
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFusogenic liposome-coated nanoparticles for rapid internalization into donor corneal endothelial tissue to enable prophylaxis before transplantation†
Thanuja
M. Y.‡
,
Suraksha S.
Tellakula‡
,
Samarth V.
Suryavanshi
,
Keerthana
G. S.
,
Chandan
Vasudev S.
and
Sudhir H.
Ranganath
 *
*
Bio-INvENT Lab, Department of Chemical Engineering, Siddaganga Institute of Technology, B. H. Road, Tumakuru 572103, India. E-mail: sudhirh@sit.ac.in; Tel: +91 816 2214038
First published on 10th November 2023
Abstract
Cold stress (hypothermia) during storage and cytokine stress due to acute allograft rejection adversely affect the donor corneal endothelium in the short term. Pharmacological pre-treatment (before transplantation) of the donor corneal endothelium or cells (propagated in vitro for cell injection therapy) with microtubule stabilizers, cold stress protectants, and other molecules is an attractive strategy to tackle damage caused by hypothermia and cytokine stress. These molecules can be delivered intracellularly to the donor corneal endothelium or cells at controlled rates for desired periods and with one-time administration using nanoparticles. However, the death-to-preservation time of donor corneas of more than 4 to 6 h significantly decreases endothelial cell density and increases the risk of microbial contamination. Therefore, we have developed fusogenic liposome-coated nanoparticles for rapid internalization of nanoparticles into cultured corneal endothelial cells and ex vivo corneal endothelial tissue. Here, we have shown that the fusogenic liposome-coated nanoparticles have the intrinsic ability to efficiently and rapidly internalize into cultured corneal endothelial cells and ex vivo corneal tissue within 3 h by possibly fusing with the cell membrane and bypassing the endocytic pathway. Lactate dehydrogenase assay showed that the internalized fusogenic liposome-coated nanoparticles did not cause cytotoxicity in endothelial cells associated with the ex vivo cornea for at least up to 2 days. Thus, fusogenic liposome-coated nanoparticles have great potential as a platform for engineering cells and endothelial tissue of donor corneas to facilitate prophylactic drug delivery during storage and after transplantation.
Introduction
Corneal blindness is the third most prevalent eye disease worldwide and leads to visual impairment. The demand for corneal transplantation is increasing every year.1 Only 1 in 70 people gets access to donor corneas for transplantation.2 Annually, more than 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 corneas are being transplanted in the USA;3 however, the failure rate of corneal transplantation is >30%.4 When the death-to-preservation-time of donor corneas is more than 4 to 6 h, a significant reduction in the corneal endothelial cell density (ECD) and higher risk of microbial contamination have been reported.5 A loss in ECD by >30% is reported within 1–2 years following corneal or endothelial transplantation leading to graft failure.4,6 In addition to donor age, other factors for rejection of the allograft and failure of transplantation include repeat transplantation, infections and neovascularization in the recipient or donor corneas.7 The loss is contributed by many factors such as cold storage of donor corneas (hypothermic stress)8,9 and stress induced by cytokines secondary to inflammation post-transplantation,10,11 and acute rejection of the allograft. ECD decreases with age, and hence donor corneas (mostly from older donors) have a propensity for rapidly reaching the threshold ECD needed to maintain hydration in the stroma.12,13 Therefore, it is critical to preserve ECD during corneal storage and safeguard corneal endothelial cells from cytokine stress after transplantation.
000 corneas are being transplanted in the USA;3 however, the failure rate of corneal transplantation is >30%.4 When the death-to-preservation-time of donor corneas is more than 4 to 6 h, a significant reduction in the corneal endothelial cell density (ECD) and higher risk of microbial contamination have been reported.5 A loss in ECD by >30% is reported within 1–2 years following corneal or endothelial transplantation leading to graft failure.4,6 In addition to donor age, other factors for rejection of the allograft and failure of transplantation include repeat transplantation, infections and neovascularization in the recipient or donor corneas.7 The loss is contributed by many factors such as cold storage of donor corneas (hypothermic stress)8,9 and stress induced by cytokines secondary to inflammation post-transplantation,10,11 and acute rejection of the allograft. ECD decreases with age, and hence donor corneas (mostly from older donors) have a propensity for rapidly reaching the threshold ECD needed to maintain hydration in the stroma.12,13 Therefore, it is critical to preserve ECD during corneal storage and safeguard corneal endothelial cells from cytokine stress after transplantation.
Cold storage (hypothermic stress)9 and cytokine stress10 secondary to acute allograft rejection damage the donor corneal endothelium in the short term. Specifically, we have shown that exposure to hypothermia induces microtubule disassembly and disruption of the perijunctional actomyosin ring (PAMR) and leads to a loss of barrier function in cultured corneal endothelial cells and ex vivo corneal endothelium.9,14 In addition, TNF-α (tumor necrosis factor-α), a pro-inflammatory cytokine secreted during allograft rejection, induces disassembly of microtubules and loss of barrier function.10 Pharmacological pre-treatment of the donor corneal endothelium with microtubule stabilizers,11 Rho kinase inhibitors,15 cold stress protectants,16 anti-apoptotic genes17 and their sustained intracellular delivery are imperative to preserve its functionality (barrier function), cell density and tackle potential adverse effects induced by hypothermia during storage and cytokine stress after transplantation.
Studies on preservation of donor corneas have shown that a tissue storage time > 12 days, status of donor's health (diabetes, etc.) and other factors result in graft failure.12,18 Alternative to using donor corneal tissues, scalable cell-based therapies (cell injection approach) using human corneal endothelial cells (HCECs) propagated in vitro15 and/or 3D bioprinted corneas19 are currently being developed. Corneal endothelial cells isolated from a single donor can be administered to up to 80 patients by in vitro expansion using an appropriate medium20 and can be administered through direct cell injection into the anterior chamber.15 Despite these promises, the limitations of cell-based therapy include unbalanced cell distribution and lower persistence and repair efficiency. Reasons include the loss of cell surface proteins during trypsinization which may reduce cell–cell and cell–extracellular matrix interactions,21 cytokine stress10,22 post transplantation and oxidative stress-induced cell death,23,24 all leading to reduced efficiency to form functional monolayers. In this context, nanotechnology-based prophylactic drug delivery to in vitro expanded donor corneal endothelial cells can overcome these limitations and facilitate successful cell therapies for corneal diseases.
Therapeutic molecules including cold stress protectants, antioxidants, anti-apoptotic or pro-survival genes, microtubule stabilizers, iron chelators, and kinase inhibitors can be delivered intracellularly at controlled rates for desired periods of time using nanoparticles. Recently, we have shown that intracellular delivery of microtubule-stabilizing drug epothilone-B using poly-L-lysine (PLL)-coated poly-L-lactic-co-glycolic acid (PLGA) nanoparticles demonstrated stabilization of microtubules with no toxicity and opposed cold stress-induced PAMR damage and ZO-1 redistribution.9 However, the uptake of these nanoparticles by corneal endothelial cells took very long (up to 24 to 48 h). Moreover, there was scant evidence of their complete internalization due to multiple factors including agglomeration and the slow and energy-dependent endocytosis of charged nanoparticles.25 Most surface modified nanoparticles are internalized by endocytosis during which they are entrapped by the plasma membrane leading to a delay in the uptake. It is noteworthy that the reported time for uptake of charged nanoparticles is significantly longer than the death-to-preservation-time of donor corneas of about 4–6 h.5,12 Thus, a nanoparticle system for rapid uptake (within 3 to 4 h) into the donor corneal endothelium (for transplantation) and donor corneal endothelial cells (for cell injection therapy) is an urgent unmet need to protect the health and functionality of the donor tissue/cells.
A process of cellular uptake which can bypass the endocytic pathway would hasten the cytosolic delivery of nanoparticles. A class of cationic liposomes called fusogenic liposomes directly fuse with the cell membrane (membrane fusion is a non-endocytic process). Fusion enhances the interaction between fusogenic liposomes and the cell membrane for delivering therapeutic molecules directly and rapidly into the cytoplasm. Hence, we hypothesized that fusogenic liposomes coated on the PLGA nanoparticle surface would hasten cellular internalization. Accordingly, in this study, we have developed a simple and novel strategy to significantly hasten the internalization kinetics of nanoparticles in the donor corneal endothelial tissue and in vitro expanded cells. We report the preparation of fusogenic liposome-coated nanoparticles and assess their internalization in ex vivo corneal tissue and cultured cells. The novelty of this approach is that we have employed the membrane fusion properties of the fusogenic liposomes to achieve rapid tissue and cellular uptake of PLGA nanoparticles.
Results and discussion
We first prepared DiO (3,3′-dioctadecyloxacarbocyanine perchlorate, green fluorescent dye)-loaded nanoparticles (DNPs) using the PLGA polymer as the carrier via the nanoprecipitation method.9 DiO was used to enable visualization of the nanoparticles in the tissue and cells using confocal fluorescence microscopy. Size, surface charge, and surface chemistry of the nanoparticles affect kinetics of cellular uptake. Thus, for the preparation of nanoparticles and to engineer them for sustained delivery of drugs and uptake by donor endothelium, we need a biodegradable and biocompatible material. PLGA is a polymer present in several FDA-approved carrier systems for delivery of drugs, proteins/peptides, genes, and siRNA26 and it is also biocompatible and biodegradable. Thus, we chose to prepare and modify PLGA nanoparticles in this study. Uniformly sized spherical DNPs were obtained by the nanoprecipitation method as shown in the TEM micrograph (Fig. 1A).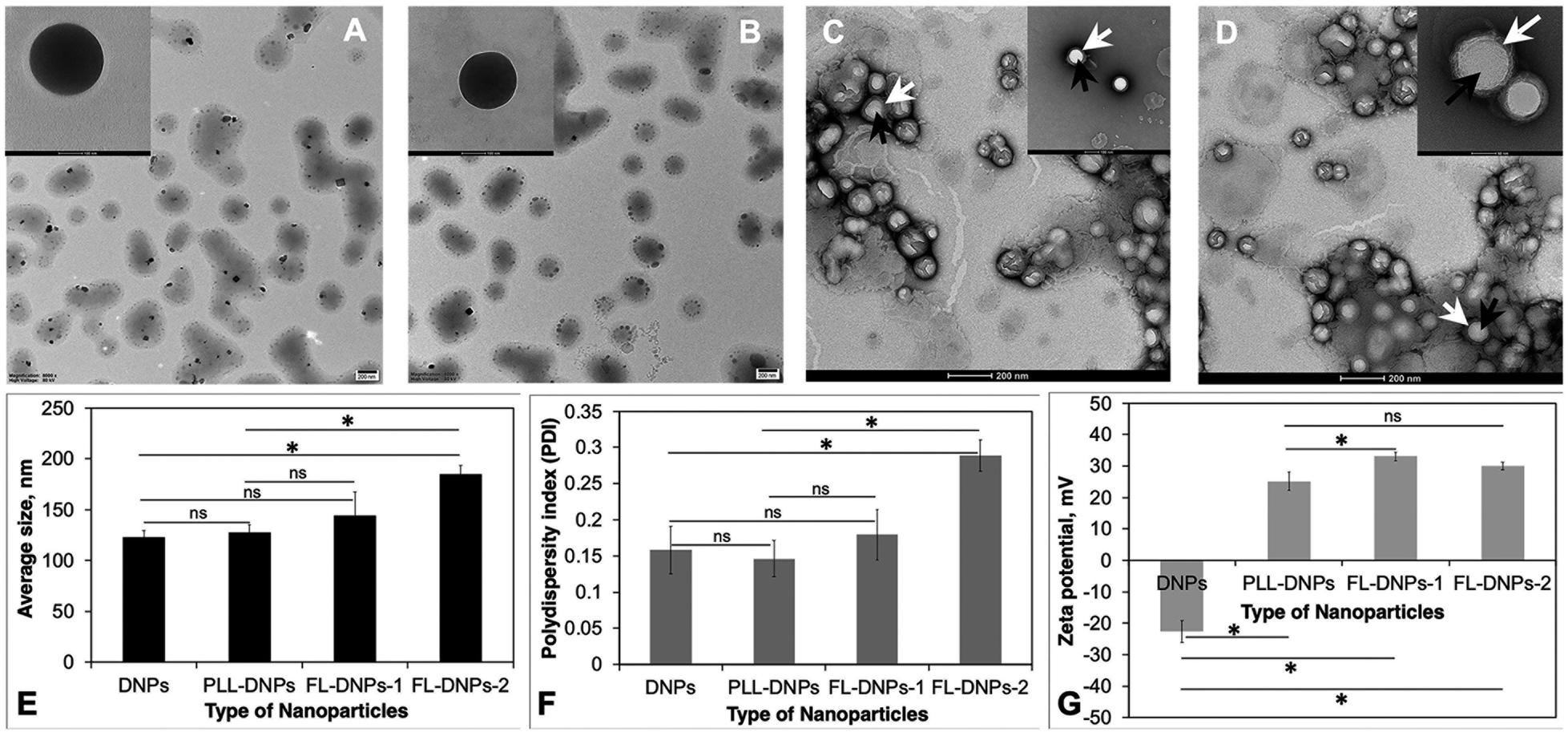 | ||
| Fig. 1 Size, size distribution, surface charge, morphology and surface coating of the nanoparticles. Representative TEM micrographs of (A) DiO-loaded PLGA nanoparticles (DNPs) (scale bar is 200 nm). Inset: the scale bar is 100 nm; (B) PLL-coated DiO-loaded PLGA nanoparticles (PLL-DNPs) (scale bar is 200 nm). Inset: the scale bar is 100 nm; (C) DiO-loaded PLGA nanoparticles coated with DOTAP + DOPE fusogenic liposomes (FL-DNPs-1) (scale bar is 200 nm). Inset: the scale bar is 100 nm; (D) DiO-loaded PLGA nanoparticles coated with DOTAP + SoyaPC fusogenic liposomes (FL-DNPs-2) (scale bar is 200 nm). Inset: the scale bar is 50 nm. Images (C) and (D) were visualized by using uranyl acetate negative staining at a resolution of 100 nm and 50 nm, respectively. (E) Average size of nanoparticles. (F) Polydispersity index (PDI) of nanoparticles. (G) Zeta potential (surface charge) of nanoparticles. Data represent the average of triplicates. * indicates statistical significance (p < 0.05) and “ns” indicates not significant (p > 0.05). | ||
In a recent study, we reported that epothilone B-loaded PLGA nanoparticles (ENPs) possessed a negative surface charge and PLL modification (PLL-ENPs) led to a positive surface charge without significantly affecting the size of the nanoparticles.9 As expected, the positive charge led to an enhanced kinetics of internalization of PLL-ENPs in cultured cells. This is an essential consideration since faster internalization will decrease the risk of loss of cell viability due to long-term exposure at 37 °C. However, our results showed that incubation for 6 to 24 h with PLL-ENPs would not be adequate to obtain significant internalization. This result has clinical implications since a death-to-preservation time of >4–6 h for donor corneas leads to a decrease in endothelial cell viability and enhances the microbial contamination risk. Therefore, we next aimed at developing a nanoparticle system which could be taken up by donor cells/tissue in about 3 h at 37 °C.
Based on this, the nanoparticles (DNPs) were surface modified using fusogenic liposomes made from DOTAP (dioleoyl-3-trimethylammonium propane) + DOPE (dioleoylphosphatidylethanolamine) (FL-DNPs-1) and DOTAP + SoyaPC (soya phosphatidylcholine) (FL-DNPs-2). We also surface modified the nanoparticles with PLL (PLL-DNPs) for comparison with fusogenic liposome-coated nanoparticles. As seen in Fig. 1B–D, these nanoparticles were also spherical in shape. Fig. 1C and D illustrate that the fusogenic liposomes were uniformly coated on the nanoparticles (white arrow represents liposomes; black arrows represent nanoparticles) with a core–shell structure. The average size of DNPs, PLL-DNPs, FL-DNPs-1, and FL-DNPs-2 measured by dynamic light scattering analysis was 122 ± 7 nm, 128 ± 7 nm, 145 ± 20 nm, and 185 ± 8 nm (Fig. 1E), respectively. Statistical analysis confirms that the modification with PLL did not significantly alter the nanoparticle size (p = 0.39). The fusogenic liposome coating on DNPs increased the nanoparticle size in the range of 25 to 60 nm. However, the increase in size is not significant for FL-DNPs-1 (p = 0.18). On the other hand, the increase in NP size is significant for FL-DNPs-2 (p = 0.0005). These data indicate that size of the surface coated nanoparticle depends on the lipid composition of the fusogenic liposomes. In comparison to PLL-DNPs, the fusogenic liposomes were bigger, but the increase was nonsignificant for FL-DNPs-1 (p = 0.29), but significant for FL-DNPs-2 (p = 0.0007).
The polydispersity indices of DNPs, PLL-DNPs, FL-DNPs-1, and FL-DNPs-2 were 0.16 ± 0.03, 0.15 ± 0.02, 0.18 ± 0.03, and 0.29 ± 0.02, respectively, suggesting monodispersity of the nanoparticles (Fig. 1F). The small values of PDI indicate that the nanoprecipitation method as well as lipid hydration followed by serial extrusion resulted in a uniform size distribution of the nanoparticles. Statistical analysis confirms that PLL coating did not significantly alter the size distribution (p = 0.69). The fusogenic liposome coating on DNPs increased the PDI. However, the increase is not significant for FL-DNPs-1 (p = 0.52). On the other hand, the increase in the PDI is significant for FL-DNPs-2 (p = 0.0055). These data indicate that size distribution of the surface coated nanoparticle depends on the lipid composition of the fusogenic liposomes. In comparison to PLL-DNPs, the fusogenic liposomes had relatively wider size distribution, but the increase was nonsignificant for FL-DNPs-1 (p = 0.28), but significant for FL-DNPs-2 (p = 0.0017). Despite an increase in the PDI upon coating, the change was well within the acceptable limit of 0.2 to 0.3, and it represents relatively homogeneous size distribution.
Zeta potential (surface charge) of DNPs, PLL-DNPs, FL-DNPs-1, and FL-DNPs-2 was −22.7 ± 3.4 mV, 25.1 ± 2.9, 33.3 ± 1.3, and 29.3 ± 1.3 respectively, indicating nanoparticle stability in suspension (Fig. 1G). More importantly, the positive surface charge of PLL-DNPs, FL-DNPs-1 and FL-DNPs-2 confirms that PLL, (DOTAP + DOPE) and (DOTAP + SoyaPC) were coated on the nanoparticle surface, respectively, since PLL and DOTAP are cationic molecules. The reversal in surface charge of all three types of coated nanoparticles was significant in comparison to that of uncoated nanoparticles (p = 0.00005 for PLL-DNPs, p = 0.000012 for FL-DNPs-1 and p = 0.000015 for FL-DNPs-2). However, the change in surface charge for PLL-DNPs was not significant against FL-DNPs-2 (p = 0.055).
Engineering in vitro cultured corneal endothelial cells with nanoparticles before exogenous administration into the recipient's eye is a futuristic approach for enhancing the efficacy of cell therapy. In this context, we investigated the internalization of nanoparticles in cultured porcine corneal endothelial cells in vitro. Our goal was to investigate if fusogenic liposome-coated DNPs are rapidly internalized into the cells in comparison to bare nanoparticles (DNPs) and PLL-DNPs. Cultured porcine corneal endothelial cells were incubated with DNPs, PLL-DNPs, FL-DNPs-1, and FL-DNPs-2 for different intervals of time (3 and 6 h). The internalization of DNPs in cultured corneal endothelial cells was then qualitatively assessed by imaging the green fluorescence of DiO (representing DNPs), red fluorescence (representing the cell membrane) and blue fluorescence (representing the nucleus) as shown in Fig. 2.
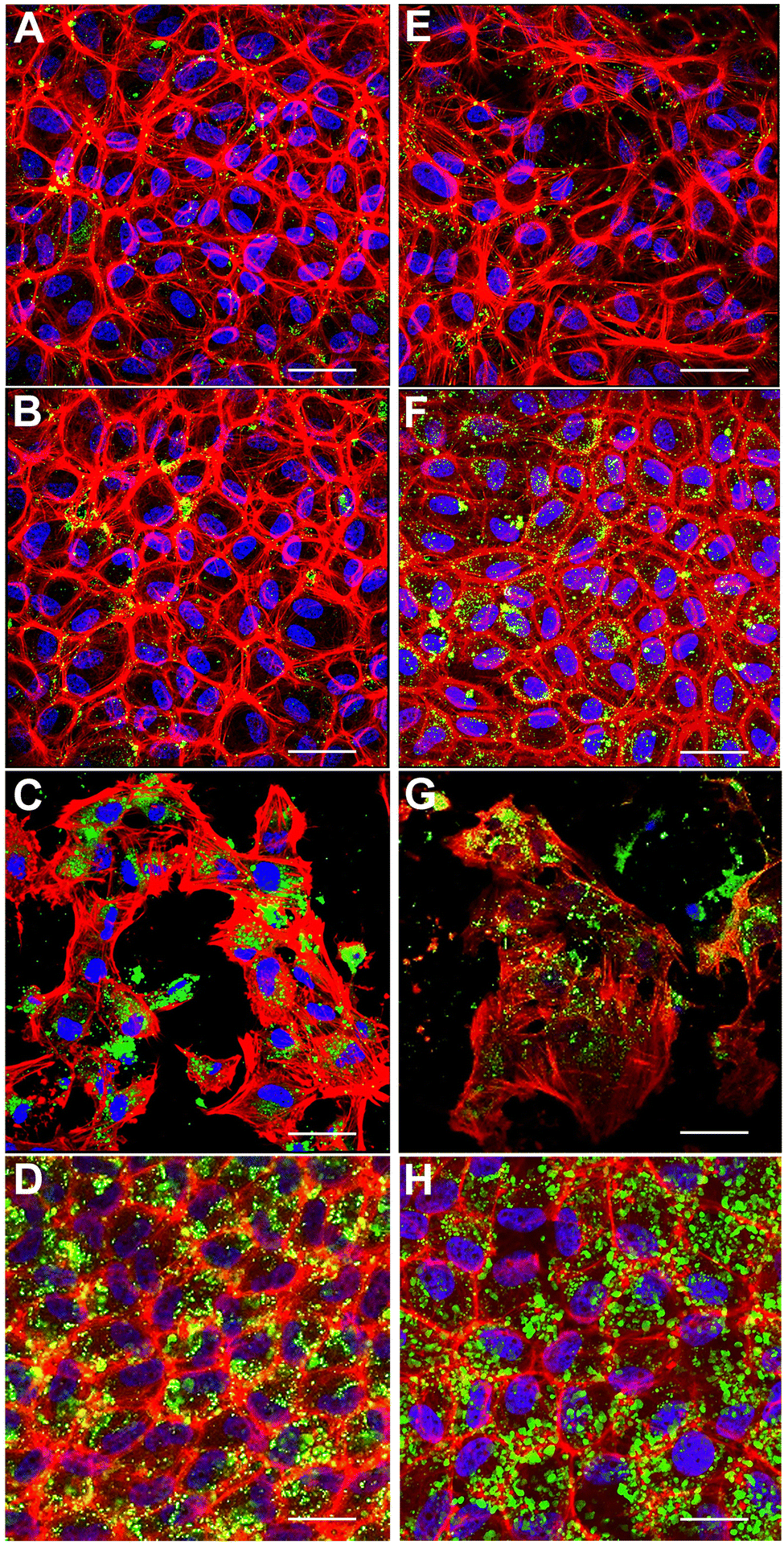 | ||
| Fig. 2 Representative confocal fluorescence microscope images of nanoparticles internalized in porcine corneal endothelial cells in vitro at 37 °C. (A) Cells incubated with DNPs for 3 h; (B) cells incubated with PLL-DNPs for 3 h; (C) cells incubated with FL-DNPs-1 for 3 h; (D) cells incubated with FL-DNPs-2 for 3 h; (E) cells incubated with DNPs for 6 h; (F) cells incubated with PLL-DNPs for 6 h; (G) cells incubated with FL-DNPs-1 for 6 h; (H) cells incubated with FL-DNPs-2 for 6 h. Red: phalloidin (cell membrane), blue: DAPI (nucleus), and green: DiO (nanoparticles). The images shown are typical of three independent experiments. The scale bar is 10 μm. | ||
Fig. 2A shows that after 3 h of incubation, few DNPs are taken up by the cells (green staining in and around the nucleus) and upon an increase in incubation to 6 h, no significant change in the number of DNPs is observed (Fig. 2E). When DNPs were coated with PLL, it improved the uptake slightly for both 3 h and 6 h of incubation as observed in Fig. 2B and F. In addition, the cells seemed to take up more PLL-DNPs when incubated for 6 h in comparison to 3 h. When the cells were incubated with FL-DNPs-1, which had DOTAP + DOPE fusogenic liposome coating, a significant increase in uptake of cells is observed (Fig. 2C and G). However, we also observed a decrease in cell viability when they were incubated with FL-DNPs-1, suggesting nanoparticle-induced cell death. In contrast, FL-DNPs-2 which contained DOTAP + SoyaPC fusogenic liposome coating exhibited significantly higher nanoparticle uptake at both 3 and 6 h of incubation (Fig. 2D and H). 6 h of incubation seemed to slightly increase the nanoparticle uptake compared to 3 h. Notably, FL-DNPs-2 did not adversely affect cell viability, as observed by the presence of a large number of intact cells.
Engineering the endothelium of ex vivo corneal tissue with nanoparticles before storage and transplantation is an actively pursued area of translational research. In this context, we investigated the internalization of various nanoparticles in the ex vivo corneal endothelium. Our goal was also to investigate if fusogenic liposome-coated DNPs are rapidly internalized into the cells of the endothelium in comparison to bare nanoparticles and PLL-DNPs. We also assessed if the nanoparticles could penetrate the endothelium and reach the stroma, which is the middle layer of the cornea. The cornea was isolated from porcine eyes and placed in an eye cup with the endothelium facing upwards. DNPs, PLL-DNPs, FL-DNPs-1 and FL-DNPs-2 were suspended in Cornisol® medium and ultrasonicated to ensure uniform suspension. Later, the corneal endothelium was incubated with these nanoparticles at a concentration of 0.8 mg mL−1 for internalization at 37 °C for 3 h and 6 h. Untreated tissue was also used in this study to compare the staining patterns of nanoparticles and the cell membrane to understand nanoparticle localization. The z-stack images shown in Fig. 3 and 4 depict a part of the corneal tissue containing the endothelial monolayer (corneal endothelium) (yellow arrows) which is about 5 to 10 μm in thickness and a part of the adjoining stroma (blue arrows) (about 30 μm deep from the apical side of the endothelium). The stroma was included in the image to understand if the nanoparticles have penetrated into it.
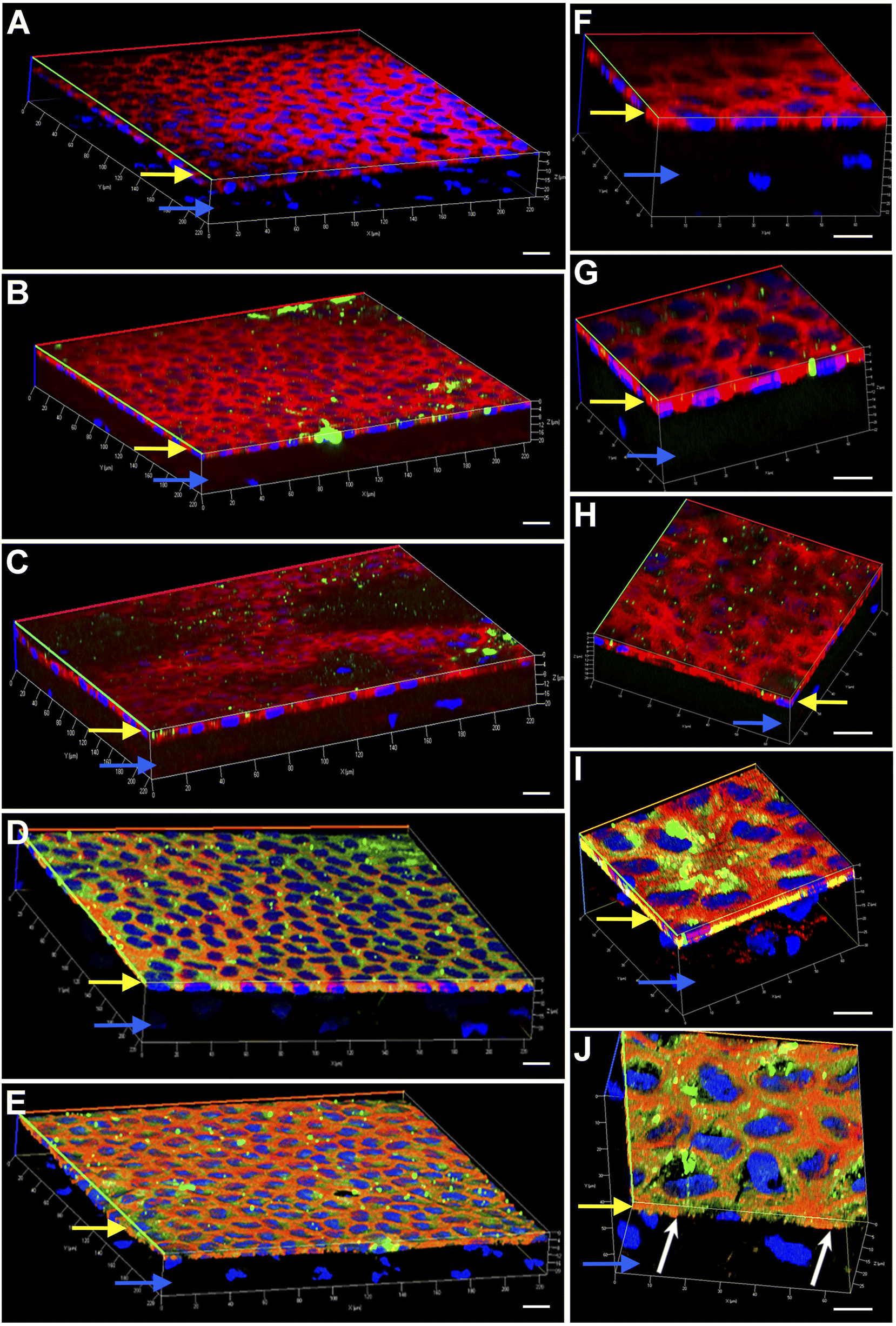 | ||
| Fig. 3 Representative 3-dimensional confocal fluorescence microscope images of nanoparticles internalized in the porcine corneal endothelium ex vivo after 3 h of incubation at 37 °C. (A) Untreated tissue; (B) tissue incubated with DNPs for 3 h; (C) tissue incubated with PLL-DNPs for 3 h; (D) tissue incubated with FL-DNPs-1 for 3 h; (E) tissue incubated with FL-DNPs-2 for 3 h. (A–E) Magnification of 63×. (F–J) Magnification of 100×. (F) Untreated tissue; (G) tissue incubated with DNPs for 3 h; (H) tissue incubated with PLL-DNPs for 3 h; (I) tissue incubated with FL-DNPs-1 for 3 h; (J) tissue incubated with FL-DNPs-2 for 3 h; red: phalloidin (cell membrane), blue: DAPI (nucleus), and green: DiO (nanoparticles). Yellow and blue arrows indicate the endothelial monolayer and the adjoining stroma, respectively in all the images. White arrows in (I) and (J) indicate the presence of nanoparticles inside the cells. The images shown are typical of three independent experiments. The scale bar is 10 μm. | ||
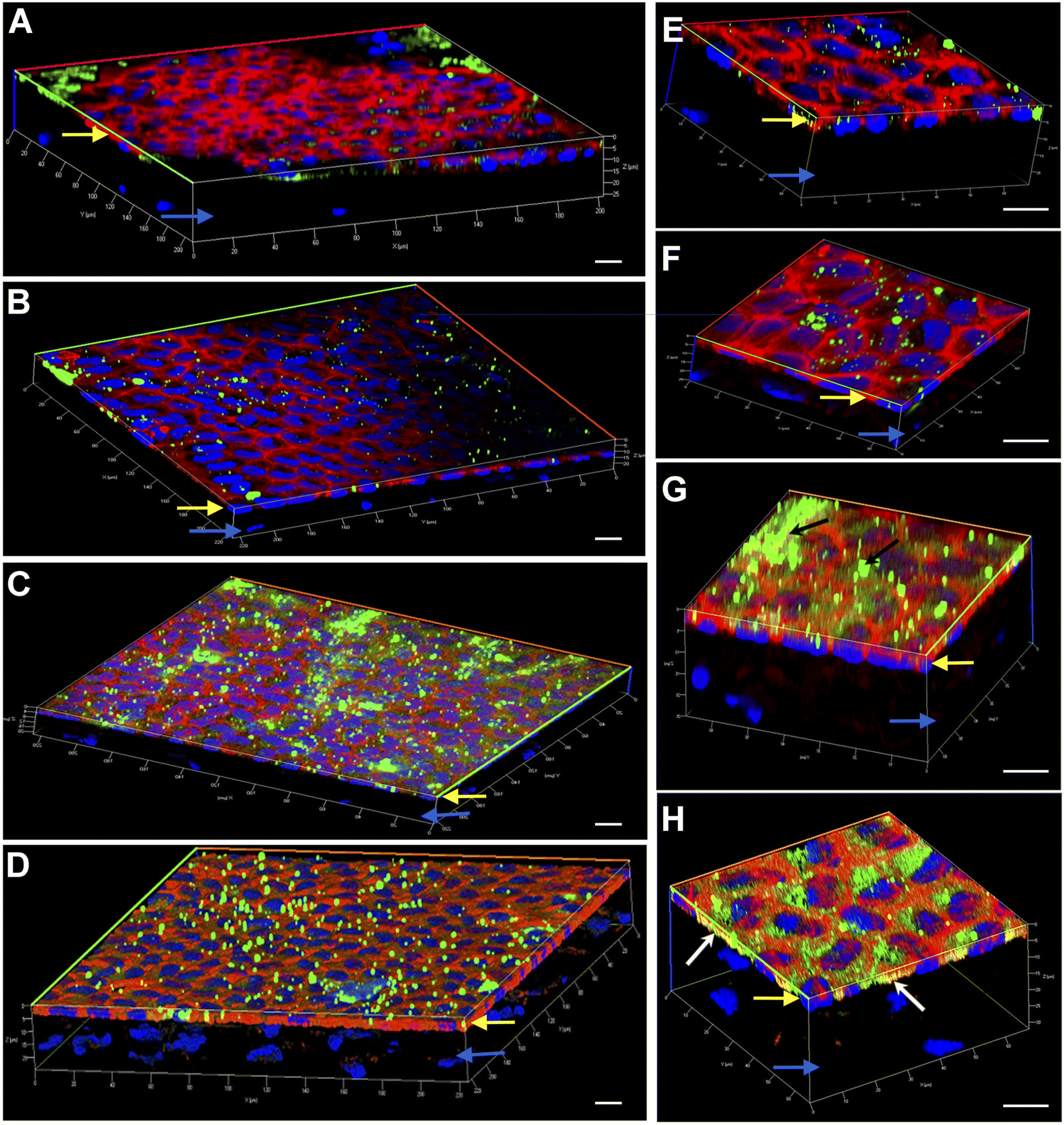 | ||
| Fig. 4 Representative 3-dimensional confocal fluorescence microscope images of nanoparticles internalized in the porcine corneal endothelium ex vivo after 6 h of incubation at 37 °C. (A) Tissue incubated with DNPs for 6 h; (B) tissue incubated with PLL-DNPs for 6 h; (C) tissue incubated with FL-DNPs-1 for 6 h; (D) tissue incubated with FL-DNPs-2 for 6 h. (A–D) Magnification of 63×. (E–H) Magnification of 100×. (E) Tissue incubated with DNPs for 6 h; (F) tissue incubated with PLL-DNPs for 6 h; (G) tissue incubated with FL-DNPs-1 for 6 h; (H) tissue incubated with FL-DNPs-2 for 6 h; red: phalloidin (cell membrane), blue: DAPI (nucleus), and green: DiO (nanoparticles). Yellow and blue arrows indicate the endothelial monolayer and the adjoining stroma, respectively in all the images. White arrows in (G) and (H) indicate the presence of nanoparticles inside the cells, while black arrows show nanoparticle aggregates on the cell membrane. The images shown are typical of three independent experiments. The scale bar is 10 μm. | ||
Fig. 3 illustrates the internalization of various nanoparticle types in ex vivo corneal tissue after 3 h of nanoparticle incubation at 37 °C. It is evident from Fig. 3 that the corneal endothelium is a hexagonal mosaic of cells of a thickness of about 5 to 10 μm (yellow arrows). The red staining of phalloidin-conjugated Texas Red which binds to the lipid bilayer of the cells indicates the cell membrane. On the other hand, a part of the stroma (blue arrows) seen here has very few cells (represented by the lack of DAPI staining or nuclei). Fig. 3A and F represent ex vivo corneas which were not subjected to any nanoparticles (control). They reveal the corneal endothelial tissue comprising the monolayer of endothelium (yellow arrows) and a part of the stroma (blue arrows). These images were taken as a reference without nanoparticles to compare them with corneas containing nanoparticles since we are interested in observing the localization of the nanoparticles. As observed in Fig. 3B and G, bare nanoparticles (DNPs) hardly penetrated into the corneal endothelium in 3 h, which is confirmed by the lack of green fluorescence merging with red. PLL-DNPs also exhibit insignificant penetration into the endothelium in 3 h (Fig. 3C and H). On the other hand, FL-DNPs-1 and FL-DNPs-2 penetrated into the corneal endothelium significantly in 3 h (Fig. 3D, E, I and J). The high intensity of green fluorescence and uniform distribution of merged green/red fluorescence (Fig. 3D and E) confirm the same. In the high magnification images (Fig. 3I and J), we can also observe that the nanoparticles are in fact internalized deeply into the cells (white arrows) and not just associated with the outer cell membrane. In addition, some nanoparticle aggregates were observed on the cell surface (Fig. 3D, E, I and J), which did not internalize into the tissue due to their larger size.
We next investigated if nanoparticle internalization can be enhanced by incubating the tissue with various nanoparticle types for 6 h. As observed in Fig. 4A, B, E and F, bare DNPs and PLL-DNPs hardly penetrated into the corneal endothelium in 6 h, but the penetration was better than that in 3 h, which is confirmed by the relatively higher number of green fluorescence spots. The localization of green spots on the top surface of the tissue also indicates the presence of nanoparticle aggregates associated with the cell membrane rather than the cytoplasm, confirming that there was minimal internalization (Fig. 4A, B, E and F). On the other hand, FL-DNPs-1 and FL-DNPs-2 penetrated into the corneal endothelium significantly in 6 h (Fig. 4C, D, G and H). The high intensity of green fluorescence and uniform distribution of merged green/red fluorescence (Fig. 4C and D) confirm the same. In the high magnification images (Fig. 4G and H), we also observe that the nanoparticles are in fact internalized by the cells. However, in comparison to 3 h, 6 h incubation led to the formation of more nanoparticle aggregates on the cell surface, indicated by black arrows (Fig. 4G), which did not internalize into the tissue due to larger size. There is also clear evidence that with 3 h and 6 h of incubation, the nanoparticles were unable to penetrate the stroma, as expected. Very interestingly, cellular toxicity was not observed in ex vivo corneas when incubated with nanoparticles, as opposed to significant toxicity in cultured cells in vitro (Fig. 2C and G).
We also estimated the extent of internalization by measuring green fluorescence intensity of z-stack images taken at various depths of the cells (except at the top and bottom layers which represent the cell membrane) using ImageJ software in both cultured cells and tissue ex vivo. This quantitative data also estimated the kinetics of internalization after 3 and 6 h of incubation (Fig. 5). Fig. 5A confirms that DNPs did not internalize into the cultured cells after 3 h and 6 h of incubation, as indicated by low fluorescence intensity values. In comparison, more PLL-DNPs were internalized after 3 h of incubation (about 5 times, p < 0.05), which further improved upon 6 h of incubation (about 8 times, p < 0.05). For PLL-DNPs, 6 h of incubation demonstrated about 4 times higher fluorescence versus 3 h of incubation, indicating a higher degree of internalization (p < 0.05). In comparison to DNPs, the DOTAP + DOPE liposome-coated DNPs (FL-DNPs-1) were internalized at an even higher level (about 25 times, p < 0.05) after 3 h and about 13 times (p < 0.05) after 6 h of incubation.
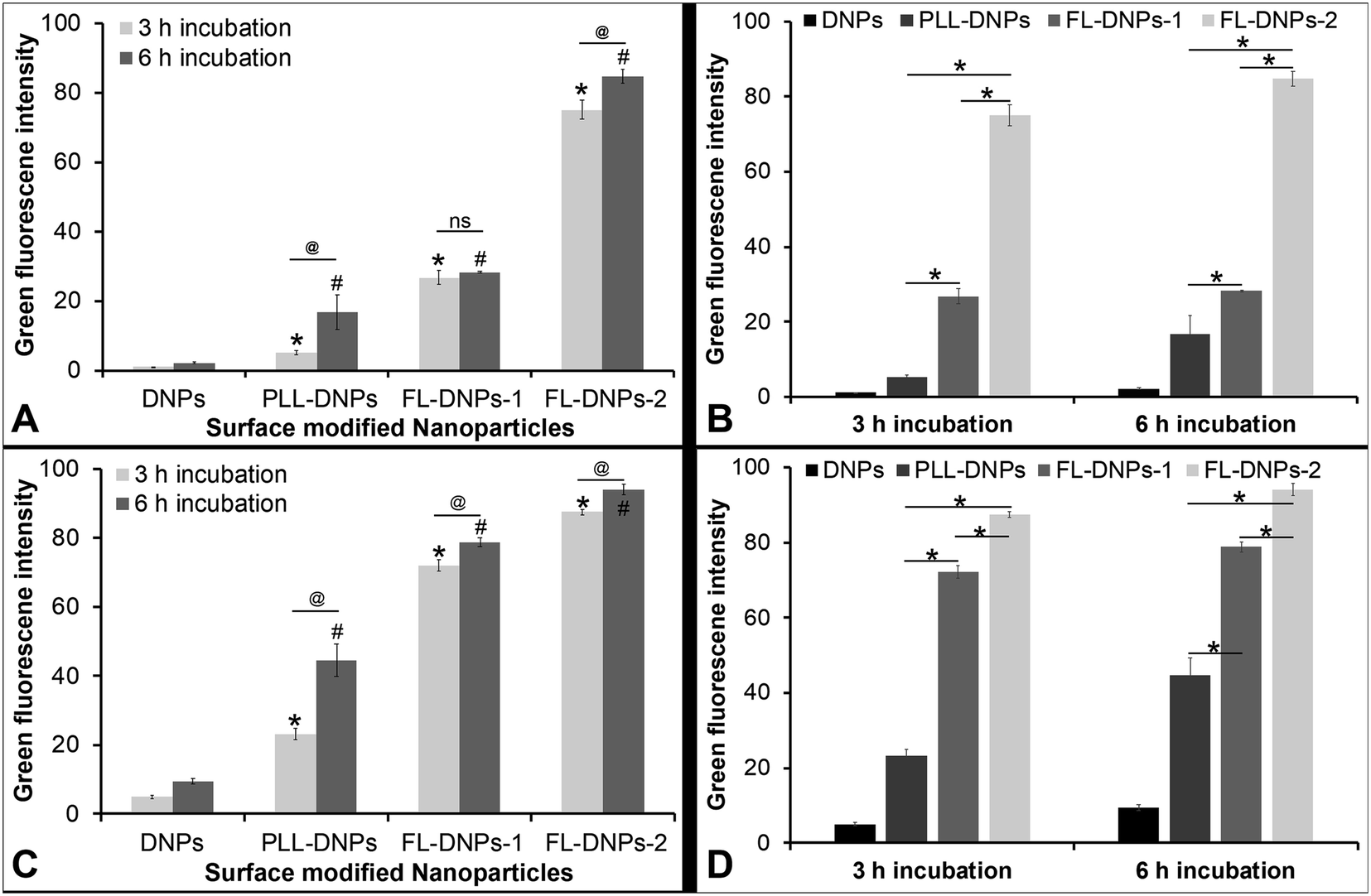 | ||
| Fig. 5 Quantification of nanoparticle internalization in cultured corneal endothelial cells and the corneal endothelium ex vivo. (A) Intensity of green fluorescence of nanoparticles inside cultured endothelial cells. * represents p < 0.05 for data against DNPs for 3 h. # represents p < 0.05 for data against DNPs for 6 h. @ represents p < 0.05. (B) Intensity of green fluorescence of nanoparticles inside cultured endothelial cells. * represents p < 0.05. (C) Intensity of green fluorescence of nanoparticles inside the corneal endothelium ex vivo. * represents p < 0.05 for data against DNPs for 3 h. # represents p < 0.05 for data against DNPs for 6 h. @ represents p < 0.05. (D) Intensity of green fluorescence of nanoparticles inside cultured endothelial cells ex vivo. * represents p < 0.05. Data represent the average of three replicate experiments. For each experiment, a minimum of five z-stack images taken from different fields of view were used. We excluded the images of the top and bottom layers in the z-stack, since we intended to quantify internalization i.e., to quantify nanoparticles which were inside the cells and not just associated with the cell membrane. All the cells in those images were included while quantifying green fluorescence intensity and deducting background fluorescence using ImageJ. Error bars represent standard deviation. “ns” represents non-significance. | ||
However, it is important to note that for FL-DNPs-1, the period of incubation did not significantly improve internalization (p > 0.05) which is confirmed by a small increase in fluorescence intensity between 3 h and 6 h incubation. Against PLL-DNPs, the internalization of FL-DNPs-1 was significantly higher i.e., about 5 times more for 3 h incubation (p < 0.05) and about 2 times more for 6 h incubation (p < 0.05), as seen in Fig. 5B. On the other hand, the DOTAP + SoyaPC liposome-coated DNPs (FL-DNPs-2) were appreciably taken up by the cells. Specifically, with reference to DNPs, the increase in fluorescence intensity for 3 h and 6 h incubation was about 70 times (p < 0.05) and 40 times (p < 0.05), respectively. Very importantly, the increase in fluorescence intensity between 3 h and 6 h for FL-DNPs-2 was not significant (p > 0.05). This suggests that 3 h of incubation is sufficient to achieve very high uptake of nanoparticles in cultured corneal endothelial cells with DOTAP + SoyaPC liposome-coated DNPs. With reference to PLL-DNPs, the increase in the uptake of FL-DNPs-2 was about 14 times (p < 0.05) and 5 times (p < 0.05) for 3 h and 6 h, respectively (Fig. 5B). In comparison to FL-DNPs-1, the increase was about 3 times (p < 0.05) for both 3 h and 6 h (Fig. 5B). This is a clear indication that FL-DNPs-2 was the best nanoparticle type that allowed rapid uptake of nanoparticles into cultured cells. The very large difference in fluorescence intensity between FL-DNPs-1 and FL-DNPs-2 is probably due to the fact that FL-DNPs-1 induced cell death in comparison to FL-DNPs-2.
In the case of corneal endothelium ex vivo, Fig. 5C and D confirm that bare DNPs were internalized into the endothelium after 3 h and 6 h of incubation. There was also a 2 fold increase in the uptake after 6 h of incubation. In comparison to DNPs, more PLL-DNPs were internalized after 3 h of incubation (about 5 times, p < 0.05), which further improved upon 6 h of incubation (about 5 times, p < 0.05). For PLL-DNPs, 6 h of incubation demonstrated about 2 times higher fluorescence versus 3 h of incubation, indicating a higher degree of internalization (p < 0.05) (Fig. 5C).
The DOTAP + DOPE liposome-coated DNPs (FL-DNPs-1) were internalized at an even higher level (about 14 times, p < 0.05) after 3 h and about 8 times (p < 0.05) after 6 h of incubation when compared to DNPs (Fig. 5C). However, with FL-DNPs-1, the period of incubation did not improve internalization notably (p < 0.05) which is confirmed by a small increase in fluorescence intensity between 3 h and 6 h of incubation. Against PLL-DNPs, the internalization of FL-DNPs-1 was significantly higher i.e., about 3 times more for 3 h incubation (p < 0.05) and about 2 times more for 6 h incubation (p < 0.05), as seen in Fig. 5D. On the other hand, the DOTAP + SoyaPC liposome-coated DNPs (FL-DNPs-2) were taken up by the cells more than all other nanoparticles. With reference to bare DNPs, the increase in fluorescence intensity for 3 h and 6 h of incubation was statistically significant at about 18 times (p < 0.05) and 10 times (p < 0.05) (Fig. 5C), respectively. With reference to PLL-DNPs, there was an appreciable increase in fluorescence intensity at about 4 times (p < 0.05) and 2 times (p < 0.05) for 3 h and 6 h, respectively (Fig. 5D). In comparison to FL-DNPs-1, the increase was also significant at about 1.2 times (p < 0.05) for both 3 h and 6 h of incubation. This is a clear indication that FL-DNPs-2 was the best nanoparticle type which allowed rapid uptake of nanoparticles into the ex vivo corneal endothelium. Very importantly, the increase in fluorescence intensity between 3 h and 6 h for FL-DNPs-2 was negligible (p < 0.05). This suggests that 3 h of incubation is sufficient to achieve very high uptake of nanoparticles in the ex vivo corneal endothelium with DOTAP + SoyaPC liposome-coated DNPs.
Finally, a cytotoxicity study was performed on the fusogenic liposome-coated nanoparticles after internalization into the ex vivo corneal endothelium. For this, the fusogenic liposome-coated nanoparticles did not have DiO (as in the internalization studies) since there was no imaging involved in this study. The nanoparticles used were: (i) (DOTAP + DOPE) liposome-coated nanoparticles (FLNP1) and (ii) (DOTAP + SoyaPC) liposome-coated nanoparticles (FLNP2). Cell viability was assessed in the ex vivo tissue by first subjecting only the endothelial surface of the ex vivo cornea (placed inside a contact lens holder) to the nanoparticles at a concentration of 0.8 mg mL−1 at 37 °C and 5% CO2 for 3 h (Fig. 6A1–6A4) and then using a lactate dehydrogenase (LDH) assay by measuring LDH release by the cells after 3, 6, 12, 24 and 48 h. LDH release correlates with the damage to the cell membrane and a decrease in cell viability. The LDH assay results showed that the viability of the endothelial cells when treated with FLNPs was not affected significantly (Fig. 6). As a positive control, Triton X-100 was used to damage the cell membrane and reduce cell viability. Thus, Triton-treated endothelial cells showed an acute and sustained LDH release owing to damage to the plasma membrane (fold change in LDH release versus untreated corneas was 2.72, 2.68, 2.45, 1.52 and 1.88 at 3, 6, 12, 24 and 48 h, respectively), which was statistically significant (p < 0.01) (Fig. 6B). As the time of incubation increases, the Triton X-100 treated corneas show lower fold change in LDH release. This could be because, as cell death occurred, the dead cells were washed off using PBS before addition of lysis solution during the LDH assay. Nevertheless, the fold change was significantly higher than in untreated corneas confirming a decrease in cell viability due to Triton treatment.
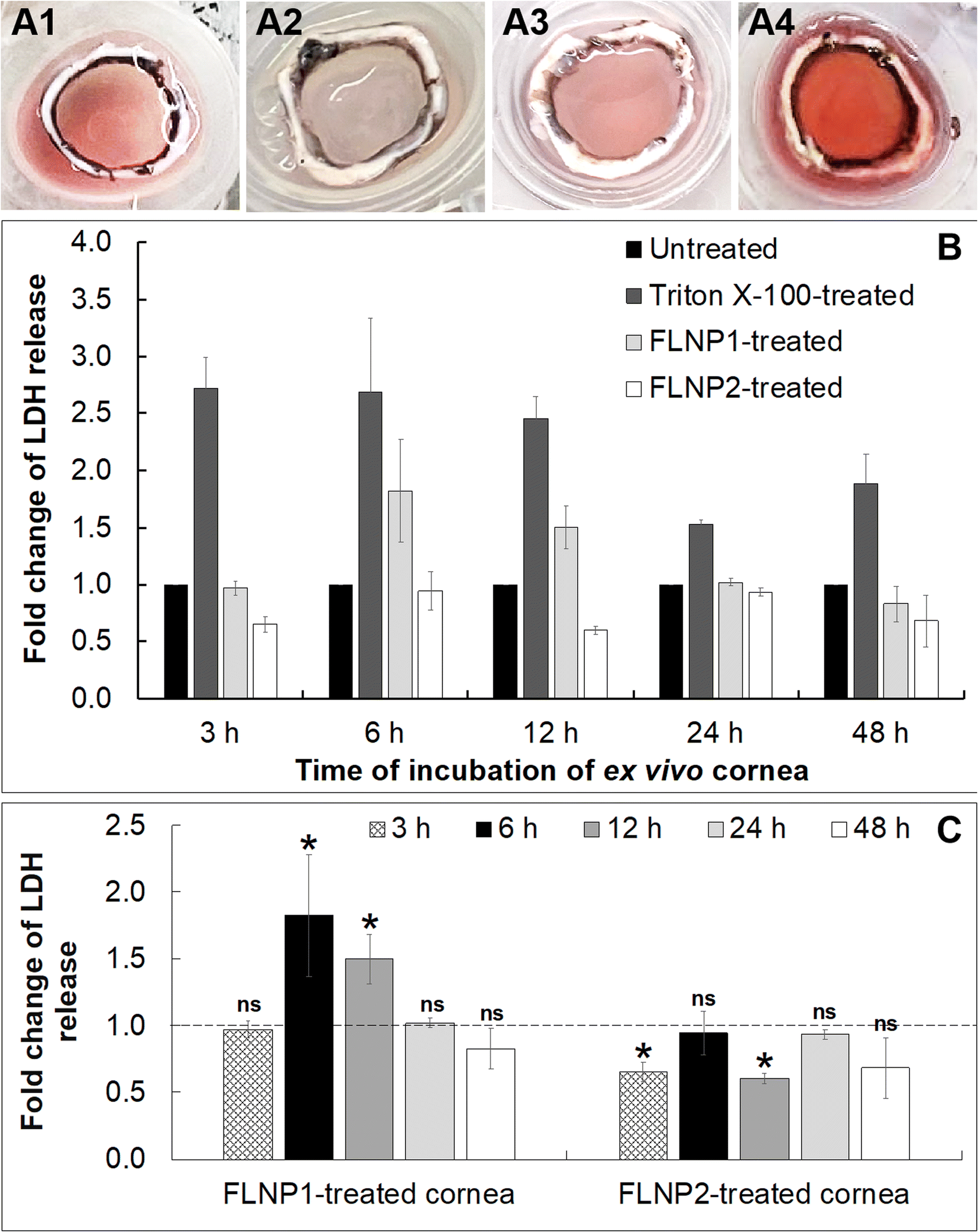 | ||
| Fig. 6 Assessment of cell viability in the corneal endothelium ex vivo after nanoparticle internalization by lactate dehydrogenase assay. Representative brightfield optical images of (A1) untreated cornea; (A2) Triton X-100 treated cornea; (A3) FLNP1-treated cornea; (A4) FLNP2-treated cornea after internalization of nanoparticles for 3 h at 37 °C and 5% CO2. (B) Fold change in lactate dehydrogenase (LDH) release versus that of untreated cornea against time (3, 6, 12, 24 and 48 h) after 3 h of nanoparticle internalization, determined by LDH assay. Untreated cells were used as the control group, and Triton X-100 treated cells were used as a positive control for maximum LDH release and a consequent decrease in cell viability. (C) Fold change of LDH release with respect to the type of fusogenic liposome-coated nanoparticles internalized for 3 h and cell viability assessed after 3, 6, 12, 24 and 48 h. * represents p < 0.01 and “ns” represents non-significance for FLNP1 and FLNP2 against untreated corneas. Data represent the average of three replicate experiments ± standard deviation, with each experiment having two corneas. | ||
On the other hand, both FLNPs did not show an appreciable increase in LDH release in comparison to untreated corneas. Notwithstanding, FLNP1 seemed to be slightly cytotoxic to the cells at 6 and 12 h (fold change in LDH release was 1.8 and 1.5 at 6 and 12 h, respectively). FLNP1 did not cause cytotoxicity (fold change was about 1 versus untreated corneas) at 3, 24 and 48 h. Meanwhile, FLNP2 did not result in significant LDH release compared to untreated corneas. The fold change in LDH release from FLNP2-treated corneas was either similar to or slightly less than that of the untreated corneas, indicating that they did not reduce cell viability (Fig. 6B). Fig. 6C summarizes the fold change in LDH release when the ex vivo corneas were treated with FLNP1 and FLNP2 after various time intervals. The results suggest that both FLNPs were biocompatible and did not cause any significant cytotoxicity. Even though FLNP1 showed higher LDH release at 6 and 12 h, it is possibly due to the very heterogeneous nature of the experiment which uses corneas from different animals which could sometimes yield an inconsistent number of endothelial cells after scrapping during LDH assay. Thus, we have considered this potential heterogeneity by choosing a more rigorous level of confidence (p < 0.01) when performing statistical analysis.
Several studies have attempted to prevent apoptosis of donor corneal endothelial cells during cold storage by supplementing the storage medium with salubrinal, iron-chelators, NOS inhibitors, poloxamers, and α-melanocyte-stimulating hormone or by delivering anti-apoptotic genes.8,16,17,27–29 Another study demonstrated that complexed ubiquinol in the corneal storage medium protected HCEC-B4G12 cells against hypothermia-induced oxidative stress and erastin-induced ferroptosis.30 However, this strategy is effective only as long as the cornea is stored. After transplantation, the donor corneal endothelium encounters cytokine stress10 secondary to allograft rejection leading to apoptosis, reduction in ECD, graft failure and loss of functionality. Any pharmacological intervention to prevent these post transplantation events can be perfomed via topical administration. However, topical drugs have pharmacokinetic limitations to reach the corneal endothelium owing to resistance from multiple layers of the cornea.31 In addition, frequent drug administrations are needed to achieve the necessary efficacy. Hence, nanoparticle-based drug delivery provides sustained release, greater drug bioavailability, and one-time administration. This can be achieved by engineering the donor corneal endothelium or donor cells before cold storage with drug-loaded nanoparticles. In other words, a one-time administration of drug- or gene-loaded nanoparticles immediately after isolation of the cornea appears to be adequate to handle both long-term cold storage and potential acute allograft rejection. Thus, in this study, nanoparticles were coated with fusogenic liposomes to enhance the internalization rate to about 3 h before hypothermic storage and maintain the viability of the corneal grafts or donor cells.
The interface between nanoparticles and the cell membrane is complex and involves many physicochemical interactions including hydrophobic, electrostatic, and steric interactions. The key factors that influence these interactions are size, surface chemistry and surface charge. Larger particles (50 to 200 nm) enter the cell through endocytic and non-endocytic pathways. The transmembrane (plasma membrane) potential of cells plays a key role in this interaction since the membrane is the first line of entry for the nanoparticles. The cell membrane being negatively charged, the surface charge of the nanoparticles is a critical driving force for entry into the cell. Positively charged nanoparticles are taken up by cells faster than negatively charged nanoparticles.9 However, positively charged nanoparticles may disrupt the integrity of the cell membrane leading to increased toxicity.32 A high positive charge causes disruption of the electron transfer chain inside the cells and increases production of reactive oxygen species leading to the disruption of the cell membrane and cell death.33 A high positive charge on nanoparticles induces cell death, while neutrally charged nanoparticles undergo slower cellular uptake compared to charged nanoparticles.34 Negatively charged nanoparticles after internalization can lead to gelation of the lipid bilayer membrane due to surface reconstruction of phospholipids and interaction with the N+ terminus of the lipid membrane. In the case of positively charged nanoparticles, the interaction with the negatively charged lipid bilayer membrane causes fluidity in the cell membrane due to interaction with the P-terminus of the lipid membrane.35,36
Molecular dynamics simulation studies on interaction between charged nanoparticles and the cell membrane demonstrated that adhesion of charged nanoparticles on the cell membrane was better than that of neutral nanoparticles.37 Cationic nanoparticles showed strong electrostatic interaction with phosphate groups of the cell membrane leading to stronger binding and increased surface tension at the membrane which results in the formation of pores in the cell membrane.38 Many researchers have demonstrated that positively charged liposomes were internalized by non-endocytic pathways (membrane fusion)39–42 while charged nanoparticles follow endocytic pathways.25 Thus, uptake of negatively charged nanoparticles was delayed. The major drawback of charged nanoparticles of delayed endocytosis (negatively charged nanoparticles) and cell membrane disruption (positively charged nanoparticles) can be overcome by avoiding the endocytic pathway. For this, the use of fusogenic liposomes has been reported. Unilamellar fusogenic liposomes rapidly fuse with the cellular plasma membrane, because fusion is driven by surface pressure gradients (lateral diffusion) due to attractive force between two opposite charges (negative charge of the cell membrane and positive charge of fusogenic liposomes). The fusion mechanism mainly occurs due to van der Waals interaction, which brings the two membranes together to undergo fusion. This mechanism is not energy dependent unlike the endocytosis pathway43 and hence fusogenic liposomes are being used to rapidly deliver DNA or proteins.44
We describe here the internalization of fusogenic liposome-coated nanoparticles (FL-DNPs) in ex vivo corneal tissue and cultured cells. The novelty of this approach is that we have harnessed the membrane fusion properties of the fusogenic liposomes to achieve rapid tissue and cellular uptake of PLGA nanoparticles. By coating fusogenic liposomes on the nanoparticle surface, we intend to rapidly deliver the polymeric nanoparticles into ex vivo corneal tissue as well as cultured corneal endothelial cells for transplantation. This study will pave the way to engineer corneal tissue and cells for therapeutic drug delivery to enhance the success of corneal transplantation. As shown earlier by us, PLL-coated PLGA nanoparticles internalized into cultured corneal endothelial cells, but it took very long (24 h).9 In comparison, here, we demonstrate a significantly faster approach of nanoparticle uptake by not only cultured corneal endothelial cells, but also the ex vivo corneal endothelium. Modifying the surface of the nanoparticles was needed to facilitate faster uptake.
To achieve this, we wanted to select the best lipid by comparing the relative biocompatibilities and fusogenic properties of natural and synthetic lipids. There is evidence that natural lipids might be more biocompatible and less cytotoxic45 when compared to synthetic lipids such as DOPE,46 despite both being neutral in charge. Thus, we employed two types of fusogenic liposomes, i.e., one made from soya-derived phosphatidylcholine (Soya-PC) which is a natural lipid and the other one made from dioleoylphosphatidylethanolamine (DOPE) which is a synthetic lipid. Studies on fusogenic liposomes have demonstrated that the presence of cationic lipids in combination with neutral lipids can strongly influence their uptake rate.42 Neutral lipids such as DOPE in combination with cationic lipids (e.g., DOTAP) have been employed to facilitate rapid delivery of therapeutic molecules inside cells via the membrane fusion process.40,41 In this study, the significant decrease in the time of nanoparticle internalization of FLNPs is probably due to the fact that the fusion of FLNPs with the cell membrane increases with decreasing the head group size of neutral lipids to form liposomes.39 Therefore, selection of lipids plays a vital role in the internalization process. Another study has indicated that DOPE is generally considered a better fusogenic lipid compared to PC. However, fusogenicity of DOPE/PC alone was insufficient for fusion with the complex cell membrane.47 Thus, their combination with cationic lipids increases the fusogenicity. Another study demonstrated that conical molecular shape of DOTAP was able to fuse with the plasma membrane with 90% efficiency in comparison to a 30% efficiency with cylindrical cationic lipids such as DMTAP (1,2-dimyristoyl-3-trimethylammonium-propane chloride) or DOEPC (1,2-dioleoyl-sn-glycero-3-ethylphosphocholine).39 Therefore, we selected DOTAP as the cationic lipid and DOPE or SoyaPC as the neutral lipid in our study.
Fusogenic liposomes were prepared by the thin film hydration method and nanoparticles were prepared by the nanoprecipitation method. Coating the fusogenic liposomes on the surface of nanoparticles was carried out during hydration and extrusion steps with uniformity (Fig. 1C and D). However, repeated extrusion is a time-consuming step; thus, we need to automate extrusion and size reduction.48 Microfluidic methods have also been developed for producing nanoliposomes and can be adopted for coating fusogenic liposomes on the surface of nanoparticles on a large scale.49 The fusogenic liposome-coated nanoparticles prepared were nano-sized (Fig. 1E) with uniform size distribution (Fig. 1F) and positive charge (Fig. 1G) to enhance the fusion process during internalization. Morphology of the fusogenic liposome coated nanoparticles was spherical and showed uniform coating on the surface of nanoparticles (Fig. 1C and D). Thus, fusogenic liposome-coated nanoparticles appear to be suitable for internalization into corneal tissue based on their potential for rapid fusion with the cell membrane.
Instead of transplanting donor tissues directly into a small number of patients, corneal endothelial cells isolated from a single donor can be administered to up to 80 patients by in vitro expansion using an appropriate medium.20 Hence, an alternative for corneal transplantation i.e., scalable cell-based therapies (cell injection approach) using human corneal endothelial cells (HCECs) propagated in vitro15 and tissue engineered constructs to meet the global demands for functional HCECs19 were developed. Cultured HCECs can be therapeutically administered through direct cell injection into the anterior chamber.15 Although cell-based therapy has raised new hopes for corneal endothelial regeneration, limitations include unbalanced cell distribution and lower persistence and repair efficiency. These limitations are mainly related to the loss of cell surface proteins during trypsinization, which are essential for cell to cell and cell to extracellular matrix interactions.21 Nanotechnology-based prophylactic drug delivery to donor corneal endothelial cells can overcome these limitations and facilitate successful cell therapies for corneal diseases.
As shown in Fig. 5, the cationic charge as well as the fusogenic properties of the coated liposomes led to a significant increase in the internalization kinetics of FL-DNPs in comparison to bare DNPs and PLL-DNPs in both cultured cells and ex vivo tissue. This is an important finding, since faster internalization will decrease the risk of loss of cell viability due to prolonged exposure at 37 °C before cold storage. Our results show that 3 h of incubation with FL-DNPs-1 or FL-DNPs-2 would be sufficient to obtain internalization (Fig. 5). These results have clinical implications since prolonged death-to-preservation time (DTPT) > 4–6 h of donor corneas leads to a decrease in endothelial cell viability and an increased risk of microbial contamination.50 The fusogenic liposomes were made of the cationic lipid DOTAP in combination with DOPE or SoyaPC. DOTAP is known to induce membrane fusion due to electrostatic interactions with the cell membrane and also its presence imparts a positive charge to liposomes. The neutral lipids DOPE or PC assist in membrane destabilization due to the low hydration of their small head groups.40,42,47 The fusion of liposomes with the cell membrane seems to have followed a non-endocytosis pathway; thus, within 3 h of incubation, a large number of FL-DNPs (shown by significantly higher fluorescence intensity) were internalized into corneal endothelial cells and ex vivo tissue. A similar response has been shown by other studies.39
DiO is a biocompatible and non-toxic fluorescent dye (at 1% w/w nanoparticle loading) as reported by us earlier to demonstrate nanoparticle internalization in other cells.9,51 In addition, at initial time points (i.e., 48 h), PLGA degradation would not have begun and hence release of surface-bound DiO would be diffusion-driven.52 Following the coating of the fusogenic liposomes, it is expected to further delay DiO release since the coating acts as a barrier to DiO diffusion. Thus, both PLGA degradation products and DiO release are highly unlikely to contribute to cytotoxicity during 48 h of incubation.
The cell viability studies (Fig. 6) confirmed that both the fusogenic liposome-coated nanoparticle formulations were well tolerated by the endothelial cells associated with ex vivo corneas for at least 48 h after internalization under physiological conditions. However, DOPE + DOTAP liposome-coated nanoparticles (FLNP1) exhibited moderate cytotoxicity at 6 and 12 h. DOPE forms an inverted hexagonal phase at acidic pH and thus releases cationic lipids into the cytoplasm. This phenomenon has been reported to result in cationic lipid cytotoxicity due to destabilization of the endosome membrane.46 Nevertheless, SoyaPC + DOTAP liposome-coated nanoparticles did not demonstrate any cytotoxicity for at least up to 48 h after internalization. SoyaPC being a natural lipid is more suitable as a liposomal carrier for drug delivery applications due to its favorable biocompatible characteristics45 compared to the synthetic lipid DOPE.
We also noted another interesting result from the LDH release study. In the case of the Triton-X-treated cornea, even though a decreasing trend of cytotoxicity is observed with time, the decrease (in fold change in LDH release or decrease in cytotoxicity) with time is not statistically significant (p > 0.01) except at 24 h, when compared to the 3 h data. For FLNP2, there is no decrease in cytotoxicity observed over time; rather no trend is observed. However, for FLNP1, a decrease in cytotoxicity is seen after 12 h. One possible reason for these results is the nature of these experiments, where we had to use corneas from different animals, whose initial endothelial cell density would not be the same for each experiment. A high degree of heterogeneity is thus expected, as we had to use different corneas for experiments related to each time point. Since the fold change in LDH release was calculated with respect to untreated corneas, and due to heterogeneity in the initial cell density of each cornea, we cannot expect to normalize fold change data equally for each time point. Thus, we think that the decrease in cytotoxicity for FLNP1 is a result of the heterogeneity in the samples.
Therefore, fusogenic liposome-coated nanoparticles can be used for delivery of therapeutic molecules for cell-based therapy and tissue transplantation. Our work has demonstrated that (SoyaPC + DOTAP)-based fusogenic liposome-coated nanoparticles could be a powerful tool for rapid internalization of nanoparticles into cultured corneal endothelial cells and ex vivo corneal tissue before preservation and transplantation without affecting cell viability.
Conclusions
The prolonged time of nanoparticle internalization (24 h to 48 h) causes damage to the donor tissue and cells and increases the risk of contamination at 37 °C. Therefore, we have developed fusogenic liposome-coated nanoparticles for very rapid internalization of nanoparticles into cultured corneal endothelial cells and ex vivo corneal endothelial tissue. Here, we have shown that the fusogenic liposome-coated nanoparticles have the intrinsic ability to efficiently and rapidly internalize within 3 h by possibly fusing with the cell membrane and bypassing the endocytic pathway. The internalization of the fusogenic liposome-coated nanoparticles in endothelial cells associated with ex vivo corneas did not cause cytotoxicity for at least up to 48 h after internalization. Thus, (SoyaPC + DOTAP)-based fusogenic liposome-coated nanoparticles have great potential as a platform for engineering cells and endothelial tissue of donor corneas to facilitate prophylactic drug delivery during storage and after transplantation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the SERB-EMR (EMR/2017/002449) research grant from DST, Government of India, and Roche Collaborative Research Fellowship from ARVO (USA) to Sudhir H. Ranganath and in part by the intramural research grant from Sree Siddaganga Education Society, Tumakuru (3150/1516). We acknowledge Mrs B. S. Suma (Bioimaging Facility, JNCASR, Bangalore, India) for confocal microscopy, Mr Arun Aravindakshan, AMRL TEM Facility, JNCASR, Bangalore, India and Mr Parthiban, EM Facility, IISc, Bangalore for TEM imaging. We are grateful to Ms Namratha, Ms T. G. Deepika and Mr K. S. Suraj (SIT, Tumakuru) for their technical help with cell viability assay and nanoparticle preparation.References
- D. Robaei and S. Watson, Clin. Exp. Ophthalmol., 2014, 42, 213–214 CrossRef PubMed.
- H. Hashemi, A. Yekta, E. Jafarzadehpur, A. Doostdar, H. Ostadimoghaddam and M. Khabazkhoob, Eye, 2017, 31, 1221–1228 CrossRef CAS PubMed.
- P. Gain, R. Jullienne, Z. He, M. Aldossary, S. Acquart, F. Cognasse and G. Thuret, JAMA Ophthalmol., 2016, 134, 167–173 CrossRef PubMed.
- Y. T. Zhu, S. Tighe, S. L. Chen, T. John, W. Y. Kao and S. C. Tseng, Curr. Ophthalmol. Rep., 2015, 3, 207–217 CrossRef PubMed.
- S. C. A. Mohamed and P. Garg, Indian J. Ophthalmol., 2016, 64, 635–638 CrossRef PubMed.
- W. Xu, Y. Tao, L. Wang and Y. Huang, Med. Sci. Monit., 2017, 23, 3649–3656 CrossRef PubMed.
- H. Lam and M. R. Dana, Int. Ophthalmol. Clin., 2009, 49, 31–41 CrossRef PubMed.
- U. Rauen, U. Kerkweg, M. C. Wusteman and H. de Groot, Cornea, 2006, 25, 68–77 CrossRef PubMed.
- M. Y. Thanuja, B. S. Suma, D. Dinesh, S. H. Ranganath and S. P. Srinivas, J. Ocul. Pharmacol. Ther., 2021, 37, 399–411 CrossRef CAS PubMed.
- M. Shivanna, G. Rajashekhar and S. P. Srinivas, Invest. Ophthalmol. Visual Sci., 2010, 51, 1575–1582 CrossRef PubMed.
- M. Shivanna and S. P. Srinivas, Exp. Eye Res., 2009, 89, 950–959 CrossRef CAS PubMed.
- J. H. Lass, B. A. Benetz, S. V. Patel, L. B. Szczotka-Flynn, R. O'Brien, A. R. Ayala, M. G. Maguire, Y. J. Daoud, M. A. Greiner, S. B. Hannush, W. B. Lee, T. F. Mauger, H. J. Menegay, M. D. Mifflin, M. B. Raizman, J. Rose-Nussbaumer, R. L. Schultze, G. A. Schmidt, A. Sugar, M. A. Terry, D. D. Verdier and G. Cornea, Preservation Time Study, JAMA Ophthalmol., 2019, 137, 185–193 CrossRef PubMed.
- Writing Committee for the Cornea Donor Study Research Group, J. H. Lass, B. A. Benetz, R. L. Gal, C. Kollman, D. Raghinaru, M. Dontchev, M. J. Mannis, E. J. Holland, C. Chow, K. McCoy, F. W. Price Jr, A. Sugar, D. D. Verdier and R. W. Beck, Ophthalmology, 2013, 120, 2428–2435 CrossRef PubMed.
- M. Y. Thanuja, S. H. Ranganath and S. P. Srinivas, J. Ocul. Pharmacol. Ther., 2022, 38, 664–681 CrossRef CAS PubMed.
- N. Okumura, S. Kinoshita and N. Koizumi, J. Ophthalmol., 2017, 2017, 2646904 Search PubMed.
- R. A. Steinhardt and J. M. Alderton, Cornea, 2006, 25, 839–844 CrossRef PubMed.
- T. A. Fuchsluger, U. Jurkunas, A. Kazlauskas and R. Dana, Hum. Gene Ther., 2011, 22, 549–558 CrossRef CAS PubMed.
- J. H. Lass, L. B. Szczotka-Flynn, A. R. Ayala, B. A. Benetz, R. L. Gal, A. J. Aldave, M. M. Corrigan, S. P. Dunn, T. L. McCall, S. Pramanik, G. O. Rosenwasser, K. W. Ross, M. A. Terry, D. D. Verdier and Writing Committee for the Cornea Preservation Time Study Group, Cornea, 2015, 34, 601–608 CrossRef PubMed.
- G. S. L. Peh, H. P. Ang, C. N. Lwin, K. Adnan, B. L. George, X. Y. Seah, S. J. Lin, M. Bhogal, Y. C. Liu, D. T. Tan and J. S. Mehta, Sci. Rep., 2017, 7, 14149 CrossRef PubMed.
- A. M. Williams and K. W. Muir, Clin. Ophthalmol., 2018, 12, 1049–1059 CrossRef PubMed.
- M. Li, J. Ma, Y. Gao and L. Yang, Cytotherapy, 2019, 21, 3–16 CrossRef CAS PubMed.
- M. Shivanna and S. P. Srinivas, Mol. Vision, 2010, 16, 1781–1790 CAS.
- C. Anupama, A. Shettar, S. H. Ranganath and S. P. Srinivas, J. Ocul. Pharmacol. Ther., 2023, 39, 70–79 CrossRef CAS PubMed.
- A. Chalimeswamy, M. Y. Thanuja, S. H. Ranganath, K. Pandya, U. B. Kompella and S. P. Srinivas, J. Ocul. Pharmacol. Ther., 2022, 38, 74–84 CrossRef CAS PubMed.
- Y. Y. Yuan, C. Q. Mao, X. J. Du, J. Z. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476–5480 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- W. L. Corwin, J. M. Baust, J. G. Baust and R. G. Van Buskirk, Cryobiology, 2011, 63, 46–55 CrossRef CAS PubMed.
- Z. Luznik, Z. Sun, H. Nakagawa, A. W. Taylor, U. V. Jurkunas, J. Yin and R. Dana, JAMA Ophthalmol., 2020, 138, 1192–1195 CrossRef PubMed.
- D. M. Meisler, T. Koeck, J. T. Connor, K. S. Aulak, B. H. Jeng, J. G. Hollyfield, D. J. Stuehr and K. G. Shadrach, Exp. Eye Res., 2004, 78, 891–894 CrossRef CAS PubMed.
- Y. W. Naguib, S. Saha, J. M. Skeie, T. Acri, K. Ebeid, S. Abdel-Rahman, S. Kesh, G. A. Schmidt, D. Y. Nishimura, J. A. Banas, M. Zhu, M. A. Greiner and A. K. Salem, Biomaterials, 2021, 275, 120842 CrossRef CAS PubMed.
- R. Agarwal, I. Iezhitsa, P. Agarwal, N. A. Abdul Nasir, N. Razali, R. Alyautdin and N. M. Ismail, Drug Delivery, 2016, 23, 1075–1091 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- S. Bhattacharjee, D. Ershov, M. A. Islam, A. M. Kämpfer, K. A. Maslowska, J. van der Gucht, G. M. Alink, A. T. Marcelis, H. Zuilhof and I. M. Rietjens, RSC Adv., 2014, 4, 19321–19330 RSC.
- H. Sun, C. Jiang, L. Wu, X. Bai and S. Zhai, Front. Bioeng. Biotechnol., 2019, 7, 414 CrossRef PubMed.
- S. Guo, Y. Liang, L. Liu, M. Yin, A. Wang, K. Sun, Y. Li and Y. Shi, J. Nanobiotechnol., 2021, 19, 32 CrossRef CAS PubMed.
- B. Peter, I. Lagzi, S. Teraji, H. Nakanishi, L. Cervenak, D. Zambo, A. Deak, K. Molnar, M. Truszka, I. Szekacs and R. Horvath, ACS Appl. Mater. Interfaces, 2018, 10, 26841–26850 CrossRef CAS PubMed.
- X. Zhang, G. Ma and W. Wei, NPG Asia Mater., 2021, 13, 52 CrossRef CAS.
- E. Okoampah, Y. Mao, S. Yang, S. Sun and C. Zhou, Colloids Surf., B, 2020, 196, 111312 CrossRef CAS PubMed.
- B. Kim, H. B. Pang, J. Kang, J. H. Park, E. Ruoslahti and M. J. Sailor, Nat. Commun., 2018, 9, 1969 CrossRef PubMed.
- S. Kube, N. Hersch, E. Naumovska, T. Gensch, J. Hendriks, A. Franzen, L. Landvogt, J. P. Siebrasse, U. Kubitscheck, B. Hoffmann, R. Merkel and A. Csiszar, Langmuir, 2017, 33, 1051–1059 CrossRef CAS PubMed.
- J. Kunisawa, T. Masuda, K. Katayama, T. Yoshikawa, Y. Tsutsumi, M. Akashi, T. Mayumi and S. Nakagawa, J. Controlled Release, 2005, 105, 344–353 CrossRef CAS PubMed.
- T. Wiedenhoeft, S. Tarantini, A. Nyul-Toth, A. Yabluchanskiy, T. Csipo, P. Balasubramanian, A. Lipecz, T. Kiss, A. Csiszar, A. Csiszar and Z. Ungvari, GeroScience, 2019, 41, 711–725 CrossRef CAS PubMed.
- J. Yang, A. Bahreman, G. Daudey, J. Bussmann, R. C. Olsthoorn and A. Kros, ACS Cent. Sci., 2016, 2, 621–630 CrossRef CAS PubMed.
- M. Hoffmann, N. Hersch, S. Gerlach, G. Dreissen, R. Springer, R. Merkel, A. Csiszar and B. Hoffmann, Int. J. Mol. Sci., 2020, 21(6), 2244 CrossRef CAS PubMed.
- A. Werlein, A. Peters, R. Ngoune, K. Winkler and G. Putz, Biochim. Biophys. Acta, 2015, 1848, 1599–1608 CrossRef CAS PubMed.
- M. C. Filion and N. C. Phillips, Biochim. Biophys. Acta, 1997, 1329, 345–356 CrossRef CAS PubMed.
- N. Hersch, B. Wolters, Z. Ungvari, T. Gautam, D. Deshpande, R. Merkel, A. Csiszar, B. Hoffmann and A. Csiszar, J. Biomater. Appl., 2016, 30, 846–856 CrossRef CAS PubMed.
- M. Vicario-de-la-Torre, M. Caballo-Gonzalez, E. Vico, L. Morales-Fernandez, P. Arriola-Villalobos, B. De Las Heras, J. M. Benitez-Del-Castillo, M. Guzman, T. Millar, R. Herrero-Vanrell and I. T. Molina-Martinez, Polymers, 2018, 10(4), 425 CrossRef PubMed.
- J. Kotoucek, F. Hubatka, J. Masek, P. Kulich, K. Velinska, J. Bezdekova, M. Fojtikova, E. Bartheldyova, A. Tomeckova, J. Straska, D. Hrebik, S. Macaulay, I. Kratochvilova, M. Raska and J. Turanek, Sci. Rep., 2020, 10, 5595 CrossRef CAS PubMed.
- A. Ranjan, S. Das and S. K. Sahu, Indian J. Ophthalmol., 2014, 62, 935–937 CrossRef PubMed.
- S. H. Ranganath, Z. Tong, O. Levy, K. Martyn, J. M. Karp and M. S. Inamdar, Stem Cell Rep., 2016, 6, 926–939 CrossRef CAS PubMed.
- J. Yoo and Y. Y. Won, ACS Biomater. Sci. Eng., 2020, 6, 6053–6062 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00535f |
| ‡ Authors have equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |