
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton conductivity dependence on the surface polymer thickness of core–shell type nanoparticles in a proton exchange membrane†
Keisuke
Tabata
 a,
Tomohiro
Nohara
a,
Haruki
Nakazaki
a,
Tsutomu
Makino
a,
Takaaki
Saito
a,
Toshihiko
Arita
*b and
Akito
Masuhara
*ac
a,
Tomohiro
Nohara
a,
Haruki
Nakazaki
a,
Tsutomu
Makino
a,
Takaaki
Saito
a,
Toshihiko
Arita
*b and
Akito
Masuhara
*ac
aGraduate School of Science and Engineering, Yamagata University, 4-3-16 Yonezawa, Yamagata 992-8510, Japan. E-mail: masuhara@yz.yamagata-u.ac.jp
bInstitute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan. E-mail: tarita@tohoku.ac.jp
cFrontier Center for Organic Materials (FROM), Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan
First published on 3rd September 2022
Abstract
The proton exchange membrane (PEM) is the main component that determines the performance of polymer electrolyte fuel cells. The construction of proton-conduction channels capable of fast proton conduction is an important topic in PEM research. In this study, we have developed poly(vinylphosphonic acid)-block-polystyrene (PVPA-b-PS)-coated core–shell type silica nanoparticles prepared by in situ polymerization and a core–shell type nanoparticle-filled PEM. In this system, two-dimensional (2D) proton-conduction channels have been constructed between PVPA and the surface of silica nanoparticles, and three-dimensional proton-conduction channels were constructed by connecting these 2D channels by filling with the core–shell type nanoparticles. The proton conductivities and activation energies of pelletized PVPA-coated core–shell type nanoparticles increased depending on the coated PVPA thickness. Additionally, pelletized PVPA-b-PS-coated silica nanoparticles showed a good proton conductivity of 1.3 × 10−2 S cm−1 at 80 °C and 95% RH. Also, the membrane state achieved 1.8 × 10−4 S cm−1 in a similar temperature and humidity environment. Although these proton conductivities were lower than those of PVPA, they have advantages such as low activation energy for proton conduction, suppression of swelling due to water absorption, and the ability to handle samples in powder form. Moreover, by using PS simultaneously, we succeeded in improving the stability of proton conductivity against changes in the temperature and humidity environment. Therefore, we have demonstrated a highly durable, tough but still enough high proton conductive material by polymer coating onto the surface of nanoparticles and also succeeded in constructing proton-conduction channels through the easy integration of core–shell type nanoparticles.
1. Introduction
Fuel cells are a promising power generation system that plays a part in clean energy advocated by the Sustainable Development Goals (SDGs) because of having many advantages such as a small environmental load and not using fossil fuels.1,2 In particular, polymer electrolyte fuel cells (PEFCs) have some advantages such as high energy-conversion efficiency, low-temperature operation, small size, and light weight. Therefore, PEFCs are expected to be applied to fuel cell vehicles and other applications.3 The proton exchange membrane (PEM) is an important component of PEFCs, because of the proton conductivity of the PEM directly linked to the PEFC performance.4–9 Nafion® is one of the most famous and most commonly used PEMs and has a high proton conductivity of 10−1 S cm−1 or more and high chemical stability, and the high acidity and high durability have resulted in high PEM performance.10,11 However, Nafion® has some weak points such as a large environmental impact, complicated preparation, and a necessity to use a platinum electrocatalyst due to the strong acidity of perfluorosulfonic acid, and there is an urgent need to develop PEMs with low acidity and ease of preparation.12Various approaches have been reported to achieve developments in Nafion® alternative PEM such as controlling of the micro-space proton-conduction channels and composing of proton conductive polymers, nanoparticles, and so on. H. Kitagawa et al.13 reported one-dimensional (1D) proton-conduction channels and J. Matsui et al.14–16 reported two-dimensional (2D) proton-conduction channels. They succeeded in forming proton-conduction channels using metal–organic frameworks or polymer nanosheet thin films. Interestingly, these proton-conduction channels achieved a high proton conductivity of up to 3.2 × 10−2 S cm−1 (60 °C and 98% relative humidity (RH)) despite their low acidity such as using carboxylic acid and phosphoric acid groups. Therefore, if the proton-conduction channels of PEMs are limited to a small space, high proton conductivity can be achieved even with weak acids because of the high density of proton-conducting sites with an ideal configuration. Furthermore, Y.-F. Lin et al.17 reported a Nafion® and functionalized-silica nanoparticle composite membrane and T. Yamaguchi et al.18 investigated filler and proton conductive polymer composite materials. By combining the proton conductive polymer with the filler, not only the physical strength but also the water retention of the composite can be improved. In addition, the result of constructing the interfaces between filler and proton conductive polymers, the composite material showed higher proton conductivity compared with the sum conductivity of filler and proton conductive polymer. Thus, the filler can seem to reduce the proton conductivity, but it has great utility value as a PEM material for precise control of the structure. Based on the above, these studies indicate that even weakly acidic polymers can exhibit high proton conductivity by controlling the proton-conduction channels; in addition, the introduction of nanoparticles with high mechanical strength into the PEM is useful for controlling the structure of the electrolyte membrane.
Recently, our group has reported proton-conductive core–shell type nanoparticles and three-dimensional (3D) proton-conduction channels constructed by filling with core–shell type nanoparticles.19,20 The core–shell type nanoparticles composed of silica nanoparticles as a core material and poly(acrylic acid)-block-polystyrene (PAA-b-PS) as the shell polymer were prepared by reversible addition–fragmentation chain transfer polymerization with particles method (RAFT PwP).21–24 The RAFT PwP is an in situ polymer coating method on the surface of core nanoparticles in which RAFT polymerization is performed in the coexistence of core particles and monomers. Based on the above in situ polymer-coating method, it can be said that we can also fabricate core–shell type nanoparticles using the free radical polymerization with particles method. On the other hand, the RAFT PwP gives merit that it is possible to coat uniformed length polymer chains, which are directly linked to the realization of the desired high proton conductivity with low activation energy.19 Then, we have successfully established 2D proton-conducting channels at the interface between the core nanoparticle surface and PAA using the RAFT PwP method. The proton conductivity of these core–shell type nanoparticles was evaluated in the pellet state, which was prepared by filling with the core–shell type nanoparticles. We have studied the influences of the amount of polymer,25 core nanoparticle size,26 and surface silanol groups of silica nanoparticles27 on the proton conductivity. As a result of these investigations, PAA-b-PS coated silica nanoparticles achieved a proton conductivity of 4.7 × 10−3 S cm−1 at 60 °C and 98% RH and a small activation energy (Ea) of 0.21 eV. In this system, the 2D proton-conduction channels of the interface between silica nanoparticles and PAA were connected to each nanoparticle, resulting in the construction of 3D long-range proton-conduction channels. The formation of such 2D proton-conduction channels has only been found by RAFT PwP, not from another coating method yet, to our knowledge. These studies on PAA-b-PS coated silica nanoparticles have indicated the usefulness of the 2D proton-conductive channel built at the interface between silica nanoparticles and PAA.
Even more, we have developed a PEM that applies these findings to a new proton-conducting polymer, poly (vinylphosphonic acid) (PVPA).28 PVPA is attracting attention as a new proton conductive polymer, and it is expected to achieve high proton conductivity derived from a high concentration of phosphate groups. The PEM consisting of PVPA-coated cellulose nanocrystals (CNCs) which were core–shell type nanoparticles showed excellent proton conductivity (>10−1 S cm−1 at 60 °C and 95% RH) comparable to that of Nafion®. Although the core–shell type nanoparticles achieved good proton conductivity by using PVPA, detailed studies such as on the thickness of PVPA, PVPA adsorption modes, and the stability due to the coating of PS were not performed.
In this study, we have investigated in detail the proton conductivity of PVPA-based core–shell silica nanoparticles and a PEM filled with them. Spherical silica nanoparticles were employed as the core material to fabricate PVPA-b-PS coated silica nanoparticles (silica@PVPA-b-PS). Additionally, we have fabricated a PEM by filling silica@PVPA-b-PS into PC, which is a thermoplastic resin with a large mechanical strength (Scheme 1).29–32 In order to evaluate the effect of PVPA coverage on proton conductivity, a thinner PVPA layer was formed on the surface of silica nanoparticles using a different solvent from that used in the CNC system. We expected that the solubility of the electroconductive polymers (oligomers) in the solvent employed strongly affects the configuration of the polymers on the surface of nanoparticles. Herein, we have succeeded in preparing highly proton conductive silica@PVPA, silica@PVPA-b-PS, and silica@PVPA-b-PS/PC membranes. The maximum proton conductivity of these core–shell type nanoparticles and membranes was 2.0 × 10−2 S cm−1, 1.3 × 10−2 S cm−1, and 1.8 × 10−4 S cm−1 at 80 °C and 95% RH, respectively. In this study, we succeeded in improving the proton conductivity by an order of magnitude or more by increasing the thickness of PVPA with the polymerization time. Moreover, by coating with PS, a hydrophobic polymer, we succeeded in providing stability of proton conductivity against changes in temperature and humidity environments.
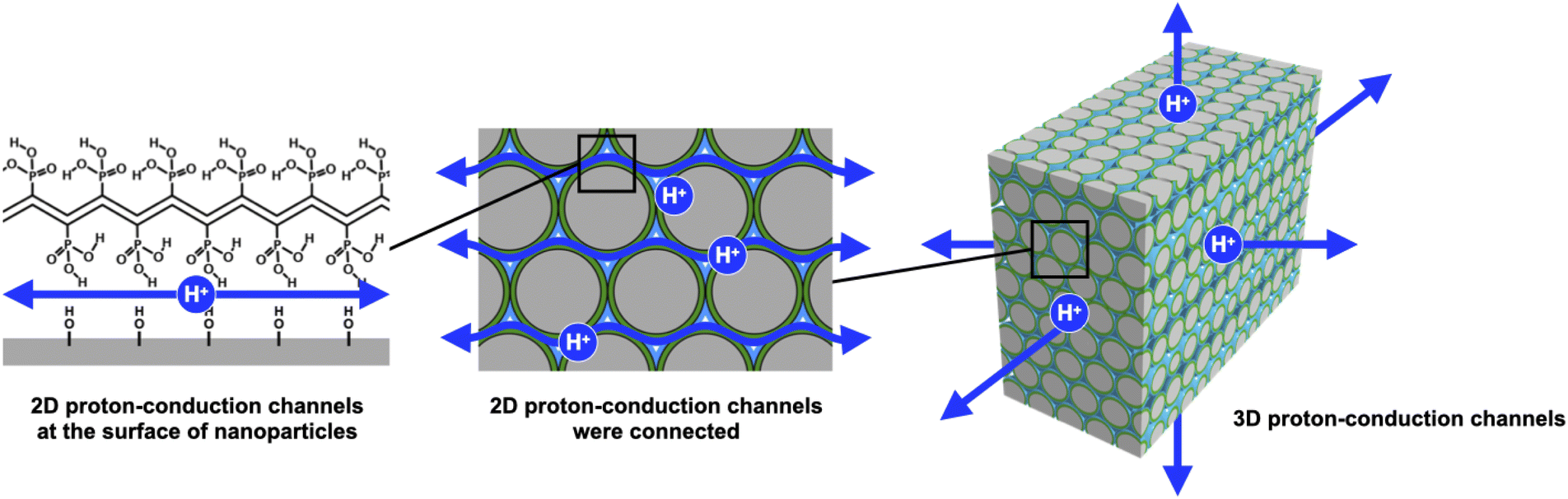 | ||
| Scheme 1 Schematic model of proton-conductive core–shell type nanoparticle-filled PEM. | ||
2. Experimental
2.1. Materials
Vinylphosphonic acid (VPA, >95.0%) and N,N-dimethylformamide (DMF, >99.5%) were purchased from Tokyo Chemical Industry Co., Ltd. 2,2′-Azobis(isobutylnitrile) (AIBN, >98.0%), styrene (>99.0%), and acetonitrile (>99.9%) were purchased from FUJIFILM Wako Pure Chemical Corporation. Bare silica nanoparticles (average particle size 235 nm determined from SEM images) were purchased from UBE EXSYMO CO., Ltd. Polycarbonate (PC) was purchased from TEIJIN Ltd.2.2. Preparation of silica@PVPA and silica@PVPA-b-PS
Firstly, O-ethyl-S-(1-ethoxycarbonyl)-ethyldithiocarbonate (X1) which is the CTA was synthesized according to a previous report, and we have succeeded in the RAFT/MADIX polymerization of PVPA by using X1 (see the ESI†). Silica@PVPA and silica@PVPA-b-PS were fabricated by RAFT PwP which performed RAFT/MADIX polymerization of PVPA under the coexistence of silica nanoparticles (Scheme 2).33,34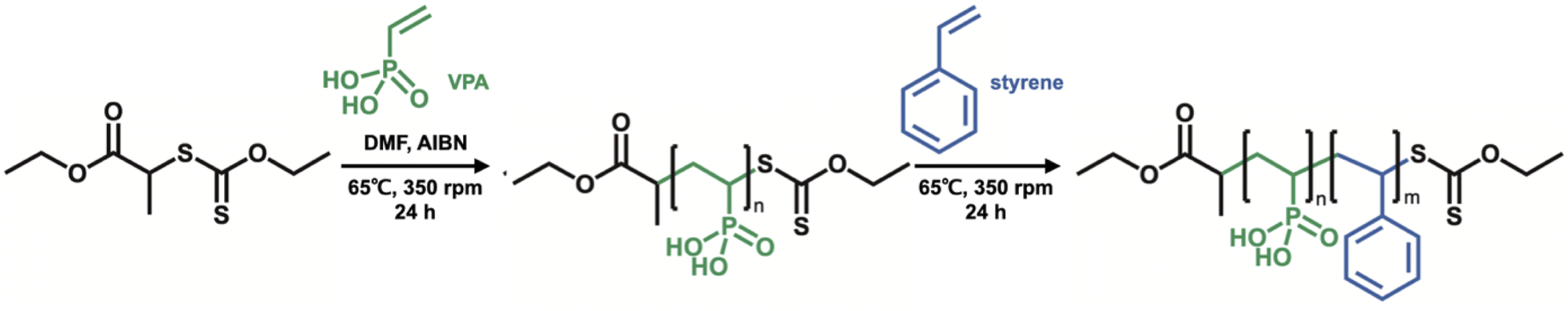 | ||
| Scheme 2 Synthesis scheme of PVPA-b-PS using X1. | ||
Silica nanoparticles (1.0 g), VPA (0.45 g, 4.17 mmol), X1 (9.3 mg, 41.8 μmol), AIBN (2.8 mg, 17.1 μmol), and DMF (3.0 g, 3.18 mL) were weighed in a test tube and irradiated with ultrasonic waves to disperse them. Later, nitrogen bubbling was performed and they were transferred to a nitrogen-filled glove box and polymerization was carried out at 65 °C with stirring. The polymerization time was set to 3, 6, 12, 24, and 48 hours in order to confirm the VPA polymerization conversion rate and the thickness of PVPA coated on the silica surface. The RAFT PwP of silica@PVPA proceeds in the following order: X1 and the radical initiator (AIBN) were added in the coexistence state of silica nanoparticles and VPA monomers, and RAFT polymerization was started by heating. After that, low molecular weight oligomers have grown in the vicinity of the nanoparticles adsorbed on the surface of the silica nanoparticles due to the decrease in solubility and the influence of the surface energy of the core particles. It was simple physical adsorption; however, the VPA monomer has a hydroxy group capable of forming a hydrogen bond, and it forms a hydrogen bond with the hydroxy group on the surface of the core nanoparticle and strongly adsorbs. In the second step, silica@PVPA-b-PS was fabricated by adding styrene monomer into the polymerization mixture of silica@PVPA. After finishing all polymerization, a large amount of acetonitrile was added to the polymerization mixture, and unreacted monomers were removed by centrifugation. The same operation was repeated 3 times for purification. Finally, the precipitate was redispersed with ethyl acetate and dried to obtain a white powder (Fig. 1(a)).
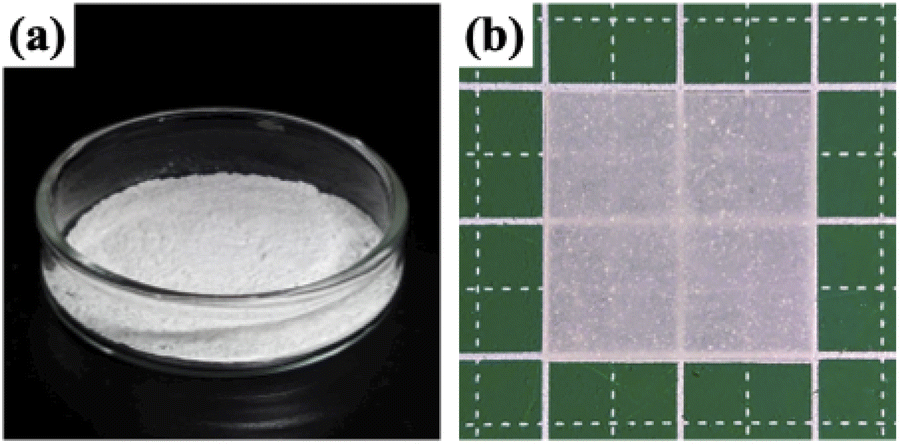 | ||
| Fig. 1 Photos of (a) silica@PVPA-b-PS nanoparticles and (b) the silica@PVPA-b-PS/PC membrane. | ||
2.3. Fabrication of the silica@PVPA-b-PS/PC membrane
The silica@PVPA-b-PS/PC membrane was obtained by a simple method such as mixing with thermoplastic resin and coating on a glass substrate.35 We selected PC as a thermoplastic resin which is flexible and has a low-glass transition temperature than the decomposition temperature of PVPA. Core-shell type nanoparticles of silica@PVPA-b-PS were dispersed, and PC was dissolved in DCM, and these dispersions and solution were mixed to prepare the coating solution. The membrane was prepared by the doctor blade method and after application, it was dried at room temperature to obtain a freestanding membrane (Fig. 1(b)). On the other hand, we also tried to fabricate silica@PVPA/PC membranes; however, the affinity between silica@PVPA and PC was too low to produce a freestanding membrane.282.4. Characterization
Field-emission scanning electron microscopy (FE-SEM, JEOL JSM-IT800 and JSM-7500F, accelerating voltage of 10 kV) measurements were performed to observe the surface morphology (magnification: ×100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Fourier transform infrared spectroscopy (FTIR, JASCO FT/IR-4700, KBr tablet method) was used to characterize silica@PVPA and silica@PVPA-b-PS. Thermal gravimetric analysis (TGA, Hitachi High-Tech Science Corporation TG-DTA/6200, 30–800 °C) measurements were performed at 10 °C min−1 under nitrogen. Ultraviolet-visible spectroscopy (UV-vis JASCO V-670) was used for identifying the CTA, which was coated onto the surface of silica nanoparticles.
000). Fourier transform infrared spectroscopy (FTIR, JASCO FT/IR-4700, KBr tablet method) was used to characterize silica@PVPA and silica@PVPA-b-PS. Thermal gravimetric analysis (TGA, Hitachi High-Tech Science Corporation TG-DTA/6200, 30–800 °C) measurements were performed at 10 °C min−1 under nitrogen. Ultraviolet-visible spectroscopy (UV-vis JASCO V-670) was used for identifying the CTA, which was coated onto the surface of silica nanoparticles.
2.5. Proton conductivity measurements
Proton conductivities were measured by the AC impedance method (HIOKI impedance analyzer IM3570, frequency range of 4.6 MHz–4.6 Hz), with samples placed in a temperature and humidity-controlled small environmental test chamber (ESPEC SH-222). The prepared core–shell type nanoparticles were shaped in the pellet state by using a tablet forming machine (∅ = 13 mm), and then fixed with Teflon® measuring cells, and the silica@PVPA-b-PS/PC membrane was also fixed with the same measurement cells. These pellets and membranes were placed in a small environmental test chamber.36,37 The proton conductivity was calculated by applying the resistance value obtained from the Cole–Cole plots to the following formula:where RS (Ω) is the resistance from the impedance data, d (cm) is the electrode distance, and S (cm2) is the electrode area.
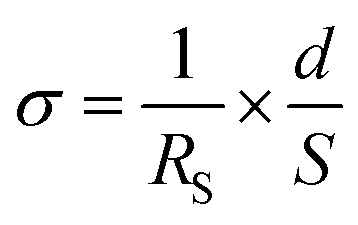
3. Results and discussion
3.1. Characterization of core–shell type nanoparticles and the silica@PVPA-b-PS/PC membrane
We have observed the surface morphology of silica@PVPA, silica@PVPA-b-PS, and silica@PVPA-b-PS/PC membranes using FE-SEM and SEM-EDX elemental mapping.Fig. S4† shows the FE-SEM images of bare silica nanoparticles, silica@PVPA, and silica@PVPA-b-PS observed at a magnification of 100000 times. The bare silica nanoparticles used in the production of core–shell type nanoparticles were spherical particles with an average particle size of 235 nm and had no irregularities on the surface. The PVPA coating on bare silica nanoparticles did not show any significant difference in the surface morphology, and also the silica@PVPA surface has remained spherical. Based on the above, the PVPA layer coated on the surface of silica nanoparticles was extremely thin compared to the CNC system.28 Here we calculated the PVPA thickness by determining the average particle size of silica@PVPA from SEM observations and subtracting the average particle size of bare silica nanoparticles (Fig. 2). The PVPA thickness correlated with the polymerization time, resulting in a successful coating of a 13.5 nm PVPA layer on silica@PVPA_48 h. The trend of increasing PVPA thickness was not linear; the coating thickness increased rapidly in the early stage of polymerization, and the change in coating thickness in the latter stage was small. From the above, it was said that PVPA is adsorbed on the surface of silica nanoparticles, which are core particles obtained by RAFT PwP.38 After the coating of PS, the core–shell type nanoparticles still maintained their spherical shape.
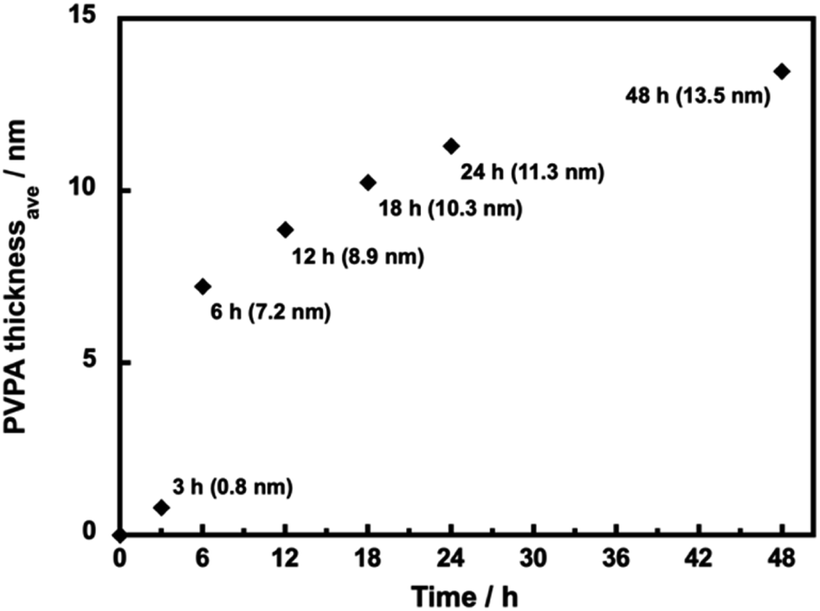 | ||
| Fig. 2 The average thickness of PVPA in silica@PVPA with various PVPA polymerization times (12–48 h) determined by SEM observation. | ||
For confirming the polymer adsorption on the surface of silica nanoparticles, SEM-EDX measurements were carried out.
From Fig. 3(a) and (b), the elemental mapping of silicon (Si) and oxygen (O) attributed to the core silica nanoparticle was strongly detected, and the phosphorus (P) from PVPA was also uniformly detected from the surface of nanoparticles. These results also supported the fact that the desired polymer was uniformly coated onto the surface of core nanoparticles by RAFT PwP and not in a mixed state of nanoparticles and polymer.39 Furthermore, FTIR measurements of each core–shell type nanoparticle were performed in order to observe the absorptions corresponding to the coated PVPA and PS (see the ESI†).
 | ||
| Fig. 3 FE-SEM and SEM-EDX images of (a) silica@PVPA_24 h, (b) silica@PVPA-b-PS, and (c) silica@PVPA-b-PS/PC membranes. | ||
Based on the evaluation of core–shell type nanoparticles so far, we will also evaluate the silica@PVPA-b-PS/PC membrane blended with PC. Note that the silica@PVPA-b-PS/PC membrane consisted of silica@PVPA-b-PS, whose PVPA polymerization time was 24 h and PC. Fig. 3(c) shows the surface and cross-section FE-SEM images of the silica@PVPA-b-PS/PC membrane. The surface SEM images and EDX mapping of the membrane showed that core–shell type nanoparticles were uniformly dispersed in the membrane, although there was some aggregation. The observed aggregation was caused by the kneading of the core–shell type nanoparticles and PC when fabricating the membrane; however, the contact points between the particles contribute to the construction of proton-conduction channels, so they do not need to be completely dispersed. In the SEM image of the cross section, the core–shell type nanoparticles were observed to be filled in the membrane as in the surface SEM image. In addition, magnified SEM images of the surface and cross section of the membrane are shown in Fig. S5.† On both the surface and cross section, silica@PVPA-b-PS was approximately filled into the PC, although some voids were present. These results suggest that the core–shell type nanoparticles were continuous in the 3D direction as well as in the through-plane direction, forming proton-conduction channels.
We can fabricate a proton conductive PVPA film by casting PVPA solution on a glass substrate; however, the film cannot remain self-standing when measuring the proton conductivity in a high humidity environment. It causes swelling of PVPA by water absorption. Therefore, constructing a PVPA thin layer on the surface of silica nanoparticles by RAFT PwP provides the advantage that PVPA can be treated as a powder.
The TGA measurement of silica@PVPA was performed in a nitrogen atmosphere from room temperature to 700 °C and a heating rate of 10 °C min−1. After that, the samples were retained for 10 minutes at 700 °C (Fig. 4 and S9†).
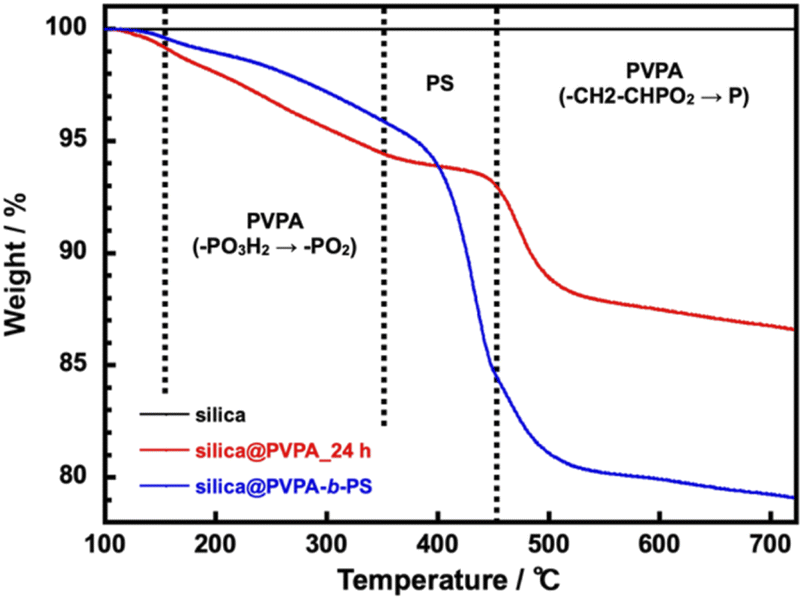 | ||
| Fig. 4 TGA curves of bare silica nanoparticles, silica@PVPA_24 h, and silica@PVPA-b-PS. | ||
The silica nanoparticles used as the core particles did not show a degradation in the measurement temperature range and the weight change after the measurement was 0%; therefore, it can be said that the weight loss of core–shell type nanoparticles was derived from coated polymers. In this system, using DMF as the polymerization solvent instead of butyl acetate (AcOBu) used in the CNC system, the PVPA layer on the surface of silica nanoparticles tended to be thin due to the different adsorption modes of PVPA oligomers (Fig. S10†). From TGA curves, phosphate group dehydration of PVPA (–PO3H2 → –PO2) occurred at 150–350 °C.40 After that a rapid weight loss of PVPA occurs at 450–500 °C, and finally, all but phosphorus decomposes.41 Note that the phosphorus remains on the surface of the silica nanoparticle surface, and the final weight loss ratio does not match the amount of PVPA adsorbed on the surface of the silica nanoparticles. Supplementally, silica@PVPA purified after polymerization before TGA measurement was white, but after TGA measurement it becomes dark green or black due to residual phosphorus atoms. Silica@PVPA_3 h showed 4.1% and silica@PVPA_48 h showed 15.2% weight degradation and the PVPA thickness derived from TGA degradation tended to increase with the polymerization time in all the samples. Hence, we have succeeded in changing the PVPA thickness with the polymerization time of PVPA as shown in Fig. S9.† Additionally, the TGA result of silica@PVPA-b-PS indicated rapid degradation attributed to PS at around 350–450 °C. Moreover, the degradation of PVPA continued to occur at around 450 °C, just after the decomposition of PS. In the experiment, we added excess styrene in the test tube, so that we succeeded in coating a sufficient amount of PS even at 12 h.
The UV-vis spectra of silica@PVPA and silica@PVPA-b-PS are shown in Fig. 5 and S11.† For the measurement, dried silica@PVPA particles were placed in a dedicated powder cell and measured using an integrating sphere unit, and also, the UV-vis spectrum of the RAFT agent (X1) was obtained.
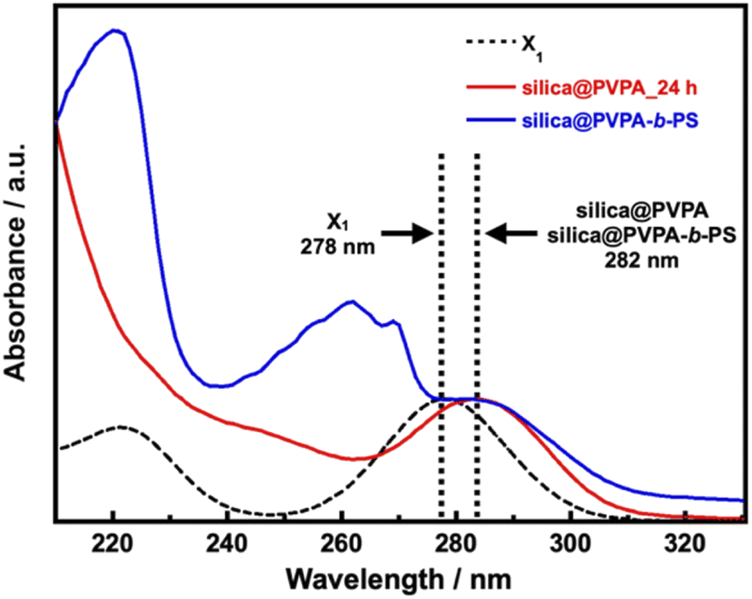 | ||
| Fig. 5 The Kubelka–Munk transformation of the reflectance curves of X1, PS, silica@PVPA_24 h, and silica@PVPA-b-PS. | ||
Then, the UV-vis spectra derived from X1 were aligned to 1.0 with the intensity of the absorption peak. X1 showed a peak of π → π* transition at 278 nm, and the prepared silica@PVPA showed an absorption peak at 281–282 nm, which is very close to X1. Since absorption derived from X1 was also observed from the surface of silica@PVPA washed after polymerization, X1 was involved in the polymerization of PVPA. Hence, it seems that silica@PVPA and silica@PVPA-b-PS were prepared via RAFT PwP. In addition, since silica@PVPA-b-PS showed absorption derived from PS at 220 and 262 nm, the coating of PS was also obvious.42
3.2. Proton conductivities of pelletized core–shell type nanoparticles and the silica@PVPA-b-PS/PC membrane
Proton conductivity was measured using the AC impedance method, and the measurement was taken every 10 °C from 20 °C to 80 °C (humidification condition was 95% RH) and proton conductivities were calculated based on the obtained Cole–Cole plots shown in Fig. S12.† Furthermore, the activation energies of proton conduction were determined using the Arrhenius type plots shown in Fig. 6. Importantly, all the prepared samples showed proton conductivity, suggesting that the 2D proton-conduction channels constructed on the surface of nanoparticles by RAFT PwP were connected to each other, resulting in the construction of 3D proton-conduction channels.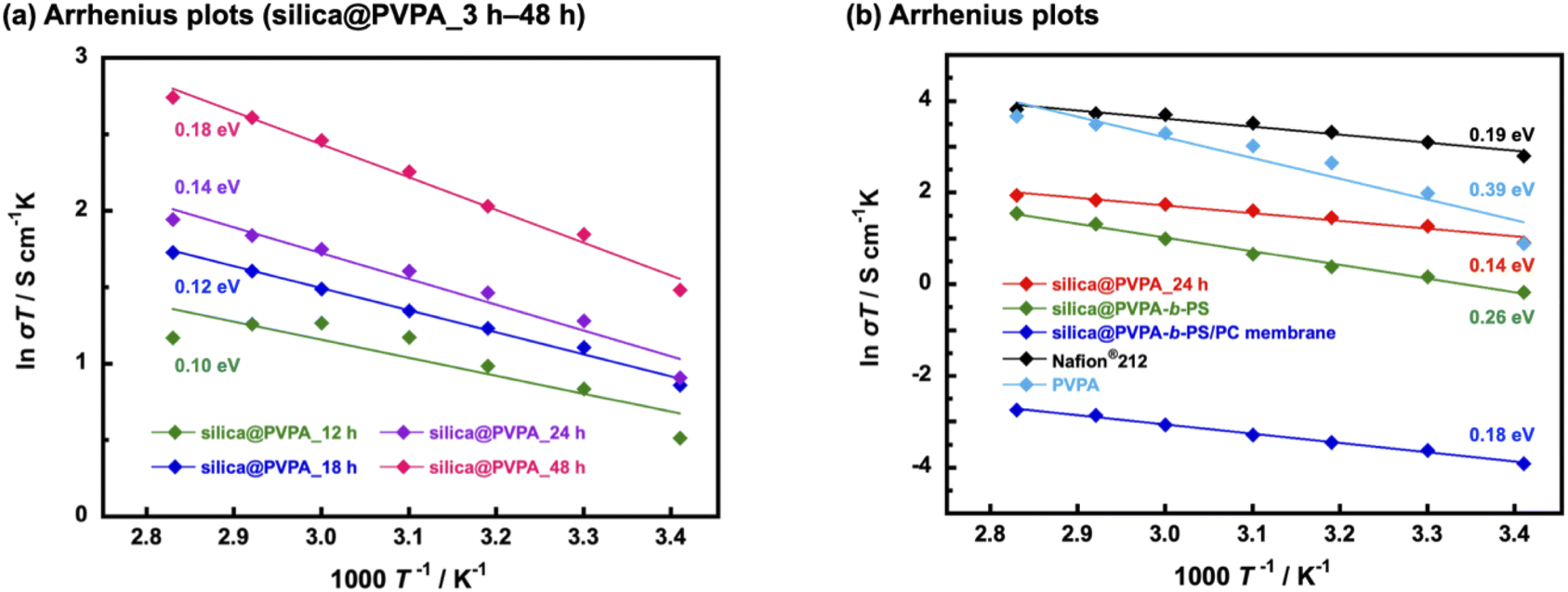 | ||
| Fig. 6 Arrhenius plot of (a) pelletized silica@PVPA with various PVPA polymerization times (12–48 h) and (b) pelletized silica@PVPA_24 h, silica@PVPA-b-PS, silica@PVPA-b-PS/PC membrane, Nafion® 212, and PVPA film. | ||
Firstly, we measured PVPA thickness dependence on the proton conductivity of silica@PVPA (Table 1 and S1†). The proton conductivity of silica@PVPA was greatly improved from 8.9 × 10−4 S cm−1 to 1.5 × 10−2 S cm−1 (20 °C and 95% RH) which depended on the thickness of coated PVPA. It suggested that the conductivity of silica@PVPA varied with the PVPA polymerization time (Fig. 6(a)). Additionally, the proton conductivity showed a correlation with the PVPA thickness in all the measured temperature ranges. Note that silica@PVPA_3 and 6 h were excluded from the discussion because the pelletized samples were brittle and no reliable data were obtained. These pellets showed lower proton conductivity and higher activation energies due to the inadequate PVPA coverage, which prevented the establishment of internal 3D proton-conduction channels between core–shell type nanoparticles.43,44 Silica@PVPA_12 h showed proton conductivity at 20–80 °C, and the proton conductivity increased with increasing temperature. On the other hand, the proton conductivity decreased at 70 °C and 80 °C despite the increase in temperature. This phenomenon can be explained by the next two studies. J. Matsui et al.16 discussed the cause of proton conductivity decrease with increasing temperature at the 2D proton-conduction channels constructed on the glass substrate. In the low-temperature range, the –COOH groups, which were proton sites, and water molecules were arranged at the optimum distance, which enables the construction of 2D proton-conduction channels. On the other hand, the proton conduction mechanism was switched from the Grotthuss-type to the Vehicle-type which is the oxonium ion diffusion mechanism according to increasing temperature. The oxonium ion diffusion mechanism requires a larger amount of energy for proton conduction than the Grotthuss-type, and the proton conductivity decreased against the temperature change. Furthermore, T. Ichikawa et al.45 also discussed the hydration ability of materials. Silica@PVPA_12 h was considered to have evaporated water molecules with increasing temperature due to the insufficient amount of PVPA coverage, even though the nanoparticle surface was covered with PVPA. From the above previous studies, it seems that silica@PVPA_12 h with insufficient PVPA coverage had difficulty in proton conduction in the high temperature range. In contrast, silica@PVPA_18–48 h had sufficient PVPA coverage to prevent the evaporation of absorbed water at higher temperatures. The proton conductivity of silica@PVPA_18–48 h improved according to the temperature increase, and no significant decrease in proton conductivity was confirmed such as in the other nanoparticles.
| Sample | σ/S cm−1 | E a/eV (95% RH) |
|---|---|---|
| Silica@PVPA_12 h | 9.1 × 10−3 (80 °C) | 0.10 |
| Silica@PVPA_18 h | 1.6 × 10−2 (80 °C) | 0.12 |
| Silica@PVPA_24 h | 2.0 × 10−2 (80 °C) | 0.14 |
| Silica@PVPA_48 h | 4.4 × 10−2 (80 °C) | 0.18 |
| Silica@PVPA-b-PS | 1.3 × 10−2 (80 °C) | 0.26 |
| Silica@PVPA-b-PS/PC membrane | 1.8 × 10−4 (80 °C) | 0.18 |
Furthermore, we discussed the activation energy of proton conduction of silica@PVPA. The activation energy of each silica@PVPA was as low as 0.10–0.18 eV. Notably, this proton conduction with low activation energy is due to the high density of PVPA coverage on the surface of nanoparticles by RAFT PwP, which results in the construction of ideal 2D proton-conduction channels. The proton conduction mechanism of these silica@PVPA samples was Grotthuss-type which was a proton hopping mechanism mediated by hydrogen bonding. According to these results, proton conduction in silica@PVPA is considered to be simultaneous conduction between silanol groups on the surface of silica nanoparticles and P–OH groups of PVPA and conduction inside PVPA.46 The activation energy of proton conduction of silica@PVPA showed a correlation with proton conductivity, increasing with increasing proton conductivity (48 h (0.18 eV) > 24 h (0.14 eV) > 18 h (0.12 eV) > 12 h (0.10 eV)). Silica@PVPA_48 h showed a relatively higher activation energy, despite which the highest proton conductivity was achieved. This suggests that, starting from silica@PVPA_12 h, which has the lowest activation energy, as the PVPA coating thickness increases, the ratio of the Vehicle-type proton conduction like the conduction of inside the PVPA was also increased. This result can be explained by the PVPA cast film on the glass substrate showing a large activation energy for proton conduction compared with core–shell type nanoparticles of silica@PVPA (Fig. 6(b)). Also considering the high activation energies of silica@PVPA_3 and 6 h, we concluded that the activation energy increases with insufficient or excessive PVPA coverage. Based on the above, take into account the activation energy increasing due to PS coating and kneading with PC, we decided that silica@PVPA_24 h was the optimum sample because of the achieved high proton conductivity, low activation energy, and lower ratio of the Vehicle-type proton conduction attributed to thin PVPA layer thickness. However, although CNC@PVPA and CNC@PVPA-b-PS achieved low activation energies of around 0.1 eV, the core–shell type nanoparticles fabricated in this system do not achieve as low activation energy and high proton conductivity as the CNC system. It seems that the effect of geometry between silica nanoparticles and CNC. Compared to spherical silica nanoparticles, rod-shaped CNCs have a larger contact area between 2D proton-conduction channels on the surface of core–shell nanoparticles, resulting in higher proton conductivity and lower activation energy.
We confirmed that the proton conductivity of silica@PVPA-b-PS and silica@PVPA-b-PS/PC membranes was lower compared with that of silica@PVPA_24 h (Fig. 6(b)). The lower proton conductivity was caused by PS and PC. Since PS does not have a proton conduction site in its structure, coating PS on the surface of silica@PVPA inhibits proton conduction between the particles, and the proton conductivity decreased. The proton conductivity at 20 °C and 95% RH fell from 8.5 × 10−3 S cm−1 (silica@PVPA_24 h) to 2.9 × 10−3 S cm−1 (silica@PVPA-b-PS). On the other hand, at 80 °C and 95% RH, silica@PVPA-b-PS showed a high proton conductivity of 1.3 × 10−2 S cm−1, which was similar to the conductivity of silica@PVPA_24 h. Based on the similar proton conductivities of silica@PVPA and silica@PVPA-b-PS, we succeeded in coating the minimum PS thickness providing affinity between the core–shell type nanoparticles and PC. A freestanding silica@PVPA-b-PS/PC membrane was prepared by kneading with PC, and the proton conductivity of the membrane significantly dropped, because of PC which also does not have proton conduction sites. Although we achieved a freestanding membrane with a very high concentration of core–shell type nanoparticles (nanoparticles/PC = 50/50 (w/w)), it had a low proton conductivity due to the large distance between the nanoparticles.47
The activation energies of these silica@PVPA-b-PS and silica@PVPA-b-PS/PC membranes were also low as those of silica@PVPA.25 These low-activation energies (<0.4 eV) suggested that the proton conduction mechanism was Grotthuss-type by proton hopping through hydrogen bonding. However. the activation energy of silica@PVPA-b-PS showed a higher value than silica@PVPA_24 h due to the lower proton conductivity in the low-temperature range. In contrast, the activation energy of the silica@PVPA-b-PS/PC membrane was close to the value of silica@PVPA_24 h. The lower activation energy of silica@PVPA-b-PS/PC membrane was caused that a large inter-particle distance compared with the pellet state, resulting in the proton conduction was hardly and the changes in proton conductivity was limited.
The humidity dependence of proton conductivities was measured at 80 °C and under various relative humidity conditions (Fig. 7(a) and Table S2†). The proton conductivity of all samples has a positive correlation with humidity environments and silica@PVPA and silica@PVPA-b-PS showed proton conductivity above 30% RH and the silica@PVPA-b-PS/PC membrane showed proton conductivity above 60% RH.48 The proton conductivity of silica@PVPA-b-PS improved from 5.8 × 10−6 S cm−1 (30% RH) to 1.5 × 10−2 S cm−1 (95% RH). Furthermore, silica@PVPA and silica@PVPA-b-PS achieved nearly the same proton conductivity at 80 °C and 95% RH, and also the conductivities of each sample were very close even with humidity change. The loss of proton conductivity between silica@PVPA_24 h and silica@PVPA-b-PS was minimal in spite of the fact that the coating of PS onto the surface of silica@PVPA provides the reproducibility of proton conductivity as discussed below. It can be said that silica@PVPA-b-PS is coated with the minimum amount of PS that can function as a protective layer. Moreover, the fact that the silica@PVPA-b-PS/PC membrane showed proton conductivity only above 60% RH suggests that water molecules strongly contribute more to proton conduction in the silica@PVPA-b-PS/PC membrane than in pelletized core–shell type nanoparticles.49,50
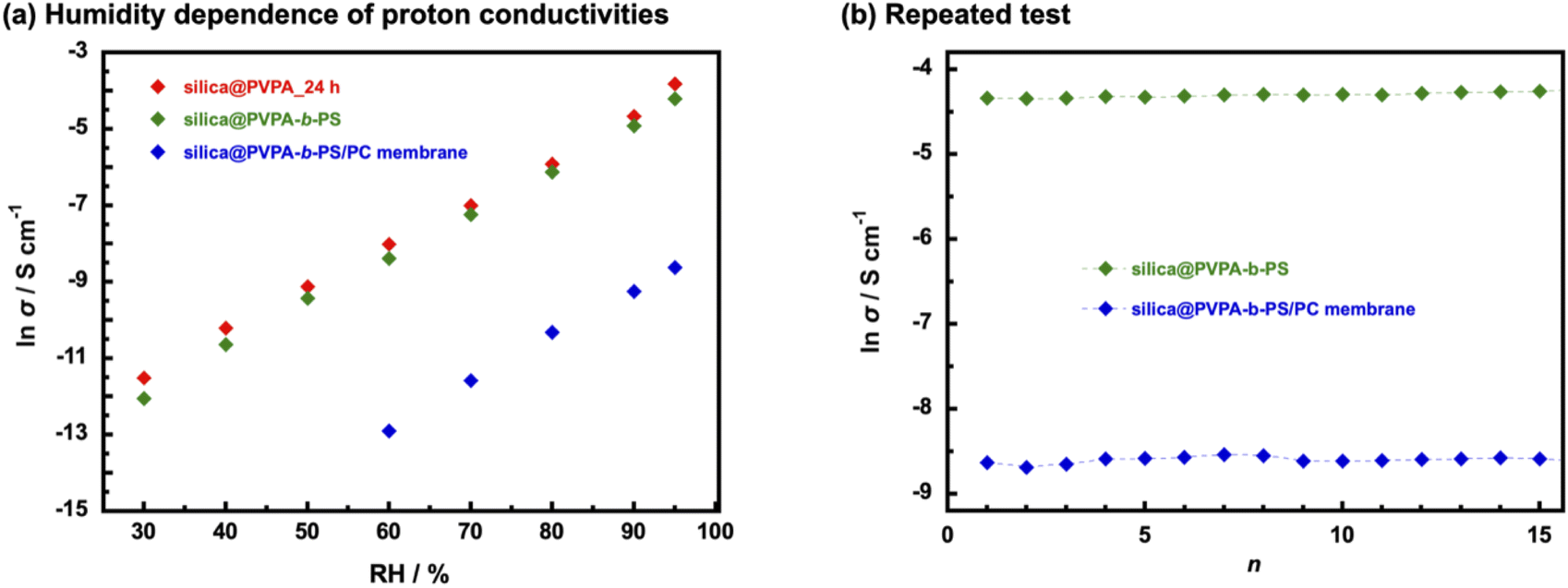 | ||
| Fig. 7 (a) Humidity dependent proton conductivities and (b) repeated test of temperature- and RH-dependent proton conductivities of pelletized silica@PVPA_24 h, silica@PVPA-b-PS, and silica@PVPA-b-PS/PC membranes. | ||
In order to evaluate the reliability of proton conductivity and establish the necessity of PS used as a protective layer of core–shell type nanoparticles, we performed temperature- and RH-dependent repeated tests. From our previous work, the core–shell type nanoparticles coated only with proton-conducting polymers could not show reproducible proton conductivity.25 It was because proton-conducting polymers are deliquescent and swell on the surface of nanoparticles, resulting in the collapse of the proton-conduction channels. Therefore, repeated tests were performed only with silica@PVPA-b-PS and silica@PVPA-b-PS/PC membranes. The tests were performed under a high-temperature and high-humidity environment (80 °C and 95% RH) and a room-temperature and low-humidity environment (20 °C and 50% RH).51 Supplementally, the environments in which the repeated test was the upper and lower limit of the controlling range of the small environmental test chamber. Fig. 7(b) shows the proton conductivity when the temperature and humidity were increased to 80 °C and 95% RH for each cycle. In contrast to core–shell type nanoparticles coated only with proton-conducting polymers, silica@PVPA-b-PS and silica@PVPA-b-PS/PC membranes showed good stability of proton conductivity in each cycle. The proton conductivity of silica@PVPA-b-PS in the first cycle was 1.3 × 10−2 S cm−1 and in the 15th cycle was 1.4 × 10−2 S cm−1 at 80 °C and 95% RH. It can be attributed to that PS suppressed the excessive moisture absorption into the PVPA, resulting in high reproducibility of proton conductivity. In our previous study, we already reported the function of PS in core–shell type nanoparticles by QCM measurement.19 Since the hydrophobic PS defines the space in the shell, PVPA can only absorb moisture in the space between the silica nanoparticles and PS. This study also supports the previous results, the 2D proton-conduction channels constructed on the surface of silica nanoparticles have remained, and the proton conductivities did not change much even after the wet/dry cycle. Similarly in the silica@PVPA-b-PS/PC membrane, PS and PC which were the hydrophobic polymers gave the hysteresis of proton conductivity.
In other words, we revealed that the stability of proton conductivity was dramatically improved by coating with a hydrophobic polymer.
4. Conclusions
A core-shell type nanoparticle-filled polymer electrolyte silica@PVPA-b-PS/PC membrane was successfully prepared by filing with PVPA-b-PS coated silica nanoparticles. We have succeeded in constructing 2D proton-conduction channels at the surface of silica nanoparticles by the RAFT PwP method and 3D proton-conduction channels by filling them with the core–shell type nanoparticles. The proton conductivity of core–shell nanoparticles and membranes was evaluated from multiple aspects such as temperature change, humidity change, and repeated tests. Both proton conductivity and activation energy of proton conduction of core–shell nanoparticles increased and it was difficult to realize both high proton conductivity and low activation energy when PVPA thickness was excessive. Among them, silica@PVPA_24 h had both high proton conductivity and low activation energy (2.0 × 10−2 S cm−1 and 0.14 eV). Moreover, the PS provided stability to the 2D proton-conduction channels due to limited space in which PVPA can absorb moisture. Although the proton conductivity of silica@PVPA-b-PS and silica@PVPA-b-PS/PC membranes was inferior to that of silica@PVPA, the proton conductivities of silica@PVPA-b-PS and silica@PVPA-b-PS/PC membranes were stable even in the 15th cycle in the repeated test. These results were important for proton conductive polymer-coated core–shell type nanoparticles and PEMs based on them.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the Cooperative Research Program of the Network Joint Research Center for Materials and Devices: Alliance for Open Innovation Bridging Human, Environment and Materials (20201076 and 20211084), and a Grant-in-Aid for Scientific Research B (JP18H01717). This work was partly supported by the Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, Grant no. JPMXP09S20NU0019.Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- D. J. Kim, M. J. Jo and S. Y. Nam, J. Ind. Eng. Chem., 2015, 21, 36–52 CrossRef CAS.
- H.-W. Wu, Appl. Energy, 2016, 165, 81–106 CrossRef CAS.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- X. Yan, H. Li, C. Lin, J. Chen, A. Han, S. Shen and J. Zhang, Sustainable Energy Fuels, 2020, 4, 772–778 RSC.
- K. Krishnan, H. Iwatsuki, M. Hara, S. Nagano and Y. Nagao, J. Mater. Chem. A, 2014, 2, 6895–6903 RSC.
- Y. Buchmüller, A. Wokaun and L. Gubler, J. Mater. Chem. A, 2014, 2, 5870–5882 RSC.
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O'Hayre, Nat. Energy, 2019, 4, 230–240 CrossRef CAS.
- Y. Nagao, Sci. Technol. Adv. Mater., 2020, 21, 79–91 CrossRef CAS PubMed.
- M. Zatoń, J. Rozière and D. J. Jones, Sustainable Energy Fuels, 2017, 1, 409–438 RSC.
- R. Wang, S. Liu, L. Wang, M. Li and C. Gao, Nanomaterials, 2019, 9, 869 CrossRef PubMed.
- V. Guccini, A. Carlson, S. Yu, G. Lindbergh, R. W. Lindström and G. Salazar-Alvarez, J. Mater. Chem. A, 2019, 7, 25032–25039 RSC.
- A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 2034–2036 CrossRef CAS.
- J. Matsui, H. Miyata, Y. Hanaoka and T. Miyashita, ACS Appl. Mater. Interfaces, 2011, 3, 1394–1397 CrossRef CAS PubMed.
- T. Sato, Y. Hayasaka, M. Mitsuishi, T. Miyashita, S. Nagano and J. Matsui, Langmuir, 2015, 31, 5174–5180 CrossRef CAS PubMed.
- T. Sato, M. Tsukamoto, S. Yamamoto, M. Mitsuishi, T. Miyashita, S. Nagano and J. Matsui, Langmuir, 2017, 33, 12897–12902 CrossRef CAS PubMed.
- Y.-F. Lin, C.-Y. Yen, C.-C. M. Ma, S.-H. Liao, C.-H. Lee, Y.-H. Hsiao and H.-P. Lin, J. Power Sources, 2007, 171, 388–395 CrossRef CAS.
- T. Ogawa, T. Aonuma, T. Tamaki, H. Ohashi, H. Ushiyama, K. Yamashita and T. Yamaguchi, Chem. Sci., 2014, 5, 4878–4887 RSC.
- K. Shito, J. Matsui, Y. Takahashi, A. Masuhara and T. Arita, Chem. Lett., 2018, 47, 9–12 CrossRef CAS.
- W. Zhou, Y. Zhang, J. Wang, H. Li, W. Xu, B. Li, L. Chen and Q. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 46767–46778 CrossRef CAS.
- T. Arita, Chem. Lett., 2013, 42, 801–803 CrossRef CAS.
- W. Peng, Y. Cai, L. Fanslau and P. Vana, Poly. Chem., 2021, 12, 6198–6229 RSC.
- T. Arita, Nanoscale, 2010, 2, 2073–2076 RSC.
- T. Arita, K.-i. Moriya, K. Minami, T. Naka and T. Adschiri, Chem. Lett., 2010, 39, 961–963 CrossRef CAS.
- K. Tabata, T. Nohara, K. Koseki, K. Umemoto, R. Sato, S. Rodbuntum, Y. Suzuki, R. Shimada, S. Asakura, T. Arita and A. Masuhara, Jpn. J. Appl. Phys., 2020, 59, SIIH01-01 CrossRef.
- T. Nohara, K. Koseki, K. Tabata, R. Shimada, Y. Suzuki, K. Umemoto, M. Takeda, R. Sato, S. Rodbuntum, T. Arita and A. Masuhara, ACS Sustainable Chem. Eng., 2020, 8, 14674–14678 CrossRef CAS.
- K. Koseki, T. Arita, K. Tabata, T. Nohara, R. Sato, S. Nagano and A. Masuhara, ACS Sustainable Chem. Eng., 2021, 9, 10093–10099 CrossRef CAS.
- T. Nohara, T. Arita, K. Tabata, T. Saito, R. Shimada, H. Nakazaki, Y. Suzuki, R. Sato and A. Masuhara, ACS Appl. Mater. Interfaces, 2022, 14, 8353–8360 CrossRef CAS.
- A. Kaltbeitzel, S. Schauff, H. Steininger, B. Bingol, G. Brunklaus, W. Meyer and H. Spiess, Solid State Ionics, 2007, 178, 469–474 CrossRef CAS.
- A. Aslan and A. Bozkurt, J. Power Sources, 2012, 217, 158–163 CrossRef CAS.
- M. Tsuksamoto, K. Ebata, H. Sakiyama, S. Yamamoto, M. Mitsuishi, T. Miyashita and J. Matsui, Langmuir, 2019, 35, 3302–3307 CrossRef CAS PubMed.
- T. Wagner, A. Manhart, N. Deniz, A. Kaltbeitzel, M. Wagner, G. Brunklaus and W. H. Meyer, Macromol. Chem. Phys., 2009, 210, 1903–1914 CrossRef CAS.
- Z. Zheng, M. Mounsamy, N. Lauth-de Viguerie, Y. Coppel, S. Harrisson, M. Destarac, C. Mingotaud, M. L. Kahn and J.-D. Marty, Poly. Chem., 2019, 10, 145–154 RSC.
- H. Yabu, S. Nagano and Y. Nagao, RSC Adv., 2018, 8, 10627–10632 RSC.
- A. Aslan, K. Gölcük and A. Bozkurt, J. Polym. Res., 2012, 19, 1–8 CrossRef CAS.
- Y. Nagao, J. Matsui, T. Abe, H. Hiramatsu, H. Yamamoto, T. Miyashita, N. Sata and H. Yugami, Langmuir, 2013, 29, 6798–6804 CrossRef CAS PubMed.
- B. Smitha, S. Sridhar and A. A. Khan, Macromolecules, 2004, 37, 2233–2239 CrossRef CAS.
- B. Özbek and Ş. Ünal, Korean J. Chem. Eng., 2017, 34, 1992–2001 CrossRef.
- R. Sato, T. Arita, R. Shimada, T. Nohara, K. Tabata, K. Koseki, K. Umemoto and A. Masuhara, Cellulose, 2020, 28, 871–879 CrossRef.
- J. Souquet-Grumey, R. Perrin, J. Cellier, J. Bigarré and P. Buvat, J. Membr. Sci., 2014, 466, 200–210 CrossRef CAS.
- C. A. Wilkie, Polym. Degrad. Stab., 1999, 66, 301–306 CrossRef CAS.
- A. Z. Wilczewska and K. H. Markiewicz, Macromol. Chem. Phys., 2014, 215, 190–197 CrossRef CAS.
- G. He, L. Nie, X. Han, H. Dong, Y. Li, H. Wu, X. He, J. Hu and Z. Jiang, J. Power Sources, 2014, 259, 203–212 CrossRef CAS.
- K. Krishnan, H. Iwatsuki, M. Hara, S. Nagano and Y. Nagao, J. Phys. Chem. C, 2015, 119, 21767–21774 CrossRef CAS.
- T. Kobayashi, Y. X. Li, Y. Hirota, A. Maekawa, N. Nishiyama, X. B. Zeng and T. Ichikawa, Macromol. Rapid Commun., 2021, 42, e2100115 CrossRef PubMed.
- L. Wang, X. Zuo, A. Raut, R. Isseroff, Y. Xue, Y. Zhou, B. Sandhu, T. Schein, T. Zeliznyak, P. Sharma, S. Sharma, B. S. Hsiao and M. H. Rafailovich, Sustainable Energy Fuels, 2019, 3, 2725–2732 RSC.
- J.-H. Won, H.-J. Lee, K.-S. Yoon, Y. T. Hong and S.-Y. Lee, Int. J. Hydrogen Energy, 2012, 37, 9202–9211 CrossRef CAS.
- Y. Ono, R. Goto, M. Hara, S. Nagano, T. Abe and Y. Nagao, Macromolecules, 2018, 51, 3351–3359 CrossRef CAS.
- H. Huang, S. Xu, L. Zhang, J. Fan, H. Li and H. Wang, Sustainable Energy Fuels, 2021, 5, 230–245 RSC.
- Y. Kim, K. Ketpang, S. Jaritphun, J. S. Park and S. Shanmugam, J. Mater. Chem. A, 2015, 3, 8148–8155 RSC.
- F. Yang, G. Xu, Y. Dou, B. Wang, H. Zhang, H. Wu, W. Zhou, J.-R. Li and B. Chen, Nat. Energy, 2017, 2, 877–883 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00450j |
| This journal is © The Royal Society of Chemistry 2022 |