
Polyclonal aptamer libraries as binding entities on a graphene FET based biosensor for the discrimination of apo- and holo-retinol binding protein 4†
Ann-Kathrin
Kissmann‡
a,
Jakob
Andersson‡
*b,
Anil
Bozdogan‡
bc,
Valerie
Amann
a,
Markus
Krämer
a,
Hu
Xing
a,
Heinz Fabian
Raber
a,
Dennis H.
Kubiczek
a,
Patrik
Aspermair
 b,
Wolfgang
Knoll
bd and
Frank
Rosenau
*a
b,
Wolfgang
Knoll
bd and
Frank
Rosenau
*a
aInstitute of Pharmaceutical Biotechnology, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: frank.rosenau@uni-ulm.de
bAIT Austrian Institute of Technology GmbH, Giefinggasse 4, 1210 Vienna, Austria. E-mail: jakob.andersson@ait.ac.at
cCEST Kompetenzzentrum für Elektrochemische Oberflächentechnologie GmbH, Viktor Kaplan Straße 2, Wiener Neustadt, Austria
dDanube Private University, Steiner Landstraße 124, 3500 Krems an der Donau, Austria
First published on 29th April 2022
Abstract
Oligonucleotide DNA aptamers represent an emergently important class of binding entities towards as different analytes as small molecules or even whole cells. Without requiring the canonical isolation of individual aptamers following the SELEX process, the focused polyclonal libraries prepared by this in vitro evolution and selection can directly be used to label their dedicated targets and to serve as binding molecules on surfaces. Here we report the first instance of a sensor able to discriminate between loaded and unloaded retinol-binding protein 4 (RBP4), an important biomarker for the prediction of diabetes and kidney disease. The sensor relies on two aptamer libraries tuned such that they discriminate between the protein isoforms, requiring no further sample labelling to detect RBP4 in both states. The evolution, binding properties of the libraries and the functionalization of graphene FET sensor chips are presented as well as the functionality of the resulting biosensor.
New conceptsCovalent surface modification of GFETs with nanoscale affinity entities like aptamers has recently been demonstrated as advantageous over non-covalent functionalization strategies. The stable and robust surfaces resulting from covalent grafting of specific binding molecules are expected to become versatile and cheap devices for biosensing applications. In this work, we show that polyclonal aptamer libraries against the two isoforms of retinol-binding protein 4 (RBP4) can be used for GFET functionalization to discriminate retinol-loaded or empty RBP4. Empty RBP4 can serve as a marker during diabetes development, which is currently not yet used in clinical routine. Thus, a respective biosensor represents not only an attractive tool for early diabetes diagnostics, but also demonstrates the power of aptamers as highly sensitive sensing interfaces of GFET-based sensors in future sensing (clinical) diagnostic platforms. We believe that due to the simplicity of library evolution and efficiency of covalent chemical grafting onto the graphene layers of GFETs will open new avenues for these electronic devices to become standard (bio-) sensors for a broad range of applications in medical diagnostics and environmental health monitoring. |
Introduction
Retinol binding protein 4 (RBP4) is a serum protein, which belongs to the lipocalin ligand binding protein family.1 It serves as a transporter in the blood for all-trans-retinol (vitamin A alcohol) and other retinoids like retinal or retinoid acid from hepatic retinoid stores to peripheral tissues of the body.2 RBP4 is expressed in the liver and circulates in blood serum,3 where normal levels of the protein can range from 40 to 60 μg mL−1 in humans.4 Under physiological conditions, mainly ligand-bound RBP4 (holo-RBP4), approximately 90% of blood RBP4,5 circulates in an 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 complex with transthyretin (TTR) (Scheme 1) and all-trans-retinol as its stability is further enhanced and RBP4 is prevented from extensive loss throughout glomerular filtration and catabolism in the kidneys.6,7 Under healthy conditions, the RBP4 expression is tightly regulated and after the transport of retinol to its target cells the remaining unbound RBP4 (apo-RBP4) is rapidly filtered out and the protein is cleared from the serum.8 However, the amount of RBP4 can be increased in the serum as a consequence of diseases like obesity,9 chronic kidney disease,10 insulin resistance,11 and type 2 diabetes mellitus.12 Depending on the level of elevation of the concentration of a certain RBP4 isoform or disbalances of RBP4 to retinol ratios, various reactions affecting the human health state can be triggered.13 Recent studies showed, that especially apo-RBP4 contributes in adipose tissue to the development of an inflammatory state, which may result in insulin resistance.14,15 Likewise, patients suffering from type 2 diabetes mellitus may exhibit moderate plasma RBP4 levels, but their level of free RBP4 in proportion to retinol is increased indicating an imbalance in ratios as a crucial factor.16 Nowadays, several enzyme-linked immunosorbent assays (ELISAs) are commercially available the for detection of RBP4 in human serum, but they lack accuracy and are insufficient for the quantification of high concentrations of RBP4.17 Hence, quantitative western blotting is used as the standard procedure for the detection of serum RBP4, especially in insulin-resistant states.12 Typically only the amount of total RBP4 is measured and no further differentiation in isoforms is made. In order to overcome these restrictions and to create a methodology allowing to discriminate between apo- and holo-RBP4 the development of specific binding molecules with distinct affinities either for the apo- or the holo-protein would be an attractive amendment of the current existing RBP4 specific diagnostic technologies.
:1:1 complex with transthyretin (TTR) (Scheme 1) and all-trans-retinol as its stability is further enhanced and RBP4 is prevented from extensive loss throughout glomerular filtration and catabolism in the kidneys.6,7 Under healthy conditions, the RBP4 expression is tightly regulated and after the transport of retinol to its target cells the remaining unbound RBP4 (apo-RBP4) is rapidly filtered out and the protein is cleared from the serum.8 However, the amount of RBP4 can be increased in the serum as a consequence of diseases like obesity,9 chronic kidney disease,10 insulin resistance,11 and type 2 diabetes mellitus.12 Depending on the level of elevation of the concentration of a certain RBP4 isoform or disbalances of RBP4 to retinol ratios, various reactions affecting the human health state can be triggered.13 Recent studies showed, that especially apo-RBP4 contributes in adipose tissue to the development of an inflammatory state, which may result in insulin resistance.14,15 Likewise, patients suffering from type 2 diabetes mellitus may exhibit moderate plasma RBP4 levels, but their level of free RBP4 in proportion to retinol is increased indicating an imbalance in ratios as a crucial factor.16 Nowadays, several enzyme-linked immunosorbent assays (ELISAs) are commercially available the for detection of RBP4 in human serum, but they lack accuracy and are insufficient for the quantification of high concentrations of RBP4.17 Hence, quantitative western blotting is used as the standard procedure for the detection of serum RBP4, especially in insulin-resistant states.12 Typically only the amount of total RBP4 is measured and no further differentiation in isoforms is made. In order to overcome these restrictions and to create a methodology allowing to discriminate between apo- and holo-RBP4 the development of specific binding molecules with distinct affinities either for the apo- or the holo-protein would be an attractive amendment of the current existing RBP4 specific diagnostic technologies.
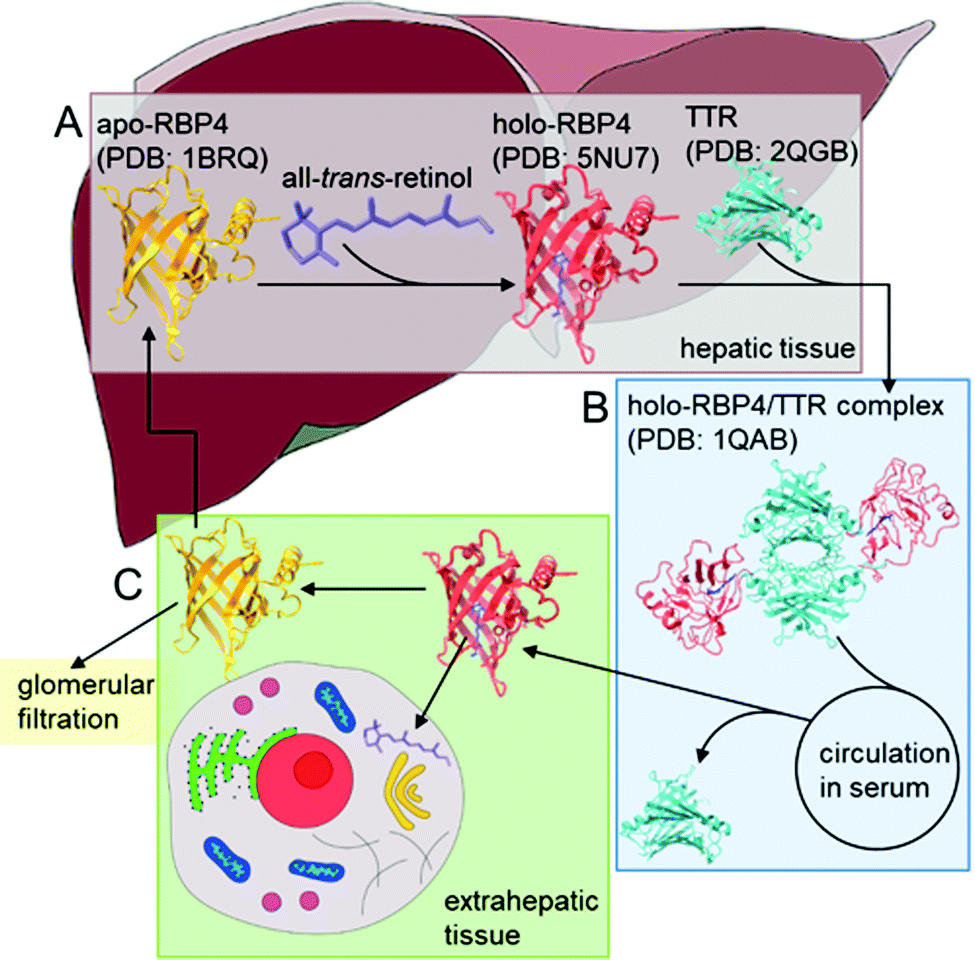 | ||
| Scheme 1 RBP4-mediated transport of all-trans-retinol in blood serum from stores in the liver to extrahepatic peripheral tissue. (A) Secretion of RBP4 from the rough endoplasmatic reticulum of hepatocytes to the blood serum. Binding of apo-RBP4 with retinol and subsequent complexation of holo-RBP4 with transthyretin (TTR) forms a 1:1:1 holo-RBP4/TTR complex. Release into the serum is mediated by retinol availability, the conformational change upon retinol-binding is supposed to trigger the release. (B) Delivery of retinol from hepatic retinoid stores to peripheral extrahepatic tissues during periods of inadequate vitamin A intake. Transport of the holo-RBP4/TTR complex in blood serum to target cells, there the complex is cleaved to release holo-RBP4. (C) RBP4-associated retinol uptake by peripheral tissue due to specific interactions of holo-RBP4 with RBP4-receptors on the surface of target cells. Free RBP4 is either cleared by glomerular filtration or returned to hepatic stores afterwards. | ||
Since their introduction more than 20 years ago, nucleic acid aptamers have become serious alternatives, which offer considerable additional technical options making them increasingly attractive for different applications compared to antibodies or antibody derivatives, as they can serve as promising binding molecules in techniques requiring highly specific detection and quantification of target molecules.18 Aptamers are single-stranded oligonucleotides like RNA or single-stranded DNA (ssDNA) with surprising physical and chemical stability, high specificity and affinity in combination with low overall immunogenicity, they can acquire different secondary and tertiary structures and are capable to bind defined targets.19 High-affinity aptamers can be evolved and isolated from large random sequence libraries. In this selection process, performed completely in vitro, called systematic evolution of ligands by exponential enrichment (SELEX), repeated rounds of target binding and PCR mediated amplification aptamers with binding affinity are successively enriched (Scheme 2).20 Additionally, to reduce the sequence space of the random nucleotide library, an initial elimination of nonspecific oligonucleotides by a counter selection using only the target carrier material prior to the selection process leads to a so called ‘counter library’ with enhanced selectivity. Counter-selections against the carrier material before each round of selection also enable increased specificity towards the target protein, since otherwise in the selection step aptamers would not only be selected against the immobilized target, but also against its carrier material. Aptamers can be selected against a wide range of target structures including proteins,21 whole cells, and microorganisms,22,23 small molecules and chemical compounds like metal ions.24 They can not only be used in diverse fields such as diagnostics,25 biomarker discovery,26 imaging agents,27 drug delivery,28 and as pharmaceutical compounds in molecular therapy,29,30 but they are also attractive molecules for the construction of binding entities in technical devices like electronic biosensors31 on which predominantly antibodies or antibody derivatives have been used.32–34
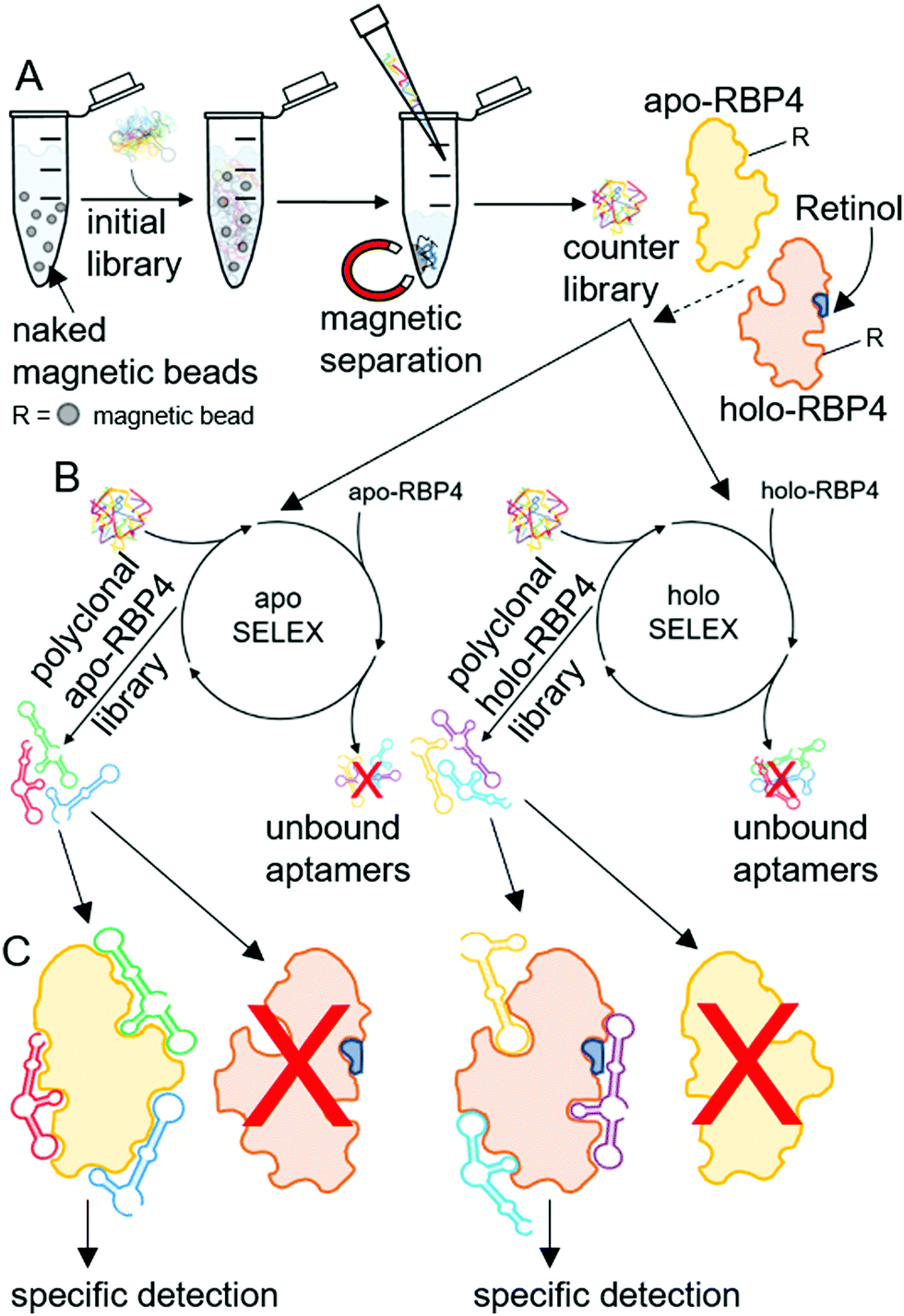 | ||
| Scheme 2 SELEX based evolution of focused aptamer libraries for specific detection of apo- or holo-RBP4. (A) Initial counter selection by incubation of an initial aptamer library (∼6 × 1014 individual aptamers), containing 40 randomized nucleotides flanked by two primer binding sites, with naked Dynabeads® M-280 tosyl-activated resulting in an aptamer ‘counter library’ with reduced amounts of aptamers with specificity against the naked beads. (B) Selection of specific polyclonal aptamer libraries by SELEX against apo- or holo-RBP4. Reduction of sequence diversity by incubating the counter selected aptamer library first with naked beads and subsequent with the target proteins (bound to Dynabeads® M-280 tosyl-activated (indicated as ‘R’)) and aptamers exhibiting an adequate three-dimensional structure bind to the targets. The remaining unbound aptamers are subsequently removed, the bound aptamers are then eluted from the target proteins, amplified by ePCR and the undesired complementary strands are removed prior to the next SELEX round. (C) Specific target detection by binding of focused aptamer libraries. Exclusive binding of polyclonal apo-RBP4 aptamers to apo-RBP4 and of holo-RBP4 aptamers to holo-RBP4. | ||
Recently, we have shown that as a significant simplification of the aptamer textbook procedures, where the overall aim is to isolate and characterize individual aptamer sequences before they are used for applications, already focused polyclonal libraries can not only be used directly after sufficient enrichment, but can even outperform single aptamers.23 Thus, the use of focused polyclonal aptamer libraries can be expected to be advantageous due to higher precision based on the larger sequence space available for productive target recognition and increased performance in sensor technologies and diagnostics.
In order to specifically detect and quantify serum RBP4 such focused polyclonal aptamer libraries were evolved in an iterative SELEX process with recombinant purified target RBP4 proteins immobilized on magnetic particles (Scheme 2) in combination with fluorescence monitoring of the success of this molecular evolution process (“FluMag-SELEX”).35 Therefore, each round of aptamer evolution started by a counter-selection step using naked magnetic beads prior to selection steps using apo- or holo-RBP4 coated magnetic beads. The aim was to create the possibility to differentiate between RBP4 isoforms (i.e. the apo- and holo-RBP4) and therefore two independent focused aptamer libraries were successfully evolved simultaneously against both apo- and holo-RBP4. The selective and precise quantification of RBP4 isoforms plays an important role in early diagnostics and in the reduction of either the level of serum apo- or holo-RBP4, as well as balancing impairments in RBP4 to retinol ratios. Exclusive specific binding of both polyclonal aptamer libraries to their dedicated targets was used for the subsequent development of aptamer-based apo-RBP4 and holo-RBP4 biosensors based on electrolyte-gated field effect transistors (EG-FETs).
EG-FET devices have been used extensively in biosensing applications, for example, to detect microRNA,36 DNA37 biomarkers for heart failure,38 cancer,39 biotin in the pM range,40 and urea.41 We have also recently developed a highly sensitive method of detecting the E7-protein for human papillomavirus implicated in carcinogenesis.31
One of the primary benefits of EG-FET sensing devices is that they do not require target labelling with fluorescent or redox probes. Both the sensor chips and the read-out equipment are low cost and therefore highly suitable for use in point-of-care settings and in low socioeconomic areas where expertise and laboratory environments are not readily available.
An EG-FET is based on the same principle as a metal oxide FET: a source and drain electrode are separated by a semiconducting channel (Scheme 3). By applying a voltage at the gate electrode, channel conductivity is changed, modulating the current flowing between the source and drain electrodes. Biorecognition elements (antibodies, antigens, proteins or aptamers) can be deposited on the channel and when the target analyte binds, the change in dielectric layers at the interface between the channel and the electrolyte changes. As a result, the mobility of charge carriers in the channel is also changed.42 At a constant gate voltage, binding events on the channel can therefore be observed as a change in the source–drain current (IDS), therefore changing the gate voltage coupling to the channel will alter the source–drain current. We deposited the previously developed aptamer libraries on reduced graphene oxide field-effect transistors (rGO-FETs, a sub-class of EG-FET devices) using a previously developed methods.31
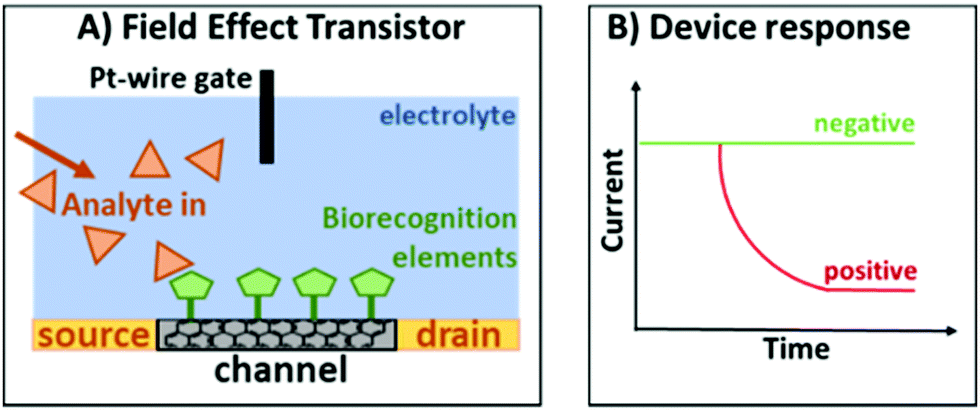 | ||
| Scheme 3 (A) Typical configuration of an EG-FET. (B) Response of the device when there is no target analyte (green) and when the target analyte is present (red). | ||
This platform also allows simultaneous optical and electronic sensing when the platinum wire gate electrode is replaced with a planar gold surface, achieving additional insight.43
Results and discussion
Recombinant RBP4 production in E. coli and purification from cell extracts
Competent cells of the expression strain E. coli BL21 (DE3) were transformed freshly prior to RBP4 over-expression and grown for 5 h after induction with IPTG, and the cell growth was monitored photometrically. After its biotechnological production, cell harvest, and cell lysis almost no RBP4 was isolated from lysate supernatants but remained in the insoluble fraction (Fig. S1A, ESI†). This recombinant protein expressed in E. coli requires a denaturation and refolding process to be active and ready for retinol/retinal binding, as the formation of inclusion bodies was described for recombinant RBP4 previously.44 It is not unusual that foreign proteins aggregate as inclusion bodies after high level over-expression in E. coli.45 Several factors like a strong promotor system or a high target gene copy number favour a higher rate of recombinant protein expression leading to inclusion bodies formation.46 Those can typically be dissolved under strongly denaturing conditions (e.g. urea) followed by incubation with the diluted denaturant in excess to enable protein refolding.47,48 Therefore, RBP4 was recovered according to Wang et al. from inclusion bodies as a soluble protein by dissolving the cell pellets in a denaturation buffer containing 8 M urea and subsequent refolding steps. It could be observed as a band with an apparent molecular mass of 21 kDa (Fig. S1A, ESI†). After all refolding steps, the resulting protein solution was submitted to size exclusion chromatography (SEC) for further purification. The collected fractions, assumed to contain purified RBP4, were analyzed by SDS-PAGE in Fig. S1C (ESI†), there single bands at a molecular mass of ∼21 kDa represented RBP4. The corresponding protein concentrations (Fig. S1B, ESI†) were calculated by the ChromLab Software and a total yield of ∼12 mg RBP4 was gained after urea extraction and purification from inclusion bodies harvested from 1 L culture.Ligand binding properties and functionality of apo-RBP4
The functionality of the purified RBP4 was further confirmed by determining its capacity to specifically bind increasing amounts of all-trans-retinal. Varying concentrations of retinal were added to a constant amount of the recombinant apo-RBP4. This mixture was incubated under exclusion of light and the quenching of intrinsic tryptophan fluorescence was monitored. The amount of apo-RBP4 in the reaction mixture decreased until only ligand-bound RBP4 was present after complexation with all-trans-retinal (Fig. S1D, ESI†). Interaction between RBP4 and retinal was confirmed, as the retinal quenched the intrinsic RBP4 tryptophan fluorescence due to energy transfer to the bound ligand. After binding, retinal is known to be incorporated into the binding cavity of the β-barrel of RBP4, where the polar groups remain solvent exposed.2 High affinity binding of all-trans-retinal to E. coli-derived RBP4 was confirmed previously by spectral analysis. There, fluorescence quenching by binding increasing concentrations of retinal was monitored demonstrating the specific interaction at a single binding site.49 In order to select aptamer libraries against apo- or holo-RBP4, first immobilization of proteins on magnetic Dynabeads M-280 tosyl-activated was performed. Covalent coupling of the tosyl-groups on the surface of the magnetic beads with primary amino groups of apo-RBP4 was performed as described by the manufacturer. To verify the coating with apo-RBP4 or holo-RBP4, the intrinsic tryptophan fluorescence emission was monitored at 340 nm with an excitation at 280 nm. Fluorescence was measured of apo-RBP4, apo- or holo-RBP4 coated beads, retinal, and naked magnetic beads (Fig. S1E, ESI†). Afterwards, to confirm retinal binding of holo-RBP4, emission was monitored at 490 nm with an excitation at 350 nm. The immobilization could be verified, as apo-RBP4 coated beads showed similar tryptophan fluorescence as single apo-RBP4 and intrinsic holo-RBP4 fluorescence was quenched after complexing apo-RBP4 immobilized on magnetic tosyl-beads with retinal. In comparison, signals gained by holo-RBP4 coated beads and by unbound retinal were similar. Uncoated naked magnetic beads exhibited no signals after fluorescence measurements, as buffers were used purely in the coating process.Evolution of aptamer libraries specific for RBP4 isoform and verification of their specificities
With the aim to develop a methodological tool for the specific quantification of serum apo- and holo-RBP4, polyclonal ssDNA aptamer libraries were evolved during an iterative FluMag-SELEX over a total of eight selection rounds. In order to differentiate between the RBP4 forms, two independent focused ssDNA aptamer libraries were selected in a simultaneous SELEX process against both RBP4 isoforms. These polyclonal aptamer libraries that bind to either apo-RBP4 or holo-RBP4 were successfully selected from a random ssDNA library containing ∼6 × 1014 individual aptamers with 40 randomized nucleotides flanked by two primer binding sites (23 nt each). The specificity was increased by early counter selection measures in which nonspecific oligonucleotides were eliminated by using empty (or “naked”) magnetic beads prior to each selection round. During the individual rounds of the FluMag-SELEX, the aptamers were Cy5-labelled via labelled PCR primers and analyzed fluorescently afterwards. To verify the evolution progress, defined amounts of aptamers (10 pmol) from each SELEX round were incubated with apo- or holo-RBP4-coated magnetic beads after the selection process. The libraries against apo-RBP4 were incubated for 30 min at 25 °C with apo-RBP4-coated magnetic beads and the holo-RBP4 libraries with holo-RBP4-coated beads and analyzed fluorescently after elution. The amount of eluted aptamers increased with each selection round indicating the progress of the enrichment process (Fig. 1A). The resulting two focused polyclonal aptamer libraries were further analyzed after the final selection round for specific target binding. The affinity binding characteristics of both final aptamer libraries were examined by analysis using apo- or holo-RBP4 coated magnetic beads as well as naked beads. Therefore, 10 pmol of round eight aptamer libraries were used for each of the RBP4 binding assays. The binding assays were performed using the apo-RBP4 library to specifically distinguish apo-RBP4 immobilized on magnetic beads from holo-RBP4 and the naked beads negative control. Then, the final polyclonal holo-RBP4 library was analyzed similar in order to specifically detect holo-RBP4. | ||
| Fig. 1 Characterization of specific RBP4 binding to Cy5-labelled aptamer libraries. (A) RBP4 ssDNA aptamer libraries enrichment. Black bars, increased binding of selected aptamers to apo-RBP4 during SELEX. Grey bars, increased binding of aptamers to holo-RBP4 during SELEX. The evolution progress was monitored using fluorescence measurement at an excitation of 635 nm with an emission at 670 nm. (B) Specificity analysis of the final polyclonal aptamer libraries. Binding of aptamers against apo-RBP4 or holo-RBP4 to both apo- and holo-RBP4 as well as to naked Dynabeads® M-280 tosyl-activated. All experiments were performed using 10 pmol aptamers, 13.6 μg apo- or holo-RBP4 and 0.68 mg magnetic beads, fluorescence was monitored at an excitation of 635 nm with an emission at 670 nm. Error bars symbolize standard deviations of measurements conducted in triplicates. P values <0.05 were considered significant. * denotes P < 0.05, ** <0.01, n.s. not significant. | ||
As expected, both aptamer libraries were confirmed to specifically and efficiently bind their dedicated targets. Only marginal signals were exhibited by the apo-RBP4 library for holo-RBP4 and magnetic beads (Fig. 1B), but high specificity was observed for the holo-RBP4 aptamer library (Fig. 1B).
In addition, the highly selective target binding of both aptamer libraries was visualized using fluorescence microscopy. Therefore, 30 pmol of each aptamer library were incubated for 30 min with RBP4-coated magnetic beads as well as with naked beads as negative controls. Afterwards, images were taken under transmitted light to visualize the magnetic beads and to detect target binding. Exclusive target binding of apo-RBP4 aptamers was observed visually, as only fluorescence signals were observed after the binding to apo-RBP4 coated magnetic beads (Fig. 2A).
 | ||
| Fig. 2 Fluorescence microscopic analysis of specific RBP4 binding to Cy5-labelled aptamer libraries. (A) Binding of apo-RBP4 aptamers to apo- and (B) holo-RBP4 as well as to (C) naked Dynabeads® M-280 tosyl-activated. (D) Binding of holo-RBP4 aptamers to apo- and (E) holo-RBP4 as well as to (F) naked Dynabeads® M-280 tosyl-activated. All experiments were performed using 30 pmol aptamers, 13.6 μg apo- or holo-RBP4 and 0.68 mg magnetic beads. Images were monitored using a Leica DMi8 coded (Leica Microsystems CMS GmbH, Wetzlar, Germany) at ×40 magnitude under transmitted light and using the Y5 filter (excitation: 590–650 nm and emission: 662–738 nm) for fluorescence imaging. | ||
As expected, beads coated with holo-RBP4 did not show any red fluorescence (Fig. 2B), indicating the differentiation between both RBP4 forms by the apo-RBP4 library. Likewise, highly specific target recognition was observed for the holo-RBP4 aptamer library. Red fluorescence signals could be observed after the binding of these aptamers to their dedicated target holo-RBP4 (Fig. 2D) and, as predicted, no signals were obtained after the incubation with apo-RBP4-coated beads (Fig. 2E). Moreover, both aptamer libraries exhibited no binding to the magnetic beads indicated by no fluorescence signals (Fig. 2C and F). These visual fluorescence signals additionally show the rapid and efficient labelling properties of both polyclonal aptamer libraries and furthermore the discriminating ability of them both between apo- and holo-RBP4.
Specific electrical sensing of apo- and holo-RBP4 using rGO-FETs
To assess the performance of aptamer libraries in biosensing applications, they were used as biorecognition element on an electrolyte-gated reduced graphene oxide field effect transistor (EG-rGO-FET). Chips were fabricated using a previously optimized protocol.50 Measurements were made in a Micrux all-in-one flow cell using an Ag/AgCl electrode as gate. Special aptamer libraries were prepared with primers bearing an amine functional group at the 5′-end. This aptamer library was then immobilized via EDC–NHS coupling on a pyrene-carboxylic acid functionalized reduced graphene oxide surface using previously published protocols (see Scheme 4).31 Analyte binding alters the charge transfer characteristics of the device which can be measured via ‘IDVG-curves’ in which the gate voltage (VG) is swept from −0.5 to 0.5 V and the source–drain current IDS is recorded. Higher voltages were not tested to avoid damaging biological components. Shifts in IDVG characteristics are caused by differences in charge carrier mobility originating from changes in the charge distribution at the channel-electrolyte interface.51 The charge distribution is altered during binding events, for example of charged analytes such as proteins and due to structural changes in the negatively charged aptamer backbone upon analyte binding.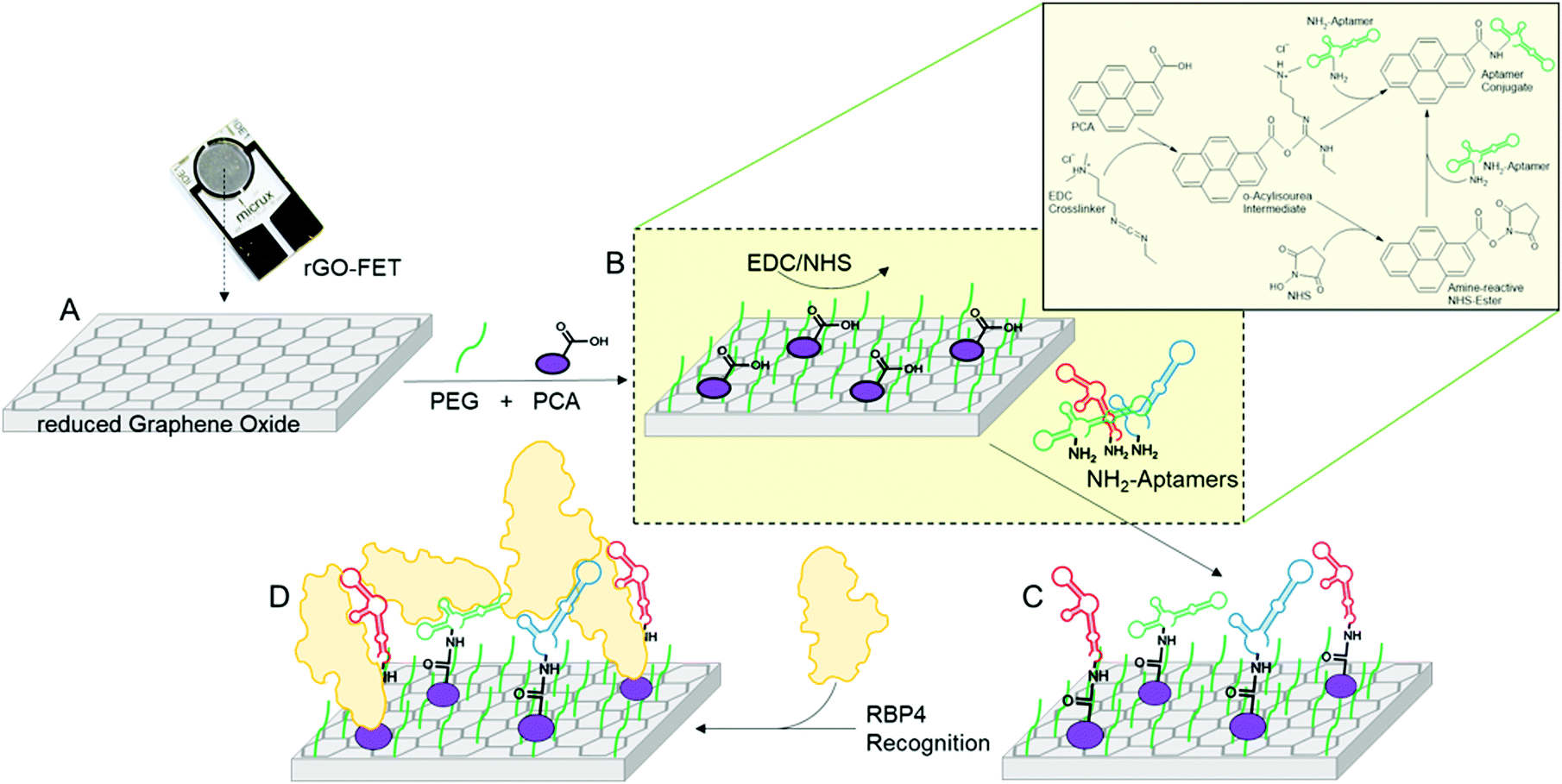 | ||
| Scheme 4 Functionalization of rGO-FETs with polyclonal ssDNA aptamer libraries and specific apo- or holo-RBP4 detection. (A) The rGO-FET were immersed into a mixture of PyPEG (PEG pre-conjugated with a PBSE) (500 μM) and 1-pyrenecarboxylic acid (PCA, 50 μM, linker) in DMSO for 12 h at room temperature to obtain a 10:1 ratio of blocking and linking agents on the biosensor's surface. (B and C) Apo- or holo-RBP4 aptamer library immobilization by first activating the carboxyl groups by immersion into a solution of EDC (15 mM)/NHS (15 mM) in 150 mM PBS solution for 30 min, followed by covalent coupling of the 5′-NH2-modified aptamer (100 nM in milliQ grade water for 40 min at 25 °C). (D) Specific affinity recognition of either apo- or holo-RBP4 by the on rGO-FET immobilized polyclonal ssDNA aptamer libraries in electrical measurements. | ||
Analyte binding can be observed in real time by monitoring IDS at a fixed gate potential during analyte addition38,39,52 or IDVG curves can be recorded after analyte addition.36,53–55 Measuring IDS in real time during analyte addition was not reliable (see Fig. S5, ESI†). We suspect that when a constant gate voltage is applied during analyte binding, a subset of the aptamer library might be misfolded and lose some or all affinity for RBP4. Instead, we recorded full IDVG curves after incubation of the chip with each analyte concentration, which allowed excellent reproducibility between devices. As the time required for either sensing method is relatively similar, IDVG – based sensing is acceptable for diagnostic purposes.
An IDVG-shift can occur both in the Dirac point (the potential at which IDS is at its minimum) and the slope of the curves, with specific interactions causing a significantly more pronounced change than non-specific interactions. IDVG curves were recorded first in 1× PBS solution as a background and after the addition of each analyte concentration (3–300 nM) with both target and non-target analyte (either apo- or holo-RBP4).
Fig. 3 shows typical device responses to specific and non-specific binding events. Additional data can be found in the ESI.†
 | ||
| Fig. 3 Top: Detection of apo-RBP4. Bottom: Detection of holo-RBP4. IDVG curves are an average of a minimum of 3 individual curves with error bars showing the standard deviation. Both devices showed significantly more pronounced shifts in their IDVG characteristics upon specific analyte binding. | ||
Slight changes in the shape and slope of the IDVG curve occurred after adding the non-target analyte (see Fig. 4), but there was no Dirac point shift. This change can be attributed to non-specific binding events of the analyte to the aptamers or PEG groups on the surface. Addition of the target analyte resulted in a more pronounced shift in the IDVG curve, with changes in both the slope and the Dirac point, indicating of stronger, specific binding events.
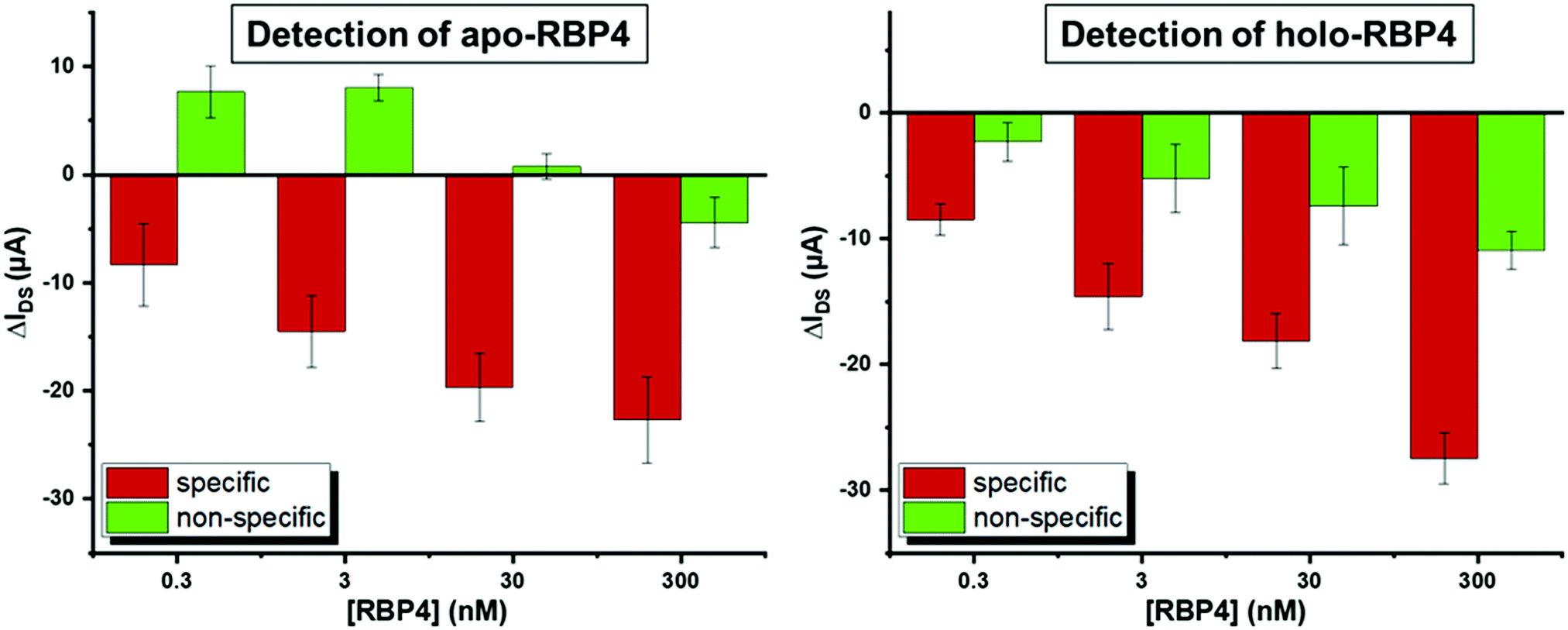 | ||
| Fig. 4 Average difference in current at −0.4 V upon addition of increasing concentrations of RBP4. Δ-Values shown are averages of a minimum of three independent experiments on different devices. Error bars show the standard deviation. | ||
The most pronounced shifts in IDVG curves upon analyte binding occurred around −0.4 V (see Fig. 3). To quantify sensor response, IDS shifts at this potential upon analyte addition were used to compare the signal change for specific and non-specific binding events. Fig. 4 shows the device response to specific and non-specific analyte binding. Both for the apo- and holo- aptamer libraries, addition of the target analyte produced a significantly stronger IDS – shift than the non-target analyte.
As the total concentration of RBP4 in human serum is approximately 1–3 μM (estimates vary depending on the method used to quantify RBP4 levels),17,56 we tested RBP4 concentrations from 0.3–300 nM to determine device performance. At all concentrations, the specific response was significantly stronger than the nonspecific signal. A small signal could also be seen upon addition of 100 pM RBP4 (see Fig. S5, ESI†), indicating a theoretical detection limit of 2 ng mL−1 RBP4, which is three orders of magnitude more sensitive than a previously reported aptamer-based RBP4 sensing method.57
As the sample matrix for RBP4 detection would be human blood plasma (HBP), we used blood plasma to test sensor passivation. Due to the high viscosity of undiluted HBP, it was impractical to test the sensor in this environment, and device response was only tested in 10% HBP or less. There was a change in device response in the presence of 10% and 1% HBP, however, rinsing with buffer returned the signal to the baseline. This could be observed both with an rGO-FET device and in a surface plasmon resonance experiment (see Fig. S6, ESI†). Thus, there was no unspecific binding potentially interfering with RBP4 detection. To determine if the presence of HBP interfered with RBP4 detection, we added 1% HBP spiked with RBP4. After rinsing, the signal did not return to baseline, indicating that while HBP was removed, RBP4 remained on the surface.
Diluting the patient sample to 10% would also reduce the concentration of RBP4 tenfold from 3 μM to 300 nM. This is well within the sensitivity range of our device, which would allow a dilution by a factor of up to 10000. Using the device in a clinical setting, a 100-fold dilution of a healthy patient sample should produce a pronounced signal with the holo-RBP4 aptamer library and little to no response in the device functionalized with the apo-library. Pathological states with increased apo-RBP4 levels should produce a decreased response in the former and an increased response in the latter. Future studies will further optimize device performance in blood and with real patient samples.
Conclusion
The evolution of focused polyclonal aptamer libraries instead of single aptamers against target molecules serves as a promising strategy in diagnostics and sensor technology. Larger sequence spaces enable a distinct and specialized diversity of binding aptamers. Multiple target recognition can be achieved generating enhanced versatility and thus increased performance in sensing and quantification of target molecules. Moreover, the great biotechnological availability of polyclonal aptamer libraries contributes to the rapid, cost-effective and easy development of novel, highly specific electronic sensors for use as powerful tools in clinical diagnostics. By functionalizing the channel of an rGO-FET device, we were able to selectively discriminate between RBP4 isoforms at physiologically relevant concentrations between 0.3 and 30 nM. We believe that these results may open a route to develop biosensors for the fast and reliable measurement of the RBP4 isoform concentrations as a novel diagnostic marker.Conflicts of interest
There are no conflicts to declare.References
- E. Esteve, W. Ricart and J. M. Fernández-Real, Diabetes Care, 2009, 32, S362–S367 CrossRef CAS PubMed.
- M. E. Newcomer and D. E. Ong, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1482, 57–64 CrossRef CAS.
- S. J. Thompson, A. Sargsyan, S. A. Lee, J. J. Yuen, J. Cai, R. Smalling, N. Ghyselinck, M. Mark, W. S. Blaner and T. E. Graham, Diabetes, 2017, 66, 58–63 CrossRef CAS PubMed.
- W. S. Blaner, Endocr. Rev., 1989, 10, 308–316 CrossRef CAS PubMed.
- C. H. Chen, T. J. Hsieh, K. Der Lin, H. Y. Lin, M. Y. Lee, W. W. Hung, P. J. Hsiao and S. J. Shin, J. Biol. Chem., 2012, 287, 9694–9707 CrossRef CAS PubMed.
- M. Kanai, A. Raz and D. S. Goodman, J. Clin. Invest., 1968, 47, 2025–2044 CrossRef CAS PubMed.
- L. Quadro, L. Hamberger, V. Colantuoni, M. E. Gottesman and W. S. Blaner, Mol. Aspects Med., 2003, 24, 421–430 CrossRef CAS PubMed.
- J. Raila, T. E. Willnow and F. J. Schweigert, J. Nutr., 2005, 135, 2512–2516 CrossRef CAS PubMed.
- K. Kelly, S. Kashyap, V. O’Leary, J. Major, P. Schauer and J. Kirwan, Obesity, 2009, 18, 663–666 CrossRef PubMed.
- C.-H. Chu, H.-C. Lam, J.-K. Lee, C.-C. Lu, C.-C. Sun, H.-J. Cheng, M.-C. Wang and M.-J. Chuang, Endocr. J., 2011, 58, 841–847 CrossRef CAS PubMed.
- Q. Yang, T. E. Graham, N. Mody, F. Preitner, O. D. Peroni, J. M. Zabolotny, K. Kotani, L. Quadro and B. B. Kahn, Nature, 2005, 436, 356–362 CrossRef CAS PubMed.
- T. E. Graham, Q. Yang, M. Blüher, A. Hammarstedt, T. P. Ciaraldi, R. R. Henry, C. J. Wason, A. Oberbach, P.-A. Jansson, U. Smith and B. B. Kahn, N. Engl. J. Med., 2006, 354, 2552–2563 CrossRef CAS PubMed.
- S. K. Frey, B. Nagl, A. Henze, J. Raila, B. Schlosser, T. Berg, M. Tepel, W. Zidek, M. O. Weickert, A. F.-H. Pfeiffer and F. J. Schweigert, Lipids Health Dis., 2008, 7, 29 CrossRef PubMed.
- H. Xu, G. T. Barnes, Q. Yang, G. Tan, D. Yang, C. J. Chou, J. Sole, A. Nichols, J. S. Ross, L. A. Tartaglia and H. Chen, J. Clin. Invest., 2003, 112, 1821–1830 CrossRef CAS PubMed.
- J. M. Olefsky and C. K. Glass, Annu. Rev. Physiol., 2010, 72, 219–246 CrossRef CAS PubMed.
- C. Erikstrup, O. H. Mortensen, A. R. Nielsen, C. P. Fischer, P. Plomgaard, A. M. Petersen, R. Krogh-Madsen, B. Lindegaard, J. G. Erhardt, H. Ullum, C. S. Benn and B. K. Pedersen, Diabetes, Obes. Metab., 2009, 11, 204–212 CrossRef CAS PubMed.
- T. E. Graham, C. J. Wason, M. Blüher and B. B. Kahn, Diabetologia, 2007, 50, 814–823 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- T. K. Sharma, J. G. Bruno and A. Dhiman, Biotechnol. Adv., 2017, 35, 275–301 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- P. Bayat, R. Nosrati, M. Alibolandi, H. Rafatpanah, K. Abnous, M. Khedri and M. Ramezani, Biochimie, 2018, 154, 132–155 CrossRef CAS PubMed.
- D. Kubiczek, N. Bodenberger and F. Rosenau, Antimicrobial Research: Novel bioknowledge and educational programs, 2017, vol. 6, pp. 368–378 Search PubMed.
- D. Kubiczek, H. Raber, N. Bodenberger, T. Oswald, M. Sahan, D. Mayer, S. Wiese, S. Stenger, T. Weil and F. Rosenau, Chem. – Eur. J., 2020, 26, 14536–14545 CrossRef CAS PubMed.
- H. Qu, A. T. Csordas, J. Wang, S. S. Oh, M. S. Eisenstein and H. T. Soh, ACS Nano, 2016, 10, 7558–7565 CrossRef CAS PubMed.
- S. S. Sekhon, P. Kaur, Y. H. Kim and S. S. Sekhon, npj 2D Mater. Appl., 2021, 5, 21 CrossRef CAS.
- A. Minopoli, B. Della Ventura, B. Lenyk, F. Gentile, J. A. Tanner, A. Offenhäusser, D. Mayer and R. Velotta, Nat. Commun., 2020, 11, 6134 CrossRef CAS PubMed.
- J. Zhang, L. P. Smaga, N. S.-R. Satyavolu, J. Chan and Y. Lu, J. Am. Chem. Soc., 2017, 139, 17225–17228 CrossRef CAS PubMed.
- T. Liang, Z. Yao, J. Ding, Q. Min, L. Jiang and J. J. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 34050–34059 CrossRef CAS PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2017, 16, 181–202 CrossRef CAS PubMed.
- G. Mahlknecht, R. Maron, M. Mancini, B. Schechter, M. Sela and Y. Yarden, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8170–8175 CrossRef CAS PubMed.
- P. Aspermair, V. Mishyn, J. Bintinger, H. Happy, K. Bagga, P. Subramanian, W. Knoll, R. Boukherroub and S. Szunerits, Anal. Bioanal. Chem., 2020, 7–11 Search PubMed.
- Y. Chen, R. Ren, H. Pu, X. Guo, J. Chang, G. Zhou, S. Mao, M. Kron and J. Chen, Sci. Rep., 2017, 7, 10974 CrossRef PubMed.
- S. Afsahi, M. B. Lerner, J. M. Goldstein, J. Lee, X. Tang, D. A. Bagarozzi, D. Pan, L. Locascio, A. Walker, F. Barron and B. R. Goldsmith, Biosens. Bioelectron., 2018, 100, 85–88 CrossRef CAS PubMed.
- G. Seo, G. Lee, M. J. Kim, S.-H. Baek, M. Choi, K. B. Ku, C.-S. Lee, S. Jun, D. Park, H. G. Kim, S.-J. Kim, J.-O. Lee, B. T. Kim, E. C. Park and S. Il Kim, ACS Nano, 2020, 14, 5135–5142 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Anal. Bioanal. Chem., 2005, 383, 83–91 CrossRef CAS PubMed.
- B. Cai, L. Huang, H. Zhang, Z. Sun, Z. Zhang and G.-J. Zhang, Biosens. Bioelectron., 2015, 74, 329–334 CrossRef CAS PubMed.
- S. Xu, S. Jiang, C. Zhang, W. Yue, Y. Zou, G. Wang, H. Liu, X. Zhang, M. Li, Z. Zhu and J. Wang, Appl. Surf. Sci., 2018, 427, 1114–1119 CrossRef CAS.
- Y.-M. Lei, M.-M. Xiao, Y.-T. Li, L. Xu, H. Zhang, Z.-Y. Zhang and G.-J. Zhang, Biosens. Bioelectron., 2017, 91, 1–7 CrossRef CAS PubMed.
- L. Zhou, H. Mao, C. Wu, L. Tang, Z. Wu, H. Sun, H. Zhang, H. Zhou, C. Jia, Q. Jin, X. Chen and J. Zhao, Biosens. Bioelectron., 2017, 87, 701–707 CrossRef CAS PubMed.
- S. Wang, M. Z. Hossain, K. Shinozuka, N. Shimizu, S. Kitada, T. Suzuki, R. Ichige, A. Kuwana and H. Kobayashi, Biosens. Bioelectron., 2020, 165, 112363 CrossRef CAS PubMed.
- T. Ono, Y. Kanai, K. Inoue, Y. Watanabe, S. Nakakita, T. Kawahara, Y. Suzuki and K. Matsumoto, Nano Lett., 2019, 19, 4004–4009 CrossRef CAS PubMed.
- C. Reiner-Rozman, M. Larisika, C. Nowak and W. Knoll, Biosens. Bioelectron., 2015, 70, 21–27 CrossRef CAS PubMed.
- P. Aspermair, U. Ramach, C. Reiner-Rozman, S. Fossati, B. Lechner, S. E. Moya, O. Azzaroni, J. Dostalek, S. Szunerits, W. Knoll and J. Bintinger, J. Am. Chem. Soc., 2020, 142, 11709–11716 CrossRef CAS PubMed.
- T. T.-Y. Wang, K. C. Lewis and J. M. Phang, Gene, 1993, 133, 291–294 CrossRef CAS PubMed.
- A. Singh, V. Upadhyay, A. K. Upadhyay, S. M. Singh and A. K. Panda, Microb. Cell Fact., 2015, 14, 41 CrossRef PubMed.
- A. Mitraki, B. Fane, C. Haase-Pettingell, J. Sturtevant and J. King, Science, 1991, 253, 54–58 CrossRef CAS PubMed.
- D. Shortle and M. S. Ackerman, Science, 2001, 293, 487–489 CrossRef CAS PubMed.
- Y. Xie, H. A. Lashuel, G. J. Miroy, S. Dikler and J. W. Kelly, Protein Expr. Purif., 1998, 14, 31–37 CrossRef CAS PubMed.
- M. S. Levin, B. Locke, N. C. Yang, E. Li and J. I. Gordon, J. Biol. Chem., 1988, 263, 17715–17723 CrossRef CAS PubMed.
- C. Reiner-Rozman, R. Hasler, J. Andersson, T. Rodrigues, A. Bozdogan, J. Bintinger and P. Aspermair, Micro Nano Lett., 2021, 16(8), 436–442 CrossRef CAS.
- C. Zhao, K. M. Cheung, I. W. Huang, H. Yang, N. Nakatsuka, W. Liu, Y. Cao, T. Man, P. S. Weiss, H. G. Monbouquette and A. M. Andrews, Sci. Adv., 2021, 7, 25–27 Search PubMed.
- B. Thakur, G. Zhou, J. Chang, H. Pu, B. Jin, X. Sui, X. Yuan, C.-H. Yang, M. Magruder and J. Chen, Biosens. Bioelectron., 2018, 110, 16–22 CrossRef CAS PubMed.
- N. Nakatsuka, K.-A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim, Y. Yang, P. S. Weiss, M. N. Stojanović and A. M. Andrews, Science, 2018, 362, 319–324 CrossRef CAS PubMed.
- S. M. Majd, A. Salimi and F. Ghasemi, Biosens. Bioelectron., 2018, 105, 6–13 CrossRef CAS PubMed.
- T.-Y. Chen, P. T.-K. Loan, C.-L. Hsu, Y.-H. Lee, J. Tse-Wei Wang, K.-H. Wei, C.-T. Lin and L.-J. Li, Biosens. Bioelectron., 2013, 41, 103–109 CrossRef CAS PubMed.
- J. M. Fernández-Real, J. M. Moreno and W. Ricart, Diabetes, 2008, 57, 1918–1925 CrossRef PubMed.
- J. L. Su, B. S. Youn, W. P. Ji, J. H. Niazi, S. K. Yeon and B. G. Man, Anal. Chem., 2008, 80, 2867–2873 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1nh00605c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |