
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of MoS2-supported Cu single-atom catalysts by machine learning potential for enhanced peroxidase-like activity†
Deting
Xu
ab,
Wenyan
Yin
 a,
Jie
Zhou
a,
Liyuan
Wu
a,
Haodong
Yao
a,
Minghui
Sun
a,
Ping
Chen
a,
Xiangwen
Deng
a and
Lina
Zhao
*ab
a,
Jie
Zhou
a,
Liyuan
Wu
a,
Haodong
Yao
a,
Minghui
Sun
a,
Ping
Chen
a,
Xiangwen
Deng
a and
Lina
Zhao
*ab
aCAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China. E-mail: linazhao@ihep.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 7th March 2023
Abstract
Two-dimensional molybdenum disulfide (2D-MoS2)-supported single atom nanomaterials with enhanced enzyme-like activities are potential substitutes for natural enzymes due to their huge specific surface areas, ease of decoration, high catalytic activity and high catalytic stability. However, their catalytic mechanism remains unclear, making the rational design of nanozymes difficult to achieve. Herein, the mechanisms have been explored to enhance the peroxidase-like activity of MoS2 for H2O2 decomposition. Global neutral network (G-NN) potentials were constructed to accurately and quickly illustrate the mechanisms of MoS2 catalysts and their surface modifications. The high peroxidase-like activity of the MoS2-supported Cu single atom catalyst with sulfur vacancy (Cu@MoS2-Vs) in acidic conditions was systematically evaluated using the trained G-NN potential and density functional theory (DFT), as well as experimental validation. Further analysis of the geometric and electronic properties of pivotal stationary structures revealed the enhanced electron transfer process for high catalytic performance with the modulation of the Cu single atom loading, sulfur vacancy engineering and the surrounding acidic and alkaline environment regulation on the MoS2 basal plane. The results also showed that Cu@MoS2-Vs in an acidic environment exhibited the highest peroxidase-like activity. This work is expected to provide broad implications for the rational design of substrate-supported single-atom catalysts with superior performance and lower cost by surface modification and acidic and alkaline environment regulation.
1. Introduction
Nanozymes, with high stability, high catalytic activity and specified active centers, have become the frontrunners to replace natural enzymes in the future. The nanomaterials used as nanozymes mainly include metal/metal oxides, metal–organic frameworks, and carbon-based materials.1 These nanozymes and their ramifications are expected to have great application potential in biomedical, chemical and environmental fields.2,3 Despite these benefits, the practical application of nanozymes still faces great challenges, particular the rational design of nanozymes with high efficiency.Two-dimensional (2D) materials have unique physical and chemical properties such as simple construction, controllable and multiple active sites, and large surfaces.4 Consequently, 2D materials are often used as potential substrates for nanozymes. Typical 2D materials include graphene,5 hexagonal boron nitride (h-BN),6 transition metal dichalcogenides (TMDs),7,8 graphitic carbon nitride (g-C3N4),9 and layered double hydroxides (LDHs).10 Among them, due to low cost, easy availability, commendable electronic properties, high stability and high catalytic activity, 2D molybdenum disulfide (2D-MoS2) is widely used for the hydrogen evolution reaction (HER)11 and enzymatic reaction.12 However, studies have confirmed that the active sites of MoS2 are mainly concentrated at edges or defect sites. The MoS2 basal plane is catalytically inert but it occupies a large area.13 In order to improve the catalytic activity of the MoS2 basal plane, various valid strategies for adding active sites have been proposed such as N-doping, surface defect engineering, and ambient microenvironment regulation.14–16
Single-atom catalysts (SACs) have been an important research field in recent years. SACs have the advantages of maximum atomic utilization efficiency, high catalytic activity, flexible and adjustable electronic structures, etc., which have been proved to further improve the catalytic activity after doping in 2D materials.8 With atomically active sites and apparent configurations, SACs have been widely used as high-performance nanozymes and hold great promise in making up for the deficiency of the 2D MoS2 inert basal plane. Moreover, SACs are conducive to uncovering the structure–activity relationships, thereby revealing the nature of high enzyme-mimicking performance at a single atomic scale.17
Merging the advantages of 2D MoS2 and SAC catalysis, 2D MoS2-supported SACs have been found to exhibit good performances in enzyme-like catalytic reactions and have, therefore, been widely used in electrocatalysis, photothermal tumor therapy, and nanozyme catalysis.18–21 There have been studies involving single-atom-based MoS2 such as Fe-doped MoS2 nanosheets modified with amine-polyethylene glycol (MoS2@SA-Fe-PEG, denoted MSFP)22 and Fe@MoS2.23 Co–MoS2,24 single Ru atom-doped MoS2 supported by carbon cloth (Ru-MoS2/CC)25 and Al single atom-doped MoS2have also been studied for enhancing the enzymatic activity of MoS2.26 As a cheap and readily available material, non-noble Cu is also expected to be applied in 2D MoS2 supported SACs. A 2D MoS2-supported Cu single atom catalyst, denoted as Cu@MoS2, has been successfully synthesized and applied as a high-performance electrocatalyst for overall water splitting by altering the surface charge distribution and electronic band structure,27 in efficient hydrogen evolution due to changes in atomic coordination structures,28 and in low-temperature CO oxidation due to the charge transfer between the SAC and the substrate.29 Like other 2D MoS2-supported nanomaterials, Cu@MoS2 is also expected to exhibit enzyme-like catalytic activity for biomedical applications, but its actual catalytic mechanism is still unclear, which makes the rational design an obstacle.
Theoretical and experimental studies were systematically implemented to explore the peroxidase-like activity of MoS2 and its ramifications by rational design. To gain insight into the mechanisms that govern the activity and selectivity of MoS2 catalysts, it is important to clarify the detailed reaction paths and electronic properties of pivotal stationary structures. DFT methods have played a significant role in optimizing the reaction paths and improving the catalytic activity at the atomic electron level in the past decade.30,31 In this regard, we systematically studied the reaction mechanism and rational design of 2D-MoS2. We constructed 2D-MoS2, MoS2-supported Cu single atom catalyst (Cu@MoS2), and MoS2-supported Cu single atom catalyst with sulfur vacancy (Cu@MoS2-Vs) and systematically explored their enzyme-like performances on the catalytic decomposition of H2O2 under different acidic and basic conditions using the global neutral network (G-NN) potential and stochastic surface walking (SSW) method. Through Cu single atom loading, sulfur vacancy engineering and acidic and alkaline microenvironment regulation, the rational design of 2D MoS2 was implemented and investigated to greatly enhance the peroxidase-like catalytic activity. Electronic structure properties were analyzed to illustrate the greatly improved peroxidase-like activity of Cu@MoS2. Furthermore, through kinetic experiments, we confirmed the enhanced performance of Cu@MoS2 with sulfur vacancy in acidic conditions. We highlighted the enhanced catalytic performance of Cu@MoS2 and anticipate that Cu@MoS2 will provide a rational strategy for the design and development of single atom support on MoS2 for important biomedical applications.
2. Computational and experimental details
2.1 SSW–NN simulation
As performed in the LASP (Large-scale Atomistic Simulation with neural network Potential) code,32 the stochastic surface walking (SSW) global optimization in combination with the neural network (NN) (SSW-NN) method was applied to build global NN potential for the Cu@MoS2 system.33 The SSW method uses an unbiased universal potential energy surface (PES) searching method to search for the global minima (GM) structure by smoothly manipulating the structural configuration from one minimum to another on the PES.34 The machine learning NN potential was generated by iterative self-learning of the plane-wave density functional theory (DFT) global PES data set generated from SSW exploration.35 The process of SSW-NN is as follows: global data set generated by SSW simulation, NN potential fitting and global optimization utilizing NN potential and, finally, the convergence standard of the training reached 6.5 meV per atom for the energy and 0.212 eV Å−1 for the force, which is accurate enough for the simulation of the system. The double-ended surface walking (DESW) method was implemented to locate the transition state (TS) and build the most favorable pathway between the reactants and products using only the first derivatives.2.2 DFT calculations
Electronic structure calculations were performed using the Vienna Ab initio Simulation Package (VASP) with considerable precision.36 All structures are relaxed within the generalized gradient approximation (GGA) method.37 As for the mutual effect, we mainly calculated the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functionals,38 and van der Waals interactions were also considered to add a correction to the DFT-D3 method.39 All calculations include spin polarization. A cutoff energy of 500 eV was employed, accompanied by a plane-wave basis set in all systems. In our calculations, a 4 × 4 × 1 MoS2 supercell was built as an initial pure configuration that is large enough for calculating the peroxidase-like processes. During structural optimization processes, a 2 × 2 × 1 k-point grid was set as the Brillouin-zone integration, and a 5 × 5 × 1 k-point grid was used for electronic structural calculations. A 15 Å vacuum space along the z-direction was set for avoiding layer-to-layer interactions. We ended the simulation at 0.01 eV Å−1 for the force convergence criterion, and the total energy convergence threshold reached 10−6 eV. The calculation of adsorption energy Eads (eV) was as follows:| Eads = Eslab-molecule − Emolecule − Eslab | (1) |
2.3 Synthesis of Cu@MoS2 nanozymes
MoS2 nanozymes with the morphology of nanosheets were synthesized by a simple hydrothermal method according to our previous report.40 Then, to obtain Cu ions-loaded Cu@MoS2 nanozymes, 10 mL (10 mg mL−1) of the obtained MoS2 nanosheets aqueous solution was mixed with 10 mg of powder CuCl2·2H2O under sonication for 10 min based on the electrostatic interaction of negatively charged MoS2 nanosheets and the positively charged Cu ions. Finally, the as-obtained Cu@MoS2 nanozymes were washed with distilled water three times via centrifugation. The loading ratio of Cu single atoms in MoS2 was 5.02%, calculated by inductively coupled plasma mass spectrometry (ICP-MS).2.4 Peroxidase-like activity and kinetic assay
The MoS2 and Cu@MoS2 can catalyze the oxidation of the colorless 3,3′,5,5′-tetramethylbenzidine (TMB) to form the blue oxidized TMB (oxTMB) with the maximum absorption peak at 652 nm in the presence of the electron acceptor H2O2. Typically, the pH-dependent peroxidase-like activity of MoS2 and Cu@MoS2 containing H2O2 (1 mM) and TMB (1 mM) in phosphate-buffered saline (PBS) buffer with different pH values (4.0, 5.5, 6.0, 7.4, 8.0) was explored and compared at 37 °C.The peroxidase-like activities of MoS2 (20 μg mL−1) and Cu@MoS2 (20 μg mL−1) were respectively tested in PBS buffer (pH 4.5, adjusted by HCl) containing different concentrations of H2O2 and TMB as substrates. The microplate reader (SpectraMax M2MDC) was used to measure the UV-vis absorbance of the blue oxTMB at a set time point after varying the concentrations of the H2O2 substrate versus another TMB substrate at 37 °C. According to the Michaelis–Menten eqn (2), the Lineweaver–Burk double-reciprocal plot of eqn (3), and the initial reaction rates (V0) calculated from the absorbance changes during the varied H2O2 or TMB concentration, kinetic parameters including Michaelis constant (Km) and the maximal reaction velocity (Vmax) can be obtained by a linear-fitting method. The Vmax is obtained when the nanozymes are saturated with the substrate and [S] is the concentration of the substrate.
 | (2) |
 | (3) |
3. Results and discussion
3.1 Enzyme-like properties of pristine MoS2
To explore the effects of the surface rational design with Cu single atom loading, sulfur vacancy engineering and acidic and alkaline environment regulation on the peroxidase-like activity of MoS2, we constructed a series of catalyst structures based on the pure monolayer MoS2 surface (Fig. 1 and S1†).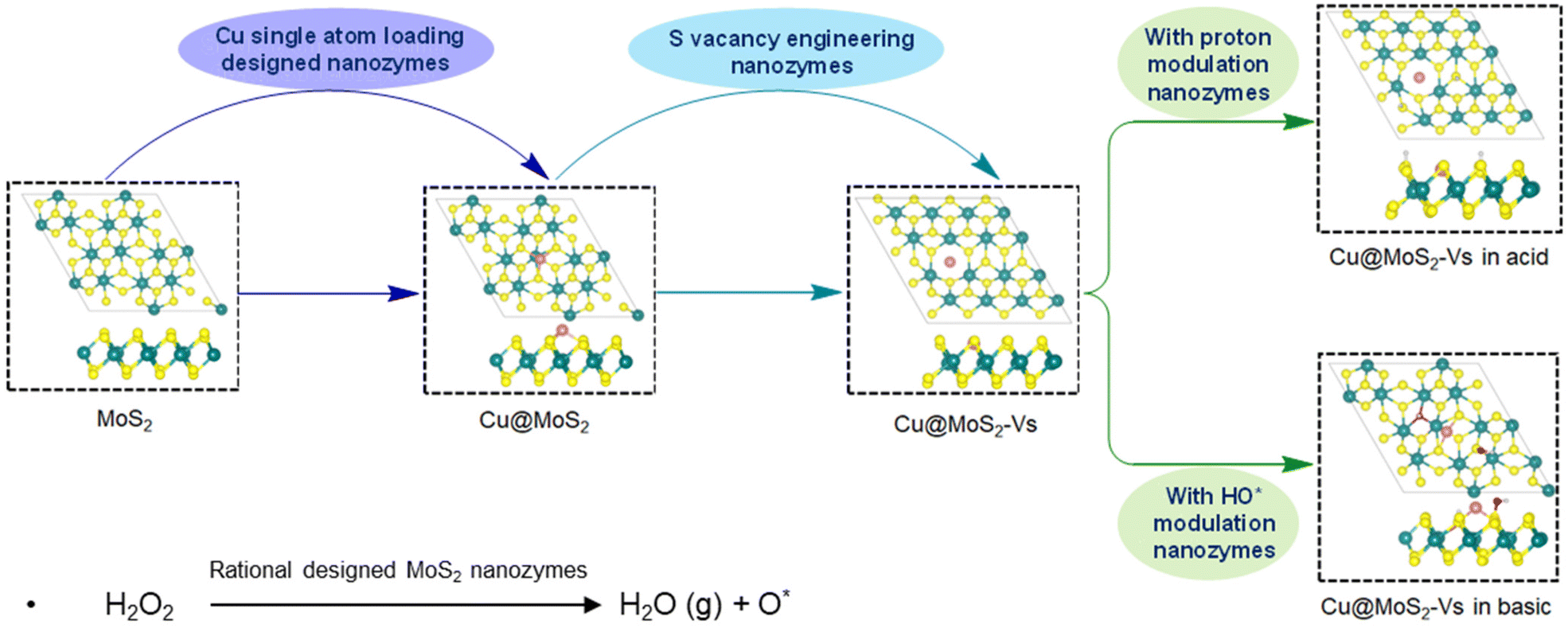 | ||
| Fig. 1 A schematic diagram of the rational design of MoS2 nanozymes to modulate peroxidase-like activity through surface modification with Cu single atom loading, sulfur vacancy and acidic and alkaline environment regulation. The spheres in green, yellow, pink, red and gray represent Mo, S, Cu, O and H atoms, respectively. | ||
As a 2D support substrate, MoS2 exhibits the potential for catalytic activities. On this basis, we systematically studied the enzyme-like activities of pure MoS2. The optimized initial configuration of the pure MoS2 surface is shown in Fig. 1. The average distance between three adjacent S atoms of MoS2 was 3.231 Å (Table S1†). Using the SSW method, we obtained a series of possible stable adsorption structures after H2O2 decomposition, including H2O2*, HO*, HOO*, H2O*, O* and proton species. Subsequently, the DESW method was used to connect the reactants with the products to find transition states and the reaction paths. Firstly, H2O2 was adsorbed parallel to the substrate with a low adsorption energy of −0.31eV (Table S2†). There was negligible electron aggregation between the H2O2 molecule and MoS2 after H2O2 adsorption, suggesting weak interaction between the H2O2 molecule and substrate (Fig. S2a†). H2O2 has two possible decomposition pathways on the pure MoS2 surface. One possible pathway is as shown in Fig. S3,† where H2O2 possibly decomposed into HOO* and H* species that needed to overcome a high energy barrier of 3.00 eV. On the other hand, the O–O bond of H2O2 will be elongated to form two HO* species, overcoming an energy barrier of 1.81 eV, which is the rate-determining step of the whole reaction path. One HO* species was transferred to an adjacent sulfur atom with an energy barrier of 1.26 eV (Fig. 2).
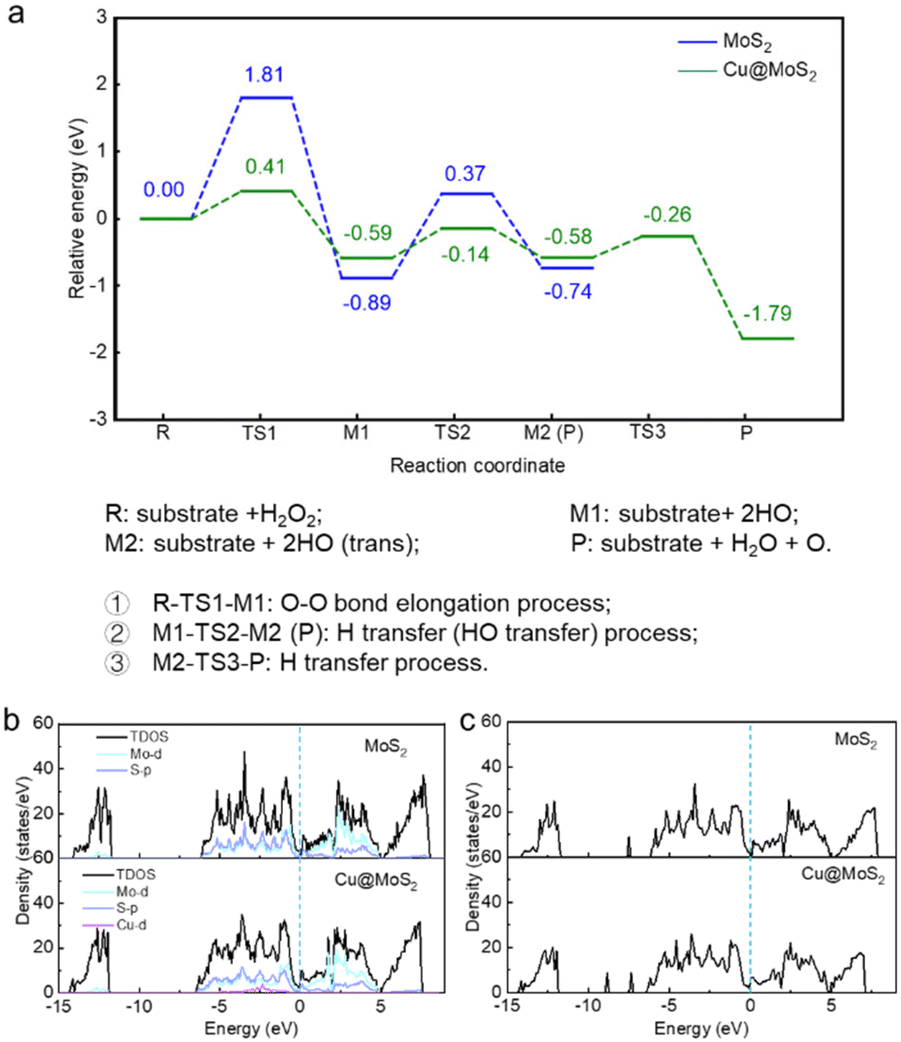 | ||
| Fig. 2 (a) Energy profile for the decomposition of H2O2 on pure MoS2 and Cu@MoS2 surfaces. The blue and green curves show the decomposition process of H2O2 on the surface of pure MoS2 and Cu@MoS2 catalysts, respectively. The numbers represent the energy (eV) at each state. The abscissa R, TS, M1, and P represent reactants, transition states, intermediates, and products, respectively. (b) TDOS and PDOS of MoS2 and Cu@MoS2 substrates. (c) TDOS of H2O2 adsorption on MoS2 and Cu@MoS2 substrates. | ||
On comparing these two reaction pathways, we concluded that the H2O2 decomposition into two HO* species needs a smaller energy barrier, which means that MoS2 prefers to demonstrate peroxidase-like activity with HO* species generation. However, the peroxidase-like activity of pure MoS2 is chemically inert at the basal plane and needs relatively higher energy to be activated. Various rational design strategies are urgently needed to improve the peroxidase-like activity to achieve better practical applications. Therefore, a series of modifications were proposed to improve the peroxidase-like performance of MoS2, including surface modification with Cu single atom loading, sulfur vacancy engineering and acidic and alkaline microenvironment regulation.
3.2 Peroxidase-like activity of Cu@MoS2
The loading of the Cu single atom on the MoS2 support can change the electronic structure and improve its peroxidase-like activity. We randomly placed a Cu single atom above a pure MoS2 support, and the SSW method was used to globally optimize all possible adsorption sites of the Cu single atom. Ultimately, we obtained the most stable MoS2-supported Cu adsorption structure (denoted as Cu@MoS2) (Fig. 1). The Cu single atom was stably adsorbed at the hollow site above the Mo atom. As Cu was adsorbed, the average distance between the three S atoms neighbouring the Cu single atom became 3.346 Å on Cu@MoS2, which was 0.115 Å longer than that of MoS2 (Table S1†). The extended distance makes it easier for the Cu single atom to attach to the slab. Simulation results showed that the adsorption energy of the Cu single atom on the MoS2 support was −3.27 eV, which indicated that the Cu single atom was stably adsorbed on Cu@MoS2 (Table S3†). Bader charge analysis was implemented to further elucidate the effects of the Cu single atom loading on the peroxidase-like activity of MoS2 (Table S4†). The charge density difference plots are shown in Fig. S4,† where the cyan and purple regions correspond to electron accumulation and depletion, respectively. Accordingly, it can be seen from the charge density difference results that there was some electron depletion on the adsorbed Cu single atom and Mo atoms. The electron accumulation between the interacting Cu single atom and three sulfur atoms significantly indicates the covalent interaction between the adsorbed Cu single atom and the MoS2 support (Fig. S4b†). The accumulation of the electron density suggests the chemical bonds formation of Cu–S bonds, which indicates that the Cu atom was chemically adsorbed on the MoS2 support. The strong interaction between Cu and the MoS2 support was also verified by the Bader charge results, where the electrons transferred from Cu to adjust S atoms with a value of −0.53|e|. The electronic property results showed that the Cu@MoS2 structure is stable, suggesting that a Cu single atom can provide a stable active site for H2O2 decomposition.Furthermore, we systematically studied the peroxidase-like activity of Cu@MoS2 catalysing the decomposition of the H2O2 substrate. As shown in Fig. 2, on the Cu@MoS2 surface, H2O2 was firstly chemically adsorbed on the Cu active site with an adsorption energy of −0.80 eV (Table S2†). As shown in Fig. S5,† we denoted the O atom of the H2O2 molecule that directly interacted with the catalyst surface as O1′, and the corresponding H atom of H2O2 that bonded with O1′ was denoted as H1′. The other O atom and H atom were respectively denoted as O2′ and H2′. After H2O2 adsorption, some electrons were localized mainly between the surface Cu atom and O1′, while there were few electrons at O1′ on Cu@MoS2, which was in line with the Bader charge results in Table 1, indicating the obvious overlap between the H2O2 molecule and the Cu single atom (Fig. S2b†). The O–O bond of H2O2 stretched to 1.498 Å, which was longer than that in the gas phase, indicating H2O2 activation. The elongation of the bond length was mostly attributed to the electron transfer from the primary bond to the newly formed bond between the H2O2 molecule and substrate. Further, H2O2 decomposed into H2O molecules and O* species after three processes of O–O bond stretching, hydroxyl migration and H transfer, which only overcame low barriers of 0.41, 0.45 and 0.32 eV, respectively, and with a high exothermic energy of 1.79 eV. It was revealed that Cu single atom loading greatly reduced the energy barrier required for the rate-determining step, resulting in enhanced reactive activation. The energy required for H2O2 decomposition on Cu@MoS2 was 1.40 eV lower than that required for the pure MoS2 surface, suggesting that Cu single atom adsorption enhanced the peroxidase-like catalytic activity of Cu@MoS2.
| Pure MoS2 | Cu@MoS2-Vs | Cu@MoS2-Vs in acid | Cu@MoS2-Vs in basic | |||||
|---|---|---|---|---|---|---|---|---|
| Before ads | Ads | Before ads | Ads | Before ads | Ads | Before ads | Ads | |
| Mo (S) average | 0.54 | 0.55 | −0.99 | −0.98 | −1.00 | −0.99 | −1.17 | −1.17 |
| Mo | −1.07 | −1.07 | — | — | — | — | — | — |
| Cu | — | — | −0.08 | −0.17 | −0.02 | −0.24 | −0.48 | −0.60 |
| O1′ | — | 0.62 | — | 0.64 | — | 0.69 | — | 0.64 |
| O2′ | — | 0.63 | — | 0.57 | — | 0.60 | — | 0.59 |
| H1′ | — | −0.61 | — | −0.62 | — | −0.65 | — | −0.62 |
| H2′ | — | −0.62 | — | −0.60 | — | −0.63 | — | −0.65 |
To gain more insight into the electronic behaviour of MoS2 with or without Cu single atom loading systems, we calculated the total density of states (TDOS) as well as the projected density of states (PDOS) as shown in Fig. 2b. Compared with that of pure MoS2, the electron distribution of the Cu@MoS2 around the Fermi level (Ef) slightly increased, which indicated that the loading of the Cu single atom enhanced the metallic property of MoS2. In addition, the TDOS of Cu@MoS2 shifted to the left, suggesting that loading the Cu single atom increased the electrons of the MoS2 system. As a result, the Cu single atom became the active site for Cu@MoS2 to catalyse the H2O2 decomposition. It can be seen from the PDOS result that Cu-d orbital and S-p orbital are strongly hybridized, indicating that the Cu atom was stably adsorbed on the MoS2 substrate. Also, we studied the DOS with respect to the Ef of H2O2 adsorption on MoS2 and Cu@MoS2 (Fig. 2c). We found that the electron energy state of H2O2 adsorbed on the Cu@MoS2 substrate was much lower than that on MoS2, indicating that the electron transfer was more likely to occur when H2O2 was adsorbed on Cu@MoS2. In summary, DOS analysis results showed that Cu@MoS2 can provide more electrons for H2O2 to promote O–O bond breaking, further enhancing the peroxidase-like catalytic activity.
3.3 The enhanced peroxidase-like performance of Cu@MoS2-Vs
The design of sulfur vacancy engineering has been widely reported to regulate the intrinsic electronic properties and induce local electronic environment redistribution, thus greatly enhancing their catalytic performance. To study the sulfur vacancy engineering effects, a sulfur atom was randomly removed from the pure MoS2 surface, and a stable configuration was obtained after SSW global optimization (Fig. S1a†). We added the Cu single atom to obtain the stable Cu@MoS2-Vs structure using the SSW method (Fig. 1). Unlike the Cu@MoS2 surface, the Cu single atom was more inclined to fill and adsorb at the S vacancy site with Cu@MoS2-Vs. The adsorption energy of the Cu single atom on the surface Cu@MoS2-Vs was −4.96 eV, indicating that the Cu single atom was stably adsorbed on Cu@MoS2-Vs. In addition, the adsorption energy was more negative than that of Cu@MoS2 with the value of −3.27 eV, indicating that the Cu single atom active site was more stable when bonded to the Cu@MoS2-Vs catalyst. Subsequently, Bader charge and charge density difference analysis were simultaneously carried out on the Cu@MoS2-Vs surface to clarify the effects of the sulfur vacancy on the peroxidase-like activity of Cu@MoS2 (Fig. S4, and Table S1†). We recorded the charge transfer between adjacent S, Mo atoms and the Cu single atom of the Cu@MoS2-Vs surface. As for Cu@MoS2-Vs, the three Mo atoms of the slab around Cu respectively transferred 0.99, 0.99 and 1.00|e|, and the Cu atom also lost some electrons correspondingly. Also, there was electron depletion on the adsorbed Cu atom and three Mo atoms on the Cu@MoS2-Vs surface. The electrons increased between Cu and Mo atoms, which suggested the strong metal–support interaction after the adsorption of the Cu atom (Fig. S4c†). Accordingly, it can be seen from the charge density difference results that there was great overlapping of the charge density on Cu@MoS2-Vs, indicating a strong coupling between Cu and the slab (Fig. S4c†). It was also shown that there was a covalent bond for electrons converging between the Cu single atom and the support. The geometric and electronic structure analysis of the surface Cu@MoS2-Vs showed that the sulfur vacancy promoted the adsorption of the Cu single atom on MoS2.The reaction pathway was calculated to explore the function of sulfur vacancy engineering in the enhanced intrinsic peroxidase-like activity of Cu@MoS2-Vs. Using the stable configurations of the reactant, product and intermediates obtained by the SSW method, we applied the DESW method to establish all possible reaction paths and their chemical bond-making/breaking transition states (TS) of H2O2 decomposition on the Cu@MoS2-Vs surface to study its peroxidase-like catalytic performance. In Fig. 3a, the catalytic cycle on the catalyst surface generally includes four stages: H2O2 adsorption, O–O bond stretching, H transfer and TMB substrate oxidation. As shown in Fig. 3b, with Cu@MoS2-Vs (energy profile in green line), H2O2 was adsorbed with the highest adsorption energy of −0.89 eV, which was due to chemisorption. It was confirmed by visualizing the charge density difference that a chemical bond was formed between H2O2 and the Cu single atom. In addition, H2′ had an electron coupling with the surface sulfur atom. The interaction between H2′–S and O1′–Cu jointly led to stronger adsorption and further activation of H2O2 (Fig. 3b). The O1′–O2′ bond length was elongated to 1.472 Å. The H2O2 molecule went through a transition state to decompose into two HO* species, which was the rate-determining step of the whole reaction and required overcoming a barrier of 0.51 eV. The subsequent H transfer process was accompanied by a nearly zero energy barrier (0.05 eV). The entire reaction process was thermodynamically favourable with a large exothermic energy of 4.13 eV. The results showed that the loading of the Cu single atom greatly reduced the energy barrier of the rate-determining step, and the sulfur vacancy was beneficial for system exothermicity. The decomposition of H2O2 into two HO* species on Cu@MoS2-Vs was both kinetically and thermodynamically favourable, which indicated that the Cu@MoS2-Vs was the most ideal peroxidase-like nanozyme as compared with pure MoS2 and Cu@MoS2.
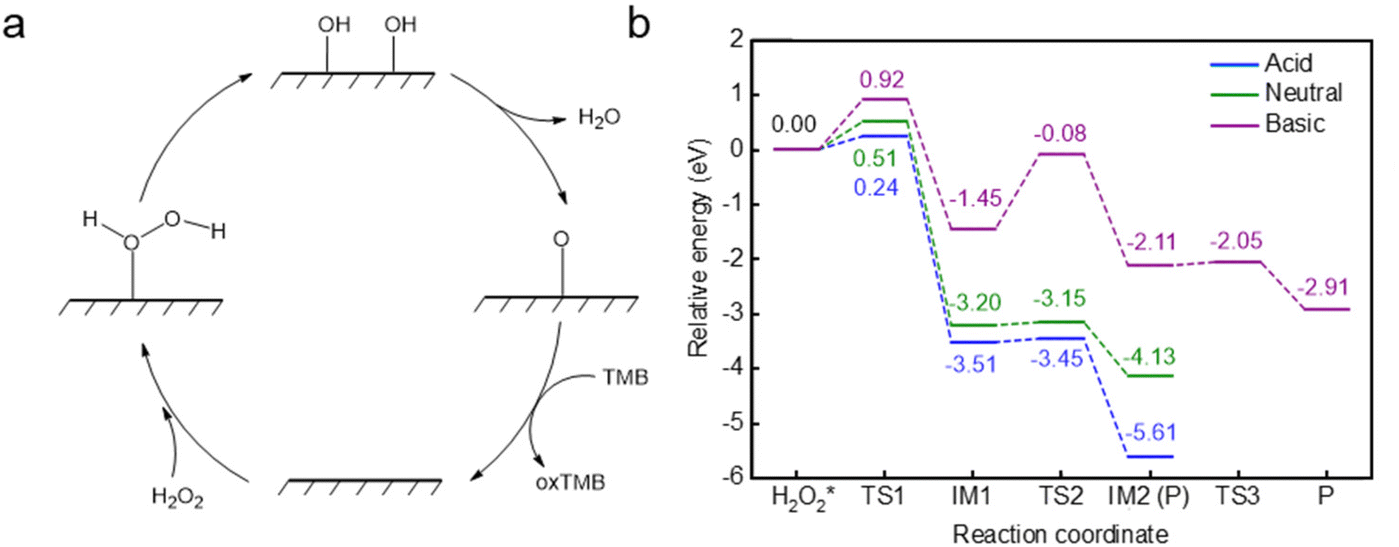 | ||
| Fig. 3 (a) Schematic diagram of the H2O2 decomposition reaction cycle on Cu @MoS2-Vs. (b) Energy profiles of Cu@MoS2-Vs in neutral, acidic and alkaline conditions. The numbers represent the energy (eV) at each state. The abscissa TS, M, and P represent transition states, intermediates and products, respectively. | ||
3.4 The acidic and alkaline environment regulation of the peroxidase-like properties of Cu@MoS2-Vs
To better simulate the peroxidase-like activity of Cu@MoS2-Vs in real physiological conditions, we investigated the effects of acidity and alkaline conditions on Cu@MoS2-Vs.The pre-adsorbed OH and H* groups on Cu@MoS2-Vs surfaces can only be advantageously developed in basic and acidic environments, respectively, which can lead to acidic and alkaline environment regulation and POD-like activities.41 Therefore, two protons and two hydroxyl groups were randomly added to Cu@MoS2-Vs to simulate acidic and alkaline environments, respectively. Firstly, stable catalyst configurations were obtained by the SSW global optimization of Cu@MoS2-Vs in neutral, acidic and basic conditions (Fig. 1). The neutral conditions have been discussed above regarding the structure and catalytic performance of Cu@MoS2-Vs (section 3.3). In acidic conditions, two protons were respectively adsorbed on sulfur atoms near the active site with H–S bond lengths of 1.362 Å. The Cu atom was stably adsorbed at the sulfur vacancy site. The average distance between the three neighbouring sulfur atoms was 0.053 Å longer than that under neutral conditions and the average distance between the three Mo atoms was 0.070 Å longer than that under neutral conditions. The elongated distance caused the steadier adsorption of the Cu single atom on Cu@MoS2-Vs. In basic conditions, one of the two HO* radicals preferred to adsorb at the sulfur vacancy. Another HO* radical preferred to adsorb on sulfur atoms. In addition, the adsorption energies of the Cu single atom were −3.74 and −2.79 eV in acidic and basic conditions, respectively, suggesting the stable adsorption of the Cu single atom in both acidic and basic conditions.
The H2O2 adsorption and decomposition abilities of Cu@MoS2-Vs under different acidic and alkaline environments were analysed through the energy profile. H2O2 was added to the surface to obtain stable adsorption configurations in different acidic and alkaline microenvironments. The energy profiles of the most favourable route for the H2O2 decomposition in acid and basic conditions are shown in Fig. 3b. Under acidic conditions, H2O2 was adsorbed steadily and obliquely on Cu single atom active sites with an adsorption energy of −0.83 eV. Then, only a 0.24 eV barrier was needed to be overcome to form two HO* groups. These HO* groups quickly combined with the H on the surface to form two H2O molecules. The entire reaction process was thermally favourable with a large exothermic energy of 6.65 eV. However, in an alkaline environment, H2O2 was chemically adsorbed on Cu single atom active sites with an adsorption energy of −1.03 eV. H2O2 decomposition needed to overcome an energy barrier of 0.92 eV to break the O–O bond. Subsequently, the two HO* groups were adsorbed on the Cu single atom and underwent two H transfer processes to form two H2O molecules and two O* species, requiring energy barriers of 1.37 and 0.06 eV, respectively. The formation of the first H2O molecule became the rate-determining step, which required overcoming larger obstacles. From the perspective of the reaction paths, the alkaline conditions are not conducive to the decomposition of H2O2 on the surface of Cu@MoS2-Vs as compared with the neutral environment. On the contrary, acidic environments not only greatly reduced the reaction energy barrier of H2O2 decomposition, but also caused the reaction to release the most heat.
3.5 The accelerated electron transfer process: the essence of high peroxidase-like performance
The low energy barrier of the rate-determining step directly reflects high catalytic activity, and the essence of the low energy barrier is reflected in the basic electronic and structural properties. The electronic structural properties were further analysed to elucidate the intrinsic origin of the high peroxidase-like activity and high-activity sites of the kinetically and thermodynamically favourable Cu@MoS2-Vs catalyst under acidic conditions.After H2O2 was adsorbed on the Cu@MoS2-Vs catalyst surface in neutral, acidic and basic conditions, the charge density difference diagram showed that there were electron transfers between the H2O2 molecule and catalysts to different degrees (Fig. 4a–c). The Cu single atoms of catalysts all had obvious bond interactions with H2O2. As shown in Table 1, Cu atoms on Cu@MoS2-Vs catalysts have different degrees of electron transfer in different acidic and alkaline environments. Under neutral conditions, the Cu atom adsorbed by the H2O2 molecule loses 0.09 electrons, while in an acidic environment, the Cu atom loses 0.22 electrons, which is 0.13 electrons more than in neutral conditions. Under basic conditions, the Cu atom loses 0.12 electrons. The O1′ atoms in different acid and alkaline environments all gained more electrons (0.64|e| in neutral and basic conditions, 0.69|e| in acidic conditions) as compared to pure MoS2 (0.62|e|). The O1′ atom gained the most electrons in acidic conditions. These results indicated that Cu single atom loading, sulfur vacancy engineering and the acidic and alkaline environment regulation can modulate the degree of electron transfer between adsorbed H2O2 molecules and catalyst surfaces. More importantly, acidic conditions can promote better electron transfer between H2O2 and the support as compared to neutral and basic conditions.
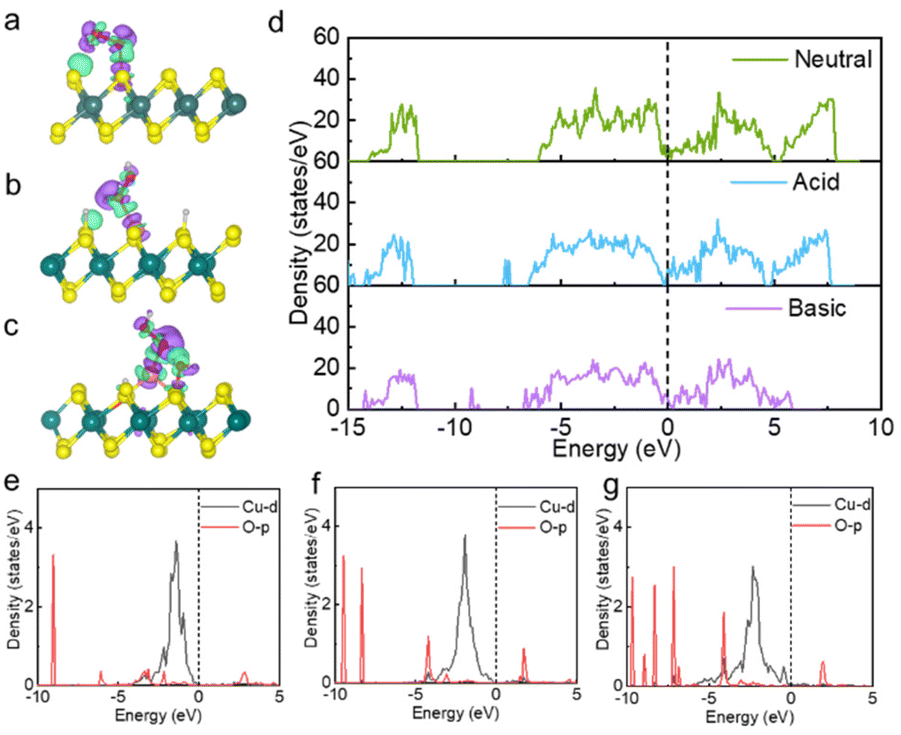 | ||
| Fig. 4 Charge density difference for H2O2 adsorption on Cu@MoS2-Vs in (a) neutral, (b) acidic and (c) basic conditions, respectively. The isosurface value of electron density was taken as 0.002 e Å−3. The cyan and purple regions for the charge density difference represent the areas of electron accumulation and depletion, respectively. (d) TDOS of Cu@MoS2-Vs in different conditions of acidity and alkalinity. (e and f) PDOS of Cu-d and O-p orbitals of Cu@MoS2-Vs in (e) neutral, (f) acidic and (g) alkaline conditions. | ||
TDOS and projected density of states (PDOS) were performed to further confirm the binding nature as well as the electronic properties of the considered systems. TDOS of Cu@MoS2-Vs substrates under different acidic and alkaline environments are shown in Fig. 4d. Under acidic conditions, there was a left shift of TDOS, indicating the increased electron properties of Cu@MoS2-Vs. Meanwhile, new peaks at −7.5 eV, and −14.8 eV were added at the valence band. In addition, the electronic state at the Ef increased, which indicated that the acidic condition increased the electrical conductivity of the catalyst. Further PDOS analysis revealed that electrical conductivity mainly resulted from the overlapping of Mo-d, S-p, and Cu-d orbitals (Fig. S6†). DOS results showed that the acid environment is conducive to the subsequent electron transfer between the H2O2 molecule and the substrates. However, the electronic density decreased on Cu@MoS2-Vs in basic conditions. The electron state density near 6.0 eV to 7.5 eV on the conduction band side disappeared and new peaks appeared at −6.7 eV, −8.9 eV and −9.2 eV on the valence band side. Cu-d and O-p orbitals were to varying degrees overlapped in neutral, acidic and alkaline environments. H2O2 molecules bonded more strongly with Cu single atom on the surface under basic conditions than neutral or acidic conditions (Fig. 4e and f).
We also analysed the relationship between the activity for H2O2 decomposition and the adsorption energy of H2O2 molecules on a range of MoS2-based surfaces. The results showed that the H2O2 dissociation was well in line with the volcano plot (Fig. S7†).42 The Cu single atom loading, sulfur defect engineering and the adjustment of the acid and alkaline environment caused the adsorption capacity of MoS2 to increase, and thus the catalytic activity increased accordingly. The too-small adsorption energy of H2O2 on the surface of pure MoS2 or the too-large adsorption energy on Cu@MoS2-Vs in alkaline environments is not conducive to catalytic activity. The acidic environment causes the Cu@MoS2-Vs surface to have the optimal adsorption energy, which led to the best catalytic efficiency.
The analysis of the geometric structure and electronic structure showed that the loading of Cu single atoms, sulfur defect engineering and regulation of acidic conditions led to the optimal adsorption energy of the H2O2 molecule, accelerated the electron transfer rate, and therefore greatly promoted the peroxidase-like enzymic activity.
3.6 Kinetic experiments on the peroxidase-like properties of MoS2 and Cu@MoS2 with sulfur vacancy in acid conditions
To experimentally verify the pH-dependent peroxidase-like catalytic activities of the as-synthesized MoS2 and Cu@MoS2 nanosheets, we performed the catalytic oxidation of the chromogenic substrate of the colorless 3,3′,5,5′-tetramethylbenzidine (TMB) in the presence of H2O2 at 37 °C (Fig. 5a). The default 0 K setting during the theoretical calculation was due to the limitations of the potential functional training methods (SSW and DESW) in LASP, but it did not affect the consistency of the theoretical and experimental results after free energy correction (Table S2†). The UV-vis absorption of the blue oxidized TMB (oxTMB) showed that both the MoS2 and Cu@MoS2 nanosheets had high peroxidase-like catalytic activities at pH 4.0 due to the easy generation of ˙OH. Particularly, the Cu@MoS2 nanosheets had a relatively higher peroxidase-like catalytic activity than the MoS2 nanosheets at pH 4.0. With the pH value changing from 5.0 to 8.0, both the MoS2 and Cu@MoS2 nanosheets suffered from reduced catalytic activity. However, the maintained value of the catalytic activity of Cu@MoS2 nanosheets was significantly higher than that of the MoS2 nanosheets.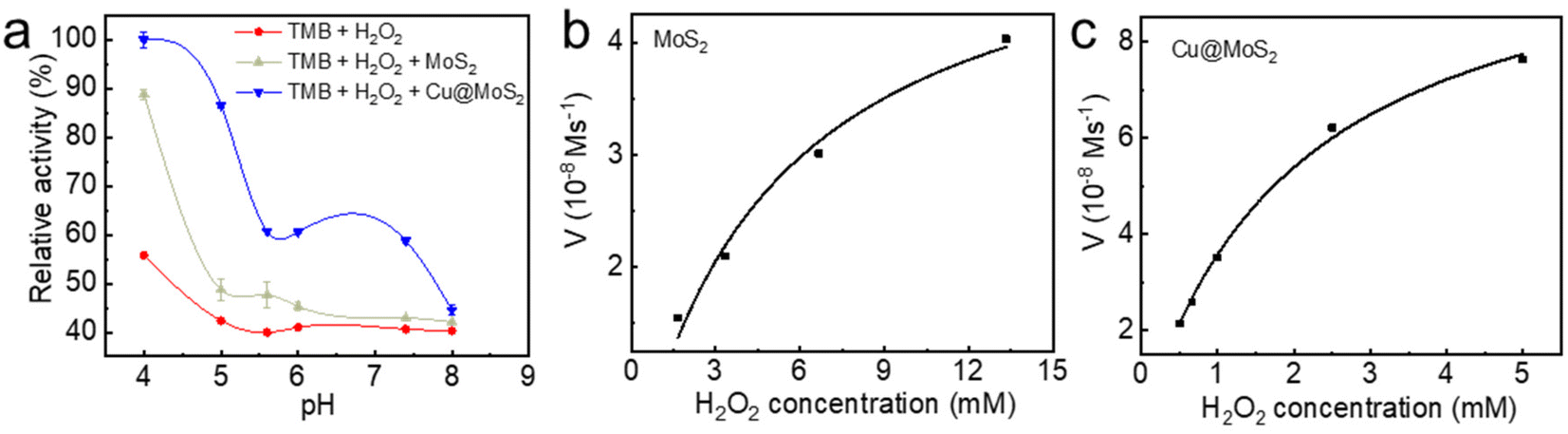 | ||
| Fig. 5 (a) Relative catalytic activity of the MoS2 and Cu@MoS2 as peroxidase mimics under different pH values at 37 °C tested by UV-vis absorption of oxTMB. The velocity of the reaction between ˙OH and the various concentrations of H2O2 in the presence of (b) MoS2 and (c) Cu@MoS2. The successful Cu loading on the MoS2 surface was well in agreement with the DFT calculations. | ||
The Michaelis–Menten kinetic graphs of MoS2 and Cu@MoS2 nanosheets were obtained using the initial rate versus the concentrations of H2O2 at pH 4.5 (Fig. 5b and c). The steady-state kinetics of MoS2 and Cu@MoS2 nanosheets matched well with the typical Michaelis–Menten steady-state kinetics model in various concentration ranges of H2O2. In Table 2, the lower Michaelis–Menten constant (Km) of the Cu@MoS2 nanosheets was 2.05 mmol L−1, suggesting the higher binding affinity of the Cu@MoS2 nanosheets for the H2O2 substrate as compared to the MoS2. Also, the maximum velocity (Vmax) of Cu@MoS2 nanosheets was calculated to be 1.09 × 10−7 mmol L−1 s−1, which was significantly higher than that of the MoS2 nanosheets. These results suggest that the H2O2 concentration-dependent oxidation kinetics of Cu@MoS2 can promote the enhancement of the peroxidase-like catalytic activity of the MoS2 in acidic conditions because of the successful Cu loading on the MoS2 surface, which was well in agreement with the DFT calculations.
| Catalyst | Substrate | K m (mmol L−1) | V max (mmol L−1 s−1) |
|---|---|---|---|
| MoS2 | H2O2 | 4.94 | 0.54 × 10−7 |
| Cu@MoS2 | H2O2 | 2.05 | 1.09 × 10−7 |
4. Conclusions
The rational design and regulation of single-atom loaded MoS2 nanozymes are the key drawbacks to be overcome for use as substitutes for natural enzymes. Through neural network potential trained by machine learning, the potential including Mo, S, Cu, O, and H elements was successfully constructed and was used to search the stable configurations and their transition states rapidly and massively to efficiently find the reaction path of H2O2 decomposition on different catalyst surfaces. The simulation results showed that the activity of the peroxidase-like nanozyme of MoS2 was increased by Cu single atom loading. In particular, the peroxidase-like activity of Cu@MoS2 modified with sulfur vacancy was greatly increased under acidic conditions. The electronic properties calculations further confirmed that the Cu single-atom loading and sulfur vacancy effect, and acidic and alkaline microenvironment regulation affected the electron transfer process of catalysts by changing their electronic properties. The results showed that Cu@MoS2-Vs under acidic conditions accelerated the electron transfer process, thus further promoting the activation of adsorbed H2O2 molecules and enhancing the peroxidase-like activity. At the same time, acidic and alkaline environments affect the density of states of the catalysts. The alkaline environment reduces the density of states of Cu@MoS2-Vs, thus limiting the electron transfer process to some extent and inhibiting the decomposition of H2O2. Furthermore, kinetics experiments demonstrated that the peroxidase-like activity was greatly improved by Cu loading and the adjustment of the acidic and alkaline environments. Our results show that surface modification with Cu single atom loading, sulfur vacancy engineering and acidic and alkaline environment regulation can significantly improve the peroxidase-like activity of MoS2 theoretically and experimentally, which provides guidance for the rational design of nanozymes in the future, and is expected to be applied in the free radical generation and scavenging.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Key Research and Development Program of China (2021YFA1200904 and 2020YFA0710700), the National Natural Science Foundation of China (31971311, 62205338), the Innovation Program for IHEP (E35457U210), Xie Jialin Foundation of IHEP (E2546EU210).References
- Z. R. Wang, R. F. Zhang, X. Y. Yan and K. L. Fan, Mater. Today, 2020, 41, 81–119 CrossRef CAS.
- Y. Y. Huang, J. S. Ren and X. G. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- J. J. X. Wu, X. Y. Wang, Q. Wang, Z. P. Lou, S. R. Li, Y. Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- C. L. Tan, X. H. Cao, X. J. Wu, Q. Y. He, J. Yang, X. Zhang, J. Z. Chen, W. Zhao, S. K. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Q. H. Weng, X. B. Wang, X. Wang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4012 RSC.
- K. L. Tan and B. H. Hameed, J. Taiwan Inst. Chem. Eng., 2017, 74, 25–48 CrossRef CAS.
- X. Wang, Y. W. Zhang, J. Wu, Z. Zhang, Q. L. Liao, Z. Kang and Y. Zhang, Chem. Rev., 2022, 122, 1273–1348 CrossRef CAS PubMed.
- C. Y. Zhi, Y. Bando, C. C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- S. W. Niu, J. Y. Cai and G. M. Wang, Nano Res., 2021, 14, 1985–2002 CrossRef CAS.
- X. Li and H. Zhu, J. Materiomics, 2015, 1, 33–44 CrossRef.
- S. Y. Cho, S. J. Kim, Y. Lee, J. S. Kim, W. B. Jung, H. W. Yoo, J. Kim and H. T. Jung, ACS Nano, 2015, 9, 9314–9321 CrossRef PubMed.
- F. M. Enujekwu, Y. Zhang, C. I. Ezeh, H. T. Zhao, M. X. Xu, E. Besley, M. W. George, N. A. Besley, H. N. Do and T. Wu, Appl. Surf. Sci., 2021, 542, 148556 CrossRef CAS.
- H. N. Kuang, Z. Y. He, M. Li, R. F. Huang, Y. Q. Zhang, X. M. Xu, L. Wang, Y. Chen and S. F. Zhao, Chem. Eng. J., 2021, 417, 127987 CrossRef CAS.
- Y. Gui, J. Shi, P. Yang, T. Li, C. Tang and L. Xu, High Voltage, 2020, 5, 454–462 CrossRef.
- L. Jiao, H. Y. Yan, Y. Wu, W. L. Gu, C. Z. Zhu, D. Du and Y. H. Lin, Angew. Chem., Int. Ed., 2020, 59, 2565–2576 CrossRef CAS PubMed.
- N. Aguilar, M. Atilhan and S. Aparicio, Appl. Surf. Sci., 2020, 534, 147611 CrossRef CAS.
- Y. D. Zhu, K. Zhao, J. L. Shi, X. Y. Ren, X. J. Zhao, Y. Shang, X. L. Xue, H. Z. Guo, X. M. Duan, H. He, Z. X. Guo and S. F. Li, ACS Appl. Mater. Interfaces, 2019, 11, 32887–32894 CrossRef CAS PubMed.
- H.-Y. Su, X. Ma and K. Sun, Appl. Surf. Sci., 2022, 597, 153614 CrossRef CAS.
- F. L. Ling, W. D. Xia, L. Li, X. J. Zhou, X. Luo, Q. Z. Bu, J. C. Huang, X. Q. Liu, W. Kang and M. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 17412–17419 CrossRef CAS PubMed.
- J. C. Yang, H. L. Yao, Y. D. Guo, B. W. Yang and J. L. Shi, Angew. Chem., Int. Ed., 2022, 61, e202200480 CAS.
- S. R. Ali and M. M. De, ACS Appl. Mater. Interfaces, 2022, 2c11245 Search PubMed.
- Y. Wang, K. Qi, S. S. Yu, G. R. Jia, Z. L. Cheng, L. R. Zheng, Q. Wu, Q. L. Bao, Q. Q. Wang, J. X. Zhao, X. Q. Cui and W. T. Zheng, Nano-Micro Lett., 2019, 11, 102 CrossRef PubMed.
- D. W. Wang, Q. Li, C. Han, Z. C. Xing and X. R. Yang, Appl. Catal., B, 2019, 249, 91–97 CrossRef CAS.
- D. L. Li, W. L. Li and J. P. Zhang, Appl. Surf. Sci., 2019, 484, 1297–1303 CrossRef CAS.
- T. H. Zhu, X. R. Gan, Z. H. Xiao, S. Dai, H. R. Xiao, S. J. Zhang, S. C. Dong, H. M. Zhao and P. F. Wang, Surf. Interfaces, 2021, 27, 101538 CrossRef CAS.
- Z. D. Li, X. X. Yang, D. He, W. H. Hu, S. Younan, Z. J. Ke, M. Patrick, X. H. Xiao, J. Huang, H. J. Wu, X. Q. Pan and J. Gu, ACS Catal., 2022, 7687–7695 CrossRef CAS.
- H. C. Huang, J. Wang, J. Li, Y. Zhao, X. X. Dong, J. Chen, G. Lu, Y. X. Bu and S. B. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 19457–19466 CrossRef CAS PubMed.
- Y. Gao, Z. W. Cai, X. C. Wu, Z. L. Lv, P. Wu and C. X. Cai, ACS Catal., 2018, 8, 10364–10374 CrossRef CAS.
- K. Y. Chen, J. K. Deng, X. D. Ding, J. Sun, S. Yang and J. Z. Liu, J. Am. Chem. Soc., 2018, 140, 16206–16212 CrossRef CAS PubMed.
- S. D. Huang, C. Shang, P. L. Kang, X. J. Zhang and Z. P. Liu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1415 CAS.
- T. Mueller, A. Hernandez and C. Wang, J. Chem. Phys., 2020, 152, 050902 CrossRef CAS PubMed.
- C. Shang and Z. P. Liu, J. Chem. Theory Comput., 2013, 9, 1838–1845 CrossRef CAS PubMed.
- S. Ma, C. Shang and Z.-P. Liu, J. Chem. Phys., 2019, 151, 050901 CrossRef.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. H. Fu, X. Zhang, L. Q. Mei, R. Y. Zhou, W. Y. Yin, Q. Wang, Z. J. Gu and Y. L. Zhao, ACS Nano, 2020, 14, 10001–10017 CrossRef CAS PubMed.
- J. N. Li, W. Q. Liu, X. C. Wu and X. F. Gao, Biomaterials, 2015, 48, 37–44 CrossRef CAS PubMed.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr07270j |
| This journal is © The Royal Society of Chemistry 2023 |