
Development of self-cooperative nanochaperones with enhanced activity to facilitate protein refolding†
Menglin
Yang
a,
Yanli
Zhang
a,
Fei
Deng
a,
Xiaohui
Wu
a,
Yujie
Chen
a,
Feihe
Ma
*b and
Linqi
Shi
 *a
*a
aKey Laboratory of Functional Polymer Materials of Ministry of Education, State Key Laboratory of Medicinal Chemical Biology, Institute of Polymer Chemistry and College of Chemistry, Nankai University, Tianjin, 300071, P.R. China. E-mail: shilinqi@nankai.edu.cn
bState Key Laboratory of Separation Membranes and Membrane Processes and School of Materials Science and Engineering, Tiangong University, Tianjin, 300387, P.R. China. E-mail: feihema@tiangong.edu.cn
First published on 6th October 2023
Abstract
Regulating protein folding including assisting de novo folding, preventing misfolding and aggregation, and facilitating refolding of proteins are of significant importance for retaining protein's biological activities. Here, we report a mixed shell polymeric micelle (MSPM)-based self-cooperative nanochaperone (self-CO-nChap) with enhanced activity to facilitate protein refolding. This self-CO-nChap was fabricated by introducing Hsp40-mimetic artificial carriers into the traditional nanochaperone to cooperate with the Hsp70-mimetic confined hydrophobic microdomains. The artificial carrier facilitates transfer and immobilization of client proteins into confined hydrophobic microdomains, by which significantly improving self-CO-nChap's capability to inhibit unfolding and aggregation of client proteins, and finally facilitating refolding. Compared to traditional nanochaperones, the self-CO-nChap significantly enhances the thermal stability of horseradish peroxidase (HRP) epicyclically under harsher conditions. Moreover, the self-CO-nChap efficiently protects misfolding-prone proteins, such as immunoglobulin G (IgG) antibody from thermal denaturation, which is hardly achieved using traditional nanochaperones. In addition, a kinetic partitioning mechanism was devised to explain how self-CO-nChap facilitates refolding by regulating the cooperative effect of kinetics between the nanochaperone and client proteins. This work provides a novel strategy for the design of protein folding regulatory materials, including nanochaperones.
 Linqi Shi | Congratulations on the 10th anniversary of Materials Horizons, and we are excited to contribute an article to this outstanding journal to show our latest work and deep understanding on how nanochaperones regulate protein folding. It is always interesting to read Materials Horizons articles, especially for the new concept section which has inspired us with many innovative ideas. In the year of 2023, we first published a paper in Materials Horizons and our partnership has grown rapidly ever since. We look forward to reading more wonderful papers published in Materials Horizons and contributing to this excellent journal for many decades to come! |
New conceptsWe demonstrate a new strategy to fabricate nanochaperones by mimicking the cooperation of co-chaperones (e.g., Hsp40) and chaperones (e.g., Hsp70). The new strategy of introducing artificial carriers into nanochaperones to mimic the cooperative effect of co-chaperones facilitates the transfer and immobilization of protein clients into the chaperone-mimetic confined hydrophobic microdomains, which significantly enhances the nanochaperone's activity for protein protection. We for the first time come up with a kinetic partitioning mechanism to explain how nanochaperones work. The artificial co-chaperones increase the rate constant of binding of protein clients to the nanochaperone (kon), thus enhancing the chaperone activity for two reasons: (i) the faster binding ensures kon is greater than the rate constant of protein aggregation (kagg) and thus effectively inhibiting aggregation, especially under harsher conditions, which is necessary for protein refolding; (ii) this promotes the immobilization of protein clients, thus inhibiting unfolding of trapped clients when heated at higher temperatures, ensuring that the rate constant of folding (kfold) of released protein clients is greater than kagg after cooling, thereby facilitating refolding. We believe that this kinetic partitioning mechanism is universal for different nanochaperone systems and should provide a direction to design new protein folding regulatory materials with enhanced chaperone activity. |
Introduction
Protein folding is paramount for regulating protein's biological activities.1 In particular, understanding the principles of protein folding has contributed to the design and synthesis of new types of materials including artificially designed proteins,2 foldamers,3 and chaperone mimetic materials.4 Among these materials, nanochaperones with natural chaperone-like activity have attracted increasing interest in recent years due to the fact that they are potent in assisting protein (re)folding,4 protein misfolding disease treatment,5–8 and protein protection and delivery.9–12 Moreover, nanochaperones also provide a way for biologists to test and deepen their understanding of protein folding.13,14 As an expanding research area, improving nanochaperone's activity and understanding their working mechanisms are challenging and long-term goals.To date, most nanochaperone systems including gold nanoparticles,15,16 nanogels17,18 and coacervates19 only simply mimic the behaviors of natural chaperones that catch their clients via hydrophobic and electrostatic interactions to inhibit irreversible aggregation and release clients to allow correct folding. Although they are efficient in preventing aggregation and some of them also help to fold, the unfolded clients adsorbed on the surface of nanoparticles can interact intermolecularly, limiting protein refolding. Natural GroEL-GroES chaperonin utilizes a confined nano-cage to completely prevent intermolecular interactions of unfolded protein clients and promotes folding.20 Inspired by this unique nanostructure, Kameta's group developed a soft nanotube hydrogel with confined space to encapsulate denatured proteins and assist refolding.21 Fujita's group utilized rigid self-assembled molecular cages to precisely encapsulate a single native protein in one cage and significantly stabilized the protein through the protein-refolding effect of the cage.22 This is a big success with promising applications in molecular biology and industry. However, in situ co-assembly of protein clients and chemical ligands was necessary for the encapsulation, which would limit their in vivo applications. Moreover, the encapsulation of protein in a closed space limits its dynamic personality and interaction with other biomacromolecules.
Self-assembled mixed shell polymeric micelle (MSPM)-based nanochaperones that were first developed by our group mimic both the structure and function of natural chaperones and have shown great potential in regulating protein folding and mediating protein delivery.4,23–27 This MSPM-based nanochaperone has a hydrophobic core and a hydrophilic mixed micellar shell that consists of hydrophilic polymers (e.g. poly(ethylene glycol) (PEG)) and stimuli-responsive polymers (e.g. poly(β-amino ester) (PAE)). The deprotonation of PAE makes PAE chains turn to hydrophobic and collapse, resulting in the formation of several individually semi-opened, soft, and confined hydrophobic microdomain nanostructures on its surface, through which to interact with protein clients to perform chaperone activities.23 This unique nanostructure has the advantage of dynamically interacting with protein clients, providing flexibility for the dynamic personality of proteins.8,24 Furthermore, this dynamical interaction ensures that nanochaperones are in their dormant state under suitable conditions and do not perturb the function of native proteins, while being activated under harsh conditions to perform chaperone activities.27 This unique nanostructure also has the advantage of trapping one or a few protein molecules in one hydrophobic microdomain, which is similar to the nano-cage of GroEL-GroES, preventing intermolecular interaction of clients efficiently. However, the trapping of protein clients by traditional non-specific nanochaperones depends on the randomly simple diffusion of clients into the hydrophobic microdomains, and the out-stretched hydrophilic shell prevents clients from colliding into the hydrophobic microdomains, which limits the chaperone activity of the MSPM-based nanochaperone (Fig. 1). For example, the non-specific nanochaperones exhibited lower protection activity for misfolding-prone proteins, especially at higher heating temperatures,24,27 and we failed to protect IgG antibody using non-specific nanochaperones. Therefore, strategies to facilitate the diffusion of clients are urgently needed to enhance the activity of MSPM-based nanochaperones and to expand their applications.
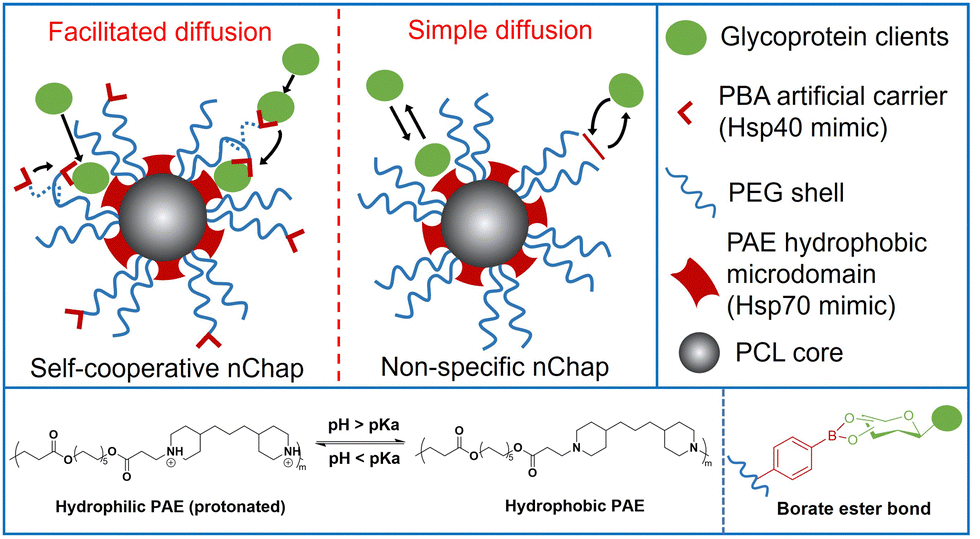 | ||
| Fig. 1 Schematic illustration of self-cooperative nChap and traditional non-specific nChap and the diffusion process of protein clients. | ||
Living cells utilize co-chaperones (e.g., Hsp40) to assist the transfer of client to chaperone (e.g., Hsp70) and promote protein folding.28,29 A process called facilitated diffusion also uses a similar functional carrier protein to facilitate the transfer of a substance across the plasma membrane.30 Inspired by these processes, here we fabricated a self-cooperative nanochaperone (self-co-nChap) by introducing an artificial carrier to the shell surface of a traditional non-specific nanochaperone to mimic the co-chaperone Hsp40, and to cooperate with the Hsp70-mimetic hydrophobic microdomains of the nanochaperone (Fig. 1). This self-co-nChap was designed for the protection of glycoproteins as they are one of the most important and commonly used products in the field of biopharmaceuticals and industrial catalysis.31,32 Furthermore, the modification of glycans on the glycoprotein's surface increases its hydrophilicity which makes it more difficult to be trapped by the nanochaperone through the short-range hydrophobic interactions. The artificial carrier is a phenylboronic acid (PBA) motif that can recognize and capture glycan on the glycoprotein's surface via a reversible covalent bond between the boronic acid group and cis-1,2-diol groups. This recognition recruits glycoprotein clients close to the nanochaperone, thus facilitating the transfer and immobilization of clients into the confined hydrophobic microdomains, which significantly enhances the chaperone activity of self-co-nChap. In particular, when HRP was heated at 95 °C for 1 h, the recovery enzyme activity of HRP was 2.3-fold higher with the protection of self-co-nChap than with the non-specific nanochaperone. Self-co-nChap also significantly improves the thermal stability of HRP when being used for industrial catalysis applications in harsh conditions. Moreover, self-co-nChap can efficiently protect immunoglobulin G (IgG) antibody from thermal denaturation, which is hardly achieved using non-specific nanochaperones. The strategy of introducing an artificial carrier to facilitate diffusion of clients into the confined hydrophobic microdomains of nanochaperone closely mimics the cooperation of co-chaperone Hsp40 with Hsp70, providing important insights for the design of new protein folding regulatory materials, including nanochaperones.
Results and discussion
Poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG114-b-PCL51; PEG-b-PCL), pH-responsive poly (amino ester)-block-poly(ε-caprolactone) (PDAE13-b-PCL55; PDAE-b-PCL), and phenylboronic acid-poly(ethylene glycol)-block-poly(ε-caprolactone) (PBA-modified PEG114-b-PCL51; PBA-PEG-b-PCL, F-PBA-PEG-b-PCL and NO2-PBA-PEG-b-PCL) were synthesized and their chemical structures were confirmed using 1H-NMR spectroscopy (Fig. S1–S3, ESI†). MSPM-1 and ten other control MSPMs (MSPM-2–11) were prepared by self-assembling amphiphilic di-block copolymers in water (Fig. S4 and Table S1, ESI†). All eleven MSPMs had narrow size distributions whose polydispersity index (PDI) was below 0.20 and hydrodynamic diameters were around 100 nm as measured by dynamic light scattering (DLS) while transmission electron microscopy (TEM) images revealed uniform spherical structures (Fig. S5 and Table S1, ESI†). The block of PDAE is pH-responsive whose acid dissociation constant (pKa) is 6.29 and it is deprotonated and hydrophobic at pH > 6.29 while turning to protonated and hydrophilic when the pH < 6.29.25 The zeta potential results confirmed the protonation and deprotonation of the PDAE chains at different pH values (Fig. S6, ESI†).HRP (molecular weight (Mw) = 40 kDa and isoelectric point (pI) = 8.9) with a glycolytic content of 18% was selected as a model glycoprotein and was incubated with MSPM-1–4 at 25 °C, pH 7.4 to evaluate the effects of the PBA-carrier on the protein adsorption. Quantitative analysis showed that 75 ± 2, 63 ± 1 and 61 ± 2 μg proteins were adsorbed to 1 mg of the F-PBA (MSPM-1), PBA (MSPM-2), and NO2-PBA (MSPM-3) modified nanochaperones, respectively, which were higher than that of non-specific nChaps (MSPM-4, 49 ± 3 μg) (Fig. 2a). The decrease of the fluorescence intensity of MSPM-1/Alizarin Red S (ARS) solution after the addition of HRP indicates the capture of proteins by the F-PBA (Fig. 2b).33 Förster resonance energy transfer (FRET) assay using HRP labeled with Cy3 and MSPM-1 labeled with Cy5 at PDAE showed a FRET peak at 670 nm, indicating that the captured HRP was transferred into the hydrophobic microdomains (Fig. 2c).34 The increased mean square radius of gyration (Rg) and decreased hydrodynamic radius (Rh) of MSPM-1/HRP measured by light scattering as a function of time (Fig. 2d) further confirmed that the captured HRP was transferred into the hydrophobic microdomains.24 Quartz Crystal Microbalance with Dissipation (QCM-D) measurements using HRP and non-glycoprotein carbonic anhydrase B (CAB, Mw = 28.9 kDa and pI = 5.9) showed that the resonance frequency attenuation (ΔF) of MSPM-1 was higher than that of MSPM-4 for the adsorption of HRP, while the ΔF of MSPM-1 and MSPM-4 was similar for the adsorption of CAB (Fig. 2e and f), suggesting that the modification of PBA on the surface of the nanochaperone enhances the adsorption of glycoproteins explicitly.
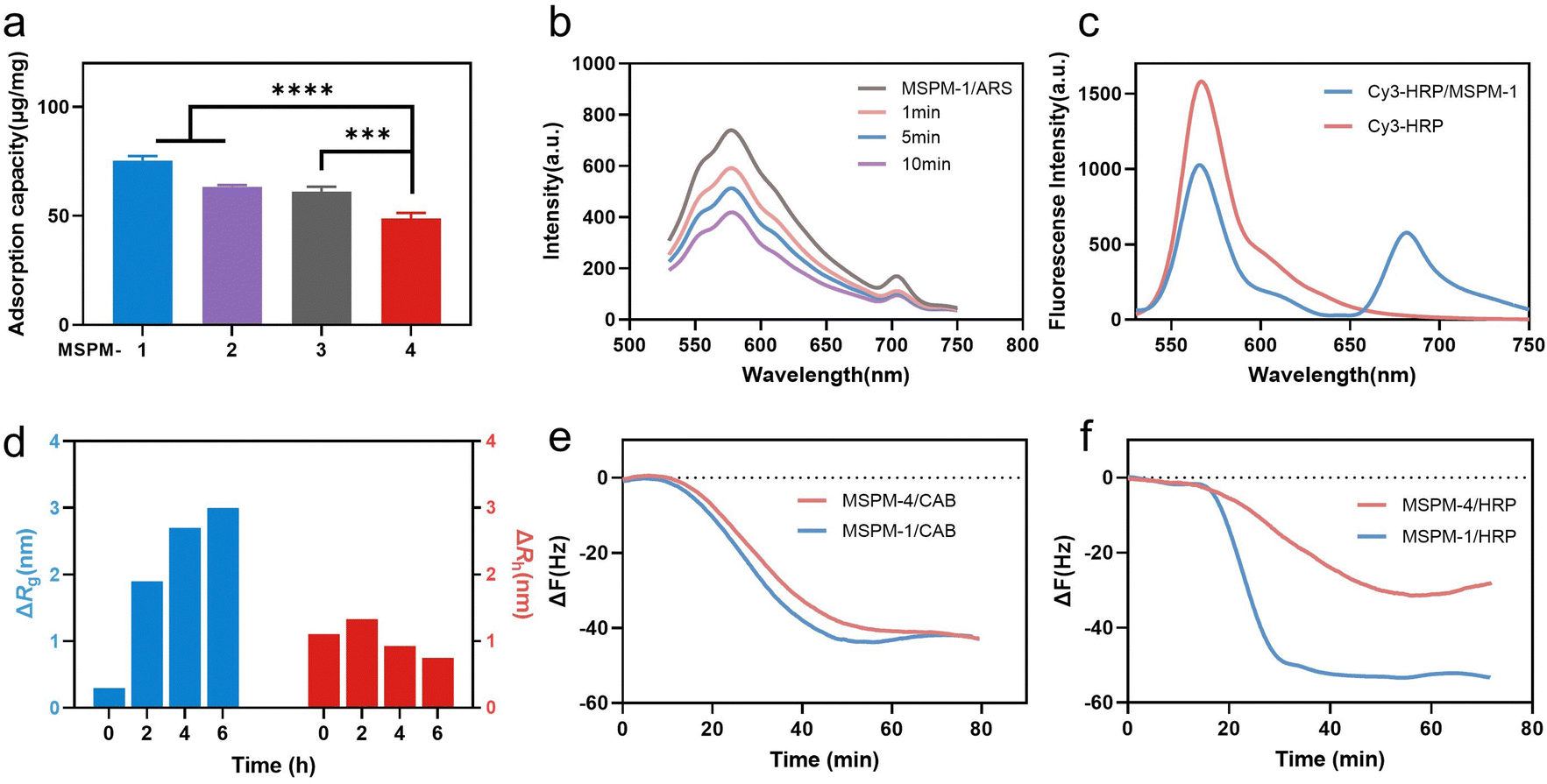 | ||
| Fig. 2 Protein adsorption by nanochaperones. (a) HRP adsorption by MSPM-1–4 after incubation of MSPMs (0.5 mg mL−1) with HRP (0.2 mg mL−1) at 25 °C, pH 7.4 for 6 h. (b) Fluorescence intensity at 586 nm of MSPM-1 and ARS in the presence of HRP. (c) Fluorescence spectra of Cy5-labeled MSPM-1 after being incubated with Cy3-labeled HRP. The spectra were obtained with an excitation wavelength of 515 nm. (d) The changes of the mean square radius of gyration (Rg) and hydrodynamic radius (Rh) of MSPM-1 (0.1 mg mL−1) after incubation with native HRP (0.02 mg mL−1) at 25 °C, pH 7.4 for different times. (e) and (f) Time courses of frequency changes of QCM responding to the addition of CAB (e) and HRP (f) with MSPM-1 and non-specific nChap (MSPM-4). Statistical significance was analyzed by one-way ANOVA with Dunnett's multiple comparisons test in (a). Significant levels are defined as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. | ||
The activity of nanochaperones on the refolding of thermal denatured HRP was first investigated by measuring the residual enzymatic activity of HRP after heating at pH 7.4, 85 °C for 1 h, followed by cooling the mixture to 4 °C and adjusting the pH to 5 for 1 h to release the denatured HRP and allow enzyme renaturation (Fig. S7, ESI†). As shown in Fig. 3a, the addition of nanochaperones significantly increased the refolding efficiency of HRP and the highest recovered enzymatic activity was 80 ± 2% with the assistance of MSPM-1 compared to that of MSPM-2 (67 ± 3%), MSPM-3 (66 ± 4%), and MSPM-4 (45 ± 1%). The best capability to facilitate diffusion of HRP was also achieved by the F-PBA-carrier (Fig. 2a). Thereby, we chose F-PBA-modified MSPM-1 and termed it as “self-co-nChap” for the following studies. Furthermore, the circular dichroism (CD) spectroscopy results confirmed that there was no significant difference between the secondary structure of the refolded HRP and the native HRP (Fig. S8, ESI†). It is particularly notable that, when being heated at pH 5.0, 85 °C for 1 h, the recovered enzymatic activity of HRP was only 18 ± 1% and 29 ± 2% with the assistance of MSPM-1 and MSPM-4, respectively (Fig. S9, ESI†), confirming the important role of the confined hydrophobic microdomain nanostructures on protein refolding.
 | ||
| Fig. 3 Nanochaperones protect proteins from thermal denaturation. (a), (b) and (c) The recovered enzyme activity of thermally denatured HRP (50 μg mL−1) in the absence (control) or presence of MSPMs (0.5 mg mL−1) after being heated at pH 7.4, 85 °C for 1 h. (d) The recovered enzyme activity of thermally denatured lysozyme (50 μg mL−1) in the absence (control) or presence of self-co-nChap (MSPM-1) or non-specific nChap (MSPM-4) (0.5 mg mL−1) after being heated at pH 7.4, 85 °C for 1 h. (e) The recovered enzyme activity of HRP and lysozyme in the absence (control) or presence of self-co-nChap (MSPM-1) (0.5 mg mL−1) after the HRP (50 μg mL−1) and lysozyme (50 μg mL−1) mixture was heated at pH 7.4, 80 °C for 1 h. (f) The residual enzyme activity of HRP (50 μg mL−1) in the absence (control) or presence of self-co-nChap (MSPM-1) or non-specific nChap (MSPM-4) (0.5 mg mL−1) after being heated at pH 7.4, 40 °C for different times. | ||
The ratio of the modification of F-PBA and the ratio between PEG-b-PCL and PDAE-b-PCL on the recovered enzymatic activity was then investigated. The residual HRP activity was first increased and then decreased on increasing the percentage of F-PBA modification from 3% to 20% (Fig. 3b and Table S1, ESI†) while the protein adsorption was gradually increased (Fig. S10a, ESI†). Similar results were obtained by changing the ratio of PEG-b-PCL/PDAE-b-PCL when fixing the percentage of F-PBA modification to 10% (Fig. 3c and Table S1, Fig. S10b, ESI†). These results revealed a sterically synergistic effect between the artificial carrier and confined hydrophobic macrodomains on enhancing the chaperone activity of self-co-nChap for glycoproteins. This was further confirmed using non-glycoprotein lysozyme (Mw = 14.0 kDa and pI = 9.3) as a control. The non-specific nChap (MSPM-4) and self-co-nChap (MSPM-1) exhibited similar chaperone activity for the protection of lysozyme such that the recovered enzymatic activity of lysozyme was 63 ± 2% with the assistance of MSPM-1 compared to that of MSPM-4 (63 ± 3%) (Fig. 3d). Moreover, when the mixture of HRP and lysozyme was heated at 85 °C for 1 h in the presence of MSPM-1, only HRP efficiently refolded with a recovered enzymatic activity of 68 ± 2% while lysozyme failed to refold (Fig. 3e). The effects of self-co-nChap and non-specific nChap on the long-term storage stability of HRP were also investigated by incubating the mixture of HRP and nanochaperones at 40 °C for three weeks. As summarized in Fig. 3f, approximately 38 ± 2% HRP activity was retained without nanochaperones after being stored for 22 days, while the retained enzyme activity was 66 ± 1% in the presence of self-co-nChap and 64 ± 2% in the presence of non-specific nChap.
It is noted that the refolding capability of self-co-nChap was only a 1.03-fold increase compared to that of non-specific nChap for long-term storage of HRP at 40 °C for 22 days while it was a 1.78-fold increase when HRP was heated at 85 °C for 1 h, suggesting that the self-co-nChap was more powerful under harsher conditions. To verify this, we further measured the residual enzymatic activity of HRP after heating HRP at different temperatures using the same procedure as before (Fig. S7, ESI†). As summarized in Fig. 4a, the refolding capability of self-co-nChap was 1.07-, 1.20-, 1.58-, and 2.25-fold increase compared to that of non-specific nChap after heating HRP at 65 °C, 75 °C, 85 °C, and 95 °C, respectively. The effect of periodic temperature changes on protein activity was then investigated as the protein products may suffer from periodic heating and cooling during storage. HRP was heated at 60 °C for 1 h followed by cooling to 25 °C for 1 h, and the heating–cooling cycle was repeated seven times. As shown in Fig. 4b, the residual enzymatic activity slightly decreased (slope: k = −2.653, half-life: t1/2 = 18 cycles) in the presence of self-co-nChap on increasing the cycle-times while decreased significantly for the group of free HRP (k = −9.266, t1/2 = 5 cycles) and non-specific nChap (k = −6.490, t1/2 = 7 cycles) (Fig. S11, ESI†). The residual enzymatic activity of HRP was 78 ± 3%, 52 ± 3%, and 34 ± 2% in the presence of self-co-nChap, non-specific nChap, and PBS, respectively after 7 cycles. In comparison, the decreased rate of the residual enzymatic activity was nearly equal for self-co-nChap (k = −1.532, t1/2 = 33 days) and non-specific nChap (k = −1.633, t1/2 = 31 days) under mild conditions for long-term storage of HRP at 40 °C (Fig. S12, ESI†).
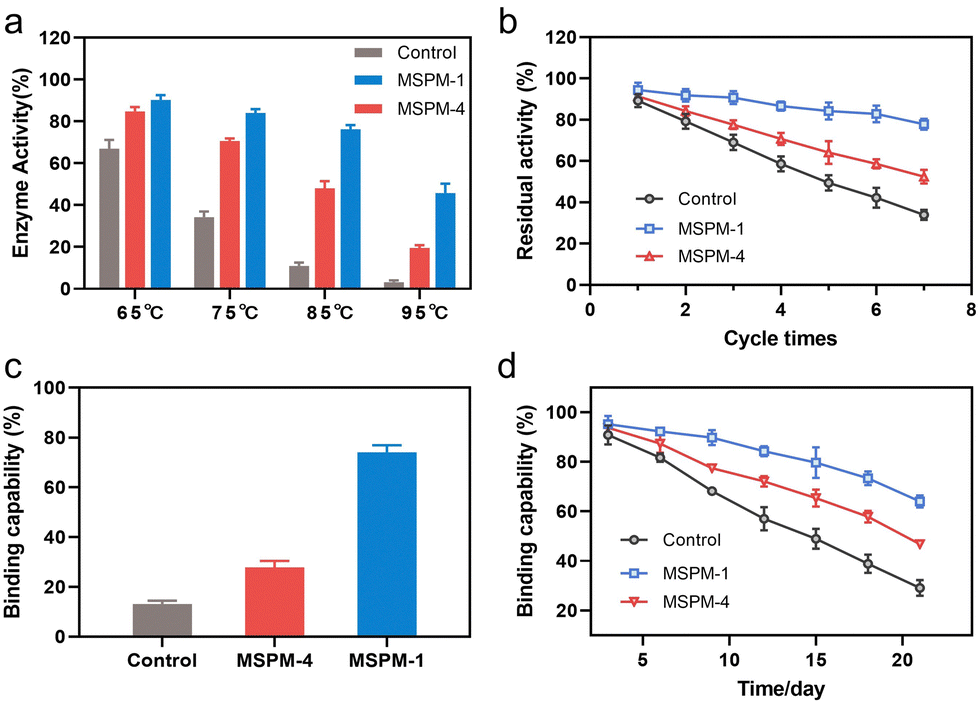 | ||
| Fig. 4 Self-co-nChap enhances the thermal stability of proteins under harsher conditions. (a) The recovered enzyme activity of HRP (0.05 mg mL−1) in the absence (control) or presence of self-co-nChap (MSPM-1) or non-specific nChap (MSPM-4) (0.5 mg mL−1) after being heated at pH 7.4 and different temperatures for 1 h. (b) The residual enzyme activity of HRP (0.05 mg mL−1) in the absence (control) or presence of self-co-nChap (MSPM-1) or non-specific nChap (MSPM-4) (0.5 mg mL−1) after cyclic heating at pH 7.4, 60 °C for 1 h and cooling to 25 °C for 1 h. (c) The residual binding capability of G-IgG (0.05 mg mL−1) in the absence (control) or presence of self-co-nChap (MSPM-1) or non-specific nChap (MSPM-4) (0.5 mg mL−1) after being heated at pH 7.4, 75 °C for 30 min. (d) The residual binding capability of G-IgG (0.05 mg mL−1) in the absence (control) or presence of self-co-nChap (MSPM-1) or non-specific nChap (MSPM-4) (0.5 mg mL−1) after being stored at pH 7.4, 40 °C for different times. | ||
Encouraged by the outstanding functioning of self-co-nChap under harsher conditions, we further investigated its activity for the protection of misfolding-prone proteins. IgG antibodies are the most widely used protein therapeutics, yet with low stability and high propensity to undergo misfolding and aggregation.35 The Goat-anti bull serum albumin (BSA) IgG polyclonal antibody (G-IgG) was used as a model protein whose activity could be measured by evaluating its binding capability against BSA using enzyme-linked immunosorbent assay (ELISA) assay.36 G-IgG in the presence or absence of nanochaperones was either heated at 75 °C for 30 min or at 40 °C for 3 weeks. As shown in Fig. 4c, the binding capability of G-IgG was only 13 ± 1% left after heating in the absence of nanochaperones while was 74 ± 3% left with the protection of self-co-nChap, which was 2.6-fold higher than that of non-specific nChap (28 ± 3%). Self-co-nChap was also more efficient in protecting G-IgG during long-term storage than non-specific nChap. The residual binding capability of G-IgG was 64 ± 2%, 47 ± 1%, and 29 ± 3% in the presence of self-co-nChap, non-specific nChap, and PBS, respectively after heating at 40 °C for 3 weeks (Fig. 4d). Moreover, the decreased rate of the residual binding capability was slower for self-co-nChap (k = −1.684, t1/2 = 31 days) than that for non-specific nChap (k = −2.525, t1/2 = 21 days) (Fig. S13, ESI†). These results suggest that self-co-nChap can efficiently protect misfolding-prone antibodies from thermal denaturation, which is hardly achieved using non-specific nanochaperones.
HRP is a highly selective and efficient biocatalyst for the removal of toxic phenolic compounds from wastewater.37 Here, we further investigated the effect of self-co-nChap on the protection of HRP in the removal of phenolic compounds from water. The effect of self-co-nChap on the reaction kinetics of HRP for degradation of phenolic compounds under mild conditions (25 °C) was first investigated by measuring the initial reaction rates using different concentrations of hydrogen peroxide (H2O2) and guaiacol as substrates. The enzyme kinetics were presented in the form of a Lineweaver–Burk plot (Fig. S14, ESI†). As listed in Table 1, the Michaelis–Menten constant (Km) for the group of HRP@self-co-nChap is 1.4-fold and 1.7-fold higher for the degradation of guaiacol and H2O2, respectively than that of free HRP, indicating that the self-co-nChap interferes with the binding of HRP to its substrate. While the maximum reaction rate (Vmax) for the group of HRP@self-co-nChap is 1.9-fold and 2.0-fold higher for the degradation of guaiacol and H2O2, respectively than that of free HRP, indicating a higher reactivity of HRP@self-co-nChap in the presence of sufficient substrate.
| Parameter | Free HRP | HRP@self-co-nChap | ||
|---|---|---|---|---|
| Guaiacol | H2O2 | Guaiacol | H2O2 | |
| K m (Mm) | 13.29 | 0.56 | 18.84 | 0.93 |
| V max (106, U g−1) | 1.62 | 1.21 | 3.05 | 2.37 |
We then investigated the removal efficiency of phenolic compounds in water by HRP and HRP@self-co-nChap at harsh temperatures, respectively. The entire reaction process was monitored until the removal efficiency was saturated at 120 min. The initial removal efficiency is defined as the percentage of phenolic compounds removed within 1 minute of the start of the reaction. The final removal efficiency is the final percentage of phenolic compounds removed after the reaction is terminated. As shown in Fig. 5a, the initial removal efficiency of phenol was firstly increased and then decreased on increasing the temperature from 20 to 65 °C either for the group of free HRP or HRP@self-co-nChap. Notably, the initial removal efficiency of phenol began decreasing at 50 °C for the group of free HRP while began dropping at 60 °C for the group of HRP@self-co-nChap, indicating that self-co-nChap enhances the thermal stability of HRP and inhibits its denaturation. This was further confirmed by the final removal efficiency of phenol by free HRP or HRP@self-co-nChap, respectively. The final removal efficiency of phenol was stable for HRP@self-co-nChap (∼90%) on increasing the temperature from 20 to 50 °C while it was gradually reduced for free HRP (Fig. 5b). Similar results were also obtained for the removal of dichlorophenol (Fig. 5c and d).
 | ||
| Fig. 5 Self-co-nChap enhances the thermal stability of HRP for the removal of phenolic compounds. (a) and (b) The initial removal efficiency (a) and the final removal efficiency (b) of phenol by free HRP or HRP@self-co-nChap (MSPM-1) at different temperatures. (c) and (d) The initial removal efficiency (c) and the final removal efficiency (d) of dichlorophenol by free HRP or HRP@self-co-nChap (MSPM-1) at different temperatures. | ||
Protein folding is thermodynamically driven and kinetically controlled.38,39 For de novo protein folding, natural chaperones promote folding by a generic mechanism of kinetic partitioning.40 Here, we expanded this kinetic partitioning mechanism to the form of protection of native proteins from thermal denaturation by nanochaperones. As shown in Fig. 6, native proteins will unfold upon heating, and the higher the temperature, the faster the protein unfolds.41 Subsequently, the partially unfolded (PU) states of proteins were represented as PU-1, PU-2, and PU-3. The rate of (re)folding of PU-1 to native states (kfold-1) is faster than that of PU-2 (kfold-2) and kfold-2 is faster than that of PU-3 (kfold-3) (kfold-1 > kfold-2 > kfold-3), while the rate of aggregation of PU-1 to aggregates (kagg-1) is slower than that of PU-2 (kagg-2) and kagg-2 is slower than that of PU-3 (kagg-3) (kagg-1 < kagg-2 < kagg-3). Refolding only proceeds efficiently when kfold is greater than kagg. Therefore, native proteins tend to unfold and aggregate at harsh temperatures without nanochaperones. The binding of protein clients to nanochaperones blocks aggregation and reduces the concentration of aggregation-prone clients. Release from the nanochaperone allows refolding. We hypothesize that increasing the rate constant of binding of protein clients to the nanochaperone (kon) will enhance its chaperone activity for two reasons: (i) the faster binding ensures kon is greater than kagg to inhibit aggregation, especially under harsher conditions which is necessary for protein refolding; (ii) this promotes the immobilization of protein clients by nanochaperones which inhibits unfolding of trapped clients when heated at higher temperatures, ensuring that kfold of the released protein clients is greater than kagg after cooling, thereby facilitating refolding. Moreover, the gradual release of unfolded clients from the hydrophobic macrodomains driven by the phase transition of pH-responsive polymers may also partially facilitate refolding, thereby increasing kfold of the released clients, which is an advantage of the unique nanostructure of the MSPM-based nanochaperone.
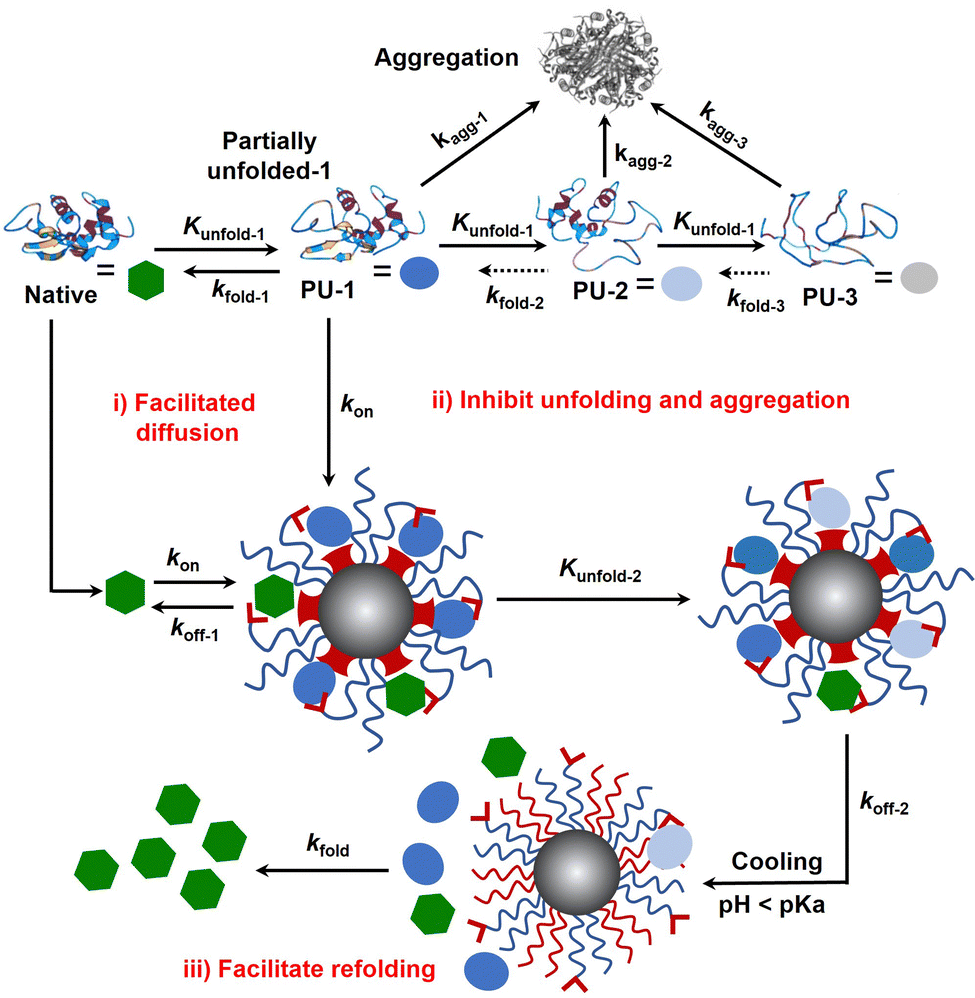 | ||
| Fig. 6 Schematic illustration of self-co-nChap facilitates refolding of thermally denatured proteins by a mechanism of kinetic partitioning of native and unfolded clients. Native protein partially unfolds during heating and the partially unfolded proteins irreversibly aggregate and lose their activity. Binding to the nanochaperone blocks aggregation and reduces the concentration of aggregation-prone clients. Release from the nanochaperone allows refolding. Compared to non-specific nChap, self-co-nChap increases the rate constant of binding of protein clients (native and unfolded) to nanochaperone (kon), ensuring that kon is faster than the rate constant of aggregation (kagg). More importantly, this promotes the trapping of protein clients to the hydrophobic macrodomains of self-co-nChap, slowing the rate of unfolding (kunfold-2) of trapped clients by a manner of immobilization, which ensures that the rate constant of folding (kfold) of released protein clients is faster than kagg after cooling, thereby facilitating refolding. The gradual release of unfolded proteins from the hydrophobic macrodomains driven by the phase transition of pH-responsive polymers may also partially facilitate refolding, increasing kfold. | ||
Since the first protein structure was published by Kendrew and co-workers in 1958,42 protein folding has become a quintessential basic science, hence deriving the research field of how to regulate protein folding. Regulating protein folding including assisting de novo folding of nascent polypeptide, protecting native proteins from unfolding and facilitating refolding, and preventing structurally unstable proteins from misfolding and aggregating, are of significant importance for both biomedical and industrial applications. In this work, we designed and fabricated a self-co-nChap for the thermal protection of glycoproteins by introducing a PBA artificial carrier to the shell surface of non-specific nanochaperone (Fig. 1). We demonstrated that compared with non-specific nChap, this artificial carrier facilitates the diffusion and trapping of native HRP into the confined hydrophobic microdomains of self-co-nChap (Fig. 2). The artificial carrier may increase the kon, thus facilitating diffusion and immobilization of clients by two mechanisms: (i) when the clients collide with the outstretched hydrophilic PEG shell, the artificial carrier facilitates translocation of clients into the hydrophobic microdomains, which is like the mechanism of chaperone network and facilitated diffusion; (ii) when the clients collide or are transferred into the hydrophobic microdomains, the artificial carrier assists the immobilization of clients on the surface of hydrophobic microdomains (Fig. 6(i)). The results of protecting native HRP from thermal denaturation confirmed the important role of this artificial carrier (Fig. 3), and it is noted that, compared to our previous works24,27, the best ratio of PEG-b-PCL/PDAE-b-PCL for thermal protection of proteins was increased from 1/6 to 1/1, which is conducive to increasing the stability of MSPMs. Furthermore, self-co-nChap was more powerful than non-specific nChap for the protection of HRP under harsher conditions, or for the protection of IgG with a higher propensity to undergo misfolding and aggregation (Fig. 4), confirming the importance of this artificial carrier-provided cooperative effect. In addition, our results showed that self-co-nChap does not affect the activity of native HRP under normal conditions (Table 1), which may be due to the fast kon and the fast rate constant of the release of native HRP to nanochaperone (koff-1) (Fig. 6(i)). Moreover, our results indicated that trapping of native HRP stabilized native HRP's structure and inhibited its unfolding (Fig. 5 and 6(ii)). This is important to ensure the kfold of the released protein clients is greater than kagg after cooling, thereby facilitating refolding (Fig. 6(iii)). Despite the above advantages of the increased kon, this may lead to a concern about the insufficient release of unfolded clients from nanochaperones, which limits protein refolding according to our previous findings.23,24 However, different from the electrostatic interactions, the diester bond formed between boronic acid and the cis-1,2-diol group of glycoprotein is reversible and can break under acidic conditions, which should have negligible effects on the rate constant of release of clients from self-co-nChap at pH 5.0 (koff-2).
Conclusions
In summary, here we come up with a novel strategy to enhance the chaperone activity of nanochaperones to protect native proteins from thermal-induced unfolding and facilitating refolding. An artificial carrier is introduced into the nanochaperone systems, for the first time mimicking the cooperative effect of co-chaperones (e.g., Hsp40) to facilitate the transfer and immobilization of clients into the Hsp70-mimetic confined hydrophobic microdomains. Moreover, we for the first time expanded the kinetic partitioning mechanism of de novo protein folding assisted by natural chaperones to the form of protection of native proteins from thermal denaturation by nanochaperones. Despite the fact that we need further study to completely illustrate the (re)folding kinetics regulated by self-co-nChap, we believe that this kinetic partitioning mechanism is universal for different nanochaperone systems and should provide a direction to design new protein folding regulatory materials with enhanced chaperone activity.Experimental section
See ESI.†Author contributions
Conceptualization, L. S. and F. M.; methodology, M. Y. and F. M.; investigation, M. Y., Y. Z., F. D., X. W., Y. C.; writing – original draft, F. M. and M. Y.; writing – review & editing, L. S. and F. M.; funding acquisition, L. S. and F. M.; supervision, L. S. and F. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (Grant Number 51933006, 52103180, and 21991081) and Haihe Laboratory of Sustainable Chemical Transformations (Project number: YYJC202102).References
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042–1046 CrossRef CAS PubMed.
- L. Cao, B. Coventry, I. Goreshnik, B. Huang, W. Sheffler, J. S. Park, K. M. Jude, I. Marković, R. U. Kadam, K. H. G. Verschueren, K. Verstraete, S. T. R. Walsh, N. Bennett, A. Phal, A. Yang, L. Kozodoy, M. DeWitt, L. Picton, L. Miller, E. M. Strauch, N. D. DeBouver, A. Pires, A. K. Bera, S. Halabiya, B. Hammerson, W. Yang, S. Bernard, L. Stewart, I. A. Wilson, H. Ruohola-Baker, J. Schlessinger, S. Lee, S. N. Savvides, K. C. Garcia and D. Baker, Nature, 2022, 605, 551–560 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- F. H. Ma, C. Li, Y. Liu and L. Shi, Adv. Mater., 2020, 32, 1805945 CrossRef CAS PubMed.
- A. B. Caballero and P. Gamez, Angew. Chem., Int. Ed., 2021, 60, 41–52 CrossRef CAS PubMed.
- N. Pradhan, K. Debnath, S. Mandal and N. R. Jana, Biomacromolecules, 2018, 19, 1721–1731 CrossRef CAS PubMed.
- F. Huang, J. Wang, A. Qu, L. Shen, J. Liu, J. Liu, Z. Zhang, Y. An and L. Shi, Angew. Chem., Int. Ed., 2014, 126, 9131–9136 CrossRef.
- X. Wu, F. Ma, B. B. Pan, Y. Zhang, L. Zhu, F. Deng, L. Xu, Y. Zhao, X. Yin, H. Niu, X. Su and L. Shi, Angew. Chem., Int. Ed., 2022, 61, e202200192 CrossRef CAS PubMed.
- T. Nishimura and K. Akiyoshi, Bioconjugate Chem., 2020, 31, 1259–1267 CrossRef CAS PubMed.
- O. Hanpanich and A. Maruyama, Biomaterials, 2020, 254, 120150 CrossRef CAS PubMed.
- D. Muraoka, N. Harada, H. Shiku and K. Akiyoshi, J. Controlled Release, 2022, 347, 175–182 CrossRef CAS PubMed.
- Y. Zhang, J. Chen, L. Shi and F. Ma, Mater. Horiz., 2023, 10, 361–392 RSC.
- A. Das, A. Yadav, M. R. P. Gupta, V. L. Terse, V. Vishvakarma, S. Singh, T. Nandi, A. Banerjee, K. Mandal, S. Gosavi, R. Das, S. R. K. Ainavarapu and S. Maiti, J. Am. Chem. Soc., 2021, 143, 18766–18776 CrossRef CAS PubMed.
- S. Sadeghi, S. Deshpande, G. Vallerinteavide Mavelli, A. Aksoyoglu, J. Bafna, M. Winterhalter, R. M. Kini, D. P. Lane and C. L. Drum, Nat. Commun., 2021, 12, 5720 CrossRef CAS PubMed.
- Y. D. Alvarez, J. A. Fauerbach, J. V. Pellegrotti, T. M. Jovin, E. A. Jares-Erijman and F. D. Stefani, Biophys. J., 2014, 106, 257a–258a CrossRef.
- G. Gao, X. Liu, Z. Gu, Q. Mu, G. Zhu, T. Zhang, C. Zhang, L. Zhou, L. Shen and T. Sun, Nano Lett., 2022, 22, 2964–2970 CrossRef CAS PubMed.
- J. M. Beierle, K. Yoshimatsu, B. Chou, M. A. A. Mathews, B. K. Lesel and K. J. Shea, Angew. Chem., Int. Ed., 2014, 126, 9275–9279 CrossRef PubMed.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS PubMed.
- N. Martin, M. Li and S. Mann, Langmuir, 2016, 32, 5881–5889 CrossRef CAS PubMed.
- M. Hayer-Hartl, A. Bracher and F. U. Hartl, Trends Biochem. Sci., 2016, 41, 62–76 CrossRef CAS PubMed.
- N. Kameta, M. Masuda and T. Shimizu, ACS Nano, 2012, 6, 5249–5258 CrossRef CAS PubMed.
- D. Fujita, R. Suzuki, Y. Fujii, M. Yamada, T. Nakama, A. Matsugami, F. Hayashi, J. K. Weng, M. Yagi-Utsumi and M. Fujita, Chemistry, 2021, 7, 2672–2683 CrossRef CAS.
- F. H. Ma, Y. An, J. Wang, Y. Song, Y. Liu and L. Shi, ACS Nano, 2017, 11, 10549–10557 CrossRef CAS PubMed.
- F. Ma, X. Wu, A. Li, L. Xu, Y. An and L. Shi, Angew. Chem., Int. Ed., 2021, 60, 10865–10870 CrossRef CAS PubMed.
- X. Li, Y. Zhang, X. Wu, J. Chen, M. Yang, F. Ma and L. Shi, Small, 2022, 18, 2203100 CrossRef CAS PubMed.
- Y. Zhang, H. Fu, J. Chen, L. Xu, Y. An, R. Ma, C. Zhu, Y. Liu, F. Ma and L. Shi, Small Methods, 2022, 2201051 Search PubMed.
- X. Liu, Y. Liu, Z. Zhang, F. Huang, Q. Tao, R. Ma, Y. An and L. Shi, Chem. – Eur. J., 2013, 19, 7437–7442 CrossRef CAS PubMed.
- D. Balchin, M. Hayer-Hartl and F. U. Hartl, Science, 2016, 353, aac4354 CrossRef PubMed.
- D. W. Summers, P. M. Douglas, C. H. I. Ramos and D. M. Cyr, Trends Biochem. Sci., 2009, 34, 230–233 CrossRef CAS PubMed.
- P. A. Gale, R. Pérez-Tomás and R. Quesada, Acc. Chem. Res., 2013, 46, 2801–2813 CrossRef CAS PubMed.
- L. Angenendt, J. H. Mikesch and C. Schliemann, Cancer Treat. Rev., 2022, 108, 102409 CrossRef CAS PubMed.
- Y. Q. Yao, Y. Q. Xu and Q. X. Li, J. Agric. Food Chem., 2022, 70, 646–655 CrossRef CAS PubMed.
- C. Li, X. Liu, Y. Zhang, J. Lv, F. Huang, G. Wu, Y. Liu, R. Ma, Y. An and L. Shi, Nano Lett., 2020, 20, 1755–1765 CrossRef CAS PubMed.
- M. Li, A. Lee, K. L. Kim, J. Murray, A. Shrinidhi, G. Sung, K. M. Park and K. Kim, Angew. Chem., Int. Ed., 2018, 57, 2120–2125 CrossRef CAS PubMed.
- E. Y. Chi, S. Krishnan, T. W. Randolph and J. F. Carpenter, Pharm. Res., 2003, 20, 1325–1336 CrossRef CAS PubMed.
- Y. Feng, H. Wang, S. Zhang, Y. Zhao, J. Gao, Y. Zheng, P. Zhao, Z. Zhang, M. J. Zaworotko, P. Cheng, S. Ma and Y. Chen, Adv. Mater., 2019, 31, 1805148 CrossRef PubMed.
- S. Liu, B. Huang, G. Zheng, P. Zhang, J. Li, B. Yang, Y. Chen and L. Liang, Int. J. Biol. Macromol., 2020, 150, 814–822 CrossRef CAS PubMed.
- R. Nassar, G. L. Dignon, R. M. Razban and K. A. Dill, J. Mol. Biol., 2021, 433, 167126 CrossRef CAS PubMed.
- D. Baker and D. A. Agard, Biochemistry, 1994, 33, 7505–7509 CrossRef CAS PubMed.
- Y. E. Kim, M. S. Hipp, A. Bracher, M. Hayer-Hartl and F. Ulrich Hartl, Annu. Rev. Biochem., 2013, 82, 323–355 CrossRef CAS PubMed.
- R. Day, B. J. Bennion, S. Ham and V. Daggett, J. Mol. Biol., 2002, 322, 189–203 CrossRef CAS PubMed.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H. Wyckoff and D. C. Phillips, Nature, 1958, 181, 662–666 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00619k |
| This journal is © The Royal Society of Chemistry 2023 |