
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper and silver nanowires for CO2 electroreduction
Andrea
Conte
 ,
Marco
Baron
,
Sara
Bonacchi
*,
Sabrina
Antonello
* and
Alessandro
Aliprandi
*
,
Marco
Baron
,
Sara
Bonacchi
*,
Sabrina
Antonello
* and
Alessandro
Aliprandi
*
University of Padova, Department of Chemistry, Via Marzolo 1, I-35131 Padova, Italy. E-mail: alessandro.aliprandi@unipd.it; sabrina.antonello@unipd.it; sara.bonacchi@unipd.it
First published on 19th January 2023
Abstract
Copper and silver nanowires have been extensively investigated as the next generation of transparent conductive electrodes (TCEs) due to their ability to form percolating networks. Recently, they have been exploited as electrocatalysts for CO2 reduction. In this review, we present the most recent advances in this field summarizing different strategies used for the synthesis and functionalization/activation of copper and silver nanowires, as well as, the state of the art of their electrochemical performance with particular emphasis on the effect of the nanowire morphology. Novel perspectives for the development of highly efficient, selective, and stable electrocatalysts for CO2 reduction arise from the translation of NW-based TCEs in this challenging field.
 Andrea Conte | Andrea Conte obtained his Master's Degree in Chemistry at the University of Padova (Italy) in Molecular Electrochemistry and Nanosystems Group under the supervision of Prof. Flavio Maran (2021), working on the synthesis and characterization of gold nanoclusters. He got a research grant under the supervision of Prof. Sara Bonacchi working on metal nanoclusters as building blocks to assemble novel hierarchical architectures. Currently, he is a PhD student at the University of Padova in Molecular Sciences under the supervision of Prof. Sara Bonacchi and Dr Marco Baron for a research project that concerns the electrocatalytic production of renewable feedstocks. |
 Marco Baron | Marco Baron received his PhD in 2014 at the University of Padova (Doctor Europaeus), working in the groups of Prof. Marino Basato and Dr Stéphane Bellemin-Laponnaz (University of Strasbourg). Subsequently, he worked as a Postdoc Researcher at the University of Padova (Prof. Cristina Tubaro and Prof. Andrea Biffis) and at the Technical University of Munich (Dr Alexander Pöthig). Since 2019, he has been an Assistant Professor in Inorganic Chemistry at the University of Padova. In 2020, he received the Flavio Bonati Award by the Organometallic Group of the Italian Chemical Society. |
 Sara Bonacchi | Sara Bonacchi obtained her PhD in Chemistry at the University of Bologna (Italy) under the supervision of Prof. Prodi (2009) working on photophysical investigations of fluorescent nanoparticles. Afterward, she joined Prof. Samorí's group at “Institut de Science et Ingenierie Supramoleculaires” in Strasbourg (France). In 2014, she was awarded the Marie Curie Intra-European Fellow for a project on hybrid organic/inorganic heterostructures. In 2018, she was appointed Assistant Professor in the Molecular Electrochemistry and Nanosystems Group, led by Prof. Maran at Department of Chemistry of University of Padova (Italy). Currently, she is an Associate Professor working on molecular photo-electro-catalysts and nanoclusters to generate valuable chemicals. |
 Sabrina Antonello | Sabrina Antonello obtained her PhD in Chemistry at the University of Padova (Italy) under the supervision of Prof. F. Maran in 2001 working on dissociative electron transfer reactions. Her thesis was awarded in 2001 by the Electrochemistry Division of the Italian Chemical Society. She carried out research as a Post-Doc and since 2015 as Associate Professor in the Molecular Electrochemistry and Nanosystems group at the Department of Chemistry of the University of Padova. Her research interests include electron transfer reaction across interfaces and through molecules, redox catalysis, electroactive monolayer-protected nanoclusters, and electrochemical microscopy techniques. |
 Alessandro Aliprandi | Alessandro Aliprandi is currently an Associate Professor at the Department of Chemical Sciences (DISC, University of Padova, Italy). He received his PhD at the Institute of Supramolecular Science and Engineering (ISIS), University of Strasbourg in 2015 under the supervision of Prof. Luisa De Cola working on the self-assembly of luminescent Pt(II) complexes. To expand his skills in the field of low-dimensional materials such as graphene and copper nanowires, he carried out his postdoctoral studies in the group of Prof. Paolo Samorì. In 2018 he was appointed as Ingénieur de Recherche (CNRS). In 2020, he was awarded with an ERC-STG. His fields of interest range from electroluminescence to chemiluminescence, photocatalysis, CO2 valorisation and electrochromism. |
Introduction
Addressing global warming is a compulsory challenge for our generation, requiring urgent action to stop the emission of CO2 to the atmosphere toward net-zero carbon emissions no later than 20501 and realize ways to remove existing carbon. Furthermore, decarbonizing global economy before irreversible damage occurs to the world and to our way of life, requires swift and worldwide efforts to find sustainable energy solutions together with policy initiatives, strengthened collaborations between companies and researchers, and effective strategies for education and communication.In this broad scenario, the combination of sustainable-renewable energy resources such as, solar and wind energy with electrochemical catalytic CO2 conversion, minimizing the high thermodynamic activation barrier of CO2 reduction and speeding up the reactions, has emerged as a pivotal strategy to obtain add-valued multi-carbon feedstocks within a pure circular economy paradigm.2 Obviously, this approach must consider the performance required to reach economic viability in terms of products, market size and price, operational costs, efficiency, stability, etc.3 The majority of existing CO2 reduction electrocatalysts can be divided into three groups, i.e., metallic, non-metallic, and molecular catalysts, each presenting advantages and drawbacks in terms of efficiency, robustness and selectivity, but their discussion is not the focus of this mini-review.4 Herein, we aim to provide an overview of a particular class of nanomaterials, metallic nanowires (NWs), which have been employed in the last decade as transparent conductive electrodes (TCE),5 as electrocatalysts for the electrochemical reduction of CO2. As TCE, the metallic NWs6 have already found application in working devices such as organic light-emitting diodes (OLEDs),7 organic photovoltaic modules,8 and electrochromic displays.9 Only recently, metallic NWs have been considered as promising materials for the CO2 electrochemical reduction reaction (CO2RR). Research endeavours have also been devoted to the selection of the most suitable metal in terms of electrochemical CO2RR efficiency, highlighting that noble metals such as Ag,10 Au,11 and Pd12 are efficient electrocatalysts for CO2 reduction with good selectivity towards CO but only Cu is of particular interest as a unique metal with remarkable selectivity towards formic acid, methane, methanol, and other multi-carbon oxygenates and hydrocarbons (denoted as C2+) due to the moderate binding energy between the Cu metal and the reaction intermediates.13–15 Considering these scientific breakthroughs, this abundant metal has been exploited to form inexpensive CuNWs as potential highly active and selective nanostructured materials for the electrochemical CO2RR at ambient temperature and pressure. Besides, AgNWs also show versatile properties as catalysts for the electrochemical CO2RR. Interestingly, the doping of these metals NWs with heteroatoms has been recently demonstrated to be an effective approach to increase the NWs' electrocatalytic efficiency,16 since they have a profound effect on the electronic structure of the metal NWs and on binding properties of the chemical species that participate in the reaction as reagents, products, or intermediates. Ultimately, alloying also plays a crucial role in increasing the wire resistance to poisoning processes.17
The importance of this mini-review is to provide a comprehensive understanding of the recent developments of the more promising metal NWs towards catalytic applications in the field of electrochemical CO2RR. In the first section, we describe the main recent methodologies to synthesize NWs, while in the second section, we present insights into the electrocatalytic performance of NWs toward the CO2RR.
Metal NWs synthetic strategies
Since the discovery of the CO2-to-C2 electroreduction on Cu electrodes by Hori and coworkers in the 1980s,13,18–21 researchers have contributed efforts to elucidate the reaction mechanisms and the synthesis and characterization of copper-based nanostructured catalytic materials with the aim of enhancing their efficiency, selectivity, and extending their lifetime. At the begging of the 90s, Prof. Hori and coworkers also recognized Ag as one of best electrocatalyst, enabling the formation of CO with a faradaic efficiency (FE) of 81.5% (E = −1.14 V), thus establishing a potential alternative to Cu for the CO2RR in terms of efficiency and catalyst cost.22 Among the various methods developed thus far, we mainly focus on the wet chemical approaches including the hydrothermal route and reduction of metal precursors, allowing the growth of seed-based NWs, which are considered facile and scalable methods to produce metal NWs, thus making them an attractive strategy for industrial applications.Synthesis of CuNWs
The preparation of CuNWs typically involves aqueous solution and cost-effective green chemicals, which represent important prerequisites for promoting their industrial application and development. A pioneering example was reported by Liu and coworkers, who described the hydrothermal synthesis of CuNWs based on the reduction of a Cu(II)–glycerol complex in highly concentrated NaOH and glycerol aqueous solutions with the assistance of sodium dodecyl benzenesulfonate (SDBS) surfactant at room temperature.23 However, the development of this synthetic method was hindered by the requirement of a high amount of NaOH (i.e., 2.4 kg of NaOH waste to produce 1 g of CuNWs)24 until 2010, when mild reducing agents and aqueous/neutral solutions were successfully developed. Different efficient synthetic strategies have been optimized to tune the NWs anisotropic growth. In particular, all these methodologies are typically based on four components: (i) water as the main solvent; (ii) copper source such as CuCl2, CuBr2, Cu(NO3)2, Cu(acac)2, and CuCl; (iii) reducing agents such as, glucose, hydrazine, and catechin;25 and (iv) capping agents such as aliphatic amines. Notably, the type of capping agent employed during the synthesis of CuNWs ensures anisotropic nanocrystalline growth, leading to the formation of elongated nanostructures instead of other geometrical shapes such as nanocubes and nanoparticles.26Hereafter, we summarize the most relevant CuNW synthetic procedures developed in the last few years. Pure oleylamine (OAm) has been used as a reducing and capping agent at the same time, but high temperature (200 °C) and inert atmosphere are required.27 Water can be used as an environment-friendly alternative to organic solvents, but Cu ions need to be properly stabilized in aqueous conditions to form a nanocrystalline material upon reduction. This requirement has been achieved in alkaline conditions and using hydrazine as a reducing agent.28 Under alkaline conditions, the use of natural amino acids as capping agents has been exploited to obtained Cu-based nanostructures. Indeed, amino acids such as lysine, tryptophan, and glycine assist the formation of well-defined CuNWs.29 However, this approach is limited by the necessity of using the highly toxic hydrazine as a reducing agent. Nowadays, the most used procedure for the synthesis of CuNWs is the hydrothermal method under neutral conditions, which is much cheaper and safer, and thus attractive for large-scale production. The optimized procedures employ glucose as a mild reducing agent together with aliphatic amines such as hexadecylamine (HDA) and octadecylamine (ODA) at temperatures slightly above 100 °C.30 Under the latter conditions, the reaction mechanism starting from a Cu(II) precursor and resulting in the formation of CuNWs has been recently elucidated by Sue and coworkers, which involves a stepwise reduction through Cu(I)–alkylamine species.26 Consequently, mild reducing agents can be used stoichiometrically with respect the Cu(II) precursor because they are necessary to promote the reduction from Cu(I) to Cu(0). Notably, the morphology of the so-obtained CuNWs can be tuned by changing the type and quantity of alkylamine. For instance, by reducing the amine quantity from four to three equivalents with respect to the Cu(II) source, the diameter of the CuNWs increases. The same behaviour is observed by changing the alkylamine from ODA to HDA; however, the formation of thicker NWs is often accompanied by the formation of impurities such as nanoparticles.26 As reported by Sue and coworkers,31 phenylenediamines can be also employed instead of alkylamines as capping agents to produce very stable CuNWs due to the thin coating layer of conducting polyphenylenediamine, thus exhibiting excellent anti-oxidation properties even in water. Overall, the Cu(II) reduction mechanism strongly depends on the temperature, where the Maillard reaction, which produces species (reductones) with strong reducing power, is the dominant mechanism only if the synthesis is carried out at low temperature and using excess glucose.30,32 Alternatively, at a temperature higher than 100 °C, the Cu(I)–alkylamine mechanism prevails when a stoichiometric amount of glucose is used,26 as shown in Fig. 1.
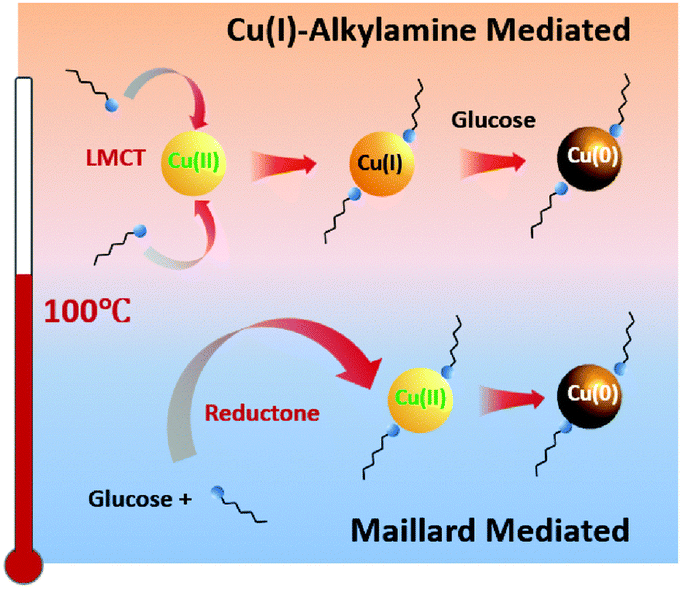 | ||
| Fig. 1 Proposed dominant mechanism for the synthesis of Cu nanowires below and above 100 °C. Reproduced with permission from ref. 26. Copyright 2020, The Royal Chemical Society. | ||
Activation of CuNWs
Importantly, CuNWs are covered by a native insulating CuxOy layer, which is formed during their hydrothermal growth, highly reducing their electrical conductivity. In the quest of forming highly conductive percolating networks for optoelectronic applications, different strategies have been reported to solve this issue. Typically, the oxide layer is removed via post-deposition treatments such as high-temperature annealing (>175 °C) in a reducing hydrogen atmosphere or annealing-free processes consisting of acid etching33 and photonic sintering.34 Recently, morphological studies combined with chemical reactivity analysis have highlighted how these types of approaches can also strongly affect the catalytic performance of CuNWs, thus opening novel paths for improved and tuneable nanostructures.35 Another important challenge in this field is related to the limited chemical stability of CuNWs once they are activated since they are prone to oxidation with consequent loss of electrical conductivity. Therefore, great efforts have been devoted to protecting CuNWs from oxidation including nickel alloying36 and the use of semiconducting metal oxides shells,37 which require cumbersome additional steps such as atomic layer deposition or the electrodeposition of a metal (Zn, Sn, or In) on the surface of the CuNW network, followed by oxidation to create a transparent oxide shell.37 Interestingly, these surface modifications have been designed to improve the oxidation resistance without losing the optical transparency and retaining the high conductivity required for TCE application, but there are no investigations on their impact on the electrocatalytic performance in the CO2 conversion into valuable chemicals or fuel.Synthesis of AgNWs
Typically, AgNWs are synthetized by polyol reduction procedure.38–40 To prevent the uncontrolled aggregation of the twinned seeds and to assure the normal growth of AgNWs, the seeds are typically stabilized as follows: (i) addition of poly-vinylpyrrolidone (PVP) to the reaction solution as a selective capping agent that can stabilize the Ag seeds on the {100} surface via physical adsorption; (ii) addition of chloride ions in the form of halide salts to the reaction, enabling electrostatic stabilization of the seeds; and (iii) regulation of the reduction rate to minimize the seed aggregation. Furthermore, it has been shown that tight control over the molar PVP/AgNO3 ratio is essential to create consistent and stable AgNWs. The use of a low PVP/AgNO3 molar ratio leads to an increase in diameter due to the fact that AgNWs grow on both the (111) and (100) facets. Alternatively, a higher PVP/AgNO3 molar ratio gradually reduces the diameter of the NWs.41 Above a PVP/AgNO3 molar ratio of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the surplus PVP molecules cover the entire surface of the Ag nanoparticles, preventing their anisotropic development into nanowires, thus generating microparticles.41 Using this approach, it has been proven that the best PVP/AgNO3 ratio is 7.5:1.
:1, the surplus PVP molecules cover the entire surface of the Ag nanoparticles, preventing their anisotropic development into nanowires, thus generating microparticles.41 Using this approach, it has been proven that the best PVP/AgNO3 ratio is 7.5:1.
Fig. 2 shows a schematic representation of the synthesis outcomes by varying the AgNO3 concentration in the aspect ratio of AgNWs by the polyol method. In this synthesis, the effect of AgNO3 concentration on the morphology of AgNWs is investigated by changing the concentration of AgNO3 without changing the PVP concentration.
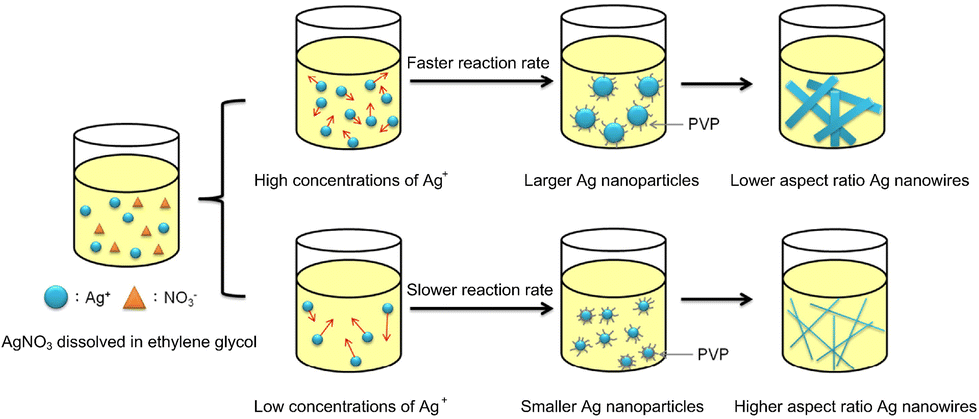 | ||
| Fig. 2 Effect of the different concentrations of silver nitrate on the silver nanowire diameter. Reproduced with permission from ref. 38. Copyright 2019, MDPI. | ||
Others critical variables that can be exploited to maximize the yield of AgNWs are the reaction time, temperature and molecular weight of PVP. Unalan and coworkers showed that the use of PVP with different length chains results in the production of AgNWs with tuneable aspect ratios.41 Alternatively, when ethylene glycol is employed as the polyol reducing agent, the reduction efficiency of Ag ions is strongly affected by the temperature due to the highly temperature dependence oxidation efficiency of this agent.
Wiley and coworkers carried out a series of Ag nanowire synthesis at various temperatures to study the dependency of the reducing ability of ethylene glycol in terms of temperature,42 showing that the longest Ag NWs with also the largest diameters (40–70 nm) were produced when the reaction was carried out at the lowest temperature (130 °C). Increasing the temperature led to shorter and thinner NWs. The diameter of the NWs is affected by a variety of other elements in addition to temperature. Zhan and coworkers described that increasing the quantity of AgNO3 induced an increase in the diameter of the AgNWs,43 while decreasing the concentration of AgNO3 but keeping the quantity of PVP constant led to the formation of AgNWs with higher aspect-ratio values.39
The presence of chloride ions also affects the generation of AgNWs by stabilizing the silver nanoparticles44 and by lowering the concentration of free silver ions in the solution via the production of AgCl, ultimately slowing the reduction of Ag(I), and then allowing the growth of NWs despite the presence of Ag nanoparticles.40 Positively, in the case of AgNWs, highly conductive percolating networks can be obtained without any post treatment process due to the absence of an insulating oxide layer.
Electrocatalytic performances for CO2 reduction
CO2 reduction is a complex multielectron and multiproton reaction, potentially yielding a wide range of products. The reduction standard potentials, E°, of the different processes range in a very narrow potential interval going from −0.2 V vs. RHE for the formation of formic acid to 0.17 V for methane and are very similar to the competitive reduction of water.45 A number of exhaustive reviews on electrochemical CO2RR on bulk metal can be found in the literature.2,3,46 In this mini-review, we focus only on the most recent outcomes in the field with metal NW-based catalysts.The H-cell is the typical divided electrochemical cell used for testing the electrocatalytic activity and selectivity of supported nanostructured materials with respect to the CO2RR. The main advantage of this type of cell refers to the presence of two compartments connected through a conductive membrane, with one containing the working and the reference electrodes and the other, the counter electrode this setup allows the conversion of a massive amount of CO2 into add-value products without the interference of anodic reaction products. As a conductive membrane, Nafion represents the most widely employed polymer although a better choice is represented by anion exchange membranes, which limit the polarization losses and detrimental proton concentration on the catholyte.47 Typically, an aqueous solution of 0.1 M in KHCO3 is employed for both voltammetric and controlled potential electrolysis (CPE) experiments. In particular, the activity of the catalyst is evaluated by cyclic voltammetry (CV) experiments, enabling the measurement of the current density (j) as the current normalized over the electroactive electrode surface at a given potential value, E, which is directly correlated with the electron transfer (ET) event promoting the CO2RR. In terms of chemical transformation, the faradaic efficiency (FE) provides information on the electrochemical CO2RR efficiency given that it is directly correlated with the charge amount consumed during the electrolysis and the concentration of products detected. Analytical tools such as gas chromatography (GC) and nuclear magnetic resonance (NMR) are the most widely employed for the qualitative and quantitative estimation of the electrogenerated gaseous and liquid products, respectively, and thus for the proper evaluation of the activity (i.e., efficiency and selectivity) of the different catalysts.48 Ionic chromatography and conductivity measurements have also been used for the estimation of charged products such as formate. FE is calculated by using, for liquid products, the equation FE = (ZnF/Q) × 100, where Z = number of electrons involved in the reaction, n = number of moles generated for any specific product, F = Faraday constant = 96485 C mol−1, and Q = total charge passed during the bulk electrolysis (C). In the case of gaseous products, other parameters such as the gas flow rate and the volume of gas produced should be considered for the calculation of FE, as reported by Nu and coworkers.49
In this framework, another important aspect to consider is the preparation of the working electrode. A simple but effective technique usually utilized for the preparation of NW-based electrodes is the spray coating of an ethanolic dispersion of NWs on selected substrates. This non-covalent approach has the advantage of being extremely versatile given that any type of substrate can be potentially explored, while preserving the electrochemical properties of the pristine NWs. Alternatively, this electrode preparation strategy poses the challenge of maximizing the physisorption adhesion between the substrate and the NWs to minimize their leaking in the solution during the electrolysis, as well as increasing the mechanical performances (i.e., the robustness to scratches), thus strengthening the overall durability. For instance, a glassy carbon electrode (diameter of about 5–10 mm) was used as a support for drop-casting a suspension of ethanol and 5% Nafion with a CuNW concentration of 2 mg mL−1. Interestingly, the authors observed that the use of Nafion resulted in enhanced stability of the as-obtained electrode without affecting the electrochemical CO2RR performance.50 Finally, it is also important to emphasize that the use of different preparation methods to assemble NW-based electrodes and different potentials for the CPE can affect both the FE and the selectivity, therefore hindering the effective comparison of the experimental data reported in the literature to date. To guide the readers in this field, some of the most relevant results involving metal and alloy NWs as electrocatalysts for the electrochemical CO2RR are summarized in Table 1 with the aim to provide a brief glance of the catalyst performance as a function of its composition. Subsequently, all the potential values refer to the reversible hydrogen electrode (RHE).
| Catalyst | Applied potential (V vs. RHE) | Current density (mA cm−2) | FE (%) | Electrolyte | Major products | Ref. |
|---|---|---|---|---|---|---|
| CuNWs | −1.01 | −17.3 | 77.40 | 0.1 M KHCO3 | C2H4 | 35 |
| PTFE–CuNW | −0.6 | 6 | 68 | 0.1 M KHCO3 | HCOOH | 76 |
| CuNWs | −1.1 | 4.25 (A g−1) | 35 (C2H4) | 0.1 M KHCO3 | C2H4, C2H6 | 84 |
| 25 (C2H6) | ||||||
| R-CuO | −1.3 | −25 | 40 (H2) | 0.1 M KHCO3 | H2, CO, HCOOH, C2H4 | 77 |
| 10 (CO) | ||||||
| 15 (HCOOH) | ||||||
| 10 (C2H4) | ||||||
| R-CuO-Nafion-60 | −1.3 | −25 | 30 (H2) | 0.1 M KHCO3 | H2, CO, HCOOH, C2H4 | 77 |
| 35 (CO) | ||||||
| 15 (HCOOH) | ||||||
| 10 (C2H4) | ||||||
| CuCuxONWs | −1.03 | −24.1 | 78.4 (C2+ products) | 0.1 M KHCO3 | C2H4 | 50 |
| Cu68Ag32NWs | −1.17 | −50 | 80 (C1+ and C2+ products) | 0.5 M KHCO3 | CH4 | 55 |
| OD-CuNWs | −0.5 | −4 | 70 | 0.1 M KHCO3 | H2, CO | 85 |
| CuO/Cu2ONWs | −0.7 | −18 | 60 (H2) | 0.1 M KHCO3 | H2, CO, HCOOH | 49 |
| 15 (CO) | ||||||
| 10 (HCOOH) | ||||||
| Cu2SbNWs | −0.90 | −6 | 86.5 | 0.1 M KHCO3 | CO | 63 |
| Cu9AgNWs | −1.2 | −2.7 | 66 (CH4) | 0.1 M KHCO3 | H2, C2H4, CH4 | 58 |
| 25 (H2) | ||||||
| 8 (C2H4) | ||||||
| CuNWs | −1.25 | −7.5 | 55 (CH4) | 0.1 M KHCO3 | CH4 | 86 |
| CuNWsPDA | −0.93 | −20 | 29 (CH4) | 0.5 M NaHCO3 | CH4 | 83 |
| Au-Bypi-CuNWs | −0.9 | −13 | 25 (CH3CHO) | 0.1 M KHCO3 | CH3CHO | 87 |
| Ag/CNWs | −0.67 | −0.23 | 100 (CO) | 0.1 M KHCO3 | CO | 68 |
| AgAu | −1.2 | −5.61 | 99.65 (CO) | 0.1 M KCl | CO | 88 |
| Ag/PTFE-PE-200NWs | −0.9 | −62 | 85 (CO) | 0.5 M NaHCO3 | H2, CO | 89 |
| 15 (H2) | ||||||
| AgNWs | −0.9 | −0.10 | 88 (H2) | 0.1 M KHCO3 | H2, CO, CH4 | 90 |
| 6 (CO) | ||||||
| 2 (CH4) | ||||||
| AgPdNWs | −0.4 | 7 | 94 | 0.1 M KHCO3 | HCOO− | 91 |
| AgNWs | −1.75 (Ag/AgCl) | 16 (CO) | 98 (CO) | 0.5 M NaHCO3 | CO, H2 | 92 |
| 1(H2) | 2 (H2) | |||||
| AgNWs | −1.75 (Ag/AgCl) | 140 (CO) | 70 (CO) | 2 M KOH | CO, H2, HCOO− | 92 |
| 25 (H2) | 10 (H2) | |||||
| 25 (HCOO–) | 10 (HCOO−) | |||||
| CuNWs | −0.7 | −10 | 65 (H2) | 0.1 M KHCO3 | H2, CO | 57 |
| 8 (CO) | ||||||
| 17 (HCOO–) | ||||||
| CuAgNWs | −0.7 | −10 | 46 (H2) | 0.1 M KHCO3 | H2, CO, HCOO− | 57 |
| 14 (CO) | ||||||
| 24 (HCOO−) | ||||||
| OD-CuNWs | −1.1 | −0.85 (CH3CH2OH) | 7.3 (CH3CH2OH) | 0.1 M KHCO3 | CH3CH2OH, CH4, C2H4 | 93 |
| −1.73 (CH4) | 20 (CH4) | |||||
| −2.35 (C2H4) | 20.1 (C2H4) | |||||
| Cu(Ag-20)20 NWs | −1.1 | −4.1 (CH3CH2OH) | 16.5 (CH3CH2OH) | 0.1 M KHCO3 | CH3CH2OH, CH4, C2H4 | 93 |
| −2.39 (CH4) | 10 (CH4) | |||||
| −3.76 (C2H4) | 14.9 (C2H4) | |||||
| CuAuNWs (Au on Cu) | −1.1 | // | 45 (CO) | 0.1 M NaHCO3, 0.5 M NaClO4 | CO, H2, HCOO− | 60 |
| 5 (HCOO−) | ||||||
| 50 (H2) | ||||||
| CuAuNWs (Cu on Au) | −1.1 | // | 55 (CO) | 0.1 M NaHCO3, 0.5 M NaClO4 | CO, H2, HCOO− | 60 |
| 5 (HCOO) | ||||||
| 40 (H2) | ||||||
| CuAgNWs (Cu on Ag) | −1.1 | /// | 45 (CO) | 0.1 M NaHCO3, 0.5 M NaClO4 | CO, H2, HCOO− | 60 |
| 10 (HCOO) | ||||||
| 45 (H2) | ||||||
| CuAgNWs (Ag on Cu) | −1.1 | // | 55 (CO) | 0.1 M NaHCO3, 0.5 M NaClO4 | CO, H2, HCOO− | 60 |
| 10 (HCOO) | ||||||
| 35 (H2) | ||||||
| CuAuNWs | −1.25 (Ag/AgCl) | −3.8 | 89 (CO) | 0.5 M KHCO3 | CO | 94 |
| Cu NWs-Bi NSs | −0.86 | −10 | 87 (HCOO−) | 0.1 M KHCO3 | HCOO− | 95 |
| CuSn | −0.9 | 30 | 78.6 | 0.1 M KHCO3 | HCOO− | 61 |
Electrocatalytic properties of CuNWs and their alloys
Although numerous metal-based nanostructured catalysts have been developed to electrochemically transform CO2 into valuable products and fuels, Cu are still considered the most promising metal to achieve this goal. Indeed, among the metals, Cu exhibits intermediate CO adsorption and relatively weak adsorption of H on its surface, guaranteeing the perfect energy balance to promote the production of C2+ products. Moreover, nanotechnology has opened the door to new approaches, enabling the tuning of the shapes and surface of Cu material nanostructures.51 Today, ad hoc CuNW synthetic protocols ensure not only a large surface area, but also fine control over the exposed facets, allowing high selectivity toward the desired reduction product. Moreover, the sophisticated design of the atomic surface structures of CuNWs and fine control over the catalytic conditions such as pH and CO2 concentration represent innovative strategies not only to control the electrochemical activation of CO2 but also to minimize the competitive reduction of water protons into hydrogen, the so-called hydrogen evolution reaction (HER). In this field, an important example is the selective production of ethylene, which is ascribed to the Cu (100) facets, enabling C–C coupling after stabilization by the adsorption of CO and CO2.45,52 Interestingly, Smith and co-workers demonstrated that the tuning of the length and density of CuNWs represents an effective strategy to modulate the reduction products selectivity.53 In particular, they showed that the increase in the length of the CuNWs is accompanied by the augmented formation of C2H4. This result was attributed to the local increase of pH due to the hindered diffusion of OH− produced during CO2 reduction away from the CuNW network structure. This basic environment favours CO dimerization, and ultimately the formation of C2+ products. More recently, Wang and co-workers highlighted the correlation between the catalytic activity of CuNWs and their length in the range of 4.6–13 μm.49The density of surface defects in terms of grain-boundaries has been also exploited to enhance the catalytic performance of CuNWs. In 2020, Goddard III, Huang and coworkers reported the electrochemical post-treatment of chemically synthesized CuNWs, enabling impressive selectivity towards C2 products by suppressing the C1 pathway.35 In particular, the authors demonstrated that the application of a very large reductive potential (−1.05 V) in the same electrolyte used for the electrochemical CO2RR governs the formation of a highly stepped surface structure with an increased density of catalytic active sites, resulting in an FE for C2H4 greater than 70% and exceptional stability for more than 200 h.
Recently, Xia and coworkers proposed an alternative approach to increase the surface roughness of CuNWs based on a synergistic chemical oxidation with O2 or H2O2. Even if with different efficiencies for these two oxidizing agents, an increased selectivity toward ethylene was observed.50 The use of a post-synthetic electrochemical treatment based on a sequence of potentiostatic pulse at different potentials to remove the capping agent has also shown to be beneficial to increase the conversion of CO2 to CO with an FE of 100% at E = −1.0 V.54
Although a variety of post-synthesis treatments have been recently implemented for CuNWs, three main aspects still need to be improved in this field for the implementation of the practical and robust electrocatalytic production of added-value products from CO2, as follows: (i) the competition between the CO2RR and HER in the highly active sites, which refers to the strong affinity of these active sites to water or electrolytes; (ii) the low CO2RR selectivity among added-value products (e.g., CO vs. HCOOH); and (iii) the long-term stability of CuNWs. In particular, the spontaneous oxidation of Cu NWs is still a major problem to be tackled given it results in the attenuation of the electrode capacitance.
Recently, some of these issues have been overcome by the insertion of proper heteroatoms in the Cu nanostructure. For example, Chen and coworkers recently highlighted a remarkable decrease in overpotential toward methane production as a direct consequence of optimized amounts of Ag interdiffused in the pristine CuNW.55 Generally, CuAg alloy NWs impart larger selectivity towards the formation of hydrocarbons and oxygenated derivatives with respect to pure CuNWs, which is attributed to the ability of Ag atoms to suppress the competitive HER.56 A promising result in this field was reported by Choi and coworkers, who achieved impressive tuneable selectivity from C2H4 to CH4 with a maximum of 72% CH4 yield at −1.17 V. The authors introduced the concept of “intimate atomic CuAg interface”, as schematically shown in Fig. 3, which was realized by means of galvanic replacement of Cu with Ag.57
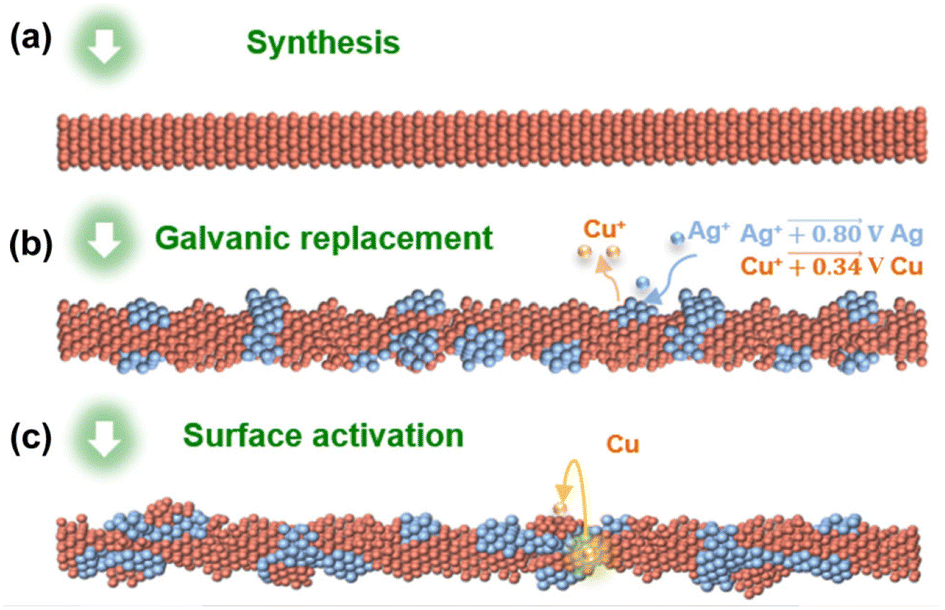 | ||
| Fig. 3 (a) Starting CuNWs. (b) Galvanic replacement on the surface. (c) Surface activation through in situ formation of CuAg assemblies during the electrochemical CO2RR. Reproduced with permission from ref. 57. Copyright 2021, Springer. | ||
This approach enables the creation of localized CuAg alloy, then overcoming the loss of the peculiar Cu ability to form C2 and C3 hydrocarbons and alcohols, which is often observed after alloy formation. Importantly, the proposed procedure can potentially be implemented by exploiting multi-metal compositions with a limited mixing degree with Cu.57
Alternatively, high selectivity toward ethanol production was recently demonstrated by exploiting an electrochemical approach, which was suitable for the production of Au-doped CuNWs.58 In particular, a potentiostatic-pulse electrodeposition procedure was used to produce CuxAuy nanowire arrays starting from a solution of Cu and Au salt, over an Au foil using an aluminium oxide mask. Interestingly, the Cu/Au ratio in the final alloy was optimized by controlling the deposition potential value and the so-obtained catalysts ensured the selective production of ethanol from CO2 with an FE of 48% at E as low as −0.5 to −0.7 V RHE. Recently, CuNWs decorated with AuNPs were grown by homo-nucleation, enabling good selectivity towards multi-carbon products. The best-performing alloy, containing only 0.7% Au, exhibited an FE for C2+ species of 65.3% and a larger current density (12.1 mA cm−2 at −1.25 V) than pure CuNWs.59
The electrodeposition of different metals has also been explored to grow bimetallic catalysts in a segmented configuration, allowing to drive different CO2 reduction product distributions as a function of the relative position of the two metals.60 Recently, the synergy between Cu activity and Sn selectivity has been also proposed for the electrochemical production of formate. Wang and coworkers61 designed, synthesized, and tested a bimetallic copper-tin based core–shell alloy@oxide nanostructure, where the CuSn core ensured efficient electrical contact with the active sites, whereas the catalytic activity and selectivity towards formate were guaranteed by the external layer of SnO2 doped with amorphous Cu. The selectivity was demonstrated to be affected by the thickness of the external shell, resulting in the generation of formate when it was thick enough (1.8 nm), whereas for thinner layers (0.8 nm), the main product was CO.
The doping of CuNWs with halides has been also tested, where iodide showed increased selectivity toward C2H6. The FE observed was 24%, a substantial increase compared to the 4% observed with the undoped CuNW. The increased C2H6 production was accompanied by a concomitant decrease in C2H4. These experimental results were corroborated by DFT calculation, showing that the presence of iodide in the Cu structure strengthened the absorption of C2H4 and accelerated its hydrogenation.62 Interestingly, the use of antimony has been exploited to obtain good selectivity for the formation of CO, as in the case of the electrodeposition of Cu2Sb on a CuNW array.63
Electrocatalytic properties of AgNWs and their alloy
The development of Ag-based electrocatalysts presents several analogies to their Cu-based counterparts. Nanostructures and surface engineering have been explored with Ag, showing a significant effect on its catalytic performance as a function of its shape and dimensions.64 In particular, a larger density of grain boundaries was observed in AgNWs with respect to other structures such as thin films and nanocubes, while the augmented efficiency in the CO2RR was correlated with the high index of exposed facets. For instance, AgNW arrays with a diameter of 200 nm were used by Yao and coworkers to obtain a large current density and an efficiency of over 90% towards CO even at a mild applied electrode potential (E = −0.49 V).65 In the same year, Liu et al. reported an FE of 99.3% at an E value of about −0.85 V, where thinner AgNWs with a diameter of 25 nm showed a better performance than the 100 nm NWs and Ag NPs synthesized using the same polyol approach.66 Interestingly, the authors also highlighted the very good durability of their catalyst setup, which maintained uniform current densities for at least 24 h. In the same year, Zhao and coworkers demonstrated that AgNWs could be efficiently used for syngas generation and an almost 80% FE was achieved at E = −0.9 V with 35 nm NW.67 The mass production of thin-diameter AgNWs can be achieved quite easily and, by simple drop-casting, electrodes with good conductivity, low contact resistance, high and sustained FE can be obtained over any substrate.67Moreover, in the case of CuNWs, the efficiency of the electrical contact between the catalysis sites and the support is known to affect the overall current density and chemical transformation rate of the process. In this framework, a very simple experimental approach based on mixing NWs with carbon powder and Nafion was exploited to support AgNWs with a diameter of 200–400 nm on a glassy carbon electrode, showing a superior catalytic performance to that of NWs with a similar diameter.68 Alternatively, the core–shell NW configuration has emerged as an effective solution to boost the catalyst/support coupling, in particular in the vertical array geometry. Li and coworkers reported the largest specific CO2RR activity (413 μA cm−2 at −0.6 V) to date and specific electrocatalytic activity toward CO production (current density 5.27 mA cm−2 at −0.6 V), which were directly correlated with the use of a silver@silver chloride core−shell nanowire array formed by the nanoimprinting technique.69 The exceptional activity and good selectivity were ascribed to the presence of Cl as subsurface atoms in the metal Ag lattice, contributing to the suppressed HER and increased activity.
Heteroatom doping or alloying represents a valuable opportunity toward the discovery of new catalytic materials displaying enhanced activity, including nanostructured Ag-based catalysts.70–72 Moreover, alloying represents an effective approach for increasing the wire resistance to poisoning processes as in the case of alloyed PdAgNWs, where Pd effectively works in term of selectivity toward the formation of formate from electrochemical CO2RR with an overpotential that is nearly zero, while it also reduces the reaction intermediates interaction due to its lower work function, thus overall increasing the catalyst lifetime. However, as a drawback, Pd suffers from rapid deactivation due to CO poisoning.73
Although the use of noble metals as catalysts has limited interest for large-scale application, they still represent proof-of-concept approaches that can pave the way for near future perspective. This is the case of AgAu core–shell NWs, which showed nearly 100% CO2-to-CO electroreduction at −1.2 V as a result of the mixing of Au and Ag in a 0.5:1 ratio to form Ag@Au0.5 alloy NWs88 (Fig. 4).
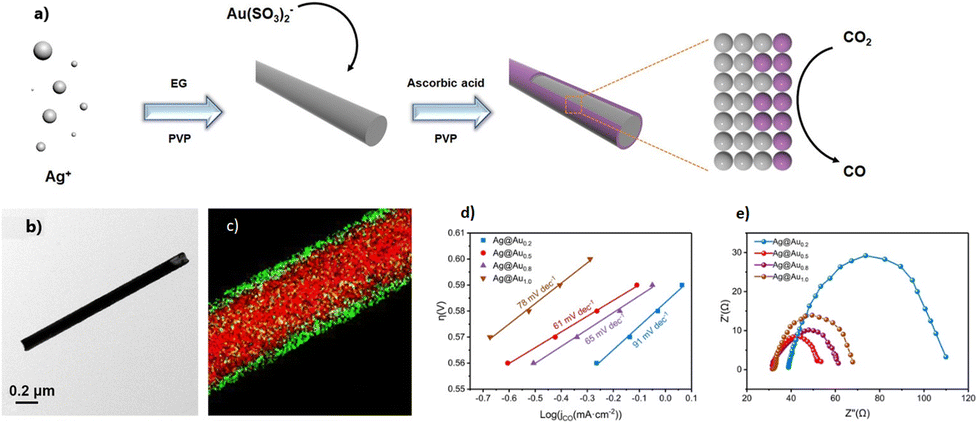 | ||
| Fig. 4 (a) Schematic preparation process of Ag@Au core–shell nanowires. (b) Representative TEM image. (c) HAADF-STEM elemental mapping. (d) Tafel slope of Ag@Au0.2, Ag@Au0.5, Ag@Au0.8 and Ag@Au1 electrocatalysts. (e) EIS characterization of Ag@Au0.2, Ag@Au0.5, Ag@Au0.8 and Ag@Au1 electrocatalysts. | ||
Interesting kinetic studies were also carried out to shed light on the nature of the electrochemical CO2RR rate-determining step. In particular, electrochemical impedance spectroscopy analysis (EIS) and Tafel plot analysis showed the lowest charge transfer resistance (Rct) and smaller slope for the Ag@Au0.5 alloy composition among others, highlighting how the latter parameter can be potentially exploited to tune the heterogeneous ET rate.
Effect of organic capping layers
In the previous sections we discussed some of the most relevant strategies to increase the catalytic performance of metal NWs such as metal doping, defect engineering and controlling exposed facets, while here we focus on the effects of organic molecules and polymers on the surface of NWs.Ligands and surfactants are not always detrimental to the catalytic activity, but it has been showed that they can drive the transformation of CO2 by tailoring the physical and chemical environment of NWs,74 thereby tuning the interactions between the metal and the reaction intermediates.75 The use of an organic layer to impart hydrophobic character to CuNWs has been reported to be an effective strategy to increase the CO2 concentration near the Cu active sites and to inhibit the HER as a result of the decreased water affinity. Polytetrafluoroethylene (PTFE) was used as a surface modifier to allow the increased diffusion of CO2 to the active layer. The so-obtained modified NWs showed good selectivity toward CO formation with an FE of 71% at −0.4 V and a formic acid selectivity at −0.6 V characterized by an FE of 68%.76 A similar effect was also obtained using a Nafion layer over CuONWs, resulting in higher selectivity toward CO, which increased from 8.72% to 43.15% at −1.3 V with suppression of the HER.77 Interestingly, the hydrophobic effect was also invoked to explain the results obtained after the functionalization of the surface of a Cu dendritic structure by a monolayer of 1-octanethiol, which enabled the observation of the strong deactivation of the HER from 71% to 10% FE and an effective increase in the generation of C2H4 and ethanol, achieving an FE of 56% and 17%, respectively, at −1.5 V. This achievement was attributed to the formation of a triple solid/liquid/gas phase boundary, a type of “lotus effect”, favouring the interaction of CO2 with the Cu surface.78
Polydopamine polymer has also been exploited to functionalize the surface of Cu, resulting in a 2.3-times increase in the CH4 generation respect to the bare CuNWs as well as resulting in enhanced stability, most probably due the synergistic effect of the phenolic group in the polymer backbone, which could stabilize the activated CO by H-bonding and the amino groups that capture and transfer protons to it, thus overall favouring the formation of CH4.79 Recently, Agapie and coworkers showed that the incorporation of polyamine on the flat or dendritic Cu surface results in an increase in the selectivity toward ethylene,80,81 while the use of N-substituted pyridinium additives enables the production of ethanol.82 Interestingly, theoretical calculations predict a path toward the use of amine-based ligands to stabilize useful intermediates for the formation of C2 oxygenated products.83
Conclusions
Synthetic protocols for the synthesis of Cu and Ag NWs in a variety of lengths and widths are well established, but they are usually exploited to assemble percolating networks that are typically used as TCE. In this mini-review, we highlighted how these NWs are currently attracting increasing interest as efficient electrocatalysts for CO2 reduction, whose efficiency, selectivity and stability can be modulated through sophisticated surface activation/functionalization. In this ambitious and competitive field, we strongly believe that all the strategies employed so far to activate CuNWs for the fabrication of TCE should be re-evaluated as nanotechnological tools for the control of the nanowire morphology, and thus ultimately the selectivity and efficiency in the CO2RR process. The recent manipulation of the CuNW stepped surfaces to favour C2 products, while suppressing the C1 pathway and hydrogen production is one of the most representative examples of this approach. We also consider that the strategies employed to protect CuNWs from corrosion may also affect the cathodic process by acting as a barrier against charged species such as hydroxonium ions, thus slowing down the HER or other parasitic reactions, while favouring the CO2RR. Considering this, Cu corrosion inhibitors, i.e., a huge family of organic and inorganic compounds that can interact with the Cu surface, can also be re-investigated in a new light not only to protect Cu from oxidation, but also to make the CO2RR more selective and efficient. Finally, we believe that the exploitation of these different but well-established approaches will give new insights into the CO2RR mechanism, paving the way for the further improvement and use of broad metal-based materials towards the electrosynthesis of valuable chemicals from CO2, and ultimately to a greener and sustainable society.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the financial support from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 949087) (A. A.); the Department of Chemical Sciences of the University of Padova [P-DiSC#01BIRD2019-UNIPD] (M. B.); the University of Padova – STARS@UNIPD Starting Grant 2019 “TimeToResponse” project (S. B.); Basell Poliolefine Italia S.r.l (A. C.).Notes and references
- V. Masson-Delmotte, A. Pirani, Y. Chen, J. B. R. Matthews, O. Yelekci, E. Lonnoy, S. L. Connors, L. Goldfarb, S. Berger, R. Yu, T. K. Maycock, P. Zhai, C. Péan, M. I. Gomis, M. Huang, B. Zhou, T. Waterfield and N. Caud, Climate Change 2021 The Physical Science Basis Working Group I Contribution to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, 2021 Search PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. R. Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- T. Sannicolo, M. Lagrange, A. Cabos, C. Celle, J. P. Simonato and D. Bellet, Small, 2016, 12, 6052–6075 CrossRef CAS PubMed.
- S. Ye, A. R. Rathmell, Z. Chen, I. E. Stewart and B. J. Wiley, Adv. Mater., 2014, 26, 6670–6687 CrossRef CAS PubMed.
- Y. Wang, P. Liu, H. Wang, B. Zeng, J. Wang and F. Chi, J. Mater. Sci., 2019, 54, 2343–2350 CrossRef CAS.
- F. Guo, P. Kubis, T. Przybilla, E. Spiecker, A. Hollmann, S. Langner, K. Forberich and C. J. Brabec, Adv. Energy Mater., 2015, 5, 1401779 CrossRef.
- A. Aliprandi, T. Moreira, C. Anichini, M. A. Stoeckel, M. Eredia, U. Sassi, M. Bruna, C. Pinheiro, C. A. T. Laia, S. Bonacchi and P. Samorì, Adv. Mater., 2017, 29, 1703225 CrossRef PubMed.
- K. Chan, Nat. Commun., 2020, 11, 5954 CrossRef CAS PubMed.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H. R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- C. Reller, R. Krause, E. Volkova, B. Schmid, S. Neubauer, A. Rucki, M. Schuster and G. Schmid, Adv. Energy Mater., 2017, 7, 1602114 CrossRef.
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- N. Han, M. Sun, Y. Zhou, J. Xu, C. Cheng, R. Zhou, L. Zhang, J. Luo, B. Huang and Y. Li, Adv. Mater., 2021, 33, 2005821 CrossRef CAS PubMed.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 897–898 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1988, 17–19 RSC.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, Chem. Lett., 1987, 1665–1668 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochimica Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Z. Liu, Y. Yang, J. Liang, Z. Hu, S. Li, S. Peng and Y. Qian, J. Phys. Chem. B, 2003, 107, 12658–12661 CrossRef CAS.
- N. C. Maji and J. Chakraborty, ACS Sustainable Chem. Eng., 2019, 7, 12376–12388 CAS.
- S. Zhao, F. Han, J. Li, X. Meng, W. Huang, D. Cao, G. Zhang, R. Sun and C. P. Wong, Small, 2018, 14, 1800047 CrossRef PubMed.
- T. Zhang, W. Y. Hsieh, F. Daneshvar, C. Liu, S. P. Rwei and H. J. Sue, Nanoscale, 2020, 12, 17437–17449 RSC.
- E. Ye, S. Y. Zhang, S. Liu and M. Y. Han, Chem. – Eur. J., 2011, 17, 3074–3077 CrossRef CAS PubMed.
- Y. Chang, M. L. Lye and H. C. Zeng, Langmuir, 2005, 21, 3746–3748 CrossRef CAS PubMed.
- J. C. Yu, F. G. Zhao, W. Shao, C. W. Ge and W. S. Li, Nanoscale, 2015, 7, 8811–8818 RSC.
- M. Kevin, G. Y. R. Lim and G. W. Ho, Green Chem., 2015, 17, 1120–1126 RSC.
- T. Zhang, F. Daneshvar, S. Wang and H. J. Sue, Mater. Des., 2019, 162, 154–161 CrossRef CAS.
- M. A. J. S. van Boekel, Nahrung-Food, 2001, 45, 150–159 CrossRef CAS PubMed.
- Y. Won, A. Kim, W. Yang, S. Jeong and J. Moon, NPG Asia Mater., 2014, 6, e132 CrossRef CAS.
- S. Ding, J. Jiu, Y. Gao, Y. Tian, T. Araki, T. Sugahara, S. Nagao, M. Nogi, H. Koga, K. Suganuma and H. Uchida, ACS Appl. Mater. Interfaces, 2016, 8, 6190–6199 CrossRef CAS PubMed.
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- A. R. Rathmell, M. Nguyen, M. Chi and B. J. Wiley, Nano Lett., 2012, 12, 3193–3199 CrossRef CAS PubMed.
- Z. Chen, S. Ye, I. E. Stewart and B. J. Wiley, ACS Nano, 2014, 8, 9673–9679 CrossRef CAS PubMed.
- Y. Shi, L. He, Q. Deng, Q. Liu, L. Li, W. Wang, Z. Xin and R. Liu, Micromachines, 2019, 10, 330 CrossRef PubMed.
- J. Y. Lin, Y. L. Hsueh, J. J. Huang and J. R. Wu, Thin Solid Films, 2015, 584, 243–247 CrossRef CAS.
- P. Zhang, I. Wyman, J. Hu, S. Lin, Z. Zhong, Y. Tu, Z. Huang and Y. Wei, Mater. Sci. Eng., B, 2017, 223, 1–23 CrossRef CAS.
- S. Coskun, B. Aksoy and H. E. Unalan, Cryst. Growth Des., 2011, 11, 4963–4969 CrossRef CAS.
- S. M. Bergin, Y. H. Chen, A. R. Rathmell, P. Charbonneau, Z. Y. Li and B. J. Wiley, Nanoscale, 2012, 4, 1996–2004 RSC.
- J. Ma and M. Zhan, RSC Adv., 2014, 4, 21060–21071 RSC.
- B. Wiley, T. Herricks, Y. Sun and Y. Xia, Nano Lett., 2004, 4, 1733–1739 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- A. Gawel, T. Jaster, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U.-P. Apfel, iScience, 2022, 25, 104011 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Wang, Y. Zhu and C. Niu, J. Phys. Chem. Solids, 2020, 144, 109507 CrossRef CAS.
- Z. Lyu, S. Zhu, M. Xie, Y. Zhang, Z. Chen, R. Chen, M. Tian, M. Chi, M. Shao and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 1909–1915 CrossRef CAS PubMed.
- S. Mitchell, R. Qin, N. Zheng and J. Pérez-Ramírez, Nat. Nanotechnol., 2021, 16, 129–139 CrossRef CAS PubMed.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., 2016, 128, 6792–6796 CrossRef.
- H. Hu, M. Liu, Y. Kong, N. Mysuru, C. Sun, M. D. J. Gálvez-Vázquez, U. Müller, R. Erni, V. Grozovski, Y. Hou and P. Broekmann, ACS Catal., 2020, 10, 8503–8514 CrossRef CAS.
- C. J. Chang, S. C. Lin, H. C. Chen, J. Wang, K. J. Zheng, Y. Zhu and H. M. Chen, J. Am. Chem. Soc., 2020, 142, 12119–12132 CrossRef CAS PubMed.
- Y. Wang, C. Niu and Y. Zhu, Nanomaterials, 2019, 9, 173 CrossRef CAS PubMed.
- C. Choi, J. Cai, C. Lee, H. M. Lee, M. Xu and Y. Huang, Nano Res., 2021, 14, 3497–3501 CrossRef CAS.
- W. Zhu, K. Zhao, S. Liu, M. Liu, F. Peng, P. An, B. Qin, H. Zhou, H. Li and Z. He, J. Energy Chem., 2019, 37, 176–182 CrossRef.
- Z. Wei, S. Yue, S. Gao, M. Cao and R. Cao, Nano Research, 2022 DOI:10.1007/s12274-023-5430-z.
- B. A. Zhang and D. G. Nocera, ChemElectroChem, 2021, 8, 1918–1924 CrossRef CAS.
- Y. Wu, X. Deng, H. Yuan, X. Yang, J. Wang and X. Wang, ChemElectroChem, 2021, 8, 2701–2707 CrossRef CAS.
- K. Xiang, Y. Liu, C. Li, X. Liu, H. Yi, L. Wu, F. Shen, M. Liu, P. Wang and H. Liu, Energy Fuels, 2021, 35, 15987–15994 CrossRef CAS.
- S. Mou, Y. Li, L. Yue, J. Liang, Y. Luo, Q. Liu, T. Li, S. Lu, A. M. Asiri, X. Xiong, D. Ma and X. Sun, Nano Res., 2021, 14, 2831–2836 CrossRef CAS.
- A. Salehi-Khojin, H. R. M. Jhong, B. A. Rosen, W. Zhu, S. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- C. Luan, Y. Shao, Q. Lu, S. Gao, K. Huang, H. Wu and K. Yao, ACS Appl. Mater. Interfaces, 2018, 10, 17950–17956 CrossRef CAS PubMed.
- S. Liu, X. Z. Wang, H. Tao, T. Li, Q. Liu, Z. Xu, X. Z. Fu and J. L. Luo, Nano Energy, 2018, 45, 456–462 CrossRef CAS.
- W. Xi, R. Ma, H. Wang, Z. Gao, W. Zhang and Y. Zhao, ACS Sustainable Chem. Eng., 2018, 6, 7687–7694 CrossRef CAS.
- L. Zeng, J. Shi, H. Chen and C. Lin, Energies, 2021, 14, 2840 CrossRef CAS.
- J. Ge, J. Long, Z. Sun, H. Feng, J. Hu, S. W. Koh, Q. Yu, J. Xiao and H. Li, ACS Appl. Energy Mater., 2019, 2, 6163–6169 CrossRef CAS.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérez-Ramírez, J. Catal., 2016, 343, 266–277 CrossRef.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- S. Singh, R. K. Gautam, K. Malik and A. Verma, J. CO2 Util., 2017, 18, 139–146 CrossRef CAS.
- N. Han, M. Sun, Y. Zhou, J. Xu, C. Cheng, R. Zhou, L. Zhang, J. Luo, B. Huang and Y. Li, Adv. Mater., 2021, 33, 2005821 CrossRef CAS PubMed.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- J. Zhang, S. Deo, M. J. Janik and J. W. Medlin, J. Am. Chem. Soc., 2020, 142, 5184–5193 CrossRef CAS PubMed.
- Y. Zhang, Z. Cai, Y. Zhao, X. Wen, W. Xu, Y. Zhong, L. Bai, W. Liu, Y. Zhang, Y. Zhang, Y. Kuang and X. Sun, Nanoscale Horiz., 2019, 4, 490–494 RSC.
- M. Wang, L. Wan and J. Luo, Nanoscale, 2021, 13, 3588–3593 RSC.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- H. Liu, K. Xiang, Y. Liu, F. Zhu, M. Zou, X. Yan and L. Chai, ChemElectroChem, 2018, 5, 3991–3999 CrossRef CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- A. Thevenon, A. Rosas-Hernández, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 58, 16952–16958 CrossRef CAS PubMed.
- Z. Han, R. Kortlever, H. Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed.
- X. Bai, L. Shi, Q. Li, C. Ling, Y. Ouyang, S. Wang and J. Wang, Energy Environ. Mater., 2022, 5, 892–898 CrossRef CAS.
- H. Zhang, Y. Zhang, Y. Li, S. Ahn, G. Tayhas, R. Palmore, J. Fu, A. A. Peterson and S. Sun, Nanoscale, 2019, 11, 12075–12079 RSC.
- Y. Wang, C. Niu, Y. Zhu, D. He and W. Huang, ACS Appl. Energy Mater., 2020, 3, 9841–9847.82 CrossRef CAS.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS PubMed.
- J. Fu, W. Zhu, Y. Chen, Z. Yin, Y. Li, J. Liu, H. Zhang, J. J. Zhu and S. Sun, Angew. Chem., 2019, 131, 14238–14241 CrossRef.
- J. Liu, Y. Wang, H. Jiang, H. Jiang, X. Zhou, Y. Li and C. Li, Chem. – Asian J., 2020, 15, 425–431 CrossRef CAS PubMed.
- D. Raciti, T. Braun, B. M. Tackett, H. Xu, M. Cruz, B. J. Wiley and T. P. Moffat, ACS Catal., 2021, 11, 11945–11959 CrossRef CAS.
- F. Hu, S. C. Abeyweera, J. Yu, D. Zhang, Y. Wang, Q. Yan and Y. Sun, Chem, 2020, 6, 3007–3021 CAS.
- N. Han, M. Sun, Y. Zhou, J. Xu, C. Cheng, R. Zhou, L. Zhang, J. Luo, B. Huang and Y. Li, Adv. Mater., 2020, 33, 2005821 CrossRef PubMed.
- M. de J. Gálvez-Vázquez, S. Alinejad, H. Hu, Y. Hou, P. Moreno-García, A. Zana, G. K. H. Wiberg, P. Broekmann and M. Arenz, Chimia, 2019, 73, 922 CrossRef PubMed.
- L. R. L. Ting, O. Piqué, S. Y. Lim, M. Tanhaei, F. Calle-Vallejo and B. S. Yeo, ACS Catal., 2020, 10, 4059–4069 CrossRef CAS.
- S. Liu, S. Yin, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Cell Rep. Phys. Sci., 2022, 3, 100869 CrossRef CAS.
- L. Li, F. Cai, F. Qi and D. Ma, J. Alloys Compd., 2020, 841, 155789 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |