
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectro-optical π-radicals: design advances, applications and future perspectives†
Petri
Murto
 and
Hugo
Bronstein
*
and
Hugo
Bronstein
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: hab60@cam.ac.uk
First published on 27th April 2022
Abstract
The unique open-shell electronic structure of neutral π-radicals has inspired research on novel molecular materials for over a century. In recent years, rapid development in carbon-centred π-radicals has resulted in new molecular electronics technologies, record breaking organic light-emitting diodes and other efficient energy applications. With the rapidly expanding literature in hand, it is not always straightforward to draw conclusions from results across different fields to aid further materials development. In this review, classification and design of neutral π-radicals are discussed and clarified in relation to their electro-optical properties and different target applications. Factors such as type of radical centre, steric effects and extent of conjugation, delocalisation and additional electron-donating and electron-withdrawing groups synergistically affect the stability, electronic structure, energetics, intra- and intermolecular spin–spin interactions as well as charge transport, optical and magnetic properties of π-radicals. Control of these properties and successful use of previously reported materials are exemplified in optics and electronics applications. Future potential and research directions are discussed in the light of remaining challenges that require critical consideration. The aim of this review is to provide an overview of the advantages and pitfalls of π-radicals and in that way help future design advances in the field.
 Petri Murto | Dr Petri Murto carried out his PhD studies in a cotutelle programme between Chalmers University of Technology, Sweden, and Flinders University, Australia. He received a double PhD degree in Materials Science and Chemistry in 2019. Later in 2019 he joined the group of Prof. Hugo Bronstein to work on luminescent neutral π-radicals at University of Cambridge, United Kingdom. In 2020 he received the prestigious Marie Sklodowska-Curie Individual European Fellowship, which has enabled him to extend his research on novel small molecular and polymeric π-radicals for use in organic light-emitting diodes. |
 Hugo Bronstein | Prof. Hugo Bronstein was born in Buenos Aires, Argentina in 1980 but grew up in London, UK. He studied Chemistry at Oxford, before going on to do a PhD at Imperial College with Prof. Charlotte Williams. He then spent a year at the University of Washington in Seattle working as a postdoc for Prof. Christine Luscombe. After this he returned to Imperial College to do a second postdoc with Prof. Iain McCulloch. He was awarded am Imperial College Junior Research Fellowship in 2012 before being appointed as a lecturer at University College London in 2013. In 2015 he was awarded an ERC starting grant and then in 2017 was appointed as a lecturer joint between the physics and chemistry departments at the University of Cambridge. He was awarded the Macro Group young research medal in 2019. |
1. Introduction
Neutral π-radicals are an intriguing class of materials where absorption (optical excitation) and emission of light (radiative relaxation) involve a spin doublet. They are inherently magnetic due to their open-shell electronic structure with an unpaired electron in their neutral ground state. The spin doublet has long been utilised in a wide range of applications spanning from molecular conductors and solid-state electronics,1,2 to molecular and polymer magnetics,3,4 spintronics5,6 and recently to organic light-emitting diodes (OLEDs),7,8 just to mention a few examples. Many π-radical structures are robust enough to allow device fabrication by solution-processing and vacuum-deposition techniques,8 and chemical coupling of radical molecules onto surfaces.1 The common feature is that optical, electronic and magnetic properties of π-radicals can be tuned with subtle changes in their conjugated environment by means of proper molecular design. On the other hand, all these functions can be modulated with an applied external stimulus, which has led to the development of magnetic-field sensitive optics,9 molecules with thermally controlled spin states and radical pair interactions,10,11 new energy transfer pathways in light-harvesting12 and OLEDs delivering record-breaking efficiencies.13Optical and electrical excitation and relaxation processes of π-radicals are critical in all of their optoelectronic applications. As for electroluminescence (EL), the spin doublet has a major benefit over conventional organic closed-shell emitters in that excitation to the doublet excited state (D1) and relaxation to the doublet ground state (D0) are totally spin-allowed processes, thereby setting the limit of internal quantum efficiency (IQE) to 100%.14 This provides a possibility to overcome the 25/75% singlet/triplet (S1/T1) exciton formation ratio in organic closed-shell molecules without leveraging the triplet excitons in delayed emission, that is, via thermally activated delayed fluorescence (TADF)15,16 and triplet–triplet annihilation (TTA).17,18 Although these nonradical systems can efficiently convert “dark” triplet excitons to “bright” singlets, long triplet lifetimes and singlet–triplet (reverse) intersystem crossing ((R)ISC) may slow their fluorescence down to hundreds of nanoseconds or even microseconds (see Fig. 1 and ref. 8 for excellent discussion). On the other hand, fast doublet emission process within nanosecond timescales (see Table S1, ESI†) is expected to reduce the chance of exciton quenching and alleviate related issues in device lifetime and efficiency, making stable π-radicals attractive candidates for use in electroluminescent applications.19,20
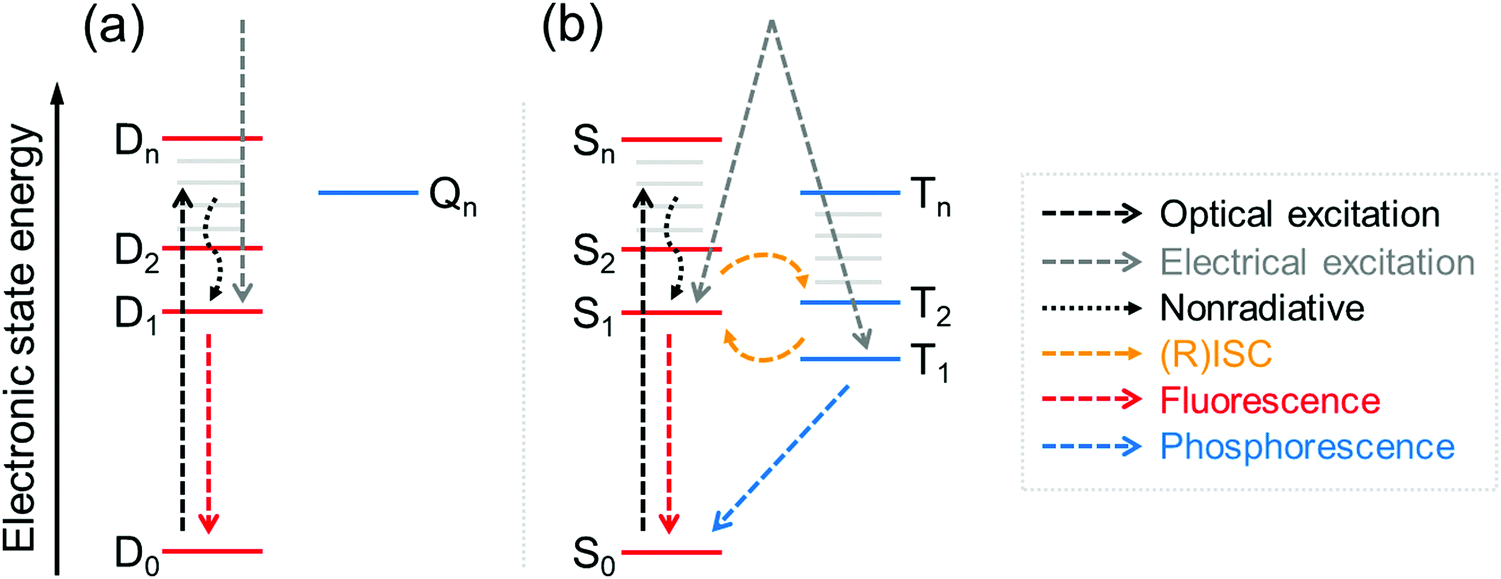 | ||
| Fig. 1 Jablonski energy diagrams illustrating optical and electrical excitations and some radiative and nonradiative relaxation pathways for organic (a) open-shell molecules showing doublet–doublet (D1–D0) fluorescence and (b) closed-shell molecules showing singlet–singlet (S1–S0) fluorescence and triplet–singlet (T1–S0) phosphorescence. | ||
Various design strategies have been applied to reach the theoretical doublet exciton formation efficiency and to utilise luminescent doublet excitons in different applications, as exemplified by selected key works in this review. The unique feature of open-shell radicals is that they are paramagnetic, unlike closed-shell molecules that are diamagnetic, which means that both their ground state and excited state can be magnetically modulated. Furthermore, two or more interacting radicals can couple either ferromagnetically or antiferromagnetically, which enables the construction of various types of intramolecular and intermolecular diradical and polyradical assemblies.21,22 Some interesting examples are highlighted in this review. An important aspect worth mentioning is the stability of the radicals. One common method to stabilise the unpaired electron has been steric protection of the radical centre with substantially bulky substituents. The spin-density and energetics of the radical can be tuned with additional electron-donating and electron-withdrawing functional groups. Another successful strategy has been extension of conjugation up to a point of fully delocalising the radical, which effectively dilutes the spin-density typically in planar molecules. Both methods and their successful uses are discussed in this review.
Carbon-centred π-radicals have been particularly successful in optoelectronic applications because of the possibility to stabilise the trivalent carbon either by steric protection or by spin-delocalisation, as mentioned above. Despite the promising progress in the field, however, it is still largely unknown how far different radical structures can be chemically modified and physically stressed without compromising their designed electro-optical properties and long-term stabilities. It may also be challenging to draw conclusions from the few success stories across different fields, in other words, how applicable are different types of π-radicals. The aim herein is to answer these questions and provide some degree of clarification as to what to consider when designing functional π-radicals.
Previously, excellent reviews have been published on various types of molecular and polymeric radicals and their use in organic electronics.23–26 Synthesis and applications of unique classes of N-heterocyclic radicals,27–30 boron containing radicals,31 graphene fragments,32–34 helicenes35 and porphyrinoids36 have been summarised elsewhere. This review focuses specifically on carbon-centred neutral π-radicals and recent advances in their design, synthesis, electro-optical properties and applications, and provides critical consideration of the challenges and prospects of future materials development. This review provides a comprehensive overview of functional π-radicals, not only those that have been proven successful in applications, but also those that feature special characteristics and potential from a stability point of view.
2. Carbon-centred π-radicals, their properties and applications
2.1. Bulky chlorophenylmethyl radicals
Gomberg's studies on triphenylmethyl (trityl) radicals in early 1900's were the first examples of carbon-centred neutral π-radicals.37,38 The structure allows some degree of delocalisation of the unpaired electron, which makes it unstable and results in dimerization of the radical in solution, as shown in Scheme 1. The dimer is stable enough to be collected as solid crystals. Various degrees of chlorine substitution at ortho-, meta- and para-positions of the trityl phenyl rings was later introduced by Ballester, Veciana et al.39–42 as a successful method to obtain radicals that are stable for several months under ambient air, light and moisture without any degradation or side reactions. Specifically, the ortho-chlorine atoms induce significant torsion of the phenyl rings about the radical centre, effectively localising the spin-density mainly at the carbon centre and simultaneously providing sufficient steric protection to prevent unwanted side reactions, for example, with ambient oxygen. The meta- and para-chlorine substitution effectively inactivate σ-coupling reactions, i.e., dimerization of the radical.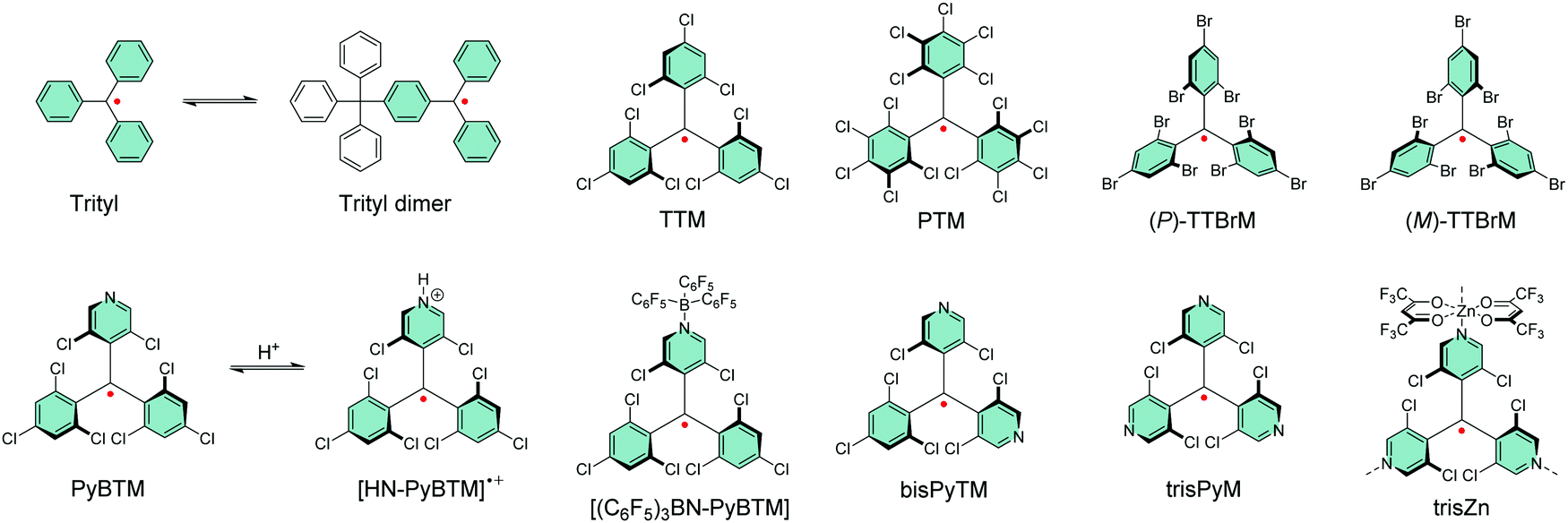 | ||
| Scheme 1 Chemical structures trityl radical and its chlorophenylmethyl derivatives. | ||
TTM and PTM are the two most common examples of chlorophenylmethyl radicals, largely due to their chemical inertness, thermal stability up to 300 °C and well-known synthetic methodology.43 The hydrogenated precursors of TTM and PTM (αH-TTM and αH-PTM) can be synthesized in a matter of hours via a single-step Friedel–Crafts reaction from chloroform and 1,3,5-trichlorobenzene or pentachlorobenzene, respectively, in the presence of AlCl3 as the Lewis acid.41 Deprotonation and subsequent one-electron oxidation of the resulting carbanions give TTM and PTM radicals in nearly quantitative yields (Scheme 2a, middle route).42 A much harsher option is to perchlorinate triphenylmethane with strong chlorinating agents in strongly acidic conditions (Scheme 2a, top route).44,45 Although the latter route can be considered as the “first-generation” method first reported in 1960's, it has been applied to the synthesis of various chlorophenylmethyl radical derivatives aiming to either tune the spin-density or introduce additional functional groups to the para-positions.46–49 The common feature is that incomplete ortho-substitution makes the radical susceptible to oxidation in air.
 | ||
| Scheme 2 Key synthetic steps for (a) TTM and PTM radicals and (b) PyBTM, bisPyTM and trisPyM radicals and their αH-precursors (DDQ = 2,3-dichloro-5,6-dicyano-p-benzoquinone, NCS = N-chlorosuccinimide). Some repeating reaction steps are omitted for clarity. | ||
TTM, PTM and their derivatives are intrinsically chiral due to the propeller-like torsion of the phenyl rings about the radical centre.50,51 The radicals exist as racemic mixtures of plus (P) and minus (M) enantiomers depending on their right- or left-handed torsion, respectively.4 This means that it is possible to obtain circularly polarized luminescence from enantiopure radicals. Veciana et al.52,53 reported the first studies of circularly polarized emission properties of (P) and (M) isomers of TTM and PTM radicals. PTM had a slightly higher racemization energy barrier as compared to TTM, although both radicals were stable against racemization at low temperatures (−20 °C in tetrachloromethane solution). Recently, brominated derivative of TTM (TTBrM) was synthesized by the same authors on the merit of increasing the steric bulk around the radical centre, which in turn increased the racemization barrier (no racemization was observed even at 60 °C temperatures).53 The significant result was that (P)-TTBrM and (M)-TTBrM could be used as stable circularly polarized emitters at room temperature, which have been discussed in detail elsewhere.8,54 From a synthetic point of view, para-bromine substitution may turn practical for functionalisation of TTBrM derivatives, as mentioned by the authors.53 However, it would be equally relevant to address the chemical inertness of the other bromine positions, which is likely to be covered in their future works. In that regard, Kuehne et al.55 used a similar strategy in their design of mixed-halide TTM derivatives that allowed site-selective radical functionalisation and polymerisation, albeit admittedly with various degrees of dehalogenation in these reactions. An alternative pathway to TTM and PTM radical functionalisation could be selective para-iodine substitution (3I-PTM, Scheme 2a, bottom route).56 Iodination has also been shown to increase the radiative rate of PTM radical in comparison to its chlorinated counterpart, which is significant when using PTM as the single emitter in the solid state (Table S1, ESI†).57 We point out that, for simplicity, herein only one isomer is included in most cases and the structures are drawn planar.
Another class of stable trityl radicals include pyridine as one or more of the aromatic rings. Nitrogen atom at para-position retains the chemical and thermal stability of PTM but ortho-nitrogen renders the radical less stable and such compound decomposes at temperatures above 200 °C.58 It goes without saying that the pyridine nitrogen atom can be protonated in the presence of strong acids, but only the para-nitrogen containing structure is stable in its protonated form without impairment of the radical character. Nishihara et al.59 have utilised this special feature in modulated luminescence and electrochemical redox reactions of PyBTM and its N-protonated counterpart [HN-PyBTM]˙+, switched by protonation with p-toluenesulfonic acid and deprotonation with triethylamine. The pyridine nitrogen allows similar coordination of B(C6F5)3 as Lewis acid to give a stable neutral radical [(C6F5)3BN-PyBTM] (Scheme 1) that features redshifted emission and increased electrochemical reduction potential as compared to PyBTM (see Table S1, ESI†).60 This example demonstrates a straight-forward method to obtain low-energy electron acceptors by reversible chemical modification of π-radicals, although their primary use may be as stimuli-responsive emissive materials. Analogous metal N-coordination complexes of PyBTM have been developed to enhance the emission efficiency and optical response to an external magnetic field.61–65 Kusamoto and Kimura66 have provided an excellent overview of related materials.
Magnetoluminescence (that is, magnetic field-sensitive luminescence) of neutral π-radicals has been observed in αH-PyBTM crystals (α-hydrogenated precursor molecule of PyBTM, Fig. 2a) doped with various concentrations of PyBTM radical by Nishihara et al.9,67 Low doping concentrations (0.1%) delivered emission selectively from PyBTM monomer, but high concentrations (10%) caused aggregation of the radicals and a broad emission profile with two main peaks at 563 and 674 nm in the solid state. Ratio of the short-wavelength monomer and long-wavelength excimer emission bands (originating from isolated radicals and aggregated radical dimers, respectively)68 was modulated under an applied magnetic field ranging from 0 to 14.5 T at 4.2 K (see Fig. 2d and Table S1, ESI†). Increasing temperature counteracted the magnetic field effect. In other words, the spin states of the radical dimers can be modulated by both magnetic field and thermal energy. In the absence of magnetic field, singlet ground state is mainly populated, whereas population of triplet ground state increases with the magnetic field (see Fig. 2b and c). As a result, the monomer emission component increased with the magnetic field (via increasing population of the triplet state associated with slower decay rate) while the excimer emission diminished slightly (see Fig. 2e and f and ref. 9 for mechanistic insights). This ground state associated magnetic field effect can be counteracted by thermal energy, as mentioned above. It is also distinctively different from closed-shell systems where only the excited state can be affected by a magnetic field. Simple π-radicals like PyBTM enable novel approaches to the control of intermolecular through-space spin–spin interactions, as they can be accessed also in the ground state.
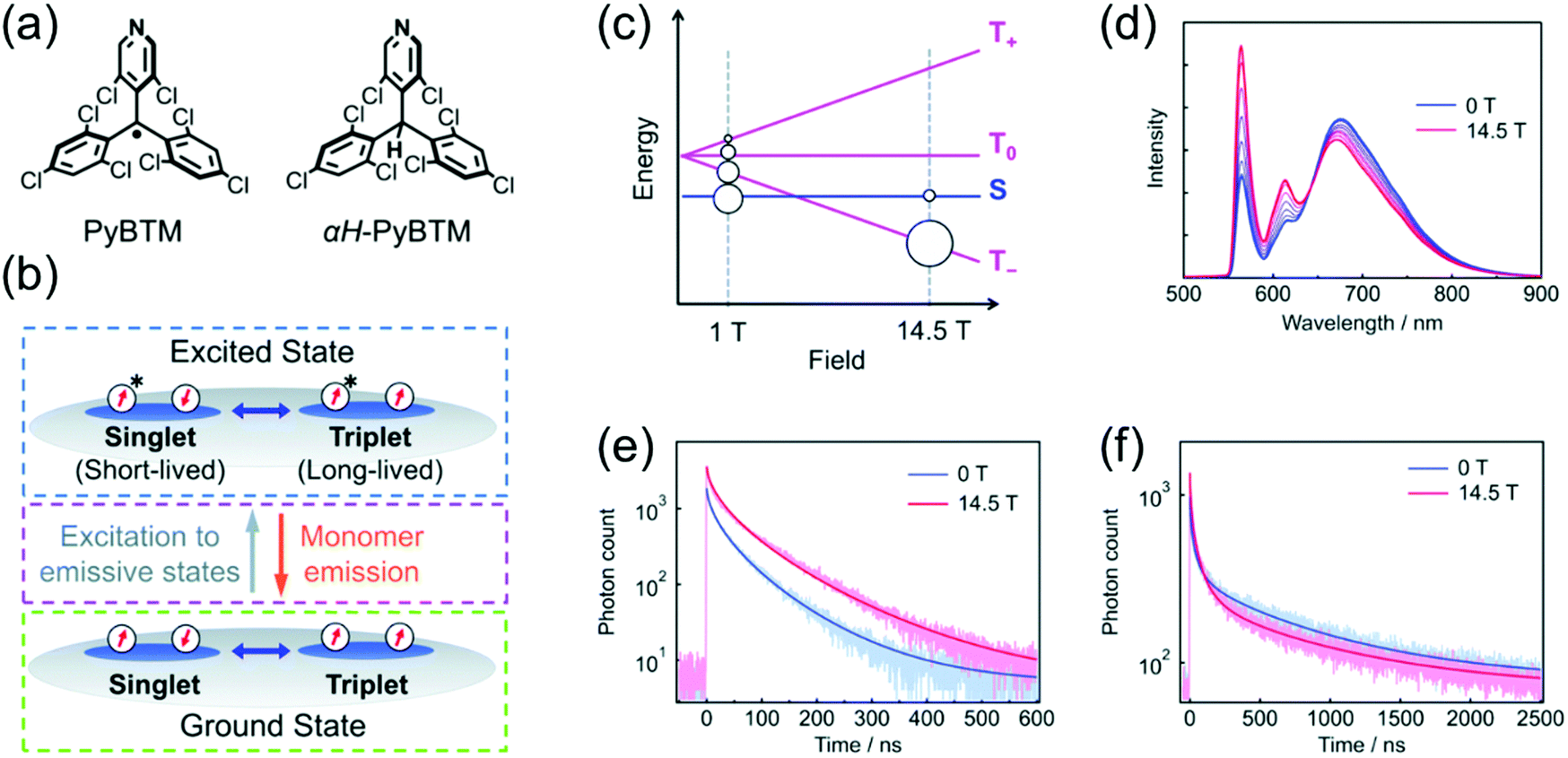 | ||
| Fig. 2 (a) Chemical structures of PyBTM and αH-PyBTM. (b) Scheme showing the changes in spin sublevel populations in the ground state (green dashed box), direct excitations to emissive states (magenta dashed box) and two excited states with different lifetimes (blue dashed box). (c) Population distribution as a function of the applied magnetic field. (d) Emission spectra at 10% doping at 4.2 K under magnetic fields of 0–14.5 T. Emission decays and corresponding fits at 4.2 K under magnetic fields of 0 and 14.5 T at (e) 563 nm and (f) 674 nm. Reproduced from ref. 9 under CC BY-NC 3.0 licence. | ||
α-Hydrogenated precursors are generally regarded as ideal host matrices for the corresponding π-radicals, because the structural similarity of the two molecules makes them compatible and ensures that the π-radical is well dispersed in the host matrix. Moreover, large spectral overlap of the host emission and π-radical absorption typically enables efficient energy transfer from the host to the π-radical, which is of particular interest for luminescence in the solid state. For example, Zhao et al.69 utilised the excimer emission of αH-PyBTM microcrystals doped with PyBTM, and they were able to control the polarization of photoluminescence (PL) via a concentration- and temperature-sensitive exciton funnelling scheme.
Two pyridine rings containing bisPyTM allows further tuning of the electro-optical properties of its singly and doubly N-protonated counterparts, but the synthesis of αH-bisPyTM precursor requires severe heating at 300 °C and results in a minimal yield of ca. 1% (Scheme 2b, middle route).70,71 In contrast, αH-PyBTM has been synthesized with practical yields of 66% or so, followed by almost quantitative conversion to a neutral π-radical (see Scheme 2b, top route).59 trisPyM incorporates three pyridine rings and it is the latest luminescent derivative in this series (Scheme 2b, bottom route).72 The strongest absorption band of trisPyM peaks in the UV-visible region (molar extinction coefficient, ε = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at 350 nm) and a weaker band tails beyond 600 nm (ε = 933 M−1 cm−1 at 518 nm, Table S1, ESI†) in dichloromethane solution, which can be regarded as typical optical behaviour of chlorophenylmethyl derived radicals. The low-energy band accordingly leads to the lowest excited state (D0 → D1 excitation). The number of pyridine rings had a direct impact on photostability. trisPyM had modest photoluminescence quantum yield (PLQY) of 0.9% at 700 nm but half-life of its emission intensity (2.2 × 104 s) was about 2–4 orders of magnitude higher than that of TTM due to its stabilised (downshifted) frontier orbital energy levels (we note that the reported half-lives also depend on the exact experimental setup, see Table S1, ESI†, and the references therein). The trigonal N-coordination chemistry of trisPyM was also utilised in the construction of a polymeric two-dimensional zinc coordination complex trisZn. For clarity, only one of the zinc coordinations is illustrated in Scheme 1 (see ref. 72 for original representation). Both trisPyM and trisZn were luminescent also in the solid state when cooled to 79 K, although their emission quickly diminished at higher temperatures of 250 K or so. Emission of neat bisPyTM and trisPyM radicals were unaffected by an applied magnetic field, but reduction of the inter-radical interactions in their respective one- and two-dimensional zinc coordination polymers (through larger spin–spin separation) facilitated solid-state magnetoluminescence at 4.2 K (Table S1, ESI†).73 This was observed as increasing emission intensity under an applied magnetic field of 15 T, but again was counteracted by added thermal energy already at 20 K. Admittedly, this seems an appealing approach to design well-structured macromolecules with tuneable optical and electronic interactions and magnetic responses for use as luminescent/nonluminescent switches. However, covalently connected polyradicals may have a major benefit over coordination polymers in that they are structurally robust, thermally stable, easily processable and thereby widely applicable in optoelectronics, as discussed later in chapter 2.5.
000 M−1 cm−1 at 350 nm) and a weaker band tails beyond 600 nm (ε = 933 M−1 cm−1 at 518 nm, Table S1, ESI†) in dichloromethane solution, which can be regarded as typical optical behaviour of chlorophenylmethyl derived radicals. The low-energy band accordingly leads to the lowest excited state (D0 → D1 excitation). The number of pyridine rings had a direct impact on photostability. trisPyM had modest photoluminescence quantum yield (PLQY) of 0.9% at 700 nm but half-life of its emission intensity (2.2 × 104 s) was about 2–4 orders of magnitude higher than that of TTM due to its stabilised (downshifted) frontier orbital energy levels (we note that the reported half-lives also depend on the exact experimental setup, see Table S1, ESI†, and the references therein). The trigonal N-coordination chemistry of trisPyM was also utilised in the construction of a polymeric two-dimensional zinc coordination complex trisZn. For clarity, only one of the zinc coordinations is illustrated in Scheme 1 (see ref. 72 for original representation). Both trisPyM and trisZn were luminescent also in the solid state when cooled to 79 K, although their emission quickly diminished at higher temperatures of 250 K or so. Emission of neat bisPyTM and trisPyM radicals were unaffected by an applied magnetic field, but reduction of the inter-radical interactions in their respective one- and two-dimensional zinc coordination polymers (through larger spin–spin separation) facilitated solid-state magnetoluminescence at 4.2 K (Table S1, ESI†).73 This was observed as increasing emission intensity under an applied magnetic field of 15 T, but again was counteracted by added thermal energy already at 20 K. Admittedly, this seems an appealing approach to design well-structured macromolecules with tuneable optical and electronic interactions and magnetic responses for use as luminescent/nonluminescent switches. However, covalently connected polyradicals may have a major benefit over coordination polymers in that they are structurally robust, thermally stable, easily processable and thereby widely applicable in optoelectronics, as discussed later in chapter 2.5.
Li et al.74 replaced one of the trichlorobenzene rings of TTM with carbazole in CzBTM (Scheme 3). This new type of neutral π-radical featured improved thermal- and photostability as a result of partial delocalisation of spin-density from the central carbon atom and phenyl rings to the extended carbazole structure. Optical excitation takes place between the carbazole unit and the radical centre, which is characterised as charge-transfer (CT) transition instead of the typical localised excitation at the carbon centre of TTM and PTM radicals. In other words, carbazole functions as an electron-rich donor (D) unit and the central carbon with an unpaired electron naturally is an electron-deficient acceptor (A) unit in the push–pull (D–A) type CzBTM radical. The latter statement is supported by the observation that TTM, PTM and their pyridine derivatives can be electrochemically reduced to their anionic forms (reversible one-electron reduction), but they appear less able to undergo reversible electrochemical oxidation reactions, much due to the high potentials required to oxidise the radicals (see Table S1, ESI†). Addition of a donor monomer is expected to stabilise the radicals by introducing a low-potential oxidation site into the D–A molecules. Replacing carbazole with its nitrogen containing derivative β-carboline in PyID-BTM increased the radiative rate (kr) and decreased the nonradiative rate (knr) of the radical, which is observed as ten-fold PLQY increasing from 2.0% at 697 nm (for CzBTM) to 19.5% at 664 nm (for PyID-BTM) in cyclohexane solution (Table S1, ESI†).75 This comparison clearly highlights the ease of tuning the electrical and optical properties of π-radicals for different target applications.
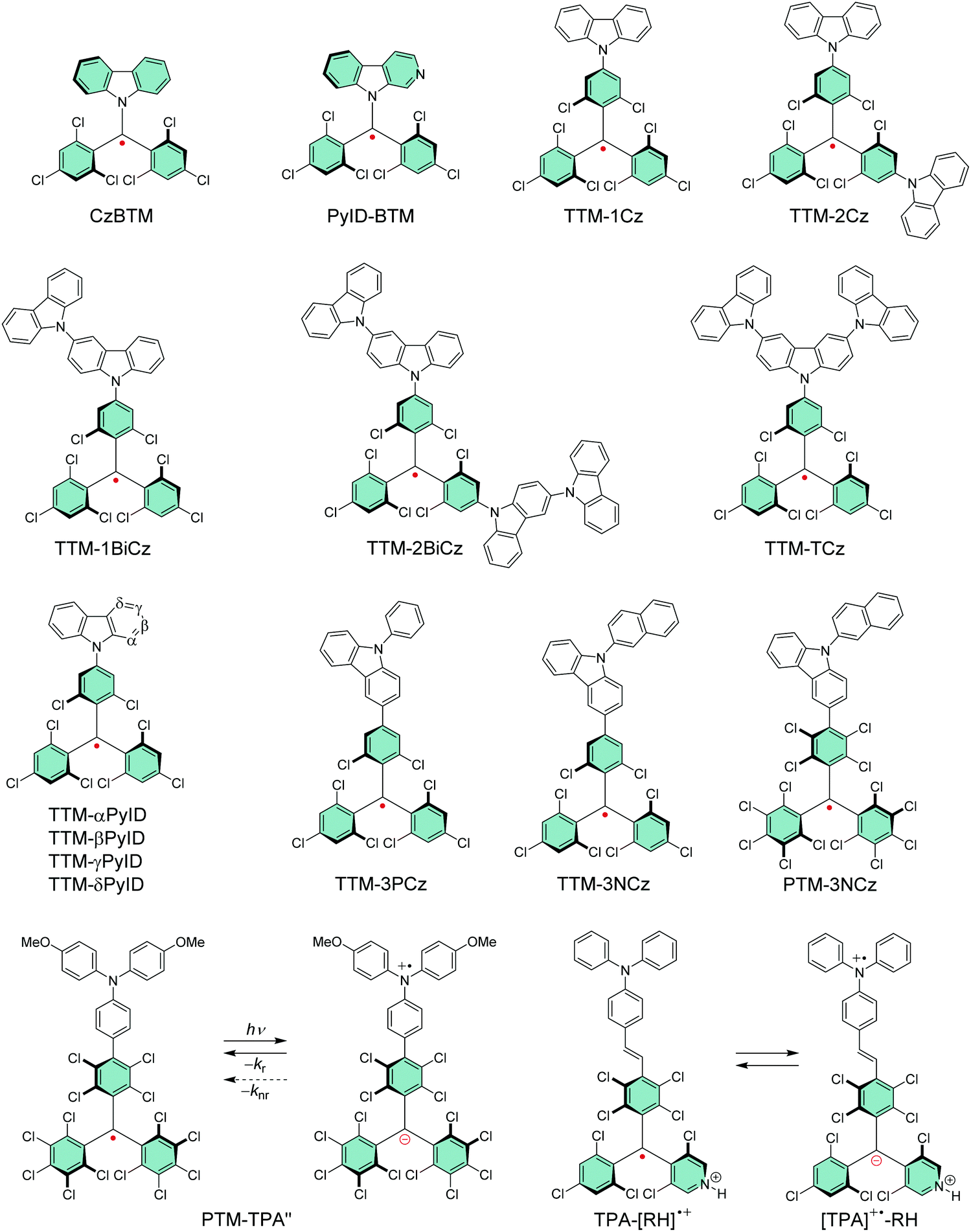 | ||
| Scheme 3 Chemical structures of chlorophenylmethyl derived D–A type radicals. | ||
Selective C–N coupling of carbazole to the para-position of TTM has opened another versatile approach to synthesize D–A type radicals, originally by Juliá et al.76–78 and later by Li et al.14,79 Various degrees of carbazole substitution with increasing electron-donating strength in TTM-1Cz, TTM-2Cz, TTM-1BiCz, TTM-2BiCz and TTM-TCz (Scheme 3) has strengthened the CT character of the radicals and redshifted their emission up to 689 nm with enhanced PLQY of 26% (for TTM-TCz) in cyclohexane solution (Table S1, ESI†).79 Kuehne et al.80 improved the PLQY of TTM-1Cz/TTM-2Cz/TTM-3Cz series up to 87% in toluene solution, while slightly blueshifting their emission wavelengths by introducing electron-withdrawing cyano groups or bromine substituents to carbazole 2,7-positions (for the latter structures S1–S3, see Scheme S1 and Table S1, ESI†). However, the above question about inertness of the bromine substituents remains. TTM-1Cz has been a pioneering compound in the application of deep-red emitting radicals in OLEDs.14,81 One reason has to be its relatively low molecular weight and good thermal stability that allow device fabrication via conventional vacuum deposition techniques. TTM-1Cz radicals feature easily tuneable and ambipolar charge transport properties (electron and hole mobilities up to 10−2 and 10−3 cm2 V−1 s−1, respectively), which is excellent indication of their suitability for organic electronics in general.82 TTM-1Cz has also been used as a luminescent sensitizer for fluorescent molecules in OLEDs and for TTA active molecules in photon upconversion (UC) by ensuring appropriate D1–S1 and D1–T1 energy levels alignment in these two systems, respectively, by Li et al.12,83 The latter example is shown in Fig. 3, where the doublet–triplet energy transfer (DTET) from TTM-1Cz to 9,10-diphenylanthracene (DPA) has been used to initiate the triplet–triplet annihilation photon upconversion (TTA-UC) process in solution, converting red light (excitation at 635 nm) into blue light (emission at 432 nm) with a large anti-Stokes energy shift of 0.92 eV. The benefit of π-radical sensitizer is that the doublet exciton can be utilised directly in TTA-UC without requiring additional singlet-to-triplet excited state ISC like in conventional closed-shell sensitizers, in other words, with significantly reduced energy losses in the process. In general, TTM derived D–A radicals typically show strong absorption close to the UV edge of visible spectrum, while their PL is characterised with large Stokes shift in the red spectral region. This feature provides a unique wide spectral window for efficient TTA-UC process with different TTA active molecules, perhaps also in the solid state by using a highly luminescent radical sensitizer and a well-suited host matrix. Teki84 has reported an in-depth discussion on the photophysics behind such systems.
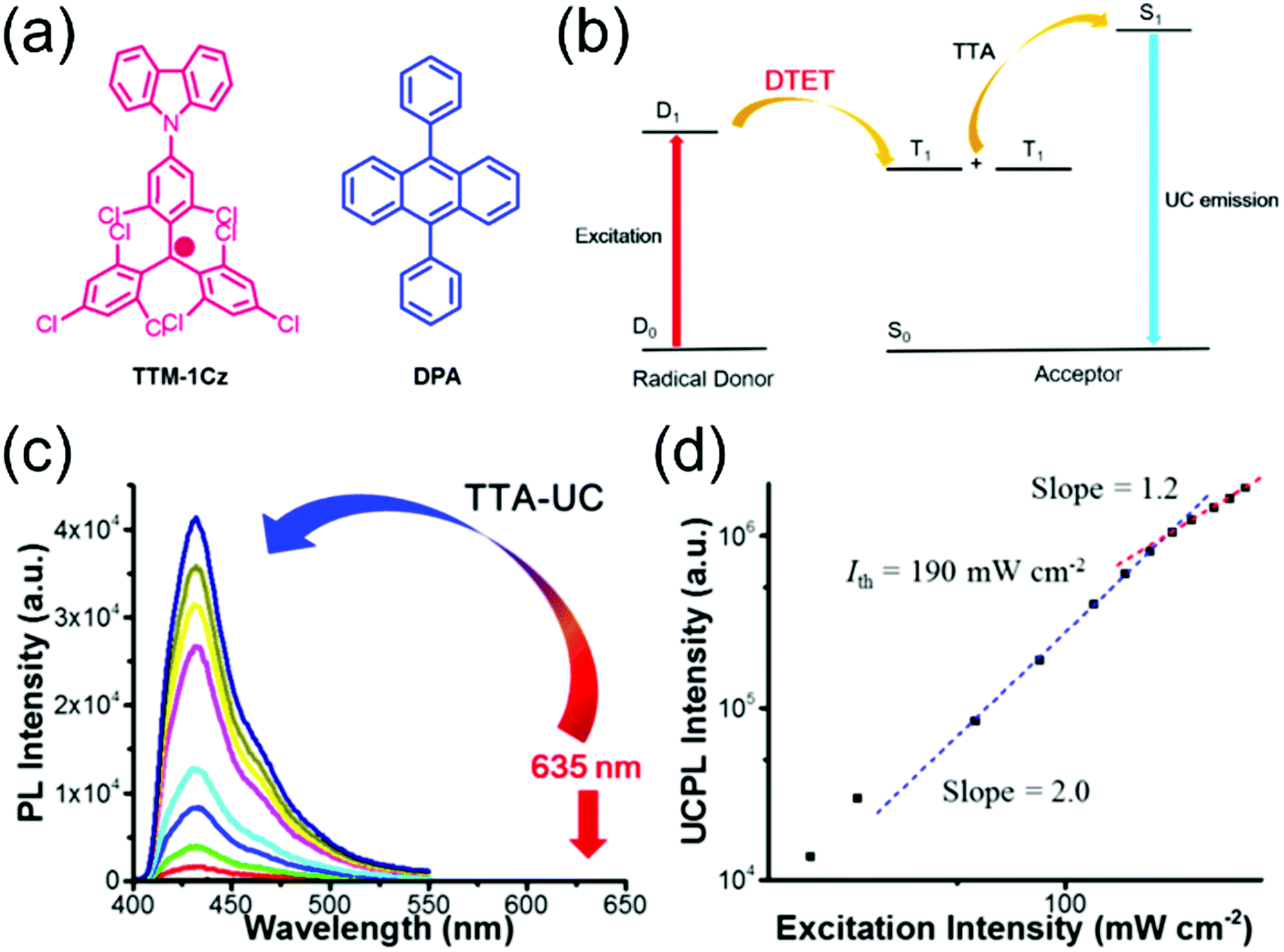 | ||
| Fig. 3 (a) Chemical structures of TTM-1Cz and DPA. (b) Illustration of TTA-based upconversion sensitized by doublet exciton. (c) Upconversion emission spectra of TTM-1Cz/DPA with different incident power density of 635 nm laser in deaerated toluene. (d) Dependence of upconversion emission intensity at 432 nm on the incident power density. The dashed lines are fitting results with slopes of 2.0 (blue) and 1.2 (red) in the low and high-power regions, respectively. Reprinted (adapted) with permission from ref. 12 Copyright 2017 American Chemical Society. | ||
Evans et al.85 designed a systematic series of TTM-xPyID radicals, where x stands for nitrogen heteroatom either on α, β, γ or δ position (see Scheme 3). The non-alternant donor broke the symmetry of the radicals and lifted the degeneracy of their frontier orbitals. The presence of nitrogen heteroatom on the donor increased the energy of the lowest excited CT state and generally decreased the nonradiative decay rates compared to TTM-1Cz.86 Degenerate optical excitations are deemed to limit the luminescent efficiency of symmetrical radicals such as TTM-1Cz (PLQY 5% at 687 nm in chloroform solution). The non-alternant design boosted the TTM-xPyID series’ PLQYs >90% and blueshifted their emission into the orange-red spectral region at 611–643 nm in chloroform solution (Table S1, ESI†). The same luminescent enhancement was also reflected in the radicals’ OLED device performance, as shown in Fig. 4. Overall, the simplicity of C–N coupling (heating in the presence of carbonate base) has enabled the design of many more D–A type radicals that have found uses mainly in OLED applications, with a minor caveat that the reaction yields a mixture of singly, doubly and triply coupled products lowering the yield of the target molecule. However, such mixtures can be easily purified with conventional chromatography techniques.87,88
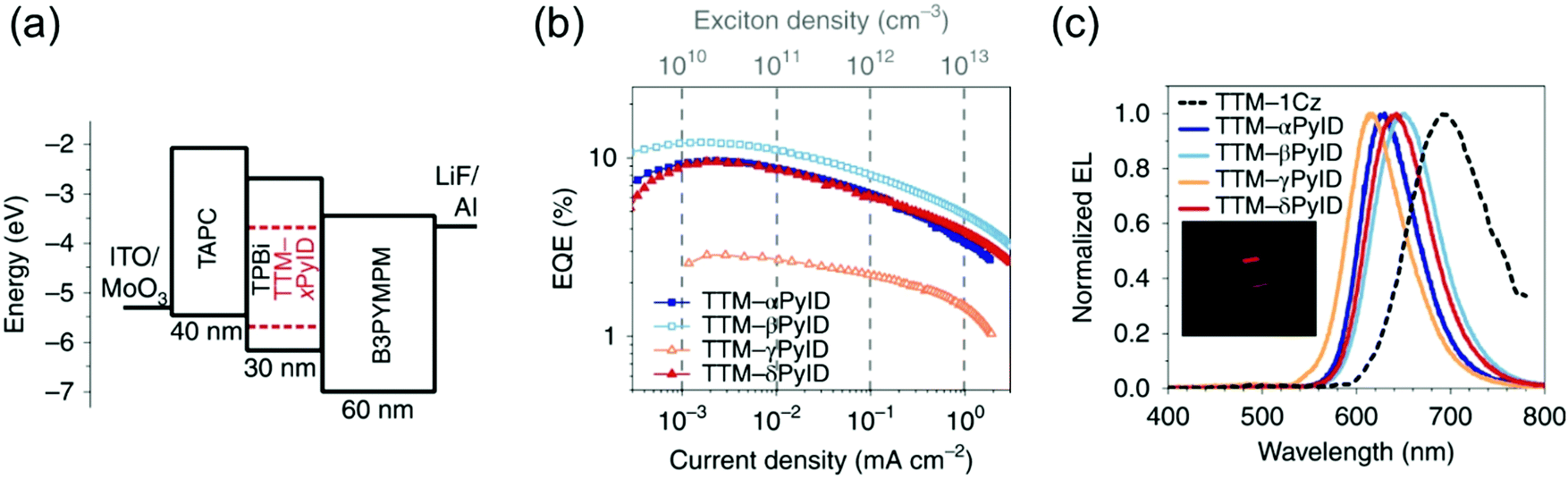 | ||
| Fig. 4 (a) Device architecture for OLEDs based on the TTM-xPyID series. (b) EQE (external quantum efficiency)–current density. (c) EL spectra. Inset: Photograph of the TTM-βPyID device. Reprinted by permission from ref. 85 under exclusive licence to Springer Nature Limited 2020. | ||
Somewhat revolutionary coupling of carbazole from its 3-position to TTM using Pd-catalysed Suzuki–Miyaura reaction afforded TTM-3PCz and TTM-3NCz radicals featuring strong CT excitation character and efficient emission of light after donor (3PCz/3NCz) to acceptor (TTM) charge transfer, as reported by Li et al.13 These radicals were the first practical examples reaching the theoretical maximum 100% formation ratio of emissive doublet excitons and IQE near unity in OLEDs, in other words, devices that are not limited by the 25 and 75% singlet/triplet exciton formation ratio of organic closed-shell emitters. This important discovery has motivated the design of related TTM and PTM derived D–A radicals with half-lives of emission intensity up to 9.1 × 106 s (for PTM-3NCz, Scheme 3, Fig. 5c, d and Table S1, ESI†), that is, five–six orders of magnitude higher than those of TTM and PTM radicals under same conditions.89,90 The greatly improved photostability is obtained by downshifting the singly occupied molecular orbital (SOMO) energy level below the highest (doubly) occupied molecular orbital (HOMO) energy levels, “so-called” non-Aufbau electronic structure (see Fig. 5a and b).91–93
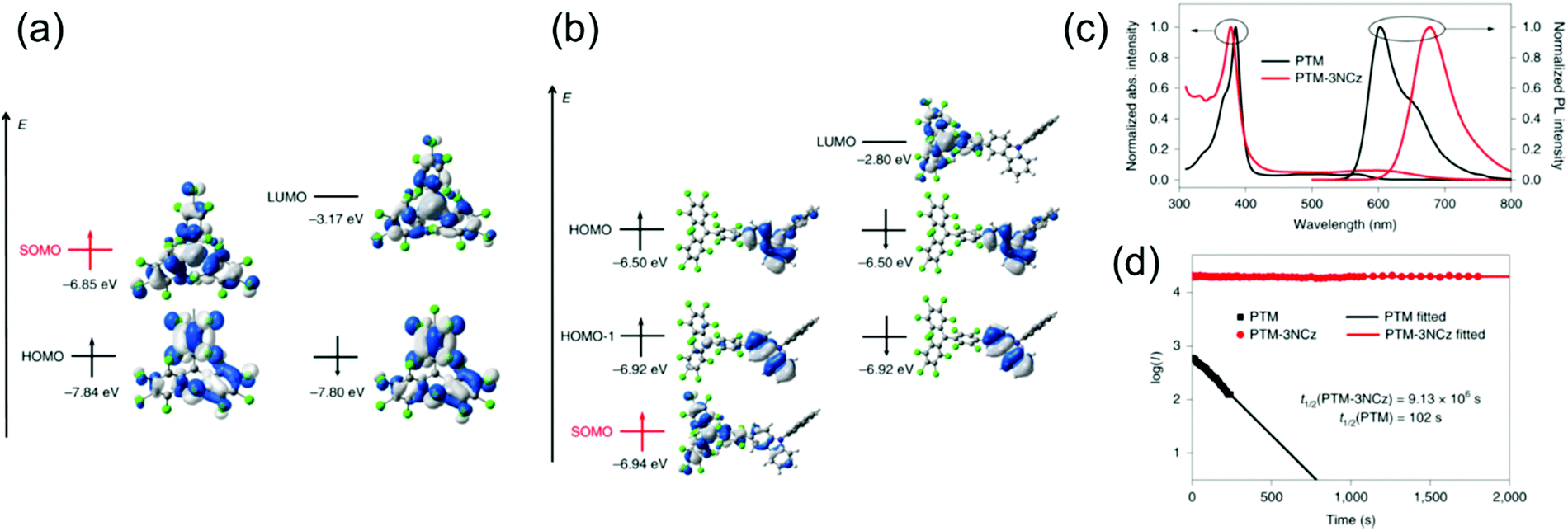 | ||
| Fig. 5 Energies and wavefunctions for the frontier molecular orbitals of (a) PTM and (b) PTM-3NCz, as calculated at the tuned ωB97XD/6-31+G(d,p) level of theory. The two colours used for the electron wavefunctions distinguish the wavefunction phases. (c) Optical absorption and fluorescence spectra of the PTM and PTM-3NCz radicals in cyclohexane solution. (d) Time dependence of the emission intensities (I) of the PTM and PTM-3NCz radicals in dilute cyclohexane under 355-nm laser radiation. Reprinted by permission from ref. 89 under exclusive licence to Springer Nature Limited 2019. | ||
Fig. 6 summarises the effect of donor group on the luminescent efficiency of TTM- and PTM-based D–A radicals. The common feature is that the rigid fused-ring carbazole units in PTM-PCz and PTM-3PCz decrease the nonradiative decay rate of the radical, as compared to the triphenylamine (TPA) substituted PTM-TPA. Although structural rigidity somewhat weakened the electron-donating ability of the donor groups, it also decreased the nonradiative back-electron transfer of the radicals and ultimately enhanced their PLQYs. On the other hand, smaller torsion angles between the donor and acceptor (i.e., radical) groups enhanced the CT character of the D–A system and, again, led to improved luminescent efficiency. These factors combined, PTM-3PCz showed the smallest calculated torsion angle (78.8°), which was reflected in the strongest experimental low-energy absorption band (2680 M−1 cm−1 at 606 nm) and highest PLQY (57% at 679 nm, Table S1, ESI†). Same trend was observed for the corresponding TTM series, albeit with decreased torsion angles. This comparison once again shows that introduction of donor substituents into TTM and PTM radicals not only enhance their PLQYs but also improve their photostabilities by orders of magnitude.90
 | ||
| Fig. 6 (a) Molecular geometries of substituted PTM radicals (DFT (UB3LYP/6-31G(d)). (b) UV-Vis absorption, (c) PL and (d) fluorescence lifetime of the radicals in cyclohexane solution (concentration: 10−5 M, excitation wavelength: 380 nm). Reproduced with permission from ref. 90 Copyright the Owner Societies 2018. | ||
Design of D–A type π-radicals has not only been adopted in OLEDs, but the same concept has been used in optically active electron transfer radicals incorporating TPA as the electron-donor (oxidation site) and PTM radical as the electron-acceptor (reduction site). Lambert et al.48,94 varied the distance between the two redox centres by coupling TPA to the radical either directly or through an ethylene or acetylene bridge. Radicals with spatially (and energetically) separated redox centres underwent optically induced donor-to-acceptor electron transfer and transition from the neutral ground state to a stable zwitterionic excited state, but with exchanged donor and acceptor characters. Further studies allowed tuning of radiative and nonradiative back-electron transfer of PTM-TPA radical by varying the donor strength of TPA unit with electron-donating and electron-withdrawing substituents.95 Electron-donating substituents (such as 4-methoxy side chains in PTM-TPA′′, Scheme 3 and Table S1, ESI†) stabilised the excited state leading to redshifted CT absorption band in the NIR region, but the same operation rendered the radical poorly emissive. Electron-withdrawing substituents reduced the rate of nonradiative back-electron transfer leading to enhanced emission from the CT state (PLQY up to 37% at 725 nm in cyclohexane solution, structure S4, Scheme S1 and Table S1, ESI†). This example highlights that doublet CT state as the lowest-energy excited state has a clear advantage over closed-shell D–A systems where relaxation to low-lying triplet states add another nonradiative pathway.95,96 Strong CT absorption in the NIR region could be utilised in molecular storage and switching devices and in solar energy conversion. PTM-based D–A radicals incorporating acetylene-bridged TPA donors undergo two-photon absorption which is another interesting feature of neutral π-radicals for potential use in organic photovoltaics.56 Winter and Breimaier97 further increased the donor strength of TPA by introducing 4-dimethylamino substituents in TPAN-PTM or by fusing the TPA group into a planar conformation with oxygen bridges in TOTA-PTM. Both operations lowered the energy gap between the neutral radical and the zwitterionic excited state, which could be observed as exceptionally redshifted (albeit relatively weak) low-energy absorption at 973 nm (3200 M−1 cm−1) and 819 nm (1600 M−1 cm−1) for TPAN-PTM and TOTA-PTM, respectively, in dichloromethane solution (see Scheme S1 and Table S1, ESI†).
Beverina et al.98 reported the first demonstration of PyBTM-based D–A structures for use as luminophores in luminescent solar concentrators (LSCs), where by default the radicals must be thermally and photochemically stable (Fig. 7). One major benefit of using π-radicals in these types of devices is their generally weak low-energy absorption bands and large Stokes shifts (Scheme S1 and Table S1, ESI†), which translate to minimal reabsorption losses allowing scale-up of thin film LSC technology. From a synthesis perspective, the authors used an intriguing water-based micellar Suzuki–Miyaura reaction motivated by sustainable chemistry and selective coupling at the para-chlorine atoms on PyBTM. Three different D–A structures, that is, doubly phenyl, 6-methoxy-2-naphthyl and 9-phenanthryl substituted PyBTM were isolated in 63, 50 and 35% yields, respectively. Micellar catalysis certainly is a promising approach to clean coupling of chlorophenylmethyl radicals, but the somewhat modest yields in this example may hinder its use in the synthesis of larger oligo- and polyradicals, especially when the solubility starts to play a larger role in the reactivity (this is probably the reason for lower yield in the case of 9-phenanthryl substituted radical). It would be interesting to see further progress on this type of radical chemistry from the authors or other groups.
 | ||
| Fig. 7 (a) Synthesis of PyBTM derived D–A type radicals ((a) reaction was performed at 80 °C). (b) Working principle of an LSC. Reproduced from ref. 98 under CC BY-NC 3.0 licence. | ||
A structural isomer of PyBTM, PyBTM′′ with a nitrogen atom on meta-position, and its D–A structured mono-, di- and triradical derivatives incorporating a TPA donor and 1–3 PyBTM′′ acceptors were introduced by Lambert et al.99 (structures S5–S7, Scheme S1 and Table S1, ESI†). Unlike the typical doublet ground- and excited states of monoradicals, diradicals were most stable in singlet state regardless of their side chain substitution. Triradicals adopted spin doublet. The general observation is that more radical centres enhanced both one-photon CT absorption and two-photon absorption bands in the NIR region. The radicals were also NIR-emissive and functional in OLEDs, demonstrating that devices can be fabricated from radicals with different spin multiplicities.99 Switching PyBTM nitrogen atom from para- to meta-position generally enhances the solid-state emission even at room temperature (see structure S8, Scheme S1 and Table S1, ESI†).100 Hattori et al.101 coupled different aryl substituents to the meta-positions of PyBTM. Electron-rich thiophene and furan rings changed the localised electronic structure of PyBTM into D–A type systems whose photostabilities were enhanced, albeit with concomitant emission quenching effects (structures S9 and S10, respectively, Scheme S1 and Table S1, ESI†).
Nishihara et al.102 applied the above discussed reversible protonation of PyBTM to electron transfer reactions in ethylene-bridged TPA-R˙ radical and its N-protonated counterparts TPA-[RH]˙+ and [TPA]˙+-RH (the latter two structures are shown in Scheme 3). Neutral TPA-R˙ radical featured SOMO–HOMO converted non-Aufbau electronic structure and stable emission from the CT state at 910 nm in cyclohexane solution. However, protonation of the radical site with trifluoromethanesulfonic acid enabled modulation of the frontier orbitals of TPA-[RH]˙+ by increasing its reduction potential, in other words, by making the radical stronger of an acceptor (see Scheme S1 and Table S1, ESI†). The SOMO–HOMO converted electronic structure was conserved but its emission was greatly suppressed in the protonated form. Addition of excess acid (>200 equiv.) triggered electron transfer from TPA to the radical site generating [TPA]˙+–RH radical. Electron transfer between these two states could be switched by changing the acid concentration, which certainly provides an interesting approach to stimulus-regulated electro-optical π-radicals and their design.
An interesting addition to the discussion herein is coupling of two PTM radicals through para- and meta-divinylbenzene bridges by Veciana et al.103,104 Such neutral diradicals have very weak or negligible electronic interaction between the two unpaired electrons because of their large through-bond separation of ca. 24 and 23 Å (see structures S11 and S12, respectively, Scheme S1 and Table S1, ESI†). We note that radical pairs with negligible or nearly negligible electron exchange interactions are also called “biradicals” in the literature, but we use a single term “diradicals” for consistency.105,106 The lowest-energy optical absorption of the diradicals is roughly twice as intense as that of their corresponding monoradical (S13, ESI†) – originating from the two radical centres, that is, two doublets – making them distinctively different from interacting diradicals discussed later in chapter 2.4. However, PTM diradicals are redox active as discussed above, and electrochemical one-electron reduction of the para-bridged molecule generates a radical/anion pair that behaves similarly to the D–A pairs discussed in this chapter. A new NIR absorption band peaking at 1400 nm originates from intramolecular electron transfer between separated anion and radical redox centres. Charge transfer in PTM anion/radical pairs has been observed even through extended oligothienylenevinylene spacers up to 53 Å.107,108 Bis(phenylene)diyne bridges suppressed inter-radical interactions but, interestingly, PTM radicals proved effective in stabilising positive charges in the bridging units by forming quinoidal ring segments.109 Electrochemical control of stable radical/radical, radical/anion and radical/cation pairs could perhaps be a versatile approach to obtain D–A radicals with switchable and strong optical responses.
2.2. Non-chlorinated anthryl, cyclopentadienyl, fluorenyl and dicyanomethyl radicals
A chlorine-free approach to stable carbon-centred π-radicals is attractive in many ways: from synthetic perspective to avoid harsh chlorination reactions and possible dehalogenation during coupling reactions or spontaneously over time, and from applications point of view considering the cost, (bio)compatibility and environmental impact of the end products. Trityl radical is unstable, as discussed in previous chapter, but increasing the steric bulk around the radical centre with anthryl structures in TAntM (Scheme 4) has provided excellent stabilisation that compares to those of TTM and PTM.40,42,110 Nishiuchi, Kubo et al.110 synthesized mesityl-protected TAntM radical (1) and its cationic form by employing a high-yielding Negishi coupling reaction. The radical was stable under ambient conditions for at least one month. It showed strong absorption in the visible spectral region but a very weak NIR band peaking at 1020 nm in dichloromethane solution (see Fig. 8 and Table S1, ESI†). In contrast, the cationic form of 1 showed an intense and broad absorption at 900 nm in trifluoroacetic acid (the cation was generated in acidic conditions from the same intermediate as the radical, see ref. 110 for details).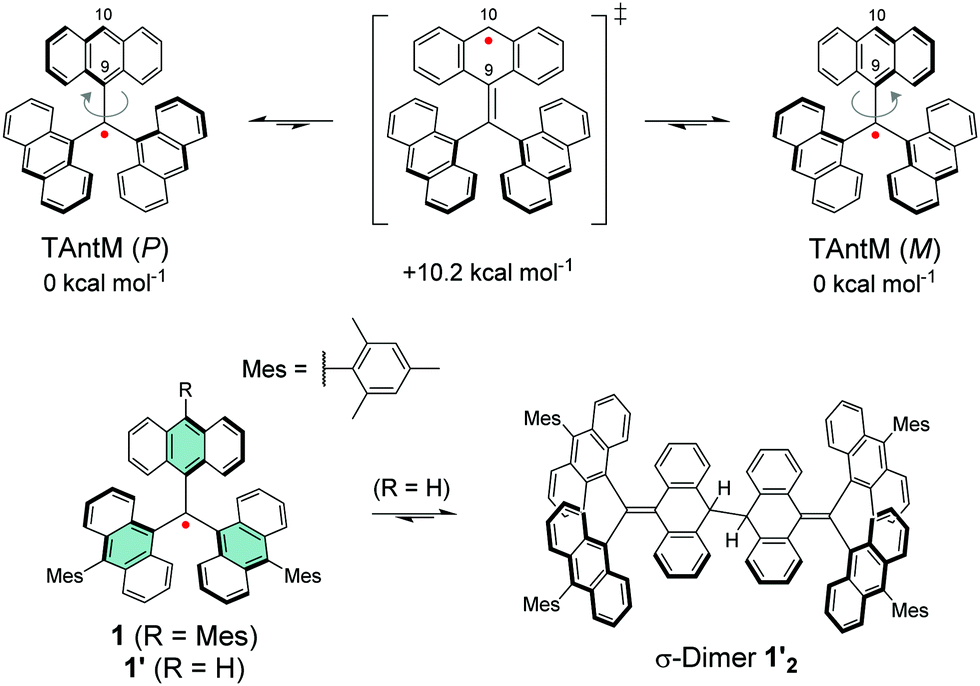 | ||
| Scheme 4 Chemical structures of anthryl derived radicals. | ||
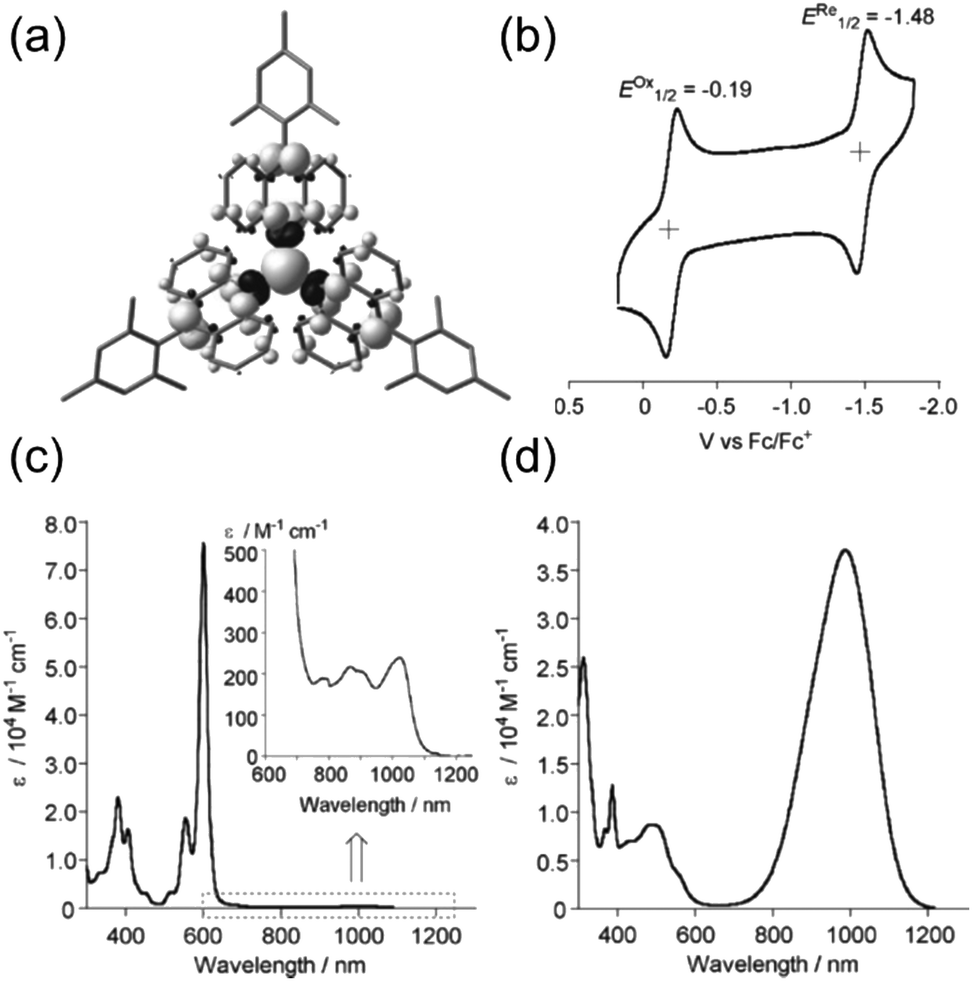 | ||
| Fig. 8 (a) Calculated spin density map of 1. The white and black surfaces represent α and β spins, respectively. (b) Cyclic voltammogram (CV) of 1. Measurement conditions: 0.1 M n-Bu4NPF6 in CH2Cl2, scan rate = 100 mV s−1. UV-Vis-NIR spectra of (c) 1 in CH2Cl2 (inset: expansion from 600 to 1250 nm) and (d) its cationic form. Reproduced with permission from ref. 110 Copyright 2018 Wiley-VCH Verlag GmbH & Co. | ||
Leaving one of the anthryl groups unprotected resulted in σ-dimerization of radical 1′ from 10-position (Scheme 4), which is distinctively different from the head-to-tail coupling of trityl radical (Scheme 1). Dimerization of 1′ was pronounced even in dilute solution (10−5–10−4 M) and 1′2 was isolated with good yield of 65%. Highly congested propeller structure of TAntM scaffold provided superior steric protection of the radical centre but the spin density was somewhat delocalised to the anthryl groups (see Fig. 8a). Hence, cutting off further conjugation by large twist angles of the mesityl groups was necessary to stabilise the radical. Another interesting observation is that the TAntM propeller transits between two helicities (P and M). Dimerization was suggested to occur readily at room temperature through a quinoidal transition state, 10.2 kcal mol−1 relative to the ground state, which would explain the differential dimerization chemistry of 1′ and trityl radicals. Equilibrium of dimerization of TAntM (and 1′) is towards the dimer because it is stabilised by 18.0 kcal mol−1 energy difference to the ground state radical. Electrochemical redox reactions are relevant for applications in electronics. The mesityl-protected radical 1 showed reversible oxidation and reduction waves (Fig. 8b), whereas oxidation of 1′2 caused dissociation of two monomeric cations.110 It would be interesting to see if the strong optical contrast between 1 and its (chemically prepared) carbocation could be obtained by stable electrochemical redox switching too, of interest for electrochromics and related applications. Latest contribution from the same authors includes a triisopropylsilyl (TIPS) ethynyl substituted TAntM radical that was stable in air for a month or so, which compares to the stability of 1 despite being less sterically congested (structure S14, Scheme S1 and Table S1, ESI†).111
Cyclopentadienyl is perhaps the simplest π-radical centre, but its high reactivity towards oxygen has hampered its use in practical applications.112–114 Kitagawa et al.115,116 introduced cyclopentadienyl radicals that were fused with two homoadamantene frameworks. Their stability was improved by the addition of bulky phenyl substituent in an allyl-like radical 2, which could be stored as crystalline solid for a week or so without decomposition in air. Perhaps in-depth optical studies will follow, although 2 was very air-sensitive in solution much like other cyclopentadienyl radicals.116
Fluorene structures have been a common base for π-radicals alongside triphenylmethyls.117–119 The simple fluorenyl radical is reactive towards oxygen but introduction of bulky groups such as phenyl and mesityl in 3 and 4, respectively, or an electron-withdrawing cyano group in 5 have greatly improved its stability by steric effects and spin delocalisation (Scheme 5).119–121 Extension of the π-system enabled the synthesis of BDPA radical and its derivatives with electron-donating and electron-withdrawing substituents at the para-position of the phenyl ring, first by Koelsch122 and later by Fox et al.123 and Swager et al.124 The spin density is effectively delocalised between the two fluorenyl groups and BDPA has been reported to be stable to oxygen in both solution and solid state.123,124 However, its photoreactivity results in various oxidation products which seriously limits the utility of the radical, especially in view of organic electronic applications. It is a shame because BDPA exhibits similarly strong NIR absorption band (ε = 1580 M−1 cm−1 at 859 nm) as the CT absorption of D–A radicals like PTM-TPA′′ (ε = 1300 M−1 cm−1 at 787 nm) and significantly stronger than that of 1 (ε = ca. 250 M−1 cm−1 at 1020 nm), although the band in visible region dominates the absorption of BDPA (see Table S1, ESI†).95,110,123 Addition of water-solubilising carboxylic acid groups in 6 and different amounts of sulfonate groups in 7 (Scheme 5) allowed dynamic nuclear polarization studies in aqueous solutions by Swager et al.124–126 The radicals were unaffected by the ionic substituents and their absorption profiles in aqueous solutions closely resembled that of BDPA in dichloromethane solution.123 BDPA radicals are commonly regarded as air-stable but, recently, Sigurdsson and Mandal127 revealed that they undergo decomposition under various conditions excluding low temperatures at −80 °C or so. Dimerization was one of the main degradation pathways. BDPA-nitroxide diradicals have been of interest in the dynamic nuclear polarization field,124,127 and addition of quaternary ammonium groups into BDPA has improved their persistence and made them water-soluble.128
 | ||
| Scheme 5 Chemical structures of cyclopentadienyl and fluorenyl derived radicals. | ||
Swager et al.129 extended the π-conjugation in a planar truxene-based radical 8. The spin density was more delocalised than in other examples discussed here so far, but large portion of the spin was localised on carbon-5 position (see Scheme 5). Introduction of mesityl groups aimed to protect the radical site but when the solution of 8 in dichloromethane was exposed to air, it decomposed completely during 24 h and its half-life was estimated to be ca. 5.8 h. However, it is noteworthy that the radical could be stored intact for several months in solid state under inert atmosphere. Structural rigidity of the truxene core enhanced the absorption of 8 (ε = 2720 M−1 cm−1 at 626 nm, Table S1, ESI†) compared to BDPA. Despite the remaining stability issues, these works stand as great examples for other fields in terms of design of electro-optical π-radicals that absorb light in the low-energy spectral region and are also water-soluble.
Kubo et al.130 improved the steric protection of fluorenyl radicals by coupling 9-anthryl group to dibenzofluorenyls in 9–11 (Scheme 5). Excessive steric congestion provided kinetic stabilisation of the radical centre, while thermodynamic stabilisation was obtained by extending the fluorene π-conjugation with additional benzene rings. Interestingly, 9 was isolated as crystalline σ-dimer 92, whereas 10 could be obtained as an unassociated monoradical. Reactivity of 11 was dependent on the environment and it could be obtained as monoradical from hexane. When recrystallized from polar solvent the radical dimerized from 10-anthryl position to give σ-dimer 112. The difference in reactivities of 9–11 arises from their different spin distributions and steric protection due to the varied positions of the benzene rings. In 9 and 10 the spin density is mainly delocalised within the dibenzofluorenyl moiety, whereas in 11 some of the spin is distributed onto the anthryl group making the 10-position particularly reactive (Fig. 9). The dimerization is reversible and the monoradical could be reproduced by dissociation of the dimer in solution. In fact, the energy difference between 11 and 112 is just 0.1 kcal mol−1 in favour of the dimer (see ref. 130 for original representation of the transition states of dimerization). Optical absorption was commonly weak in the NIR region. 9 displayed a very weak lowest-energy band peaking at 1220 nm in dichloromethane solution (ε = ca. 100 M−1 cm−1) whereas for 11 the band was barely detectable (see Table S1, ESI†), in both cases due to weak electronic transitions from the naphthalene delocalised orbitals to the fluorenyl radical centre. 10 showed totally different optical activity as the lowest-energy transition originates from CT transition from anthryl group to dibenzofluorenyl, which was observed as strong and blueshifted absorption at 669 nm (ε = 2100 M−1 cm−1, Table S1, ESI†). Although 10 showed superior persistence towards dimerization, it was more easily oxidized than the other radicals. Hence, half-lives of 9, 10 and 11 in toluene solution open to air in dark were 7, 3.5 and 43 days, respectively. This is clear improvement to the stabilities of above discussed fluorene derived radicals, but significant design efforts are still needed to obtain truly robust derivatives and to utilise fluorenyl radicals in meaningful electro-optical applications.
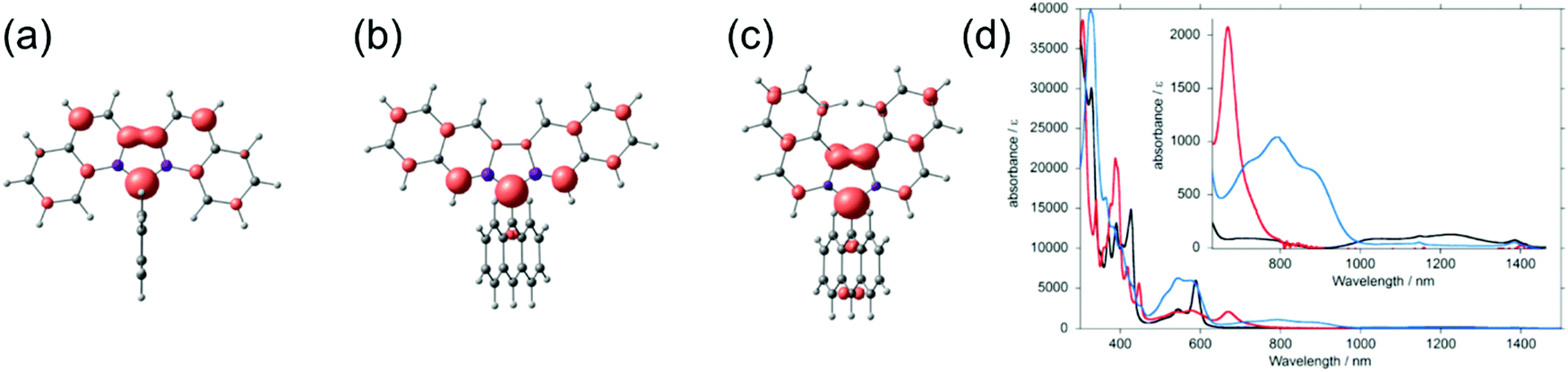 | ||
| Fig. 9 Spin density maps of (a) 9, (b) 10 and (c) 11 calculated with UBLYP/6-31G**//UB3LYP/6-31G** method. Red and purple surfaces represent α and β spin densities drawn at 0.004 e a.u.−3 level, respectively. (d) UV-Vis-NIR spectra (1.0 × 10−4 M in CH2Cl2, room temperature) of 9 (black line), 10 (red line), and 11 (blue line). Inset shows a magnified view in the NIR region. Reprinted (adapted) with permission from ref. 130 Copyright 2014 American Chemical Society. | ||
Porphyrins are known for their strong optical absorption which has been utilised in controlled spin dynamics of meso-coupled BDPA-zinc porphyrins.131 Partial delocalisation of spin density of fluorenyl radical onto meso-coupled smaragdyrin has been another promising approach from optics perspective. An interesting observation is that 12 featured two reversible electrochemical reduction reactions at remarkably high potentials (see Table S1, ESI†), implying low energies of its unoccupied frontier orbitals that would be easily accessible for n-type doping. In fact, chemical reduction (and oxidation) reactions could be optically observed as switching between strongly NIR-absorbing neutral radical 12 (above 30000 M−1 cm−1 at 824 nm) and its weaker absorbing anionic (and cationic) forms, respectively, as estimated from the absorption spectra in Table S1 (ESI†).132 Although the negative charge of the reduced species was assigned mainly to the fluorenyl unit (while the positive charge of the oxidised species was mainly distributed through the smaragdyrin macrocycle), the electrochemical reduction potential of 12 is by far higher (less negative) than for any other fluorenyl radical discussed in this chapter. Half-life of the radical in solution was close to 10 days under ambient air and light, that is, without any protection from photooxidation.132 Some degree of π-delocalisation may well be the key to obtain fluorene derived radicals that are chemically and optically stable under ambient conditions, as exemplified with few promising structures in this chapter.
Winter et al.133–139 and Seki et al.10,140,141 have taken a different approach and utilised the stabilising effect of electron-withdrawing dicyanomethyl group in their design of carbon-centred aryl dicyanomethyl radicals. Unsubstituted aryl dicyanomethyls undergo irreversible head-to-tail dimerization, but para-substitution is an effective method to prevent such coupling, as first introduced by Seki et al.140,141 and later studied by Winter et al.133,137 The para-substituted radical 13 is remarkably thermally and air stable (for 12 months or so) and interestingly it can be reversibly switched between dissociated radical and associated σ-dimer forms by heating and cooling the sample, respectively, within a temperature range of 5–95 °C in toluene solution. Stronger electron-donating substituents (such as dimethylamino or methoxy groups, Scheme 6) stabilise the radical leading to weaker bonding interaction, whereas weaker electron-donors or electron-withdrawing substituents (such as phenyl or cyano groups, Scheme 6) lead to stronger tendency to form σ-dimer 132.133
 | ||
| Scheme 6 Chemical structures of dicyanomethyl derived radicals. | ||
Julolidine skeleton 14 (Scheme 6) locks the amino nitrogen in a planar conformation, effectively increasing the spin delocalisation within the radical and suppressing its σ-dimerization. However, on cooling 14 undergoes reversible head-to-tail π-dimerization with very weak bonding interaction (π-dimer 142 is stabilised by less than 1 kcal mol−1versus the dissociated radical).137,139–141 Winter et al.137 pointed out that although both σ-dimers and π-dimers are diamagnetic and electron paramagnetic resonance (EPR) silent, only σ-dimerization breaks the π-conjugation. As a result, σ-dimers are generally weaker absorbers in the visible spectral range than their parent radicals, whereas π-dimers typically feature strong absorption from the visible to the NIR region. Seki et al.10 have utilised this property in their design of dicyanomethyl dyes bearing a coplanar triphenylamine skeleton and showing thermochromic operation, that is, optical switching between strongly NIR-absorbing radicals and their weakly absorbing σ-dimers. An interesting feature is that the two states showed weak visible absorption and almost no perceptible colour change, especially in the case of O2DP radical (Fig. 10a). The reversible switching of the fused triphenylamine containing O2DP correlates with the smaller negative dimerization enthalpy and reduced Mulliken atomic spin density on the central carbon of the dicyanomethyl group (ρc). A compound with lower ρc tends to form a weaker intermolecular C–C bond, in line with our discussion above.140,141 In fact, the calculated ρc of O2DP (0.426, Fig. 10b) was slightly smaller than that of 14 forming π-dimer (0.438).10 This comparison may be helpful for the design of other π-radicals that undergo desired/undesired σ- or π-dimerization.
 | ||
| Fig. 10 (a) Variable-temperature UV-Vis-NIR absorption spectra of O2DP radical (1.0 × 10−4 M in toluene). The jagged spectral shapes around 1150 nm are due to the artefacts (see ref. 10 and the supporting information therein). (b) Plot of the change in experimental enthalpy upon dimerization vs. calculated ρc of para-amino-substituted radicals [UB3LYP/6-31G(d)], including the chemical structures of O2DP and related dicyanomethyl radicals. Reprinted (adapted) with permission from ref. 10 Copyright 2019 American Chemical Society. | ||
Winter et al.134 took another interesting step towards radicals that are tethered together with an aliphatic side chain at the ortho-position (with varying alkyl chain lengths, see Scheme 6). Unpaired diradicals 15 and 16 featured closely similar dimerization behaviour than their monoradical counterparts 13 and 14 in solution, respectively, but tethered diradicals like 15 can be switched between the two states with better temperature response also in the solid state in air.136 The latest strategy to stabilise aryl dicyanomethyls electronically involves conjugation of formally antiaromatic substituents to the radicals, so as to allow mixing of the high-energy antiaromatic π-orbitals with the radical SOMO and formation of a low-energy zwitterionic state. This stabilisation effectively reduced the dimerization association constant of the radical. Moreover, radical 17 (containing an oxygen-bridged phenoxazine donor) differed from the typical head-to-head dimerization mode of aryl dicyanomethyls; it preferably formed an ortho–ortho ring σ-dimer (Fig. 11a) because mixing of the zwitterionic state caused significant spin delocalisation into the naphthyl ring. Theoretical calculations further suggested that the ortho–ortho dimer of 17 is energetically stabilised by attractive London dispersion forces not present in the head-to-head dimer (see Fig. 11b).138 These observations link back to Chapter 2.1, where similar oxygen-bridged donor structure was used to stabilise a D–A type TOTA-PTM radical, but without evident mixing of a zwitterionic state.97
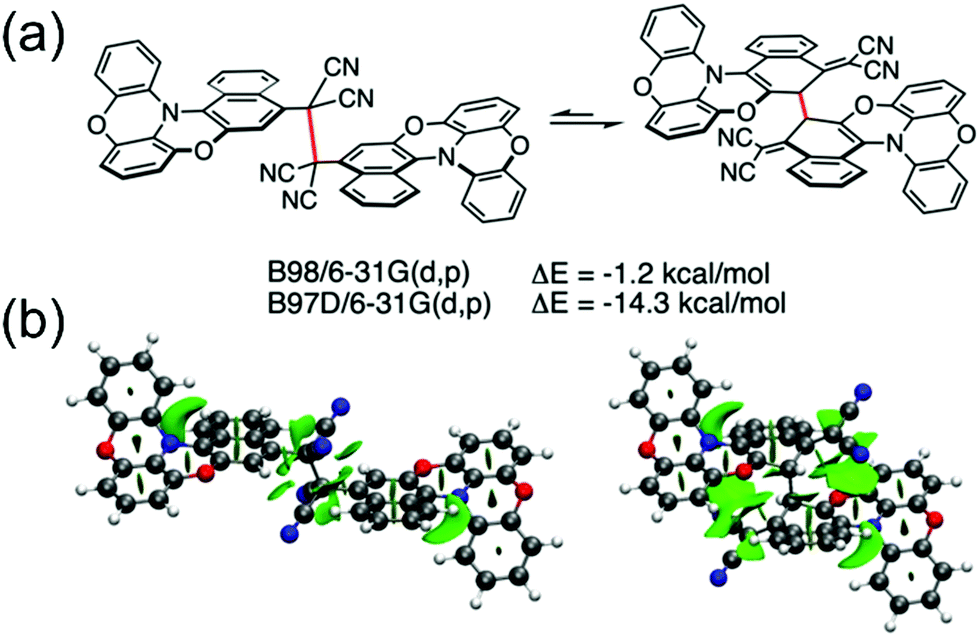 | ||
| Fig. 11 (a) Computed relative electronic energies in gas phase for the head-to-head (left) and ortho–ortho (right) σ-dimer of 17 with the same functional, but one (B97D) including a dispersion correction. (b) Non-covalent interactions plot showing dispersion interactions (isovalue = 0.5; −5 to 5 a.u.) derived from the reduced density gradient showing larger dispersion interactions for the ortho–ortho dimer. Favourable dispersion interactions are shown with green and unfavourable steric interactions with red. Reproduced with permission from ref. 138 Copyright 2021 Wiley-VCH GmbH. | ||
Other dicyanomethyl substituted structures like carbazole and indolo[3,2-b]carbazole diradicals undergo reversible formation/dissociation of σ-bonded cyclic aggregates (σ-tetramers and σ-dimers) in a similar fashion on cooling and heating, respectively. Their special feature is that the dissociation can be also achieved mechanically in the solid state by applying pressure in the few GPa range, as studied by Delgado et al.142–144 Otsuka et al.145 tethered tetraarylsuccinonitrile in a polystyrene chain to suppress the recombination of its dissociated radical form. Mechanical grinding of the solid sample generated a pair of dicyanomethyl radicals (dissociation ratio was approximately 5%) that were stable in air and showed unusually high-energy emission in the yellow spectral region. The stabilisation effect was lost in solution due to reversible recombination of the radicals. On the other hand, the reversible switching was successfully utilised as a visual sign of stress caused by polymer crystallization (yellow fluorescence was observed because of micro-mechanical dissociation of dicyanomethyl radicals during crystallization of polycaprolactone) by Otsuka and co-workers.146 Reversible switching between open-shell radical and closed-shell σ-dimer (or π-dimer) may find uses in thermochromic, mechanochromic and other stimuli-responsive applications with optical and magnetic contrast, which is also an appealing and, to the best of our knowledge, an unexplored aspect in the field of organic optoelectronics. However, significant research efforts are still needed to merge the superior stability of chlorinated radicals like TTM family, the reversible electrochemical redox reactions of sterically congested radicals like TAntM derivatives and the strong NIR absorption of delocalised/less protected radicals like porphyrins and smaragdyrins, but in a non-chlorinated metal-free structure. We next address some of these aspects and take a closer look at spin-delocalised radicals, their dimerization modes and optical absorption and redox properties.
2.3. Spin-delocalised carbon radicals
Effective conjugation has been the basis of materials design for organic electronics, but unlike sterically protected carbon-centred radicals, delocalised π-radicals are prone to dimerise by σ-bond formation.147,148 Phenalene is a commonly used planar platform where the unpaired electron is stabilised by delocalisation over the SOMO orbital over six identical carbon atoms, as schematically illustrated by the Hückel molecular orbital model in Scheme 7.149,150 The spin-delocalised structure provides thermodynamic stabilisation, but phenalenyl radical exists in equilibrium with its σ-dimer in solution and the radical undergoes rapid oxidation in air without extensive steric protection or electronic stabilisation.149,151,152 Steric protection (e.g., by means of tert-butyl substitution in 18), addition of heteroatoms (one or more nitrogen atoms in the π-system) or electron-withdrawing substituents (such as cyano or carbonyl groups) are required to isolate the radical in the solid state.150–155 Positioning of the substituents affects the spin-delocalisation and aromaticity of phenalenyl radicals; α-cyano or carbonyl substitution seems to provide superior stabilisation by extending the anti-aromatic (allylic) character of the radical centre. Bulky substituents on β-carbon atoms have less effect on the electronic structure (the 2,5,8-positions are nodes in the SOMO, see Scheme 7) but they provide kinetic stabilisation inhibiting the energetically favoured σ-dimerization.22,156–158 It is interesting to note here that while steric hindrance effectively suppresses σ-dimerization of phenalenyl radicals (making π-dimerization favourable), the same operation makes σ-dimerization favourable for planar dicyanomethyl radicals (suppressing π-dimerization), as discussed in the previous chapter.139 | ||
| Scheme 7 Chemical structures of phenalenyl and triangulene derived radicals (R groups are omitted from the resonance structures for clarity). | ||
Phenalenyls are further stabilised by formation of π-dimers in their crystalline state, and the intermolecular interactions are also beneficial in obtaining enhanced (strongly allowed) optical absorptions. For example, the low-energy absorption band of dissociated phenalenyl radical (peaking at 595 nm) in dichloromethane solution was enhanced up to 200-fold as a result of π–π stacking at low temperatures (Fig. 12a and Table S1, ESI†) as demonstrated by Kochi et al.22 Close stacking of two paramagnetic radicals forms a π-dimer where the SOMOs are partially shared between the two molecules making the dimer diamagnetic (closed-shell), yet strongly absorptive due to the allowed intermolecular HOMO–LUMO transitions (HOMO and LUMO are formed by bonding and antibonding combinations of the overlapping SOMOs of the two radicals, see Fig. 12b). On the other hand, inter-radical distance defines if the π-dimer is characterised as a closed-shell species or an open-shell diradical. The interplanar separation in this case was estimated to be 3.1 Å, which corresponds to about 25% diradical character and 75% bonding character (lower part of Fig. 12b), suggesting a weak 12-centered bond between the two phenalenyls.22 Konar et al.159 have added further discussion on so-called “magnetic bistability” of phenalenyls and other planar radicals, whose switching between paramagnetic and diamagnetic species can be modulated by external stimuli (temperature, pressure, light, electric field, etc.). Cimpoesu et al.160,161 have provided recent computational insights into spin–spin interactions in phenalenyl dimers.
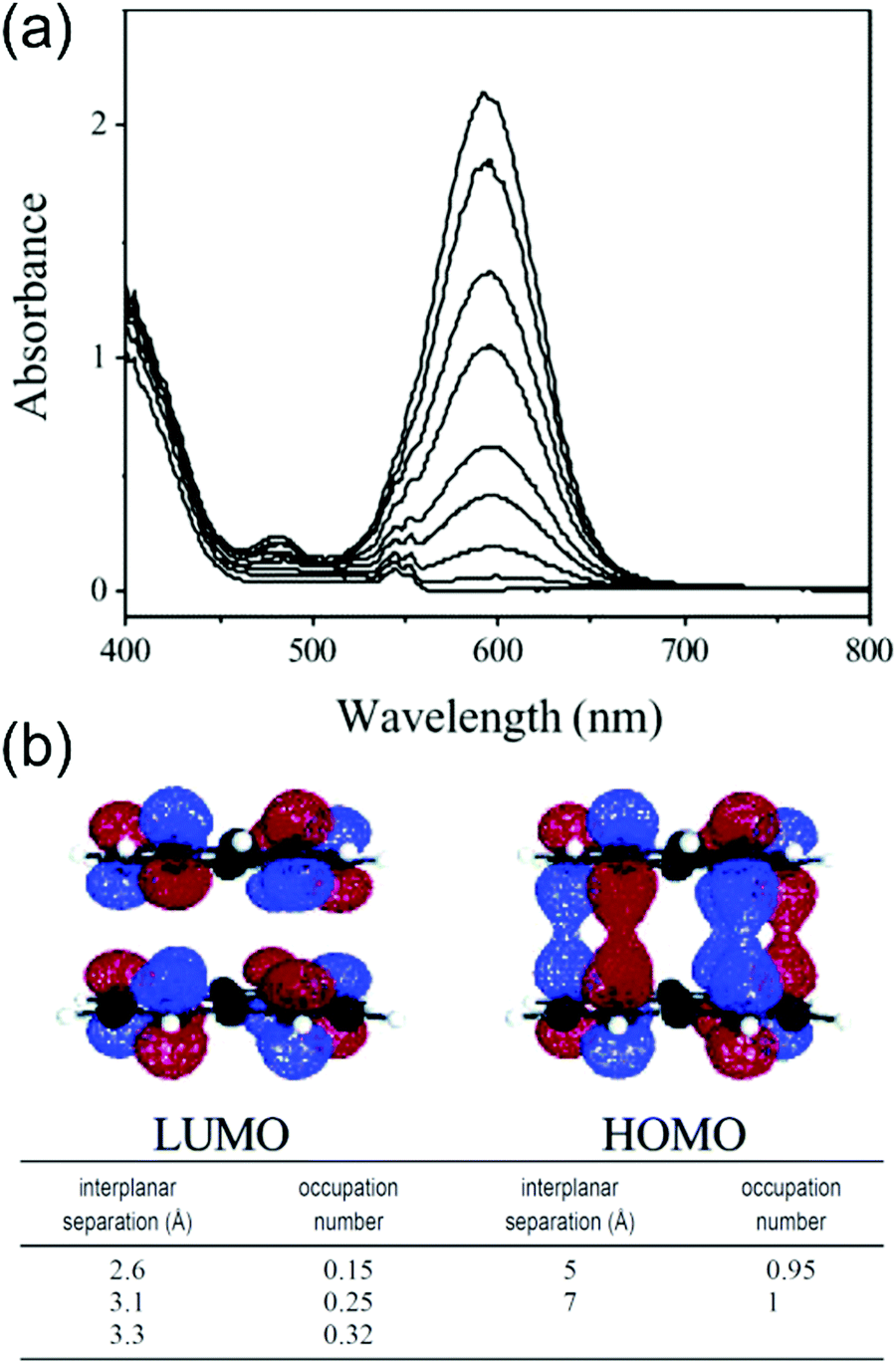 | ||
| Fig. 12 (a) Temperature-modulated spectral changes of the solution of phenalenyl radical (5 mM in CH2Cl2). Temperature from 278 K (bottom) to 191 K (top). (b) HOMO and LUMO of phenalenyl π-dimer. LUMO occupation numbers calculated for phenalenyl π-dimer at different geometries (occupation number of 1.0 corresponds to a pure diradical, whereas a value of 0.0 indicates a closed-shell species with no diradical character). Reprinted (adapted) with permission from ref. 22 Copyright 2004 American Chemical Society. | ||
Kubo et al.162–165 have studied the dimerization chemistry of phenalenyl radicals incorporating various bulky substituents and revealed that the formation of π-dimers can proceed through two pathways. One is direct π–π stacking of two radical monomers with no or very low energy barrier. The other is a stepwise mechanism where initial formation of a σ-dimer leads to a π-dimer via a higher energy barrier. The dimerization modes of the diamagnetic dimers could be observed experimentally by 1H NMR spectroscopy (Fig. 13). The choice of substituents play a key role in determining the strength of stacking and whether phenalenyl predominantly forms eclipsed (fully superimposed) or staggered (60° rotated) π-dimers (see Fig. 13 and ref. 162–164 for detailed representation of the dimerization modes of methyl-substituted phenalenyls). Perchlorophenalenyl radical is an interesting exception in this class of materials. It forms molecular stacks but the spacing within the stacks (3.78 Å) is too large to allow good SOMO–SOMO overlap, thus preventing dimerization.166,167 Control of π-dimerization of phenalenyls may be beneficial for tuning the inter-radical electrical, optical and magnetic interactions and obtaining other stacking modes like π-trimers for use in molecular electronics.
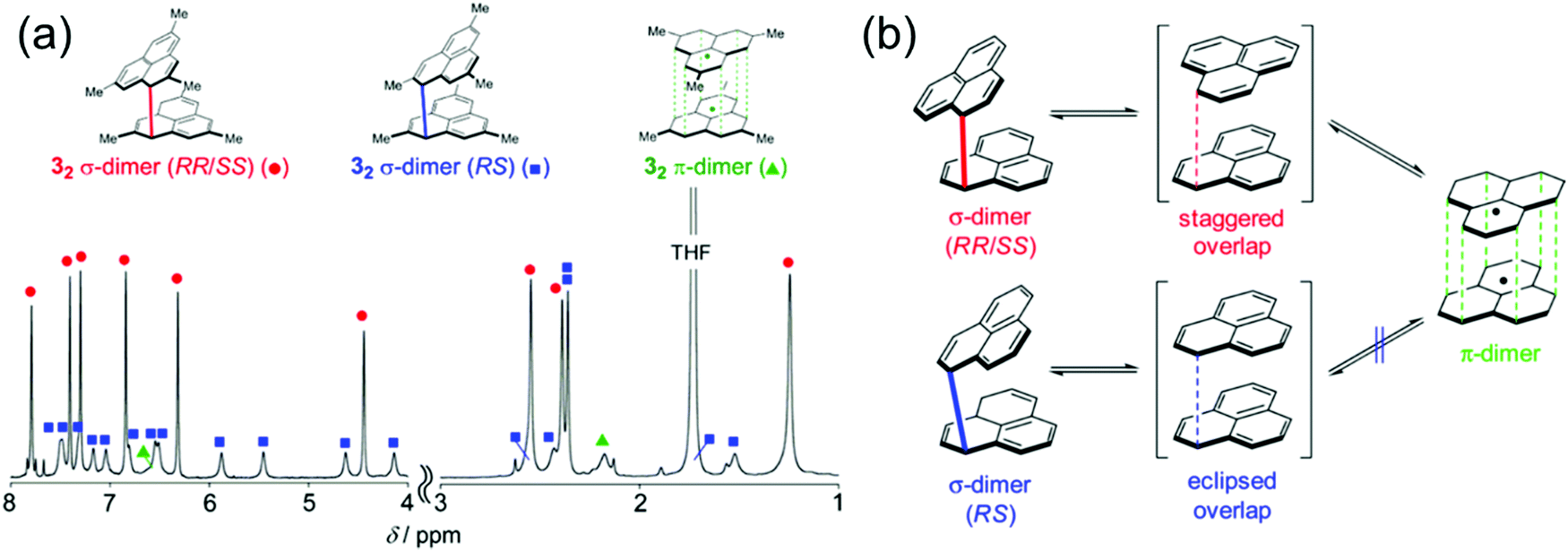 | ||
| Fig. 13 (a) 1H NMR spectrum of methyl-substituted phenalenyl dimers measured in degassed THF-d8 at 173 K. The aromatic and olefinic region (8–4 ppm) is magnified 3 times relative to the aliphatic region (3–1 ppm) for ease of visualization. (b) Dynamic Exchange between the dimers (methyl groups are omitted for clarity). Reprinted (adapted) with permission from ref. 162 Copyright 2016 American Chemical Society. | ||
Characteristics of other phenalenyl derivatives like 19 and 20, developed by Morita et al.,154,169 and larger planar radicals like triangulenes are in many ways analogous to the smaller phenalenyls. Morita et al.170–178 have utilised the added stability of trioxotriangulene (TOT) and dicyanomethyl substituted triangulene 21 (Scheme 7), which have four and eight reversible redox states, respectively, in a narrow potential range due to their degenerate LUMOs that are also energetically close to their SOMO. These radicals are electrically conductive and air-stable in the solid state, and even more so in their corresponding anionic forms (π-stacked crystals of 21 and TOT decompose at 195 and >300 °C in air, respectively), making them very attractive for use as rechargeable energy storage materials in organic batteries.170,171,178,179 TOT featured a strong NIR absorption band at 834 nm (ca. 2000 M−1 cm−1 at 300 K) in chloroform solution and the absorption was further enhanced on cooling (above 9000 M−1 cm−1 at 215 K) as a result of π-dimerization (Fig. 14a and b and Table S1, ESI†). TOT crystallized as columnar stacks in the solid state (Fig. 14c) and interdimer π–π interactions redshifted its absorption to 1134 nm.168 A general observation is that orbital overlap between the π-dimers decreased the excitation energy and redshifted the absorption of extended “polymer-like” π-stacked crystals. However, the crystals only absorbed light along the column direction. Another interesting observation is that substitution at the α-positions of TOT had a negligible effect on its electronic spin structure (due to small SOMO coefficients of the neutral radical at the α-positions) but α-hydroxy groups led to intramolecular hydrogen bonding to the adjacent carbonyl oxygens where some of the SOMO was delocalised. This in turn reduced the SOMO energy and affected the redox properties of the radical (see structures S15 and S16, Scheme S1 and compare to the redox potentials of TOT in Table S1, ESI†). Intramolecular hydrogen bonding also enhanced molecular planarity and strong intermolecular π-stacking, which was observed as redshifted low-energy absorption band peaking at 1416 nm in the solid state.177 Similar formation of diamagnetic π-dimers was recently reported for a silyl substituted TOT derivative by Morita et al.180 Self-assembly of the radical by polycondensation of the silyl groups gave rise to microporous organosilanes that gained additional stabilisation by spin delocalisation throughout the porous structure. Porous radical assemblies with reversible multi-stage redox reactions may find uses, for example, as very high charge-density materials in energy storage, and it would be exciting to see future progress towards this direction.
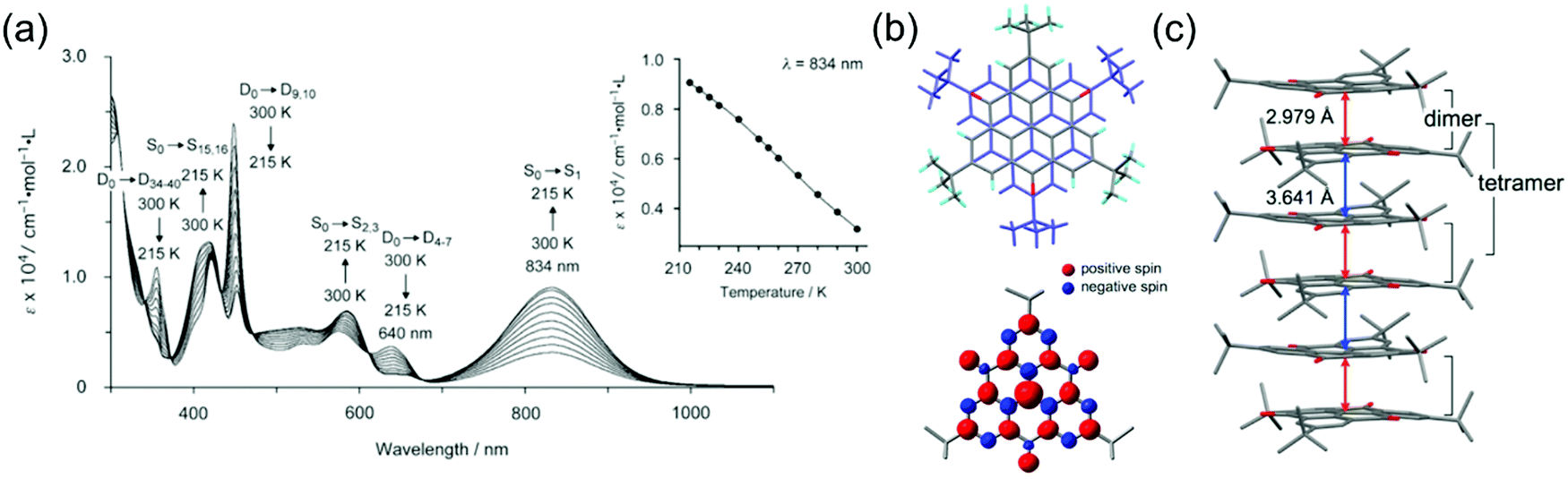 | ||
| Fig. 14 (a) Temperature-dependent UV-Vis spectra of TOT in CHCl3 solution (8.25 × 10−5 M). Assignments by the TD-DFT calculation of monomer (doublet, D) and dimer (singlet, S) are shown. Inset in (a) shows the plot of temperature dependence of the absorbance at 834 nm. (b) Crystal structure and calculated spin-density distribution of TOT. Red and blue regions denote positive and negative spin densities, respectively. (c) Overlap pattern of the 1D column and the column structure, where red and blue arrows designate intradimer and interdimer distances between Cb atoms, respectively (Cb stands for atoms which possesses the largest spin population). Reproduced from ref. 168 under CC BY 4.0 licence. | ||
Extension of the planar structure can be used as an effective tool to tune the intramolecular distribution of spin density and aromaticity, i.e., the number of so-called Clar's π-sextets,181,182 which allows the control of reactive sites and overall stability of spin-delocalised radicals.183 Haddon, Hirao and Kubo150 have designed sterically unprotected radical 22 where the unpaired electron can delocalise between two interconnected phenalene moieties. The SOMO of 22 (and its n-butyl substituted derivative 23, Scheme 7) was shown to distribute over 12 carbon atoms instead of six carbons in phenalenyl radical. Highly spin-delocalised structures 22/23 provided additional thermodynamic stabilisation that effectively suppressed their σ-dimerization. The n-butyl side chain improved the solubility of 23 allowing its purification in air and isolation as crystalline (non-bonded) π-dimers. Half-life of the radical in toluene solution was 60 h when exposed to air in the dark. The low-energy absorption band of 23 redshifted to 1355 nm in toluene solution, which can be expected from its rigid and planar structure, but the absorption as dissociated monoradical was significantly stronger (ε = 7910 M−1 cm−1, Table S1, ESI†) than that of above discussed spin-delocalised monoradicals or spin-localised radicals covered in chapters 2.1. and 2.2.150 The most recent addition to this series is a homoconjugated phenalenyl radical trimer and its monoradical counterpart reported by Kubo et al.184 (structures S17 and S18, respectively, Scheme S1 and Table S1, ESI†). Although the trimer was characterised behaving magnetically as a monoradical with an unpaired electron localised on one phenalenyl and the other two phenalenyls coupled through space antiferromagnetically, an optical benefit was observed as a new absorption band at 685 nm in dichloromethane solution (ca. 4000 M−1 cm−1 at room temperature, Table S1, ESI†) involving charge-transfer transitions between the phenalenyls. This through-space interaction was only effective in solution due to structural distortion in the solid state. Replacing the phenalenyls with similarly homoconjugated perylenes (with full delocalisation of the spin density) significantly enhanced the optical absorption, as reported earlier by the same authors,185 and it may be a good reference when designing novel spin-delocalised radicals with a strong optical response. This example leads us to the discussion of intramolecular spin–spin interactions in delocalised and localised diradicals in the next chapter.
2.4. Diradicals and their resonance forms
Chemical coupling of two radical centres in a planar structure is an effective method to design diradicals with controlled intramolecular spin interactions. Previously, Abe186 has reported an extensive summary of different classes of diradicals and Hoffmann et al.187 have provided an in-depth discussion on their reactivity and related classification. Shortly rehearsing these works (we sincerely recommend reading them and the references therein for thorough background of diradicals), diradicals can be categorised as localised and delocalised. Our interest is mainly on delocalised diradicals that are divided into Kekulé type structures like para-quinodimethane (p-QDM) and ortho-quinodimethane (o-QDM) and non-Kekulé type structures such as meta-quinodimethane (m-QDM).188 We describe these structures as resonance between closed-shell and open-shell forms shown in Scheme 8. Antiaromatic molecules like cyclobutadiene (CBD) and pentalene with 4n π-electrons are generally unstable, as opposed to aromatic compounds following the Hückel's rule189–191 with 4n + 2 π-electrons. Antiaromatic closed-shell structures can be classified as delocalised diradicals, although their classification varies in the literature.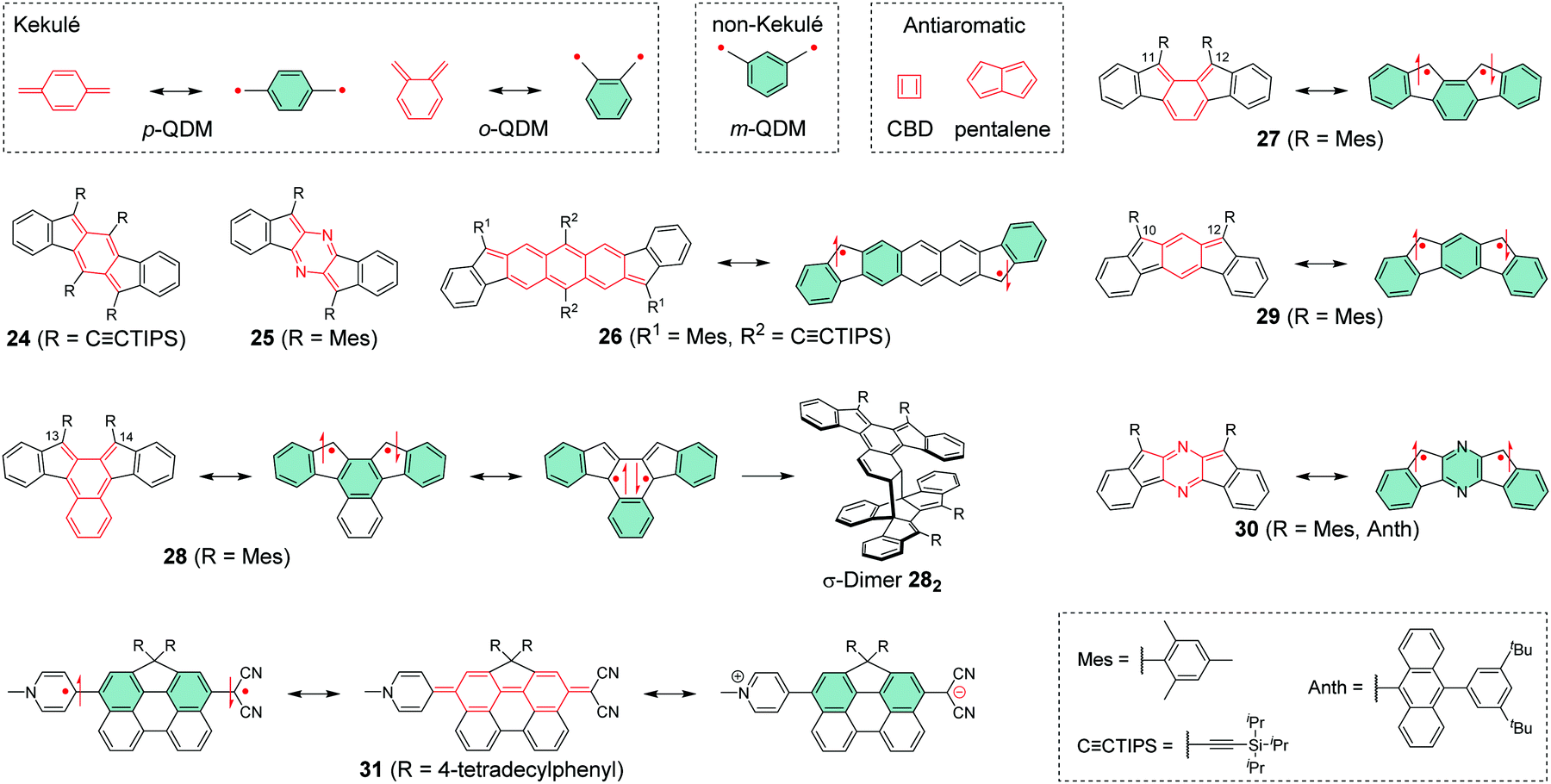 | ||
| Scheme 8 Delocalised diradicals and their closed-shell and open-shell resonance structures (R groups are omitted from some resonance structures for clarity). | ||
While monoradicals have a spin quantum number of S = ½ (an unpaired electron), there are two possible states, spin-paired singlet (S = 0) or spin-parallel triplet (S = 1), as the ground state for diradicals. This is one of the main defining features of diradicals. It could be expected from Hund's rule192,193 that for a diradical the triplet ground state (two degenerate SOMOs with unpaired spins) is energetically more stable than the spin-paired singlet ground state. However, Hund's rule may not apply to all diradicals. Some examples of manipulating the singlet and triplet ground states are discussed in this chapter. On the other hand, the singlet ground state of a diradical is stabilised when the two SOMOs are energetically separated (such systems are also called “diradicaloids” in the literature, but we stick to a single term “diradicals” in our discussion). The ground state configuration of diradicals determines their magnetic properties. Triplet diradicals are ferromagnetic and EPR active (NMR silent) in their ground state, similar to paramagnetic monoradicals, whereas singlet diradicals are antiferromagnetic and EPR silent (NMR active). An interesting note here is that for a singlet (triplet) diradical, triplet (singlet) is the lowest excited state. With a low singlet–triplet energy gap, the excited state is thermally accessible making singlet diradicals EPR active and triplet diradicals EPR silent. As the singlet–triplet energy gap affects the population of singlet and triplet species at different temperatures, it also affects the contribution of singlet and triplet excitations to the optical absorption of diradicals.194,195 Although thermal and optical excitations of diradicals are sometimes mixed and thereby inconsistently reported in the literature, our goal herein is to highlight some of the previously published molecules and thereby motivate more research efforts in the field. Lee et al.196 and Rajca et al.197 have discussed design strategies for high-spin molecules (total spin, S ≥ 1) from computational and synthetic points of view, respectively. High-spin molecules have great potential in magnetics and spintronics, but they are mostly experimental and meaningful applications for example in organic electronics have been hampered by their limited stabilities. We focus on some intriguing examples of low-spin (open-shell singlet) and high-spin (open-shell triplet) diradicals and their closed-shell quinoidal resonance structures, optical, electrical and magnetic properties as well as stabilities and prospective applications.
Haley et al.199–203 have developed various indenofluorene and fluorenofluorene structures and generalised the effect of electron delocalisation on the diradical character of different isomers. The common feature is that they exhibit relatively low HOMO–LUMO energy gaps, absorption reaching to the NIR region and reversible electrochemical redox reactions, which are attractive properties for any optoelectronic material.204,205 For example, indeno[1,2-b]fluorene derivative 24 is a closed-shell ground state molecule because the antiaromatic p-QDM core (coloured red in Scheme 8) lacks sufficient stabilisation of the diradical form. However, extension of the central core to a larger anthracene unit in diindeno[b,i]anthracene (26) stabilises the diradical by moderate aromatization of the quinoidal structure (shown as three Clar's π-sextets in Scheme 8 and the aromatic ring current in Fig. 15, see also structures S19–S23, Scheme S2 and Table S1, ESI†).198,206 In the ground state, the closed-shell structure is in resonance with the aromatic open-shell diradical. 26 showed typical 1H NMR characteristics of a singlet diradical, that is, broadening of the aromatic signals on heating from 298 to 423 K as a result of increasing population of the triplet excited state (the singlet–triplet energy gap, –4.2 kcal mol−1, was relatively small and thermally accessible). Thermally accessible triplet state is significant in that it enables reversible switching on and off the magnetic response of a stable diradical. 26 also stabilised electrochemically injected and extracted charges as it underwent two reversible reduction and oxidation reactions via its closed-shell and open-shell resonance structures, respectively (Table S1, ESI†). In other words, it behaves both as an aromatic electron-donor and as a quinoidal electron-acceptor. This amphoteric redox behaviour was also reflected in its balanced electron and hole mobilities in organic field-effect transistors (OFETs, mobilities were in the range of 10−3–10−2 cm2 V−1 s−1). 26 showed strong optical absorption peaking at 690 nm (above 40000 M−1 cm−1) and another weaker band extending to 900 nm, both originating from transitions to the low-energy singlet excited states. This example highlights the potential of (singlet) diradicals and their substantially strong absorption as compared to (doublet) monoradicals discussed in previous chapters. Combined with straightforward synthesis and stability in the crystalline solid state (half-life in dichloromethane solution open to air was 64 days), 26 undoubtedly is an excellent example of a robust material for real-life applications.198,206
 | ||
| Fig. 15 (a) Isosurface (yellow) and current density vectors (green lines) calculated by ACID (anisotropy of the induced current density) for the open-shell singlet state of 26. The general aromatic current is indicated by the superimposed clockwise magenta arrows (some of the side chains have been truncated or omitted from the structure for calculation purposes). (b) NICS-XY (nucleus-independent chemical shift) scans of the open-shell singlet and triplet states of 26 skeleton show an increase in aromaticity for the thermally accessible triplet state. Capital letters (top) refer to the rings in the model structure and variable r describes the distance along the dashed red path. Reprinted by permission from ref. 198 Copyright 2016 Macmillan Publishers Limited. | ||
Tobe et al.207–209 have designed related structures with varying singlet diradical characters. For example, indeno[2,1-a]fluorene derivative 27 is based on an o-QDM skeleton where the spin density is mainly delocalised. Positions C11 and C12 exhibit the largest α- and β-spin densities (indicated by the “up” and “down” arrows, respectively, in Scheme 8).207 Bulky mesityl groups are needed to provide sufficient steric protection and stabilisation of the two radical sites. Solution of 27 in dichloromethane can be stored under ambient air and light for a week or so. The solution showed two low-energy absorption bands at 730 nm (ε = 790 M−1 cm−1) and 537 nm (ε = 15200 M−1 cm−1, Table S1, ESI†). It is noteworthy that 27 and related structures are totally non-emissive. All in all 27 was characterised with weak diradical character that did not affect its NMR signals even at elevated temperatures.207
Extension of conjugation by a larger naphthalene ring in 28 resulted in stronger antiaromatic singlet diradical character as compared to 27.209 In the case of 28, some of the spin density is distributed in the naphthalene moiety, making the molecule susceptible to reacting with oxygen from the inner naphthalene carbons rather than the mesityl-protected 13- and 14-carbons. 28 was air-stable in the crystalline solid state but in dichloromethane solution it rapidly reacts with oxygen with a half-life of 77 min. The oxidation reaction can be observed as a complete disappearance of its strong low-energy absorption at 697 nm (initial ε = 12800 M−1 cm−1, see Table S1, ESI†). Under inert atmosphere, the diradical showed reversible broadening of 1H NMR signals at high temperatures due to increasing population of thermally excited triplet state. Both 27 and 28 can be regarded thermally stable up to 100 °C. However, prolonged heating of 28 (80 °C, 48 h) resulted in irreversible cyclo-dimerization to give 282 in nearly quantitative yield.209
A m-QDM derived indeno[2,1-b]fluorene 29 showed triplet excited state species already at room temperature.208 Its 1H NMR spectrum could be resolved on cooling down to 180 K as a result of increasing occupation of the singlet ground state (the singlet–triplet gap was estimated to be −4.2 kcal mol−1). EPR measurements showed opposite trend as the signal of the paramagnetic species increased at higher temperatures. Also 29 decomposed over time despite the steric protection of the 10- and 12-positions. It is a shame because the diradical showed intense absorption at 638 nm (above 35000 M−1 cm−1) and another weak low-energy singlet excitation band spanning between 800–2000 nm in dichloromethane solution (see Table S1, ESI†).208 Such low-energy absorption would be very attractive for various light-harvesting applications utilising the entire NIR spectrum, albeit with possible contamination by absorption of thermally excited triplet species.
Wang et al.11 modified the spin density distribution of 29 with an electron-deficient pyrazine unit. This operation increased the spin density on the meta-pyrazine core in 30 (coloured red in Scheme 8) and resulted in triplet diradical as the dominant ground state resonance structure (illustrated by the two “up” arrows in Scheme 8). 30 displayed significant aromatic character in the pyrazine ring and, including the two outer benzene rings, the diradical was represented as exhibiting three Clar's π-sextets (also seen from the aromatic current in Fig. 16). In contrast, its para-quinoidal derivative 25 showed a dominant antiaromatic closed-shell ground state structure (compare to 24, Scheme 8). This difference is also observed in the optical absorption of the two molecules. The low-energy absorption of 25 peaked at 535 nm, whereas 30 showed significantly redshifted absorption extending to 1200 nm (the latter attributed to the diradical resonance structure, see Table S1, ESI†). Opposite to the singlet diradical 29, 30 showed increasing EPR signal intensity on cooling from 294 to 100 K. In other words, 30 exhibited inverted singlet–triplet energy levels with 1.8 and 1.0 kcal mol−1 energy gap (that is, for mesityl and 9-anthryl substituted diradicals, respectively) and near-full occupation of the triplet ground state. Incorporation of electron-withdrawing substituents into the central ring seems to be an excellent method to increase the spin density of the radical centres and in that way obtain stable high-spin diradicals. That said, the half-life of 30 was 22 days under sunlight and 28 days under darkness making it among the most stable diradicals reported to date, as the authors rightfully stated.11
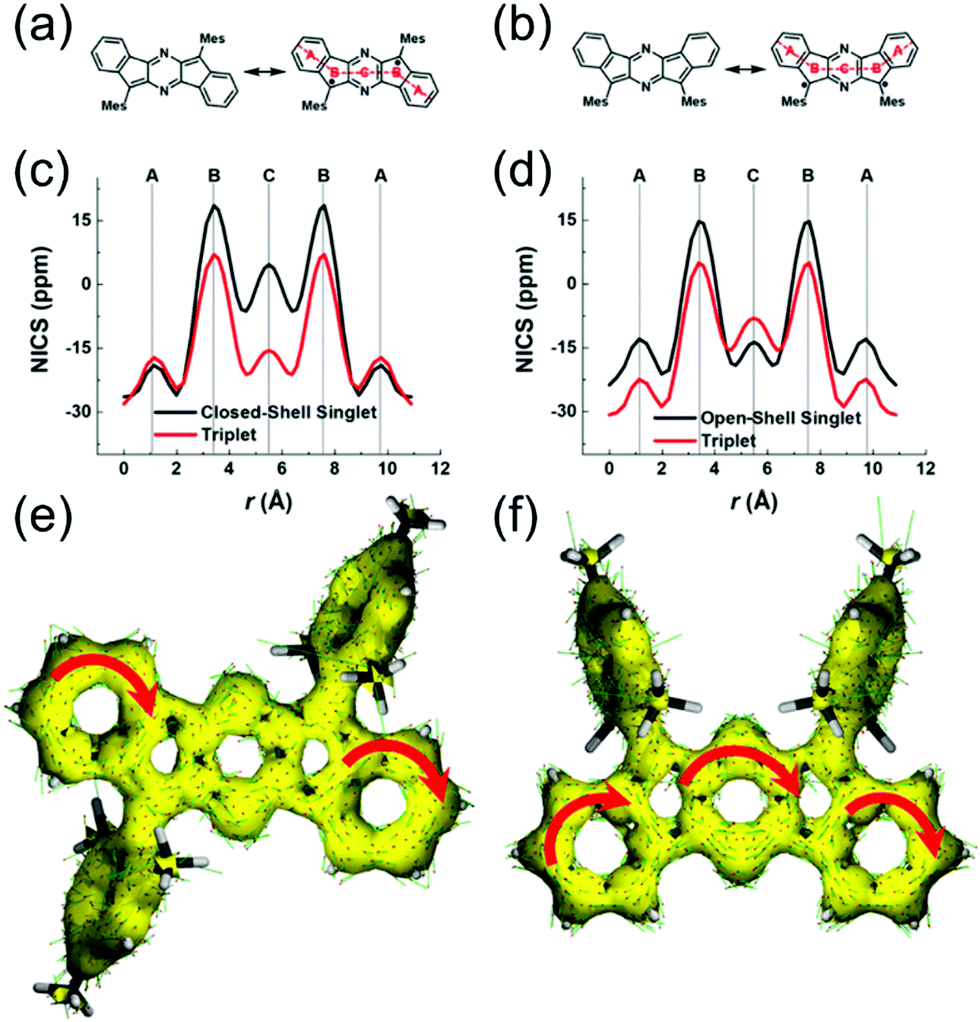 | ||
| Fig. 16 Comparison of the resonance structures of (a) 25 and (b) 30. NICS-XY scans of (c) 25 and (d) 30. ACID plots of (e) closed-shell singlet 25 and (f) open-shell triplet 30 at an isosurface (yellow) value of 0.04. The general aromatic current is indicated by the superimposed clockwise red arrows. Reproduced with permission from ref. 11 Copyright 2020 Wiley-VCH GmbH. | ||
Bisphenalenyls are another class of diradicals where the two phenalenyls are separated by p-QDM core. But unlike the peripheral benzene rings in related diradicals like 24, the two phenalenyl moieties provide thermodynamic stabilisation of the singlet diradical resonance structure by means of delocalisation of the odd electrons. Diradical character can be enhanced by separating the two radical sites with a naphthalene spacer. The works of Kubo et al.210–213 stand as excellent examples of neutral bisphenalenyl diradicals showing narrow and intense absorption at 750 nm or longer (see structures S24–S26, Scheme S2 and Table S1, ESI†). Extensive spin-delocalisation and strong intermolecular spin–spin interactions between bisphenalenyls has provided a range of NIR-absorbing diradicals with almost non-existing absorption in the visible spectral region, making them distinctively different from the phenalenyl monoradicals and homoconjugated phenalenyls discussed in chapter 2.3. They may be of interest for various NIR light-harvesting applications, but challenges in obtaining truly stable diradicals remain. Chen et al.214,215 have based their design on the same concept and synthesized heteroatoms containing electroactive and luminescent bisphenalenyls that are stable in their cationic forms. On the other hand, m-QDM core has been used in the design of nickel porphyrin diradicals with triplet ground state by Song et al.216 Added stability is obtained by effective delocalisation of the spin density in two porphyrin rings, and the diradical can be stored for several months in the solid state in air (half-life in 1,2-dichlorobenzene was about 28 days when heated at 80 °C under ambient air).
Graphene fragments have been appealing design platforms for planar radicals; some examples were discussed in the previous chapter 2.3.32,183,218–220 Kubo et al.221,222 reported teranthene and quarteranthene molecules which exhibited open-shell singlet diradical ground states. Wu et al.223 extended the synthetic series of rylene oligomers up to dodecarylene (containing 12 fused naphthalene units) with small HOMO–LUMO energy gaps and a promise – or possibility – to deliver polymeric graphene nanoribbons with metallic or half-metallic properties. Perhaps even more relevant to the discussion herein is the design of a quinoidal D–A type perylene molecule 31 that can resonate between a closed-shell quinoidal form, a closed-shell zwitterionic form and an open-shell singlet diradical form (see Scheme 8).217 All three resonance forms were present in the ground state but delicate balance between them could be tuned by the polarity of the solvent, thereby allowing modulation of the optical response and electrochemical redox properties. The special feature was that 31 showed negative solvatochromism due to a large contribution of the open-shell electronic structure in less polar solvents (ε = 25860 M−1 cm−1 at 822 nm in dichloromethane solution) and stabilisation of the zwitterionic form in more polar solvents (ε = 15030 M−1 cm−1 at 654 nm in DMSO solution), as shown in Fig. 17 and Table S1 (ESI†).
 | ||
| Fig. 17 (a) Electronic absorption spectra of 31 in different solvents. (b) Optimized geometry with dipole moment (left) and the calculated electrostatic potential map (right) of 31 in gas phase. Reproduced with permission from ref. 217 Copyright the Royal Society of Chemistry and the Chinese Chemical Society 2019. | ||
Thiele's,225 Chichibabin's226 and Müller's227 hydrocarbons are classical examples of diradicals where the two radical centres are separated by one (32), two (33) and three benzene rings (34), respectively, as shown in Scheme 9.228–230 Chichibabin's derivatives have been of particular interest in the field because resonance between their open-shell diradical and closed-shell quinoidal forms involves some degree of distortion of the molecular geometries, which allows tuning of their ground state and excited state properties.231,232 Wu et al.224 have utilised this feature in benzannulated Chichibabin's structures 35 and 36, where the central benzene rings are extended to larger anthracene units (Scheme 9). Although the two compounds are structurally similar and their quinoidal forms are stabilised by gaining another two Clar's π-sextets when converted from diradicals to quinoids, 35 was a closed-shell molecule but 36 exhibited an open-shell ground state. The latter is due to thermodynamic stabilisation of the fluorenyl units in the orthogonal conformation as compared to the contorted quinoidal form (Fig. 18, top panel). The diradical lacked 1H NMR signals even when cooled down to 173 K (singlet–triplet energy gap was estimated to be 0.3 kcal mol−1) indicating population of its paramagnetic triplet ground state. Another interesting observation for the discussion herein is that 36 was extremely stable. No decomposition was observed in solution or in the solid state when stored under ambient air and light for months, which was attributed to the stabilising effect of the fluorenyl units (SOMOs of α- and β-spins in the triplet ground state were delocalised to the fluorenyl moieties, see Fig. 18). 36 showed weak absorption tail in the NIR region, but both 35 and 36 could be chemically oxidized to stable dications for the realisation of stronger NIR bands (two oxidation states at relatively low potentials enabled removal of the two unpaired electrons, see Table S1, ESI†). Introduction of dimethylamino substituents into 35 resulted in a rather peculiar ammonium salt with two chlorine counterions (structure S27, Scheme S2, ESI†).233 The substitution added to the resonance of the two radical electrons, enhancing switching between triplet excited diradical and quinoidal forms on heating and cooling, respectively. The switching could be observed visually as diminishing and intensifying absorption at 642 nm (peaking close to 400000 M−1 cm−1 at room temperature, see Table S1, ESI†), and it stands as one of the rare examples of luminescent (neutral) diradicals, which promises great use of Chichibabin's structures in thermochromic and thermomagnetic applications. Such diradical design may well deliver the desired chemical, optical and electrochemical stability requirements for use in organic electronics as well.
 | ||
| Scheme 9 Closed-shell and open-shell resonance structures of Thiele's, Chichibabin's and Müller's hydrocarbons and related derivatives (R groups are omitted from some resonance structures for clarity). | ||
 | ||
| Fig. 18 Calculated geometric structures (with part of the bonds labelled with length in Å) and the frontier molecular orbitals of the closed-shell (CS) and open-shell (OS) forms of 36 both in singlet and in triplet states. The bottom panel shows the spin density distributions of the singlet and triplet open-shell forms of 36. Reprinted (adapted) with permission from ref. 224 Copyright 2012 American Chemical Society. | ||
Building upon the above-discussed works, others have developed Thiele's, Chichibabin's and Müller's N-heterocyclic,235 acyclic diaminocarbene236 and molecular cage237 analogues on the merit of tuning the diradical characters, singlet–triplet energy gaps, absorption spectra, redox properties and magnetic responses. One could argue that all diradicals discussed in this chapter can be considered as derivatives of the classical hydrocarbons, where the central benzene rings are either freely rotating or fused in a planar conformation. Stępień et al.234 have merged the two concepts and improved the aromatic stabilisation of a Chichibabin's derivative diindeno[a,i]phenanthrene (37, Scheme 9), whose singlet diradical character is enhanced by four Clar's π-sextets. The α- and β-spin densities were spatially separated and localised at the five-membered rings in both singlet and triplet states further stabilising the diradical. However, the aromaticity of the central phenanthrene core increased in the triplet state resulting in an easily accessible paramagnetic excited state (singlet–triplet energy gap was −1.3 kcal mol−1), which was seen as broadening of 1H NMR signals on heating from 220 K to room temperature and above (see Fig. 19a). The main absorption band peaked at 600 nm (ε = 41000 M−1 cm−1, Table S1, ESI†) with a low-energy band tailing to 1050 nm in dichloromethane solution. 37 was also stable as a monoradical cation but its dication and dianion forms were unstable, making it distinctively different from other Chichibabin's structures like 35 and 36. Perhaps the most interesting observation is that 37 was stable in dichloromethane solution for 25 days or so, but the choice of solvent greatly affected its stability. Solid sample was stable up to 110 °C, but excessive heating resulted in irreversible oligomerisation of hydrogenated structures of sizes up to n = 15, as shown in Fig. 19b. The authors pointed out that such “hydrogen-scrambling” oligomerisation may be relevant as a prospective method of synthesizing open-shell polymeric materials,234 which links to our later discussion in chapter 2.5.
 | ||
| Fig. 19 (a) Temperature-dependent 1H NMR spectra of 37 (CDCl3, 600 MHz). The signals were assigned at 220 K using 2D spectroscopic methods (see ref. 234 for details). (b) Thermally induced oligomerization of 37. Reagents and conditions: (a) argon, pressure tube, 240 °C, 14 h. Reproduced from ref. 234 under CC BY-NC 3.0 licence. | ||
The classical diradical concept can be simplified to the smallest possible diradical, that is, an ethylene group where spin–spin interactions of the two bonding electrons can be weakened by elongated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and substantial torsion angles. Perchlorinated and benzannulated 9,9′-bifluorenylidenes are examples of sterically congested ethylene structures whose thermally accessible triplet excited states can be regarded as diradials.238,239 Kubo et al.240,241 stabilised twisted bianthrone (38) by aliphatic side chain substitution. The molecule exists as a mixture of twisted and folded forms, which was characterised as two different optical absorption bands at 757 and 444 nm in dichloromethane solution, respectively. The twisted form (see Scheme 9 and ref. 241 for original representation) exhibited CC bond length of 1.429 Å and torsional angle of ca. 57° in the solid state, which decreased the singlet–triplet energy gap to −5.7 kcal mol−1. Also, the electrochemical redox potentials of the twisted form were comparable to the above-discussed quinoidal diradicals (see Table S1, ESI†). Twisted 38 was interpreted as an intermediate state between CC double bond and bond dissociation, retaining its bonding interaction as a singlet diradical but allowing thermal excitation to the triplet excited state, as illustrated in Scheme 9.241 Twisted CC diradicals are still far less studied than the larger quinoidal diradicals, but we speculate that they might find uses in various (transient) energy harvesting processes.
C double bond and substantial torsion angles. Perchlorinated and benzannulated 9,9′-bifluorenylidenes are examples of sterically congested ethylene structures whose thermally accessible triplet excited states can be regarded as diradials.238,239 Kubo et al.240,241 stabilised twisted bianthrone (38) by aliphatic side chain substitution. The molecule exists as a mixture of twisted and folded forms, which was characterised as two different optical absorption bands at 757 and 444 nm in dichloromethane solution, respectively. The twisted form (see Scheme 9 and ref. 241 for original representation) exhibited CC bond length of 1.429 Å and torsional angle of ca. 57° in the solid state, which decreased the singlet–triplet energy gap to −5.7 kcal mol−1. Also, the electrochemical redox potentials of the twisted form were comparable to the above-discussed quinoidal diradicals (see Table S1, ESI†). Twisted 38 was interpreted as an intermediate state between CC double bond and bond dissociation, retaining its bonding interaction as a singlet diradical but allowing thermal excitation to the triplet excited state, as illustrated in Scheme 9.241 Twisted CC diradicals are still far less studied than the larger quinoidal diradicals, but we speculate that they might find uses in various (transient) energy harvesting processes.
2.5. Polyradicals
Two interacting spin centres discussed in previous chapter leads us to structures with more than two SOMOs close to each other in energy in the same molecule, giving rise to either interacting or noninteracting spins, herein referred to as polyradicals. We note that term “polyradicaloids” is often used in the literature for molecules with more than two radical centres, whereas for conjugated polymers used in the organic optoelectronics field the number of repeating units, n > 10, is commonly used as a definition for polymeric materials. Given that some borderline cases are also discussed in this chapter, we use a single term “polyradicals” for consistency. We have excluded so-called “small or zero bandgap” polymers from our discussion.242,243 Although they are composed of quinoidal (or proquinoidal) structures with open-shell character and high-spin properties, they do not fall in with our focus on π-radicals whose spin(s) can be stabilised at one (or more) specific carbon centre(s) by gaining a number of Clar's π-sextets. Plentiful examples of such diradical systems were discussed in the previous chapter. In this chapter, we discuss different approaches to chemically couple radicals in conjugated and non-conjugated macromolecules.Since the summaries of magnetic polyradicals two–three decades ago by Rajca,3,21 the group has reported various calix[4]arene-based macromolecules with up to 36 triarylmethyl radical centres, tuned ferromagnetic and antiferromagnetic couplings between the spin centres and very high ground-state spins (average S = 5–13, see structures 39–41 in Fig. 20).244–247 Ferromagnetically coupled radicals are appealing for polymer magnetics and superconductors but they require rigorous synthesis and handling at low temperatures. Their ferromagnetic character decreases above cryogenic temperatures due to thermal excitation to low spin states, while exposure to room temperature leads to irreversible thermal decomposition, typically after 30 min or so. Incorporation of TTM and PTM radicals into conjugated backbones has made polyradicals stable at room temperature also in the solid state, and thereby practical for use in photonics and electronics. Some early examples are acetylene- and ethylene-bridged PTM polyradicals where strong ferromagnetic coupling is absent because of spatial separation of the radical centres, in other words, due to negligible spin–spin interactions. This also meant that PTM polyradicals were practically insulators with no measurable electrical conductivities.248
 | ||
| Fig. 20 Chemical structures of high-spin polyradicals 39 (S = 5–6), 40 (S = 7–9) and 41 (S = 11–13) as organic spin clusters. The average spins are below the expected values (component spins 7/2, 6/2, 4/2 and 1/2 shown below the chemical structures) due to evident antiferromagnetic couplings and chemical defects. Reprinted (adapted) with permission from ref. 246 Copyright 2004 American Chemical Society. | ||
Based on the works of Veciana group,249–251 others have reported several approaches to obtain PTM polyradicals and macrocycles from one-dimensional copper and cobalt coordination polymers of para-carboxylic substituted PTM radical to chemically coupled two-dimensional nanosheets and covalent organic frameworks252–254 and supramolecular arrangement of PTM radicals thorough hydrogen bonding,255,256 and recently to self-assemblies of chiral PTM tetraradical cages via imine condensation.257 The common feature is that the magnetism can be tuned by controlling the intramolecular ferromagnetic and antiferromagnetic interactions of the radical centres, while the bulky PTM groups effectively minimise additional through-space intermolecular magnetic interactions.255 On the contrary, Zhao et al.258 reported a platinum-PTM metallacage where the two radical centres were linked relatively close (ca. 15 Å) to allow through-space spin–spin interactions and thermal modulation of the spin state. Yang et al.259 have provided an overview of the recent progress on the development of supramolecular radical cages and their applications.
Lambert et al.260 applied the D–A design strategy (discussed in chapter 2.1) to the synthesis of polyradical 42 consisting of alternating TPA donor and PTM acceptor units (Scheme 10). The polymer was synthesized by Horner–Wadsworth–Emmons reaction261,262 of PTM bisphosphonate and TPA dialdehyde in basic reaction condition at room temperature, yielding a number-average molecular weight (Mn) of 11 200 g mol−1. Like the D–A systems discussed in chapter 2.1, electron transfer from TPA sites to PTM centres could be optically induced and this transition is observed as CT absorption band peaking at 820 nm (ε = 6500 M−1 cm−1) in toluene solution (see Table S1, ESI†). Intensity of the CT band was comparable to that of a corresponding D–A monomer,94,95 also in the solid state, meaning that the electron transfer was confined to only one D–A repeating unit in the polymer backbone (additional evidence is provided in ref. 260). It is highly relevant for photonic and electronic applications that 42 underwent reversible electrochemical oxidation and reduction of the TPA and PTM sites, respectively (Table S1, ESI†). This was also reflected in balanced ambipolar charge transport in films in OFETs (electron and hole mobilities were both 3 × 10−5 cm2 V−1 s−1). Low mobilities were attributed to the amorphous solid state, because the bulky propeller-like TPA and PTM sites prevented closed stacking of the polymer chains. Spin interactions in the ground state are also expected to be weak because the PTM sites are even more separated by the TPA moieties than by the ethylene- and acetylene-bridges in the above discussed PTM polyradicals. It is worth mentioning that solid polymer and polymer in toluene solution were stored for several months and all characterisation was performed under ambient conditions (the polymer film was even annealed at 110 °C). This clearly highlights the added stability of neutral radicals when incorporated into conjugated backbones.
 | ||
| Scheme 10 Chemical structures of TTM, PTM and fluorenyl based polyradicals. | ||
Scherf et al.263 utilised the stability of D–A radicals in electrochemical polymerisation of di- and tricarbazolyl substituted PTM to obtain polyradicals poly-D˙ and poly-T˙, respectively (Scheme 10). In contrast to chemical approach where the α-hydrogenated polymer needs to be deprotonated in basic conditions followed by one-electron oxidation by an electron-acceptor dopant to obtain a neutral polyradical (same procedure as for neutral monoradicals), electrochemically oxidative C–C coupling of the radical monomers gave the polyradicals directly without further reactions. It is worth mentioning here that electropolymerisation in general is less scalable than chemical synthesis, but it could be practical for direct deposition of polyradical thin films as sensors for magnetoelectronics and related applications. Polymerisation of the dicarbazolyl radical gave a linear polymer (shown in the ESI in ref. 263) whereas the tricarbazolyl radical resulted in a three-dimensional structure that is expected to be microporous. Both poly-D˙ and poly-T˙ films showed strongest absorption in the UV-visible but CT interactions between the carbazole moieties and the PTM centre were observed at 800 nm or beyond. An interesting feature of these polymers is that they could be chemically or reversibly electrochemically reduced to air-stable anionic forms, poly-D− and poly-T−, accompanied by a substantial change in colour and optical absorption profile, as shown in Fig. 21. In contrast, molecular PTM carbanions are known to oxidize instantly to give neutral π-radicals if not stabilised in an inert environment (this feature is commonly used for the benefit of the synthesis of PTM radicals, see Scheme 2). Neutral polyradicals, poly-D˙ and poly-T˙, were both paramagnetic as characterised by EPR spectroscopy. We assume that electron transfer was confined to one repeating unit and the polyanionic species were stabilised by multiple charge-separated zwitterionic states, similar to that observed in the corresponding monoradicals (and comparable to the PTM-TPA radicals discussed in chapter 2.1).
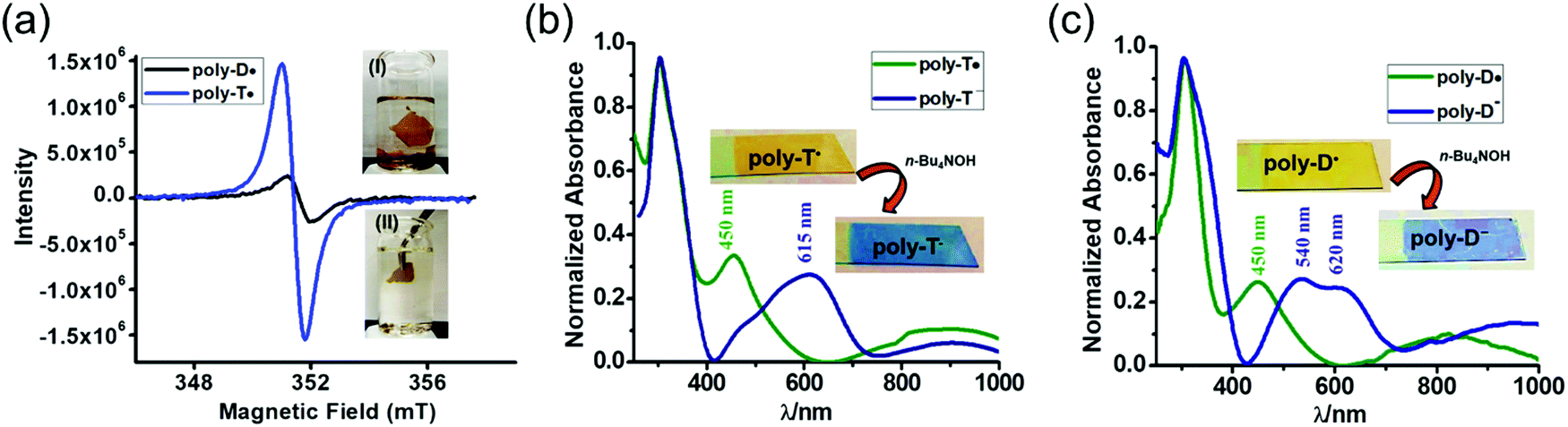 | ||
| Fig. 21 (a) EPR spectra of poly-T˙ (blue) and poly-D˙ (black) thin films (the insets show photographs of free-standing thin films of (I) poly-T˙ and (II) poly-D˙ dispersed in CH2Cl2). Normalized UV−Vis absorption spectra of (b) poly-T˙, poly-T− and (c) poly-D˙, poly-D− thin films before and after immersing poly-T˙ or poly-D˙ films with n-Bu4NOH in methanol (the insets show photographs of thin film-coated ITO electrodes). Reprinted (adapted) with permission from ref. 263 Copyright 2019 American Chemical Society. | ||
Li et al.264,265 coupled TTM-1Cz (see chapter 2.1) as a pendant substituent in a nonconjugated polystyrene backbone and reported the first luminescent polyradical PS-CzTTM (Scheme 10). The polymer was synthesized via radical polymerisation of vinylbenzyl-substituted monomer and styrene, followed by conversion of the α-hydrogenated carbon centres to neutral radicals. Only small amount of TTM-1Cz was incorporated into the polymer (ratio of styrene, n, and radical, m, repeating units was 292:5) so that the radical concentration was less than 2%. In other words, the radical centres were expected to be statistically distributed and well-separated in the polymer backbone (Mn 38700 g mol−1), although approximately three out of four TTM-1Cz sites were successfully chemically converted to neutral radicals and the rest remained α-hydrogenated. PS-CzTTM was paramagnetic at room temperature, as expected with negligible spin–spin interactions based on the molecular design. The polymer was also highly luminescent in both solution and solid-state film, because the random ordering prevented strong aggregation quenching of the radicals’ emission. As a result, PS-CzTTM showed notably efficient deep-red emission peaking at 694 nm in a solid-state film (PLQY 25%, Table S1, ESI†). Another important merit of radical emitters is their photostability. Half-life of emission intensity of PS-CzTTM in cyclohexane solution was 1.6 × 104 s, which is about 300 times higher than that measured for TTM monoradical under the same conditions. Despite the low radical concentration, the spin density was mainly localised at the radical sites delivering emission purely from the doublet excited state even when the temperature was varied between 10–300 K. The emission process was maintained in OLEDs when using PS-CzTTM as the emitter (see Fig. 22). However, the nonconjugated polystyrene host is not an ideal charge transport matrix and additional host material was needed to increase the EQE from 1.0% (for neat PS-CzTTM) to 3.0% (for PS-CzTTM in TPBi matrix, so-called “host-guest” system) with emission peaking at 685 nm in both cases.264,265
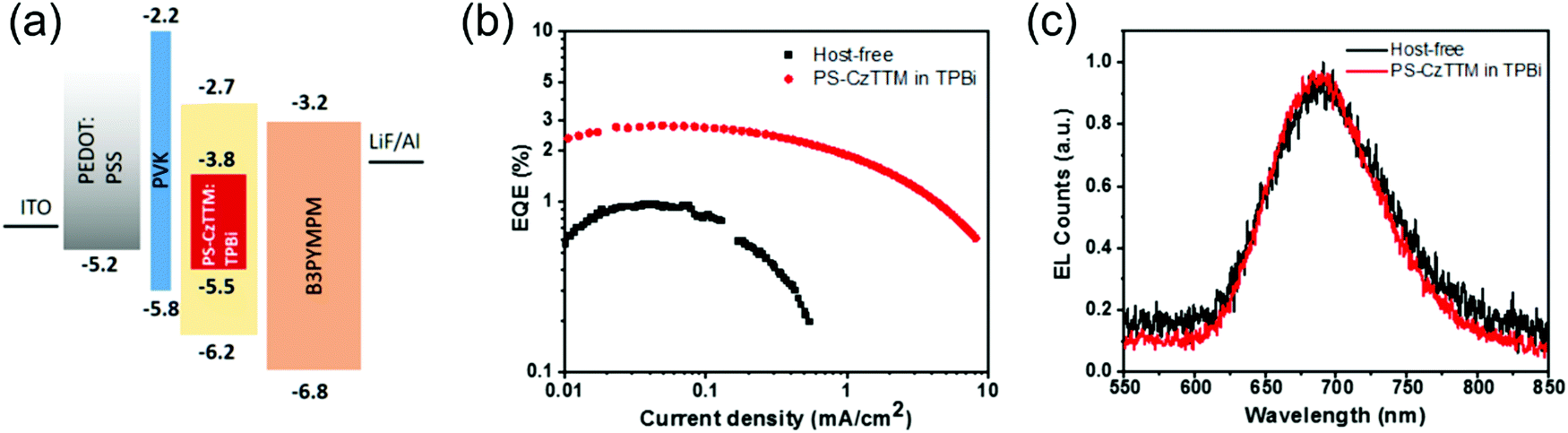 | ||
| Fig. 22 (a) Schematic diagram of the device structure of PS-CzTTM-based OLED. (b) EQE of host-free and host-guest OLEDs versus current density. (c) EL spectra of both host-guest and host-free OLEDs at 12 V operating voltage. Reprinted (adapted) with permission from ref. 264 Copyright 2020 American Chemical Society. | ||
Wu et al. have reported various types of low-spin fluorenyl oligomers,266 macrocycles267,268 and dendrons269 that exhibit polyradical characters with intramolecular antiferromagnetic spin–spin interactions, that is, resonance between open-shell polyradical and closed-shell quinoidal forms. Linear 3,6-linked fluorenyl oligomers (FR-n, Scheme 10) are interesting examples that were synthesized via multiple Pd-catalysed Suzuki reaction steps, followed by conversion to neutral radicals. The chain length had a significant impact on their optical, electrochemical and magnetic properties, which may be relevant for the design of other oligo/polyradicals as well. Oligomers with even number of repeating units (n = 2 dimer, n = 4 tetramer and n = 6 hexamer shown in Scheme 10) had an open-shell singlet ground state, whereas structures with odd number or repeating units (n = 1 monomer, n = 3 trimer and n = 5 pentamer) had a doublet ground state. In all cases, the unpaired electron density was mainly localised at the fluorenyl units, thereby requiring kinetic stabilisation by bulky anthryl groups. Energy gaps from the low-spin ground states to the lowest high-spin excited states (that is, singlet–triplet gap for even number of repeating units and doublet–quartet gap for odd number with n ≥ 2) were low enough to be thermally accessible at room temperature. This was observed as sharpening of the 1H NMR signals (and weakening of EPR signals) on cooling, in line with the characteristics discussed in chapter 2.4. Perhaps the most interesting observation for the discussion herein is that the optical absorption was systematically intensified and redshifted as the number of repeating units increased, with the low-energy tail extending beyond 1000 nm when n ≥ 2 (for FR-6, ε = 26 400 M−1 cm−1 at 739 nm in toluene solution, see Table S1, ESI†). The oligomers could be reversibly electrochemically oxidized to the corresponding cationic (for n = 1), dicationic (for n = 2), tricationic (for n = 3), etc. forms, which was observed as electrochromic changes of the spectral profiles.266
Linear fluorenyl oligomers were obtained via stepwise coupling, as mentioned above, but one-pot Suzuki polycondensation of 3,6-difunctionalised monomers yielded macrocyclic structures (FR-MCn) up to a hexamer (n = 4 and 6 shown in Scheme 10). Macrocycles with odd number of fluorenyls (n = 3 and 5) were synthesized via Yamamoto coupling. Various resonance forms are expected to contribute to the ground state of the macrocycles, and each variation involves gain or loss of two Clar's π-sextets. Macrocyclization of fluorenyls resulted in antiaromatic cyclic π-conjugated systems (for FR-MC4, seen as counterclockwise ring current containing 36 π-electrons, Fig. 23a) and to some extent enhanced absorption (for FR-MC4, close to 60000 M−1 cm−1 at 710 nm, Fig. 23b). Further extension of the macrocycles caused substantial twisting of the conjugated backbone and weakening of the optical absorption (see Table S1, ESI†). The torsional twisting was counteracted by inclusion of acetylene bridges between the fluorenyl units (via Pd-catalysed Stille coupling) resulting in nearly planar macrocycles. Such macrocyclic polyradicals attained lower energy gaps between the low-spin ground state and the high-spin excited state, as compared to their linear fluorenyl counterparts.268
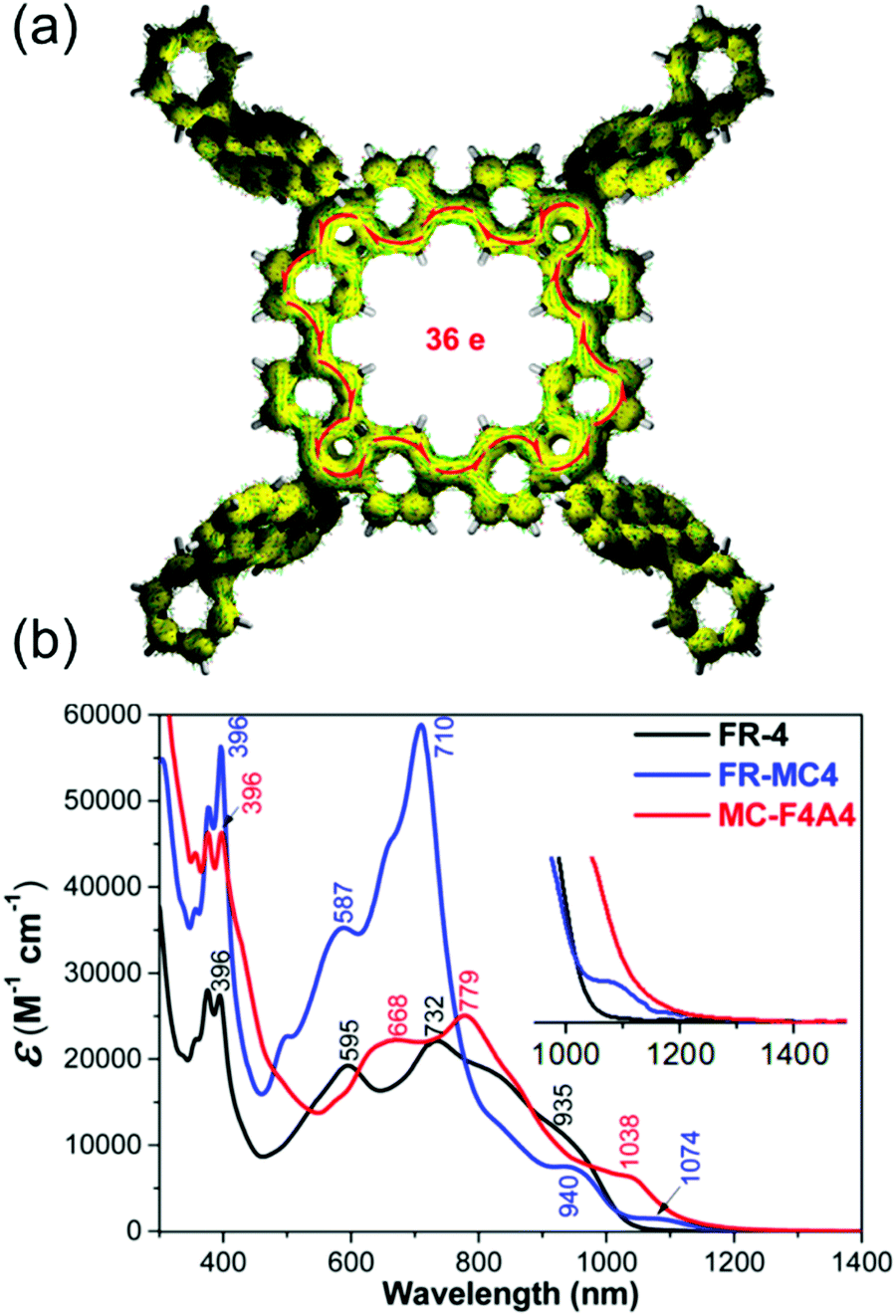 | ||
| Fig. 23 (a) Calculated ACID plots of FR-MC4. The red arrows indicate the counterclockwise paratropic ring current flow. (b) Comparison of the UV-Vis-NIR absorption of oligomers with same number of fluorenyl units: linear FR-4, macrocyclic FR-MC4 and acetylene-bridged MC-F4A4 (see ref. 268) in toluene solution. Insets are the magnified onset absorption bands. Reprinted (adapted) with permission from ref. 268 Copyright 2017 American Chemical Society. | ||
Other examples of macrocyclic polyradicals include coronoid structures displaying so-called “annulene-within-annulene” aromaticity (43, Fig. 24, in which the inner and outer conjugation circuits are formally fully decoupled) or full conjugation around the system 44, as reported by Wu et al.271,272 and Stępień et al.273,274 The common feature in these structures is that the interactions of adjacent spins are equivalent. Stępień and co-workers270 added to this series a structure 45 that combines the classical Chichibabin's and Schlenk's275 hydrocarbons and creates a system that alternates between two types of spin interactions, as illustrated in the bottom panel of Fig. 24. Chichibabin conjugation is represented as an even-electron pathway between the interacting spins (compare to diradical 37 discussed in chapter 2.4), whereas Schlenk conjugation features an odd-electron path (similar to diradical 29, Scheme 8). The structure can adopt multiple open-shell and closed-shell forms, but 45 was characterised as an open-shell singlet ground state with significant contribution from a hexaradical that is stabilised by nine Clar's π-sextets. This is also observed as odd-electron density localised mainly on the m-QDM subunits (see Fig. 24 and ref. 270 for further theoretical and experimental details). Moreover, 45 exhibited multiple thermally accessible high-spin states with uniform (calculated) energy caps of ca. 1.0 kcal mol−1 between consecutive multiplicities, meaning that these states were already populated at room temperature. Although 45 had a low-energy absorption edge extending deep into the NIR region (ca. 2000 nm in chloroform solution, Table S1, ESI†), it is worth mentioning that its stability was limited, and the compound degraded under ambient conditions in a couple of days.
 | ||
| Fig. 24 Chichibabin (red arrows) and Schlenk conjugation (blue arrows) in the fully open-shell forms of coronoid structures 43–45. The unpaired electrons are represented as red p-orbitals. Calculated odd-electron density of singlet ground state of 45 (0.002 a.u. isosurface). Reproduced with permission from ref. 270 Copyright 2021 Wiley-VCH GmbH. | ||
Swager et al.276 designed BDPA-based polymers where the radical centres were integrated into conjugated backbones and separated either by smaller thiophene spacer (in RP-T, Fig. 25a) or larger bithiophene or thienothiophene spacers. Stille polycondensation and additional radical conversion step (deprotonation–oxidation) yielded polymers with reasonably high Mn ranging between 18–39 kg mol−1. Incorporating radicals into conjugated backbones made the polymers more conductive as compared to their nonradical analogues. The difference was pronounced in the corresponding electrochemically oxidized (p-doped) forms. The radical conversions were incomplete in all three polymers (EPR analysis revealed spin concentration ranging between 17–39 mol-%) but the polymers showed ambipolar redox activities and decrease of EPR signal intensities during in situ electrochemical oxidation and reduction to cationic and anionic forms, respectively, as illustrated in Fig. 25). Higher degrees of radical conversion resulted in insoluble polymers likely due to cross-linking or intermolecular stacking of the polymer chains, or combination of both. This polymer series was also demonstrated as promising magneto-optic materials with Faraday effect (that is, rotation of polarization direction when light travels through the material). Direction of the Faraday rotation could be controlled by conversion between diamagnetic α-hydrogenated polymer (P–T) and paramagnetic polyradical (RP–T). Uncertainty about stability of BPDA-based polyradicals may still be of concern for practical applications in solid-state electronics, and therefore it will be interesting to see future research directions from the Swager group124–126,276 and others on these types of weakly NIR-absorbing but redox active paramagnetic polymers (see structures RP-BT and RP-TT, Scheme S2 and Table S1, ESI†).
 | ||
| Fig. 25 (a) Chemical structures of RP-T and its anionic and cationic forms (R = OC10H21). (b–e) EPR spectroelectrochemistry of P-T and RP-T coated on Pt wire as the working electrode (0.1 M Bu4NPF6 in CH3CN was used as the electrolyte, Pt wire as the counter electrode, and Ag/AgNO3 as the reference electrode). Scan rate: 10 mV s−1. Ferrocene was used as external standard. CV and EPR were taken in a flat cell quartz EPR tube. (b) CVs and (c) EPRs of negative scan between −1.03 and −0.13 V. (d) CVs of positive scan between −1.03 and 0.87 V and (e) EPRs of positive scan between −1.03 and 0.97 V. Reprinted (adapted) with permission from ref. 276 Copyright 2018 American Chemical Society. | ||
3. Future perspectives
When discussing about neutral π-radicals, the focus is often on their stability – likewise in this review – although debate about rightful use of the word stable regularly arises in the literature. Already more than four decades ago, Ingold148 made an important point saying that “the word stable should only be used to describe a radical so persistent and so unreactive to air, moisture, etc., under ambient conditions that the pure radical can be handled and stored in the lab with no more precautions than would be used for the majority of commercially available organic chemicals”. It still makes an excellent reference today. Paton and co-workers277 added to this discussion and described stability quantitatively as a combination of thermodynamic and kinetic factors, which we recognise as electronic resonance stabilisation and steric protection, respectively. Two-dimensional analysis of these descriptors revealed that stable radicals appeared in a distinct region of the parameter space, while most of the known radical structures were identified quantitatively unstable, also worth reading in the light of novel carbon-centred π-radicals. However, in some cases the question about stability may not be so relevant after all, as we have discussed in this review. Instead, new questions may arise, for example, about utilisation of short-lived radical states in different energy/charge transfer processes in light-emitting and energy harvesting applications without necessarily having to rely on long-lived doublet or higher-spin state materials. In other words, understanding the radical-like excited states of closed-shell molecules and tuning their singlet–triplet energy gaps may provide answers to both stability and lifetime issues. Over a decade ago, Ikeda et al.278,279 introduced a concept where emission of light was obtained from thermally or electronically excited triplet trimethylenemethane diradical. Other promising and more recent examples were discussed in the previous chapters.Neutral π-radicals typically exhibit a low-energy excited state, which translates to absorption and emission of light in the far-red/NIR spectral region. Blueshifting radical emission into the visible red-green-blue is an attractive thought as it would enable the use of doublet emission in various light-emitting technologies without the limitations caused by singlet and triplet excited states.13,14 Increasing the excited CT state energy of D–A radicals has been demonstrated as a promising strategy to obtain visible emission,85,280 and it may well lead to further structural design delivering even higher-energy emission. Tuning the electron-donating strength of the donor moiety and electron-accepting strength of the radical unit, torsion between the donor and the acceptor and rigidity of the molecule have been the common tools to increase the radiative fluorescence rate and to reduce the nonradiative decay rate. Plentiful examples were discussed in this review. On the other hand, could the radical function as a donor if coupled with a suited acceptor? Wasielewski et al.281 reported a step somewhat in that direction by coupling BDPA radical as a third spin in D–A type electron transfer system (see structure S28, Scheme S2, ESI†). Covalently connected neutral π-radical increased the rate of singlet–triplet ISC following photoexcitation up to picosecond timescale, which may be of great interest for solar energy harvesting applications. Strong NIR absorption would be highly attractive for energy harvesting, but π-radicals often show weak low-energy absorption bands arising from excitations to the lowest excited doublet states. Some strategies to enhance the NIR absorption were discussed in the previous chapters, such as delocalisation of the spin density in planar molecules,150 close π–π interactions,168 enhancing the CT character48,56,90 and via spin–spin interactions of two or more radical centres.210,268 Examples of π-radical based solar cells have been reported.282 However, to the best of our knowledge, a broadly applicable design is still missing and this gap will certainly motivate further research in the field.
As for diradicals, this review focused specifically on intramolecular spin–spin interactions in molecular diradicals, but intermolecular diradical couplings deserve attention as well; they are important in defining the magnetic properties of macromolecular systems. Different from the diamagnetic σ- and π-dimers of planar radicals discussed in chapters 2.2 and 2.3, Zhao et al.283 reported examples of ferromagnetic diradical assemblies where two fluorene radicals were connected by a [2.2]paracyclophane unit (see structures S29–S32, Scheme S2 and Table S1, ESI†). Depending on the type of connection either through aromatic rings or 9-carbon positions of the two fluorenyls, the coupling could be switched from antiferromagnetic (singlet diradical, coupling through π–π stacking) to ferromagnetic (triplet diradical, through-space coupling), respectively. Although the stability of the latter system was merely a few hours under nitrogen atmosphere and the low singlet–triplet energy gap (ca. 0.1–0.2 kcal mol−1) gave rise to weak ferromagnetic coupling nature, this work highlights the significance of relatively long-range (<10 Å) through-space radical interactions with minimal SOMO overlap, which could be used more broadly in the design of ferromagnetic systems. On the other hand, this work also highlights that ferromagnetic diradicals (and polyradicals) are commonly unstable, which has limited the development of related applications. Obtaining truly stable and isolable high-spin materials has long been a “hot” research topic,3,5,186,197 and it will certainly continue to be so in the future. Emerging computational tools that better describe the electronic states (and the singlet–triplet energy gaps) of diradicals may turn useful in that regard.187,284 On the other hand, considering practical use of mono-, di- and polyradicals as semiconductors and luminescent, magnetic, etc. materials, their properties in the solid state (as opposed to solution or gas phase calculations) are highly relevant and worth considering when designing novel π-radicals, as pointed out recently by Li and Dong.285
Beyond molecular materials, this review (and the summary in Table S1, ESI†) indicates that the number of functional polyradicals is far less than the amount of published monoradicals or diradicals. Presumably, one reason is that some unwanted side reactions may be difficult to control during various chemical coupling reactions, even though extended conjugation has been shown to provide additional stabilisation of the spin centres. Without a doubt, structural defects (including incomplete conversion of precursor polymers to polyradicals) are one of the main factors affecting the optical, electrical and magnetic properties of conjugated polymers. Chemical reactions are typically carried out on α-hydrogenated radical precursors but obtaining stable delocalised radicals has been a great challenge from synthetic point of view. Despite the remaining challenges, however, conjugated polymers commonly deliver many benefits over small molecules, such as low-cost solution-based processing and possibility to couple radicals into desired host polymer backbones as intramolecular dopants. Therefore, more synthetic efforts are needed to obtain high molecular weight, highly conductive and defect-free polyradicals that are specificaly tailored for light-absorbing, light-emitting, charge-transport and energy storage applications. Closely related to our discussion, quinoidal conjugated polymers with open-shell character have emerged as an alternative to the classical polyradicals; excellent summaries have been reported recently.242,243
Environmental aspects of materials used in organic (opto)electronics often appear no more than curiosities in the research of high performance, and the same applies to π-radicals. Although many of the π-radical structures have been proven stable and thereby pose no immediate threat to the environment in terms of free radical-mediated reactions, their synthesis often involves harsh chemistry (see Scheme 2 as an example). That said, neutral π-radicals can be made water soluble,125,126,128,286,287 and to some extent also green and biocompatible.288–290 Examples of using renewable resources as material platforms have been reported, such as disaccharide, D-maltose, coupled with TTM and PTM to improve the solubility of redox active radicals.288 Cellulose and other biopolymers have been functionalised with free radicals for use in energy applications but, to the best of our knowledge, the same has not yet been attempted with π-radicals.291–293 Non-toxic NIR-emitters are attractive for use as photosensitizers for in vivo photodynamic therapy and bioimaging applications. Li et al.290 utilised the doublet emission of D–A type PTM-TPA radical and reported the first π-radical nanoparticles for use in photodynamic therapy. Considering these examples, and various types of inorganic and organic substrates that have been functionalised with covalently coupled neutral π-radicals,294–297 they may also find uses as photoactive open-shell materials in skin- and tissue-mounted electronics for sensing and related applications. It would be pleasing to see stronger considerations of environmental and biocompatibility aspects in the “next-generation” π-radicals.
4. Conclusions
Although we summarised and discussed a large number of structures and promising concepts, only few of the most sterically congested π-radicals have been successfully demonstrated in organic optoelectronics applications. Chlorophenylmethyl radicals (TTM, PTM, etc.) were the first long-lived π-radical derivatives reported almost exactly half a century ago, and despite the recent progress presented in this review, they are still the basis of the state-of-the-art π-radicals used in optics, electronics and magnetics.39,298 TTM and PTM based D–A systems and their heteroatom derivatives have been particularly successful in optoelectronic applications delivering stable emission from the doublet excited state and facilitating record-breaking efficiencies in OLEDs, undoubtedly because of their superior steric protection and energetic stabilisation associated with kinetic and thermodynamic effects, respectively. Debate on inertness of the chlorine substituents on chlorophenylmethyl radicals regularly arises in the literature, and many of the published works correctly report variable degrees of dehalogenation during radical functionalisation (synthesis) and application (device operation). However, no widely applicable approach has been reported to tackle these issues. In that perspective, making non-chlorinated π-radicals bright would be an appealing research topic in the future. Although chlorine-free radicals have not been able to deliver such high luminescent performances thus far, they have introduced other important functions to π-radicals. Non-chlorinated radicals have shown excellent electrochemical stabilities with strong optical contrasts and multiple reversible redox states. Their optical absorption in the low-energy deep red/NIR spectral region has been enhanced by delocalising the radical in planar conjugated molecules. Strong absorption profile is highly desirable for harvesting the entire solar spectrum from visible to NIR and – importantly – π-radicals have been demonstrated to facilitate related energy transfer processes with reduced energy losses. Stabilisation of two or more unpaired electrons in diradicals and polyradicals has opened the possibility to control the open-shell singlet, triplet and higher spin states with thermal energy. Development of stable high-spin materials and their utilisation in optoelectronics would certainly open a new chapter in the field, perhaps even as metal-like conductors. However, truly polymeric radicals (n ≫ 10) are still scarce, whether based on interacting or noninteracting spin centres.299 More design approaches are needed for functional polyradicals with controlled intra- and intermolecular spin interactions for use in optoelectronics, charge-transport, energy storage and related applications, possibly with magnetic responses. It remains to be seen if this review motivates more research in the field and leads to the development of electro-optical π-radicals towards unexpected directions.Author contributions
P. M. conceptualization, visualization and writing original draft. H. B. conceptualization, supervision and reviewing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 891167.References
- M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506–2527 CrossRef CAS PubMed
.
- M. Souto, C. Rovira, I. Ratera and J. Veciana, CrystEngComm, 2017, 19, 197–206 RSC
- A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutierrez, M. Mas-Torrent, J. Veciana, H. S. J. van der Zant, C. Rovira, E. Burzuri and N. Crivillers, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed
- A. Obolda, M. Zhang and F. Li, Chin. Chem. Lett., 2016, 27, 1345–1349 CrossRef CAS
- J. M. Hudson, T. J. H. Hele and E. W. Evans, J. Appl. Phys., 2021, 129, 180901 CrossRef CAS
- S. Kimura, S. Kimura, K. Kato, Y. Teki, H. Nishihara and T. Kusamoto, Chem. Sci., 2021, 12, 2025–2029 RSC
- K. Okino, D. Sakamaki and S. Seki, ACS Mater. Lett., 2019, 1, 25–29 CrossRef CAS
- Z.-Y. Wang, Y.-Z. Dai, L. Ding, B.-W. Dong, S.-D. Jiang, J.-Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2021, 60, 4594–4598 CrossRef CAS PubMed
- J. Han, Y. Jiang, A. Obolda, P. Duan, F. Li and M. Liu, J. Phys. Chem. Lett., 2017, 8, 5865–5870 CrossRef CAS PubMed
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed
- J. Kido and Y. Iizumi, Appl. Phys. Lett., 1998, 73, 2721–2723 CrossRef CAS
- D. Y. Kondakov, T. D. Pawlik, T. K. Hatwar and J. P. Spindler, J. Appl. Phys., 2009, 106, 124510 CrossRef
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed
- C. Zhao and L. Duan, J. Mater. Chem. C, 2020, 8, 803–820 RSC
- A. Rajca, Chem. – Eur. J., 2002, 8, 4834–4841 CrossRef CAS
- D. Small, V. Zaitsev, Y. Jung, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 13850–13858 CrossRef CAS PubMed
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS PubMed
- T. Kubo, Molecules, 2019, 24, 665 CrossRef PubMed
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Adv. Mater., 2020, 32, 1908015 CrossRef CAS PubMed
- Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CAS
- B. D. Koivisto and R. G. Hicks, Coord. Chem. Rev., 2005, 249, 2612–2630 CrossRef CAS
- Y. Kim and E. Lee, Chem. – Eur. J., 2018, 24, 19110–19121 CrossRef CAS PubMed
- F. J. M. Rogers, P. L. Norcott and M. L. Coote, Org. Biomol. Chem., 2020, 18, 8255–8277 RSC
- Y. Ji, L. Long and Y. Zheng, Mater. Chem. Front., 2020, 4, 3433–3443 RSC
- Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed
- T. Kubo, Chem. Rec., 2015, 15, 218–232 CrossRef CAS PubMed
- J. Su, M. Telychko, S. Song and J. Lu, Angew. Chem., Int. Ed., 2020, 59, 7658–7668 CrossRef CAS PubMed
- F. Tani, M. Narita and T. Murafuji, ChemPlusChem, 2020, 85, 2093–2104 CrossRef CAS PubMed
- D. Shimizu and A. Osuka, Chem. Sci., 2018, 9, 1408–1423 RSC
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef
- M. Gomberg, J. Am. Chem. Soc., 1901, 23, 496–502 CrossRef
- M. Ballester, J. Riera-Figueras, J. Castaner, C. Badfa and J. M. Monso, J. Am. Chem. Soc., 1971, 93, 2215–2225 CrossRef CAS
- M. Ballester, Acc. Chem. Res., 1985, 18, 380–387 CrossRef CAS
- M. Ballester, J. Riera, J. Castañer, C. Rovira and O. Armet, Synthesis, 1986, 64–66 CrossRef CAS
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Castaner, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608–5616 CrossRef CAS
- M. Ballester, J. Castaner, J. Riera, J. Pujadas, O. Armet, C. Onrubia and J. A. Rio, J. Org. Chem., 1984, 49, 770–778 CrossRef CAS
- M. Ballester, C. Molinet and J. Castañer, J. Am. Chem. Soc., 1960, 82, 4254–4258 CrossRef CAS
- J.-L. Muñoz-Gómez, I. Marín-Montesinos, V. Lloveras, M. Pons, J. Vidal-Gancedo and J. Veciana, Org. Lett., 2014, 16, 5402–5405 CrossRef PubMed
- M. Ballester, J. Castaner, J. Riera, A. Ibanez and J. Pujadas, J. Org. Chem., 1982, 47, 259–264 CrossRef CAS
- M. Ballester, J. Veciana, J. Riera, J. Castaner, C. Rovira and O. Armet, J. Org. Chem., 1986, 51, 2472–2480 CrossRef CAS
- A. Heckmann and C. Lambert, J. Am. Chem. Soc., 2007, 129, 5515–5527 CrossRef CAS PubMed
- V. Lloveras, F. Liko, J. L. Muñoz-Gómez, J. Veciana and J. Vidal-Gancedo, Chem. Mater., 2019, 31, 9400–9412 CrossRef CAS
- J. Veciana, C. Rovira, N. Ventosa, M. I. Crespo and F. Palacio, J. Am. Chem. Soc., 1993, 115, 57–64 CrossRef CAS
- J. Irurre, J. Santamaría and M. C. González-rego, Chirality, 1995, 7, 154–157 CrossRef CAS
- P. Mayorga Burrezo, V. G. Jiménez, D. Blasi, I. Ratera, A. G. Campaña and J. Veciana, Angew. Chem., Int. Ed., 2019, 58, 16282–16288 CrossRef CAS PubMed
- P. Mayorga-Burrezo, V. G. Jiménez, D. Blasi, T. Parella, I. Ratera, A. G. Campaña and J. Veciana, Chem. – Eur. J., 2020, 26, 3776–3781 CrossRef CAS PubMed
- T. Zhao, J. Han, P. Duan and M. Liu, Acc. Chem. Res., 2020, 53, 1279–1292 CrossRef CAS PubMed
- L. Chen, M. Arnold, R. Blinder, F. Jelezko and A. J. C. Kuehne, RSC Adv., 2021, 11, 27653–27658 RSC
- X. Wu, J. O. Kim, S. Medina, F. J. Ramírez, P. Mayorga Burrezo, S. Wu, Z. L. Lim, C. Lambert, J. Casado, D. Kim and J. Wu, Chem. – Eur. J., 2017, 23, 7698–7702 CrossRef CAS PubMed
- C.-H. Liu, E. Hamzehpoor, Y. Sakai-Otsuka, T. Jadhav and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 23030–23034 CrossRef CAS PubMed
- L. Julia, M. Ballester, J. Riera, J. Castaner, J. L. Ortin and C. Onrubia, J. Org. Chem., 1988, 53, 1267–1273 CrossRef CAS
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef CAS PubMed
- T. Kusamoto, S. Kimura, Y. Ogino, C. Ohde and H. Nishihara, Chem. – Eur. J., 2016, 22, 17725–17733 CrossRef CAS PubMed
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2015, 54, 3731–3734 CrossRef CAS PubMed
- T. Kusamoto, Y. Hattori, A. Tanushi and H. Nishihara, Inorg. Chem., 2015, 54, 4186–4188 CrossRef CAS PubMed
- Y. Hattori, T. Kusamoto, T. Sato and H. Nishihara, Chem. Commun., 2016, 52, 13393–13396 RSC
- S. Kimura, S. Kimura, H. Nishihara and T. Kusamoto, Chem. Commun., 2020, 56, 11195–11198 RSC
- Y. Hattori, R. Kitajima, R. Matsuoka, T. Kusamoto, H. Nishihara and K. Uchida, Chem. Commun., 2022, 58, 2560–2563 RSC
- T. Kusamoto and S. Kimura, Chem. Lett., 2021, 50, 1445–1459 CrossRef CAS
- S. Kimura, T. Kusamoto, S. Kimura, K. Kato, Y. Teki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 12711–12715 CrossRef CAS PubMed
- K. Kato, S. Kimura, T. Kusamoto, H. Nishihara and Y. Teki, Angew. Chem., Int. Ed., 2019, 58, 2606–2611 CrossRef CAS PubMed
- Z. Zhou, C. Qiao, J. Yao, Y. Yan and Y. S. Zhao, J. Mater. Chem. C, 2022, 10, 2551–2555 RSC
- T. Itoh, A. Takada, K. Hirai and H. Tomioka, Org. Lett., 2005, 7, 811–814 CrossRef CAS PubMed
- S. Kimura, A. Tanushi, T. Kusamoto, S. Kochi, T. Sato and H. Nishihara, Chem. Sci., 2018, 9, 1996–2007 RSC
- S. Kimura, M. Uejima, W. Ota, T. Sato, S. Kusaka, R. Matsuda, H. Nishihara and T. Kusamoto, J. Am. Chem. Soc., 2021, 143, 4329–4338 CrossRef CAS PubMed
- S. Kimura, R. Matsuoka, S. Kimura, H. Nishihara and T. Kusamoto, J. Am. Chem. Soc., 2021, 143, 5610–5615 CrossRef CAS PubMed
- X. Ai, Y. Chen, Y. Feng and F. Li, Angew. Chem., Int. Ed., 2018, 57, 2869–2873 CrossRef CAS PubMed
- A. Abdurahman, Y. Chen, X. Ai, O. Ablikim, Y. Gao, S. Dong, B. Li, B. Yang, M. Zhang and F. Li, J. Mater. Chem. C, 2018, 6, 11248–11254 RSC
- V. Gamero, D. Velasco, S. Latorre, F. López-Calahorra, E. Brillas and L. Juliá, Tetrahedron Lett., 2006, 47, 2305–2309 CrossRef CAS
- D. Velasco, S. Castellanos, M. López, F. López-Calahorra, E. Brillas and L. Juliá, J. Org. Chem., 2007, 72, 7523–7532 CrossRef CAS PubMed
- S. Castellanos, D. Velasco, F. López-Calahorra, E. Brillas and L. Julia, J. Org. Chem., 2008, 73, 3759–3767 CrossRef CAS PubMed
- S. Dong, A. Obolda, Q. Peng, Y. Zhang, S. Marder and F. Li, Mater. Chem. Front., 2017, 1, 2132–2135 RSC
- L. Chen, M. Arnold, Y. Kittel, R. Blinder, F. Jelezko and A. J. C. Kuehne, Adv. Opt. Mater., 2022, 2102101 CrossRef CAS
- A. Obolda, X. Ai, M. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35472–35478 CrossRef CAS PubMed
- M. Reig, C. Gozálvez, V. Jankauskas, V. Gaidelis, J. V. Grazulevicius, L. Fajarí, L. Juliá and D. Velasco, Chem. – Eur. J., 2016, 22, 18551–18558 CrossRef CAS PubMed
- Y. Chen, L. Yang, Y. Huang, A. Obolda, A. Abdurahman, Z. Lu and F. Li, J. Phys. Chem. Lett., 2019, 10, 48–51 CrossRef CAS PubMed
- Y. Teki, Chem. – Eur. J., 2020, 26, 980–996 CrossRef CAS PubMed
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224–1229 CrossRef CAS PubMed
- E. Cho, V. Coropceanu and J.-L. Brédas, J. Mater. Chem. C, 2021, 9, 10794–10801 RSC
- Y. Gao, A. Obolda, M. Zhang and F. Li, Dyes Pigm., 2017, 139, 644–650 CrossRef CAS
- Y. Gao, W. Xu, H. Ma, A. Obolda, W. Yan, S. Dong, M. Zhang and F. Li, Chem. Mater., 2017, 29, 6733–6739 CrossRef CAS
- H. Guo, Q. Peng, X.-K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J.-L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed
- S. Dong, W. Xu, H. Guo, W. Yan, M. Zhang and F. Li, Phys. Chem. Chem. Phys., 2018, 20, 18657–18662 RSC
- R. B. Mallion and D. H. Rouvray, Mol. Phys., 1978, 36, 125–128 CrossRef CAS
- G. Gryn'ova, M. L. Coote and C. Corminboeuf, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 440–459 Search PubMed
- L. Abella, J. Crassous, L. Favereau and J. Autschbach, Chem. Mater., 2021, 33, 3678–3691 CrossRef CAS
- A. Heckmann, C. Lambert, M. Goebel and R. Wortmann, Angew. Chem., Int. Ed., 2004, 43, 5851–5856 CrossRef CAS PubMed
- A. Heckmann, S. Dümmler, J. Pauli, M. Margraf, J. Köhler, D. Stich, C. Lambert, I. Fischer and U. Resch-Genger, J. Phys. Chem. C, 2009, 113, 20958–20966 CrossRef CAS
- M. Holzapfel and C. Lambert, J. Phys. Chem. C, 2008, 112, 1227–1243 CrossRef CAS
- S. Breimaier and R. F. Winter, Eur. J. Org. Chem., 2021, 4690–4700 CrossRef CAS
- S. Mattiello, F. Corsini, S. Mecca, M. Sassi, R. Ruffo, G. Mattioli, Y. Hattori, T. Kusamoto, G. Griffini and L. Beverina, Mater. Adv., 2021, 2, 7369–7378 RSC
- Y. Hattori, E. Michail, A. Schmiedel, M. Moos, M. Holzapfel, I. Krummenacher, H. Braunschweig, U. Müller, J. Pflaum and C. Lambert, Chem. – Eur. J., 2019, 25, 15463–15471 CrossRef CAS PubMed
- R. Matsuoka, S. Kimura and T. Kusamoto, ChemPhotoChem, 2021, 5, 669–673 CrossRef CAS
- Y. Hattori, S. Tsubaki, R. Matsuoka, T. Kusamoto, H. Nishihara and K. Uchida, Chem. – Asian J., 2021, 16, 2538–2544 CrossRef CAS PubMed
- A. Tanushi, S. Kimura, T. Kusamoto, M. Tominaga, Y. Kitagawa, M. Nakano and H. Nishihara, J. Phys. Chem. C, 2019, 123, 4417–4423 CrossRef CAS
- J. Bonvoisin, J.-P. Launay, C. Rovira and J. Veciana, Angew. Chem., Int. Ed. Engl., 1994, 33, 2106–2109 CrossRef
- C. Rovira, D. Ruiz-Molina, O. Elsner, J. Vidal-Gancedo, J. Bonvoisin, J.-P. Launay and J. Veciana, Chem. – Eur. J., 2001, 7, 240–250 CrossRef CAS
- P. Muller, Pure Appl. Chem., 1994, 66, 1077–1184 CrossRef
- G. P. Moss, P. A. S. Smith and D. Tavernier, Pure Appl. Chem., 1995, 67, 1307–1375 Search PubMed
- C. Franco, P. M. Burrezo, V. Lloveras, R. Caballero, I. Alcón, S. T. Bromley, M. Mas-Torrent, F. Langa, J. T. López Navarrete, C. Rovira, J. Casado and J. Veciana, J. Am. Chem. Soc., 2017, 139, 686–692 CrossRef CAS PubMed
- P. Mayorga Burrezo, C. Franco, R. Caballero, M. Mas-Torrent, F. Langa, J. T. López Navarrete, C. Rovira, J. Veciana and J. Casado, Chem. – Eur. J., 2018, 24, 3776–3783 CrossRef CAS PubMed
- P. Mayorga-Burrezo, F. Bejarano, J. Calbo, X. Zhao, J. A. De Sousa, V. Lloveras, M. R. Bryce, E. Ortí, J. Veciana, C. Rovira and N. Crivillers, J. Phys. Chem. Lett., 2021, 12, 6159–6164 CrossRef CAS PubMed
- T. Nishiuchi, S. Aibara and T. Kubo, Angew. Chem., Int. Ed., 2018, 57, 16516–16519 CrossRef CAS PubMed
- T. Nishiuchi, D. Ishii, S. Aibara, H. Sato and T. Kubo, Chem. Commun., 2022, 58, 3306–3309 RSC
- H. Sitzmann and R. Boese, Angew. Chem., Int. Ed. Engl., 1991, 30, 971–973 CrossRef
- H. Sitzmann, H. Bock, R. Boese, T. Dezember, Z. Havlas, W. Kaim, M. Moscherosch and L. Zanathy, J. Am. Chem. Soc., 1993, 115, 12003–12009 CrossRef CAS
- N. Jux, Y. Rubin and K. Holczer, Angew. Chem., Int. Ed. Engl., 1996, 35, 1986–1990 CrossRef CAS
- T. Kitagawa, K. Ogawa and K. Komatsu, J. Am. Chem. Soc., 2004, 126, 9930–9931 CrossRef CAS PubMed
- K. Ogawa, K. Komatsu and T. Kitagawa, J. Org. Chem., 2011, 76, 6095–6100 CrossRef CAS PubMed
- M. K. Kalinowski and Z. R. Grabowski, Trans. Faraday Soc., 1966, 62, 926–934 RSC
- D. R. Dalton and S. A. Liebman, J. Am. Chem. Soc., 1969, 91, 1194–1199 CrossRef CAS
- E. A. Chandross and C. F. Sheley, J. Am. Chem. Soc., 1968, 90, 4345–4354 CrossRef CAS
- E. Font-Sanchis, C. Aliaga, K. S. Focsaneanu and J. C. Scaiano, Chem. Commun., 2002, 1576–1577, 10.1039/B203158B
- E. Font-Sanchis, C. Aliaga, E. V. Bejan, R. Cornejo and J. C. Scaiano, J. Org. Chem., 2003, 68, 3199–3204 CrossRef CAS PubMed
- C. F. Koelsch, J. Am. Chem. Soc., 1957, 79, 4439–4441 CrossRef CAS
- D. T. Breslin and M. A. Fox, J. Phys. Chem. C, 1993, 97, 13341–13347 CrossRef CAS
- E. L. Dane, T. Maly, G. T. Debelouchina, R. G. Griffin and T. M. Swager, Org. Lett., 2009, 11, 1871–1874 CrossRef CAS PubMed
- E. L. Dane and T. M. Swager, J. Org. Chem., 2010, 75, 3533–3536 CrossRef CAS PubMed
- O. Haze, B. Corzilius, A. A. Smith, R. G. Griffin and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 14287–14290 CrossRef CAS PubMed
- S. Mandal and S. T. Sigurdsson, Chem. – Eur. J., 2020, 26, 7486–7491 CrossRef CAS PubMed
- S. Mandal and S. T. Sigurdsson, Chem. Commun., 2020, 56, 13121–13124 RSC
- D. K. Frantz, J. J. Walish and T. M. Swager, Org. Lett., 2013, 15, 4782–4785 CrossRef CAS PubMed
- Y. Tian, K. Uchida, H. Kurata, Y. Hirao, T. Nishiuchi and T. Kubo, J. Am. Chem. Soc., 2014, 136, 12784–12793 CrossRef CAS PubMed
- M. T. Colvin, A. L. Smeigh, E. M. Giacobbe, S. M. M. Conron, A. B. Ricks and M. R. Wasielewski, J. Phys. Chem. A, 2011, 115, 7538–7549 CrossRef CAS PubMed
- H. Kalita, T. Y. Gopalakrishna and J. Wu, Org. Lett., 2018, 20, 445–448 CrossRef CAS PubMed
- J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497–6501 CrossRef CAS PubMed
- R. Zhang, J. P. Peterson, L. J. Fischer, A. Ellern and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 14308–14313 CrossRef CAS PubMed
- J. P. Peterson and A. H. Winter, J. Am. Chem. Soc., 2019, 141, 12901–12906 CrossRef CAS PubMed
- J. P. Peterson, R. Zhang and A. H. Winter, ACS Omega, 2019, 4, 13538–13542 CrossRef CAS PubMed
- J. P. Peterson, A. Ellern and A. H. Winter, J. Am. Chem. Soc., 2020, 142, 5304–5313 CrossRef CAS PubMed
- R. Zhang, A. Ellern and A. Winter, Angew. Chem., Int. Ed., 2021, 60, 25082–25088 CrossRef CAS PubMed
- R. Zhang, A. Ellern and A. H. Winter, J. Org. Chem., 2022, 87, 1507–1511 CrossRef CAS PubMed
- T. Kobashi, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2016, 55, 8634–8638 CrossRef CAS PubMed
- K. Okino, S. Hira, Y. Inoue, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2017, 56, 16597–16601 CrossRef CAS PubMed
- D. Wang, C. Capel Ferrón, J. Li, S. Gámez-Valenzuela, R. Ponce Ortiz, J. T. López Navarrete, V. Hernández Jolín, X. Yang, M. Peña Álvarez, V. García Baonza, F. Hartl, M. C. Ruiz Delgado and H. Li, Chem. – Eur. J., 2017, 23, 13776–13783 CrossRef CAS PubMed
- I. Badía-Domínguez, A. Pérez-Guardiola, J. C. Sancho-García, J. T. López Navarrete, V. Hernández Jolín, H. Li, D. Sakamaki, S. Seki and M. C. Ruiz Delgado, ACS Omega, 2019, 4, 4761–4769 CrossRef
- I. Badía-Domínguez, M. Peña-Álvarez, D. Wang, A. Pérez Guardiola, Y. Vida, S. Rodríguez González, J. T. López Navarrete, V. Hernández Jolín, J. C. Sancho García, V. García Baonza, R. Nash, F. Hartl, H. Li and M. C. Ruiz Delgado, Chem. – Eur. J., 2021, 27, 5509–5520 CrossRef PubMed
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC
- S. Kato, S. Furukawa, D. Aoki, R. Goseki, K. Oikawa, K. Tsuchiya, N. Shimada, A. Maruyama, K. Numata and H. Otsuka, Nat. Commun., 2021, 12, 126 CrossRef CAS PubMed
- D. H. Reid, Tetrahedron, 1958, 3, 339–352 CrossRef CAS
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS
- L. Beer, S. K. Mandal, R. W. Reed, R. T. Oakley, F. S. Tham, B. Donnadieu and R. C. Haddon, Cryst. Growth Des., 2007, 7, 802–809 CrossRef CAS
- T. Kubo, Y. Katada, A. Shimizu, Y. Hirao, K. Sato, T. Takui, M. Uruichi, K. Yakushi and R. C. Haddon, J. Am. Chem. Soc., 2011, 133, 14240–14243 CrossRef CAS PubMed
- K. Goto, T. Kubo, K. Yamamoto, K. Nakasuji, K. Sato, D. Shiomi, T. Takui, M. Kubota, T. Kobayashi, K. Yakusi and J. Ouyang, J. Am. Chem. Soc., 1999, 121, 1619–1620 CrossRef CAS
- V. Zaitsev, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Org. Chem., 2006, 71, 520–526 CrossRef CAS PubMed
- Y. Morita, T. Aoki, K. Fukui, S. Nakazawa, K. Tamaki, S. Suzuki, A. Fuyuhiro, K. Yamamoto, K. Sato, D. Shiomi, A. Naito, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2002, 41, 1793–1796 CrossRef CAS PubMed
- S. Nishida, K. Kariyazono, A. Yamanaka, K. Fukui, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Chem. – Asian J., 2011, 6, 1188–1196 CrossRef CAS PubMed
- A. Ueda, H. Wasa, S. Suzuki, K. Okada, K. Sato, T. Takui and Y. Morita, Angew. Chem., Int. Ed., 2012, 51, 6691–6695 CrossRef CAS PubMed
- Y. Morita, J. Kawai, K. Fukui, S. Nakazawa, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Org. Lett., 2003, 5, 3289–3291 CrossRef CAS PubMed
- Y. Morita, S. Nishida, J. Kawai, T. Takui and K. Nakasuji, Pure Appl. Chem., 2008, 80, 507–517 CrossRef CAS
- P. Li, S. Wang, Z. Wang, C. Zheng, Y. Tang, Q. Yang, R. Chen and W. Huang, J. Mater. Chem. C, 2020, 8, 12224–12230 RSC
- A. Paul, A. Gupta and S. Konar, Cryst. Growth Des., 2021, 21, 5473–5489 CrossRef CAS
- A. M. Toader, C. M. Buta, B. Frecus, A. Mischie and F. Cimpoesu, J. Phys. Chem. C, 2019, 123, 6869–6880 CrossRef CAS
- M. C. Buta, B. Frecus, M. Enache, I. Humelnicu, A. M. Toader and F. Cimpoesu, J. Phys. Chem. A, 2021, 125, 6893–6901 CrossRef CAS PubMed
- K. Uchida, Z. Mou, M. Kertesz and T. Kubo, J. Am. Chem. Soc., 2016, 138, 4665–4672 CrossRef CAS PubMed
- K. Uchida, Y. Hirao, H. Kurata, T. Kubo, S. Hatano and K. Inoue, Chem. – Asian J., 2014, 9, 1823–1829 CrossRef CAS PubMed
- Z. Mou, K. Uchida, T. Kubo and M. Kertesz, J. Am. Chem. Soc., 2014, 136, 18009–18022 CrossRef CAS PubMed
- K. Uchida, S. Ito, M. Nakano, M. Abe and T. Kubo, J. Am. Chem. Soc., 2016, 138, 2399–2410 CrossRef CAS PubMed
- P. A. Koutentis, Y. Chen, Y. Cao, T. P. Best, M. E. Itkis, L. Beer, R. T. Oakley, A. W. Cordes, C. P. Brock and R. C. Haddon, J. Am. Chem. Soc., 2001, 123, 3864–3871 CrossRef CAS PubMed
- P. A. Koutentis, R. C. Haddon, R. T. Oakley, A. W. Cordes and C. P. Brock, Acta Crystallograph. Sect. B, 2001, 57, 680–691 CrossRef CAS PubMed
- Y. Ikabata, Q. Wang, T. Yoshikawa, A. Ueda, T. Murata, K. Kariyazono, M. Moriguchi, H. Okamoto, Y. Morita and H. Nakai, npj Quantum Mater., 2017, 2, 27 CrossRef
- S. Nishida, J. Kawai, M. Moriguchi, T. Ohba, N. Haneda, K. Fukui, A. Fuyuhiro, D. Shiomi, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Chem. – Eur. J., 2013, 19, 11904–11915 CrossRef CAS PubMed
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef CAS PubMed
- A. Ueda, H. Wasa, S. Nishida, Y. Kanzaki, K. Sato, D. Shiomi, T. Takui and Y. Morita, Chem. – Eur. J., 2012, 18, 16272–16276 CrossRef CAS PubMed
- A. Ueda, H. Wasa, S. Nishida, Y. Kanzaki, K. Sato, T. Takui and Y. Morita, Chem. – Asian J., 2013, 8, 2057–2063 CrossRef CAS PubMed
- Y. Morita, T. Murata, A. Ueda, C. Yamada, Y. Kanzaki, D. Shiomi, K. Sato and T. Takui, Bull. Chem. Soc. Jpn., 2018, 91, 922–931 CrossRef CAS
- T. Murata, C. Yamada, K. Furukawa and Y. Morita, Commun. Chem., 2018, 1, 47 CrossRef
- H. Ito, T. Murata, T. Miyata, M. Morita, R. Tsuji and Y. Morita, ACS Omega, 2019, 4, 17569–17575 CrossRef CAS PubMed
- T. Murata, M. Yokoyama, A. Ueda, Y. Kanzaki, D. Shiomi, K. Sato, T. Takui and Y. Morita, Chem. Lett., 2020, 49, 95–98 CrossRef CAS
- T. Murata, Y. Yamamoto, A. Ueda, T. Ise, D. Shiomi, K. Sato, T. Takui and Y. Morita, J. Org. Chem., 2021, 86, 10154–10165 CrossRef CAS PubMed
- T. Murata, S. Nakanishi, H. Nakayama, H. Ito, M. Morita, R. Tsuji and Y. Morita, ACS Appl. Energy Mater., 2022, 5, 1218–1225 CrossRef CAS
- H. Ito, T. Murata, M. Fujisaki, R. Tsuji and Y. Morita, ChemSusChem, 2021, 14, 1377–1387 CrossRef CAS PubMed
- T. Murata, N. Asakura, R. Tsuji, Y. Kanzaki, K. Sato, T. Takui and Y. Morita, Chem. – Eur. J., 2022, e202104447, DOI:10.1002/chem.202104447
- E. Clar and M. Zander, J. Chem. Soc., 1958, 1861–1865, 10.1039/JR9580001861
- M. Solà, Front. Chem., 2013, 1, 22 Search PubMed
- P. Ravat, O. Blacque and M. Juríček, J. Org. Chem., 2020, 85, 92–100 CrossRef CAS PubMed
- T. Kodama, M. Aoba, Y. Hirao, S. M. Rivero, J. Casado and T. Kubo, Angew. Chem., Int. Ed., 2022, e202200688, DOI:10.1002/anie.202200688
- T. Kodama, S. Miyazaki and T. Kubo, ChemPlusChem, 2019, 84, 599–602 CrossRef CAS PubMed
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed
- T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. De Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291–11351 CrossRef CAS PubMed
- A. Kekulé and A. Franchimont, Ber. Dtsch. Chem. Ges., 1872, 5, 908–910 CrossRef
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef
- E. Hückel, Z. Phys., 1931, 72, 310–337 CrossRef
- E. Hückel, Z. Phys., 1932, 76, 628–648 CrossRef
- F. Hund, Z. Phys., 1925, 33, 345–371 CrossRef CAS
- F. Hund, Z. Phys., 1925, 34, 296–308 CrossRef CAS
- Z. Chen, W. Li, Y. Zhang, Z. Wang, W. Zhu, M. Zeng and Y. Li, J. Phys. Chem. Lett., 2021, 12, 9783–9790 CrossRef CAS PubMed
- Z. Feng, Y. Chong, S. Tang, Y. Fang, Y. Zhao, J. Jiang and X. Wang, Chem. Sci., 2021, 12, 15151–15156 RSC
- D. Cho, K. C. Ko and J. Y. Lee, Int. J. Quantum Chem., 2016, 116, 578–597 CrossRef CAS
- N. M. Gallagher, A. Olankitwanit and A. Rajca, J. Org. Chem., 2015, 80, 1291–1298 CrossRef CAS PubMed
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS PubMed
- D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127–1130 CrossRef CAS PubMed
- B. D. Rose, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Org. Lett., 2012, 14, 2426–2429 CrossRef CAS PubMed
- A. G. Fix, P. E. Deal, C. L. Vonnegut, B. D. Rose, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 15, 1362–1365 CrossRef CAS PubMed
- J. J. Dressler, Z. Zhou, J. L. Marshall, R. Kishi, S. Takamuku, Z. Wei, S. N. Spisak, M. Nakano, M. A. Petrukhina and M. M. Haley, Angew. Chem., Int. Ed., 2017, 56, 15363–15367 CrossRef CAS PubMed
- H. Hayashi, J. E. Barker, A. Cárdenas Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S.-I. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142, 20444–20455 CrossRef CAS PubMed
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed
- J.-i Nishida, S. Tsukaguchi and Y. Yamashita, Chem. – Eur. J., 2012, 18, 8964–8970 CrossRef CAS PubMed
- G. E. Rudebusch, G. L. Espejo, J. L. Zafra, M. Peña-Alvarez, S. N. Spisak, K. Fukuda, Z. Wei, M. Nakano, M. A. Petrukhina, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 12648–12654 CrossRef CAS PubMed
- A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906–6910 CrossRef CAS PubMed
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed
- H. Miyoshi, S. Nobusue, A. Shimizu, I. Hisaki, M. Miyata and Y. Tobe, Chem. Sci., 2014, 5, 163–168 RSC
- T. Kubo, A. Shimizu, M. Sakamoto, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 6564–6568 CrossRef CAS PubMed
- T. Kubo, A. Shimizu, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Org. Lett., 2007, 9, 81–84 CrossRef CAS PubMed
- A. Shimizu, M. Uruichi, K. Yakushi, H. Matsuzaki, H. Okamoto, M. Nakano, Y. Hirao, K. Matsumoto, H. Kurata and T. Kubo, Angew. Chem., Int. Ed., 2009, 48, 5482–5486 CrossRef CAS PubMed
- A. Shimizu, T. Kubo, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Hirao, K. Matsumoto, H. Kurata, Y. Morita and K. Nakasuji, J. Am. Chem. Soc., 2010, 132, 14421–14428 CrossRef CAS PubMed
- C. M. Wehrmann, R. T. Charlton and M. S. Chen, J. Am. Chem. Soc., 2019, 141, 3240–3248 CrossRef CAS PubMed
- C. M. Wehrmann, M. Imran, C. Pointer, L. A. Fredin, E. R. Young and M. S. Chen, Chem. Sci., 2020, 11, 10212–10219 RSC
- K. Wang, P. Liu, F. Zhang, L. Xu, M. Zhou, A. Nakai, K. Kato, K. Furukawa, T. Tanaka, A. Osuka and J. Song, Angew. Chem., Int. Ed., 2021, 60, 7002–7006 CrossRef CAS PubMed
- W. Zeng and J. Wu, Mater. Chem. Front., 2019, 3, 2668–2672 RSC
- Q. Deng and J.-D. Chai, ACS Omega, 2019, 4, 14202–14210 CrossRef CAS PubMed
- M. E. Sandoval-Salinas, A. Carreras and D. Casanova, Phys. Chem. Chem. Phys., 2019, 21, 9069–9076 RSC
- J. Li, S. Sanz, J. Castro-Esteban, M. Vilas-Varela, N. Friedrich, T. Frederiksen, D. Peña and J.
I. Pascual, Phys. Rev. Lett., 2020, 124, 177201 CrossRef CAS PubMed
- A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 CrossRef CAS PubMed
- A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430–1437 CrossRef CAS PubMed
- W. Zeng, H. Phan, T. S. Herng, T. Y. Gopalakrishna, N. Aratani, Z. Zeng, H. Yamada, J. Ding and J. Wu, Chem, 2017, 2, 81–92 CAS
- Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 CrossRef CAS PubMed
- J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463–1470 CrossRef
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef CAS
- E. Müller and H. Pfanz, Ber. Dtsch. Chem. Ges., 1941, 74, 1051–1074 CrossRef
- G. J. Sloan and W. R. Vaughan, J. Org. Chem., 1957, 22, 750–761 CrossRef CAS
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004–6011 CrossRef CAS PubMed
- G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef CAS PubMed
- K. Fukuda, Y. Suzuki, H. Matsui, T. Nagami, Y. Kitagawa, B. Champagne, K. Kamada, Y. Yamamoto and M. Nakano, ChemPhysChem, 2017, 18, 142–148 CrossRef CAS PubMed
- S. Escayola, M. Callís, A. Poater and M. Solà, ACS Omega, 2019, 4, 10845–10853 CrossRef CAS PubMed
- C. Jiang, Y. Bang, X. Wang, X. Lu, Z. Lim, H. Wei, S. El-Hankari, J. Wu and Z. Zeng, Chem. Commun., 2018, 54, 2389–2392 RSC
- M. A. Majewski, P. J. Chmielewski, A. Chien, Y. Hong, T. Lis, M. Witwicki, D. Kim, P. M. Zimmerman and M. Stępień, Chem. Sci., 2019, 10, 3413–3420 RSC
- D. Rottschäfer, N. K. T. Ho, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 CrossRef PubMed
- A. Maiti, S. Chandra, B. Sarkar and A. Jana, Chem. Sci., 2020, 11, 11827–11833 RSC
- Y. Ni, F. Gordillo-Gámez, M. Peña Alvarez, Z. Nan, Z. Li, S. Wu, Y. Han, J. Casado and J. Wu, J. Am. Chem. Soc., 2020, 142, 12730–12742 CrossRef CAS PubMed
- E. Molins, C. Miravitlles, E. Espinosa and M. Ballester, J. Org. Chem., 2002, 67, 7175–7178 CrossRef CAS PubMed
- C. Wentrup, M. J. Regimbald-Krnel, D. Müller and P. Comba, Angew. Chem., Int. Ed., 2016, 55, 14600–14605 CrossRef CAS PubMed
- Y. Hirao, Y. Hamamoto, N. Nagamachi and T. Kubo, Phys. Chem. Chem. Phys., 2019, 21, 12209–12216 RSC
- Y. Hamamoto, Y. Hirao and T. Kubo, J. Phys. Chem. Lett., 2021, 12, 4729–4734 CrossRef CAS PubMed
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC
- X. Ji and L. Fang, Polym. Chem., 2021, 12, 1347–1361 RSC
- A. Rajca, K. Lu and S. Rajca, J. Am. Chem. Soc., 1997, 119, 10335–10345 CrossRef CAS
- A. Rajca, J. Wongsriratanakul, S. Rajca and R. Cerny, Angew. Chem., Int. Ed., 1998, 37, 1229–1232 CrossRef CAS PubMed
- A. Rajca, J. Wongsriratanakul and S. Rajca, J. Am. Chem. Soc., 2004, 126, 6608–6626 CrossRef CAS PubMed
- S. Rajca, A. Rajca, J. Wongsriratanakul, P. Butler and S.-M. Choi, J. Am. Chem. Soc., 2004, 126, 6972–6986 CrossRef CAS PubMed
- V. M. Domingo, J. Castaner, J. Riera, L. Fajari and A. Labarta, Chem. Mater., 1995, 7, 314–323 CrossRef CAS
- N. Roques, D. Maspoch, A. Datcu, K. Wurst, D. Ruiz-Molina, C. Rovira and J. Veciana, Polyhedron, 2007, 26, 1934–1948 CrossRef CAS
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. M. Hernández, F. Lloret, J. Tejada, C. Rovira and J. Veciana, Inorg. Chem., 2007, 46, 1627–1633 CrossRef CAS PubMed
- N. Roques, N. Domingo, D. Maspoch, K. Wurst, C. Rovira, J. Tejada, D. Ruiz-Molina and J. Veciana, Inorg. Chem., 2010, 49, 3482–3488 CrossRef CAS PubMed
- Y. Yang, C. Liu, X. Xu, Z. Meng, W. Tong, Z. Ma, C. Zhou, Y. Sun and Z. Sheng, Polym. Chem., 2018, 9, 5499–5503 RSC
- S. Wu, M. Li, H. Phan, D. Wang, T. S. Herng, J. Ding, Z. Lu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8007–8011 CrossRef CAS PubMed
- Y. Jiang, I. Oh, S. H. Joo, O. Buyukcakir, X. Chen, S. H. Lee, M. Huang, W. K. Seong, J. H. Kim, J.-U. Rohde, S. K. Kwak, J.-W. Yoo and R. S. Ruoff, ACS Nano, 2019, 13, 5251–5258 CrossRef CAS PubMed
- D. Maspoch, L. Catala, P. Gerbier, D. Ruiz-Molina, J. Vidal-Gancedo, K. Wurst, C. Rovira and J. Veciana, Chem. – Eur. J., 2002, 8, 3635–3645 CrossRef CAS
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, G. Vaughan, J. Tejada, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 1828–1832 CrossRef CAS PubMed
- T. Jiao, H. Qu, L. Tong, X. Cao and H. Li, Angew. Chem., Int. Ed., 2021, 60, 9852–9858 CrossRef CAS PubMed
- Z. Liu, Z. Zhang, T. Li and W. Zhao, Organometallics, 2021, 40, 2379–2383 CrossRef CAS
- B. Huang, L. Mao, X. Shi and H.-B. Yang, Chem. Sci., 2021, 12, 13648–13663 RSC
- D. Reitzenstein, T. Quast, F. Kanal, M. Kullmann, S. Ruetzel, M. S. Hammer, C. Deibel, V. Dyakonov, T. Brixner and C. Lambert, Chem. Mater., 2010, 22, 6641–6655 CrossRef CAS
- L. Horner, H. Hoffmann and H. G. Wippel, Chem. Ber., 1958, 91, 61–63 CrossRef CAS
- W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS
- V. S. Mothika, M. Baumgarten and U. Scherf, ACS Appl. Nano Mater., 2019, 2, 4832–4841 CrossRef CAS
- Q. Gu, A. Abdurahman, R. H. Friend and F. Li, J. Phys. Chem. Lett., 2020, 11, 5638–5642 CrossRef CAS PubMed
- A. Abdurahman, Q. Peng, O. Ablikim, X. Ai and F. Li, Mater. Horiz., 2019, 6, 1265–1270 RSC
- X. Lu, S. Lee, J. O. Kim, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Z. Lim, Z. Zeng, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2016, 138, 13048–13058 CrossRef CAS PubMed
- S. Das, T. S. Herng, J. L. Zafra, P. M. Burrezo, M. Kitano, M. Ishida, T. Y. Gopalakrishna, P. Hu, A. Osuka, J. Casado, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 7782–7790 CrossRef CAS PubMed
- X. Lu, S. Lee, Y. Hong, H. Phan, T. Y. Gopalakrishna, T. S. Herng, T. Tanaka, M. E. Sandoval-Salinas, W. Zeng, J. Ding, D. Casanova, A. Osuka, D. Kim and J. Wu, J. Am. Chem. Soc., 2017, 139, 13173–13183 CrossRef CAS PubMed
- J. Wang, G. Kim, M. E. Sandoval-Salinas, H. Phan, T. Y. Gopalakrishna, X. Lu, D. Casanova, D. Kim and J. Wu, Chem. Sci., 2018, 9, 3395–3400 RSC
- B. Prajapati, D.-K. Dang, P. J. Chmielewski, M. A. Majewski, T. Lis, C. J. Gómez-García, P. M. Zimmerman and M. Stępień, Angew. Chem., Int. Ed., 2021, 60, 22496–22504 CrossRef CAS PubMed
- C. Liu, M. E. Sandoval-Salinas, Y. Hong, T. Y. Gopalakrishna, H. Phan, N. Aratani, T. S. Herng, J. Ding, H. Yamada, D. Kim, D. Casanova and J. Wu, Chem, 2018, 4, 1586–1595 CAS
- X. Lu, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Q. Jiang, C. Liu, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 13052–13056 CrossRef CAS PubMed
- M. Stępień, Chem, 2018, 4, 1481–1483 Search PubMed
- H. Gregolińska, M. Majewski, P. J. Chmielewski, J. Gregoliński, A. Chien, J. Zhou, Y.-L. Wu, Y. J. Bae, M. R. Wasielewski, P. M. Zimmerman and M. Stępień, J. Am. Chem. Soc., 2018, 140, 14474–14480 CrossRef PubMed
- W. Schlenk and M. Brauns, Ber. Dtsch. Chem. Ges., 1915, 48, 661–669 CrossRef CAS
- P. Wang, S. Lin, Z. Lin, M. D. Peeks, T. Van Voorhis and T. M. Swager, J. Am. Chem. Soc., 2018, 140, 10881–10889 CrossRef CAS PubMed
- S. Sowndarya, P. C. St. John and R. S. Paton, Chem. Sci., 2021, 12, 13158–13166 RSC
- H. Namai, H. Ikeda, Y. Hoshi, N. Kato, Y. Morishita and K. Mizuno, J. Am. Chem. Soc., 2007, 129, 9032–9036 CrossRef CAS PubMed
- H. Ikeda, J. Photopolym. Sci. Technol., 2008, 21, 327–332 CrossRef CAS
- E. Cho, V. Coropceanu and J.-L. Brédas, J. Mater. Chem. C, 2021, 9, 10794–10801 RSC
- N. E. Horwitz, B. T. Phelan, J. N. Nelson, M. D. Krzyaniak and M. R. Wasielewski, J. Phys. Chem. A, 2016, 120, 2841–2853 CrossRef CAS PubMed
- X. Wan, X. Lv, G. He, A. Yu and Y. Chen, Eur. Polym. J., 2011, 47, 1018–1030 CrossRef CAS
- H. Han, D. Zhang, Z. Zhu, R. Wei, X. Xiao, X. Wang, Y. Liu, Y. Ma and D. Zhao, J. Am. Chem. Soc., 2021, 143, 17690–17700 CrossRef CAS PubMed
- Y. Horbatenko, S. Sadiq, S. Lee, M. Filatov and C. H. Choi, J. Chem. Theory Comput., 2021, 17, 848–859 CrossRef CAS PubMed
- S. Dong and Z. Li, J. Mater. Chem. C, 2022, 10, 2431–2449 RSC
- N. Roques, D. Maspoch, K. Wurst, D. Ruiz-Molina, C. Rovira and J. Veciana, Chem. – Eur. J., 2006, 12, 9238–9253 CrossRef CAS PubMed
- L. Chen, L. Wu, X. Tan, A. Rockenbauer, Y. Song and Y. Liu, J. Org. Chem., 2021, 86, 8351–8364 CrossRef CAS PubMed
- J. A. Mesa, A. Velázquez-Palenzuela, E. Brillas, J. Coll, J. L. Torres and L. Juliá, J. Org. Chem., 2012, 77, 1081–1086 CrossRef CAS PubMed
- M. Hou, X. Lu, Z. Zhang, Q. Xia, C. Yan, Z. Yu, Y. Xu and R. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 44316–44323 CrossRef CAS PubMed
- X. Cui, G. Lu, S. Dong, S. Li, Y. Xiao, J. Zhang, Y. Liu, X. Meng, F. Li and C.-S. Lee, Mater. Horiz., 2021, 8, 571–576 RSC
- J. Qu, F. Z. Khan, M. Satoh, J. Wada, H. Hayashi, K. Mizoguchi and T. Masuda, Polymer, 2008, 49, 1490–1496 CrossRef CAS
- J. Qu, R. Morita, M. Satoh, J. Wada, F. Terakura, K. Mizoguchi, N. Ogata and T. Masuda, Chem. – Eur. J., 2008, 14, 3250–3259 CrossRef CAS PubMed
- K. Zhang, M. J. Monteiro and Z. Jia, Polym. Chem., 2016, 7, 5589–5614 RSC
- C. Simão, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. M. Artés, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359–364 CrossRef PubMed
- M. R. Ajayakumar, C. Moreno, I. Alcón, F. Illas, C. Rovira, J. Veciana, S. T. Bromley, A. Mugarza and M. Mas-Torrent, J. Phys. Chem. Lett., 2020, 11, 3897–3904 CrossRef CAS PubMed
- J. A. de Sousa, F. Bejarano, D. Gutiérrez, Y. R. Leroux, E. M. Nowik-Boltyk, T. Junghoefer, E. Giangrisostomi, R. Ovsyannikov, M. B. Casu, J. Veciana, M. Mas-Torrent, B. Fabre, C. Rovira and N. Crivillers, Chem. Sci., 2020, 11, 516–524 RSC
- F. J. Berger, J. A. de Sousa, S. Zhao, N. F. Zorn, A. A. El Yumin, A. Quintana García, S. Settele, A. Högele, N. Crivillers and J. Zaumseil, ACS Nano, 2021, 15, 5147–5157 CrossRef CAS PubMed
- I. Ratera, J. Vidal-Gancedo, D. Maspoch, S. T. Bromley, N. Crivillers and M. Mas-Torrent, J. Mater. Chem. C, 2021, 9, 10610–10623 RSC
- Y. Tan, S.-N. Hsu, H. Tahir, L. Dou, B. M. Savoie and B. W. Boudouris, J. Am. Chem. Soc., 2022, 144, 626–647 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Summary of optical and electrochemical redox properties of π-radicals, additional π-radical structures. See DOI: https://doi.org/10.1039/d1tc05268c |
| This journal is © The Royal Society of Chemistry 2022 |