
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enolate SNAr of unactivated arenes via [(η6-arene)RuCp]+ intermediates†
Luke J.
Williams
a,
Yunas
Bhonoah
b and
James W.
Walton
 *a
*a
aDepartment of Chemistry, University of Durham, Lower Mountjoy, Durham, DH1 3LE, UK. E-mail: james.walton@durham.ac.uk
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berks RG42 6EY, UK
First published on 21st June 2022
Abstract
A series of complexes of the general formula [(η6-arene)RuCp][PF6] (where arene = aryl halides or nitroarenes) were synthesised and the arene ring was found to be reactive towards an intermolecular nucleophilic aromatic substitution (SNAr) reaction with a series of cyclic 1,3-diones. Competition experiments indicated that leaving group ability of the aryl halides and nitroarenes went in the order of F ≫ NO2 > Cl > Br. Following SNAr, the arene rings were liberated quantitatively via a rapid photolysis reaction (<15 min).
The π-coordination of an arene to a metal fragment has significant activating effects on the ring, due to the strongly electron-withdrawing nature of the metal fragment. Such reactivity enhancements include increased electrophilicity of the ring and reduced pKa of both the aromatic and benzylic protons (Fig. 1).1 As a result, many different metal-mediated aromatic transformations have been developed,2 including arene C–H-activation3–5 and nucleophilic aromatic substitution (SNAr).6–8 Several metal–ligand frameworks can be used to activate arene rings in this way, although Cr(CO)3 is most commonly used.1,9,10 One advantage of the Cr(CO)3 system is its synthesis under mild conditions from inexpensive starting materials. However, a limitation of such systems is that removal of the aromatic ring from Cr following its transformation is difficult and usually requires oxidation, which results in the metal fragment being destroyed.10 As a result, Cr-based systems are limited to stoichiometric arene transformations, and are therefore inefficient, with toxic Cr waste being formed. Instead, it is desirable for aromatic transformations to be catalytic in the activating metal, though this process is inherently difficult to achieve since a stronger metal-arene π interaction increases the reactivity of the ring but also reduces the ability of the system to undergo arene exchange, which is a key step of the catalytic process. One metal capable of achieving this balance is ruthenium, which has previously been used to catalyse a variety of different reactions via transient η6 complexes, including SNAr fluorination,11 SNAr amination,12–15 hydroaminations16,17 and benzylic functionalisation.18 We have shown π-arene complexes of Ru can be reactive towards nucleophilic trifluoromethylation7 and C–H activation.5 In this study, we report a novel SNAr reaction of cyclic enolates towards [(η6-arene)RuCp]+ complexes, resulting in formation of new aromatic C–C bonds. Moreover, we show liberation of the free arene and recovery of the activating complex [(NCMe)3RuCp]+via a mild photolysis reaction. Overall, we show SNAr of unactivated aryl halides and nitro aryls with a broad range of enolate nucleophiles, with effective recovery of the activating Ru complex. The reaction products, 2-aryl-1,3-diones, have emerged as a promising new class of herbicidal acetyl-CoA carboxylase (ACCase) inhibitors,19 making efficient synthesis of such compounds highly sought after by agrochemical companies.
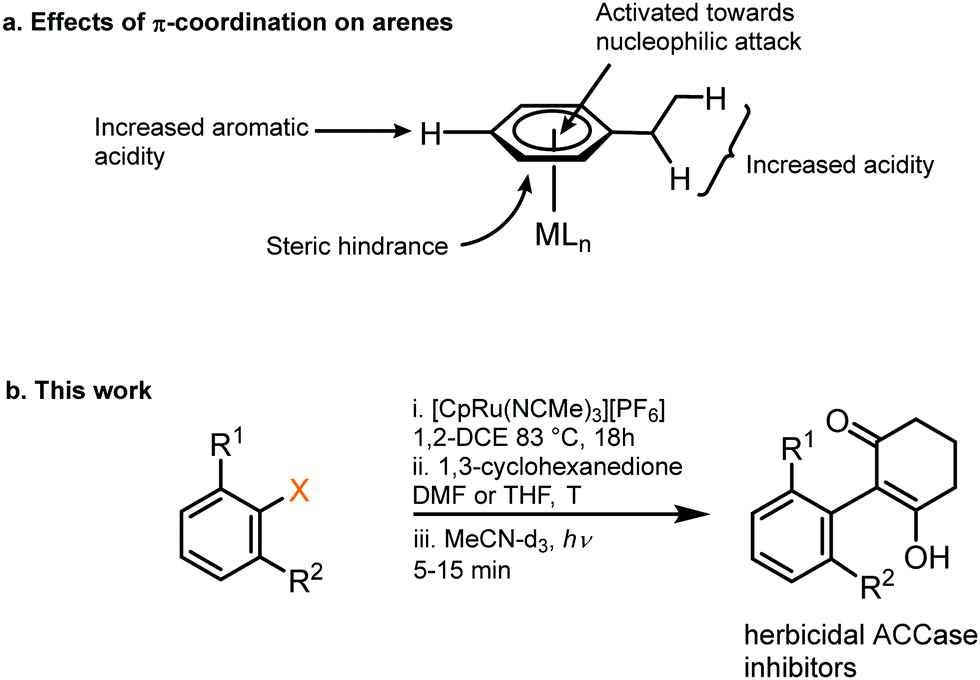 | ||
| Fig. 1 (a) Activating effects on an aromatic ring from π-coordination to a metal centre. (b) An intermolecular SNAr reaction facilitated by temporary coordination to Ru, outlined in this work. | ||
1. Synthesis of Ru sandwich complexes: to investigate the feasibility of SNAr with enolates, a series of Ru sandwich complexes 1a–3c were synthesised by heating the precursor complex, [(NCMe3)RuCp]+, in dichloroethane in the presence of the appropriate arene. The series of complexes include variation in the aryl leaving group (F, Cl, NO2) and number of adjacent methyl groups (zero, one or two) to explore the influence of sterics on reactivity. All complexes were formed in high yield (80–95%, Scheme 1) and formation and purity of each was confirmed by heteronuclear NMR spectroscopy, mass spectrometry and elemental microanalysis. In addition, single crystals of complexes 1a, 1b, 2b, 3b and 3c were grown via the vapour diffusion of diethyl ether into a concentrated acetone solution of metal complex. X-Ray diffraction analysis shows all complexes having the expected structure, with pseudo-linear geometry about the Ru centre (ESI†).
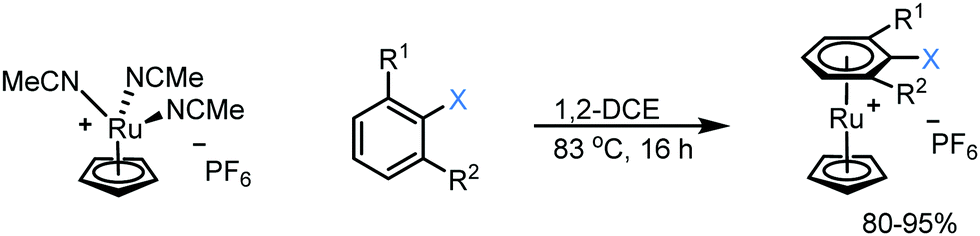 | ||
| Scheme 1 Synthesis of complexes 1–3 [Complex 1: R1, R2 = H, X = F (1a), Cl (1b), NO2 (1c); Complex 2: R1 = Me, R2 = H, X = F (2a), Cl (2b), NO2 (2c); Complex 3: R1, R2 = Me, X = F (3a), Cl (3b), NO2 (3c)]. | ||
2. Enolate-based S N Ar: initial conditions and optimisation: initially, to achieve the desired SNAr reaction, complex 1a was reacted in the presence of 1,3-cyclohexanedione and potassium carbonate in DMF at 40 °C (Table 1, entry 1). Following purification, analysis by multinuclear NMR spectroscopy confirmed an arene transformation, as evidenced by shifts in the aromatic signals and appearance of new signals corresponding to the reacted dione nucleophile, as well as a disappearance of the aromatic fluorine signal in the 19F NMR spectrum. The mass spectrum also showed a molecular ion with the predicted mass of the product complex, confirming substitution had taken place. Further analysis of the 1H NMR spectrum revealed that the nucleophile reacts exclusively at the carbon atom of the enolate and the reaction product exists in the enol form (as shown in Table 1 figure), as indicated by the absence of the proton in the 2-position of the dione. Having confirmed reaction of aryl fluorides can proceed, the leaving group was changed to Cl (1b) and NO2 (1c), and each complex showed comparable reactivity toward the SNAr reaction as 1a (entries 2–4). Solvent optimisation for the SNAr was performed on reactions with complex 1c (entries 4–11). Several solvents led to quantitative conversion, with THF emerging as the most convenient, due to its low boiling point compared with DMF. Following these studies, the effect of adding one (complexes 2a–c) and two (complexes 3a–c) methyl groups ortho to the leaving group was investigated (entries 12–26). Addition of one methyl group ortho to the substitution site appears to slightly hinder the reaction, and the reaction required elevated temperature of 70 °C to go to completion (entry 13). Under these conditions, complex 2a, was observed to undergo a hydrolysis to the aryl alcohol side product, presumably due to adventitious water present in the reaction mixture.
|
|
||||
|---|---|---|---|---|
| Entry | Starting complex | Solvent | Temperature (°C) | Conversion (%) |
| 1 | 1a | DMF | 40 | >99 |
| 2 | 1b | DMF | 40 | >99 |
| 3 | 1b | CH2Cl2 | RT | >99 |
| 4 | 1c | DMF | 40 | >99 |
| 5 | 1c | DMF | 0 | >99 |
| 6 | 1c | CH2Cl2 | RT | >99 |
| 7 | 1c | THF | RT | >99 |
| 8 | 1c | 1,4-Dioxane | RT | 25 |
| 9 | 1c | EtOH | RT | 0 |
| 10 | 1c | MeCN | RT | >99 |
| 11 | 1c | 2-Methyl THF | RT | 50 |
| 12 | 2a | DMF | 60 | 83 |
| 13 | 2b | DMF | 70 | >99 |
| 14 | 2b | THF | 65 | 76 |
| 15 | 2c | DMF | 70 | >99 |
| 16 | 3a | DMF | 80 | 60 |
| 17 | 3b | DMF | 80 | 76 |
| 18 | 3b | DMF | 90 | 87 |
| 19 | 3b | NMP | 90 | 85 |
| 20 | 3b | DMF | 120 | 67 |
| 21 | 3b | 1,2-DCE | 83 | 0 |
| 22 | 3b | 1,4-Dioxane | 90 | 0 |
| 23 | 3b | EtOH | 78 | 0 |
| 24 | 3b | MeCN | 82 | 17 |
| 25 | 3b | 2-Methyl THF | 80 | Trace |
| 26 | 3c | DMF | 90 | 60 |

As expected, adding a second ortho-methyl group meant the temperature had to be further raised to 90 °C (entry 18) where the reaction went to around 90% conversion, with complex 3b performing slightly better than 3c and complex 3a, again, undergoing a hydrolysis side reaction. A solvent screen with complex 3b indicated that DMF was the optimal solvent for more sterically hindered arenes (entries 19–26). A variety of different bases were also screened (see ESI†), including inorganic salts and organic amines, although all were outperformed by K2CO3.
3. Leaving group competition experiments: a series of competition experiments were performed to establish which leaving group is favoured in complexes with more than one potential leaving group available (Fig. 2). It was found that fluoride proved a better leaving group than both chloride and nitro (Fig. 2A and B). Substitution of a nitro group was favoured over chloride (Fig. 2C) and chloride leaving group was favoured over bromide (Fig. 2D). These results are in line with the relative polarity of the C–X bond and follow the classic order of reactivity for SNAr reactions. Next, the arene 2,5-dichlorotoluene was tested, and the reaction showed a preference for the less hindered chloride (Fig. 2E). The final competition experiment involved an arene with the best performing leaving group, fluoride, hindered by two ortho-methyl groups, and the worst performing leaving group bromide, which was unhindered (Fig. 2F). After exposure to SNAr conditions at 90 °C, analysis of NMR spectra and mass spectrometry indicated that substitution had exclusively taken place at the bromide position, and the fluorine remained on the ring. These data imply that steric hindrance has a more significant effect on the SNAr reaction than leaving group ability, and that substitution will show preference for a less hindered substitution site.
 | ||
| Fig. 2 Leaving group competition experiments. Conditions: (i) [CpRu(NCMe)3][PF6], 1,2-DCE, 83 °C, 18 h; and (ii). Cyclohexanedione (1 eq.), K2CO3, DMF, RT (A–D and F) or 70 °C (E) or 90 °C (F). | ||
4. Dione scope: finally, to determine the versatility of the SNAr reaction with respect to the nucleophile, a selection of diones were investigated under the optimised reaction conditions for complex 2b (Table 2). Generally, the reaction conditions were tolerant of many functional groups tested here, including ester/amide groups, cyclic ethers/amines and spiro-bicyclic frameworks. Of the poorer performing diones, cyclopentanedione (entry 9) went to 89% conversion and 2,2′,6,6′-tetramethyl tetrahydropyran-3,5-dione (entry 8) went to 65%, likely due to the four methyl groups close to the deprotonation site, meaning the SNAr reaction may have been hindered by sterics. The sole aliphatic dione tested was acetylacetone (entry 10), which reached a conversion of around 72%. Interestingly, the 1H NMR spectrum of this reaction indicated the presence of an uncoordinated aromatic impurity, implying that the acetylacetone may have displaced some of the bound arene during the reaction and coordinated to the Ru centre as a bidentate ligand.
|
|
|||
|---|---|---|---|
| Entry | Dione | Conversion (yield)/% | Photolysis time/mind |
| a No isolated yield, due to impurities. b Free arene observed. c Photolysis incomplete. d All photolysis reactions quantitative, unless otherwise stated. | |||
| 1 |

|
>99 (69) | 10 |
| 2 |
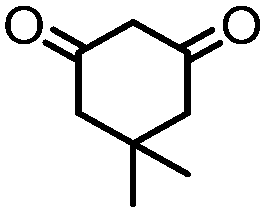
|
>99 (61) | 10 |
| 3 |
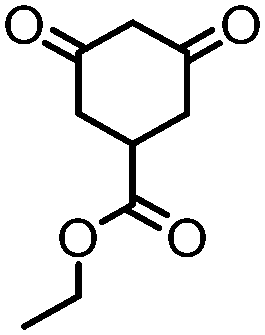
|
>99 (63) | 5 |
| 4 |

|
>99 (68) | 15 |
| 5 |

|
>99 (70) | 5 |
| 6 |
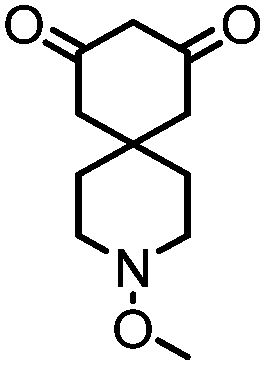
|
>99 (63) | 5 |
| 7 |
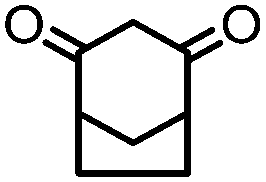
|
>99 | 5 |
| 8 |
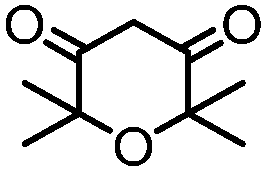
|
65a | 15 |
| 9 |
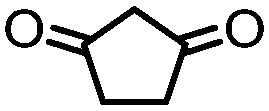
|
89a | 15 |
| 10 |

|
72b | >120c |
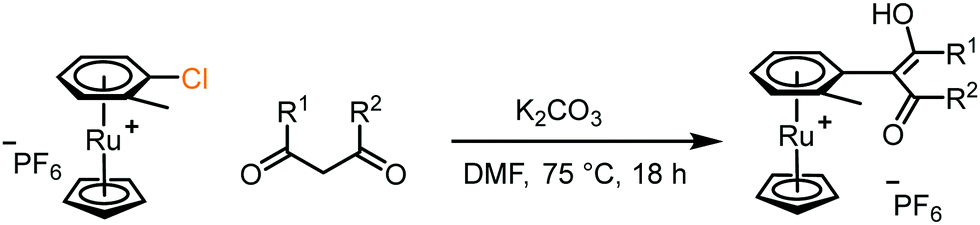
5. Photolytic liberation of free arenes: following SNAr, liberation of the functionalised arene from the RuCp+ centre was achieved by dissolving the coordinated product in deuterated acetonitrile and subjecting it to ultraviolet light irradiation (Fig. 3 and Table 2). Initial experiments were carried out using a commercial nail salon lamp (365 nm, 298 K, 36 W, 5 cm from bulb); the reaction was monitored by 1H NMR spectroscopy which showed quantitative formation of free arene after 24 hours. Irradiation by 254 nm light from the same light source produced the free arene in 48 hours. Using an alternative light source, a Penn photoreactor M2 (365 nm), the photolysis reaction was repeated and reached completion in a much shorter time of 10 minutes. Under these optimised conditions, each reaction product from the dione scope was subjected to the arene photolysis reaction to liberate the free arene, typically giving quantitative liberation in less than 15 min. Importantly, the Ru species formed after photolysis is the MeCN-d3 analogue of the initial complex used to synthesise sandwich compounds 1–3, highlighting the possibility to recycle the activating Ru complex for further use.
 | ||
| Fig. 3 Photolysis (365 nm, d3-MeCN, 298 K) of the SNAr product of complex 2b, showing complete conversion within 10 minutes. | ||
6. One-pot reaction and outlook: in a further investigation, a one-pot reaction was attempted, where sandwich complex synthesis, SNAr and subsequent photolysis to regenerate the initial Ru complex all take place in a single reaction process, without isolation or purification of intermediates (Scheme 2). After reaction in deuterated acetonitrile, 1H NMR spectroscopy was used to verify formation of the product at each stage, and a final conversion over three steps of around 23% was calculated. This one-pot process, which regenerates the activating Ru complex, shows the potential for future studies into a process fully catalytic in the Ru fragment. The low yield is attributed to poor SNAr conversion in MeCN (e.g., Table 1 entry 24) and our ongoing work looks to optimise this process.
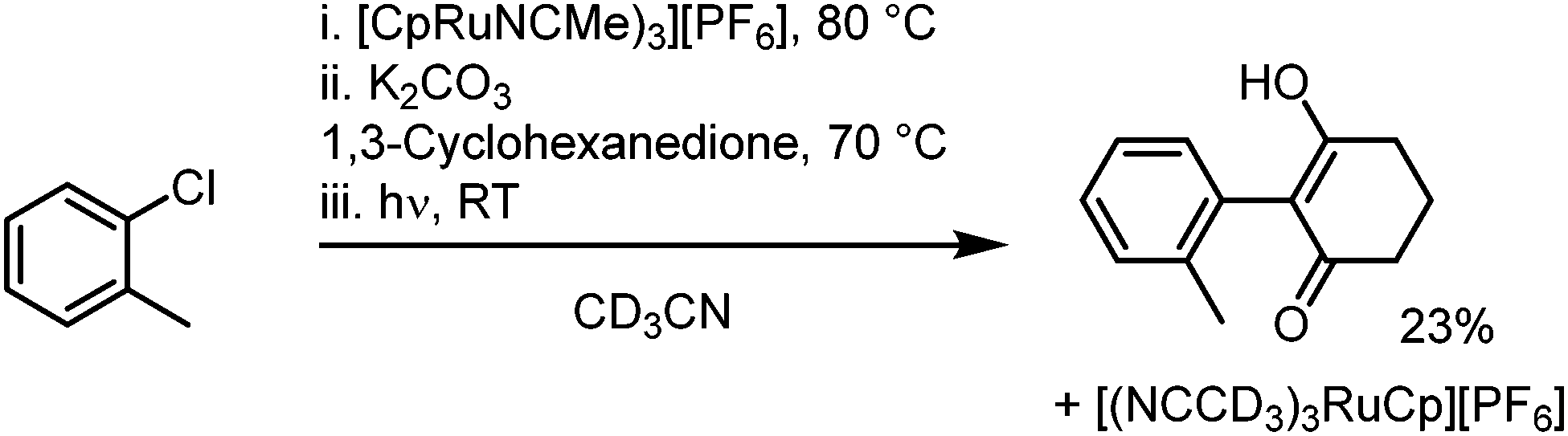 | ||
| Scheme 2 One-pot SNAr of 2-chlorotoluene. | ||
To summarise, a new intermolecular C–C bond forming SNAr reaction between cyclohexanedione-derived enolates and unactivated aryl halides and nitroarenes has been established. The reaction proceeds via formation of the [(η6-arene)RuCp]+ intermediate complexes and has been optimised for systems with zero, one or two methyl groups hindering the leaving group. Although leaving group competition experiments revealed fluoride to be the best leaving group, the chloro analogue of the complex was used for most experiments, due to its better resistance to hydrolysis side reactions. A wide variety of cyclic diones were successfully tested for their reactivity as nucleophiles, whereas one aliphatic dione appeared to undergo a side reaction in which it coordinated to the Ru centre, displacing the arene. Finally, a procedure for the rapid liberation and generation of the MeCN-d3 analogue of the activating Ru complex was optimised, indicating the potential for the SNAr to be made catalytic in Ru. Overall, we have shown an efficient method to produce important herbicidal acetyl-CoA carboxylase inhibitor components, starting from unactivated arenes, via a novel method that could readily be expanded to many other industrially-relevant syntheses.
We thank Dimitry S. Yufit for help with X-ray crystallography structures.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. W. Walton and L. A. Wilkinson, Organomet. Chem., 2019, 42, 125–171 CAS.
- L. J. Williams, Y. Bhonoah, L. A. Wilkinson and J. W. Walton, Chem. – Eur. J., 2021, 27, 3650–3660 CrossRef CAS PubMed.
- P. Ricci, K. Krämer, X. C. Cambeiro and I. Larrosa, J. Am. Chem. Soc., 2013, 135, 13258–13261 CrossRef CAS PubMed.
- M. Batuecas, J. Luo, I. Gergelitsová, K. Krämer, D. Whitaker, I. J. Vitorica-Yrezabal and I. Larrosa, ACS Catal., 2019, 5268–5278 CrossRef CAS PubMed.
- L. A. Wilkinson, J. A. Pike and J. W. Walton, Organometallics, 2017, 36, 4376–4381 CrossRef CAS.
- F. Christopher Pigge, J. J. Coniglio and S. Fang, Organometallics, 2002, 21, 4505–4512 CrossRef.
- J. A. Pike and J. W. Walton, Chem. Commun., 2017, 53, 9858–9861 RSC.
- R. C. Cambie, S. J. Janssen, P. S. Rutledge and P. D. Woodgate, J. Organomet. Chem., 1991, 420, 387–418 CrossRef CAS.
- S. Takemoto and H. Matsuzaka, Tetrahedron Lett., 2018, 59, 697–703 CrossRef CAS.
- P. E. Kündig, ed., Transition Metal Arene Pi-Complexes in Organic Synthesis and Catalysis, Springer, 1st edn, 2004 Search PubMed.
- A. I. Konovalov, E. O. Gorbacheva, F. M. Miloserdov and V. V. Grushin, Chem. Commun., 2015, 51, 13527–13530 RSC.
- J. W. Walton and J. M. J. Williams, Chem. Commun., 2015, 51, 2786–2789 RSC.
- M. Otsuka, K. Endo and T. Shibata, Chem. Commun., 2010, 46, 336–338 RSC.
- Q. K. Kang, Y. Lin, Y. Li and H. Shi, J. Am. Chem. Soc., 2020, 142, 3706–3711 CrossRef CAS PubMed.
- B. Mueller and N. Schley, Dalton Trans., 2020, 49, 10114–10119 RSC.
- M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702–2703 CrossRef CAS PubMed.
- M. Otsuka, H. Yokoyama, K. Endo and T. Shibata, Org. Biomol. Chem., 2012, 10, 3815–3818 RSC.
- S. Takemoto, E. Shibata, M. Nakajima, Y. Yumoto, M. Shimamoto and H. Matsuzaka, J. Am. Chem. Soc., 2016, 138, 14836–14839 CrossRef CAS PubMed.
- T. Smejkal, V. Gopalsamuthiram, S. K. Ghorai, A. M. Jawalekar, D. Pagar, K. Sawant, S. Subramanian, J. Dallimore, N. Willetts, J. N. Scutt, L. Whalley, M. Hotson, A. M. Hogan and G. Hodges, Org. Process Res. Dev., 2017, 21, 1625–1632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2170169–2170175. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc02508f |
| This journal is © The Royal Society of Chemistry 2022 |