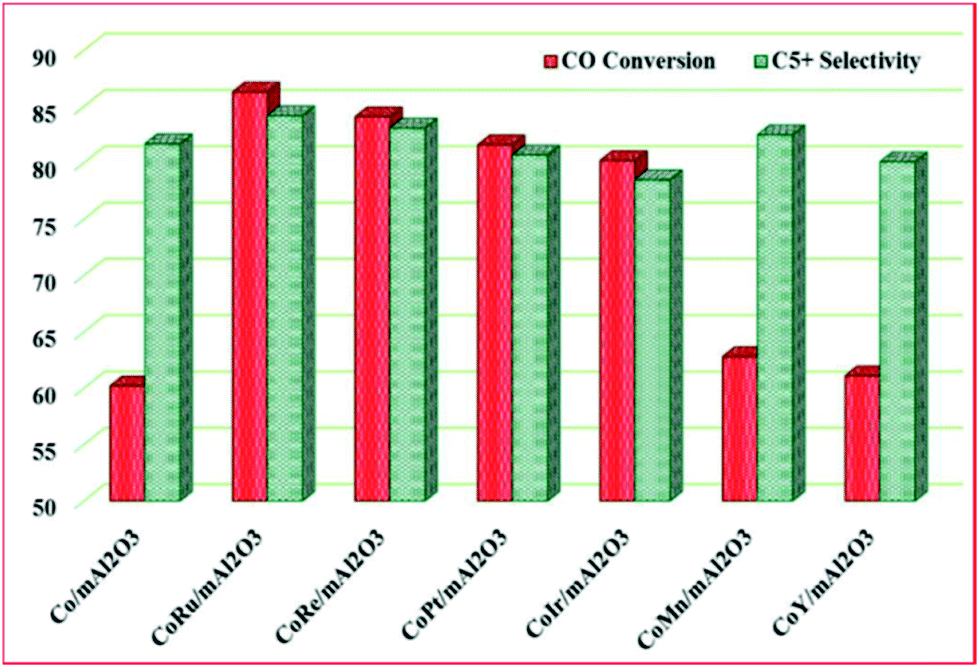 | ||
| Fig. 15 The effect of metals promotion on the activity of a 15 wt% Co catalyst supported on mesoporous alumina. Reproduced from ref. 128a with permission from Elsevier, Copyright 2017. | ||
3.3.2.1 Adding zirconia. The incorporation of ZrO2 into Co/SBA-15 for FTS was investigated by Mu et al.195 They showed that zirconia has a strong influence on SBA-15 supports (due to enhanced acidity) and the reaction rate, by creating an active interface with Co. This increased the cobalt–support interaction, which inadvertently increased the catalytic activity, by facilitating CO dissociation.
Synthetic incorporation of ZrO2 into Co/SBA-15 was also investigated by Liu et al.196 They used ZrO2 as a promoter for the reduction of Co complexes supported on SBA-15. Varying amounts of ZrO2 (0–20%) in SBA-15 supports were impregnated with 15 wt% Co, which resulted in a promotional effect on the CO conversion values obtained. The activity then decreased, with a further increase in ZrO2 content; but the selectivity for C5–11 hydrocarbons increased, which might be due to a direct impact of zirconia facilitating an increase in CO dissociation. The catalytic activity was affected by the number of available active sites, and partly dependent on the reducibility of the metal and its dispersion. The researchers concluded that zirconia promotion increased the strength of the cobalt–support interaction, resulting in high dispersion, but low reducibility, which led to lower conversion for ZrO2 (>5 wt%) and more selectivity to CH4. The results showed: chemical grafting to be the most effective method and ZrO(NO3)2 as an effective precursor; the promotional effect of ZrO2 is only effective at an optimal percentage. The effect of 10 wt% zirconium addition [by wet impregnation (i), precipitation (p), isomorphous substitution (s), and chemical graft (g) methods] on a 15 wt% Co/SBA-15 catalyst is shown in Fig. 16. The addition of Zr not only improved the conversion but also limited the selectivity to CH4.
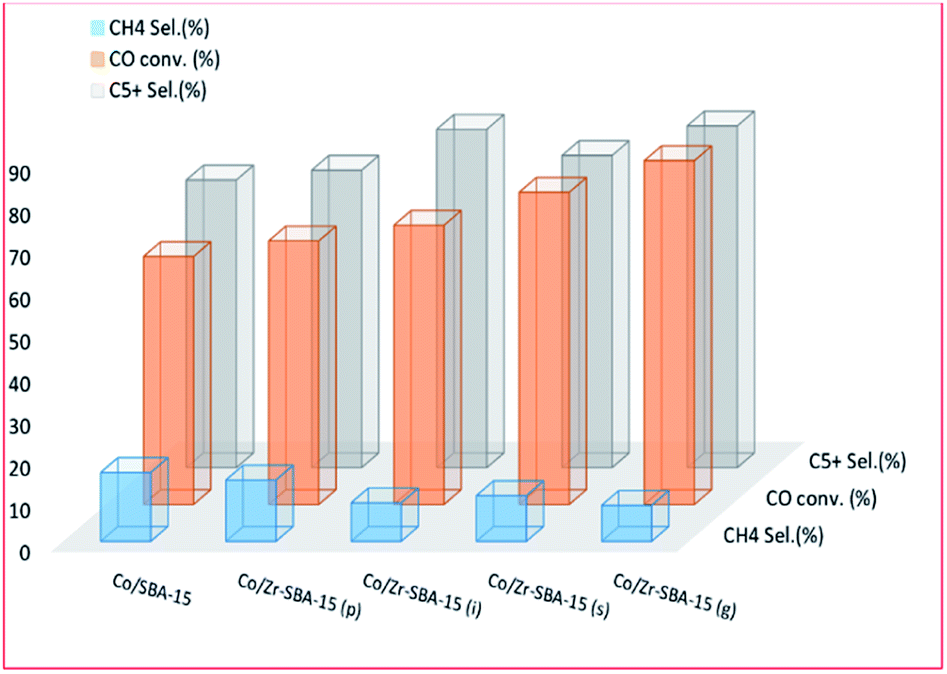 | ||
| Fig. 16 The effect of ZrO2 addition [by wet impregnation (i), precipitation (p), isomorphous substitution (s), and chemical graft (g) methods] on catalyst activity, adapted from ref. 196. | ||
Hong et al.208 investigated the effect of zirconia promotion on large and small-pored silica-supported cobalt catalysts in the FTS reaction. The catalysts were prepared by sequential incipient impregnation with cobalt nitrate and zirconyl nitrate. Modification with zirconia had no significant effect on the reducibility of a Co-based SBA-15 catalyst, in terms of catalytic performance. Tao et al.193,194 also studied the effect of incorporating Zr (1:20 Zr:Si ratio) into SBA-15, on the morphology, activity and selectivity of cobalt-based FT catalysts. The zirconia was incorporated in situ and post-synthesis into the SBA-15 support, using wetness impregnation. However, they highlighted that the hexagonal structure of SBA-15 was better preserved when using in situ isomorphic incorporation than when using wetness impregnation. The catalytic activity was found to increase with an increase in Zr content. The activity initially increased with a Zr:Si ratio of 1:100 to 1:20, and decreased with an increase in ratio to 1:3. The catalyst preparation method was also seen to affect the CO conversion and hydrocarbon yield. The catalysts prepared by isomorphic substitution were more active than those prepared by impregnation. A further increase in Zr content led to the gradual destruction of the uniform hexagonal tubular array of the SBA-15 support; a decrease in pore volume and surface area; and a stronger Co–Zr interaction. This resulted in low CO conversion, due to a lower amount of reduced cobalt specie. An increase in Zr content led to an increase in the crystallite size, higher C5+ selectivity and a decrease in catalyst dispersity.
In their study on the effect of incorporating Zr on a Co (5 and 15 wt%) SBA-15 catalyst for FTS, Li et al.199 found improved selectivity to C5+ hydrocarbons, due to the interaction between the reaction intermediates and the Lewis acid of ZrO2. The catalysts were prepared by sequential impregnation, co-impregnation and in situ synthesis methods. The authors found that the catalyst stability and turnover frequency were also improved by the addition.
3.3.2.2 Adding alumina. The incorporation of Al into SBA-15, as support for a 10 wt% Co-based FT catalyst, was investigated at atmospheric pressure by Sibianu and colleagues.202 The AlSBA-15 (with Si/Al molar ratio of 20) catalyst was impregnated with cobalt, manganese and calcium nitrates, in a 10
:1:1 molar ratio, before reduction. The CO conversion was found to have increased with an increase in temperature and the hydrogen content of the feed gas, while CO2 selectivity increased with temperature, but decreased with enrichment of the H2 content of the feed gas.
The effect of Ti and Al addition via direct synthesis to SBA-15 as support for cobalt-based FT catalysts was investigated by Lualdi et al.203 The SBA-15 material was doped with Ti and Al (5 and 10 wt%), and further impregnated with 12 wt% Co. The authors detected a correlation between the channel length and the extent of CO-diffusion limitations, by adding external water to the reaction.
Kim et al.204 studied the modification of SBA-15 by Al (with Si/Al molar ratios 5, 7 and 10) for cobalt-based FT catalysts. The authors found that the catalyst acid sites increased with the addition of Al to SBA-15, and produced middle distillate range products. The cobalt–support interaction also increased and affected the activity of the catalyst for the FTS. The catalyst with a Si/Al mole ratio of 5 exhibited higher conversion and less selectivity to CH4 hydrocarbons. This was attributed to the strength of the metal–support interactions, as shown in Fig. 17. However, the catalyst with a Si/Al mole ratio of 10, displayed a higher (isomer + alkene)/alkane ratio for C5+ compared to the other catalysts, which was attributed to a large B-acid and L-acid site ratio on the catalyst.
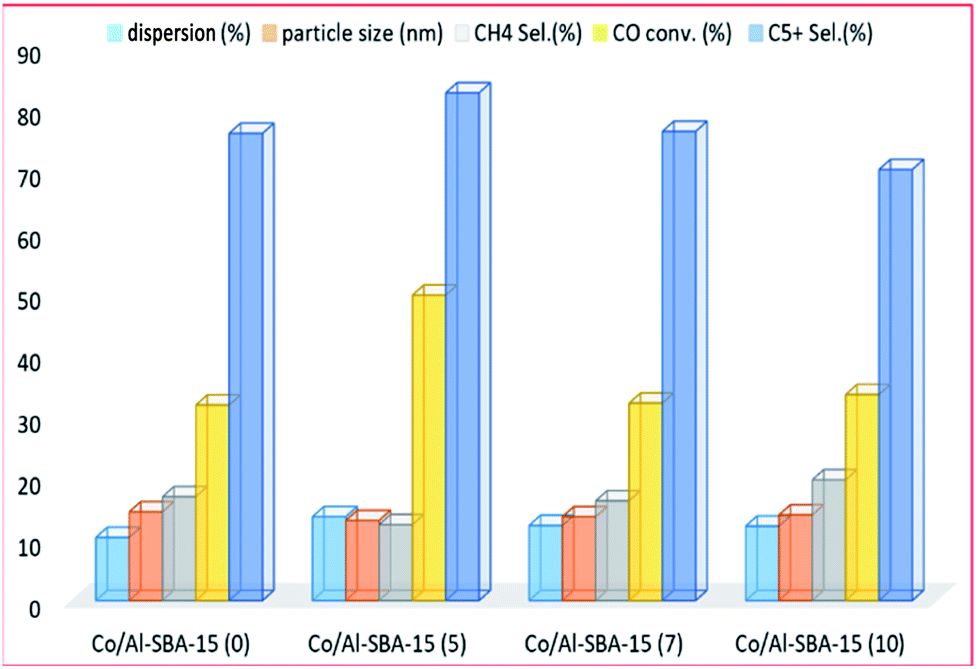 | ||
| Fig. 17 The effect of alumina addition (with Si/Al molar ratios of 0, 5, 7 and 10) on the catalyst activity, adapted from ref. 204. | ||
3.3.2.3 Other modifications and materials. Early investigations were conducted by Kim and co-workers205 into the silylation of silica-based materials to enhance the reducibility of cobalt oxide species in the mesoporous silica (see Fig. 18). The silylation was carried out using hexamethyldisilazane (HMDS) and the material retained its mesoporous structure afterwards. Larger cobalt particles were formed with an increase in HMDS loading, leading to enhanced reducibility of cobalt oxides species on an SBA-15 supported catalyst; an increase in chain growth probability; increased catalytic performance; and increased selectivity to higher hydrocarbons.
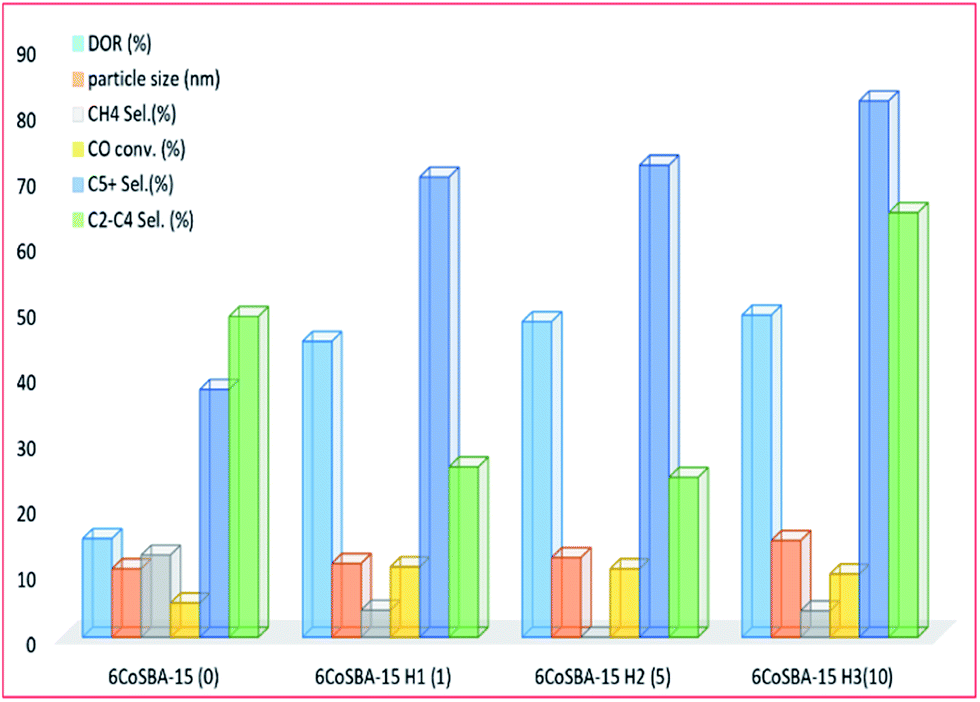 | ||
| Fig. 18 The effect of silylation (with 0, 1.03, 5.16, and 10.33 mmol g−1 SBA-15) on a 6% Co-SBA-15 catalyst activity, adapted from ref. 205. | ||
Yuanyuan et al.206 also investigated the effect of silylation of SBA-15 (using TMCS), on the reducibility of cobalt oxides supported on SBA-15, and the catalyst was evaluated for the FT reaction. Varying degrees of silylation was employed to make 10 wt% Co-SBA-15 catalyst via wetness impregnation. The increase in silylation resulted in higher reducibility of the cobalt complex and CO conversion, which was similar to Kim's findings. The increase in selectivity was ascribed to an increase in cobalt particle size.
Jia et al.207 also investigated the effect of silylating SBA-15 (using HMDS), on the FTS catalytic activity of supported 20 wt% Co. The silylation was done prior-to and post-metal loading, with the post-metal loaded catalyst having the lowest activity and higher selectivity for CH4. Silylation before impregnation weakened the interaction between Co and the support, and it was believed that it had enhanced the reducibility of the cobalt oxide species, and hence the catalytic activity, compared to the post impregnated catalyst. This lower rate and higher selectivity for methane for the post-metal loaded catalyst was ascribed to both low reducibility of cobalt oxide specie and steric congestion caused by the grafted silica, which inhibits both the re-adsorption of α-alkenes and chain growth.
Loc et al.200 studied the effect of modifying SBA-15 with trimethylbenzene (TMB) for cobalt-based catalysts in the FT reaction. The catalysts were prepared by impregnation with cobalt nitrate and cobalt acetate, with cobalt loading of 10, 15 and 20 wt%. The addition of TMB led to pore widening of the SBA-15 support, thereby improving the dispersion and enhanced reducibility of the cobalt oxide, which led to improved efficiency for the catalysts. The addition of ruthenium weakened the cobalt and support interaction, thereby improving catalyst reducibility, and production of C5+ hydrocarbons.
Qiu and colleagues211 recently synthesised SBA-15 with penetrable pores as the support for 15 wt% Co-based FT catalysts. The novel penetrable-pore SBA-15 materials were synthesised using amino-ended hyperbranched polyamide (AEHPA) and P123 as co-templates, which imprinted mesoporous cavities into the mesostructure. The catalysts were prepared by conventional wet impregnation, and displayed improved reducibility of the cobalt species, leading to higher CO conversion and C5+ selectivity. Pardo-Tarifa et al.212 also recently synthesised and used 3D mesocellular pore structured materials (MCM-41 and SBA-15) as the support for a 12 wt% Co-based FT catalyst, prepared by incipient wetness impregnation. The materials were modified using swelling agents triisopropylbenzene (TIPB) and 1,3,5-trimethylbenzene (TMB) to expand the pores during synthesis. The growth of cobalt oxide particles and metal–support interaction was affected by the pore size and structure, which, in turn, affected the metal dispersion and reducibility of the catalyst. The better activity of the Co/PE-SBA-15 catalyst was attributed to a higher degree of reduction of the cobalt species, as well as the open network in the 3D structure pores of the silica support, which reduced reactants and product diffusion limitations.
ZSM-5/SBA-15 composite material was evaluated as support for Co (15 wt%) catalysts in the FTS reaction, by Wu et al.224 The composites were prepared by physically mixing ZSM-5 and SBA-15 of variable proportions and impregnating it with Co, to the catalysts. Although increasing the ZSM-5 composite ratio from 0–20% did not have a significant effect on the CO conversion, the CH4 selectivity decreased while the C5+ selectivity increased. The authors attributed the high catalytic activity displayed by the 20% ZSM-5 catalyst to high Co dispersion and the larger pore size of the catalyst, which afforded an optimal acid site density. However, it was observed that a further increase in the proportion of ZSM-5 caused a decrease in CO conversion and C5+ product selectivity, with a concomitant increase in selectivity to lower hydrocarbons.
Liu et al.129 studied nitrogen-rich mesoporous carbon (NMC) as a suitable support for 40 wt% iron FT catalyst, denoted as 40Fe/Nx. The large pore volumes and specific surface areas of NMCs necessitated high loadings and proper dispersions of iron, while the nitrogen-containing groups boosted the basicity of the catalysts. The observed presence of weak metal–support interactions was instrumental to the enhanced reduction, carburization of iron, and further the FTS activities. The nitrogen-containing group's acted as anchoring sites of iron to improve the dispersion, and also helped to suppress the selectivity to methane and increase the selectivity to lower olefins in the FTS reaction, at the same time.
Vosoughi et al.128b in another work studied the impact of the impregnation solvent, such as ethanol (E), acetone (A) and water (W), as well as the loading effect on the stability, activity and selectivity of cobalt based mesoporous alumina catalyst. They found the organic solvent to better retain the textural properties of the synthesized mesoporous alumina. The 15Co(E)/m-Al2O3 catalyst, showed higher CO conversion and hydrocarbon yield compared to the commercial γ-Al2O3 catalyst. The stable mesoporous alumina supported catalyst (15Co(E)/m-Al2O3) exhibited higher FT performance as compared to γ-alumina supported cobalt catalyst. More so, by increasing the cobalt loading, the authors were able to enhance the selectivity towards C5+ HCs, and suppress methane formation, which is attributed to the increased CO hydrogenation activity.
Also, Sadek, et al.130 studied the effect of porosity size and acidity on Co-containing microporous and mesoporous beta zeolite catalysts for the FT reaction, shown in Fig. 19. The mesoporous beta zeolite catalyst (Red-Me-CoSiBeta) performed better compared to the other catalysts, with CO conversion of 91% and liquid products selectivity of 88.9%. They ascribed this high activity to the weaker metal–support interaction with the lager cobalt nanoparticles. More so, the reduced acidity in the samples leads to improved resistance to carbon deposition and increased catalyst stability in the FT reaction.
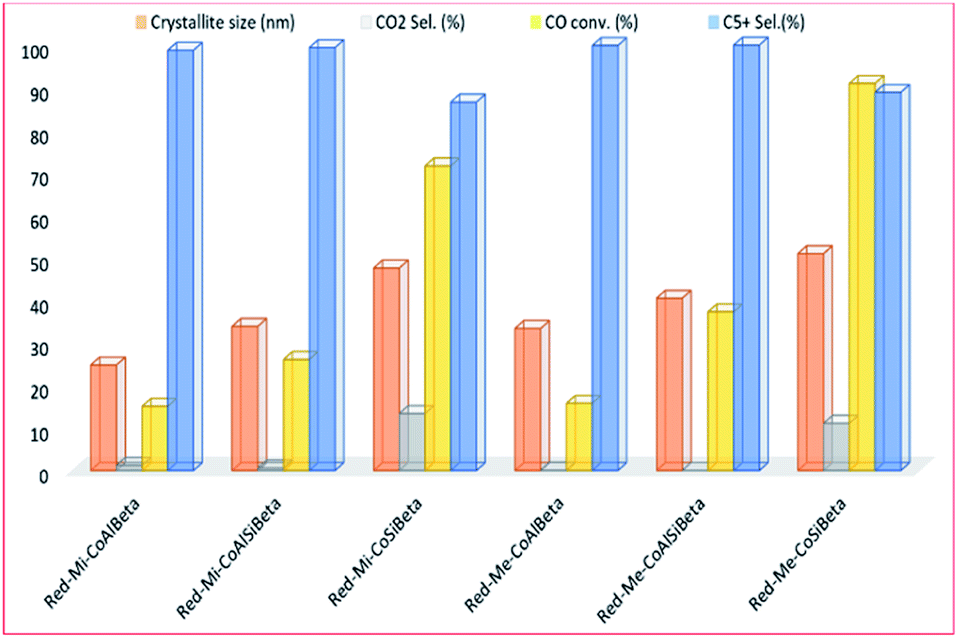 | ||
| Fig. 19 The activity and selectivity of the zeolite catalysts (with Si/Al molar ratios of 0, 5, 7 and 10) on the catalyst activity, adapted from ref. 130. | ||
Wei et al.131 whilst using 3D mesoporous cellular silica foams (MCF) investigated the activity of 20 wt% Co-based FT catalysts for the FT reaction. The 3D MCF comprises pore networks, in which large spherical cell pores (6–42 nm) are interconnected with small window pores (5–11 nm). This arrangement affords an open pore structure that favours the accessibility of reactants and products, as well as the enhancement of mass transfer in the reactions. The bifunctional catalyst was prepared by assembling ZMS-5 seeds into the 3D mesoporous MCF, to improve on the surface acidity and stability of the material (MCF-Z), which in turn, influenced the reactivity of the cobalt bifunctional catalyst. Compared with Co/MCF catalyst, the Co/MCF-Z catalysts showed higher activity and improved durability in FTS. Besides, by increasing the quantity of ZSM-5 seed, the authors observed a gradual increase in the activity of Co/MCF-Z catalysts, followed by a decreased. However, the Co/MCF-Z catalyst with the highest ZSM-5 seed content (nominal molar ratio, Al/Si = 1.5%) showed the lowest deactivation rate, ascribed to increased metal–support interaction, and the superior stability of the MCF-Z support.
Li et al.224 used zeolite-based micro-capsule catalyst (with mesoporous nano-silica shell and HY (zeolite composition SiO2/Al2O3 = 5.2) core) as support for 10 wt% cobalt based FT catalyst (Co/HY@SiO2), to tune the FTS product distribution. The micro-capsule catalyst offered the spatial confinement effects for intermedium products polymerization zone, necessitating the formation of long-chain hydrocarbons (which were hydrocracked by acidic HY core), consequently tuning the products selectivity. The silica shell through the spatial confinement effects accordingly improved the selectivity to C5–11 hydrocarbons. Li et al.133 used cobalt nanoparticles supported on mesoporous Y-type zeolites (Ymeso) to achieve the integrated tuneable synthesis of liquid fuels through FT technology. The control of the porosity and acidic properties of the zeolites was employed in tuning the type of liquid product formed. The study revealed that the porosity and acidic properties of Ymeso zeolites were instrumental, and played key roles in tuning the product distribution. Rai et al.134 studied the activity of mesoporous γ-alumina (with isolated silica sites) as support for 16 wt% cobalt catalyst. The Si doping significantly contributed to a controlled Brönsted acidity at the surface of the catalyst. This led to appropriate cracking, to produce middle distillate hydrocarbons and the suppressed formation of long-chain waxy hydrocarbons. The selectivity to CH4 was suppressed due to better heat and mass transfer.
In all of the above-mentioned cases, various aspects of the catalyst structure were exploited, to improve the activity of the catalysts, as more active sites were made available for catalysis.
Table 3 shows the properties and hydrocarbon selectivity for a modified mesoporous material-based catalyst used in FTS. The different modifications (material or structural) applied to the catalysts impacted on their activity, depending on the ratio of addition. Hence, in order to achieve optimal activity for a catalyst and obtain the desired products, the right balance is required between the active metal, support and the promoter material.
| Catalyst | CO conversion | Textural properties of the support, and catalystb | CH4 selectivity | C5+ selectivity | Comments | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| D p (nm) | V p (cm3 g−1) | BET (m2 g−1) | ||||||
| a CH – conventional heating, HDMS – hexamethyldisilazane, IW – incipient wetness, IWI – incipient wetness impregnation, MW – microwave, PS – post-synthesis, SMI – support–metal interaction, SS – supercritical solvent. b Textural properties of the catalyst. | ||||||||
| 1% Ru–20% Co SBA-15 | 55 | 7/11 [b0.33–6.9] | 1–1.3 [b0.4–0.9] | 504–957 [b375–412] | 9.5–13.5 | 72.5–81.9 | The catalyst was synthesised by IW. FTS reaction was carried out in a fixed bed, at: T = 220 °C; P = 2.0 MPa; H2/CO = 2; GSHV = 6.9 Lsyngas (gcat h)−1; mass = 1 g diluted with SiC. The Co0 crystallite size = 8.6–10.4 nm (XRD), 10.2–17.8 nm (H2 chemisorption); dispersion = 5.6–9.8%; reduction degree = 85–∼100%. CTY (TOS > 8 h) = 149–285 (10−3 molCO gCo−1 h−1) | 186 |
| 17.7% Co/Al-MCM-41 | 38.5 | 3.2 [b3.2 fresh (3.4 used)] | 0.93 [b0.66 fresh (0.23 used)] | 860 [b625 fresh (212 used)] | 38.5 | 74.4 | The impregnation method was used to prepare the catalyst. It was evaluated in microactivity reference equipment, at reaction conditions: T = 250 °C; P = 1 MPa; H2/CO = 2; GSHV = 0.15 mol kg−1 s−1; catalyst mass = 450 mg; TOS = 70 h. Co3O4 crystallite diameter = 18.1 nm; dispersion = 5.3%; reduced fraction = 44%; alpha value = 0.72; CO2 selectivity = 0.4 | 185 |
| 20% Co/INT-MM1 | 40.3 | 2.6 [b3.1 fresh (3.6 used)] | 0.66 [b0.5 fresh (0.15 used)] | 730 [b BET 584 fresh (170 used)] | 40.3 | 71.8 | Evaluation of the catalyst was at iso conversion ∼40%, reaction conditions: T = 250 °C; P = 1 MPa; H2/CO = 2; GSHV = 0.15 mol kg−1 s−1; Co0 crystallite size 20.3 nm; dispersion = 4.7%; reduced fraction = 36%; alpha value = 0.7; CO2 selectivity = 0.7 | 185 |
| 3.25–3.99% Ru/SBA-15 | 19.3–41.78 | 3.7–13.3 [b3.6–13.1] | 0.53–1.94 [b0.38–1.78] | 536.6–676.8 [b402–628.7] | 22.56–38.62 | 44.15–60.3 | Prepared by incipient wetness impregnation and ethylene glycol reduction. The SBA-15 was modified using trimethyl silane (TMS) and Ru was introduced by complexation, by APTS. Fixed bed reaction conditions: P = 1.0 MPa; T = 235 °C; H2/CO = 2; flow = 0.5 SL h−1 g−1; mass = 100 mg mixed with 1 g carborundum. Particle size = 2.8–5.4 nm (TEM), 3.8–5.7 nm (H2 chemisorption). Ru time yield = 28.89–50.33 (mmolCO gRu−1 h−1) | 188 |
| 10% Co/ZrSBA-15 | 17.62–26.61 | 6.3–8.5 | 0.53–1.1 | 226.3–515.9 | 7.45–14.37 | 79.01–86.71 | Prepared by IWI and isomorphic substitution of Zr on SBA-15 (Zr/Si ratios 1:3–100). The reaction was carried out in a fixed bed, at: T = 230 °C; P = 1 MPa; H2/CO = 2; SV = 8 SL g−1 h−1; catalyst mass = 500 mg. Corrected metal cluster diameter = 10.3–22 nm; corrected dispersion = 6.96–14.55%; reduction degree = 37.48–58.47% |
193 |
| 10% Co/ZrSBA-15 | 19.28–33.53 | 5.7–17.6 | 0.81–2.07 | 355.4–572.6 | 6.71–10.83 | 82.41–85.47 | Prepared by IWI and isomorphic substitution of Zr on SBA-15 in ratios 1:3–100. The reaction was carried out in a fixed bed, at: T = 230 °C; P = 1 MPa; H2/CO = 2; SV = 8 SL g−1 h−1; catalyst mass = 500 mg. Corrected metal cluster diameter = 6.7–19.2 nm; corrected dispersion = 3.67–8.45%; reduction degree = 48.15–52.02% |
194 |
| 15% Co/N-SBA-15 | 42.3–56.7 | 5.1–5.2 [b5.1] | 0.88–0.91 [b0.71/0.82] | 775.8–792.5 [b600.5–645.8] | 11.8–13.8 | 73.4–78.3 | Support synthesized AEHPA, to imprint mesoporous cavities into the silica structure (AEHPA/P123 mass ratios, 1 and 2.5). The catalyst was prepared using the IWI method, and FTS reaction was carried out in a fixed bed reactor, at: T = 240 °C; P = 1 MPa; H2/CO = 2; GHSV = 8000 mL (gcat h)−1; mass = 100 mg mixed with 300 mg carborundum. Particle size = 9.5–10.6 nm (Co3O4), 7.1–8 nm (Co); dispersion 12–13.5%; alpha value = 0.78. Amino-ended hyper-branched polyamide (AEHPA) was used as the auxiliary template to create penetrable pores | 211 |
| 12% Co/PE-MCM-41, *12% Co/PE-SBA-15 | 5.7–29.1/*18.5–28.5 | 6.3/(*29.3, window 5) [b6.8/*29.0 (7.1 window)] | 1.8/*1.9 [b1.4/*1.8] | *860/900 [b*576/633] | 8.7–10.1/*7.2–11.1 | 80–84.6/*73.6–85.2 | The catalyst was prepared by IWI. FTS reaction conditions: T = 210 °C; P = 2 MPa; H2/CO = 2; GHSV = 1600–6000 (SBA-15: 4800–6000) mL (gcat h)−1 NTP; mass = 1 g with 5 g SiC. Particle size (after reduction): Co3O4 = 9.4/*14.7 nm, Co0, 7/*11.1 nm (XRD), 6.1/*7.5 nm (chemisorption); corrected dispersion = 5.6/*7.4%; reduction degree = 43/*58% CO2 selectivity = 0.4–0.7/*0.8–1.2. Window = diameter between the pores. The 3D structured pores reduced the diffusion limitations of the reactants and products | 212 |
| 0.05–0.5% Ru/5/*30% Co/SBA-15 | 6.9–8.9/*33.4–37.7 | 6.46 [b*6.15–6.16] | 1.37 [b*0.67–0.68] | 897 [b*467–476] | 33.9–39.6/*6.83–8.47 | 53.6–60.7/*81.7–84.2 | The IWI method was used to prepare the catalyst. It was evaluated under FTS reaction conditions in a tubular fixed bed reactor, at: T = 210 °C; P = 2 MPa; H2/CO = 2; GHSV = 8000 h−1 (8 SL h−1 g−1); 0.5 g with 5 g carborundum. Corrected metal cluster diameter = *26.4–28.7 nm; cobalt particle diameter = *11.5–13; dispersion = *3.6–3.9%; DOR = *53.4–60.7%; CO2 selectivity = 0.6–1.2/*0.02–0.8 | 215 |
| 20% (CoMnCa; 10:1:1) Al SBA-15 |
∼7–43 | 6.29 [b3.94 ] | 1.25 [b*0.76] | 955 [b*459] | ∼60–83 | ∼2.4–7.5 | The Co-impregnation method was employed in catalyst synthesis. Reaction was carried out in a fixed bed reactor at: T = 250–350 °C; P = 0.1 MPa; H2/CO = 1–2; GHSV = 300 h−1; CO2 selectivity = ∼5–29. Optimal reaction condition was obtained at 325 °C, and a CO:H2 ratio of 1.25 |
202 |
| 12% Co/(5/10% Ti)/SBA-15 | 8.6–43.6/8.6–40 (*5% Ti, 60.3/46.2 and 10% Ti 51.6/44.9, with water addition of 20% & 33%, respectively) | 10.2/11.2 [b3.5/4] | 1.29/1.19 [b0.43/0.58] | 907/894 [b606/633] | 7.5–13.9/8.7–13.5 (*5% Ti: 4/3.4 and 10% Ti: 4.4/3.5, with water addition of 20% & 33%, respectively) | 74.1–85.9/75.2–84.2 (*5% Ti: 91.4/92.5, 10% Ti: 90.4/91.8, water addition of 20% & 33%, respectively) | The catalyst was synthesized by IWI. Fixed bed FT synthesis reaction conditions: T = 200 °C; P = 2 MPa; H2/CO = 2; GHSV = 5143 h−1; mass = 700 mg diluted with 5 g SiC. Co0 diameter 12/10 nm (XRD), 20/18 nm (chemisorption); dispersion 4.3/4.7%, corrected 4.8/5.2%; reduction degree = 89/90%; TOF (after 10 hours on stream) = 0.063/0.061 s−1; O/P ratio (C3) = 1.9–3.4/1.8–3.2 (*5% Ti, 2.7/3.4 and 10% Ti, 2.1/4.0, with water addition of 20% & 33%, respectively) | 203 |
| 12% Co/(5/10% Al)/SBA-15 | 16.6–49.9/Al 10%, 14.1–41.8 (*Al 5%, 44.2/34.4, and Al 10%, 51.6/44.9, with the addition of 20% & 33% water, respectively) | 9.7/11.2 [b*3.8/4.8] | 1.2/1.39 [b0.54/0.78] | 920/1010 [b638/715] | 11.6–15.8/11.5–17.5 (*Al 5%, 6.2/5.0, and Al 10%, 6.1/4.8, with the addition of 20% & 33% water, respectively) | 69.6–75.5/66.7–76 (*Al 5%, 84.2/86.3, and Al 10%, 84.9/87.3, with the addition of 20% & 33% water, respectively) | The catalyst was synthesized using the IWI method. Fixed bed FT synthesis reaction conditions: T = 200 °C; P = 2 MPa; H2/CO = 2; GHSV = 8857/6214 h−1; mass = 700 mg diluted with 5 g SiC. Co0 diameter 8 nm (XRD), 10 nm (chemisorption); dispersion = 7.8 corrected 10.1%, and/7.6 corrected to 9.9% for Al 5% and Al 10% respectively; reduction degree = 77%; TOF (after 10 hours on stream) = 0.065/0.057 s−1; O/P ratio (C3) = 1.8–2.6/1.7–2.6 (*Al 5%, 3/3.9, and Al 10%, 2.8/3.9, with the addition of 20% & 33% water, respectively). Conversion & C5+ selectivity increased significantly with H2O addition for Ti catalyst, but not as much for the others. The addition of Al resulted in a less reducible catalyst, with respect to SBA-15 | 203 |
| 10% Co/0.2% Pd SBA-15 | Rate = 0.7-*1.1 (×10−2 molCO gcat−1 h−1) | 10.6/*11.2 [b8.8/*10.8] | 1.45/*1.48 [b0.85/*0.83] | 633/*487 [b397/*307] | 26.1/*27.8 | 56.4/*54.8 | Supports were synthesized conventionally and with the addition of decane (as the swelling agent to control the pore size, *M). The catalyst was then synthesised using the co-impregnation method. FTS (fixed bed) reaction conditions: T = 230 °C; P = 0.5 MPa; H2/CO = 2; mass = 1 g. Co3O4 metal crystal size 10.5/*9.7 nm. O/P ratio = 0.07/*0.03 (C2–C4); alpha value = 0.74/*0.71; CO2 selectivity = 0. The SBA-15(M) catalyst displayed enhanced activity due to shorter cavity length and the presence of macropores, which enhanced the diffusion of reactants, intermediates and products. The SBA-15(M) bimetallic catalyst displayed higher conversion. The presence of Pd enhanced the reducibility of cobalt oxides | 191 |
| 0.3% Ru/10% Co/MCM41 (0.3% Ru/10% Co/SBA-15) | 4.7/(*7.8–8.9) | 3.4/(4.4–*5.7) [b(4.4 –*6.1)] | 1.05/(0.92–*1.13) [b0.31/(0.50 –*0.69)] | 935/*1029 (1180) [b441/*519 (439)] | 11.9/(*9.3–10.1) | 60/(73.2 –*76.3) | The catalyst was prepared by IWI and FTS was conducted in a fixed bed, at: T = 190 °C; P = 0.1 MPa; H2/CO = 2; GHSV = 1800 mL (g h)−1; mass = 500 mg. Co3O4 crystallite diameter = 6.4/(8.8–*9.2) nm, (XRD), 2.1/(*8.7) nm (XPS); dispersion = 47.7/*14.7% (XPS); CTY = 2/(3.3–*3.8) (10−4 s−1); alpha value = 0.74/(0.79–*0.81). *(The SBA-15 catalyst was synthesized with 5 times the concentration of HCl). Ru promotion of smaller pored catalysts led to an increased FT reaction rate, but less on the larger pore catalyst | 208 |
| 10% Co/2–10% Zr/SBA-15 (10% Co/2–10% Zr/MCM-41) | 17.7 (4.8) | 7.7 (3.4) [b7.2–7.5 (2.8)] | 1.13 (1.05) [b0.79–0.91 (0.54–0.65)] | 935 (1189) [b516–613 (498–608)] | 21.1 (33) | 55 (31.4) | The support was prepared by sequential impregnation. FT reaction conditions (in a fixed bed tubular micro reactor): T = 200 °C; P = 2 MPa; H2/CO = 2; GHSV = 18000 mL (g h)−1; mass = 500 mg. CO3O4 crystallite size 9.7–10.2 (9.8–13.3) nm, (from XRD); dispersion 9.4–10.8 (7.1–9.8)% (from propene chemisorption); TOF = 4.2–4.5 (2.4–3.8) × 10−2 s−1; FT reaction rate = 4.2–4.5 (2.4–3.8) × 10−4 s−1 |
197 |
| 20% Co (0.5% Ru) SBA-15 | 39–40 | 7.1 [b*6.6] | 0.79 [b0.71] | 641 [b385] | 4.3–18.6 | 72.1–80.9 | Support was synthesised by *MW, as well as CH, while synthesis of the catalyst was achieved by wet and co-impregnation methods. FTS was carried out in a slurry (stirred batch 500 mL autoclave) reactor at: T = 240 °C; P = 2 MPa; H2/CO = 1; mass = 3 g; mechanical agitation of 800 rpm. Average Co3O4 crystallite = 16.1 nm. O/P ratio = 0.9–1.5. Reducibility was said to increase with Ru content, but no data was provided | 209 |
| 5% Co (0–0.5% Ru) SBA-15 | 6.51–20.51 | 7.7 | 1.41 | 860 | 19.17–49.39 | 42.43–77.14 | The catalyst was prepared by IWI. Fixed bed reaction conditions: T = 230 °C; P = 1 MPa; H2/CO = 2; GHSV = 3000 h−1; mass = 3 g mixed with 36 g carborundum and 80 g glass beads. Average Co3O4 diameter = 11.5–13.4 nm (XRD); DOR = 17.8–28.7%; TOF = 27.1–30.8 (10−3) s−1. Alpha value = 0.79/0.81 | 179 |
| 15% Co 5% Zr–SBA-15 | *80–93 | *7.9/9.1 [b3.5–*3.8] | *1.13/1.18 [b0.23–0.32 (*0.29)] | *588/831 [b277–339] | 15–*28 | *52–62 | The catalyst was prepared by *IWI, sequential impregnation and co-impregnation. Fixed bed reaction conditions: T = 240 °C; P = 2 MPa; H2/CO = 2; GHSV = 1000 h−1. Co3O4 crystallite size = *13–15 nm; dispersion = 6.3–*7.5%; reduction degree = *74–90%; TOF = 0.04–0.1 (×10−3) s−1 @ CO conversions of 5–10% & GSVH of 10000–18000 h−1. Alpha value = *0.78–0.84; CO2 selectivity = *3–14. Higher selectivity of C12–C22 hydrocarbons (53%) was displayed by the catalyst prepared by means of sequential impregnation (Co/Zr/SBA-15), with α value up to 0.84, which was attributed to higher reducibility of the cobalt species |
199 |
| 15% Co/5% Zr/SBA-15 | ||||||||
| 15% Co 5% Zr/SBA-15 | ||||||||
| Co/Al-SBA-15 (Al: 0–10) | 32.06–49.59 | 4.7 [b6.2–10.4] | 1.03 [b1.03–1.14] | 867 [b415–732] | 12.32–19.6 | 69.98–82.44 | The catalyst was prepared by dry impregnation (DI). FTS reaction was carried out in a fixed bed reactor, at: T = 230 °C; P = 2 MPa; H2/CO = 2; WHSV = 10000 mL gcat−1 h−1; mass = 300 mg. Co3O4 crystallite size = 9.4–12.5 nm; Co0 13–14.4 nm (after reduction); dispersion = 10.08–13.62%; O/P ratio = 0.43–1.21. Si/Al molar ratio (0–10) |
204 |
| 15% Co-SBA-15/(*ZSM-5) | 70.5–90.6 | 2.4(*1.6) [b2.2–3] | 0.28/*(0.16) [b0.15–0.3] | 486/(*398) [b273–396] | 13.3–21.4 | 68.8–79.7 | The composite support was prepared by physically mixing various proportions of SBA-15 and ZSM-5, before incorporating Co via IWI. FTS reaction was carried out in a fixed bed reactor, at: T = 240 °C; P = 2 MPa; H2/CO = 2; WHSV = 1000 h−1; mass = 1 g mixed with 1.5 mL quartz. Co3O4 crystallite size = 12.1–26 nm; dispersion = 4.9–10.6%; reduction degree = 48.3–76.5%; CO2 selectivity = 18.5 | 224 |
| 40Fe/N0 | 86.3–92.6 | 11.5–15.1 [b0.57–1.35] | 2.01–3.62 [b0.88–1.24] | 697–960 [b202–396] | 11.6–24.7 | 25.0–53.3 | 40Fe/Nx catalysts were prepared by ultrasonic assisted IWI with different nitrogen content, [40Fe/N0, nitrogen content 0 (wt%); 40Fe/N1, nitrogen content 8.3 (wt%); 40Fe/N2, nitrogen content 16.5 (wt%)]; fixed bed, 260 °C. NMC prepared with diff nitrogen content. T = 230 °C; P = 1 MPa; H2/CO = 1; GHSV = 505–1720 h−1. Crystallite size = 12.2–15.8 nm; dispersion = 6.1–7.9%; reduction degree = 60.0–79.7%; CO2 selectivity = 1.4–3.6% | 129 |
| 40Fe/N1 | ||||||||
| 40Fe/N2 | ||||||||
| 15Co(E)/γ-Al2O3 | 39.3–74.5 | 11.8(*7.7) [b7.5–11.6] | 1.6(*0.79) [b0.57–1.35] | 410(*280) [b221–342] | 13.2–16.7 | 79.4–84.6 | The effect of metal promotion study with catalysts prepared by IWI using ethanol (E), acetone (A) and water (W) solvents. Fixed bed reaction conditions: T = 230 °C; P = 2 MPa; H2/CO = 2; GHSV = 505–1720 h−1. Crystallite size = 12.2–15.8 nm; dispersion = 6.1–7.9%; reduction degree = 60.0–79.7%; CO2 selectivity = 1.4–3.6% | 128b |
| 15Co(E)/m-Al2O3 | ||||||||
| 15Co(A)/m-Al2O3 | ||||||||
| 15Co(W)/m-Al2O3 | ||||||||
| 22.5Co(E)/m-Al2O3 | ||||||||
| 30Co(E)/m-Al2O3 | ||||||||
4. Concluding remarks and perspectives
4.1. Concluding remarks
An adequate understanding of the structure–performance relationship could lead to the development of improved, efficient and selective FT catalysts. This review discussed the potential role that ordered mesoporous materials could play to improve the possible and desirable FT products, by fostering and understanding of the complex systems at the atomic and molecular level.Mesoporous materials, as supports for the FTS reaction, have very important unique properties, such as highly ordered mesopores, hydrothermal stability, thick walls, uniform pore size distribution and high surface area. Due to the ease of morphology modification, they afford the controllability of dispersity, reducibility, crystallite size, adsorption, and conductive and catalytic properties.
Different templating pathways are generally used for the synthesis of both silica and non-silica based mesoporous materials (such as carbon, zeolite, alumina etc.). These approaches include hard templating, soft templating, in situ templating, multiple templating, reticular chemistry guiding approach, template free packing method, etc.
The product distribution of the FT reaction strongly depends on the density, the size of the active species and the structure of the support. Larger pores give higher reducibility and larger metal cluster sizes, while it decreases both the dispersion of the active atoms and the surface area. In addition, larger pores enhance the selectivity of the long chain hydrocarbons. A shape selectivity effect occurs during FTS due to the influence of the structure on the CO diffusion barrier and the removal rate of the reaction intermediates.
The various modifications can be achieved through pore tuning (over a wide range), functionalization/promotion (with Al, Zr, Ti, etc.) or immobilisation of other active elements (such as Ru, Re, Mn, Pd, etc.) into their porous structure. Structural modifications using oxides (MnO and ZrO2), TMB, HMDS, AEHPA, contribute to better reducibility of the catalysts and mass transfer of the reactants and products, which in turn, facilitate the catalytic performance of FTS.
In summary, the review shows how exploitation of the porosity of mesoporous materials and several catalysts structural features, can be used to influence the catalytic properties and selectivity toward desired FTS products. This development will contribute to the production of environmentally clean fuels, waxes and chemicals, by improving efficiency and selectivity towards desirable and necessary products. The improved efficiency will have a positive impact on sustainability with regards to the energy generated from the process.
4.2. Insights/perspectives
The product distribution of FTS is extremely complex as it covers a range of hydrocarbon products from C1 to C100+. The size of the reaction intermediates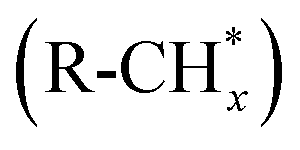 could be tuned directly by the support structure as this sets the pore size and the pore length, which influences the final product spectrum. Therefore, a uniform structure of the support is able to facilitate the selectivity to a certain range of the hydrocarbon products (such as gasoline-range, diesel-range, jet fuel-range, or hard wax-range). However, most of the studies reported the effect of the structure on the selectivity of the products in a wide range (such as C5+). More research is recommended to focus on the effect of the nature of the porosity on narrow ranges of the products.
could be tuned directly by the support structure as this sets the pore size and the pore length, which influences the final product spectrum. Therefore, a uniform structure of the support is able to facilitate the selectivity to a certain range of the hydrocarbon products (such as gasoline-range, diesel-range, jet fuel-range, or hard wax-range). However, most of the studies reported the effect of the structure on the selectivity of the products in a wide range (such as C5+). More research is recommended to focus on the effect of the nature of the porosity on narrow ranges of the products.
Although mesopores provide high surface area with suitable dispersion and reducibility of the active species, the macropores facilitate mass transfer as they play a role in reducing the internal diffusion limitation. Therefore, more research on the combination of mesoporous/macroporous materials as supports for the FTS reaction is also recommended. In addition, the investigation of the mass and heat transfer during FTS using mesoporous material supported iron and cobalt catalysts provides useful information for the design of the FT reactor and the optimization of the FTS process.
Some of the mesoporous materials exhibit high CH4 selectivity and low C5+ selectivity, especially for iron-based catalyst. More research is necessary in areas such as development of new non-silica based mesoporous materials and synthesizing more advanced structures for FT reaction by 3D printing technology.
Besides, the production of value-added building block chemicals, such as C2–C4 olefins and oxygenates, via FTS is an attractive method to cut the dependence of fossil fuels. However, how to improve the activity and selectivity of the light olefins and oxygenates are still challenging work. Structural promotion using the mesoporous metal oxides as supports, combined with chemical promotion has the potential to catalyse the syngas conversion to the production of the building block chemicals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to acknowledge the support from the University of Johannesburg, and the University of South Africa, IDEAS.References
- R. A. van Santen, I. M. Ciobîcă, E. van Santen and M. M. Ghouri, Adv. Catal., 2011, 54, 127–187 CAS.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128(12), 3956–3964 CrossRef CAS.
- N. O. Elbashir, D. B. Bukur, E. Durham and C. B. Roberts, Proceedings of the 1st Annual Gas Processing Symposium, ed. H. Alfadala, G. V. Rex Reklaitis and M. M. El-Halwagi, 2009 Search PubMed.
- (a) M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS; (b) M. E. Dry, Appl. Catal., A, 2004, 276, 1 CrossRef CAS.
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS.
- B. Jager and R. Espinoza, Catal. Today, 1995, 23, 17–28 CrossRef CAS.
- R. B. Anderson, R. A. Friedel and H. H. Storch, J. Chem. Phys., 1951, 19, 313–319 CrossRef CAS.
- C. H. Bartholomew, Catal. Lett., 1990, 7, 303–315 CrossRef CAS.
- Y. Yao, D. Hildebrandt, D. Glasser and X. Liu, Ind. Eng. Chem. Res., 2010, 49, 11061–11066 CrossRef CAS.
- A. P. Steynberg, Stud. Surf. Sci. Catal., 2004, 152, 1–63 CrossRef CAS.
- A. N. Pour, S. Taghipoor, M. Shekarriz, S. M. K. Shahri and Y. Zamani, J. Nanosci. Nanotechnol., 2009, 9, 4425–4429 CrossRef CAS.
- A. N. Pour, M. R. Housaindokht, S. F. Tayyari and J. Zarkesh, J. Nat. Gas Chem., 2010, 19, 284–292 CrossRef CAS.
- A. N. Pour, M. R. Housaindokht, S. T. Faramarz, J. Zarkesh and M. R. Alaei, J. Nat. Gas Sci. Eng., 2010, 2, 61–68 CrossRef CAS.
- A. N. Pour, M. R. Housaindokht, J. Zarkesh and S. T. Faramarz, J. Nat. Gas Sci. Eng., 2010, 2, 79–85 CrossRef CAS.
- A. N. Pour, M. R. Housaindokht, S. T. Faramarz and J. Zarkesh, J. Nat. Gas Chem., 2010, 19, 107–116 CrossRef CAS.
- A. N. Pour, M. R. Housaindokht, S. F. Tayyari, J. Zarkesh and M. R. Alaei, J. Mol. Catal. A: Chem., 2010, 330, 112–120 CrossRef.
- A. N. Pour, M. Zare and Y. Zamani, J. Nat. Gas Chem., 2010, 19(1), 31–34 CrossRef CAS.
- M. Ojeda, R. Nabar, A. U. Nilekar, A. Ishikawa, M. Mavrikakis and E. Iglesia, J. Catal., 2010, 272, 287–297 CrossRef CAS.
- G. P. van der Laan and A. A. C. M. Beenackers, Catal. Rev. - Sci. Eng., 1999, 41, 255–318 CrossRef CAS.
- M. J. A. Tijmensen, A. P. C. Faaij, C. N. Hamelinck and M. R. M. van Hardeveld, Biomass Bioenergy, 2002, 23, 129 CrossRef CAS.
- R. W. R. Zwart and H. Boerrigter, Energy Fuels, 2005, 19, 591 CrossRef CAS.
- Z. P. Liu and P. A. Hu, J. Am. Chem. Soc., 2002, 124, 11568–11569 CrossRef CAS.
- Value of several thousand square meters per H. Jahangiri, J. Bennett, P. Mahjoubi, K. Wilson and S. Gua, Catal. Sci. Technol., 2014, 4, 2210–2229 RSC.
- C. Perego, R. Bortolo and R. Zennaro, Catal. Today, 2009, 142, 9–16 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- P. Biloen, J. W. Helle and W. M. H. Sachtler, J. Catal., 1979, 58, 95–107 CrossRef CAS.
- V. Ponec, Catal. Rev. - Sci. Eng., 1978, 18, 151–171 CrossRef CAS.
- (a) T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, J. Catal., 2006, 243, 199–211 CrossRef CAS; (b) T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, Appl. Catal., A, 2006, 311, 66–75 CrossRef CAS; (c) T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, Appl. Catal., A, 2006, 308, 19–30 CrossRef CAS.
- Z.-J. Wang, S. Skiles, F. Yang, Z. Yan and D. W. Goodman, Catal. Today, 2012, 181, 75–81 CrossRef CAS.
- C. Liu, B. Zou, A. J. Rondinone and Z. J. Zhang, J. Phys. Chem. B, 2000, 104, 1141–1145 CrossRef CAS.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- B. G. Johnson, C. H. Bartholomew and D. W. Goodman, J. Catal., 1991, 128, 231–247 CrossRef CAS.
- H. Hayashi, L. Z. Chen, T. Tago, M. Kishida and K. Wakabayashi, Appl. Catal., A, 2002, 231, 81–89 CrossRef CAS.
- X. Li, B. Zhong, S. Peng and Q. Wang, Catal. Lett., 1994, 23, 245–250 CrossRef CAS.
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and F. Villain, J. Phys. Chem. B, 2001, 105, 9805–9811 CrossRef CAS.
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and V. L. Zholobenko, J. Catal., 2002, 206, 230–241 CrossRef CAS.
- K. Kaneko, J. Membr. Sci., 1994, 96, 59–89 CrossRef CAS.
- R. C. Reuel and C. H. Bartholomew, J. Catal., 1984, 85, 78–88 CrossRef CAS.
- A. Barbier, A. Tuel, I. Arcon, A. Kodre and G. A. Martin, J. Catal., 2001, 200, 106–116 CrossRef CAS.
- F. Fischer and H. Tropsch, Brennst.-Chem., 1923, 4, 276–285 CAS.
- A. M. Saib, M. Claeys and E. van Steen, Catal. Today, 2002, 71, 395–402 CrossRef CAS.
- K. Cheng, J. Kang, S. Huang, Z. You, Q. Zhang, J. Ding, W. Hua, Y. Lou, W. Deng and Y. Wang, ACS Catal., 2012, 2, 441–449 CrossRef CAS.
- T. Koh, H. M. Koo, T. Yu, B. Lim and J. W. Bae, ACS Catal., 2014, 4, 1054–1060 CrossRef CAS.
- A. J. Plomp, H. Vuori, A. O. Krause, K. P. Jong and J. H. Britter, Appl. Catal., A, 2008, 351, 9–15 CrossRef CAS.
- C. Tang, H.-F. Wang, J.-Q. Huang, W. Qian, F. Wei, S.-Z. Qiao and Q. Zhang, Electrochem. Energy Rev., 2019, 2, 332–371 CrossRef CAS.
- S. L. Suib, Chem. Rec., 2017, 17, 1–16 CrossRef.
- S. S. Park and C.-S. Ha, Adv. Funct. Mater., 2017, 1703814 Search PubMed.
- L. Travaglini, P. Picchetti, A. Del Giudice, L. Galantini and L. De Cola, Microporous Mesoporous Mater., 2019, 279, 423–431 CrossRef CAS.
- S. Kumar, M. M. Malik and R. Purohit, Mater. Today: Proc., 2017, 4, 350–357 Search PubMed.
- P. Qiu, B. Ma, C.-T. Hung, W. Li and D. Zhao, Acc. Chem. Res., 2019, 52, 2928–2938 CrossRef CAS.
- L. Liu, X. Yang, Y. Xie, H. Liu, X. Zhou, X. Xiao, Y. Ren, Z. Ma, X. Cheng, Y. Deng and D. Zhao, Adv. Mater., 2020, 1906653 CrossRef CAS.
- D. Schneider, D. Mehlhorn, P. Zeigermann, J. Kärger and R. Valiullin, Chem. Soc. Rev., 2016, 45, 3439–3467 RSC.
- G. Du, Y. Xu, S. Zheng, H. Xue and H. Pang, Small, 2019, 1804600 CrossRef.
- C. B. Contreras, O. Azzaroni and G. J. A. A. Soler-Illia, in Comprehensive Nanoscience and Nanotechnology, ed. D. Andrews, T. Nann and R. Lipson, Academic Press, United States, 2018, p. 17 Search PubMed.
- X.-P. Fu, Q.-K. Shen, D. Shi, K. Wu, Z. Jin, X. Wang, R. Si, Q.-S. Song, C.-J. Jia and C.-H. Yan, Appl. Catal., B, 2017, 211, 176–187 CrossRef CAS.
- M. G. Salameh, Appl. Energy, 2003, 75(1–2), 33–42 CrossRef.
- M. de Wit, M. Junginger, S. Lensink, M. Londo and A. Faaij, Biomass Bioenergy, 2010, 34(2), 203–217 CrossRef.
- K. Sunde, A. Brekke and B. Solberg, Forest Policy Econ., 2011, 13(8), 591–602 CrossRef.
- G. R. Timilsina and A. Shrestha, Energy, 2011, 36(4), 2055–2069 CrossRef.
- S. K. Beaumont, Phys. Chem. Chem. Phys., 2014, 16, 5034–5043 RSC.
- N. R. Shiju and V. V. Guliants, Appl. Catal., A, 2009, 356, 1–17 CrossRef CAS.
- E. Iglesia, S. L. Soled, J. E. Baumgartner and S. Reyes, J. Catal., 1995, 153, 108–122 CrossRef CAS.
- F. Masini, C. E. Strebel, D. N. McCarthy, A. U. F. Nierhoff, J. Kehres, E. M. Fiordaliso, J. H. Nielsen and I. Chorkendorff, J. Catal., 2013, 308, 282–290 CrossRef CAS.
- C. Zhang, K.-W. Jun, K.-S. Ha, Y.-J. Lee and S. C. Kang, Environ. Sci. Technol., 2014, 48, 8251–8257 CrossRef CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- B. Sun, K. Xu, L. Nguyen, M. Qiao and F. F. Tao, ChemCatChem, 2012, 4, 1498–1511 CrossRef CAS.
- W. J. Wang and Y. W. Chen, Appl. Catal., 1991, 77, 223–233 CrossRef CAS.
- B. Ernst, S. Libs, P. Chaumette and A. Kiennemann, Appl. Catal., A, 1999, 186, 145–168 CrossRef CAS.
- J.-I. Yang, J. H. Yang, H.-J. Kim, H. Jung, D. H. Chun and H.-T. Lee, Fuel, 2010, 89, 237–243 CrossRef CAS.
- J. Bao, G. H. Yang, C. Okada, Y. Yoneyama and N. Tsubaki, Appl. Catal., A, 2011, 394, 195–200 CrossRef CAS.
- A. Y. Krylova, A. A. Panin, A. S. Lyadov, S. A. Sagitov, V. I. Kurkin and Y. G. Kryazhev, Pet. Chem., 2011, 51, 317–323 CrossRef CAS.
- H. Y. Suo, S. G. Wang, C. H. Zhang, J. Xu, B. S. Wu, Y. Yang, H. W. Xiang and Y. W. Li, J. Catal., 2012, 286, 111–123 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- D. Zhao, J. Sun, Q. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275–279 CrossRef CAS.
- B.-H. Min, E.-Y. Jeong, M. Thommes and S.-E. Park, Chem. Commun., 2011, 47, 4673–4675 RSC.
- S. Shen, Y. Deng, G. Zhu, D. Mao, Y. Wang, G. Wu, J. Li, X. Liu, G. Lu and D. Zhao, J. Mater. Sci., 2007, 42, 7057–7061 CrossRef CAS.
- M. C. A. Fantini, J. R. Matos, L. C. C. da Silva, L. P. Mercuri, G. O. Chiereci, E. B. Celer and M. Jaroniec, Mater. Sci. Eng., B, 2004, 112, 106–110 CrossRef.
- A.-H. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793–1805 CrossRef CAS.
- Y. Li, W. Zhang, L. Zhang, Q. Yang, Z. Wei, Z. Feng and C. Li, J. Phys. Chem. B, 2004, 108, 9739–9744 CrossRef CAS.
- Y. Zou, X. Zhou, J. Ma, X. Yang and Y. Deng, Chem. Soc. Rev., 2019, 49, 1173–1208 RSC.
- T. Galy, D. Mu, M. Marszewski and L. Pilon, Comput. Mater. Sci., 2019, 157, 156–167 CrossRef CAS.
- Z. Teng, W. Li, Y. Tang, A. Elzatahry, G. Lu and D. Zhao, Adv. Mater., 2018, 1707612, DOI:10.1002/adma.201707612.
- E. P. Barret, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73(1), 373–380 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 4, 603–619 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Niemark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquérol and K. S. W. Sing, Pure Appl. Chem., 2015, 87(9–10), 1051–1069 CAS.
- N. Rahmat, A. Z. Abdullah and A. R. Mohamed, Am. J. Appl. Sci., 2010, 7(12), 1579–1586 CrossRef CAS.
- L. B. McCusker, F. Liebau and G. Engelhardt, Microporous Mesoporous Mater., 2003, 58, 3–13 CrossRef CAS.
- F. Liebau, Microporous Mesoporous Mater., 2003, 58, 15–72 CrossRef CAS.
- J. M. Fedeyko, D. G. Vlachos and R. F. Lobo, Microporous Mesoporous Mater., 2006, 90, 102–111 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. T. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- I. A. Aksay, M. Trau, S. Manne, I. Honma, N. Yao, L. Zhou, P. Fenter, P. M. Eisenberger and S. M. Gruner, Science, 1996, 273, 892–898 CrossRef CAS.
- S. Schächt, Q. Huo, I. G. Voigt-Martin, G. D. Stucky and F. Schüth, Science, 1996, 273, 768–771 CrossRef.
- S. Mann and G. A. Ozin, Nature, 1996, 382, 313–318 CrossRef CAS.
- S. A. Davis, S. L. Burkett, N. H. Mendelson and S. Mann, Nature, 1997, 385, 420–423 CrossRef CAS.
- S. Mann, Chem. Mater., 1997, 9, 2300–2310 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schüth and G. D. Stucky, Nature, 1994, 368, 317–321 CrossRef CAS.
- M. Trau, N. Yao, E. Kim, Y. Xia, G. M. Whitesides and I. A. Aksay, Nature, 1997, 390, 674–676 CrossRef CAS.
- H. Yang, N. Coombs and G. A. Ozin, Adv. Mater., 1997, 9, 811–814 CrossRef CAS.
- E.-P. Ng, H. Abdullahi, F. Khoerunnisa, P. Maireles-Torres, H. L. Lee, F. Adam, T. C. Ling and K.-L. Wong, Microporous Mesoporous Mater., 2020, 297, 110016 CrossRef CAS.
- Q. Zhang, K. Cheng, J. Kang, W. Deng and Y. Wang, ChemSusChem, 2014, 7, 1251–1264 CrossRef CAS.
- S. Polarz and M. Antonietti, Chem. Commun., 2002, 2593–2604 RSC.
- R. Martín-Aranda and J. Čejka, Top. Catal., 2009, 53, 141–153 CrossRef.
- A. Sayari, Chem. Mater., 1996, 8, 1840–1852 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS.
- E. Molina, M. Mathonnat, J. Richard, P. Lacroix-Desmazes, M. In, P. Dieudonné, T. Cacciaguerra, C. Gérardin and N. Marcotte, Beilstein J. Nanotechnol., 2019, 10, 144–156 CrossRef CAS.
- M. Hartmann, C. Bischof, Z. Luan and L. Kevan, Microporous Mesoporous Mater., 2001, 44–45, 385–394 CrossRef CAS.
- P. Tian, J. Blanchard, K. Fajerwerg, M. Breysse, M. Vrinat and Z. Liu, Microporous Mesoporous Mater., 2003, 60, 197–206 CrossRef CAS.
- H. B. Jervis, M. E. Raimondi, R. Raja, T. Maschmeyer, J. M. Seddon and D. W. Bruce, Chem. Commun., 1999, 2031–2032 RSC.
- V. Hulea, D. Brunel, A. Galarneau, K. Philippot, B. Chaudret, P. J. Kooyman and F. Fajula, Microporous Mesoporous Mater., 2005, 79, 185–194 CrossRef CAS.
- S. K. Jana, H. Takahashi, M. Nakamura, M. Kaneko, R. Nishida, H. Shimizu, T. Kugita and S. Namba, Appl. Catal., A, 2003, 245, 33–41 CrossRef CAS.
- A. Corma, M. S. Grand, A. V. Gonzalez and A. V. Orichilles, J. Catal., 1996, 159, 375–382 CrossRef CAS.
- Z. A. Alothman, Materials, 2012, 5, 2874–2902 CrossRef CAS.
- R. Huirache-Acuña, R. Nava, C. L. Peza-Ledesma, J. Lara-Romero, G. Alonso-Núñez, B. Pawelec and E. M. Rivera-Muñoz, Materials, 2013, 6, 4139–4167 CrossRef.
- N. Linares, A. E. Sepúlveda, M. C. Pacheco, J. R. Berenguer, E. Lalinde, C. Nájera and J. Garcia-Martinez, New J. Chem., 2011, 35, 225–234 RSC.
- C. Lawrence, W. Thielemans and R. Mokaya, J. Mater. Chem., 2010, 20, 320–325 RSC.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS.
- (a) F. Schüth, Chem. Mater., 2001, 13, 3184–3195 CrossRef; (b) F. Schüth and W. Schmidt, Adv. Mater., 2002, 14, 629–638 CrossRef; (c) F. Schüth, Angew. Chem., Int. Ed., 2003, 42, 3604–3622 CrossRef.
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS.
- Y. Han and D. Zhang, Curr. Opin. Chem. Eng., 2012, 1, 129–137 CrossRef CAS.
- A. S. Poyraz, W. A. Hines, C.-H. Kuo, N. Li, D. M. Perry and S. L. Suib, J. Appl. Phys., 2014, 115, 114309 CrossRef.
- L. U. Okonye, K. Jalama, A. Hosaka, C. Watanabe and R. Meijboom, Environ. Prog. Sustainable Energy, 2019, 28(4), 13079, DOI:10.1002/ep.13079.
- J. P. Thielemann, F. Girgsdies, R. Schlogl and C. Hess, Nanotechnol, 2011, 2, 110–118 CAS.
- L. A. Cano, A. A. Garcia Blanco, G. Lener, S. G. Marchetti and K. Sapag, Catal. Today, 2017, 282, 204–213 CrossRef CAS.
- C. F. Toncón-Leal, S. Amaya-Roncancio, A. A. García Blanco, M. S. Moreno and K. Sapag, Top. Catal., 2019, 62, 1086–1095 CrossRef.
- J. S. Albuquerque, F. O. Costa and B. V. S. Barbosa, Catal. Lett., 2019, 149, 831–839 CrossRef CAS.
- (a) V. Vosoughi, S. Badoga, A. K. Dalai and N. Abatzoglou, Fuel Process. Technol., 2017, 162, 55–65 CrossRef CAS; (b) V. Vosoughi, A. K. Dalai, N. Abatzoglou and Y. Hu, Appl. Catal., A, 2017, 547, 155–163 CrossRef CAS.
- G. Liu, Q. Chen, E. Oyunkhand, S. Ding, N. Yamane, G. Yang, Y. Yoneyama and N. Tsubaki, Carbon, 2018, 130, 304–314 CrossRef CAS.
- R. Sadek, K. A. Chalupka, P. Mierczynski, J. Rynkowski, Y. Millot, L. Valentin, S. Casale and S. Dzwigaj, Catal. Today, 2019, 354, 109–122 CrossRef.
- L. Wei, Y. Zhao, Y. Zhang, C. Liu, J. Hong, Q. Xiao, H. Xiong and J. Li, ChemCatChem, 2017, 9(20), 3895–3903 CrossRef CAS.
- W. Li, Y. He, H. Li, D. Shen and R. Yang, Catal. Commun., 2017, 98, 98–101 CrossRef CAS.
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787–793 CrossRef CAS.
- A. Rai, M. G. Sibi, S. A. Farooqui, M. Anand, A. Bhaumik and A. K. Sinha, ACS Sustainable Chem. Eng., 2017, 5, 7576–7586 CrossRef CAS.
- X. He and D. Antonelli, Angew. Chem., Int. Ed., 2002, 41, 214–229 CrossRef CAS.
- A. S. Poyraz, C.-H. Kuo, S. Biswas, C. K. King’ondu and S. L. Suib, Nat. Commun., 2013, 4, 2952, DOI:10.1038/ncomms3952.
- Q. Huo, D. I. Margolese and G. D. Stucky, Chem. Mater., 1996, 8, 1147–1160 CrossRef CAS.
- S. L. Burkett, S. D. Sim and S. Mann, J. Chem. Soc., Chem. Commun., 1996, 1367–1368 RSC.
- C. E. Fowler, B. Lebeau and S. Mann, J. Chem. Soc., Chem. Commun., 1998, 1825–1826 RSC.
- M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285–3295 CrossRef CAS.
- L. Mercier and T. J. Pinnavaia, Chem. Mater., 2000, 12, 188–196 CrossRef CAS.
- G. J. d. A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef.
- G. J. d. A. A. Soler-Illia, E. L. Crepaldi, D. Grosso and C. Sanchez, Curr. Opin. Colloid Interface Sci., 2003, 8, 109–126 CrossRef CAS.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- B. Viswanathan and B. Jacob, Catal. Rev. - Sci. Eng., 2005, 47, 1–82 CrossRef CAS.
- D. T. On, D. Desplantier-Giscard, C. Danumah and S. Kaliaguine, Appl. Catal., A, 2001, 222, 299–357 CrossRef.
- A. Stein, Adv. Mater., 2003, 15, 763–775 CrossRef CAS.
- D. Liu, L. Zhang and P. Lv, Microporous Mesoporous Mater., 2020, 297, 110056 CrossRef CAS.
- L. L. Chen, Z. Y. Zhang, N. Qi, B. Zhou, S. P. Deng, Z. Q. Chen, Y. C. Wu and X. F. Tang, Microporous Mesoporous Mater., 2020, 296, 109969, DOI:10.1016/j.micromeso.2019.109969.
- T. Sun, N. Shan, L. Xu, J. Wang, J. Chen, A. A. Zakhidov and R. H. Baughman, Chem. Mater., 2018, 30, 1617–1624 CrossRef CAS.
- J. He, L. Zhang, L. Jin and B. Liu, Front. Chem., 2018, 7, 22, DOI:10.3389/fchem.2019.00022.
- J. Wang, Q. Ma, Y. Wang, Z. Li, Z. Li and Q. Yuan, Chem. Soc. Rev., 2018, 47, 8766–8803 RSC.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1(6), 16023, DOI:10.1038/natrevmats.2016.23.
- J. Wei, Z. Sun, W. Luo, Y. Li, A. A. Elzatahry, A. M. Al-Enizi, Y. Deng and D. Zhao, J. Am. Chem. Soc., 2017, 139, 1706–1713 CrossRef CAS.
- M. Gómez, J. Pizarro, X. Castillo, A. Ghisolfi, C. Díaz, M. de Lourdes Chávez and D. Cazorla-Amorós, Microporous Mesoporous Mater., 2020, 299, 110085 CrossRef.
- T. Zhao, A. Elzatahry, X. Li and D. Zhao, Nat. Rev. Mater., 2019, 4, 775–791 CrossRef CAS.
- M. Zheng, H. Tang, L. Li, Q. Hu, L. Zhang, H. Xue and H. Pang, Adv. Sci., 2018, 5, 1700592, DOI:10.1002/advs.201700592.
- E. Doustkhah, J. Lin, S. Rostamnia, C. Len, R. Luque, X. Luo, Y. Bando, K. C.-W. Wu, J. Kim, Y. Yamauchi and Y. Ide, Chem.–Eur. J., 2018, 24, 1–23 CrossRef.
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 CrossRef.
- E. Prouzet and T. J. Pinnavaia, Angew. Chem., Int. Ed. Engl., 1997, 36, 516–518 CrossRef CAS.
- C. Chen, H. Li and M. E. Davis, Microporous Mater., 1993, 2, 17–26 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CrossRef CAS.
- D. Khushalani, A. Kuperman and G. A. Ozin, Adv. Mater., 1995, 7, 842–846 CrossRef CAS.
- T. Yasmin and K. Müller, J. Chromatogr. A, 2011, 1218, 6464–6475 CrossRef CAS.
- K. Kosuge and P. S. Singh, Chem. Mater., 2001, 13, 2476–2482 CrossRef CAS.
- Y. Ma, L. Qi, J. Ma, Y. Wu, O. Liu and H. M. Cheng, Colloids Surf., A, 2003, 229, 1–8 CrossRef CAS.
- A. Davidson, Curr. Opin. Colloid Interface Sci., 2002, 7, 92–106 CrossRef CAS.
- M. Kruk, M. Jaroniec, C. H. Ko and R. Ryoo, Chem. Mater., 2000, 12, 1961–1968 CrossRef CAS.
- M. Mesa, L. Sierra, B. Lopez, A. Ramirez and J. L. Guth, Solid State Sci., 2003, 5, 1303–1308 CrossRef CAS.
- B. M. Mogudi, P. Ncube and R. Meijboom, Appl. Catal., B, 2016, 198, 74–82 CrossRef CAS.
- G. Haddad, B. Chen and J. G. Goodwin, J. Catal., 1996, 161, 274–281 CrossRef CAS.
- F. Rohr, O. Lindvag, A. Holmen and E. Blekkan, Catal. Today, 2000, 58, 247–254 CrossRef CAS.
- M. Rothaemel, K. F. Hanssen, E. A. Blekkan, D. Schanke and A. Holmen, Catal. Today, 1997, 38, 79–84 CrossRef CAS.
- R. Ravishankar, M. M. Li and A. Borgna, Catal. Today, 2005, 106, 149–153 CrossRef CAS.
- Y. Ohtsuka, T. Arai, S. Takasaki and N. Tsubouchi, Energy Fuels, 2003, 17, 804–809 CrossRef CAS.
- A. Yakubov, M. G. Kutty, S. P. Lee, M. S. Shaharun, S. B. A. Hamid and V. Piven, Adv. Mater. Res., 2012, 364, 70–75 Search PubMed.
- J. J. Rodrigues, L. A. Lima, R. Bechara, S. W. Lima, M. G. F. Rodrigues and F. A. N. Fernandes, Braz. J. Pet. Gas, 2011, 5, 149–157 Search PubMed.
- J. J. Rodrigues, F. A. N. Fernandes and M. G. F. Rodrigues, Appl. Catal., A, 2013, 468, 32–37 CrossRef CAS.
- J. Li and Q. Cai, Catal. Commun., 2008, 9, 2003–2006 CrossRef.
- L. A. Cano, M. V. Cagnoli, N. A. Fellenz, J. F. Bengoa, N. G. Gallegos, A. M. Alvarez and S. G. Marchetti, Appl. Catal., A, 2010, 379, 105–110 CrossRef CAS.
- L. A. Cano, M. V. Cagnoli, J. F. Bengoa, A. M. Alvarez and S. G. Marchetti, J. Catal., 2011, 278, 310–320 CrossRef CAS.
- M. Oschatz, W. S. Lamme, J. Xie, A. L. Dugulan and K. P. de Jong, chemcatchem, 2016, 8, 2846–2852 CrossRef CAS.
- A. Martínez, C. López, F. Márquez and I. Díaz, J. Catal., 2003, 220, 486–499 CrossRef.
- A. Y. Khodakov, Catal. Today, 2009, 144, 251–257 CrossRef CAS.
- O. González, H. Pérez, P. Navarro, L. C. Almeida, J. G. Pacheco and M. Montes, Catal. Today, 2009, 148, 140–147 CrossRef.
- G. Prieto, A. Martinez, R. Murciano and M. A. Arribas, Appl. Catal., A, 2009, 367, 146–156 CrossRef CAS.
- M. Bartolini, J. Molina, J. Alvarez, M. Goldwasser, P. P. Almao and M. J. P. Zurita, J. Power Sources, 2015, 285, 1–11 CrossRef CAS.
- H. Xiong, Y. Zhang, K. Liew and J. Li, J. Mol. Catal. A: Chem., 2008, 295, 68–76 CrossRef CAS.
- Y. Wang, M. Noguchi, Y. Takahashi and Y. Ohtsuka, Catal. Today, 2001, 68, 3–9 CrossRef CAS.
- H. Li, J. Li, H. Ni and D. Song, Catal. Lett., 2006, 110, 71–76 CrossRef CAS.
- N. Osakoo, R. Henkel, S. Loiha, F. Roessner and J. Wittayakun, Catal. Commun., 2014, 56, 168–173 CrossRef CAS.
- Y. Lu, P. Zhou, J. Han and F. Yu, RSC Adv., 2015, 5, 59792–59803 RSC.
- C. Tao, J. Li, Y. Zhang and K. Y. Liew, J. Mol. Catal. A: Chem., 2010, 331, 50–57 CrossRef CAS.
- C. Tao, J. Li and K. Y. Liew, Sci. China: Chem., 2010, 53, 2551–2559 CrossRef CAS.
- S. Mu, D. Li, B. Hou, L. Jia, J. Chen and Y. Sun, Energy Fuels, 2010, 24, 3715–3718 CrossRef CAS.
- Y. Liu, K. Murata and K. Okabe, Chem. Lett., 2008, 37, 984–985 CrossRef CAS.
- J. Hong, W. Chu, P. A. Chernavskii and A. Y. Khodakov, Appl. Catal., A, 2010, 382, 28–35 CrossRef CAS.
- Z. Li, J. Wu, J. Yu, D. Han, L. Wu and J. Li, J. Mol. Catal. A: Chem., 2016, 424, 384–392 CrossRef CAS.
- Y. Jiang, T. Fu, J. Lu and Z. Li, Energy Chem., 2013, 22, 506–511 CrossRef CAS.
- L. C. Loc, L. K. Trieu, H. S. Thoang, H. T. M. Trang, N. M. Huan and B. T. Huong, Int. J. Nanotechnol., 2013, 10, 313–333 CrossRef CAS.
- D. J. Kim, B. C. Dunn, F. Huggins, G. P. Huffman, M. Kang, J. E. Yie and E. M. Eyring, Energy Fuels, 2006, 20, 2608–2611 CrossRef CAS.
- T. I. Sibianu, D. Berger, C. Matei and I. Calinescu, Sci. Bull. - Univ. "Politeh." Bucharest, Ser. B, 2016, 78, 39–46 CAS.
- M. Lualdi, G. Di Carlo, S. Lögdberg, S. Järås, M. Boutonnet, V. La Parola, L. F. Liotta, G. M. Ingo and A. M. Venezia, Appl. Catal., A, 2012, 443–444, 76–86 CrossRef CAS.
- N. Y. Kim, J.-S. Jung, J. S. Lee, E. H. Yang, G. H. Hong, S. S. Lim, Y. S. Noh, J. L. Hodala, K. Y. Lee and D. J. Moon, Res. Chem. Intermed., 2016, 42, 319–334 CrossRef CAS.
- D. J. Kim, B. C. Dunn, P. Cole, G. Turpin, R. D. Ernst, R. J. Pugmire, M. Kang, J. M. Kim and E. M. Eyring, Chem. Commun., 2005, 20, 1462–1464 RSC.
- S. Yuanyuan, L. Kongyong and L. Jinlin, Chin. J. Catal., 2009, 30, 1091–1095 CrossRef.
- L. Jia, L. Jia, D. Li, B. Hou, J. Wanga and Y. Sun, J. Solid State Chem., 2011, 184, 488–493 CrossRef CAS.
- J. Hong, P. A. Chernavskii, A. Y. Khodakov and W. Chu, Catal. Today, 2009, 140, 135–141 CrossRef CAS.
- J. J. Rodrigues, G. Pecchi, F. A. N. Fernandes and M. G. F. Rodrigues, J. Nat. Gas Chem., 2012, 21, 722–728 CrossRef CAS.
- J. J. Rodrigues, L. A. Lima, G. M. De Paula and M. G. F. Rodrigues, Mater. Sci. Forum, 2014, 798–799, 100–105 Search PubMed.
- Y. Qiu, S. Chen, C. Wang, R. Lyu, D. Zhang, C. Liu, Y. Zhang and J. Li, New J. Chem., 2017, 1–3 Search PubMed.
- F. Pardo-Tarifa, V. Montes, M. Claure, S. Cabrera, H. Kusar, A. Marinas and M. Boutonnet, Synth. Catal.: Open Access, 2017, 2, 11 Search PubMed.
- Q. Cheng, Y. Tian, S. Lyu, N. Zhao, K. Ma, T. Ding, Z. Jiang, L. Wang, J. Zhang, L. Zheng, F. Gao, L. Dong, N. Tsubaki and X. Li, Nat. Commun., 2018, 9, 3250 CrossRef.
- H. Li, B. Hou, J. Wang, X. Huang, C. Chen, Z. Ma, J. Cui, L. Ji, D. Sun and D. Li, Catal. Sci. Technol., 2017, 7, 3812–3822 RSC.
- H. Xiong, Y. Zhang, S. Wang, K. Liew and J. Li, Fuel Process. Technol., 2009, 90, 237–246 CrossRef CAS.
- B. Sun, M. Qiao, K. Fan, J. Ulrich and F. F. Tao, ChemCatChem, 2011, 3, 542–550 CrossRef CAS.
- H. Li, S. Wang, F. Ling and J. Li, J. Mol. Catal. A: Chem., 2006, 244, 33–40 CrossRef CAS.
- A. Y. Khodakov, R. Bechara and A. Griboval-Constant, Appl. Catal., A, 2003, 254, 273–288 CrossRef CAS.
- D. J. Kim, B. C. Dunn, M. Kang, J. E. Yie, S.-H. Kim, J. Gasser, E. Fillerup, L. Hope-Weeks and E. M. Eyring, Stud. Surf. Sci. Catal., 2007, 685–688 CrossRef.
- A. Martínez, C. López, F. Márquez and I. Díaz, J. Catal., 2003, 220, 486–499 CrossRef.
- J. W. Bae, S. M. Kim, S. H. Kang, K. V. R. Chary, Y. J. Lee, H. J. Kim and K. W. Jun, J. Mol. Catal. A: Chem., 2009, 311, 7–16 CrossRef CAS.
- N. N. Madikizela-Mnqanqeni and N. J. Coville, J. Mol. Catal. A: Chem., 2005, 225, 137–142 CrossRef CAS.
- J. Panpranot, S. Kaewkun, P. Praserthdam and J. G. Goodwin, Catal. Lett., 2003, 91, 95–102 CrossRef CAS.
- L. Wu, Z. Li, D. Han, J. Wu and D. Zhang, Fuel Process. Technol., 2015, 134, 449–455 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |