
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrated eutectic electrolytes for high-performance Mg-ion batteries†
Yunpei
Zhu
a,
Xianrong
Guo
b,
Yongjiu
Lei
a,
Wenxi
Wang
a,
Abdul-Hamid
Emwas
b,
Youyou
Yuan
b,
Yao
He
b and
Husam N.
Alshareef
 *a
*a
aMaterials Science and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23900-6955, Saudi Arabia. E-mail: husam.alshareef@kaust.edu.sa
bCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
First published on 26th January 2022
Abstract
Aqueous Mg-ion batteries are a promising electrochemical energy storage technology. However, Mg2+ ions interact strongly with electrolyte molecules and electrode materials, resulting in insufficient ionic conductivity and solid-state diffusion, and consequently limited cycling stability and rate capability. Herein, we design an aqueous Mg-ion battery chemistry involving a hydrated eutectic electrolyte, an organic molecule anode, and a copper hexacyanoferrate (CuHCF) cathode. This hydrated eutectic electrolyte features a three-dimensional percolating hydrogen bond network formed by water molecules, which facilitates fast Mg2+ transport in the electrolyte. Moreover, the suppression of water activity in the hydrated eutectic electrolyte can efficiently improve the cycling performance of the organic molecule anode by prohibiting the dissolution issue. After coupling with the open-framework CuHCF cathode, the resultant full battery delivers a wide operating voltage of 2.2 V, an energy density of 52.2 W h kg−1, and a decent low-temperature electrochemical performance. The electrolyte and electrode chemistries proposed in this work show an alternative way to develop low-cost, safe, and high-performance Mg battery technologies.
Broader contextDeveloping alternative energy storage technologies for state-of-the-art Li-ion batteries is one of the main objectives of the battery research community. Generally, the lack of suitable electrolytes and compatible electrode materials have limited the performance improvement (e.g., rate performance, cycling stability), which is especially true for “hard” Mg2+ (large charge-to-diameter ratio). Herein, we designed a hydrated eutectic electrolyte formulation based on low-cost, non-toxic, and earth abundant elements. This hydrated eutectic electrolyte features a three-dimensional percolating hydrogen bond network that is constructed by water molecules. This can suppress water activity and facilitate Mg2+ transport. These features enable a stable and high-rate organic molecule anode and copper hexacyanoferrate cathode. Importantly, the resultant full battery shows a promising energy density of 52.2 W h kg−1, which is competitive to lead-acid batteries. This work shows a new way to design high-performance multivalent-ion batteries featuring multiple advantages for grid-scale storage and micropower systems. |
Introduction
Currently, the rapid market growth of electrochemical energy storage in our society calls for the development of new battery chemistries beyond state-of-the-art Li-ion batteries.1–3 Among various post-Li-ion batteries, rechargeable Mg-ion batteries (MIBs) using a bivalent Mg2+ charge carrier show great potential for applications in grid-scale energy storage of intermittent renewable energy (e.g., solar, wind) and micropower systems.4,5 This potential is stimulated by the intrinsic advantages of the Mg element, including high earth abundance (1.94% for Mg vs. 0.002% for Li), low cost, and environmental friendliness.4–7 However, the current MIBs still show insufficient electrochemical performance like poor rate performance and low cycling stability.4,8,9 The performance bottleneck is intrinsic to the bivalent nature of Mg2+ ions, i.e., Mg2+ has twice the charge density compared to Li+ since they have a similar cationic radius. As a result, ion disassociation from the electrolytes and solid-state cation diffusion, which are two essential steps in classical intercalation chemistry, show sluggish kinetics at room temperature.4–10 Additionally, serious structural changes of electrode materials upon Mg2+ intercalation result in low cyclability.11–13 Therefore, designing new electrolytes and electrode materials has been the key to achieving performance improvement of rechargeable MIBs.The development of organic electrolytes for MIBs has been an important research topic, which typically hinges on the presence of chloride to ensure decent ionic conductivity and reversibility of Mg plating/stripping.5,15,16 However, serious challenges are associated with these chlorides, such as a high disassociation energy of the Mg–Cl bond, chemical corrosivity, moisture sensitivity, toxicity, and flammability. Recently, organic chloride-free electrolytes have been discovered (e.g., boron cluster-based Mg(CB11H12)2 dissolved in glyme) to overcome some of the challenges caused by chlorides (e.g., corrosivity, water stability),17,18 though high viscosity and strong coordination between Mg2+ and glyme lead to low ionic conductivity and sluggish kinetics. These limitations of organic electrolytes have motivated researchers to develop aqueous MIBs.19–21 Aqueous electrolytes eliminate the typical disadvantages of organic electrolytes (e.g., toxicity, flammability) and show additional advantages of low cost, high ionic conductivity, and easy handling. In this line of thought, various cathode and anode materials have been evaluated in different aqueous electrolytes (e.g., Mg(NO3)2, MgSO4). Nevertheless, many reported aqueous Mg battery chemistries only focused on the half-cell configurations.14,21–25 For the full aqueous MIBs,19,20,26,27 there are still limited Mg2+-host materials available, while the narrow electrochemical stability window (1.23–1.80 V) of the traditional aqueous electrolytes and the possible incompatibility between electrolytes and electrode materials (e.g., electrode dissolution and decomposition) could lead to low operation voltage, low energy density, and poor cycling stability. Therefore, it is critical to design a new aqueous Mg2+ electrolyte featuring high electrode compatibility and an expanded stability window.
Herein, we propose a full aqueous MIB utilizing an organic anode of perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA), an open-framework copper hexacyanoferrate (CuHCF) cathode, and a hydrated eutectic electrolyte consisting of Mg(NO3)2·6H2O and acetamide. The resultant full MIB shows a high voltage of 2.2 V, a high energy density of 52.2 W h kg−1, and increased cycling stability (65.3% capacity retention after 1000 cycles). The improved electrochemical performance towards Mg2+ storage is related to the synergistic contributions from the proposed electrolyte and electrodes. On the one hand, the presence of water in the hydrated eutectic electrolyte can ensure high ionic conductivity, while the lack of free water molecules suppresses the dissolution of PTCDA during charge and discharge. On the other hand, the non-diffusion-controlled PTCDA anode and open-framework CuHCF cathode can facilitate Mg2+ (de)intercalation without significant structural changes, ensuring good rate performance and cycling stability.
Results and discussions
Compatibility of hydrated eutectic electrolytes with organic molecule electrodes
For grid-scale energy storage or micropower systems, the battery materials, especially electrode materials, should feature low cost, Earth-abundant elements, and environmental friendliness. A promising family of such materials is organic electrode materials,28–30 which consist of electrochemically redox-active organic compounds like molecules and polymers. Organic molecules feature a high specific capacity and energy density as well as a non-diffusion controlled redox reaction mechanism, though their cycling stability is significantly limited by their fast dissolution upon cycling in conventional aqueous electrolytes and even organic electrolytes.31–33 Making organic molecules into the corresponding polymers can efficiently prohibit the dissolution issue.19,20,34,35 However, the introduction of electrochemically inactive linkers could lead to much decreased specific capacity compared to the original molecules, while the sloping charge–discharge curves would result in a decreased average operating potential. These two factors together decrease the polymer electrode energy density. Furthermore, the synthesis difficulties of polymer electrodes (e.g., polymerization reaction, thermal activation) make them unfavorable for large-scale applications. Given the above discussions, we believe organic molecules are a good starting point for designing stable aqueous MIBs with high energy density and power density. One strategy to realize this goal is to design efficient aqueous electrolytes to inhibit the dissolution issue.The dissolution issue of organic molecule electrodes is related to the abundance of free water in conventional aqueous electrolytes. Recently, several strategies have been developed to decrease the number of free water molecules or suppress the activity of water, such as “water-in-salts”,31,36–38 hydrate melts,39,40 “water-in-ionomers”,41 and “molecular crowding”.42 The strategies of “water-in-salts” and hydrate melts depend on the formation of super-concentrated aqueous electrolytes, in which water molecules are mostly localized in the first solvation sheath of cations, thus minimizing the number of free water molecules.31,36–40 However, the utilization of highly concentrated salts (e.g., fluorinated salts) causes toxicity and cost concerns. Differently, the methods of “water-in-ionomers” and “molecular crowding” rely on the strong interaction between polymers and water molecules,41,42 thereby realizing a decreased amount of water molecules at the free state. Nonetheless, a high water content is required (>30%) to generate the electrolyte of “water-in-ionomers”,41 while the “molecular crowding” approach leads to insufficient ionic conductivity (0.8 mS cm−1).42 Noticeably, both the “water-in-ionomers” and “molecular crowding” strategies are developed based on the high solubility of salts in water. Accordingly, halogenated salts (e.g., fluorinated salts) are still needed, similar to the methodologies of “water-in-salts” and hydrate melts. These studies inspire us to develop an eco-friendly and cost-efficient aqueous electrolyte with a limited amount of free water and high ionic conductivity. This electrolyte should enable stable and high-rate operation of organic molecules for aqueous MIBs.
In fact, water molecules show a bipolar nature, which endows them with the capability to perform as chemical building blocks for eutectic electrolytes.37,39,43 This has been proven by designing hydrated eutectic electrolytes for stable and efficient aqueous Zn-ion batteries.32 In the hydrated eutectic electrolytes, it is feasible to isolate water molecules from each other through rationally controlling the atomic content of water. This is critical to suppress the water activity for stable operation of organic electrodes.32 In the classical eutectic electrolytes, the metal cations (e.g., Mg2+, Zn2+, Al3+) are Lewis acids, while anions and organic compounds (e.g., amines) are Lewis bases. Upon mixing metallic salts with organic compounds, the Lewis acid–base interaction induces the formation of eutectic frameworks.44,45 Noticeably, the bipolar nature of water molecules ensures the miscibility in the conventional eutectic electrolytes. We presume that besides the Lewis acid–base interaction between cations and compounds/anions, water molecules can readily interact with metal cations (e.g., Mg2+) through judiciously controlling the water content. This hydrated eutectic framework resembles the state-of-the-art electrolytes of “water-in-salts” and hydrate melts.36–40 This feature indicates the capability of the hydrated eutectic framework in suppressing water activity by decreasing the amount of free water. Notably, the hydrated eutectic electrolytes show additional advantages like nontoxicity, low cost, and facile preparation.
Herein, we design a hydrated eutectic electrolyte for MIBs. This electrolyte features a precisely controlled hydration level through a facile formulation, i.e., mixing Mg(NO3)2·6H2O with a neural compound of acetamide (Fig. 1a). Compared to other Mg2+ salts (e.g., magnesium triflate, magnesium chloride), Mg(NO3)2·6H2O shows the advantages of low cost, non-toxicity, non-corrosiveness, and decent solubility, and capability to enable a wide voltage window. Making metal nitrates into working aqueous electrolytes can provide new strategies in designing aqueous rechargeable batteries since electrochemical nitrate reduction has been proven to be the main reason that limits the application of metal nitrates in aqueous rechargeable batteries.46 Noticeably, no additional water is used except the crystalline water of Mg(NO3)2·6H2O. Indeed, developing this hydrated eutectic electrolyte is partially motivated by the existence of abundant free water molecules for the saturated aqueous Mg(NO3)2 electrolyte, as proven by our previous work.46 The abundant free water molecules in the saturated aqueous Mg(NO3)2 electrolyte lead to the dissolution and fast capacity degradation of the PTCDA electrode (Fig. S1, ESI†). Even in the saturated aqueous Mg(NO3)2 electrolyte, the molar ratio of water to Mg2+ (nH2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nMg2+) is still larger than 12 and the solvation sheaths of Mg2+ ions are still dominated by water molecules. These are strong indications that the existence of a large amount of the clusters of free water molecules are detrimental to the cycling stability of organic molecule electrodes (e.g., PTCDA).19,31 Currently, limited information is available regarding the control over the hydrated Mg2+ species and the surrounding hydrogen-bonded localized structure of water molecules. Upon introducing acetamide, the water content of the electrolytes can be decreased by decreasing the molar ratio of Mg2+ to acetamide (nMg2+:nacetaimde) (Fig. 1b). This provides the feasibility to tailor the hydrated structure of Mg2+ and suppress the water activity.
:nMg2+) is still larger than 12 and the solvation sheaths of Mg2+ ions are still dominated by water molecules. These are strong indications that the existence of a large amount of the clusters of free water molecules are detrimental to the cycling stability of organic molecule electrodes (e.g., PTCDA).19,31 Currently, limited information is available regarding the control over the hydrated Mg2+ species and the surrounding hydrogen-bonded localized structure of water molecules. Upon introducing acetamide, the water content of the electrolytes can be decreased by decreasing the molar ratio of Mg2+ to acetamide (nMg2+:nacetaimde) (Fig. 1b). This provides the feasibility to tailor the hydrated structure of Mg2+ and suppress the water activity.
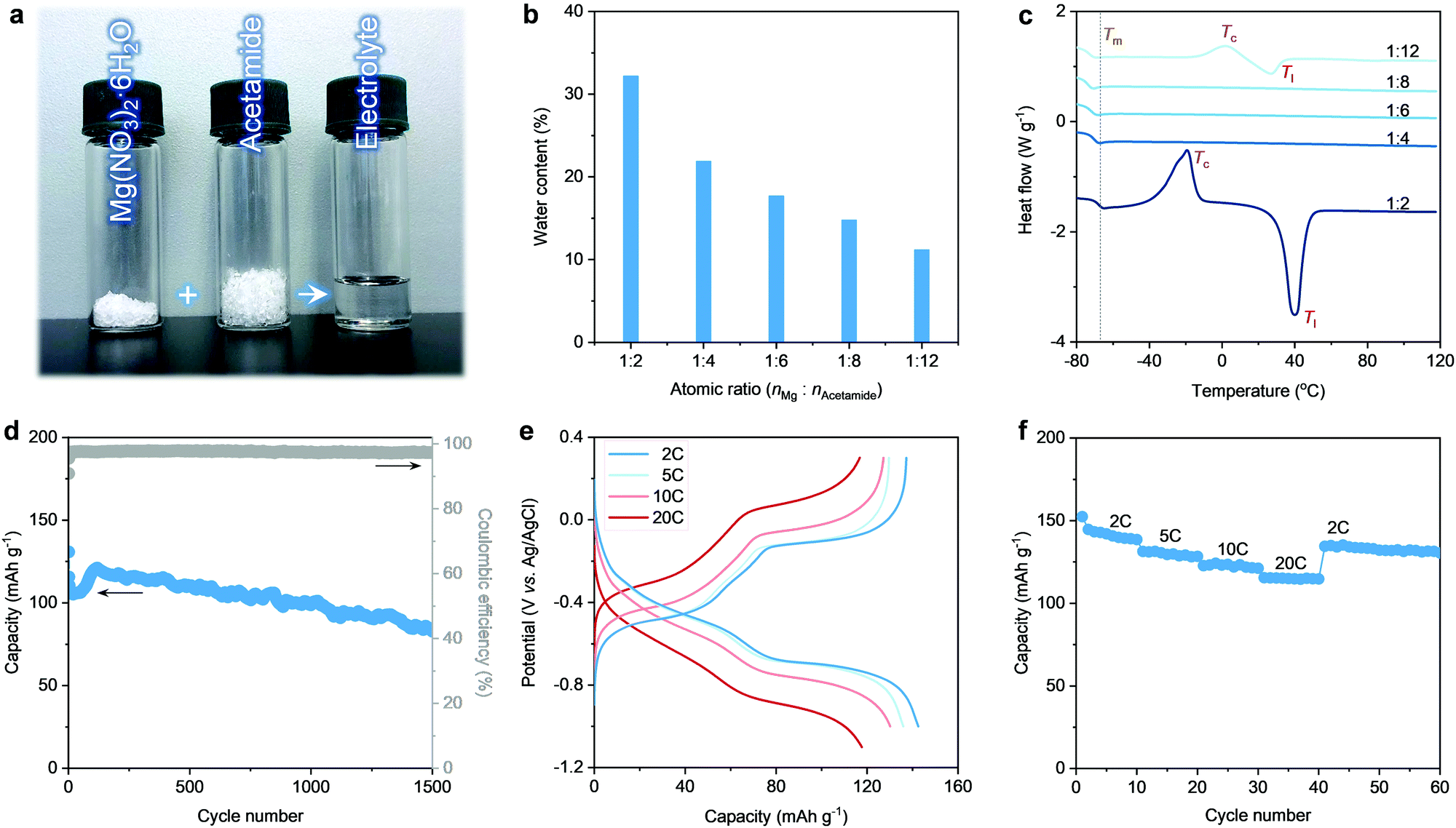 | ||
| Fig. 1 Characterization of the electrolytes and the electrochemical performance of the PTCDA anode. (a) Stoichiometric amounts of Mg(NO3)2·6H2O and acetamide to make the hydrated eutectic electrolyte with nMg2+:nacetamide = 1:8. (b) Summary of the water content in the electrolyte using different molar ratios of Mg(NO3)2·6H2O and acetamide. (c) DSC curves of the electrolytes. Electrochemical performance of the PTCDA anode using the hydrated eutectic electrolyte (nMg2+:nacetamide = 1:8): (d) cycling stability test at 10 C, (e) galvanostatic charge–discharge curves at different rates, and (f) rate capability test. | ||
Acetamide is taken as a logical choice to produce hydrated eutectic electrolytes because of its wide availability, moderate Lewis basicity, and decent polarity to dissolve different types of salts.47,48 Noticeably, bipolar water molecules are able to form hydrogen bonding with the nitrogen or oxygen atoms of the acetamide molecules, resulting in the reduction of melting point (Tm).49,50 In this line of consideration, the carbonyl and amine groups of acetamide ensure a high tendency towards hydration, which can enrich the intermolecular interaction. This is a prerequisite for yielding a eutectic liquid system, as confirmed by the transparent and homogeneous solution after mixing acetamide with Mg(NO3)2·6H2O (Fig. 1a). The hydrated eutectic solution remains stable in the liquid state without forming any precipitates even after ten months (Fig. S2, ESI†).
Differential scanning calorimetry (DSC) was conducted to illustrate the thermal properties of the prepared electrolytes (Fig. 1c). When nMg2+:nacetamide is too low (1:12) or too high (1:2), the DSC curves reveal a high liquidus temperature (Tl) (>20 °C). Additionally, an exothermic peak corresponds to the crystallization temperature (Tc), which should be related to the presence of abundant free water clusters at high nMg2+:nacetamide or abundant acetamide clusters at low nMg2+:nacetamide. The excess water or acetamide clusters do not participate in the eutectic network. In contrast, a medium nMg2+:nacetamide leads to a very low Tm, which is the typical feature for the eutectic electrolytes.44,45 Note that the lowest Tm of −76 °C can be observed at nMg2+:nacetamide = 1:8. The significantly decreased Tm originates from the charge delocalization between the components. Featuring nMg2+:nacetamide = 1:8, the hydrated eutectic solution shows well-balanced physical properties (Fig. S3, ESI†), including high ionic conductivity (5.5 mS cm−1, even comparable to the organic electrolytes of commercial Li-ion batteries51 and state-of-the-art “water-in-salts” electrolytes36–38), low viscosity (67.1 mPa s), and a low water content (14.8 wt%, Fig. 1b). Noticeably, the hydrated eutectic electrolyte shows an extended electrochemical stability window of 2.7 V (Fig. S4, ESI†), even comparable to state-of-the-art aqueous electrolytes like “water-in-salts” (2.7–3.2 V),31,36–38 hydrate melts (2.5–3.2 V),39,40 water-in-ionomer (1.8–2.6 V),41 and molecular crowding (2.5–3.2 V).42 This further confirms the efficiency of the hydrated eutectic strategy in suppressing water activity and in designing new aqueous electrolytes that are typically difficult to achieve through classical strategies (e.g., “water-in-salts”). Therefore, this eutectic formulation, without introducing additional water, is used as the electrolyte for aqueous MIBs.
First, we evaluated the electrochemical performance of the PTCDA anode towards Mg2+ storage using a conventional three-electrode setup (see the Experimental Details in the ESI†). Here, an organic PTCDA material is selected as the anode because of: (1) the commercial availability and low cost, and (2) the nanorod morphology and the monoclinic structure with an open framework that can together facilitate Mg2+ diffusion (Fig. S5 and S6, ESI†). As shown in Fig. 1d, the PTCDA electrode shows decent cycling stability, corresponding to a capacity retention of 65.5% after 1500 cycles at the current density of 10 C. The cycling stability achieved in the eutectic electrolyte with nMg2+:nacetamide = 1:8 shows a pronounced improvement compared to that realized in saturated Mg(NO3)2 electrolyte (Fig. S1, ESI†) and other eutectic formulations (Fig. S7, ESI†). This is related to the minimum of free water molecules in the hydrated eutectic electrolyte, which can efficiently prohibit the dissolution of PTCDA upon cycling.31,52 In addition, the PTCDA anode exhibits superior rate performance at 2–20 C (Fig. 1e), delivering 142 mA h g−1 at a low current rate of 2 C and 120 mA h g−1 at a high current rate of 20 C. This indicates that at a low current density, each formula unit of PTCDA follows a 2-electron transfer reaction mechanism through the non-diffusion-controlled enolization reaction (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ↔ C–O−), thus leading to nearly half of the theoretical capacity (4-electron reaction). Noticeably, the non-diffusion-controlled reaction mechanism enables the high rate capability.
O ↔ C–O−), thus leading to nearly half of the theoretical capacity (4-electron reaction). Noticeably, the non-diffusion-controlled reaction mechanism enables the high rate capability.
Instead of a proton, Mg2+ has been proven to be the cation stored at the PTCDA anode because of the neutral pH condition of the hydrated eutectic electrolytes (suppressed water activity) and overlapped charge–discharge curves even after decreasing the pH of the electrolytes (Fig. S8, ESI†). The stages in the galvanostatic charge–discharge (GCD) curves are associated with reversible phase changes of PTCDA upon Mg2+ (de)interclation, as confirmed by the previous study.25 The phase changes are induced by the unique squeezing deformation (i.e., reversible contraction and expansion) of the PTCDA anode.25 This structural flexibility further ensures the cycling stability of PTCDA upon repeated Mg2+ intercalation/extraction. The rate capability tests of the PTCDA anode further reveal its structural stability to withstand large current tests (Fig. 1f). These results highlight the capability of our hydrated eutectic electrolytes in enabling a stable and high-rate electrochemical Mg2+ storage performance when using the organic molecule electrode of PTCDA. This is intimately related to the physicochemical state changes of water molecules in the eutectic electrolyte systems.32,53
Spectroscopic characterization of the hydrated eutectic electrolytes
We next performed spectroscopic analysis using different techniques to study the physicochemical states of water molecules in the eutectic electrolytes, including Fourier-transform infrared (FT-IR), Raman, and liquid-state nuclear magnetic resonance (NMR) spectroscopy. As shown in the FT-IR spectra (Fig. 2a), the intensity of the O–H stretching bands in the range of 3000–3800 cm−1 is significantly suppressed when introducing acetamide, indicating that the hydrogen-bonding network of water clusters is severely disrupted.54 Similar results can also be found in the Raman spectra (Fig. S9, ESI†). A shoulder peak at ∼3680 cm−1 appears and intensifies when decreasing nMg2+:nacetamide. These peaks are typically the characteristics of water molecules bonded to metal ion centers.55 The appearance of this signal indicates the strong interaction between H2O and Mg2+ in the eutectic electrolytes, signifying the existence of H2O into the solvation sheaths of Mg2+. This can be further confirmed by the 17O NMR spectra (Fig. 2b). In comparison to the water reference (∼0 ppm), the 17O (H2O) resonance signal of the eutectic electrolytes shows an upfield shift. The peak broadening is associated with slightly decreased T2 relaxation time,37,54 which in turn reveals the low viscosity of the hydrated eutectic electrolytes (Fig. S3, ESI†). Furthermore, the 17O (acetamide) and 17O (NO3−) show slight shifts with the decrease of nMg2+:nacetamide. These results reveal that the solvation structures of different ions and neutral acetamide molecules strongly interpenetrate, which is the typical feature of eutectics.
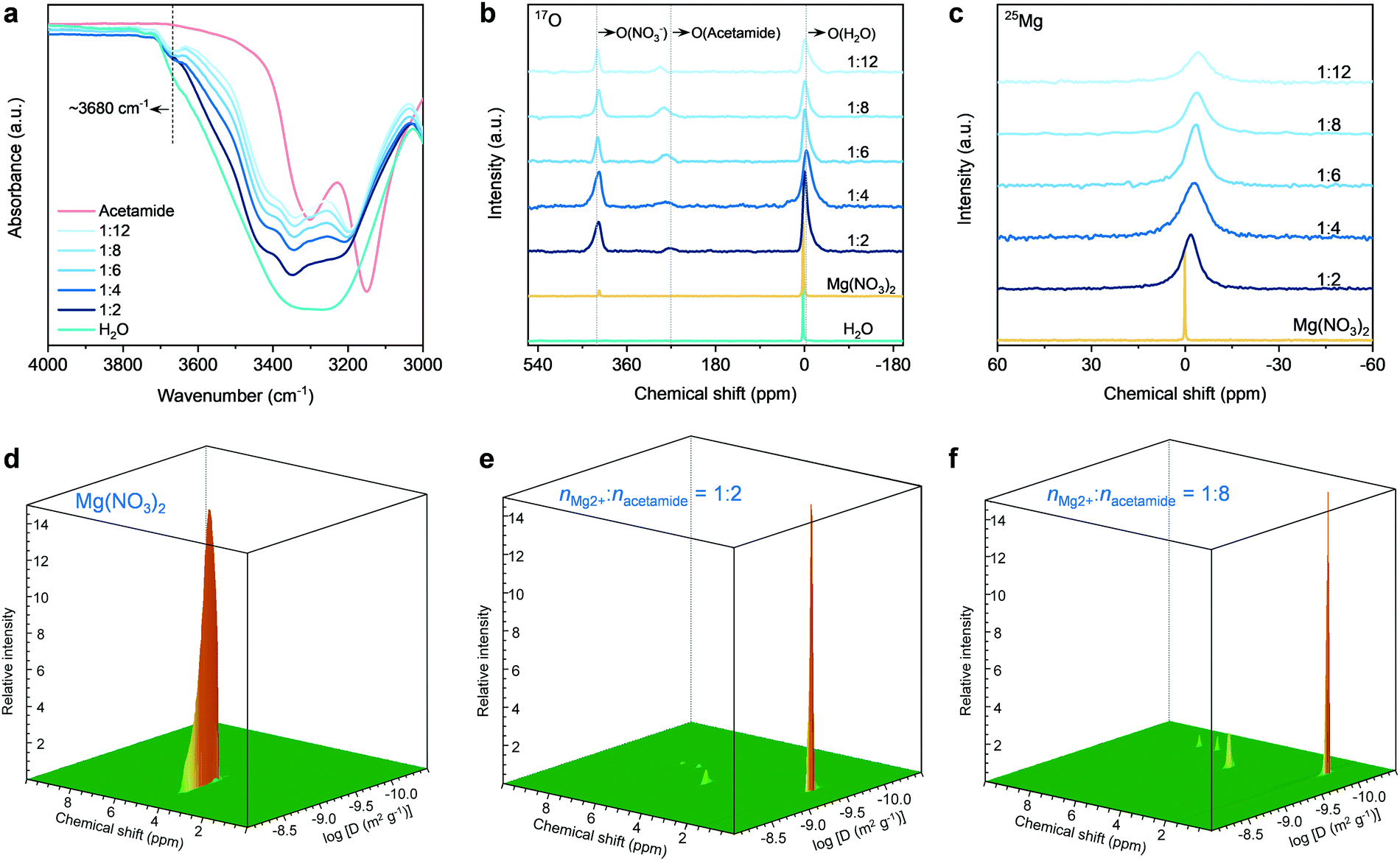 | ||
| Fig. 2 Spectroscopic characterizations of the electrolytes. (a) FT-IR, (b) liquid-state 17O NMR, and (c) liquid-state 25Mg NMR spectra. PFT-NMR contour spectra of (d) 1 M Mg(NO3)2, (e) the electrolyte with nMg2+:nacetamide = 1:2, and (f) the electrolyte with nMg2+:nacetamide = 1:8. | ||
More specifically, the Lewis acidic Mg2+ can interact with the lone-pair electrons of oxygen atoms in water or acetamide.36–40 A suppressed interaction between Mg2+ and H2O leads to the shielding effect of 17O (H2O), as shown by the upfield shift for the hydrated eutectics. The concentration increase of acetamide results in a more pronounced upfield shift of 17O (H2O). This indicates that the water molecules around Mg2+ ions are gradually replaced by the acetamide molecules. Importantly, the line width of the 25Mg resonance signal increases marginally along with an upfield peak shift, which suggests the increased complexation and interaction between Mg2+ and electron-rich groups (e.g., CO in acetamide). Based on the above spectroscopy and DSC results of the eutectic electrolytes, it is reasonable to infer that the acetamide molecules gradually substitute water molecules and penetrate into the solvation shells of Mg2+ ions in the hydrated eutectic electrolytes, which can minimize the number of free water molecules. Moreover, the steric-hindrance effect caused by NO3− dictates the identity of solution species, as reflected by the less significant shift of 17O (NO3−) compared to other oxygen species (Fig. 2b).
According to previous studies,3,36–42 the water molecules in the electrolytes designed by “water-in-salts”, hydrate melts, “water-in-ionomers”, and “molecular crowding” can ensure the decent ionic conductivity (<10 mS cm−1) of the resultant electrolytes. As proven by previous studies experimentally and theoretically,56–58 the decent ionic conductivity of these electrolytes results from an unusual nano-heterogeneity in compositions, which can decouple cations from the Coulombic traps of the anions in the solvation shells. Eventually, a three-dimensional (3D) percolating water-cation network is formed, which can efficiently liberate cations for fast ion transport in the electrolytes. This further reveals that a continuous hydrogen-bonding network constructed by water molecules in the solvation sheaths of cations is critical to ensure the ionic diffusion. For our hydrated eutectic electrolytes, the NO3− anions show much weaker interaction compared to water and acetamide, delivering low Coulombic traps for fast Mg2+ transport through the Mg2+–H2O network. The well-structured Mg2+–H2O network has been illustrated by the FT-IR, Raman, and NMR. The NO3− anions are actually located in the second solvation shells of Mg2+ ions, as confirmed by the following molecular dynamics (MD) simulations (to be discussed shortly) and stricter analysis of the eutectic electrolytes. Accordingly, the evaluation of the diffusion of water in the eutectic electrolytes would be valuable to study the percolation and continuity of the Mg2+–H2O, the prerequisite for high ionic conductivity and rate capability of aqueous MIBs.36–42,56–58
We conducted 1H pulsed-field gradient NMR (PFG-NMR) spectroscopy to investigate the diffusivity of water in the electrolytes (Fig. 2d–f and Fig. S10, ESI†). For the control electrolyte of 1 M Mg(NO3)2 (M: molar Lelectrolyte−1), the 3D PFG-NMR spectrum shows only one 1H signal at the chemical shift of 4.7 ppm (Fig. 2d and Fig. S11, ESI†), corresponding to the hydrogen of water molecules. A high diffusion coefficient of 1.2 × 10−9 m2 s−1 for water molecules can be achieved. In contrast, an obviously decreased diffusion coefficient of 7.9 × 10−10 m2 s−1 is observed for the electrolyte with nMg2+:nacetamide = 1:2 (Fig. 2e). The reduction of water diffusivity is the result of a gradually suppressed hydrogen-bonding network,59 as confirmed by the previous FT-IR, Raman, and NMR spectroscopic analyses. At a eutectic formulation of nMg2+:nacetamide = 1:8, the diffusivity of water molecules is detected to be 2.1 × 10−10 m2 s−1 (Fig. 2f). In accordance with previous studies,56–58 the vehicular motion of cation–(H2O)n dominate the cation transport through the 3D cation–H2O network. The high diffusivity of water molecules in our hydrated eutectic electrolyte ensures the fast vehicular motion of Mg2+–(H2O)n and thus the high ionic conductivity.
Since the acetamide molecules start entering the solvation shells of Mg2+ ion with increasing acetamide content, we pay additional attention to the diffusivity changes of acetamide molecules. At a low content of acetamide (nMg2+:nacetamide = 1:2) (Fig. 2e), the three new 1H resonance signals (1.6, 6.4, and 7.2 ppm; see detailed analysis in Fig. S9, ESI†) that are assigned to acetamide show that acetamide diffuses slightly (8.5 × 10−11 m2 s−1) slower than water, which is due to the bulkier nature for the acetamide molecules. The diffusivity of acetamide slightly decreases with its content increase in the electrolytes (Fig. 2f and Fig. S10, ESI†). This is because the acetamide molecules replace the water molecules in the solvation sheaths of Mg2+ ions, while the increased Coulombic traps by Mg2+ slows down the transport of the acetamide molecules. Our hydrated eutectic electrolytes are different from the classical electrolytes of “water-in-salts” and hydrate melts, in which anions in the solvation sheaths of cations deliver much stronger Coulombic traps. This solvation structure difference, initiated by the neutrally charged acetamide molecules, endows fast Mg2+ transport and high ionic conductivity, even competitive with the ionic conductivity of “water-in-salts” and hydrate melt electrolytes for aqueous Li-ion batteries.
Molecular dynamics (MD) simulations of the hydrated eutectic electrolytes
Next, we conducted MD simulations to gain theoretical insights into the structures of the electrolytes. Fig. 3 shows the MD simulation boxes and the corresponding radial distribution functions (RDF). At a molar ratio of nMg2+:nacetamide = 1:2, the solvation sheaths of Mg2+ ions are dominated by water molecules (Fig. S12, ESI†), and the water molecules interact with each other to generate a percolating hydrogen bond network (Fig. 3a). Upon increasing the content of acetamide (nMg2+:nacetamide = 1:8), the Mg2+ ions start interacting with the Lewis-basic oxygen atoms of the acetamide molecules (Fig. 3b and Fig. S13, ESI†). Previous studies confirm that this solvation structure can modify the location of the lowest unoccupied molecular orbital (LUMO) of the electrolyte, which can efficiently suppress water activity from a thermodynamics point of view.36,39,54 Notably, the 3D percolating hydrogen bond framework can be maintained, providing a continuous pathway for fast Mg2+ transport. However, a further increase of the acetamide content (nMg2+:nacetamide = 1:12) induces the generation of acetamide clusters (Fig. 3c and Fig. S14, ESI†), which breaks the extended hydrogen bond network and thus delivers decreased ionic conductivity.56,57 Noticeably, an extended hydrogen bond network of H2O–acetamide starts to form upon decreasing nMg2+:nacetamide, which is critical to suppress water activity (Fig. S15–S17, ESI†).
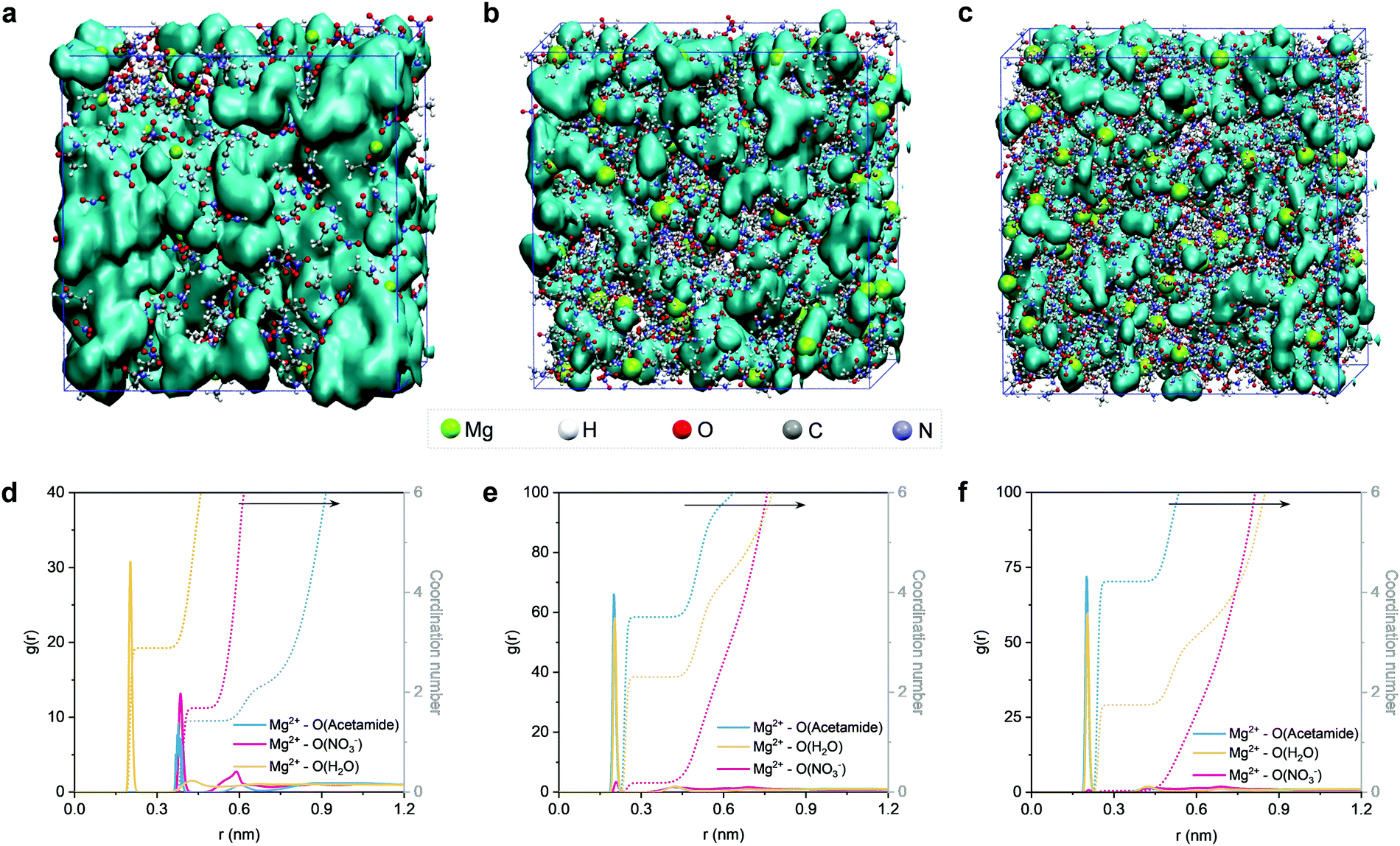 | ||
| Fig. 3 MD simulation results of the electrolytes with different molar ratios of Mg(NO3)2·6H2O and acetamide. Snapshots of MD simulation boxes featuring (a) nMg2+:nacetamide = 1:2, (b) nMg2+:nacetamide = 1:8, and (c) nMg2+:nacetamide = 1:12. The hydrogen bond networks of H2O–H2O are highlighted with a light colored surface. RDFs plots of Mg2+–O and the corresponding coordination environments of Mg2+ ion in the electrolytes with (d) nMg2+:nacetamide = 1:2, (e) nMg2+:nacetamide = 1:8, and (f) nMg2+:nacetamide = 1:12. | ||
RDF analysis can provide more detailed information about the solvation environment of Mg2+ ions in the electrolytes. According to the RDF plots in Fig. 3d–f, we find that the first solvation sheath (<0.3 nm) of Mg2+ is gradually dominated by the acetamide molecules upon decreasing nMg2+:nacetamide. Based on the statistical analysis of MD simulations, the first solvation sheath of Mg2+ (nMg2+:nacetamide = 1:8) contains ∼4 acetamide molecules and ∼2 water molecules, while the NO3− anions remain in the second solvation sheath (Fig. 3e). This simulation result is consistent with the aforementioned liquid-state NMR analysis. We note that the electrolyte composition with nMg2+:nacetamide = 1:12 shows a similar solvation structure. On the other hand, more pronounced oscillations beyond 0.3 nm can be observed for the electrolyte composition of nMg2+:nacetamide = 1:2 (Fig. 3d). A flattening effect of the second solvation shell can be identified when increasing the molar ratio of acetamide. The flattening phenomenon is generally interpreted as the structure-breaking effect.59,60 This is one sign that the fluctuations of water molecules around the Mg2+ ions become different upon changing nMg2+:nacetamide. Through the PFG-NMR technique, we have confirmed that the molar ratio increase of acetamide causes a reduction in water diffusivity. Therefore, we attribute the flattening effect to the decreased diffusivity of water molecules after partially breaking the hydrogen bond network. However, the maintenance of a 3D percolating hydrogen bond network ensures the fast Mg2+ diffusion in the electrolyte through the vehicular motion mechanism of Mg2+–H2O, in a similar manner to that of the super-concentrated aqueous electrolytes for Li-ion batteries.56,57,61 Our halide-free strategy is different from the previous strategies,36,39,40 which involve a very high concentration of expensive and toxic fluorine-containing salts to suppress the activity of water.
The aforementioned spectroscopy analysis and theoretical simulations show a limited capability to illustrate the equilibrium species and provide direct structural information of the electrolytes. To achieve this, we produced single crystals from the electrolytes and analyzed them using single-crystal X-ray diffraction (XRD) (see the Experimental details in the ESI†).32,62 The single-crystal structure analysis reveals that water molecules around the Mg2+ ion are gradually replaced by acetamide molecules upon increasing the content of acetamide (Fig. S18–S20 and Tables S1–S3, ESI†). At the eutectic formulation of nMg2+:nacetamide = 1:8, the Mg2+ ion is coordinated by six oxygen atoms (i.e., two water molecules and four acetamide molecules), while the cation charge is balanced by two persistent NO3− anions. Because of the smaller molecular size of water compared with acetamide, the repulsive interaction between water and acetamide would be weak. This effect makes several water molecules remain in the primary coordination sphere of Mg2+. Note that the single-crystal results of the electrolytes are consistent with the MD simulations.
Full battery performance
Based on the above discussions of low-cost and efficient electrolytes and PTCDA anode, it is necessary to develop a cathode that also features availability, low cost, nontoxicity, and good electrochemical performance. Herein, CuHCF is chosen as the cathode to pair with the eutectic electrolytes and PTCDA anode because:20,22,63,64 (1) CuHCF has a 3D open framework that can facilitate the intercalation of bivalent Mg2+; (2) the abundant structural water molecules can shield the charge of “hard” Mg2+ ions and thus facilitate their diffusion; (3) the Prussian Blue analogs (PBAs) (e.g., CuHCF) are usually prepared using mild co-precipitation methods (see the Experimental Details in the ESI†), representing a low-cost and potentially scalable synthesis technique; and (4) compared to other PBAs, CuHCF features a relatively higher working potential, thereby ensuring improved energy density. We further decreased the particle size of CuHCF to shorten the diffusion pathway of Mg2+ and to increase the rate capability (Fig. S21–S23, ESI†). Ex situ XRD patterns evidence the reversible intercalation of Mg2+ within the CuHCF cathode (Fig. S24, ESI†). GCD plots reveal the decent rate capability of the CuHCF cathode: 61.2 mA h g−1 at 0.5 C and 37.8 mA h g−1 at 20 C (Fig. 4a). In addition, the CuHCF cathode delivers a stable cycling performance, i.e., 91.5% capacity retention after 2000 cycles at 10 C (Fig. 4b).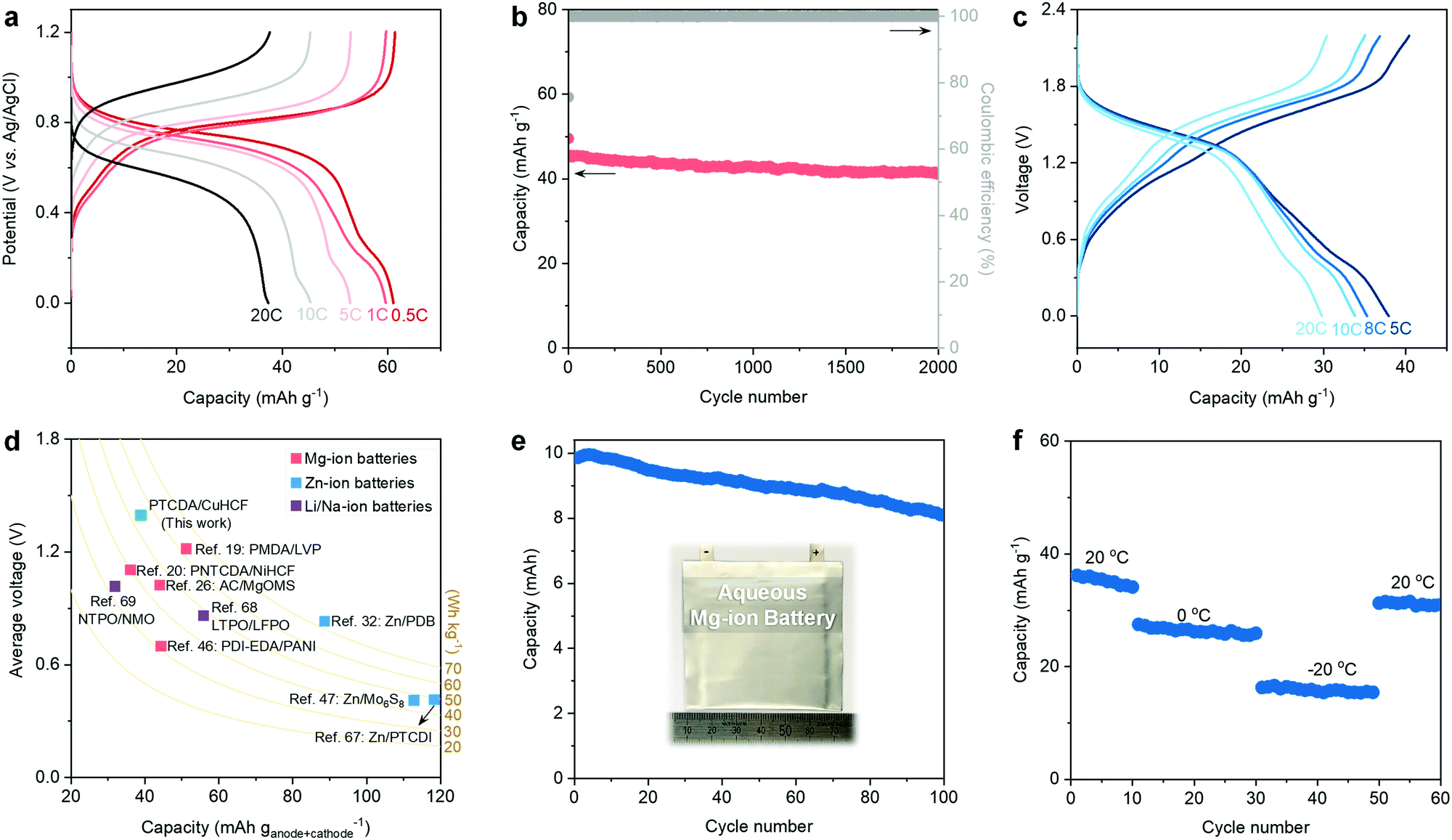 | ||
| Fig. 4 Electrochemical performance of the CuHCF cathode and the full aqueous Mg-ion battery (electrolyte formulation: nMg2+:nacetamide = 1:8). (a) Galvanostatic charge–discharge curves and (b) cycling performance test of the CuHCF cathode at 10 C. (c) Galvanostatic charge–discharge curves of the full MIB. (d) Electrochemical performance comparison of different aqueous battery chemistries, including energy density (based on the total mass of active anode and cathode), capacity, and average voltage. (e) Cycling performance of the pouch-type MIB at 5 C. The inset of panel (e) shows the photo of the MIB pouch cell with a capacity of 10 mA h. (f) Cycling performance of the full MIB at different temperatures. | ||
The stable operation of the PTCDA anode and the CuHCF cathode in the hydrated eutectic electrolyte further prompted us to assemble a full aqueous Mg-ion battery. We notice that previous studies have demonstrated the feasibility and efficiency of utilizing a CuHCF cathode and PTCDA or PTCDA analogue anode for aqueous Mg-ion batteries in different electrolytes.25,65,66 Nevertheless, the electrolytes used in these previous works have still not been optimized, leading to insufficient electrochemical performance (e.g., low Coulombic efficiency, poor cycling stability, relatively narrow working voltage window). The GCD curves of our battery chemistry demonstrate that the full battery can work stably in a wide voltage window of 2.2 V (Fig. 4c). The full battery shows a capacity of 38.1 mA h g−1 (based on the total mass of active anode and cathode) at 5 C and an average voltage of 1.38 V, as well as a decent capacity of 29.8 mA h g−1 at 20 C. Ragone plots reveal the competitive performance of our full battery to the state-of-the-art aqueous MIBs (Fig. S25, ESI†). As shown in Fig. 4d, we compared the electrochemical performance of this full MIB with previous aqueous MIBs,19,20,26,46 Zn-ion batteries (e.g., Zn–organic batteries),32,47,67 Li-ion batteries,68 and Na-ion batteries.69 Our full MIB shows a competitive performance (e.g., energy density and average voltage) compared with the state-of-the-art aqueous mobile-ion batteries.
Furthermore, the full battery can be stability cycled at 8 C, showing a capacity retention of 65.3% after 1000 cycles (Fig. S26, ESI†). In addition, we evaluate the electrochemical performance of a 10 mA h pouch cell of our aqueous Mg-ion battery chemistry (Fig. 4e). A capacity retention of 81.1% can be realized after 100 cycles. Note that our hydrated eutectic electrolyte remains stable even after 50 cycles of the full battery (Fig. S27, ESI†). Inspired by the low melting point (−76 °C) of the hydrated eutectic electrolyte, we further evaluate the low-temperature performance of the full MIB. Fig. 4f shows that our proposed full MIB can perform well at low temperatures: 26.3 mA h g−1 at 0 °C and 15.8 mA h g−1 at −20 °C. The capacity recovers to 31.9 mA h g−1 when the temperature is increased to room temperature. The decent cycling stability at different temperatures indicates the advantage of the hydrated eutectic electrolytes, which can potentially enable rechargeable aqueous MIBs working in a wide temperature range.
In view of the above discussions, the proposed MIB chemistry shows some unique features and advantages. First, the hydrated eutectic electrolyte shows a high ionic conductivity (5.5 mS cm−1) at room temperature and a low melting point, thus enabling a decent electrochemical performance towards Mg2+ even at a low temperature. This superior performance is related to the 3D percolating hydrogen bond network generated by water molecules in the hydrated eutectic formulation. The hydrated eutectic electrolyte consists of low-cost, low environmental impact, and halide-free compositions, thus ensuring the advantages in cost and safety (see Table S4 (ESI†) for the cost comparisons of different electrolytes). Second, both the cathode and anode are low-cost materials made of earth-abundant elements. The CuHCF cathode can be synthesized at room temperature through a straightforward and scalable wet chemistry method. The PTCDA anode is commercially available because of its wide applications as decorative paints. Third, the rational design of a hydrated eutectic electrolyte endows the PTCDA anode and the CuHCF cathode with decent cycling stability and rate capability. The dissolution issue of the organic-molecule PTCDA can be efficiently suppressed during cycling due to the suppression of water activity in the hydrated eutectic electrolyte. Last but not least, the elemental abundance of the key materials in our battery chemistry in the Earth's crust is much higher than those of classical batteries, such as lead-acid, vanadium-based redox flow, and nickel-metal hydride batteries. All these together reveal the potential of the proposed full MIB for large-scale or micropower applications, wherein energy density is outweighed by the above-discussed parameters.
Currently, aqueous rechargeable MIBs are still underdeveloped compared to other aqueous battery chemistries. One of the bottlenecks is related to the much more “hard” nature (high charge-to-radius ratio) of Mg2+ compared to the Li+ benchmark. This can induce a significant polarization of the electrode material framework and very strong cation–cation repulsion, thus resulting in sluggish solid-state diffusions and severe structural changes of the electrodes. All these together make it more challenging to develop suitable intercalation electrodes of MIBs, which feature large capacities, stable redox potentials, and long-term cyclability. We notice that the PTCDA anode and the CuHCF cathode used in this work hold large potential in storing other earth-abundant cations (e.g., Zn2+, Ca2+, Al3+). This is rationalized by the intrinsic nature of an open framework and suitable redox potentials for these two electrode materials. On the other hand, our hydrated eutectic electrolyte strategy can potentially enable other high-performance mobile-ion battery chemistries (e.g., Ca2+, Al3+) using PTCDA and CuHCF.
Although a decent energy density of 52.2 W h kg−1 is achieved through our current battery chemistry, we realize that further improvement of energy density will depend on the development of new intercalation electrodes. For the PBA cathode, the doping strategy is commonly used to adjust the capacity and redox potential.22,31,63 Doping other active transition metals (e.g., Mn) into the CuHCF framework can potentially increase the capacity without sacrificing the intrinsic high redox potential of CuHCF. For the anode, materials with open frameworks, higher capacity, and even lower redox potentials would further enhance the energy density of aqueous MIBs. Organic anodes represent a promising candidate because of their controllable compositions and electronic structures through precise molecular engineering.28,29
Conclusions
In summary, we have developed a hydrated eutectic electrolyte by coupling the hydrated Mg(NO3)2 salt exclusively with acetamide. The electrolyte formulation features low cost, nontoxicity, and convenient operation. More importantly, the solvation structure of Mg2+ can be precisely regulated by controlling the molar ratio of hydrated salt and acetamide. At the eutectic formulation, the primary solvation sheath of Mg2+ is dominated by acetamide and partially occupied by water molecules, as confirmed by different spectroscopic characterizations and molecular dynamics simulations. This unique solvation structure decreases the number of water molecules at the free state and provides a three-dimensional hydrogen bond network for high ionic conductivity. These features can sufficiently improve the cycling stability of the organic molecule anode of PTCDA by suppressing the severe dissolution issue, which has been challenging to achieve utilization of the conventional or concentrated aqueous electrolytes. Upon coupling with the nanostructured cathode of CuHCF, the full battery shows a decent energy density, rate capability, cyclability, and low-temperature electrochemical performance. This work presents promising battery chemistry for grid-scale energy storage and micropower systems. A step beyond can be extended to other aqueous multivalent-ion batteries through designing rational hydrated eutectic electrolytes and electrode materials.Author contributions
H. N. A. supervised the project. Y. Z. and H. N. A. conceived the idea. Y. Z. designed the experiments and conducted the experiments of material syntheses, electrochemical tests, and material characterizations including XRD, FT-IR, Raman, and DSC. X. G. performed the theoretical simulations and conducted the PFG-NMR characterizations. Y. L. conducted TEM, and W. W. performed electrochemical tests. Y. Z., Y. Y. and Y. H. conducted single-crystal XRD and analysis. X. G. and A.-H. E. performed the NMR characterization. The manuscript was written by Y. Z. and was revised by the other co-authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this work was supported by King Abdullah University of Science Technology (KAUST).Notes and references
- M. S. Whittingham, Chem. Rev., 2020, 120, 6328–6330 CrossRef CAS PubMed.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- C. P. Grey and D. S. Hall, Nat. Commun., 2020, 11, 6279 CrossRef CAS PubMed.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef CAS.
- J. L. Andrews, A. Mukherjee, H. D. Yoo, A. Parija, P. M. Marley, S. Fakra, D. Prendergast, J. Cabana, R. F. Klie and S. Banerjee, Chemistry, 2018, 4, 564–585 CrossRef CAS.
- M. Li, J. Lu, X. Ji, Y. Li, Y. Shao, Z. Chen, C. Zhong and K. Amine, Nat. Rev. Mater., 2020, 5, 276–294 CrossRef CAS.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC.
- Z. Guo, S. Zhao, T. Li, D. Su, S. Guo and G. Wang, Adv. Energy Mater., 2020, 10, 1903591 CrossRef CAS.
- M. M. Huie, D. C. Bock, E. S. Takeuchi, A. C. Marschilok and K. J. Takeuchi, Coord. Chem. Rev., 2015, 287, 15–27 CrossRef CAS.
- Y. Zhu, G. Huang, J. Yin, Y. Lei, A.-H. Emwas, X. Yu, O. F. Mohammed and H. N. Alshareef, Adv. Energy Mater., 2020, 10, 2002128 CrossRef CAS.
- Y. Xu, X. Deng, Q. Li, G. Zhang, F. Xiong, S. Tan, Q. Wei, J. Lu, J. Li, Q. An and L. Mai, Chemistry, 2019, 5, 1194–1209 CrossRef CAS.
- G. S. Gautam, P. Canepa, W. D. Richards, R. Malik and G. Ceder, Nano Lett., 2016, 16, 2426–2431 CrossRef PubMed.
- K. W. Nam, S. Kim, S. Lee, M. Salama, I. Shterenberg, Y. Gofer, J.-S. Kim, E. Yang, C. S. Park, J.-S. Kim, S.-S. Lee, W.-S. Chang, S.-G. Doo, Y. N. Jo, Y. Jung, D. Aurbach and J. W. Choi, Nano Lett., 2015, 15, 4071–4079 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- Y. Li, S. Guan, H. Huo, Y. Ma, Y. Gao, P. Zuo and G. Yin, Adv. Funct. Mater., 2021, 31, 100650 Search PubMed.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900–7904 CrossRef CAS PubMed.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- F. Wang, X. Fan, T. Gao, W. Sun, Z. Ma, C. Yang, F. Han, K. Xu and C. Wang, ACS Cent. Sci., 2017, 3, 1121–1128 CrossRef CAS PubMed.
- L. Chen, J. L. Bao, X. Dong, D. G. Truhlar, Y. Wang, C. Wang and Y. Xia, ACS Energy Lett., 2017, 2, 1115–1121 CrossRef CAS.
- X. Sun, V. Duffort, B. L. Mehdi, N. D. Browning and L. F. Nazar, Chem. Mater., 2016, 28, 534–542 CrossRef CAS.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed.
- Y. Mizuno, M. Okubo, F. Hosono, T. Kudo, K. Ohishi, A. Okazawa, N. Kojima, R. Kurono, S.-I. Nishimura and A. Yamada, J. Mater. Chem. A, 2013, 1, 13055–13059 RSC.
- J. Song, M. Noked, E. Gillette, J. Duay, G. Rubloff and S. B. Lee, Phys. Chem. Chem. Phys., 2015, 17, 5256–5264 RSC.
- I. A. Rodríguez-Pérez, Y. Yuan, C. Bommier, X. Wang, L. Ma, D. P. Leonard, M. M. Lerner, R. G. Carter, T. Wu, P. A. Greaney, J. Lu and X. Ji, J. Am. Chem. Soc., 2017, 139, 13031–13037 CrossRef PubMed.
- H. Zhang, K. Ye, K. Zhu, R. Cang, X. Wang, G. Wang and D. Cao, ACS Sustainable Chem. Eng., 2017, 5, 6727–6735 CrossRef CAS.
- H. Zhang, K. Ye, K. Zhu, R. Cang, J. Yan, K. Cheng, G. Wang and D. Cao, Chem. – Eur. J., 2017, 23, 17118–17126 CrossRef CAS PubMed.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS.
- X. Yin, S. Sarkar, S. Shi, Q.-A. Huang, H. Zhao, L. Yan, Y. Zhao and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908445 CrossRef CAS.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed.
- T. Liu, K. C. Kim, B. Lee, Z. Chen, S. Noda, S. S. Jang and S. W. Lee, Energy Environ. Sci., 2017, 10, 205–215 RSC.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015, 2, 1500124 CrossRef PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- D. P. Leonard, Z. Wei, G. Chen, F. Du and X. Ji, ACS Energy Lett., 2018, 3, 373–374 CrossRef CAS.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- Q. Zheng, S. Miura, K. Miyazaki, S. Ko, E. Watanabe, M. Okoshi, C.-P. Chou, Y. Nishimura, H. Nakai, T. Kamiya, T. Honda, J. Akikusa, Y. Yamada and A. Yamada, Angew. Chem., Int. Ed., 2019, 58, 14202–14207 CrossRef CAS PubMed.
- X. He, B. Yan, X. Zhang, Z. Liu, D. Bresser, J. Wang, R. Wang, X. Cao, Y. Su, H. Jia, C. P. Grey, H. Frielinghaus, D. G. Truhlar, M. Winter, J. Li and E. Paillard, Nat. Commun., 2018, 9, 5320 CrossRef CAS PubMed.
- J. Xie, Z. Liang and Y.-C. Lu, Nat. Mater., 2020, 19, 1006–1011 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- C. Zhang, L. Zhang and G. Yu, Acc. Chem. Res., 2020, 53, 1648–1659 CrossRef CAS PubMed.
- Y. Zhu, J. Yin, A.-H. Emwas, O. F. Mohammed and H. N. Alshareef, Adv. Funct. Mater., 2021, 31, 2107523 CrossRef CAS.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed.
- M. Angell, C.-J. Pan, Y. Rong, C. Yuan, M.-C. Lin, B.-J. Hwang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 834–839 CrossRef CAS PubMed.
- M. A. Krestyaninov, E. G. Odintsova, A. M. Kolker and M. G. Kiselev, J. Mol. Liq., 2018, 264, 343–351 CrossRef CAS.
- F. D’Amico, B. Rossi, G. Camisasca, F. Bencivenga, A. Gessini, E. Principi, R. Cucini and C. Masciovecchio, Phys. Chem. Chem. Phys., 2015, 17, 10987–10992 RSC.
- R. Younesi, G. M. Veith, P. Johansson, K. Edströmb and T. Vegge, Energy Environ. Sci., 2015, 8, 1905–1922 RSC.
- C. Han, J. Zhu, C. Zhi and H. Li, J. Mater. Chem. A, 2020, 8, 15479–15512 RSC.
- O. Borodin, J. Self, K. A. Persson, C. Wang and K. Xu, Joule, 2020, 4, 69–100 CrossRef CAS.
- Y. Zhu, J. Yin, X. Zheng, A.-H. Emwas, Y. Lei, O. F. Mohammed, Y. Cui and H. N. Alshareef, Energy Environ. Sci., 2021, 14, 4463–4473 RSC.
- J. Zhao, J. Zhang, W. Yang, B. Chen, Z. Zhao, H. Qiu, S. Dong, X. Zhou, G. Cui and L. Chen, Nano Energy, 2019, 57, 625–634 CrossRef CAS.
- O. Borodin, L. Suo, M. Gobet, X. Ren, F. Wang, A. Faraone, J. Peng, M. Olguin, M. Schroeder, M. S. Ding, E. Gobrogge, A. von, W. Cresce, S. Munoz, J. A. Dura, S. Greenbaum, C. Wang and K. Xu, ACS Nano, 2017, 11, 10462–10471 CrossRef CAS PubMed.
- J. Lim, K. Park, H. Lee, J. Kim, K. Kwak and M. Cho, J. Am. Chem. Soc., 2018, 140(46), 15661–15667 CrossRef CAS PubMed.
- Z. Li, R. Bouchal, T. Mendez-Morales, A.-L. Rollet, C. Rizzi, S. Le Vot, F. Favier, B. Rotenberg, O. Borodin, O. Fontaine and M. Salanne, J. Phys. Chem. B, 2019, 123, 10514–10521 CrossRef CAS PubMed.
- Y. Ding, A. A. Hassanali and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3310–3315 CrossRef CAS PubMed.
- M. V. Fernández-Serra and E. Artacho, J. Chem. Phys., 2004, 121, 11136–11144 CrossRef PubMed.
- K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki, W. Shinoda, K. Hashimoto, K. Ueno, Y. Umebayashi and M. Watanabe, J. Phys. Chem. B, 2018, 122(47), 10736–10745 CrossRef CAS PubMed.
- A. Du, Z. Zhang, H. Qu, Z. Cui, L. Qiao, L. Wang, J. Chai, T. Lu, S. Dong, T. Dong, H. Xu, X. Zhou and G. Cui, Energy Environ. Sci., 2017, 10, 2616–2625 RSC.
- L. Xue, Y. Li, H. Gao, W. Zhou, X. Lu, W. Kaveevivitchai, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 2164–21670 CrossRef CAS PubMed.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed.
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, K. Oh-ishi, A. Okazawa, N. Kojima, R. Kurono, S. Nishimura and A. Yamada, J. Mater. Chem. A, 2013, 1, 13055–13059 RSC.
- Z. Zhang, Y. Li, G. Zhao, L. Zhu, Y. Sun, F. Besenbacher and M. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 40451–40459 CrossRef PubMed.
- N. Liu, X. Wu, Y. Zhang, Y. Yin, C. Sun, Y. Mao, L. Fan and N. Zhang, Adv. Sci., 2020, 7, 2000146 CrossRef CAS PubMed.
- J.-Y. Luo, W.-J. Cui, P. He and Y.-Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee03691b |
| This journal is © The Royal Society of Chemistry 2022 |