
Egg-yolk core–shell mesoporous silica nanoparticles for high doxorubicin loading and delivery to prostate cancer cells†
Steffi
Tiburcius
a,
Kannan
Krishnan
a,
Linta
Jose
a,
Vaishwik
Patel
a,
Arnab
Ghosh
b,
C. I.
Sathish
 a,
Judith
Weidenhofer
c,
Jae-Hun
Yang
a,
Nicole M.
Verrills
b,
Ajay
Karakoti
*a and
Ajayan
Vinu
*a
a,
Judith
Weidenhofer
c,
Jae-Hun
Yang
a,
Nicole M.
Verrills
b,
Ajay
Karakoti
*a and
Ajayan
Vinu
*a
aGlobal Innovative Centre for Advanced Nanomaterials, Faculty of Engineering and Built Environment, The University of Newcastle, Callaghan, 2308, NSW, Australia. E-mail: ajay.karakoti@newcastle.edu.au; ajayan.vinu@newcastle.edu.au
bSchool of Biomedical Sciences and Pharmacy, Faculty of Health and Medicine, The University of Newcastle, Callaghan, 2308, NSW, Australia
cHunter Medical Research Institute (HMRI), New Lambton Heights, 2305, NSW, Australia
First published on 30th March 2022
Abstract
Mesoporous silica-based nanoparticles (MSNs) have gained rapid interest as a drug delivery system (DDS) and demonstrated their versatility in delivering drugs for the treatment of various cancers. However, the drug loading efficiency of MSNs is low and is usually improved by improving textural properties through complicated synthesis methods or by post synthesis modification of the surface that can result in the loss of surface area and modify its drug release properties. In this study, we report a direct single-step synthesis of MSNs with a unique egg-yolk core–shell morphology, large pore volume and a hydrophilic surface, decorated with nitrogen rich surface functionalities for increasing its drug loading capacity. This combination of excellent textural properties and surface functionalisation was achieved by a simple soft templating method using dual surfactants and the silica sources assisted by employing either triethylamine (TEA) or triethanolamine (TEO) as the hydrolysis agent. The morphology and well-ordered mesoporous structure can simply be tuned by changing the pH of the synthesis medium that affects the self-assembly mechanism of the micelles. HRTEM image of samples clearly revealed an egg-yolk core–shell morphology with a thin mesoporous silica shell. The optimised MSN samples synthesized at a pH of 11 using either TEA or TEO depicted a higher doxorubicin (Dox) loading capacity of 425 μg mg−1 and 481 μg mg−1 respectively, as compared to only 347 μg mg−1 for MSN samples due to the uniform distribution of nitrogen functionalities. The anticancer activity of Dox loaded MSNs evaluated in two different prostate cancer cell lines (PC-3 and LNCaP) showed a higher cytotoxicity of the drug loaded on optimised MSN samples as compared to pristine MSNs without affecting the cellular uptake of the particles. These results suggest that the unique single-step synthesis and functionalisation method resulted in successfully achieving higher drug loading in egg-yolk core–shell nitrogen functionalised MSNs and could be implemented as an effective carrier of chemotherapeutic drugs.
1. Introduction
Prostate cancer (PCa) is the sixth leading cause of death and the second most diagnosed form of cancers among men.1 Its overall high incidence and high mortality rate have prompted researchers to look for better and alternative methods of treatments. Among many treatment options, androgen deprivation therapy (ADT) is the most trusted treatment regime for patients with metastasis or castration resistant PCa. Patients who do not respond to ADT are given systemic chemotherapy. However, the non-specific and non-targeted nature of chemotherapeutic drugs often leads to the development of a number of side effects.2,3 This increases patient non-compliance and inability to stick to a chemotherapeutic treatment regime which adversely affects the 5-year survival rate.4–6 Such challenges in the administration of chemotherapy to PCa patients are currently addressed by the encapsulation of chemotherapeutic drugs within nanomaterial-based drug delivery systems to improve the efficacy and therapeutic window of the drugs and reduce the unwanted side effects.7Until now, a series of nanomaterials such as polymers, lipids and porous silica particles have been used as a platform for the delivery of chemotherapeutic drugs, such as doxorubicin (Dox), to different types of cancer cells.8–14 For example, Yuan et al. reported on the use of PEGylated solid lipid nanoparticles to enhance the bioavailability of Dox following its oral administration.15 Recently, Mu et al. used a combination of programmed cell death ligand-1 (PDL-1), siRNA and Dox loaded on polydopamine nanoparticles for targeting PCa bone metastases.16 Polymeric micelles have also been used for delivering Dox for targeted treatment of bone metastatic PCa.17 Although polymeric particles and liposomes have been widely used for drug delivery applications, the low loading threshold of the drugs in these platforms due to poor specific surface areas and the lack of porosity limits their performance in cancer treatment. Therefore, porous nanomaterials including mesoporous silica nanoparticles (MSNs) have found widespread use in drug delivery applications owing to their excellent textural features such as a large specific surface area, hydrophilic surface, nanoscale porosity, adjustable pore size and pore volume, high drug loading capacity, and biocompatibility.18–22 In particular, the ordered meso channels of MSNs help not only in enhancing the adsorption of the drugs, but also, in the release of the drugs by a simple adjustment of the functional groups on the surface. It should also be noted that MSNs have been shown to dissolve and clear from the human body within 2–3 weeks of their systemic administration.22–25 Therefore, extensive studies have been conducted on the synthesis of MSNs with tunable pore sizes and surface areas and their application in the delivery of various chemotherapeutic drugs to the PCa.26–32 MSNs are also being extensively used for the delivery of proteins such as cytochrome-c, amino acids, vitamins, lysozyme and heme proteins.33–42
For example, eccentric mesoporous silica nanoclusters have been designed for pH-responsive Dox delivery accompanied by sustained drug release.43 Similarly, capping of Dox-loaded MSNs with β cyclodextrin for ultrasound image-guided intravascular and chemo-sonodynamic therapy has also been reported.44 Using an end-cap design, Moreira et al. were successful in loading up to 77% Dox using pH-responsive on-demand drug delivery.45 On the other hand, Silveira et al. reported on reducing the cardiotoxicity of Dox by developing Dox loaded MSNs in thermo-reversible hydrogels, which exhibited high antitumour activities, prolonged drug release and reduced cardiotoxicity.46 While it is essential to develop a drug delivery system that can fulfil various criteria such as high cellular internalisation, prolonged circulation time and targeted delivery, achieving a high drug loading within the cargo is one of the most desirable features of a drug delivery system.
A high drug loading in MSNs can be achieved by changing the morphology with high textural properties with open structures as they offer a high pore volume. Surface functionalisation of MSNs with amine or carboxyl groups is the second most popular method of increasing drug loading. However, the current synthesis approaches for controlling the morphology and the surface functional groups of MSN require complicated synthesis protocols, the combination of special surfactants or long secondary processing and post-synthetic chemical treatments. These processes are extremely time-consuming and tedious and lead to a reduction in the surface area and pore size or complete collapse of the structure or a loss in the overall morphology of the MSNs, affecting their drug loading capacity and cellular internalisation. In addition, secondary processing also increases the process costs and reduces the overall scalability of MSNs for practical drug delivery applications. Therefore, the development of MSNs with controlled morphology and functional groups for high drug loading and its controlled release for the treatment of PCa is needed.
In this work, we report the direct synthesis of egg-yolk core–shell MSNs with a hydrophilic surface, decorated with nitrogen functionalities through simple self-assembly of dual surfactants and silica sources assisted by either triethylamine (TEA) or triethanolamine (TEO) as the hydrolysis agent instead of ammonium hydroxide. Non-ionic pluronic surfactant is used as the core template and cetyltrimethylammonium bromide as the mesopore structure-directing agent. Interestingly, in this unique in situ functionalisation process, TEA and TEO offer three different functions: (a) act as the hydrolysis agent, (b) morphology directing agent to achieve a yolk–shell structure, and (c) provide a uniform distribution of nitrogen groups on the surface of the MSNs to increase their affinity for chemotherapeutic drugs. The resulting MSN materials exhibit a high specific surface area, large pore volume, highly dispersed nitrogen on the wall surface, and egg-yolk core–shell morphology. It is observed that single step synthesis and functionalisation increases the Dox loading from 34% for non-functionalised MSNs to 42% for MSNs synthesised with TEA and 48% for MSNs prepared with TEO. The uniform distribution of nitrogen and the unique egg-yolk–shell morphology of MSNs prepared with TEA or TEO facilitate the high Dox loading capacity due to the large pore volume generated by hollow mesopores and the large hydrophilicity due to the nitrogen functionalities. The high drug loading in modified MSNs is directly reflected in the higher cytotoxicity exerted by the administration of the same amount of TEA and TEO modified MSNs on the PC-3 and LNCaP cells as compared to the bare MSNs without affecting the cellular internalisation. The single-step synthesis and functionalisation of ordered MSNs reported in this paper is one of the most sustainable methods of the production of egg-yolk core–shell MSNs that can be broadly applicable for increasing the drug loading capacity of MSNs for a variety of drug delivery applications.
2. Experimental section
2.1. Materials
Chemicals such as non-ionic Pluronic F-127 (EO106PO70EO106), tetrahydrofuran (THF), cetyltrimethylammonium bromide (CTAB), ammonia solution, tetraethyl orthosilicate (TEOS), TEA, TEO, dimethyl sulfoxide (DMSO), (3-aminopropyl) triethoxysilane (APTES), and ethanol were obtained from Sigma-Aldrich and used without further purification. The chemotherapeutic drug, Dox (CAS 25316-40-9) 99%, was purchased from ChemUniverse and used without further purification.2.2. Synthesis of core–shell mesoporous silica nanoparticles (MSN@SiO2) and APTES functionalised MSNs (NH2-MSN@SiO2)
For the synthesis of MSN@SiO2, 0.1 g F-127 was mixed with 20 mL THF in a polypropylene (PP) bottle and stirred for 1 h (Solution A). Simultaneously, 0.2 g CTAB, 80 mL water, and 3 mL ammonia solution were mixed in another PP bottle and stirred for 1 h (Solution B). Then, solutions A and B were combined and stirred for a further 30 minutes. After thoroughly mixing, the resultant solution was diluted by adding 160 mL of ethanol and stirred for another 4 h, followed by the dropwise addition of 0.6 mL TEOS. This final solution was stirred for another 24 h and centrifuged at 4500 rpm for 20 min. The precipitate was washed several times for the complete removal of unreacted surfactants, and the obtained precipitate was dried in a vacuum overnight. The obtained white precipitate was calcined at 550 °C for 12 h under a N2 flow followed by 12 h in air for the complete removal of surfactants.The prepared MSN@SiO2 was functionalised with amine groups to understand the effect of amine functionalisation on the drug adsorption and its release. NH2-MSN@SiO2 was prepared through a post-synthesis grafting method. Initially, 1 g of MSN@SiO2 was mixed in 50 mL ethanol for 4 h to enhance the surface silanol groups and dried overnight. Dried MSN@SiO2 was dispersed in 100 mL anhydrous toluene and refluxed at 110 °C with 193 μL of APTES under a nitrogen flow for 24 h. After refluxing, the final solution was filtered and washed with 50 mL methanol and deionised water and dried for further use. NH2-MSN@SiO2 was also used for fluorescent labelling of nanoparticles with cyanine 5 for flow cytometry studies.
2.3. Synthesis of nitrogen functionalised egg-yolk core–shell mesoporous silica nanoparticles MSN@SiO2
For the synthesis of nitrogen functionalised MSN@SiO2, the synthesis procedure was similar to the synthesis of MSN@SiO2 except that the ammonia solution in solution B was replaced with TEA or TEO. A series of samples were prepared at different solution pH, 10.9, 11, 11.2, and 11.5, by varying the amount of TEA or TEO. Once the pH stabilised, the solution was stirred for 4 h followed by dropwise addition of 0.6 mL TEOS and left stirring for another 24 h. The resultant mixture was centrifuged at 4500 rpm for 20 min, and the precipitate was dried in a vacuum overnight. The dried powder was calcined for 12 h at 550 °C under N2 followed by 12 h in air for crystallisation and complete removal of surfactants. The samples prepared using TEA or TEO are denoted as TE-@MS-x or TO-@MS-x, respectively, where x denotes the pH of the solution. Scheme 1 represents the synthesis procedure and application in cellular studies.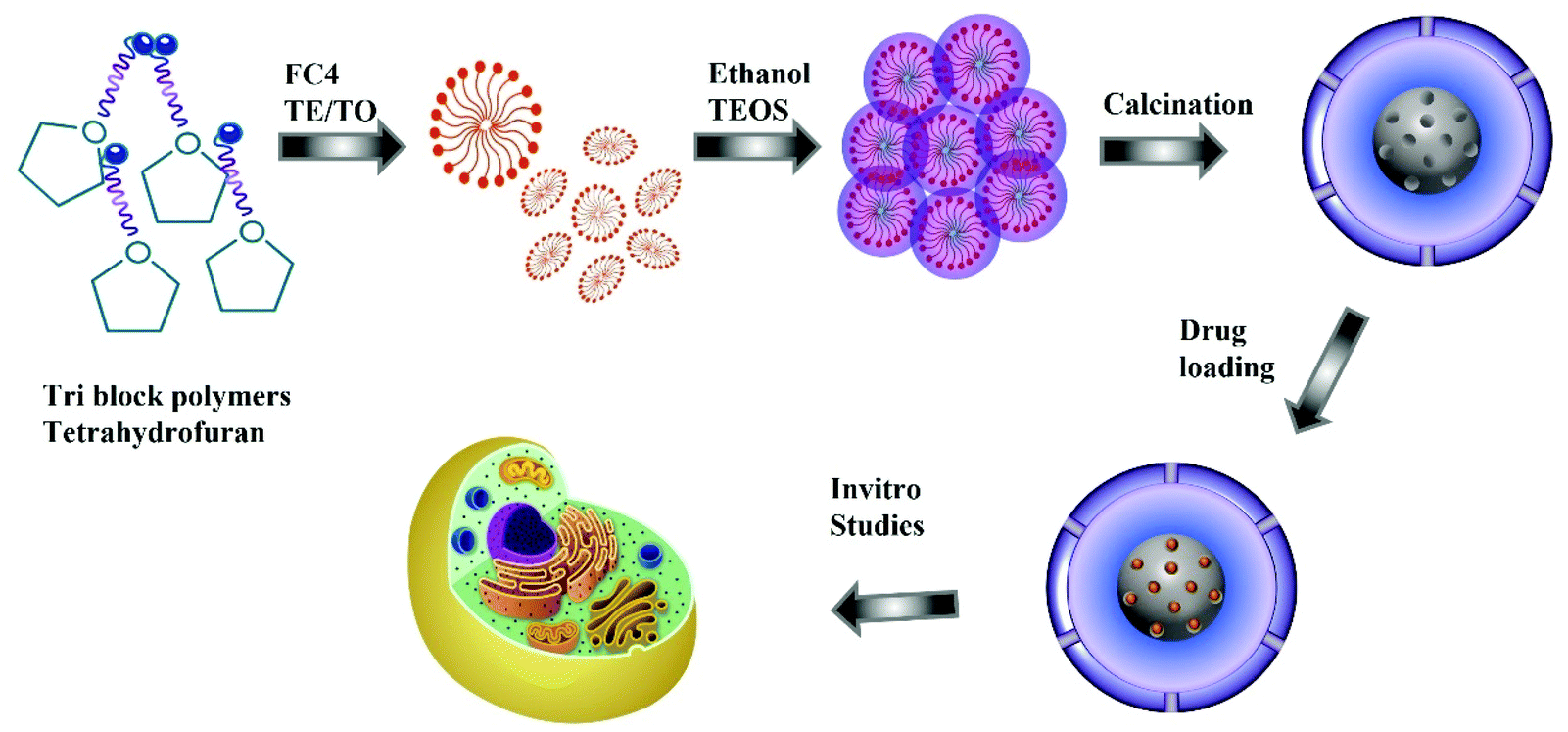 | ||
| Scheme 1 Schematic representation of MSN synthesis and drug delivery. | ||
2.4. Characterisation
Low-angle powder X-ray diffraction (LA-XRD) patterns of the synthesised MSN powders were recorded using a PANalytical Empyrean diffractometer with a Cu Kα (λ = 1.5406 Å) radiation source operating at 40 kV and 40 mA in the Bragg–Brentano geometry, with a fixed divergence slit of 1/16° and a scan rate of 0.0017° s−1 was used for low angle measurements (0.7–5°). For surface area analysis, the samples were degassed under vacuum at 200 °C for 12 h. A Micromeritics ASAP 2024 surface analyser was utilised for nitrogen adsorption analysis. The BET equation and the BJH method were used to estimate the surface areas and the pore sizes, respectively. The zeta potential of the prepared samples was analysed using a Malvern Zetasizer Pro. The structural morphology of the prepared materials was investigated using a scanning electron microscope (SEM), JEOL FEM 7001 fitted with an energy dispersive spectrometer (EDS), and the elemental distribution was observed by elemental mapping. Thermogravimetric analysis (TGA) of the MSN samples and Dox loaded MSN was performed with a heating rate of 10 °C min−1 under an airflow rate of 25 mL min−1. FT-IR spectra were measured using an FT-IR 400 system spectrometer to understand the presence of surface functional groups.2.5. Dox loading studies
For loading Dox, a 1 mg mL−1 solution was prepared using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PBS/DMSO mixture. Prior to loading, different MSN samples were dried overnight at 150 °C and 20 mg of the MSN@SiO2 sample was added to 10 mL Dox solution. This mixture was stirred slowly at room temperature for 24 h in the dark for facilitating drug adsorption. Drug loading experiments for MSN@SiO2 were continued for 48 and 72 h. As the adsorption reached equilibrium after 24 h, all further drug loading experiments with NH2-MSN@SiO2, TE-@MS-x, and TO-@MS-x were conducted for 24 h. The drug-loaded MSNs were recovered from the drug solution by centrifugation followed by washing with deionised water to remove any loosely adsorbed drug molecules. The supernatant was analysed by UV-Vis spectroscopy to estimate the amount of drug loaded in the MSNs. The drug-loaded MSNs were freeze-dried and stored in the dark for further studies.
:1 PBS/DMSO mixture. Prior to loading, different MSN samples were dried overnight at 150 °C and 20 mg of the MSN@SiO2 sample was added to 10 mL Dox solution. This mixture was stirred slowly at room temperature for 24 h in the dark for facilitating drug adsorption. Drug loading experiments for MSN@SiO2 were continued for 48 and 72 h. As the adsorption reached equilibrium after 24 h, all further drug loading experiments with NH2-MSN@SiO2, TE-@MS-x, and TO-@MS-x were conducted for 24 h. The drug-loaded MSNs were recovered from the drug solution by centrifugation followed by washing with deionised water to remove any loosely adsorbed drug molecules. The supernatant was analysed by UV-Vis spectroscopy to estimate the amount of drug loaded in the MSNs. The drug-loaded MSNs were freeze-dried and stored in the dark for further studies.
2.6. Dox release studies
Drug release from the MSN samples was analysed in two separate buffers – phosphate buffer at pH 7.4 and acetate buffer at pH 5 to stimulate the normal physiological conditions and the tumour microenvironment. For measuring the drug release, a dialysis bag with MWCO – 8000 Daltons was filled with drug-loaded MSN samples in the selected buffer solution and suspended in the corresponding release medium. The dialysis bag was gently shaken at 100 rpm at 37 °C under dark conditions to facilitate drug release. 3 mL of release media was taken out at specific intervals to quantify the amount of the drug released by measuring the absorbance at 490 nm. The release experiments were carried out for 72 h.2.7. Cyanine 5.5 NHS ester labelling
Fluorescent labelling of NH2-MSN@SiO2 with Cyanine 5 was carried out for studying their cellular uptake using flow cytometry. For functionalisation, 20 mg NH2-MSN@SiO2 was treated with 60 μL Cyanine 5.5 NHS ester (1 mg mL−1) in 5 mL PBS (7.4) and stirred at low speed at 0 °C for 2 h. This solution was centrifuged and washed with PBS followed by deionised water and freeze-dried for further studies. The absorbance and fluorescence spectra were measured to confirm the Cyanine labelling of the MSNs.2.8. In vitro cytotoxicity evaluation
LNCaP and PC-3 cells were used for testing the cytotoxicity of MSN@SiO2 and drug loaded TE-@MS-x and TO-@MS-x. LNCaP and PC-3 cells were cultured in RPMI-1640 (supplemented with 10% FBS) and Ham's F 12 nutrient medium (supplemented with 10% FBS) respectively at 37 °C under 5% CO2. The in vitro cytotoxicity of the pristine MSN@SiO2 and the Dox loaded MSN@SiO2, TE-@MS-x and TO-@MS-x was measured by the resazurin assay. The cells were seeded at a density of 5 × 103 cells per well in a 96 well plate in 100 μL of complete growth medium and incubated for 24 h. The pristine MSN@SiO2 and the Dox loaded MSN@SiO2, TE-@MS-x, and TO-@MS-x at different concentrations (50, 100, 200, 300, 400, and 500 μg mL−1) were added to the cells and placed in the incubator. The cells without any treatment were used as a control and SDS was used as a positive control for cytotoxicity. After 6 h, the cells were supplied with complete growth media. After 24 and 48 h of incubation, 20 μL resazurin was added to each well and incubated for 6 h at 37 °C. The fluorescence from the live cells was measured using a microplate reader at 590 nm using a 570 nm excitation wavelength.2.9. Cellular uptake studies
For the measurement of cellular uptake, pristine MSN@SiO2 were labelled with cyanine 5.5 NHS ester while Dox was used as the fluorescent tag for comparing the uptake of Dox loaded MSN@SiO2, TE-@MS-x and TO-@MS-x. PC-3, LNCaP, and RWPE-1 were seeded at 1 × 105 density in 6 well plates in 1 mL complete growth media and incubated for 48 h at 37 °C. The cells were then treated with different concentrations of either Dox or cyanine 5.5 labelled MSNs. The cells without any treatment were used as a control. These cells were incubated for 24 h. They were then washed three times in PBS, trypsinised, and resuspended in PBS. The suspension obtained was then analysed using a flow cytometer (BD FACS Canto II).For imaging the uptake of MSN@SiO2, and the Dox loaded MSN@SiO2, TE-@MS-x, and TO-@MS-x, the cells were seeded at a density of 1 × 105 cells per well in 6 well plates lined with coverslips in 1 mL complete growth media and incubated for 48 h at 37 °C. They were then treated with different concentrations of Dox-loaded MSNs and Cy 5.5 labelled MSNs. After 12 h of incubation, the cells were washed with PBS three times and fixed with paraformaldehyde. The cells were labelled with the Cytopainter cell plasma membrane staining kit, orange fluorescence (Abcam ab 219941), and incubated for about 20 min at 37 °C, in a 5% CO2 incubator. The stained cells were washed with complete growth media and fixed with 4% paraformaldehyde solution for 15 min. The cells were fixed using a ProLong™ Gold Antifade Mountant containing DAPI (Invitrogen) and examined using a fluorescence microscope (Olympus DP80).
2.10. Statistical analysis
The differences between the groups in cytotoxicity tests were determined by one-way analysis of variance (ANOVA) by the Holm–Sidak method. SigmaPlot was used for the statistical analysis of data. Significant differences among the groups were considered at p < 0.05 or less.3. Results and discussion
3.1. Characterisation
Powder X-ray diffraction analysis was conducted to analyse the structural order of the prepared MSN@SiO2, NH2-MSN@SiO2, TE-@MS-x and TO-@MS-x samples as it plays a critical role in determining the loading capacity and the release characteristics of the materials. The low angle powder XRD patterns of the calcined MSN@SiO2, NH2-MSN@SiO2, TE-@MS-x, and TO-@MS-x are shown in Fig. 1A, B, and C. It was observed that MSN@SiO2 showed an intense broad peak at 2θ ∼2.4° and a small broad peak at ∼4.1° corresponding to the (100) and (110) reflections, respectively, indicative of an ordered structure of the MSNs with a 2D hexagonal p6mm pore structure (Fig. 1A). The observation of both low angle and higher angle peaks after amine functionalization confirms that the mesostructural order is preserved even after the functionalisation of MSN@SiO2 with APTES. However, it was noted that the intensity of both low and higher reflection is much lower as compared to those of the parent MSN@SiO2. The large reduction in the intensity cannot be interpreted as the reduction in the structural order. It is believed that the presence of amine groups on the wall structure and the solvent molecules used for the functionalization process reduces the density between pure silica walls and mesopores and the amine functionalised silica and the mesopores. This may be responsible for the reduction in the intensity of the lower and higher order peaks. The structural order of TE-@MS-x and TO-@MS-x prepared at different solution pH was also analysed by low angle powder XRD and the results are shown in Fig. 1B and C. As can be seen in Fig. 1B and C, the structural order can be tuned by simply changing the pH of the synthesis medium by varying the amount of TEA or TEO. It was also observed that the TE-@MS-x samples except for TE-@MS-11.5 exhibited a major peak at a low angle and the higher order peaks at a high angle, indicating that they have an ordered mesoporous structure. However, the shape of the XRD pattern for TE-@MS-11.5 is completely different and only one broad peak at a low angle was observed, suggesting that the mesoporous structure of the sample is partially collapsed. This is attributed to the slow hydrolysis and the lack of stability of the silanol groups at higher pH which significantly affects the self-assembly process of TEOS with the CTAB. It was also observed that the peak corresponding to the (100) reflection shifted to a lower angle for TE-@MS-11 and TE-@MS-11.2, indicating that the unit cell constant which is directly related to the pore diameter of the materials with the hexagonal pore structure is increased. A similar observation was made for TO-@MS-x where the ordered structure of MSNs is preserved for TO-@MS-10.9, TO-@MS-11, and TO-@MS-11.2 while the mesoporous structure partially collapsed for TO-@MS-11.5 (Fig. 1C). The unit cell constant of TO-@MS-11.2 is higher than that of TO-@MS-10.9, revealing that the pH of the synthesis mixture plays a critical role in controlling the structural order and the unit cell constant of the materials. These results suggest that fine control of pH of the synthesis medium is required for achieving a well-ordered mesoporous structure for both TE-@MS-x and TO-@MS-x samples.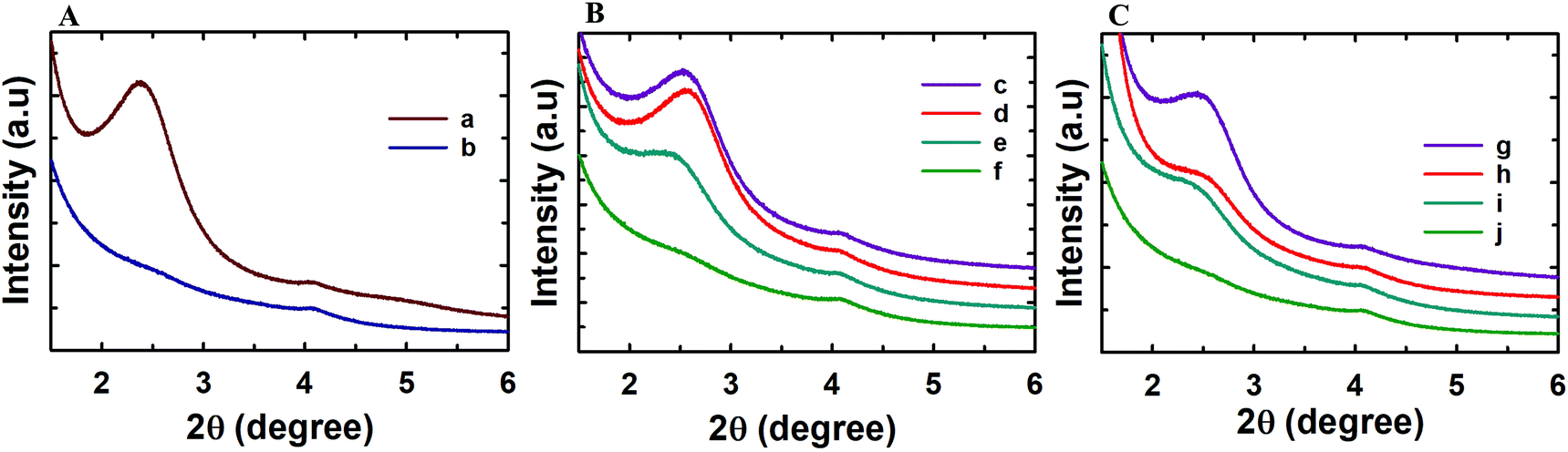 | ||
| Fig. 1 (A) Low angle powder XRD patterns of (a) MSN@SiO2 and (b) NH2-MSN@SiO2; (B) TE-@MS-x (c) TE-@MS-10.9, (d) TE-@MS-11.0, (e) TE-@MS-11.2 and (f) TE-@MS-11.5; and (C) TO-@MS-x, (g) TO-@MS-10.9, (h) TO-@MS-11, (i) TO-@MS-11.2 and (j) TO-@MS-11.5. | ||
The textural properties of the TE-@MS-x and TO-@MS-x samples were analysed by nitrogen adsorption–desorption isotherms and the results were compared with those of MSN@SiO2 and NH2-MSN@SiO2. Fig. 2A shows the nitrogen adsorption isotherms of MSN@SiO2 and NH2-MSN@SiO2, whereas Fig. 2B and C display the nitrogen adsorption–desorption isotherms of TE-@MS-x and TO-@MS-x samples, respectively. All the samples show a typical type IV adsorption isotherm based on the IUPAC classification, indicating that the samples are mesoporous in nature. MSN@SiO2 and NH2-MSN@SiO2 samples show nitrogen adsorption–desorption isotherms typical of MCM-41 samples depicting a sharp nitrogen adsorption until about P/P0 = 0.3. MSN@SiO2 does not show any hysteresis following the appearance of a knee at P/P0 = 0.3 and extending throughout the range of the plateau. The absence of the hysteresis loop in these samples is indicative of regular non-intersecting cylindrical pores, similar to that observed for MCM-41 mesoporous materials which have one-dimensional cylindrical mesopores.47 The capillary condensation step is sharp and observed at a low relative pressure, indicating that the mesopore diameter of the materials is small and uniform. On the other hand, the amount of nitrogen adsorbed significantly decreases after amine functionalisation. This may be attributed to the covering of a part of the pores with the APTES molecules. It is interesting to note that the isotherm of NH2-MSN@SiO2 displays a large hysteresis loop due to the blockage of the pore entrance by the APTES molecules. When the nitrogen adsorption isotherms of the TE-@MS-x and TO-@MS-x samples are compared, it can be seen that the solution pH significantly affects the amount of nitrogen adsorbed at a lower relative pressure which determines the specific surface area of the samples, and the sharpness of the capillary condensation step (Fig. 2B and C). For example, the isotherms of the TE-@MS-x and TO-@MS-x samples synthesized at pH 10.9, 11 and 11.2 are type IV without any hysteresis loop whereas TE-@MS-11.5 and TO-@MS-11.5 display an isotherm with a broad capillary condensation step at the higher relative pressure. It should be noted that the amount of nitrogen adsorbed decreases with increasing solution pH for both TE-@MS-x and TO-@MS-x samples. The absence of a well-developed knee, which is typically observed for typical ordered mesoporous materials, for TE-@MS-11.5 and TO-@MS-11.5, reveals that the mesostructure of the sample is partially collapsed. This is quite consistent with the XRD data for these samples.
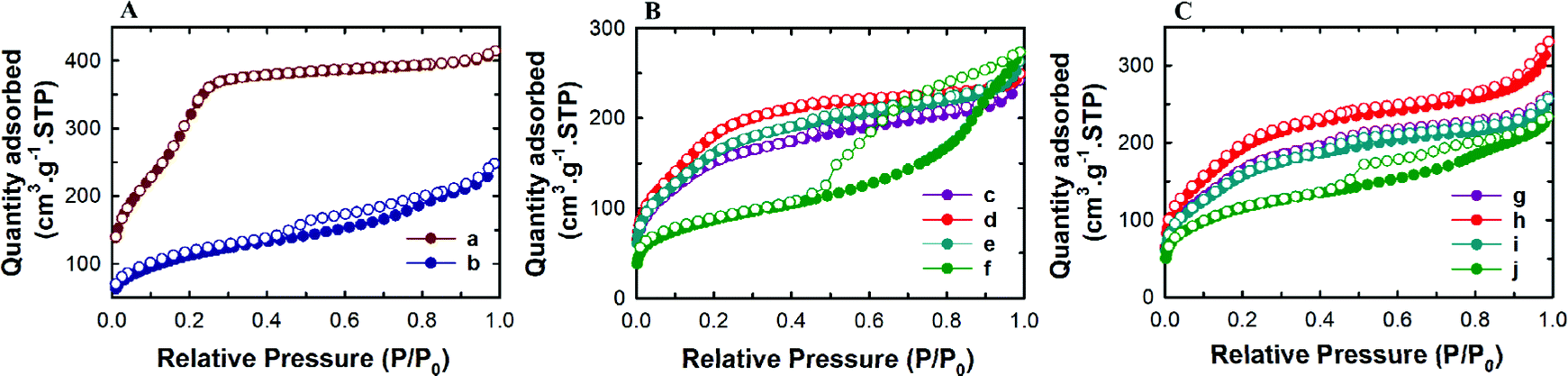 | ||
| Fig. 2 (A) Nitrogen adsorption–desorption isotherms of (a) MSN@SiO2 and (b) NH2-MSN@SiO2; (B) (c) TE-@MS-10.9, (d) TE-@MS-11.0, (e) TE-@MS-11.2 and (f) TE-@MS-11.5; and (C) TO-@MS-x, (g) TO-@MS-10.9, (h) TO-@MS-11, (i) TO-@MS-11.2 and (j) TO-@MS-11.5. | ||
The textural parameters such as the pore diameter, pore volume and the specific surface area of all the samples are given in Table 1S and SI.1.† Among the samples prepared, pristine MSN@SiO2 shows the highest specific surface area of 1230 m2 g−1 and a corresponding high pore volume of 0.69 cm3 g−1, indicative of rich mesoporous features with a uniform pore size. However, a reduction in the specific surface area from 1230 m2 g−1 to 411 m2 g−1 is observed for NH2-MSN@SiO2. This reduction in the specific surface area is not the reflection of the disorder in the mesostructure as the XRD data of the samples do not show any change in the structural order. It is believed that APTES groups block the pore channels after functionalisation which significantly affects the specific surface area and the specific pore volume of NH2-MSN@SiO2. The surface area of TE-@MS-x and TO-@MS-x samples is relatively lower as compared to the MSN@SiO2 samples probably due to the incorporation of nitrogen functionalities on the surface of silica which affects the smooth condensation of silanol groups in the wall structure. It is also believed that the addition of TEA or TEO may increase the volume of the micelles as these molecules may contribute to increase either the hydrophobic moieties of the surfactant or the repulsion between the formed CTAB micelles. The hydrophobic part of the TEA or TEO molecules may occupy the core part of the micelles whereas the hydrophilic amine groups of these molecules may occupy the interface, affecting the condensation of the silica species between the self-assembled hexagonal micellar assembly. This also caused a huge reduction in the specific surface area of TE-@MS-x and TO-@MS-x samples as compared to pristine MSN@SiO2. The specific surface area of TE-@MS-10.9 is 569 m2 g−1. When the pH of the synthesis mixture is increased, the specific surface area increases to 698 for TE-@MS-11 and then decreases to 315 m2 g−1 for TE-@MS-11.5. On the other hand, the specific surface area of TO-@MS-x increases from 627 to 736 m2 g−1 as the pH is increased from 10.9 to 11. When the pH is increased further to 11.5, the specific surface area decreases to 422 m2 g−1. Interestingly, among the TE-@MS-x, TE-@MS-11 exhibits the highest specific surface area due to the well-ordered structure while TE-@MS-11.5 registers the lowest specific surface area due to partially collapsed porous structure as well as the large mesopore diameter. A similar trend was observed for the TO-@MS-x series of samples and TO-@MS-11 exhibits the highest specific surface area. Considering the well-ordered structure and the optimal specific surface area and pore volume, TE-@MS-11 and TO-@MS-11 are chosen for the drug adsorption and drug release studies.
HRSEM was used to further examine the surface texture, morphology, and uniformity in the size distribution of the MSN samples synthesised using different procedures. The SEM images of MSN@SiO2 and NH2-MSN@SiO2 samples shown in Fig. SI.2A and B† displayed uniform spherical nanoparticles in the size range of 350–400 nm. The uniform spherical morphology with the surface texture typical of the mesoporous particles is clearly visible in the SEM images, suggesting that the selected synthesis methods yield particles with fairly uniform size distribution. It was also observed from Fig. SI.2B† that APTES functionalisation did not change the morphology or the surface texture of the nanoparticles. On the other hand, TE-@MS-x and TO-@MS-x samples showed uniform particles, but the size of these particles varied significantly when the pH of the solution was varied. The size of the particles is in the range of 425–530 nm at pH 10.9, 430–500 nm at pH 11.0, and 380–440 nm at pH 11.2. It should be noted that the particle size of TE-@MS-x samples is much larger than that of MSN@SiO2 and NH2-MSN@SiO2. In comparison, the TO-@MS-x samples exhibited a smaller size (Fig. 3D) as compared to all MSN samples. The smallest particles were observed at pH 10.9 with a size range of 270–320 nm while the size increased to 440–490 nm and 390–470 nm at pH 11.0 and 11.2, respectively. Even though the smallest particles were observed at pH 10.9 for TO-@MS-11, the sheet like features resulting from the partial loss in morphology was also observed for this sample. On the other hand, TE-@MS-11.5 and TO-@MS-11.5 synthesised at pH 11.5 depicted a complete loss in morphology with few spherical nanoparticles clustered together in large agglomerates (Fig. SI.2C and D†). As the uniform morphology and high surface texture are beneficial for drug loading and release, TE-@MS-11 and TO-@MS-11 which exhibited uniform morphology without any sheet like structure were chosen for the drug delivery applications.
 | ||
| Fig. 3 SEM images of TE-@MS-x synthesised at different solution pH: (A) TE-@MS-10.9, (B) TE-@MS-11, and (C) TE-@MS-11.2 and TO-@MS-x synthesised at different solution pH: (D) TO-@MS-10.9, (E) TO-@MS-11, and (F) TO-@MS-11.2. | ||
To confirm the internal mesoporous structure and surface morphology, the selected samples were analysed by high resolution transmission electron microscopy (HRTEM) studies and the images are presented in Fig. 4 and ESI.3.† It is also observed that pH has a huge effect on the morphology of both TE-@MS-x and TO-@MS-x samples. Fig. 4B shows that the morphology of TE-@MS-11.0 is quite different from MSN@SiO2. HRTEM image of TE-@MS-11.0 clearly reveals the egg-yolk core–shell morphology with thin mesoporous silica layers in the outer shells of the particles. Large mesopores are seen between the silica core and the outer mesoporous silica walls. This unique morphology is generally difficult to achieve in a single step synthesis, but we have demonstrated for the first time that this unique morphology can be achieved with the addition of a small amount of TEA in the synthesis mixture.48–54 This unique porous morphology offers both macropores and small mesopores which may offer large space for the large drug molecules. It should also be noted that the hollow space in the particle can be finely tuned by the simple adjustment of the amount of TEA in the synthesis mixture ‘B’. As the pH of the synthesis medium increases from 10.9 to 11.2, TE-@MS-x shows a transition from large core thin shell to egg-yolk core–shell to small core and large shell. When different particles of TE-@MS-11.0 are analysed, it can be observed that the pore size and the shell thickness of the particles are uniform. These particles are different from core–shell MSN as the hollow space is created between the silica core and the mesoporous silica walls which are beneficial for the adsorption of a large amount of drug molecules. A similar morphology is also observed for TO-@MS-x however, the hollow interior is not that well developed for these samples and can be tuned by simply changing the pH value similar to the one observed for TE modified MSNs. The formation of such unique yolk–shell structures is decided by the pH of the medium controlled by TEA or TEO. At first, the spherical micelles of F127 are formed and when CTAB is added to this solution, the electrostatic interaction between positively charged micelle CTA+ and silicate facilitates the formation of a self-assembled mesophase on the surface of the F127 spherical micelles, forming spherical morphology. The pH of the solution plays a critical role in controlling the interaction between the silanol groups and the CTA+ ions due to the difference in the stability of the silanol species at different solution pH values, which was controlled by the addition of TEA or TEO instead of ammonia. At solution pH 11, it is assumed that the large TEA molecules may interact with the hydrophobic core of the F127 micelles and interact with the unreacted silanol groups due to the high stability of the silanol groups at this particular pH, forming the silica yolk inside the core–shell structure. When CTAB is added, the electrostatic interaction between positively charged micelle CTA+ and silicate facilitates the formation of a self-assembled mesophase on the surface of the F127 spherical micelles, forming spherical morphology with a clear egg-yolk structure. Since, the solution pH is the key for controlling the self-condensation of silica species, the formation of the yolk structure occurs only at a specific pH that differs for both TEA and TEO due to the difference in their sizes and basicity.
 | ||
| Fig. 4 HRTEM images of TE-@MS-x synthesised at different solution pH: (A) TE-@MS-10.9, (B) TE-@MS-11, (C) TE-@MS-11.2 and TO-@MS-x synthesised at different solution pH: (D) TO-@MS-10.9, (E) TO-@MS-11 and (F) TO-@MS-11.2. | ||
The presence of nitrogen functionality on the surface of the MSN samples was characterised by using a CHNS analyser. It was observed that while MSN samples did not show any nitrogen, TE-@MS-11 and TO-@MS-11 showed 0.1 and 0.2 wt% nitrogen, respectively, suggesting that the surface was functionalised with the nitrogen functionality. It must also be noted that the APTES functionalised sample MSN-NH2 showed 1.74 wt% nitrogen, depicting a significantly higher nitrogen content than the TE-@MS-11 and TO-@MS-11 samples. The distribution of nitrogen in the MSN was also analysed by SEM-EDS and the representative images of TE-@MS-11 and TO-@MS-11 are depicted in Fig. SI.4.† A uniform distribution of nitrogen is clearly observed from the images overlapping with silicon and oxygen confirming the advantage of using TEO and TEA in a single-step synthesis and functionalisation process.
3.2. Dox loading and release studies
Dox is a standard first line of chemotherapy drug and was selected in this study as a model drug owing to its therapeutic action in prostate cancer and its fluorescence properties that allow its imaging inside the cells. The effect of the unique single-step synthesis and surface functionalisation method on the drug loading and release efficacy was analysed and compared with MSN@SiO2 and NH2-MSN@SiO2. TE-@MS-11 and TO-@MS-11 were used for comparing the drug loading and release characteristics of the modified silica as well as cell viability and uptake studies, as these samples depicted the best combination of high surface area and ordered porous structure, and egg-yolk core–shell morphology without the presence of sheet like particles. The Dox loaded MSN@SiO2, NH2-MSN@SiO2, TE-@MS-11, and TO-@MS-11 are labelled as @MSN-Dox, NH2-@MSN-Dox, TE-@MS-Dox and TO-@MS-Dox, respectively. It was found that TE-@MS-Dox and TO-@MS-Dox depicted higher drug loading as compared to MSN@SiO2 and NH2-@MSN-Dox, even though the specific surface area of TE-@MS-Dox and TO-@MS-Dox is lower than those of MSN@SiO2 and NH2-@MSN-Dox. MSN@SiO2 registers a Dox loading of 347 μg mg−1 of the adsorbent whereas NH2-MSN@SiO2 exhibits a drug loading capacity of 317 μg mg−1. The surface area of NH2-MSN@SiO2 is 411 m2 g−1 which is significantly lower than that of the pristine MSN@SiO2 (1230 m2 g−1) that results in reduced drug loading. The drug loading was normalised by the surface area to understand the effect of surface functionalisation on the drug loading capacity. It was noted that drug loading per unit surface area is higher for NH2-MSN@SiO2 (771 μg m−2) as compared to MSNs (282 μg m−2), suggesting that the modification of surface with amine groups increases the amount of drug adsorbed per unit surface area of the material. In comparison, the Dox loading capacities of TE-@MS-11 and TO-@MS-11 are 425 μg mg−1 and 481 μg mg−1, respectively, clearly suggesting the superiority of the single step synthesis and functionalisation process over the drug loading capacity of MSNs. Even though the specific surface area of TE-@MS-11 and TO-@MS-11 is significantly lower than MSN@SiO2, the drug loading per unit surface area of these samples is 608 μg m−2 and 577 μg m−2, respectively. It should be noted that the drug loading per unit surface area of these samples is more than twice that of pristine MSN@SiO2 and comparable to that of NH2-MSN@SiO2. A high drug adsorption per unit surface area on TE-@MS-11 and TO-@MS-11 samples clearly indicates that even though the amount of nitrogen in these samples is significantly less than that in NH2-MSN@SiO2, the single-step synthesis and functionalisation process results in a uniform distribution of nitrogen functionalities on the surface of the core–shell MSNs and the hollow space with a large pore volume that help in adsorbing a large amount of the drug. It is noted that the zeta potential of TE-@MS-11 and TO-@MS-11 is in a similar range from −29 to −34 mV to MSN@SiO2 (−26.4) (Table 2S†) and hence the higher drug adsorption cannot be ascribed to the electrostatic attraction. The high drug adsorption also suggests that Dox may also bind strongly to the surface of the MSNs that may be released slowly over a longer duration.The drug release characteristics of MSNs was measured in two different buffer systems (PBS at pH 7.4 and acetate at pH 5.0) to mimic the physiological pH conditions of normal cells and slightly acidic conditions of the cancer cells. The diffusion of the drug from the MSN surface is influenced by the pH, rate of degradation of silica and the bonding strength between the drug and the MSN surface. The drug release kinetics was measured by following the drug release from various samples of MSNs over a period of 72 h and the rate of drug release with time is depicted in Fig. 5A and B. It can be observed that the Dox release from various MSN samples is higher in acidic buffer as compared to the physiological pH. For example, the drug release of TE-@MS-Dox is 9% and 28% while TO-@MS-Dox showed a release of 12.5% and 28% of the total Dox loaded at pH 7.4 and pH 5 respectively, within 72 h. Higher release of Dox from various substrates at acidic pH has partially been attributed to the protonation of its amine groups that partially increases its hydrophilicity.55 The increased hydrophilicity results in an increased drug–solvent interaction that competes with the drug–adsorbent interaction and the balance between the two dictates the release rate. Among all samples, NH2-@MSN-Dox showed the lowest Dox release in acidic pH after 72 h that can be ascribed to the stronger drug–adsorbent interaction between the amine functionalised surface of MSNs and Dox, resulting in a lower release.
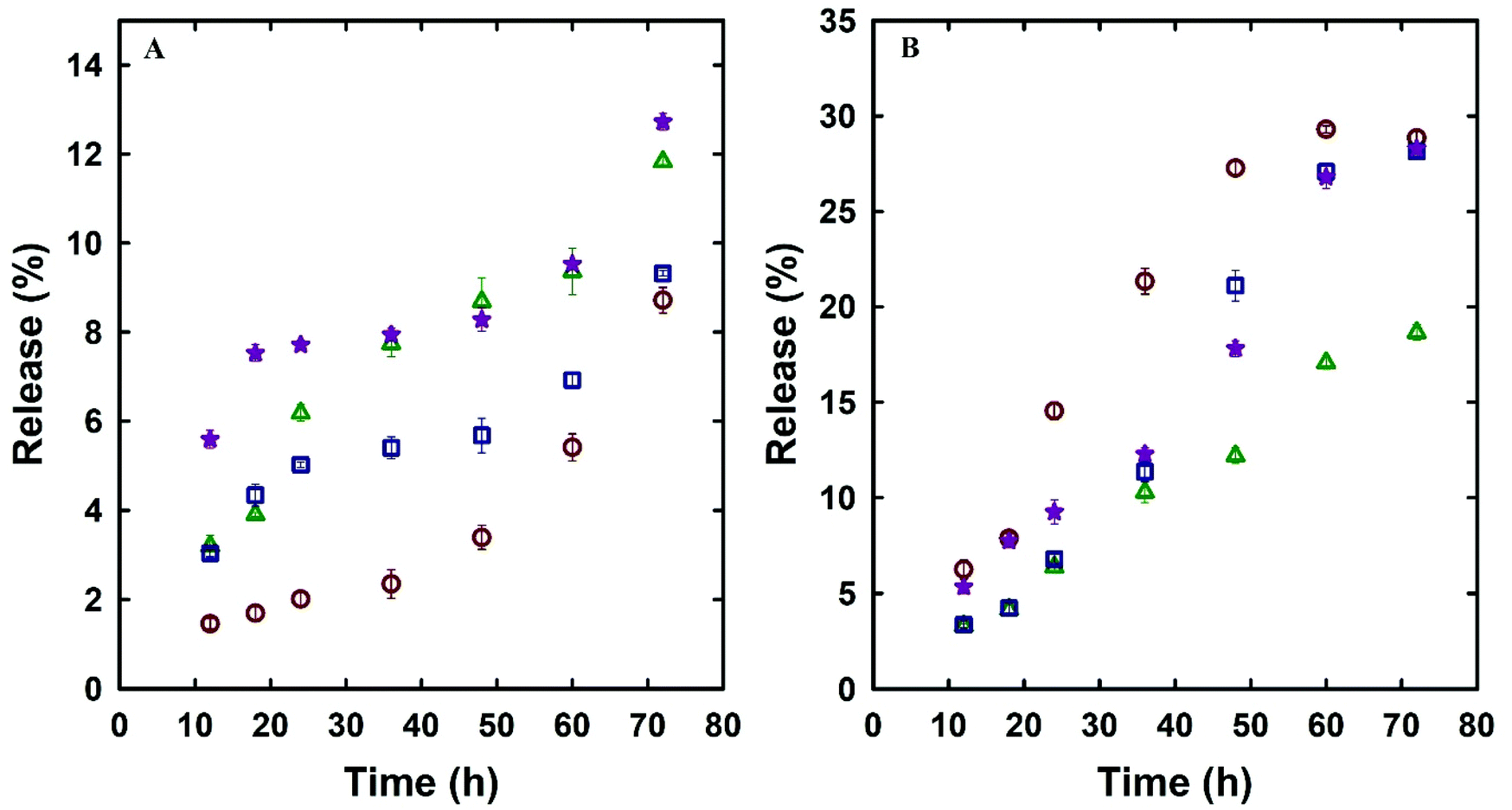 | ||
Fig. 5 Dox release profile of MSN@SiO2 ( ), NH2-MSN@SiO2 ( ), NH2-MSN@SiO2 ( ), TE-@MS-11 ( ), TE-@MS-11 ( ), TO-@MS-11 ( ), TO-@MS-11 ( ) (A) pH 7.4 and (B) pH 5.0. ) (A) pH 7.4 and (B) pH 5.0. | ||
The release rate and profile is also dictated by the pore structure, pore diameter, particle size, and the structural order as they dictate access of the solvent to the surface of the mesoporous silica as well as the diffusion of the drug molecules from the pores to the bulk media.56 It can be observed from Fig. 5A and B that the drug release rate and pattern in acidic and neutral buffers of various MSN samples is quite similar among various MSN samples, suggesting that the release is affected only by the pH of the buffer system. Since the structural order and pore size is similar among these MSN samples and hence the rate of drug release is not affected drastically among these samples. The drug release profiles of @MSN-Dox, TE-@MS-Dox and TO-@MS-Dox samples at pH 7.4 depicted an initial low drug release within the first 12 h (<6%) followed by a plateau up to 48 h and further higher release between 50 and 72 h. The initial drug release can be ascribed to the drug molecules attached to the surface of the MSNs while the drug release after 48 h may be ascribed to the slow disintegration of the MSN surface. It is also noted that the @MSN-Dox sample shows the minimum drug release within the first 12 h while the TO-@MS-Dox sample shows the highest drug release at pH 7.4, suggesting that the interaction of Dox with the nitrogen functionalities is not as strong as expected from the result of the highest drug loading in TO-@MS-Dox. This is important as it suggests that the MSN surface is optimally modified to increase the drug adsorption from a uniform distribution of TEO over a large surface of MSN.
The maximum drug release of ∼13% was demonstrated by TO-@MS-Dox and NH2-@MSN-Dox in neutral pH. In acidic pH, @MSN-Dox, TE-@MS-Dox and TO-@MS-Dox showed more than 2.5 times drug release after 72 h as compared to neutral pH. The release profile demonstrated a sustained drug release with less than 10% of the drug released in the first 24 h followed by a sustained release for the next 24–36 h followed by a plateau. The acidic pH reduces the dissolution of MSN and hence the drug release rate did not increase after 48–60 h, as observed for MSN samples in near neutral pH. This is an important result because the higher release at acidic pH is critical as most of the cancer cells are in a weakly acidic environment and our prepared materials can target the cancer cells with the help of an increased drug release rate in the acidic environment. It must be noted that even though the total percentage of drug released from TE-@MS-Dox and TO-@MS-Dox is similar to MSN samples, TE-@MS-11 and TO-@MS-11 depicted a higher drug loading capacity of 425 μg mg−1 and 481 μg mg−1 respectively compared to only 347 μg mg−1 for MSN samples. Therefore, the overall drug quantity released from TE-@MS-Dox and TO-@MS-Dox is higher as compared to pristine @MSN-Dox. This may be attributed to the large pore diameter and the unique egg-yolk core–shell morphology with a large hollow space which assists in improving the drug release performance. The slow, sustained and a larger amount of drug release from the TE-@MS-Dox and TO-@MS-Dox samples as compared to pristine @MSN-Dox and NH2-@MSN-Dox suggests that single step synthesis and surface modification of MSNs with nitrogen functionalities with the help of TEA and TEO depicts higher drug loading without compromising the release characteristics as compared to post synthesis modification of MSNs.
3.3. Cellular uptake studies
The cellular uptake studies were carried out both by flow cytometry as well as fluorescence microscopy using PC-3 cells. The nanoparticles were labelled with fluorescent dyes as discussed in the respective Materials and methods section for analysis by these methods. Flow cytometry is a cell sorting and analysis technique that detects the fluorescence or scattering from cells. Usually, the nanoparticle uptake by cells is determined by observing the increase in the side scattering signal (SSC) with increasing uptake of nanoparticles due to an increase in the granularity of the cells.57,58 @MSN-Dox, TE-@MS-Dox, and TO-@MS-Dox were incubated with PC-3 cells for 24 h at a concentration of 100 μg mL−1 for internalisation studies. Fig. 6A shows the SSC histograms of PC-3 cells after 24 h of incubation with @MSN-Dox, TE-@MS-Dox, and TO-@MS-Dox. These data reveal a high count from the SSC channel when compared to the untreated cells (Fig. 6A, d (black line)). Note that the signal intensity of the forward scatter remained unchanged. The high-count rate observed in the SSC channel confirmed the successful uptake of all Dox loaded MSNs in the PC-3 cells. Similar results were obtained for the internalisation of all Dox loaded MSN samples in the LNCaP cells as well (ESI. 5A–C†). In addition, the concentration dependent uptake of pristine MSN@SiO2 (50–300 μg mL−1) was also analysed using flow cytometry. It was found that the SSC signal intensity increases with the increase in the concentration of nanoparticles as both the number of nanoparticles internalised by the cells as well as the number of cells with nanoparticle uptake increase (Fig. 6A, d). The mean fluorescence intensity from cells incubated with 300 μg mL−1 MSN@SiO2 is significantly higher than that from the cells exposed to 50 μg mL−1 MSN@SiO2 suggesting that pristine MSNs are internalised by the cells in a concentration dependent manner. However, the localisation of these nanoparticles within the cells cannot be determined by these flow cytometry studies, and hence fluorescence microscopy analysis was conducted to ascertain the internalisation of MSNs in cells.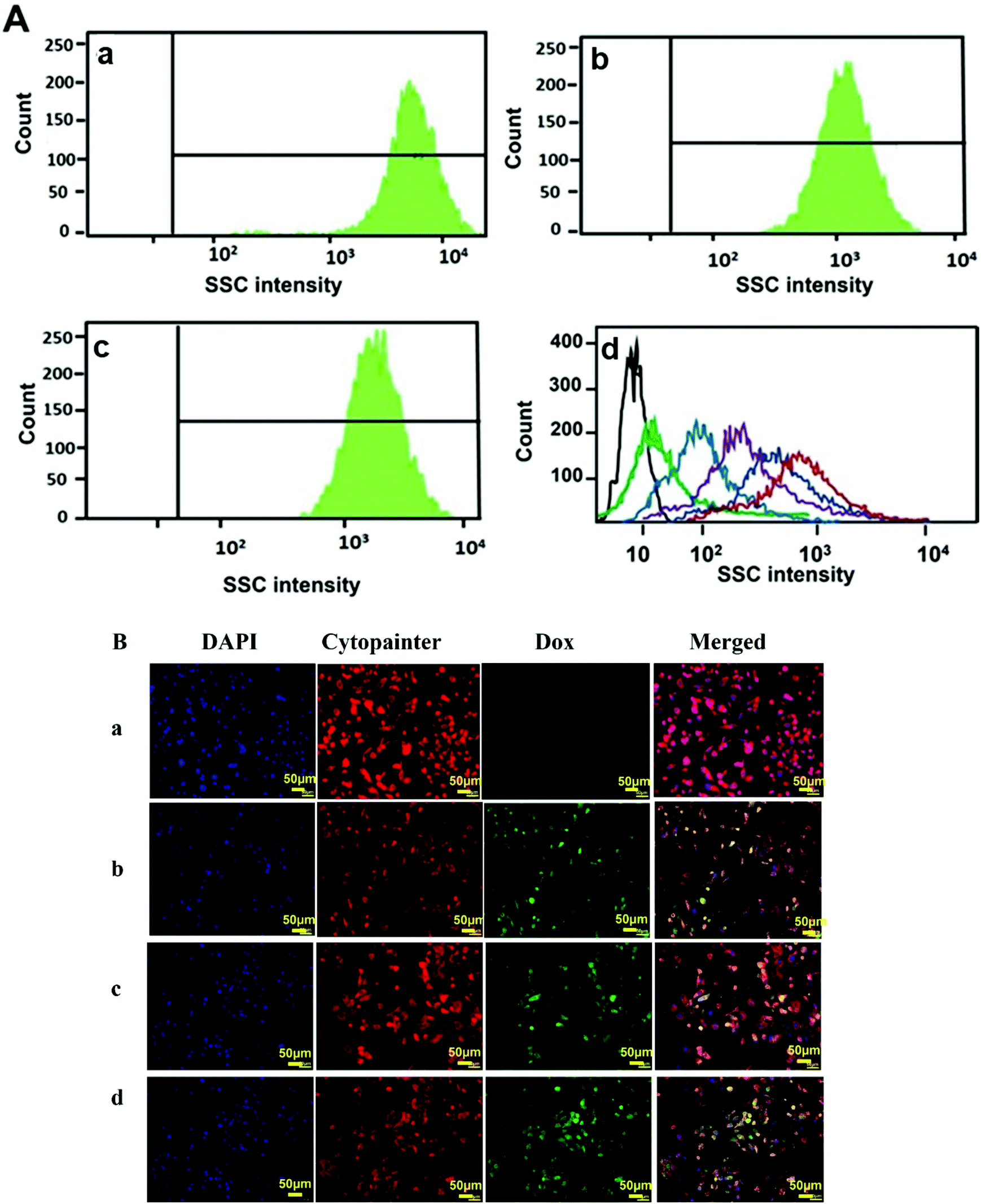 | ||
| Fig. 6 Cellular uptake studies by (A) analysing the side scattering intensity (SSC) histograms of (a) MSN@SiO2-Dox, (b) TE-@MS-Dox, and (c) TO-@MS-Dox. (d) MSN@SiO2 concentrations 50–300 μg mL−1 (cells (black), 50 (green), 100 (light blue), 150 (purple), 200 (dark blue), 300 (red) μg mL−1) and (B) fluorescence microscopy imaging of (a) PC-3 (b) @MSN-Dox, (c) TE-@MS-Dox, and (d) TO-@MS-Dox. The blue colour denotes the DAPI stained nucleus, red depicts the cytopainter stained plasma membrane and cytoplasm, green denotes Dox and the merged image depicts co-localisation. | ||
Fig. 6Ba–d depicts the internalisation of @MSN-Dox, TE-@MS-Dox and TO-@MS-Dox in PC-3 cells. For imaging, the cell nuclei were labelled with 4,6-diamidino2-phenylindole (DAPI) while the cytoplasm was labelled with Cytopainter. The natural fluorescence of Dox was used as a label for imaging the nanoparticles. The fluorescence images were collected for PC-3 cells treated with different MSN samples at a concentration of 100 μg mL−1 for 12 h. The internalisation of Dox and its intracellular release can be observed by the changes in the distribution and intensity of the fluorescence signal from Dox. The fluorescence images confirm the internalisation of Dox loaded nanoparticles within PC-3 cells as observed by the fluorescence from the Dox loaded MSNs (Fig. 6B panel b, c, and d corresponding to @MSN-Dox, TE-@MS-Dox, and TO-@MS-Dox, respectively) while no Dox fluorescence is observed from the untreated PC 3 cells (Fig. 6B panel a).
Based on the co-localisation with DAPI and Cytopainter, it is observed that the internalised MSN nanoparticles were mainly localised within the cytoplasm of the cells, though it is difficult to ascertain based on the fluorescence images whether the Dox is present within the lysosome or has been released into the cytoplasm.59 However, it is known that the nanomaterials are released from the endosomal/lysosomal compartments following the proton sponge effect, mechanical swelling or the membrane destabilisation methods within a few hours of internalisation.60 It was recently shown using amine functionalised polystyrene nanoparticles that the nanoparticles escaped the lysosomes within 6 h and a similar behaviour is expected from the amine and nitrogen functionalised MSN samples in this study.61 The cell internalisation studies confirm that the uptake of MSNs and Dox loaded MSNs do not change with surface modification and a large proportion of the nanoparticles is taken up inside the cells within 12–24 h that will facilitate the release of Dox within the cells. These results confirm that the single step synthesis and functionalisation of MSNs not only facilitate a higher drug loading within TE-@MS-11 and TO-@MS-11 but also show cellular internalisation comparable to the pristine MSNs.
3.4. In vitro cytotoxicity evaluation
The in vitro cell viability of bare and functionalised MSNs before and after Dox loading was analysed using PC-3 and LNCaP cells at 24 and 48 h of incubation. The cell viability was analysed by the resazurin assay with untreated cells used as a baseline for estimating the cell viability. The cytotoxicity of free Dox exposed to PC-3 and LNCaP cells in complete media is shown in the ESI. 6a and b.† It is observed that Dox shows similar dose dependent toxicity towards both LNCaP and PC-3 cells. The IC 50 of free Dox in 24 h is ∼50 μg mL−1 which is significantly higher than those reported in other studies although these studies were conducted in serum free media or PBS as compared to the current studies conducted in complete media.62 However, the IC 50 of free Dox in 48 h was found to be ∼6–12 μg mL−1 for LNCaP and PC-3 cells in agreement with the reported values.63 It is also observed that the cell viability at 48 h shows a gradual decrease from 50% ± 0.6 at 6 μg mL−1 to 20% ± 0.6 at 300 μg mL−1 Dox. The cell viability of MSN@SiO2, TE-@MS-11 and TO-@MS-11-treated LNCaP and PC-3 cells is shown in Fig. 7(a, b, c and d), respectively. It can be observed that both bare MSN@SiO2 and TE-@MS-11 samples are highly biocompatible even at a concentration of 100 μg mL−1 with more than 90% cell viability after 48 h of incubation in both the cell lines, revealing almost negligible cytotoxicity of these unique nanoparticles. However, TO-@MS-11 samples showed a concentration dependent decrease in the cell viability especially at higher concentrations, though the cell viability was higher than ∼70% ± 0.8 even after 48 h of incubation.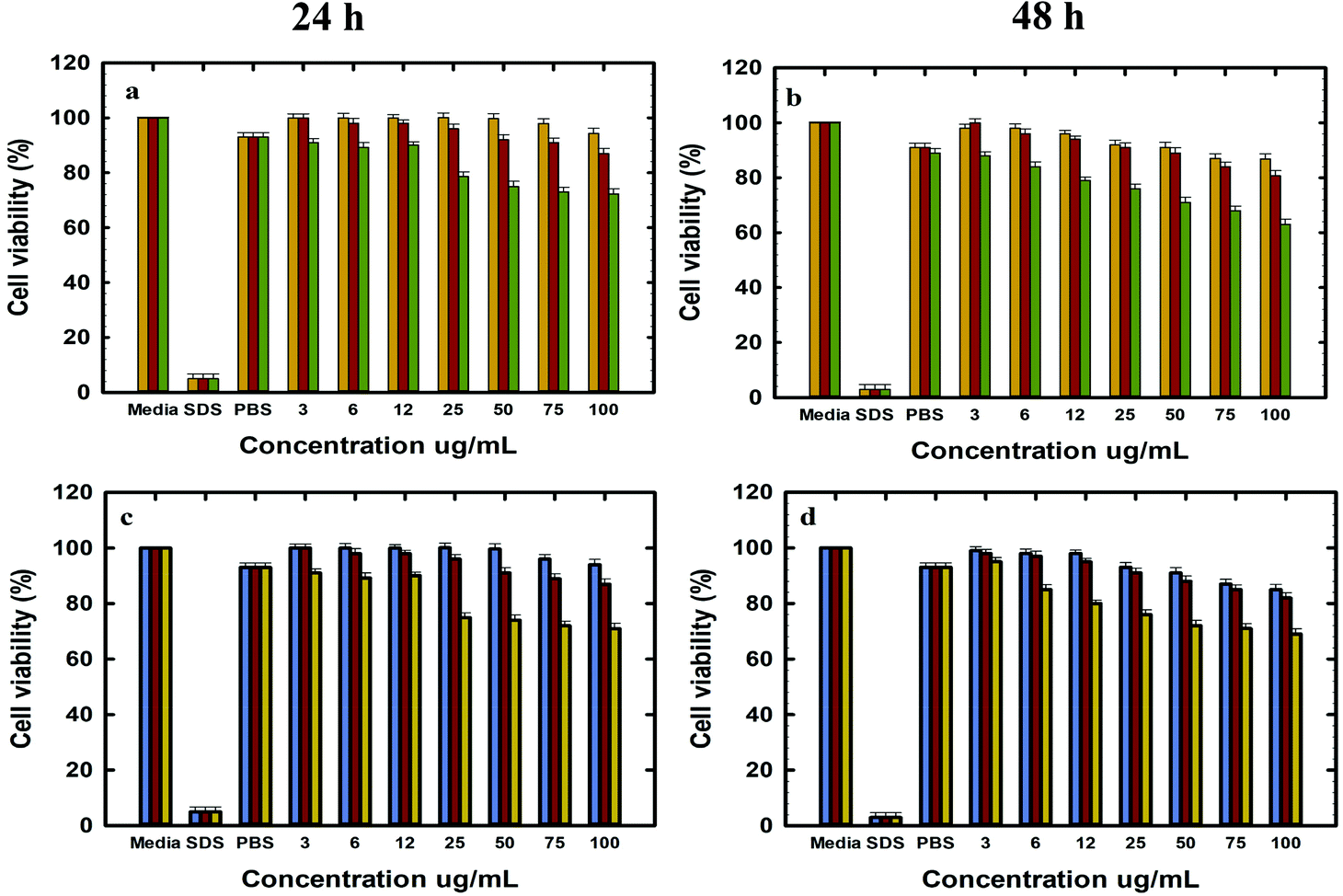 | ||
| Fig. 7 Invitro cytotoxicity studies in LNCaP cells at (A) 24 h and (B) 48 h with MSN@SiO2 (orange), TE-@MS-11 (red), and TO-@MS-11 (green); and in PC-3 cells at (C) 24 h and (D) 48 h with MSN@SiO2 (blue), TE-@MS-11 (red) and TO-@MS-11 (yellow). | ||
The results of cell viability of LNCaP, PC-3 treated with @MSN-Dox, TE-@MS-Dox and TO-@MS-Dox for 24 and 48 h are shown in Fig. 8A, B, C and D, respectively. It is observed that @MSN-Dox, TE-@MS-Dox and TO-@MS-Dox samples showed higher cytotoxicity as compared to the bare MSN@SiO2 towards both PC-3 and LNCaP cells. The higher cytotoxicity in these samples can be ascribed to the cytotoxic action of the Dox released from the MSN samples. The IC 50 of TE-@MS-Dox and TO-@MS-Dox after 48 h of incubation with the samples was ∼100 μg mL−1 for LnCaP cells while the particles were highly cytotoxic to PC3 cells, at all concentrations used in the current study within 48 h. LNCaP cells treated with @MSN-Dox showed minimal cytotoxicity in 24 h though the cytotoxicity increased in a concentration dependent manner reflecting in ∼20% ± 0.8 cell inhibition from 100 μg mL−1 of MSN.
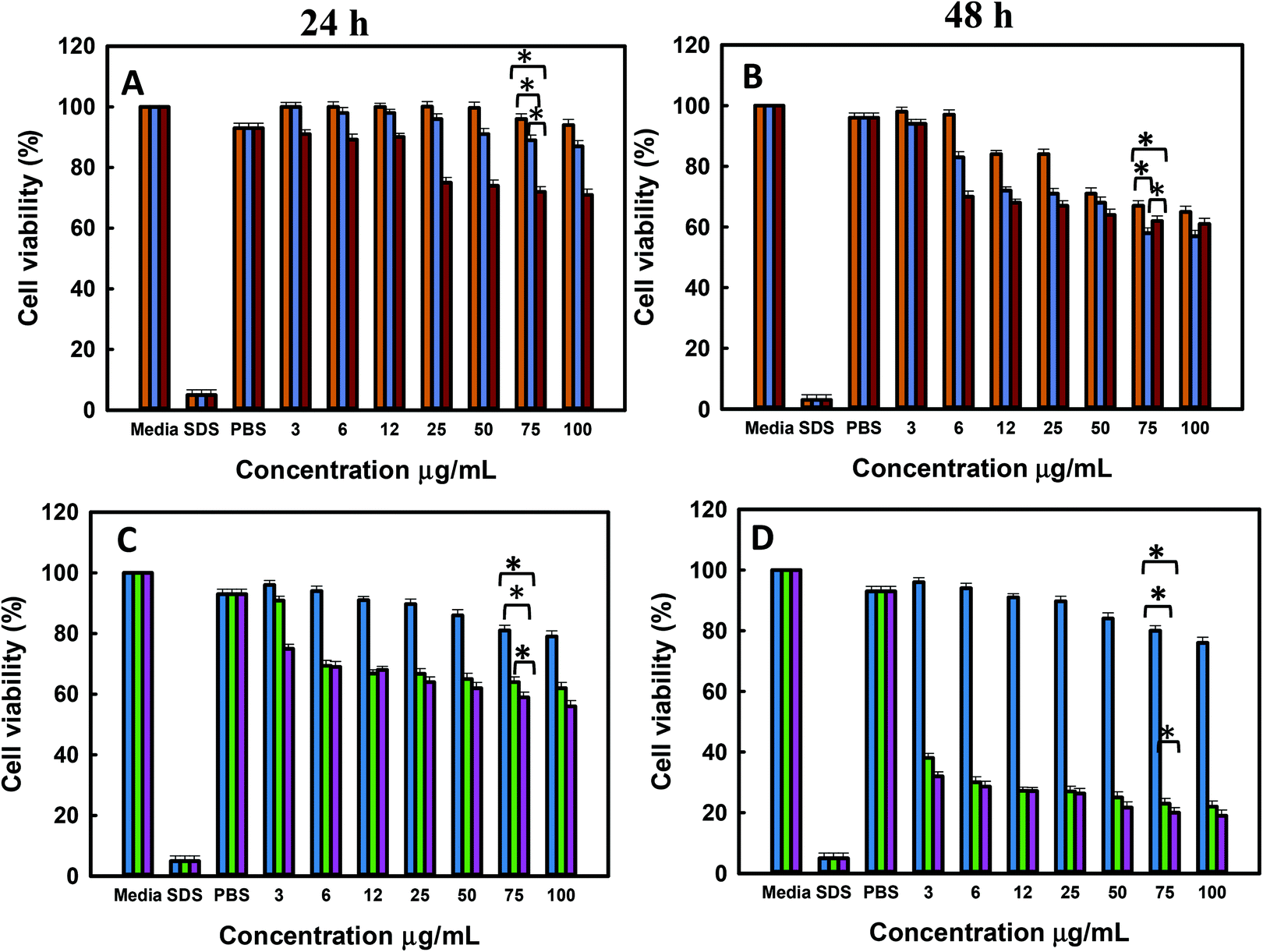 | ||
| Fig. 8 Invitro cytotoxicity studies (A) and (B) LNCaP at 24 h and 48 h with MSN@SiO2-Dox (orange), TE-@MS-Dox (blue), and TO-@MS-Dox (red); (C) and (D) PC-3 at 24 h and 48 h with MSN@SiO2-Dox (blue), TE-@MS-Dox (green), and TO-@MS-Dox (purple) (p < 0.05). | ||
However, both TE-@MS-Dox and TO-@MS-Dox showed higher cytotoxicity with >30–40% ± 1.2 cell inhibition within 24 h of their exposure to LNCaP cells at 100 μg mL−1. Similar studies are reported on cell type dependent cytotoxicity of doxorubicin.64–67 The higher cell inhibition could possibly be ascribed to the higher loading of Dox in TE-@MS-Dox (425 μg mg−1) and TO-@MS-Dox (481 μg mg−1) as compared to the bare MSN@SiO2 (347 μg mg−1). Based on the cell free drug release pattern in acidic pH, it can be calculated that a maximum of 4.9, 4.4, 3.6 and 7.8, 11.6, 8.9 μg mL−1 of Dox can be released from the 100 μg mL−1 MSN samples (@MS-Dox, TE-@MS-Dox and TO-@MS-Dox, respectively) given to the cells in 24 and 48 h, respectively.
Thus, within 24 h TE-@MS-Dox and TO-@MS-Dox are expected to release similar amounts of Dox as compared to @MSN-Dox while a slightly higher amount of Dox can be released within 48 h. The difference in the Dox released in situ within the cell free conditions suggests that either TE-@MS-Dox and TO-@MS-Dox are more efficient in delivering the Dox to the cells or that the drug release behaviour in cell free release buffer is not an absolute indicator of the Dox release behaviour inside the cells. The difference in the cytotoxicity could also be understood on the basis of the transportation of the nanoparticles. As most nanoparticles are transported via the endo-lysosomal pathway, they are exposed to a lower pH conditions within the lysosomes. The time spent by @MSN-Dox samples within the lysosomal compartment will enhance the drug released within the lysosomes at acidic pH and may be released in the cytoplasm either by membrane leakage or by the disruption of lysosomes. The differences in the drug release could also be ascribed to the differences in the time spent by TE-@MS-Dox, TO-@MS-Dox, and @MSN-Dox in the lysosomes owing to differences in the size and surface functionality of particles. Various studies have reported on the effect of structural parameters such as size, shape and morphology of MSNs on their anticancer effects.68–74 The synthesis method used in this work resulted in materials with different size and morphologies that could result in different internal structures, available surface area and the amount of drug released. It was reported that the intracellular delivery of drugs could also differ based on the loading capacity and was found to be greater in mesoporous particles with defined egg yolk shell structure as compared to core–shell silica.75 Even though the drug release studies were carried out in the serum free buffers to compare the respective release characteristics of different samples, the final drug release from the MSNs in cell culture medium (or inside the cells) is affected significantly by the surface area, pore network and presence of proteins on the surface. Thus, the relatively low cytotoxicity of @MSN-Dox as compared to other MSN samples could be ascribed to the influence of its physico-chemical properties on its biological behaviour. The high surface area of MSN-SiO2 could result in higher adsorption of proteins from the serum that can change its uptake as well as drug release behaviour compared to TE-@MS-Dox and TO-@MS-Dox. It is possible that the difference in the adsorbed proteins could promote their exocytosis from the cells resulting in poor cytotoxicity of @MSN-Dox. Such exocytosis of MSNs have been reported previously and the final uptake and release may occur after endocytosis following the surface erosion.76 Similarly, a thicker protein corona can form on @MSN-Dox compared to other MSNs (due to the high surface area of MSN@SiO2) that can reduce the overall drug release owing to the presence of a thicker diffusion barrier, blocking the access to mesopores and reduced dissolution of silica from the surface and the pore.77 From the cytotoxicity results, it is clear however that administration of Dox loaded MSNs is better than administration of free Dox to the cells at the same concentration, as MSN particles are more efficient in delivering the Dox to the cells due to their effective internalisation as compared to the free Dox. This can significantly reduce the side effects caused by the use of excess free Dox for chemotherapy treatment.
The cytotoxicity exerted by TE-@MS-Dox and TO-@MS-Dox is significantly better in PC-3 cells as compared to the LNCaP cells. It can be observed from Fig. 8C and D that the cytotoxicity from the administration of 100 μg mL−1 of the TE-@MS-Dox and TO-@MS-Dox showed only 20% ± 0.9 cell viability (80% inhibition) in 48 h. In comparison, the administration of free Dox at 200 μg mL−1 resulted in 30% cell viability (70% inhibition) as compared to <10 μg mL−1 Dox expected to be released from TE-@MS-Dox and TO-@MS-Dox samples in 48 h. The incredibly high cell death from the small amount of drug released directly in cells suggests that TE-@MS-Dox and TO-@MS-Dox are able to deliver Dox more effectively to the cancer cells as compared to administration of free Dox and @MSN-Dox. The results obtained from the resazurin based cytotoxicity assay correlate well with the cell internalisation studies suggesting that a higher uptake of nanoparticles resulted in direct delivery of Dox inside the cells, resulting in higher cytotoxicity as compared to free Dox. It also suggests that the TE-@MS-Dox and TO-@MS-Dox can deliver Dox more efficiently to the cancer cells as compared to bare MSN@SiO2.
4. Conclusions
In this study, we designed and synthesized egg-yolk core–shell MSNs with uniformly distributed nitrogen functionalities on the surface by using TEA and TEO as a hydrolysis agent. It was found that the addition of a small amount of novel hydrolysis agents during the dual surfactant template-based MSN synthesis process results in creating a unique egg-yolk-core–shell morphology in a single step. This is by far the simplest method reported for the synthesis of egg-yolk core–shell MSNs. The TEA or TEO modified MSNs showed high specific surface area, large pore volume and the presence of highly dispersed nitrogen functionalities on the wall surface. The uniform distribution of nitrogen assisted in achieving a higher Dox loading in TE-@MS-11 (425 μg mg−1) and TO-MS@11 (481 μg mg−1) as compared to MSN@SiO2 (347 μg mg−1) or NH2-MSN@SiO2 (317 μg mg−1). The drug release from both TE-@MS-11 and TO-@MS-11 was two times higher in acidic buffer and significantly higher than NH2-MSN@SiO2 depicting the superiority of single step surface functionalisation process reported in this study. The cellular uptake study showed that both pristine and TEA or TEO functionalised MSNs were internalised by the cells within 12 h. The cytotoxicity studies in LNCaP and PC-3 cells showed higher cytotoxicity from TE-@MS-Dox and TO-@MS-Dox as compared to @MSN-Dox, clearly indicating the superiority of functionalised MSNs in exerting cytotoxicity of the Dox in the cancer cells. The single-step synthesis and functionalisation of egg-yolk core–shell MSNs with high surface area and large pore volume reported in this study is one of the simplest methods for increasing the drug loading capacity of MSNs. The combination of macro and mesopores owing to the unique egg-yolk core–shell morphology will significantly increase the range of drugs, antibodies, and proteins that can be loaded on these MSNs for a variety of drug delivery applications.Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
We acknowledge the support from Ms Nicole Cole (Analytic and Biomolecular Research Facility) for the flow cytometry studies and Dr Huiming Zhang (Electron Microscopy and X-ray Unit) for the TEM imaging and Mr Nithinraj Panangattu Dharmarajan for SEM-EDS imaging.References
- R. B. Hayes, R. G. Ziegler, G. Gridley, C. Swanson, R. S. Greenberg, G. M. Swanson, J. B. Schoenberg, D. T. Silverman, L. M. Brown and L. M. Pottern, Cancer Epidemiol. Prev. Biomarkers, 1999, 8, 25–34 CAS.
- E. Pérez-Herrero and A. Fernández-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef PubMed.
- Q. Wu, Z. Yang, Y. Nie, Y. Shi and D. Fan, Cancer Lett., 2014, 347, 159–166 CrossRef CAS PubMed.
- A. M. Bode and Z. Dong, npj Precis. Oncol., 2018, 2, 1–6 CrossRef PubMed.
- Z. Li, Y. Chen, Y. Yang, Y. Yu, Y. Zhang, D. Zhu, X. Yu, X. Ouyang, Z. Xie and Y. Zhao, Front. Bioeng. Biotechnol., 2019, 7, 293 CrossRef PubMed.
- K. Nurgali, R. T. Jagoe and R. Abalo, Front. Pharmacol., 2018, 9, 245 CrossRef PubMed.
- R. Bagherifar, S. H. Kiaie, Z. Hatami, A. Ahmadi, A. Sadeghnejad, B. Baradaran, R. Jafari and Y. Javadzadeh, J. Nanobiotechnol., 2021, 19, 110 CrossRef PubMed.
- K. T. Al-Jamal, W. T. Al-Jamal, J. T.-W. Wang, N. Rubio, J. Buddle, D. Gathercole, M. Zloh and K. Kostarelos, ACS Nano, 2013, 7, 1905–1917 CrossRef CAS PubMed.
- M. G. Antoniraj, C. S. Kumar and R. Kandasamy, Colloid Polym. Sci., 2016, 294, 527–535 CrossRef CAS.
- Y. Cheng, J. Hao, L. A. Lee, M. C. Biewer, Q. Wang and M. C. Stefan, Biomacromolecules, 2012, 13, 2163–2173 CrossRef CAS PubMed.
- G. D. Kang, S. H. Cheon and S.-C. Song, Int. J. Pharm., 2006, 319, 29–36 CrossRef CAS PubMed.
- M. Peller, L. Willerding, S. Limmer, M. Hossann, O. Dietrich, M. Ingrisch, R. Sroka and L. H. Lindner, J. Controlled Release, 2016, 237, 138–146 CrossRef CAS PubMed.
- Y. Zhao, W. Ren, T. Zhong, S. Zhang, D. Huang, Y. Guo, X. Yao, C. Wang, W.-Q. Zhang and X. Zhang, J. Controlled Release, 2016, 222, 56–66 CrossRef CAS PubMed.
- M. Dadsetan, K. E. Taylor, C. Yong, Ž. Bajzer, L. Lu and M. J. Yaszemski, Acta Biomater., 2013, 9, 5438–5446 CrossRef CAS PubMed.
- H. Yuan, C.-Y. Chen, G.-h. Chai, Y.-Z. Du and F.-Q. Hu, Mol. Pharm., 2013, 10, 1865–1873 CrossRef CAS PubMed.
- X. Mu, M. Zhang, A. Wei, F. Yin, Y. Wang, K. Hu and J. Jiang, Nanoscale, 2021, 13, 8998–9008 RSC.
- M. Long, X. Liu, X. Huang, M. Lu, X. Wu, L. Weng, Q. Chen, X. Wang, L. Zhu and Z. Chen, J. Controlled Release, 2021, 334, 303–317 CrossRef CAS PubMed.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang and T. Jiang, Nanomedicine: Nanotechnology, Biol. Med., 2015, 11, 313–327 CAS.
- L. Wu, M. Wu, Y. Zeng, D. Zhang, A. Zheng, X. Liu and J. Liu, Nanotechnology, 2014, 26, 025102 CrossRef PubMed.
- R. Tapec, X. J. Zhao and W. Tan, J. Nanosci. Nanotechnol., 2002, 2, 405–409 CrossRef CAS PubMed.
- S. Hosseinpour, L. J. Walsh and C. Xu, J. Mater. Chem. B, 2020, 8, 9863–9876 RSC.
- R. M. Sábio, A. B. Meneguin, A. Martins dos Santos, A. S. Monteiro and M. Chorilli, Microporous Mesoporous Mater., 2021, 312, 110774 CrossRef.
- Y. Y. Tan, P. K. Yap, G. L. X. Lim, M. Mehta, Y. Chan, S. W. Ng, D. N. Kapoor, P. Negi, K. Anand, S. K. Singh, N. K. Jha, L. C. Lim, T. Madheswaran, S. Satija, G. Gupta, K. Dua and D. K. Chellappan, Chem.-Biol. Interact., 2020, 329, 109221 CrossRef CAS PubMed.
- H. Zhou, Q. Li, X. Cheng, C. Zhang, J. Sun, L. Du, J. Cao, Y. Liu and P. Huang, J. Mater. Chem. B, 2020, 8, 9251–9257 RSC.
- K. Ahmad, E. J. Lee, S. Shaikh, A. Kumar, K. M. Rao, S. Y. Park, J. O. Jin, S. S. Han and I. Choi, Semin. Cancer Biol., 2021, 69, 325–336 CrossRef CAS PubMed.
- J. Chen, Y. Yang, D. Xu, J. Li, S. Wu, Y. Jiang, C. Wang, Z. Yang and L. Zhao, Biochem. Biophys. Res. Commun., 2021, 540, 83–89 CrossRef CAS PubMed.
- M. K. M. Esfahani, S. E. Alavi, P. J. Cabot, N. Islam and E. L. Izake, Pharmaceutics, 2021, 13(10), 1605 CrossRef CAS PubMed.
- C. M. Liu, G. B. Chen, L. H. Lin, J. B. Zhang, S. M. Guo and M. X. Sheng, Colloids Surf., A, 2021, 621, 126592 CrossRef CAS.
- T. Popova, B. Tzankov, C. Voycheva, I. Spassova, D. Kovacheva, S. Tzankov, D. Aluani, V. Tzankova and N. Lambov, J. Drug Delivery Sci. Technol., 2021, 62, 102340 CrossRef CAS.
- M. A. Rahim, N. Jan, S. Khan, H. Shah, A. Madni, A. Khan, A. Jabar, S. Khan, A. Elhissi, Z. Hussain, H. C. Aziz, M. Sohail, M. Khan and H. E. Thu, Cancers, 2021, 13, 1–52 CrossRef PubMed.
- M. R. Villegas, V. Lopez, V. Rodríguez-García, A. Baeza and M. Vallet-Regí, Journal, 2021, 2275, 341–361 CAS.
- A. Vinu, M. Miyahara and K. Ariga, J. Nanosci. Nanotechnol., 2006, 6, 1510–1532 CrossRef CAS PubMed.
- A. Vinu, M. Miyahara, K. Z. Hossain, M. Takahashi, V. V. Balasubramanian, T. Mori and K. Ariga, J. Nanosci. Nanotechnol., 2007, 7, 828–832 CrossRef CAS PubMed.
- A. Vinu, V. Murugesan and M. Hartmann, J. Phys. Chem. B, 2004, 108, 7323–7330 CrossRef CAS.
- A. Vinu, V. Murugesan, O. Tangermann and M. Hartmann, Chem. Mater., 2004, 16, 3056–3065 CrossRef CAS.
- M. Miyahara, A. Vinu, K. Z. Hossain, T. Nakanishi and K. Ariga, Thin Solid Films, 2006, 499, 13–18 CrossRef CAS.
- M. Miyahara, A. Vinu and K. Ariga, Mater. Sci. Eng., C, 2007, 27, 232–236 CrossRef CAS.
- Q. Ji, M. Miyahara, J. P. Hill, S. Acharya, A. Vinu, S. B. Yoon, J. S. Yu, K. Sakamoto and K. Ariga, J. Am. Chem. Soc., 2008, 130, 2376–2377 CrossRef CAS PubMed.
- A. Vinu, N. Gokulakrishnan, V. V. Balasubramanian, S. Alam, M. P. Kapoor, K. Ariga and T. Mori, Chem. – Eur. J., 2008, 14, 11529–11538 CrossRef CAS PubMed.
- Q. M. Ji, S. Acharya, J. P. Hill, A. Vinu, S. B. Yoon, J. S. Yu, K. Sakamoto and K. Ariga, Adv. Funct. Mater., 2009, 19, 1792–1799 CrossRef CAS.
- M. P. Kapoor, A. Vinu, W. Fujii, T. Kimura, Q. H. Yang, Y. Kasama, M. Yanagi and L. R. Juneja, Microporous Mesoporous Mater., 2010, 128, 187–193 CrossRef CAS.
- L. Chen, L. Li, L. Zhang, S. Xing, T. Wang, Y. A. Wang, C. Wang and Z. Su, ACS Appl. Mater. Interfaces, 2013, 5, 7282–7290 CrossRef CAS PubMed.
- Y. J. Ho, C. H. Wu, Q. F. Jin, C. Y. Lin, P. H. Chiang, N. Wu, C. H. Fan, C. M. Yang and C. K. Yeh, Biomaterials, 2020, 232, 119723 CrossRef CAS PubMed.
- A. F. Moreira, V. M. Gaspar, E. C. Costa, D. De Melo-Diogo, P. Machado, C. M. Paquete and I. J. Correia, Eur. J. Pharm. Biopharm., 2014, 88, 1012–1025 CrossRef CAS PubMed.
- C. P. Silveira, L. M. Apolinário, W. J. Fávaro, A. J. Paula and N. Durán, ACS Biomater. Sci. Eng., 2016, 2, 1190–1199 CrossRef CAS PubMed.
- C. Salmas, V. Stathopoulos, P. Pomonis, H. Rahiala, J. Rosenholm and G. Androutsopoulos, Appl. Catal., A, 2001, 216, 23–39 CrossRef CAS.
- L. Dai, Q. Zhang, H. Gu and K. Cai, J. Mater. Chem. B, 2015, 3, 8303–8313 RSC.
- R. Mirbagheri, D. Elhamifar and M. Shaker, Sci. Rep., 2021, 11, 1–15 CrossRef PubMed.
- J.-C. Song, F.-F. Xue, X.-X. Zhang, Z.-Y. Lu and Z.-Y. Sun, Chem. Commun., 2017, 53, 3761–3764 RSC.
- Z. Teng, J. Zhang, W. Li, Y. Zheng, X. Su, Y. Tang, M. Dang, Y. Tian, L. Yuwen and L. Weng, Small, 2016, 12, 3473–3473 CrossRef.
- J. Xu, X. Wang, Z. Teng, G. Lu, N. He and Z. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 24440–24449 CrossRef CAS PubMed.
- Q. Yue, J. Li, Y. Zhang, X. Cheng, X. Chen, P. Pan, J. Su, A. A. Elzatahry, A. Alghamdi and Y. Deng, J. Am. Chem. Soc., 2017, 139, 15486–15493 CrossRef CAS PubMed.
- S. Zhou, M. Maeda, M. Kubo and M. Shimada, Chem. Lett., 2021, 50, 1475–1478 CrossRef CAS.
- Y. Wang and Z. Xu, RSC Adv., 2016, 6, 314–322 RSC.
- F. Qu, G. Zhu, H. Lin, W. Zhang, J. Sun, S. Li and S. Qiu, J. Solid State Chem., 2006, 179, 2027–2035 CrossRef CAS.
- R. Zucker, E. Massaro, K. Sanders, L. Degn and W. Boyes, Cytometry, Part A, 2010, 77, 677–685 CrossRef CAS PubMed.
- J. Park, M. K. Ha, N. Yang and T. H. Yoon, Anal. Chem., 2017, 89, 2449–2456 CrossRef CAS PubMed.
- T. Andrian, R. Riera, S. Pujals and L. Albertazzi, Nanoscale Adv., 2021, 3, 10–23 RSC.
- S. A. Smith, L. I. Selby, A. P. Johnston and G. K. Such, Bioconjugate Chem., 2018, 30, 263–272 CrossRef PubMed.
- F. Wang, A. Salvati and P. Boya, Open Biol., 2018, 8, 170271 CrossRef PubMed.
- L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CrossRef CAS.
- E. Tsakalozou, A. M. Eckman and Y. Bae, Biochem. Res. Int., 2012, 2012, 832059 Search PubMed.
- V. Bagalkot, O. C. Farokhzad, R. Langer and S. Jon, Angew. Chem., Int. Ed., 2006, 45, 8149–8152 CrossRef CAS PubMed.
- Y. A. Ivanenkov, A. E. Machulkin, A. S. Garanina, D. A. Skvortsov, A. A. Uspenskaya, E. V. Deyneka, A. V. Trofimenko, E. K. Beloglazkina, N. V. Zyk and V. E. Koteliansky, Bioorg. Med. Chem. Lett., 2019, 29, 1246–1255 CrossRef CAS PubMed.
- D. Kim, Y. Y. Jeong and S. Jon, ACS Nano, 2010, 4, 3689–3696 CrossRef CAS PubMed.
- M. K. Yu, D. Kim, I. H. Lee, J. S. So, Y. Y. Jeong and S. Jon, Small, 2011, 7, 2241–2249 CrossRef CAS PubMed.
- M. Gary-Bobo, O. Hocine, D. Brevet, M. Maynadier, L. Raehm, S. Richeter, V. Charasson, B. Loock, A. Morère and P. Maillard, Int. J. Pharm., 2012, 423, 509–515 CrossRef CAS PubMed.
- S. N. Harun, H. Ahmad, H. N. Lim, S. L. Chia and M. R. Gill, Pharmaceutics, 2021, 13, 150 CrossRef CAS PubMed.
- J. Li, X. Du, N. Zheng, L. Xu, J. Xu and S. Li, Colloids Surf., B, 2016, 141, 374–381 CrossRef CAS PubMed.
- K. O. Paredes, D. Díaz-García, V. García-Almodóvar, L. L. Chamizo, M. Marciello, M. Díaz-Sánchez, S. Prashar, S. Gómez-Ruiz and M. Filice, Cancers, 2020, 12, 187 CrossRef CAS PubMed.
- Z. Tao, B. B. Toms, J. Goodisman and T. Asefa, Chem. Res. Toxicol., 2009, 22, 1869–1880 Search PubMed.
- O. Taratula, O. B. Garbuzenko, A. M. Chen and T. Minko, J. Drug Targeting, 2011, 19, 900–914 CrossRef CAS PubMed.
- Y. Xu, P. Claiden, Y. Zhu, H. Morita and N. Hanagata, Sci. Technol. Adv. Mater., 2015, 045006 CrossRef PubMed.
- J. Tao, M. Dang, X. Su, Q. Hao, J. Zhang, X. Ma, G. Lu, Y. Zhang, Y. Tian and L. Weng, J. Colloid Interface Sci., 2018, 527, 33–39 CrossRef CAS PubMed.
- N. Iturrioz-Rodríguez, M. Á. Correa-Duarte, R. Valiente and M. L. Fanarraga, Pharmaceutics, 2020, 12, 487 CrossRef PubMed.
- E. Bindini, Z. Chehadi, M. Faustini, P.-A. Albouy, D. Grosso, A. Cattoni, C. Chanéac, O. Azzaroni, C. Sanchez and C. Boissiere, ACS Appl. Mater. Interfaces, 2020, 12, 13598–13612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr00783e |
| This journal is © The Royal Society of Chemistry 2022 |