
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper binding and protein aggregation: a journey from the brain to the human lens
Yanahi
Posadas
 a,
Carolina
Sánchez-López
a and
Liliana
Quintanar
*ab
a,
Carolina
Sánchez-López
a and
Liliana
Quintanar
*ab
aCenter for Research in Aging, Center for Research and Advanced Studies (Cinvestav), Mexico City, 14330, Mexico
bDepartment of Chemistry, Center for Research and Advanced Studies (Cinvestav), Mexico City, 07350, Mexico. E-mail: lilianaq@cinvestav.mx
First published on 17th October 2023
Abstract
Metal ions have been implicated in several proteinopathies associated to degenerative and neurodegenerative diseases. While the molecular mechanisms for protein aggregation are still under investigation, recent findings from Cryo-EM point out to polymorphisms in aggregates obtained from patients, as compared to those formed in vitro, suggesting that several factors may impact aggregation in vivo. One of these factors could be the direct binding of metal ions to the proteins engaged in aggregate formation. In this opinion article, three case studies are discussed to address the question of how metal ion binding to a peptide or protein may impact its conformation, folding, and aggregation, and how this may be relevant in understanding the polymorphic nature of the aggregates related to disease. Specifically, the impact of Cu2+ ions in the amyloid aggregation of amyloid-β and amylin (or IAPP- islet amyloid polypeptide) are discussed and then contrasted to the case of Cu2+-induced non-amyloid aggregation of human lens γ-crystallin proteins. For the intrinsically disordered peptides amyloid-β and IAPP, the impact of Cu2+ ion binding is highly dependent on the relative location of the metal binding site and the hydrophobic regions involved in β-sheet folding and amyloid formation. Further structural studies of how Cu2+ binding impacts amyloid aggregation pathways and the molecular structure of the final amyloid fibril, both, in vitro and in vivo, will certainly shed light into the molecular origins of the polymorphisms observed in diseased tissue. Finally, contrasting these cases to that of Cu2+-induced non-amyloid aggregation of γ-crystallins, it is evident that, although the impact in aggregation – and the nature of the aggregate – may differ in each system, at the molecular level there is a competition between metal ion coordination and the stability of β-sheet structures. Considering the importance of the β-sheet fold in biology, it is fundamental to understand the energetics and molecular details behind such competition. This opinion article aims to highlight future research directions in the field that can help tackle the important question of how metal ion binding may impact protein folding and aggregation and how this relates to disease.
 Yanahi Posadas | Yanahi Posadas studied Pharmaceutical Industrial Chemistry at the National School of Biological Sciences (IPN) and did her Master and PhD studies in Pharmacology at Cinvestav, under the tutorship of professors Liliana Quintanar and Claudia Perez Cruz. During her PhD studies, Yanahi Posadas characterized the effect of amyloid-β on copper coordination to the prion protein, which is relevant for Alzheimer's disease. Currently, she is doing her postdoctoral research with Prof. José Segovia, at the Department of Physiology, Biophysics and Neuroscience in Cinvestav, where she is studying the coordination chemistry and interactions between copper, prion protein and the N-methyl-D-aspartate receptor (NMDAR), as well as copper binding to glucose regulatory hormones. |
 Carolina Sánchez-López | Carolina Sánchez-López received a bachelor's degree in chemistry from the Autonomous University of the State of Mexico and did her PhD in Chemistry at Cinvestav with Prof. Liliana Quintanar, studying metal–protein interactions with the prion protein and the IAPP peptide. In her postdoctoral research, she focused on structural biology studies by NMR with Prof. Andres Binolfi at the Institute of Molecular and Cell Biology of Rosario in Argentina. Since 2023, she has been as a Visiting Professor at the Center for Research in Aging in Cinvestav. Her research interests include the metal coordination chemistry of hormones and peptides involved in degenerative diseases. |
 Liliana Quintanar | Liliana Quintanar studied chemistry at the National University of Mexico (UNAM) and did her PhD in chemistry at Stanford University with Prof. Edward Solomon. After postdoctoral studies in neurobiology with Prof. Massieu at UNAM, Liliana joined the Department of Chemistry at Cinvestav in 2005, where she has established a strong research program, studying metal–protein interactions that are relevant in neurodegenerative (Alzheimer's, Parkinson's and prion diseases) and degenerative (diabetes and cataracts) diseases. She has been awarded the L’Oreal-UNESCO-AMC For Women in Science fellowship, the Fulbright-García Robles fellowship and the Mexican Academy of Sciences research award. |
Introduction
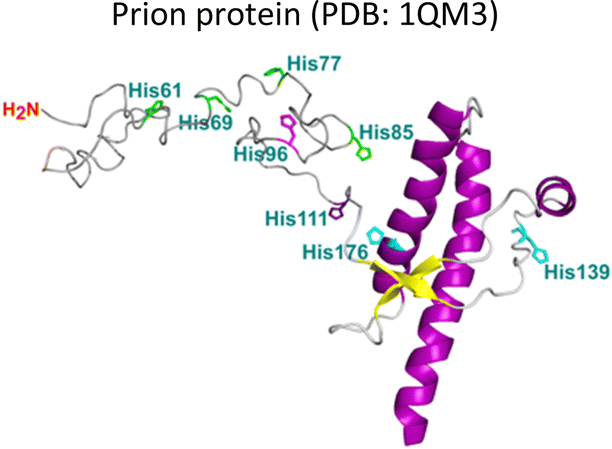
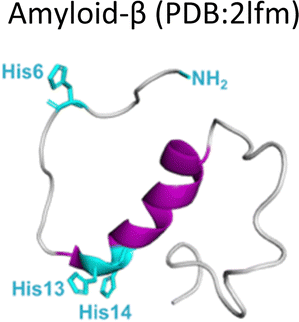
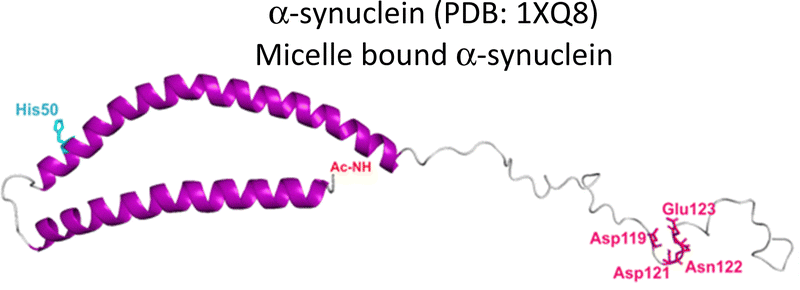
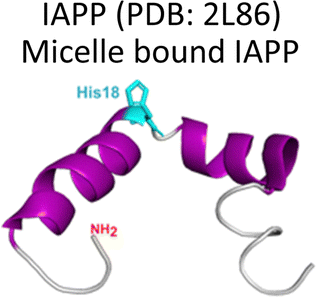
Several degenerative and neurodegenerative diseases are associated with protein aggregation and are collectively named as proteinopathies.1 One of the most studied examples is Alzheimer's disease, which is characterized by the formation of extracellular plaques composed of amyloid fibrils of the amyloid-β peptide and by the presence of neurofibrillary tangles composed of tau protein aggregates.2 Other neurodegenerative proteinopathies include Parkinson's disease, where its characteristic Lewy bodies are formed of the α-synuclein protein;3 and prion diseases that are associated with formation of aggregates of the scrapie isoform of the prion protein.4 But not all proteinopathies concern the brain; type 2 diabetes is associated with accumulation in pancreas of amyloid aggregates formed by amylin or human amyloid polypeptide (IAPP),5 a peptide hormone that is normally co-released with insulin.6–8Table 1 lists the most common proteinopathies that are associated with degenerative disorders. While most of them involve formation of amyloid aggregates, some others do not: cataract disease is thought to involve non-amyloid aggregation of human lens crystallins, which figure among the most stable proteins in the human body.9| Proteina | Disease | Protein aggregation | Metal–protein interaction | Associated functions |
|---|---|---|---|---|
| a Residues involved in metal ion binding are highlighted on each protein. References: Prion protein,18–20 amyloid-β,18,21–24 α-synuclein,25 Huntingtin,18 IAPP,26,27 Crystallin.26,28–30 | ||||
|
Prion diseases | Amyloid | Monomer: Cu2+, Cu+, Mn2+, Zn2+ | Metal ion homeostasis |
| CJD | Synaptic transmission | |||
| Neuronal Plasticity | ||||
| Excitability | ||||
| Neuritogenesis | ||||
| Myelin maintenance | ||||
| Neuroprotection | ||||
|
Alzheimer's disease | Amyloid | Monomer: Cu2+, Cu+, Fe2+, Zn2+ | Unknown |
| Cerebral amyloid angiopathy | Aggregates: Cu2+ | |||
|
Parkinson's disease | Amyloid | Monomer: Ca2+, Ni2+, Co2+, Fe3+, Fe2+, Zn2+, Mn2+, Cu2+, Cu+ | Uptake, storage, recycling of neurotransmitter vesicles |
| Auxiliary chaperone in the synapses | ||||
| Maintenance of dopamine levels | ||||

|
Huntington's disease | Amyloid | Monomer: Cu2+ | Transcription |
| RNA splicing | ||||
| Endocytosis | ||||
| Cell trafficking | ||||
| Cellular homeostasis | ||||
|
Type 2 Diabetes | Amyloid | Monomer: Cu2+, Zn2+ | Related to glucose metabolism. |
| Aggregates: Cu2+ | Regulation of food intake and body weight | |||
| Renal filtration | ||||

|
Cataracts | Non-amyloid | Monomers/Aggregates: Cu2+, Cu+, Zn2+, Hg2+, Pb2+, Cd2+ | Maintaining transparency of the human lens. |
Interestingly, in most cases the proteins involved in degenerative disorders are intrinsically disordered proteins (IDP), as is the case of the amyloid-β peptide, tau, IAPP and α-synuclein.10–14 In some cases, the protein may have a structured region and an IDP region, as is the case of the human prion protein.15 In contrast, for the case of cataract disease, the crystallin proteins involved in aggregation display a stable and well-defined structure, rich in β-sheet structure.16
The amyloid aggregation of these IDP proteins have been extensively studied in the last decades, and a very general mechanism has been proposed.17 Amyloid aggregation is thought to involve a conformational change in the monomer that leads to a β-sheet rich structure and the formation of oligomers, which in turn can grow into protofibrils. Protofibrils can combine with other protofibrils and/or incorporate more monomers to grow into amyloid fibrils, as shown in Fig. 1.17 Alternatively, many recent studies point to the ability of these proteins to undergo liquid–liquid phase separation (LLPS), forming liquid droplets that can eventually lead to the formation of amyloid fibrils (Fig. 1).17
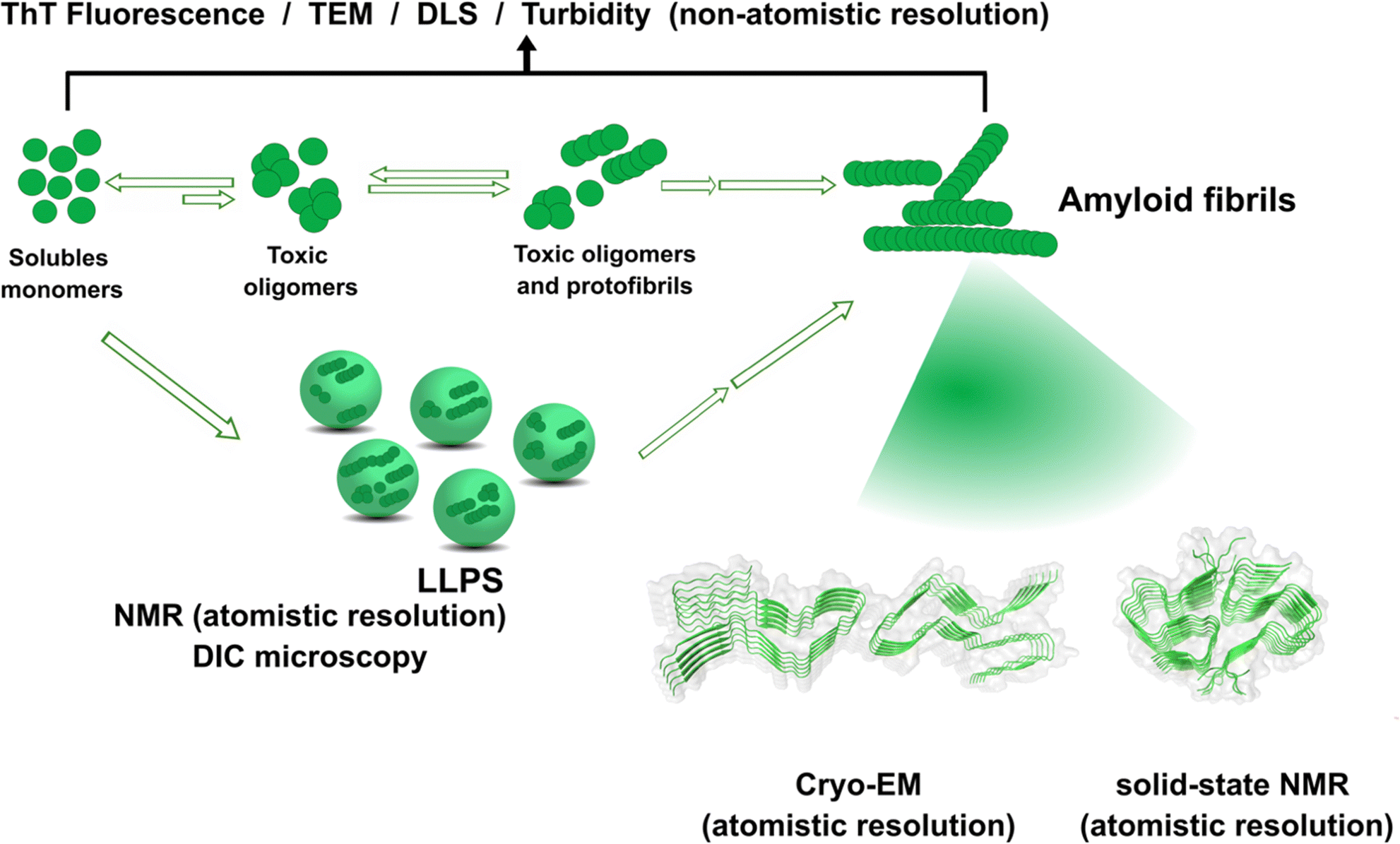 | ||
| Fig. 1 Proposed mechanisms for protein amyloid aggregation and biophysical techniques to study them. Techniques with non-atomistic resolution include: thioflavin T (ThT) Fluorescence, TEM, dynamic light scattering (DLS) and turbidity assays. Techniques with atomistic resolution include: Cryo-EM and solid-state NMR. Liquid–liquid phase separation (LLPS) as an alternative mechanism for protein aggregation and its study by NMR and Differential Interference Microscopy (DIC). Cryo-EM example of structure of prion fibrils (PDB: 6LNI) and solid-state NMR example of amyloid-β fibrils (PDB: 5KK3).31,32 | ||
Although cytotoxicity was ascribed to amyloid fibrils, evidence from the last decade suggests that oligomers are also toxic species.33,34 Particularly, for the case of amyloid-β, it has been demonstrated that oligomers can activate toxic signaling pathways.35,36 Nonetheless, the importance of amyloid fibrils and oligomeric species in the progression of neurogenerative diseases is still under discussion. Unfortunately, structural determination of oligomeric species has proven to be quite challenging,37 preventing the elucidation of the general mechanism shown in Fig. 1 at the molecular level. In contrast, amyloid aggregates have been successfully characterized by transmission electron microscopy TEM), and most recently, their structural description at the molecular level has been achieved by solid-state NMR, and Cryo-Electron Microscopy (Cryo-EM) Fig. 1.31,32,38,39 An interesting finding from recent Cryo-EM studies is that amyloid fibrils found in brains of AD patients display diverse morphologies that contrast with those obtained from in vitro studies40,41 among which there is also diversity due to different preparation methods;42 suggesting that several factors may be involved in the aggregation process and impact the final morphology of amyloid aggregates in vivo. One of these factors could be the direct binding of metal ions to the proteins engaged in aggregate formation. The focus of this opinion article is the question of how metal ion binding may impact protein folding and aggregation and how this may be relevant in understanding the diverse output observed in the related degenerative and neurodegenerative diseases.
As summarized in Table 1, most proteinopathies are also associated with the loss of essential metal ion homeostasis.18,21,26,28–30,43 In Alzheimer's disease, copper is decreased in degenerated tissue,44–46 increased in the serum,47,48 and accumulated in amyloid plaques.49–52 Similarly, copper is elevated in serum of diabetic patients and is raised in lenses with cataracts.28–30,53 In contrast, in Parkinson's disease a copper deficiency is reported for substantia nigra,54 while copper levels do not change in serum and cerebrospinal fluid (CSF).55 Interestingly, the proteins that aggregate in these degenerative diseases display metal binding sites for some essential transition metal ions (Table 1).18,56–60 Metal binding to these proteins (Table 1) has been associated with production of reactive oxygen species (ROS) and oxidative damage;61,62 however, it might also have a functional role.18 For example, extracellular Cu2+ binding to prion protein is thought to be functional for copper sensing and transport,63 as well as part of a neuroprotective mechanism that modulates the neuroreceptor NMDAR, important for memory.18,64 On the other hand, intracellular Cu1+ binding to α-synuclein may be important to stabilize the α-helix conformation of its N-terminal region,65 which would be important for its interaction with membranes.66 Additionally, it has been proposed that α-synuclein forms a ternary complex with the copper chaperone Atox1, protecting α-synuclein from aggregation.43 In contrast to these examples, the functional role of copper and zinc binding to the amyloid-β peptide (and the amyloid precursor protein – APP) remains unknown, in spite of extensive studies of these metal–peptide interactions in the past two decades.18,67 Whatever is the functional role of these metal–protein interactions, it is clear that in the context of the disease, they become aberrant to the point that these essential metal ions end up accumulated in the protein aggregates found in diseased tissue. A clear example of this phenomenon is the accumulation of copper, iron and zinc in amyloid plaques from AD brains.50,51 In order to understand this phenomenon, it would be of value to gain insight into the impact of metal ion binding in protein folding and aggregation. In this opinion article, we will discuss three case studies to illustrate what has been learned on copper binding and protein aggregation and provide an outlook for future research directions in the field. Specifically, the impact of Cu2+ ions in the amyloid aggregation of the amyloid-β and the amylin (or IAPP) peptides will be discussed and then contrasted to the case of Cu2+-induced aggregation of human lens γ-crystallin proteins.
Copper and amyloid-β peptide aggregation
Amyloid-β is a peptide of 39–42 amino acids derived from the amyloid precursor protein (APP)—a transmembrane protein highly expressed in neurons.69 Amyloid-β is produced upon cleavage of APP by β-secretase at the extracellular domain and the γ-secretase complex at its transmembrane domain.70 The N-terminal region of amyloid-β contains hydrophilic amino acids that derive from the extracellular domain of APP, while the C-terminal part of the peptide contains mainly hydrophobic amino acids that were originally located at the transmembrane region of APP (Fig. 2A).18 In aqueous environments, the N- and C-terminal ends of amyloid-β are intrinsically disordered, whereas residues from His13 to Asp23 adopt an α-helical structure (Table 1).13 Amyloid-β binds Cu2+ and Cu1+ using the hydrophilic residues at its N-terminal region, while the C-terminal hydrophobic residues are essential for β-sheet formation, oligomerization and fibrillization (Fig. 2A).18,71–73 Cu2+ ions bound to amyloid-β monomers display two coordination modes at physiological pH: Mode I that involves the NH2 group, one oxygen atom, and two His residues (His6 and His13 or 14); while in Mode II, a His residue is replaced by a deprotonated amide from the peptide backbone.67 Even though several reports support the formation of Mode I and Mode II, recent X-ray absorption studies suggest that Cu2+ bound to the amyloid-β monomer forms a complex mixture made of more than two species.74 The equilibrium between these species at physiological pH and the aggregation propensity of amyloid-β are the major challenges to determine the metal-binding affinity; dissociation constants (Kd) from 0.01 to 10 nM have been reported.75 EPR studies show that amyloid-β aggregates can form both, Mode I and Mode II, depending on the Cu2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amyloid-β ratio,22 being Mode I the most abundant species at 1:1 ratio.23 Interestingly, the redox potential suggests that Mode I, but not Mode II, can be reduced by physiological relevant reducing agents and promote ROS production.76 Amyloid-β reduces Cu2+ to Cu+ in the presence of ascorbate, yielding a di-tyrosine dimer that might promote fibril fragmentation in amyloid-β aggregates.24 Although spectroscopic features of Cu2+ bound to amyloid-β are similar between monomers and aggregates, experimental results suggest that Cu2+ binding affinity of amyloid-β aggregates is two-fold higher than that of the monomer.77 Additionally, N-truncated forms of amyloid-β found in amyloid plaques display even higher Cu2+ affinity (Kd = 30 fM).78,79
:amyloid-β ratio,22 being Mode I the most abundant species at 1:1 ratio.23 Interestingly, the redox potential suggests that Mode I, but not Mode II, can be reduced by physiological relevant reducing agents and promote ROS production.76 Amyloid-β reduces Cu2+ to Cu+ in the presence of ascorbate, yielding a di-tyrosine dimer that might promote fibril fragmentation in amyloid-β aggregates.24 Although spectroscopic features of Cu2+ bound to amyloid-β are similar between monomers and aggregates, experimental results suggest that Cu2+ binding affinity of amyloid-β aggregates is two-fold higher than that of the monomer.77 Additionally, N-truncated forms of amyloid-β found in amyloid plaques display even higher Cu2+ affinity (Kd = 30 fM).78,79
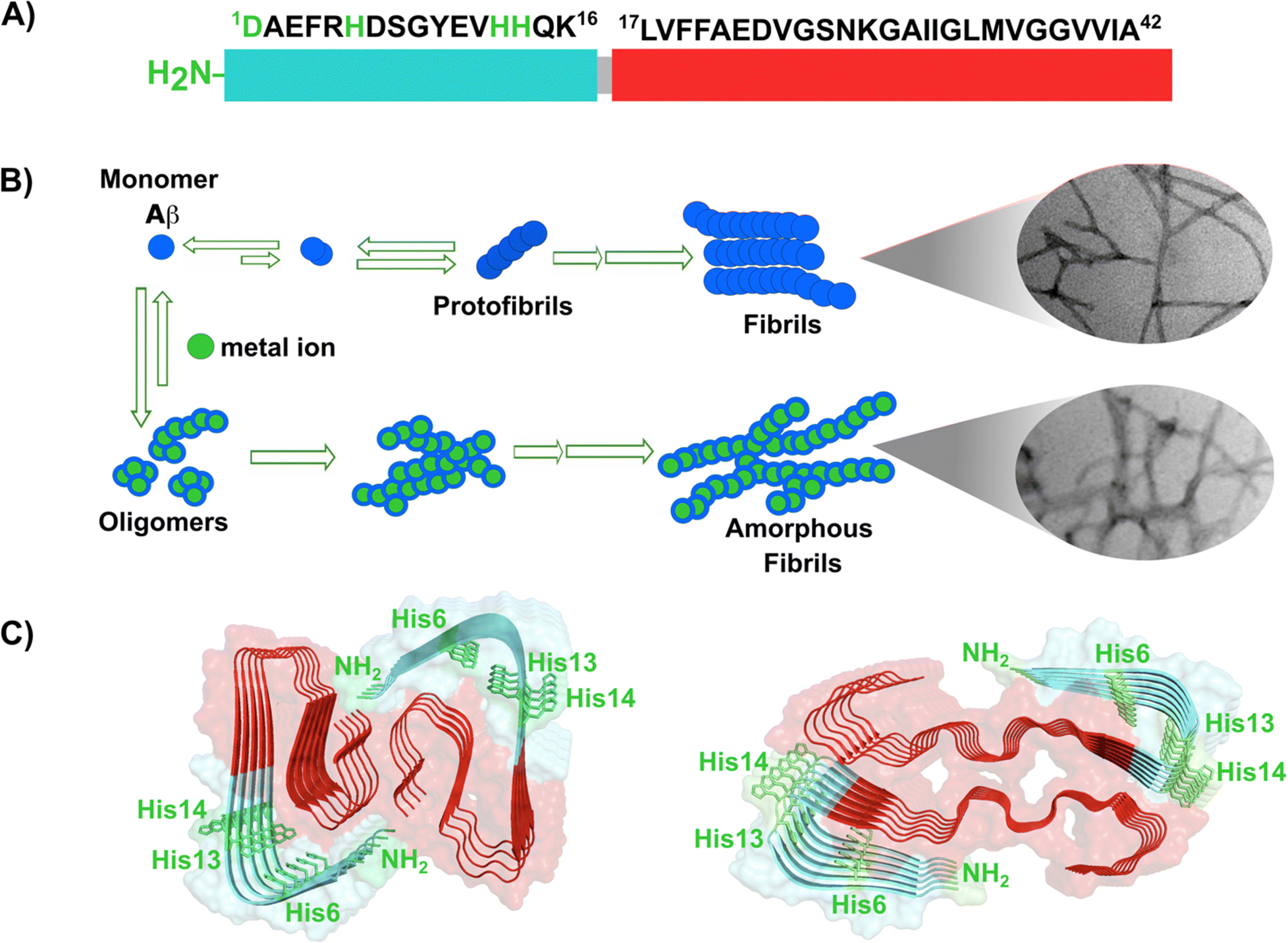 | ||
| Fig. 2 Amyloid-β peptide and its aggregation. (A) Amino acid sequence of human amyloid-β peptide, highlighting in red the hydrophobic region at C-terminal. Residues in green are involved in metal ion coordination. (B) Proposed mechanism for amyloid-β aggregation and TEM images of fibrils in the absence (up) or presence of copper ions (down). (C) Left: Cryo-EM amyloid-β fibrils formed with the recombinant peptide (PDB: 5oqv). Right: Amyloid-β fibril purified from meningeal Alzheimer's brain tissue (PDB: 6SHS).40,41,68 | ||
In the absence of metal ions, amyloid-β forms highly ordered fibrils (Fig. 2B up), where its hydrophilic region and the amino acids involved in Cu2+-coordination are embedded in a β-sheet (Fig. 2C, left).40 The effect of Cu2+ in amyloid-β aggregation has been extensively studied using different experimental approaches, including ThT fluorescence. However, due to the ability of Cu2+ ions to quench ThT fluorescence, the effect of Cu2+ on amyloid-β aggregation should be studied by combining different biophysics techniques. For instance, ThT studies indicate that Cu2+ inhibits amyloid-β aggregation, but studies by other biophysical techniques show that Cu2+ ions do not delay, nor accelerate amyloid-β aggregation, but favors a different pathway that involves formation of less ordered and inter-crossed fibrils (Fig. 2B down),68 suggesting that Cu2+ ions promote the formation of fibrils with a distinct arrangement.42,68,80,81 Unfortunately, there is no Cryo-EM structure of amyloid-β fibrils grown in vitro in the presence of Cu2+ ions. However, it has been recently demonstrated that meningeal fibrils accumulate more copper in comparison with parenchymal fibrils,49,82 and a Cryo-EM structure of fibrils from meningeal Alzheimer's brain tissue has been reported.41 These fibrils are highly polymorphous in comparison with those formed with the recombinant amyloid-β.40,41 Interestingly, in meningeal fibrils, His 13 and His 14 (involved in Cu2+ binding) are not part of a β-sheet, they are in a disordered loop between two β-sheets, suggesting that there are other factors that could impact amyloid-β aggregation in vivo.41 Hence, one could hypothesize that Cu2+ binding to amyloid-β aggregates, as those found in meningeal Alzheimer's brain tissue, is the cause for the distinct morphology where the copper binding residues are not engaged in the β-sheet. Metal ion binding could certainly impede such engagement and promote a different aggregation pathway that leads to the formation of fibrils with a distinct morphology, as observed in vitro. This picture would be consistent with the copper accumulation observed in these amyloid-β aggregates from meningeal Alzheimer's brain tissue.
Overall, Cu2+ ions do not delay, nor accelerate the aggregation of amyloid-β, since the hydrophobic residues engaged in aggregation are not part of the metal-binding site. However, Cu2+ binding could impact the maturation of the fibril and the involvement of the hydrophilic region in the final β-sheet structure of the aggregate, as illustrated in Fig. 2C. Although the impact of Cu1+ ions in aggregation is less studied, a recent report suggests that they accelerate primary nucleation.83 Further structural studies of how Cu2+ and Cu1+ binding to amyloid-β impacts its aggregation pathways and the molecular structure of the final amyloid fibril, both, in vitro and in vivo, will certainly shed light into this problem and perhaps explain the diverse morphology of amyloid aggregates in diseased brains.
Copper and IAPP amyloid aggregation
Amylin or islet amyloid polypeptide (IAPP) is a 37-amino acid peptide that is co-secreted with insulin from the pancreatic β-cells. The formation of amyloid aggregates of IAPP is one of the hallmarks of diabetes type 2. The impact of Cu2+ and Zn2+ ions in the amyloid aggregation of IAPP has been studied in vitro, finding that these metal ions inhibit or delay amyloid fibril growth (Fig. 3).27,84–87 His18 in human IAPP (hIAPP) is an anchoring site for metal ion binding and it is located right in the middle of the hydrophobic region of the peptide (Fig. 3A).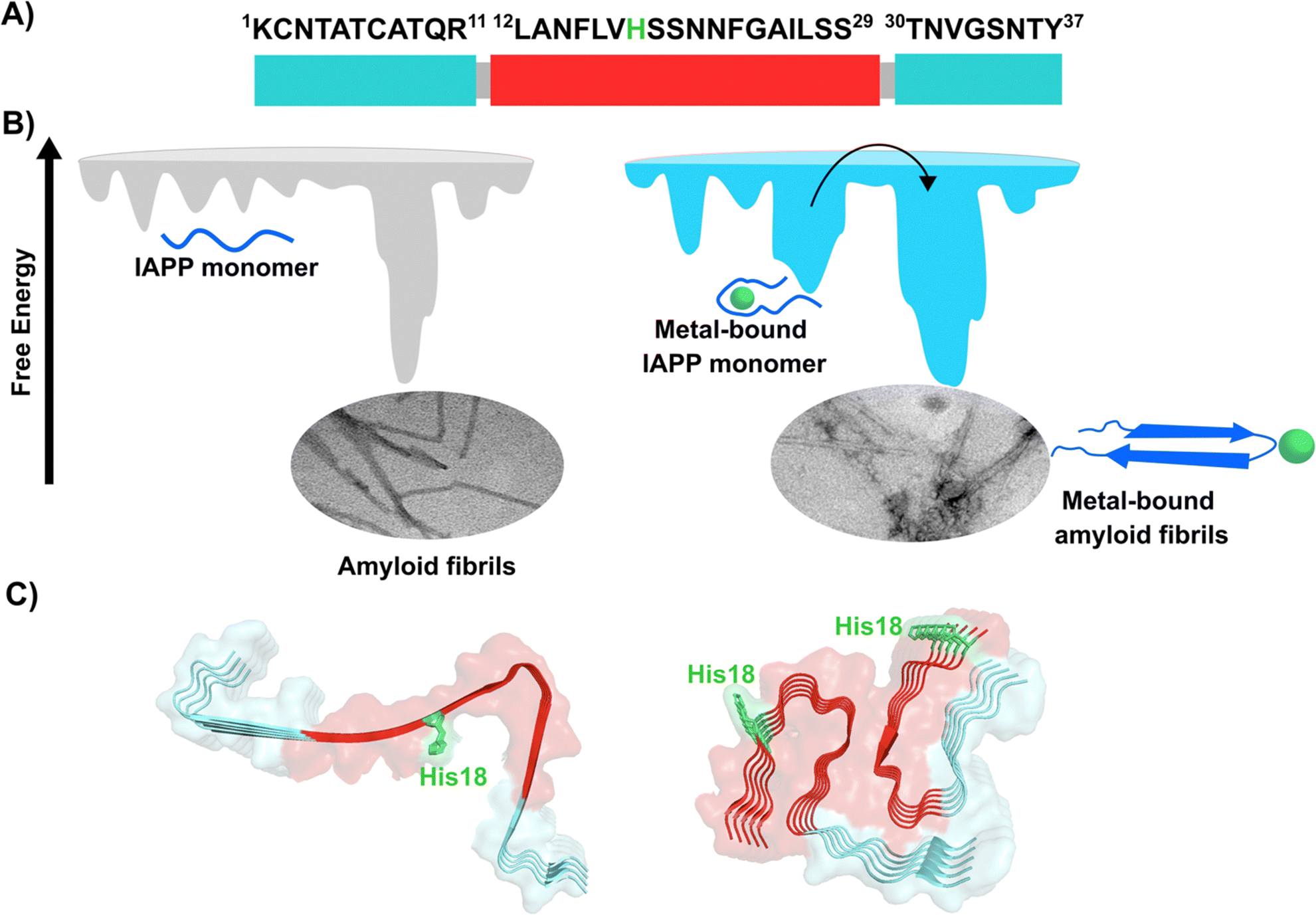 | ||
| Fig. 3 IAPP aggregation. (A) Amino acid sequence of human IAPP peptide, highlighting in red its hydrophobic region. His18, in green, is the site involved in metal ion coordination. (B) Cartoons for free energy landscapes for metal-free hIAPP and Cu(II)-bound hIAPP. TEM images of fibrils in absence (left) and presence of metal ion (right). (C) Cryo-EM structures of fibrils of hIAPP derived from a synthetic peptide (PDB:7YKW, left) and fibrils seeded with isolated aggregates from pancreas of diabetic patients (PDB: 7M61, right).27,92,93 | ||
hIAPP is considered an intrinsically disordered peptide, as it does not adopt a compact globular structure in its monomeric form. However, IAPP does not possess a classic random coil structure. Nuclear magnetic resonance (NMR) studies show that monomeric IAPP forms a transient helical structure at residues 5–20 at the N-terminal; while a more persistent helical structure has been observed when hIAPP is associated to model membranes, forming two alpha-helices at regions 5–17 and 20–27, with a helix-kink-helix arrangement (see Table 1).88–91 hIAPP is highly amyloidogenic in vitro, and its ability to form amyloid fibrils has been ascribed to the hydrophobic residues in the region 20–29; although other fragments from the 10–19 region also display amyloidogenic properties. Several structures for hIAPP amyloid fibrils have been proposed, based on solid state NMR, X-ray diffraction studies of microcrystals and Cryo-EM (Fig. 3C).90,92–94 The structural features of amyloid fibrils seeded by patient-extracted fibrils (Fig. 3C, right) are distinct from those of fibrils grown in vitro from the synthetic peptide (Fig. 3C, left); suggesting that there are additional factors that impact amyloid fibril morphology in vivo and in a pathological context. One of the origins of this polymorphism could be the impact of metal ion binding in the peptide aggregation.
Cu2+ ion binding to hIAPP involves coordination to His1887,95–97 and deprotonated amides from the peptide backbone that follows this residue in the sequence; the metal coordination sphere is completed by the hydroxyl group or the backbone carbonyl of Ser20.56 However, recent studies using hIAPP(1–19) suggest that Cu2+ binds to His18 and the residues towards the N-terminal domain;97 while coordination of the N-terminal amine has also been proposed in studies using hIAPP(1–37).98 Moreover, different Cu2+-binding affinities of hIAPP have been reported, with Kd values ranging from 11.2 nM to 2.2 μM.87,99 Despite the controversy regarding Cu2+ coordination to monomeric IAPP, there is consensus that Cu2+ ions bind to His18 and delay hIAPP amyloid fibril formation, yielding less structured aggregates, as compared to those formed in the absence of the metal ion (Fig. 3B). This is consistent with Cu2+ binding at the anchoring His18, which is found in a β-hairpin of IAPP, affecting the pathway associated with the formation of β-sheets and consequently disrupting fibril formation.27,86 Moreover, electron paramagnetic resonance (EPR) studies show that the resulting Cu2+ species bound to hIAPP aggregates formed in the presence of the metal ion is completely different from Cu2+ bound to the monomeric peptide. Likewise, direct Cu2+ binding to pre-formed hIAPP amyloid fibrils display a distinct metal coordination than that observed for the monomeric hIAPP.27 Hence, it is clear the structure of hIAPP amyloid fibrils (Fig. 3C) cannot accommodate the metal ion in the same coordination mode as the monomeric peptide.
A hallmark of copper binding to IAPP is the formation of toxic oligomers that cause impairment of pancreatic β-cells integrity.87,96,100 These oligomers have a higher random coil component than β-sheet.87 Indeed, it has been proposed that Cu2+ binding to hIAPP stabilizes different peptide conformations that accommodate the preferred metal coordination in the monomer, but that prevent or compete with the formation of the β-sheet structure that is required for fibril growth. In other words, the metal-stabilized conformers encounter a higher energetic barrier for the conformational changes required to form the amyloid structure (Fig. 3B). This is a direct competition between metal coordination and β-sheet formation, since the former requires a particular flexible conformation of the backbone amide groups in the vicinity of His18, which is not present in the latter. In contrast to the case of amyloid-β peptide, Cu2+ binding impacts the amyloid aggregation of IAPP more drastically, since the metal binding site is located right in the hydrophobic region that engages in the formation of the β-sheet structure, and hence, impacts fibril growth significantly.
Copper and γ-crystallin non-amyloid aggregation
Lens γ-crystallins are among the more stable proteins in the human body and are composed of a double Greek key β-sheet fold structure (Fig. 4).9 The formation of non-amyloid aggregates of γ-crystallins contribute to light-scattering in a lens with cataract disease.9 UV damage, posttranslational modifications like truncations and deamidations, and oxidative damage related to late age are thought to cause destabilization of γ-crystallins and formation of partially folded intermediates that are prone to non-amyloid aggregation via a domain-swapping mechanism.101 Recently, it was discovered that essential metal ions, such as Cu2+ and Zn2+, can also cause non-amyloid aggregation of human γ-crystallins in vitro.57,58,60,102 Importantly, epidemiological studies implicate metals as a potential etiological agent for cataract disease, while cataractous lenses can contain higher levels of copper and zinc, as compared to healthy lenses.28–30,53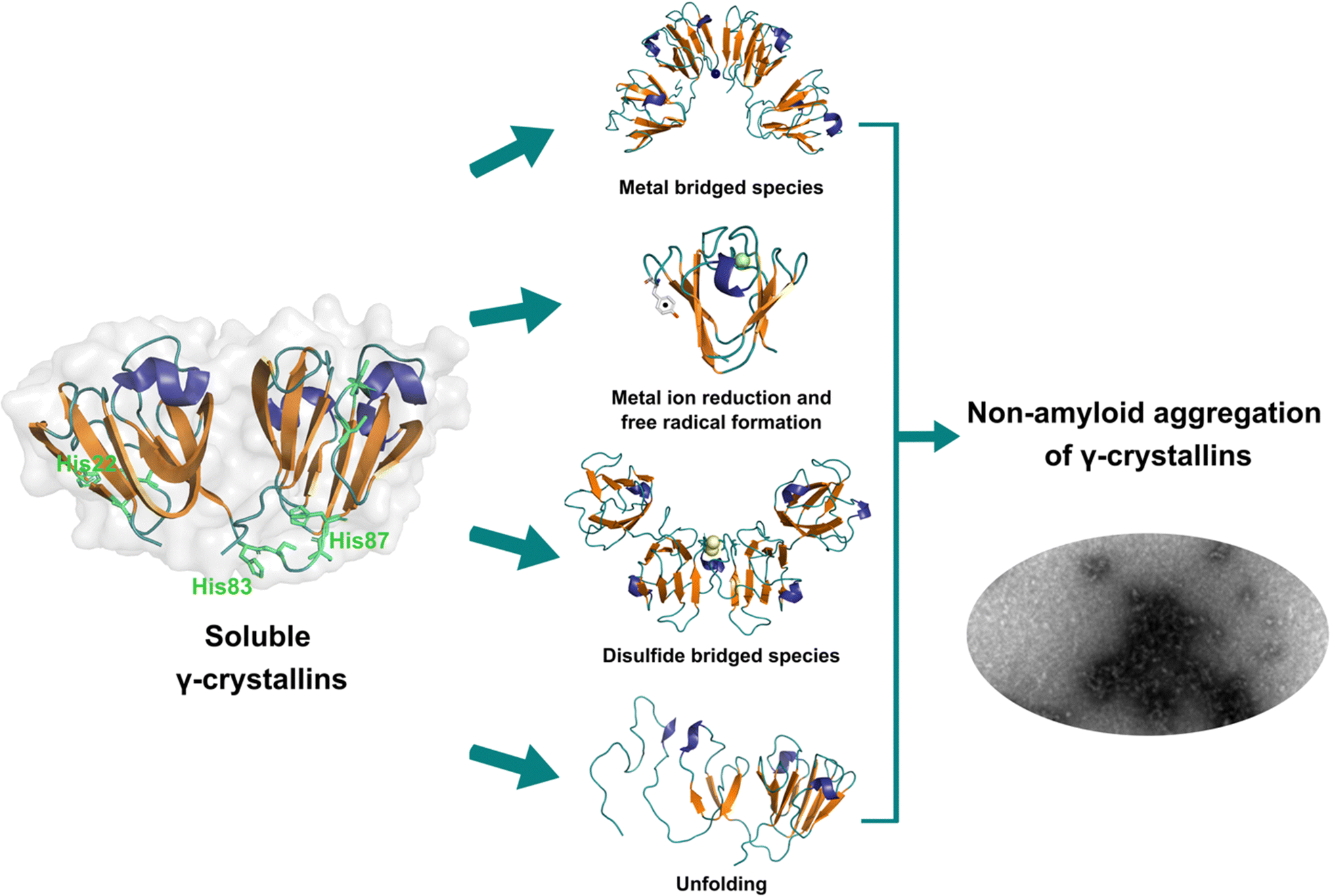 | ||
| Fig. 4 Non-amyloid aggregation of γ-crystallins. Mechanisms of metal ion-induced aggregation: metal bridged species, metal ion reduction and free radical formation, disulfide bridged species and unfolding. PDB: 1 HK0.16,57 | ||
While the mechanism of Zn2+-induced aggregation involves mostly the formation of metal-bridged species,58 the mechanism for Cu-induced aggregation of γ-crystallins is more complex and it involves: metal-bridging, formation of disulfide-bridged dimers and oligomers, protein unfolding, and Cu2+ reduction to Cu+ at expense of protein oxidation (Fig. 4).57,102 Altogether these events lead to the formation of large non-amyloid aggregates of γ-crystallins with copper ions. This is true at least for human γD-, γC- and γS-crystallins, the more abundant lens γ-crystallins.
EPR and X-ray absorption spectroscopy (XAS) studies have shown that these γ-crystallins display at least two Cu2+ binding sites, while the proteins can reduce Cu2+ to Cu+ under aerobic conditions and in the absence of a reducing agent. Cu2+ binding affinities of γ-crystallins have been determined by isothermal titration calorimetry and are best described with Kd values in the micromolar to nanomolar range; while Cu1+ binding constants have not been determined, they are estimated to be in the femtomolar range to assure Cu2+ reduction to Cu+ under aerobic conditions.57 XAS studies demonstrate the presence of at least two Cu+ binding sites in γ-crystallins. Putative copper binding sites in human γD-crystallin involve: His22/Cys18 residues at the N-terminal, His84/His88 at the inter-domain loop, and Cys108/Cys110 at the C-terminal domain.57,60
The unfolding event induced by Cu2+ ions involves loss of β-sheet structure at the N-terminal domain, and formation of partially folded intermediates that are prone to aggregation (Fig. 4).57,60 Cu2+ binding at the N-terminal domain (His22/Cys18) or the interdomain loop (His84/88) may be causing destabilization of one of the β-strands and subsequent unfolding as the metal ion recruits ligands to complete its coordination sphere. In this sense, Cu2+-induced aggregation of γ-crystallins involves an effect in β-sheet structure that can be compared to the case of copper and IAPP: in the latter Cu2+ binding prevents or competes with the formation of β-sheet structure in an intrinsically disordered peptide, while in the former Cu2+ binding causes destabilization of a β-sheet, causing local disorder and formation of partially unfolded intermediates that are prone in aggregation (Fig. 5).
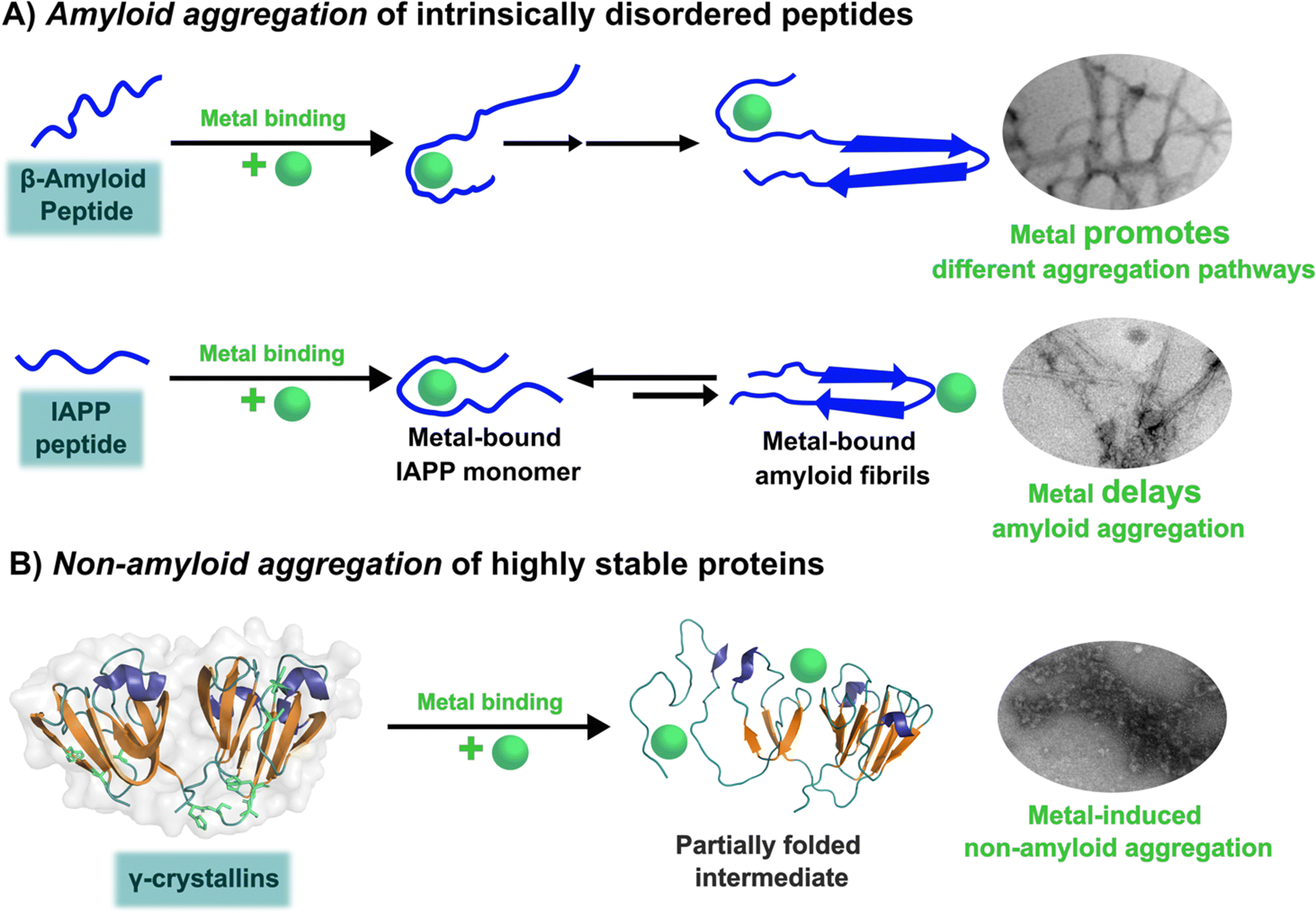 | ||
| Fig. 5 Metal ion binding and its effect on the amyloid (A) and non-amyloid (B) aggregation. | ||
Concluding remarks
Three case studies of the impact of copper binding in peptide/protein aggregation have been discussed. For the intrinsically disordered peptides amyloid-β and IAPP, the impact of Cu2+ ion binding is highly dependent on the relative location of the metal binding site and the hydrophobic regions involved in β-sheet formation and amyloid aggregation (Fig. 5A). In the amyloid-β peptide, the Cu2+ binding site is located at the hydrophilic N-terminal region, and it does not interfere with β-sheet formation, but it causes large oligomerization and results in intercrossed amyloid fibrils with distinct morphology. In contrast, in hIAPP the metal binding site is located right in the middle of the hydrophobic region engaged in β-sheet structure, hence Cu2+ binding competes directly with the formation of β-sheets and inhibits fibril growth significantly; in fact, Cu2+ bound to hIAPP aggregates displays a distinct coordination from that of the monomeric peptide. Beyond the molecular details, the impact of metal ion binding in the amyloid aggregation of amyloid-β and IAPP may be one of the factors that contribute to the observed polymorphism of the aggregates observed in the corresponding disease. To probe this hypothesis, further structural studies of aggregates grown in the presence of metal ion in vitro and correlating studies of metal ion content and in vivo aggregate structures associated to disease are required.Finally, one can contrast the case of hIAPP with that of metal-induced non-amyloid aggregation of structural lens γ-crystallins, in the sense that, what is at stake is a direct competition between Cu2+ coordination and β-sheet structure. Copper-induced aggregation of γ-crystallins involves metal binding and partial loss of β-sheet folding at the N-terminal domain of a well-structured protein. The metal ion must be anchoring at a site, where the recruitment of a ligand to complete its preferred coordination sphere may be tearing apart one of its β-sheets, causing formation of partially folded species that are prone to non-amyloid aggregation (Fig. 5B). On the other hand, Cu2+ coordination to the intrinsically disordered hIAPP peptide occurs at its hydrophobic region, and it involves backbone ligands in a way that it imposes conformations that are not compatible with the formation of the β-sheet structure; the net result being that Cu2+ binding inhibits amyloid aggregation. Although the impact in aggregation is quite the opposite, these two cases illustrate nicely that the strength of metal ion coordination can compete with the stability of β-sheet structures. Being this fold one of the most important ones in protein folding that confers stability, and also a key component for amyloid aggregation, one could argue that the fact that metal ion coordination can destabilize and/or impede the formation of β-sheet structures in peptides and proteins may have important implications for any biological system that involves metal–protein interactions. Further biophysical and chemical studies are required to understand the thermodynamic and molecular details behind such competitions between metal ion coordination and β-sheet structure stability.
Even though degenerative diseases share some characteristics, such as loss of metal homeostasis and aggregation of metal-binding proteins, there are no general rules. Each degenerative disease has specific features: copper is accumulated in the serum of diabetic103 and Alzheimer's patients47,48 but not in Parkinson's;55 copper accelerates the aggregation of γ-crystallin,57 inhibits the aggregation of IAPP,27 and modifies the amyloid-β aggregation pathway.68 Therefore, a general strategy targeting copper in degenerative diseases is not feasible. For each disease, the specific metal-binding properties of the proteins involved should be considered, such as their metal-binding affinity, the possible functional role of the interaction metal–protein, and the effect of the metal ion in protein aggregation. In this context, understanding the biophysical details of the impact of the metal ion in protein aggregations is critical for designing specific therapeutic strategies that target copper in degenerative diseases.
Author contributions
Y. P., C. S.-L. and L. Q. conceptualized the article. Y. P. managed the bibliography. C. S.-L. designed and created the figures. Text writing was mostly done by L. Q. with contributions from Y. P. and C. S.-L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank intramural support from Center for Research and Advanced Studies (Cinvestav). Authors would like to thank CONACYT for a PhD fellowship (to Y.P.) and previous research funding (to L.Q.) through grants 221134 and PN2076. L.Q. would also like to thank present and past members of her research group and collaborators, who have contributed to this research topic.References
- T. A. Bayer, Eur. Neuropsychopharmacol., 2015, 25, 713–724 CrossRef CAS PubMed.
- M. A. DeTure and D. W. Dickson, Mol. Neurodegener., 2019, 14, 32 CrossRef PubMed.
- K. Mensikova, R. Matej, C. Colosimo, R. Rosales, L. Tuckova, J. Ehrmann, D. Hrabos, K. Kolarikova, R. Vodicka, R. Vrtel, M. Prochazka, M. Nevrly, M. Kaiserova, S. Kurcova, P. Otruba and P. Kanovsky, NPJ Parkinsons Dis., 2022, 8, 3 CrossRef PubMed.
- S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13363–13383 CrossRef CAS PubMed.
- C. A. Jurgens, M. N. Toukatly, C. L. Fligner, J. Udayasankar, S. L. Subramanian, S. Zraika, K. Aston-Mourney, D. B. Carr, P. Westermark, G. T. Westermark, S. E. Kahn and R. L. Hull, Am. J. Pathol., 2011, 178, 2632–2640 CrossRef CAS PubMed.
- P. Westermark, C. Wernstedt, E. Wilander and K. Sletten, Biochem. Biophys. Res. Commun., 1986, 140, 827–831 CrossRef CAS PubMed.
- H. C. Fehmann, V. Weber, R. Goke, B. Goke and R. Arnold, FEBS Lett., 1990, 262, 279–281 CrossRef CAS PubMed.
- S. E. Kahn, D. A. D'Alessio, M. W. Schwartz, W. Y. Fujimoto, J. W. Ensinck, G. J. Taborsky Jr. and D. Porte Jr., Diabetes, 1990, 39, 634–638 CrossRef CAS PubMed.
- E. Serebryany and J. A. King, Prog. Biophys. Mol. Biol., 2014, 115, 32–41 CrossRef CAS PubMed.
- O. Coskuner-Weber, O. Mirzanli and V. N. Uversky, Biophys. Rev., 2022, 14, 679–707 CrossRef CAS PubMed.
- J. A. Williamson and A. D. Miranker, Protein Sci., 2007, 16, 110–117 CrossRef CAS PubMed.
- I. T. Yonemoto, G. J. Kroon, H. J. Dyson, W. E. Balch and J. W. Kelly, Biochemistry, 2008, 47, 9900–9910 CrossRef CAS PubMed.
- S. Vivekanandan, J. R. Brender, S. Y. Lee and A. Ramamoorthy, Biochem. Biophys. Res. Commun., 2011, 411, 312–316 CrossRef CAS PubMed.
- C. A. Waudby, C. Camilloni, A. W. Fitzpatrick, L. D. Cabrita, C. M. Dobson, M. Vendruscolo and J. Christodoulou, PLoS One, 2013, 8, e72286 CrossRef CAS PubMed.
- R. Zahn, A. Liu, T. Luhrs, R. Riek, C. von Schroetter, F. Lopez Garcia, M. Billeter, L. Calzolai, G. Wider and K. Wuthrich, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 145–150 CrossRef CAS PubMed.
- A. Basak, O. Bateman, C. Slingsby, A. Pande, N. Asherie, O. Ogun, G. B. Benedek and J. Pande, J. Mol. Biol., 2003, 328, 1137–1147 CrossRef CAS PubMed.
- M. Vendruscolo, Expert Opin. Drug Discovery, 2023, 18, 881–891 CrossRef CAS PubMed.
- Y. Posadas, V. E. López-Guerrero, T. Arcos-López, R. I. Sayler, C. Sánchez-López, J. Segovia, C. Perez-Cruz and L. Quintanar, Comprehensive Inorganic Chemistry III, 2023, pp. 575–628 DOI:10.1016/b978-0-12-823144-9.00115-1.
- Y. Posadas, V. E. Lopez-Guerrero, J. Segovia, C. Perez-Cruz and L. Quintanar, Curr. Opin. Chem. Biol., 2022, 66, 102098 CrossRef CAS PubMed.
- M. A. Wulf, A. Senatore and A. Aguzzi, BMC Biol., 2017, 15, 34 CrossRef PubMed.
- C. Esmieu, D. Guettas, A. Conte-Daban, L. Sabater, P. Faller and C. Hureau, Inorg. Chem., 2019, 58, 13509–13527 CrossRef CAS PubMed.
- S. Jun and S. Saxena, Angew. Chem., Int. Ed., 2007, 46, 3959–3961 CrossRef CAS PubMed.
- W. A. Gunderson, J. Hernandez-Guzman, J. W. Karr, L. Sun, V. A. Szalai and K. Warncke, J. Am. Chem. Soc., 2012, 134, 18330–18337 CrossRef CAS PubMed.
- M. Gu, D. C. Bode and J. H. Viles, Sci. Rep., 2018, 8, 16190 CrossRef PubMed.
- N. Gonzalez, T. Arcos-Lopez, A. Konig, L. Quintanar, M. Menacho Marquez, T. F. Outeiro and C. O. Fernandez, J. Neurochem., 2019, 150, 507–521 CrossRef CAS PubMed.
- A. Fukunaka, M. Shimura, T. Ichinose, O. B. Pereye, Y. Nakagawa, Y. Tamura, W. Mizutani, R. Inoue, T. Inoue, Y. Tanaka, T. Sato, T. Saitoh, T. Fukada, Y. Nishida, T. Miyatsuka, J. Shirakawa, H. Watada, S. Matsuyama and Y. Fujitani, Sci. Rep., 2023, 13, 3484 CrossRef CAS PubMed.
- L. Rivillas-Acevedo, C. Sanchez-Lopez, C. Amero and L. Quintanar, Inorg. Chem., 2015, 54, 3788–3796 CrossRef CAS PubMed.
- V. K. Srivastava, N. Varshney and D. C. Pandey, Acta Ophthalmol., 1992, 70, 839–841 CrossRef CAS PubMed.
- O. Cekic, Br. J. Ophthalmol., 1998, 82, 186–188 CrossRef CAS PubMed.
- J. Dawczynski, M. Blum, K. Winnefeld and J. Strobel, Biol. Trace Elem. Res., 2002, 90, 15–23 CrossRef CAS PubMed.
- L. Q. Wang, K. Zhao, H. Y. Yuan, Q. Wang, Z. Guan, J. Tao, X. N. Li, Y. Sun, C. W. Yi, J. Chen, D. Li, D. Zhang, P. Yin, C. Liu and Y. Liang, Nat. Struct. Mol. Biol., 2020, 27, 598–602 CrossRef PubMed.
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed.
- R. Cascella, A. Bigi, N. Cremades and C. Cecchi, Cell. Mol. Life Sci., 2022, 79, 174 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. D. Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- K. S. Abd-Elrahman, A. Albaker, J. M. de Souza, F. M. Ribeiro, M. G. Schlossmacher, M. Tiberi, A. Hamilton and S. S. G. Ferguson, Sci. Signaling, 2020, 13, eabd2494 CrossRef CAS PubMed.
- J. W. Um, A. C. Kaufman, M. Kostylev, J. K. Heiss, M. Stagi, H. Takahashi, M. E. Kerrisk, A. Vortmeyer, T. Wisniewski, A. J. Koleske, E. C. Gunther, H. B. Nygaard and S. M. Strittmatter, Neuron, 2013, 79, 887–902 CrossRef CAS PubMed.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J. E. Shea, N. V. Dokholyan, A. De Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesne, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Chem. Rev., 2021, 121, 2545–2647 CrossRef CAS PubMed.
- M. R. Sawaya, M. P. Hughes, J. A. Rodriguez, R. Riek and D. S. Eisenberg, Cell, 2021, 184, 4857–4873 CrossRef CAS PubMed.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fandrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- L. Gremer, D. Scholzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schroder, Science, 2017, 358, 116–119 CrossRef CAS PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fandrich, Nat. Commun., 2019, 10, 4760 CrossRef PubMed.
- M. G. M. Weibull, S. Simonsen, C. R. Oksbjerg, M. K. Tiwari and L. Hemmingsen, J. Biol. Inorg. Chem., 2019, 24, 1197–1215 CrossRef CAS PubMed.
- P. Wittung-Stafshede, Essays Biochem., 2022, 66, 977–986 CrossRef CAS PubMed.
- M. A. Deibel, W. D. Ehmann and W. R. Markesbery, J. Neurol. Sci., 1996, 143, 137–142 CrossRef CAS PubMed.
- S. Magaki, R. Raghavan, C. Mueller, K. C. Oberg, H. V. Vinters and W. M. Kirsch, Neurosci. Lett., 2007, 418, 72–76 CrossRef CAS PubMed.
- J. Xu, S. J. Church, S. Patassini, P. Begley, H. J. Waldvogel, M. A. Curtis, R. L. M. Faull, R. D. Unwin and G. J. S. Cooper, Metallomics, 2017, 9, 1106–1119 CrossRef CAS PubMed.
- S. Bucossi, M. Ventriglia, V. Panetta, C. Salustri, P. Pasqualetti, S. Mariani, M. Siotto, P. M. Rossini and R. Squitti, J. Alzheimer's Dis., 2011, 24, 175–185 CAS.
- D. D. Li, W. Zhang, Z. Y. Wang and P. Zhao, Front. Aging Neurosci., 2017, 9, 300 CrossRef CAS PubMed.
- X. Zhu, T. W. Victor, A. Ambi, J. K. Sullivan, J. Hatfield, F. Xu, L. M. Miller and W. E. Van Nostrand, Metallomics, 2020, 12, 539–546 CrossRef CAS PubMed.
- H. Wang, M. Wang, B. Wang, M. Li, H. Chen, X. Yu, K. Yang, Z. Chai, Y. Zhao and W. Feng, Metallomics, 2012, 4, 1113–1118 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS PubMed.
- P. Lei, S. Ayton and A. I. Bush, J. Biol. Chem., 2021, 296, 100105 CrossRef CAS PubMed.
- I. Konz, B. Fernandez, M. L. Fernandez, R. Pereiro, H. Gonzalez-Iglesias, M. Coca-Prados and A. Sanz-Medel, Anal. Bioanal. Chem., 2014, 406, 2343–2348 CrossRef CAS PubMed.
- K. M. Davies, S. Bohic, A. Carmona, R. Ortega, V. Cottam, D. J. Hare, J. P. Finberg, S. Reyes, G. M. Halliday, J. F. Mercer and K. L. Double, Neurobiol. Aging, 2014, 35, 858–866 CrossRef CAS PubMed.
- S. Mariani, M. Ventriglia, I. Simonelli, S. Donno, S. Bucossi, F. Vernieri, J. M. Melgari, P. Pasqualetti, P. M. Rossini and R. Squitti, Neurobiol. Aging, 2013, 34, 632–633 CrossRef CAS PubMed.
- C. Sanchez-Lopez, R. Cortes-Mejia, M. C. Miotto, A. Binolfi, C. O. Fernandez, J. M. Del Campo and L. Quintanar, Inorg. Chem., 2016, 55, 10727–10740 CrossRef CAS PubMed.
- G. Palomino-Vizcaino, N. Schuth, J. A. Dominguez-Calva, O. Rodriguez-Meza, E. Martinez-Jurado, E. Serebryany, J. A. King, T. Kroll, M. Costas and L. Quintanar, J. Am. Chem. Soc., 2023, 145, 6781–6797 CrossRef CAS PubMed.
- J. A. Dominguez-Calva, C. Haase-Pettingell, E. Serebryany, J. A. King and L. Quintanar, Biochemistry, 2018, 57, 4959–4962 CrossRef CAS PubMed.
- J. A. Dominguez-Calva, M. L. Perez-Vazquez, E. Serebryany, J. A. King and L. Quintanar, J. Biol. Inorg. Chem., 2018, 23, 1105–1118 CrossRef CAS PubMed.
- L. Quintanar, J. A. Dominguez-Calva, E. Serebryany, L. Rivillas-Acevedo, C. Haase-Pettingell, C. Amero and J. A. King, ACS Chem. Biol., 2016, 11, 263–272 CrossRef CAS PubMed.
- P. Lei, S. Ayton and A. I. Bush, J. Biol. Chem., 2021, 296, 100105 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- M. J. Pushie, I. J. Pickering, G. R. Martin, S. Tsutsui, F. R. Jirik and G. N. George, Metallomics, 2011, 3, 206–214 CrossRef CAS PubMed.
- S. A. Black, P. K. Stys, G. W. Zamponi and S. Tsutsui, Front. Cell. Dev. Biol., 2014, 2, 45 Search PubMed.
- M. C. Miotto, A. A. Valiente-Gabioud, G. Rossetti, M. Zweckstetter, P. Carloni, P. Selenko, C. Griesinger, A. Binolfi and C. O. Fernandez, J. Am. Chem. Soc., 2015, 137, 6444–6447 CrossRef CAS PubMed.
- C. C. Jao, B. G. Hegde, J. Chen, I. S. Haworth and R. Langen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19666–19671 CrossRef CAS PubMed.
- E. Atrian-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 375, 38–55 CrossRef PubMed.
- M. Marquez, L. M. Blancas-Mejia, A. Campos, L. Rojas, G. Castaneda-Hernandez and L. Quintanar, Metallomics, 2014, 6, 2189–2192 CrossRef CAS PubMed.
- E. Montagna, M. M. Dorostkar and J. Herms, Front. Mol. Neurosci., 2017, 10, 136 CrossRef PubMed.
- R. J. O'Brien and P. C. Wong, Annu. Rev. Neurosci., 2011, 34, 185–204 CrossRef PubMed.
- J. Shearer and V. A. Szalai, J. Am. Chem. Soc., 2008, 130, 17826–17835 CrossRef CAS PubMed.
- P. Faller, C. Hureau and G. L. Penna, Acc. Chem. Res., 2014, 47, 2252–2259 CrossRef CAS PubMed.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed.
- K. L. Summers, K. M. Schilling, G. Roseman, K. A. Markham, N. V. Dolgova, T. Kroll, D. Sokaras, G. L. Millhauser, I. J. Pickering and G. N. George, Inorg. Chem., 2019, 58, 6294–6311 CrossRef CAS PubMed.
- B. Alies, E. Renaglia, M. Rozga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85, 1501–1508 CrossRef CAS PubMed.
- L. G. Trujano-Ortiz, F. J. Gonzalez and L. Quintanar, Inorg. Chem., 2015, 54, 4–6 CrossRef CAS PubMed.
- D. Jiang, L. Zhang, G. P. Grant, C. G. Dudzik, S. Chen, S. Patel, Y. Hao, G. L. Millhauser and F. Zhou, Biochemistry, 2013, 52, 547–556 CrossRef CAS PubMed.
- M. Mital, N. E. Wezynfeld, T. Fraczyk, M. Z. Wiloch, U. E. Wawrzyniak, A. Bonna, C. Tumpach, K. J. Barnham, C. L. Haigh, W. Bal and S. C. Drew, Angew. Chem., Int. Ed., 2015, 54, 10460–10464 CrossRef CAS PubMed.
- E. Portelius, N. Bogdanovic, M. K. Gustavsson, I. Volkmann, G. Brinkmalm, H. Zetterberg, B. Winblad and K. Blennow, Acta Neuropathol., 2010, 120, 185–193 CrossRef CAS PubMed.
- A. Abelein, Acc. Chem. Res., 2023, 56, 2653–2663 CrossRef CAS PubMed.
- Y. Yi and M. H. Lim, RSC Chem. Biol., 2023, 4, 121–131 RSC.
- S. Wang, Z. Sheng, Z. Yang, D. Hu, X. Long, G. Feng, Y. Liu, Z. Yuan, J. Zhang, H. Zheng and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12415–12419 CrossRef CAS PubMed.
- N. Sasanian, D. Bernson, I. Horvath, P. Wittung-Stafshede and E. K. Esbjorner, Biomolecules, 2020, 10, 924 CrossRef CAS PubMed.
- B. Ward, K. Walker and C. Exley, J. Inorg. Biochem., 2008, 102, 371–375 CrossRef CAS PubMed.
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC.
- H. Li, E. Ha, R. P. Donaldson, A. M. Jeremic and A. Vertes, Anal. Chem., 2015, 87, 9829–9837 CrossRef CAS PubMed.
- S. J. C. Lee, T. S. Choi, J. W. Lee, H. J. Lee, D. G. Mun, S. Akashi, S. W. Lee, M. H. Lim and H. I. Kim, Chem. Sci., 2016, 7, 5398–5406 RSC.
- J. R. Brender, K. Hartman, R. P. Nanga, N. Popovych, R. de la Salud Bea, S. Vivekanandan, E. N. Marsh and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 8973–8983 CrossRef CAS PubMed.
- R. P. Nanga, J. R. Brender, S. Vivekanandan and A. Ramamoorthy, Biochim. Biophys. Acta, Gene Regul. Mech., 2011, 1808, 2337–2342 CrossRef CAS PubMed.
- P. Cao, P. Marek, H. Noor, V. Patsalo, L. H. Tu, H. Wang, A. Abedini and D. P. Raleigh, FEBS Lett., 2013, 587, 1106–1118 CrossRef CAS PubMed.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795–826 CrossRef CAS PubMed.
- D. Li, X. Zhang, Y. Wang, H. Zhang, K. Song, K. Bao and P. Zhu, iScience, 2022, 25, 105705 CrossRef CAS PubMed.
- Q. Cao, D. R. Boyer, M. R. Sawaya, R. Abskharon, L. Saelices, B. A. Nguyen, J. Lu, K. A. Murray, F. Kandeel and D. S. Eisenberg, Nat. Struct. Mol. Biol., 2021, 28, 724–730 CrossRef CAS PubMed.
- R. Gallardo, M. G. Iadanza, Y. Xu, G. R. Heath, R. Foster, S. E. Radford and N. A. Ranson, Nat. Struct. Mol. Biol., 2020, 27, 1048–1056 CrossRef CAS PubMed.
- M. Rowinska-Zyrek, Dalton Trans., 2016, 45, 8099–8106 RSC.
- A. Sinopoli, A. Magri, D. Milardi, M. Pappalardo, P. Pucci, A. Flagiello, J. J. Titman, V. G. Nicoletti, G. Caruso, G. Pappalardo and G. Grasso, Metallomics, 2014, 6, 1841–1852 CrossRef CAS PubMed.
- E. Dzien, D. Dudek, D. Witkowska and M. Rowinska-Zyrek, Sci. Rep., 2022, 12, 425 CrossRef CAS PubMed.
- M. Seal and S. G. Dey, Inorg. Chem., 2018, 57, 129–138 CrossRef CAS PubMed.
- E. C. Lee, E. Ha, S. Singh, L. Legesse, S. Ahmad, E. Karnaukhova, R. P. Donaldson and A. M. Jeremic, Phys. Chem. Chem. Phys., 2013, 15, 12558–12571 RSC.
- Y. P. Yu, P. Lei, J. Hu, W. H. Wu, Y. F. Zhao and Y. M. Li, Chem. Commun., 2010, 46, 6909–6911 RSC.
- P. Das, J. A. King and R. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10514–10519 CrossRef CAS PubMed.
- K. W. Roskamp, S. Azim, G. Kassier, B. Norton-Baker, M. A. Sprague-Piercy, R. J. D. Miller and R. W. Martin, Biochemistry, 2020, 59, 2371–2385 CrossRef CAS PubMed.
- Q. Qiu, F. Zhang, W. Zhu, J. Wu and M. Liang, Biol. Trace Elem. Res., 2017, 177, 53–63 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |