
Ligand-mediated electron transport channels enhance photocatalytic activity of plasmonic nanoparticles†
Gayatri
Joshi‡
 ,
Rajesh
Kashyap‡
,
Kalyani
Patrikar‡
,
Anirban
Mondal
* and
Saumyakanti
Khatua
*
,
Rajesh
Kashyap‡
,
Kalyani
Patrikar‡
,
Anirban
Mondal
* and
Saumyakanti
Khatua
*
Chemistry Discipline, Indian Institute of Technology Gandhinagar, Palaj, Gujarat-382055, India. E-mail: khatuask@iitgn.ac.in; amondal@iitgn.ac.in; gayatri.joshi@iitgn.ac.in; kashyaprajesh@iitgn.ac.in; kalyani.p@iitgn.ac.in
First published on 9th October 2023
Abstract
Photoexcitation of noble metal nanoparticles creates surface plasmons which further decay to form energetic charge carriers. These charge carriers can initiate and/or accelerate various chemical processes at nanoparticle surfaces, although the efficiency of such processes remains low as a large fraction of these carriers recombine before they can reach the reaction sites. Thus efficient utilization of these charge carriers requires designing nanostructures that promote the separation of charges and their transport toward the reaction sites. Here we demonstrate that covalently bound surface-coating ligands with suitable orbital alignment can provide electron transport channels boosting hot electron extraction from a gold nanostructure leading to a huge enhancement in the rate of hydrogen evolution reaction (HER) under NIR excitation. A (p)Br-Ph-SH substituted gold nanoprism (AuTP) substrate produced ∼4500 fold more hydrogen compared to a pristine AuTP substrate under 808 nm excitation. Further experimental and theoretical studies on a series of substituted benzene-thiol bound AuTP substrates showed that the extent of the ligand-mediated HER enhancement depends not only on the polarity of the ligand but on the interfacial orbitals interactions.
Introduction
Plasmonic nanoparticles have emerged as potential photocatalysts owing to their strong yet tunable absorption profile across a broad solar spectrum and their exceptional photochemical stability.1–16 Photoexcitation of these nanoparticles creates surface plasmons which subsequently decay to produce highly energetic charge carriers, commonly referred to as hot carriers. These charge carriers then reach the nanoparticle's surface and can initiate and enhance a broad range of chemical reactions near the nanoparticle. However, the efficiency of such plasmon-driven chemical processes remains low mainly due to the fast charge carrier recombination dynamics in metals that typically happens within a few hundreds of femtoseconds to a few tens of picoseconds and a large fraction of the charge carriers recombine before they can reach the reaction sites.17,18 The efficient utilization of these charge carriers requires designing plasmonic nanostructures that facilitate the separation of charges and their transfer toward the reaction sites. Typically, metal nanoparticles are combined with a semiconductor1,2,4,12,19–28 or another metal4,11,15,29–33 in so-called antenna-reactor constructs to achieve better charge carrier separation and utilization. The Schottky barrier at the metal–semiconductor interface acts as an electron filter and allows the hot electrons with sufficiently high energy to enter the semiconductor layer, providing a better chance to participate in a chemical reaction on the semiconductor surface.7,15,27,34,35 On the other hand, the presence of another supporting metal near the plasmonic nanoparticle may facilitate channeling energy or charge carriers into the supporting metal where the chemical reactions occur.15,17,26,27,34,35 However, the fabrication procedures often are complicated and expensive, limiting their scope for large-scale applications.It is known that organic ligands with suitable electronic structure could facilitate interfacial charge transfer at the metal–ligand or semiconductor-ligand interface.36–51 Further, some covalently bound ligands are also known to participate in the plasmon-decay process allowing the electrons to decay into an available ligand orbital.52,53 Such a decay process, known as chemical interface damping, is also reported to depend on the chemical structure of the ligand.53 However, the prospect of using suitable organic ligands to efficiently separate and transport the plasmonic hot carriers to boost a chemical reaction remains less explored. Only recently, Lu et al. reported that the co-adsorbed p-mercaptophenylboronicacid molecules could enhance the photocatalytic activity of silver nanoparticles by facilitating hot electron extraction from the nanoparticle.54 The efficiency of such a process was further related to the energy of the lowest unoccupied molecular orbital of the ligand. A few studies also reported enhancement photocatalytic activities of plasmonic nanostructures in the presence of thiolated ligands.41–43,55–59 However, the enhancements reported in these studies are related to favorable ligand–reactant interactions.41,42,56
Here we investigated the effect of simple organic ligands on the separation and utilization of the plasmonic hot electrons generated via resonant excitation of triangular prism-shaped gold nanoparticles (AuTP). We synthesized plasmonic substrates containing AuTP functionalized with a series of p-substituted benzene thiol (X-Ph-SH, where X = NO2, F, Cl, Br, H, OH, OCH3) ligands and studied their effectiveness in driving hydrogen evolution reaction (HER) from water at neutral pH under NIR (808 nm) illumination. We found that the photocatalytic HER rate strongly depends on the nanoparticle's surface capping ligand. Bare and NO2/F/Cl substituted p-benzene thiol coated AuTPs produced only a minuscule amount of hydrogen while HER rate increased by approximately 4500-fold for Br substituted p-benzene thiol ligand. The HER rate generally increases in the following order: bare ∼ Cl ∼ F ∼ NO2 < H < OH < OCH3 < Br. First, principle computations were performed on the systems to explore the microscopic factors governing HER rates. It was found that surface dipole moment and orbital interactions between nanoparticle and ligand were crucial factors determining HER rate. Subsequently, a function was developed that modeled the correlation of these properties with the HER rate.
Results & discussion
Gold triangular nanoprisms (AuTP) were synthesized in acetonitrile medium by following our previously published method (ESI Experimental details†).3 The AuTP in acetonitrile displayed a dipole peak at 752 nm and a quadrupole peak at 588 nm, as shown in Fig. S1.† The SEM analysis confirms the formation of AuTP with an average edge length of 45 ± 7 nm (Fig. 1b). The AuTPs were further attached to glass substrates via dip-coating for 2 h. The AuTP-coated glass substrate appeared blue (photograph shown in Fig. 1a, inset), having a broad extinction spectrum covering the visible and NIR wavelengths (Fig. 1a). The LSPR peak was found to be at 790 nm. The substrate's SEM images (Fig. 1b and c) showed uniform deposition of AuTP monolayer over a large area.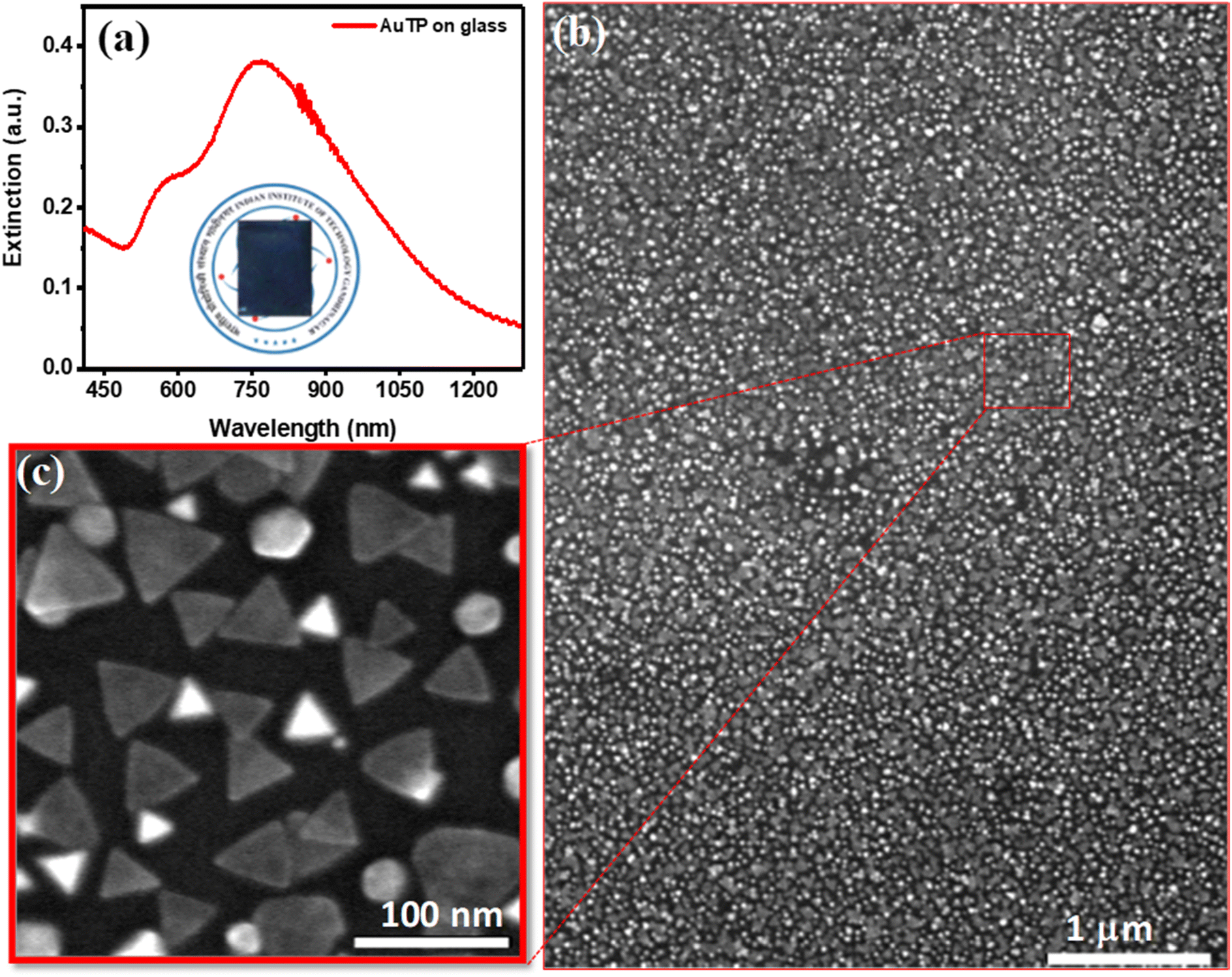 | ||
| Fig. 1 (a) UV-visible extinction spectra of AuTP attached to the glass substrate. The inset shows the photo of the AuTP functionalized glass substrate. (b) Corresponding SEM image of the AuTP functionalized on ITO substrate. The scale bar is 1 μm. (c) The zoom-out SEM image of AuTP. The scale bar is 100 nm. | ||
The AuTP-coated glass substrates were rinsed with ethanol and were subsequently incubated in 5 mM p-substituted benzene thiol (X-Ph-SH, X = NO2, F, Cl, Br, H, OH, and OCH3) solution in ethanol for 12 h for surface functionalization (Fig. 2a). Fig. 2b represents the UV-visible extinction spectra of AuTP substrate before and after the attachment of H, OH, OCH3, Br, F, NO2, and Cl substituted benzene thiol ligands. A red-shift of AuTP LSPR wavelengths was observed for all benzene-thiols, although the extent of red-shift depends on the ligands and increases as follows: OCH3 < Br < H < Cl < OH < F < NO2. SEM images were taken before and after the surface functionalization revealed no significant change in the sizes and/or shapes of the AuTP (Fig. S2†). Further, surface-enhanced Raman spectroscopy was performed to confirm the presence of different ligands on AuTP. The SERS analysis of the substrate before and after the ligand incubation is shown in Fig. 2c. The C–S stretch vibration (∼1070–1080 cm−1) and the C–C ring stretching (∼1560–1580 cm−1) observed for all samples after ligand functionalization confirms the ligand attachment onto the surface of AuTP.60 Based on the p-substitute group, a slight shift in C–S and C–C str, and shoulder peaks were observed (Fig. 2c). For example, a single peak at 1075 cm−1 and a C–C ring str mode at 1578 cm−1 were observed for F-substituted benzene thiols (Fig. 2c, orange, shaded area). On the other hand, for the Br substituted benzene thiol, the C–S peak was located at 1066 cm−1 with a shoulder at 1078 cm−1 and C–C str mode at 1559 cm−1 (Fig. 2c, pink). The Cl-substituted benzene thiol had a characteristic triple peak at 1061, 1083, and 1095 cm−1, and the C–C stretching mode at 1566 cm−1 (Fig. 2c, purple).61 H, OH OCH3 and NO2 substituted benzene thiols displayed C–S vibration at 1069 cm−1, 1076 cm−1, 1078 cm−1, and 1079 cm−1 and the C–C str at 1573 cm−1, 1588 cm−1, 1584 cm−1, and 1571 cm−1, respectively (Fig. 2c).
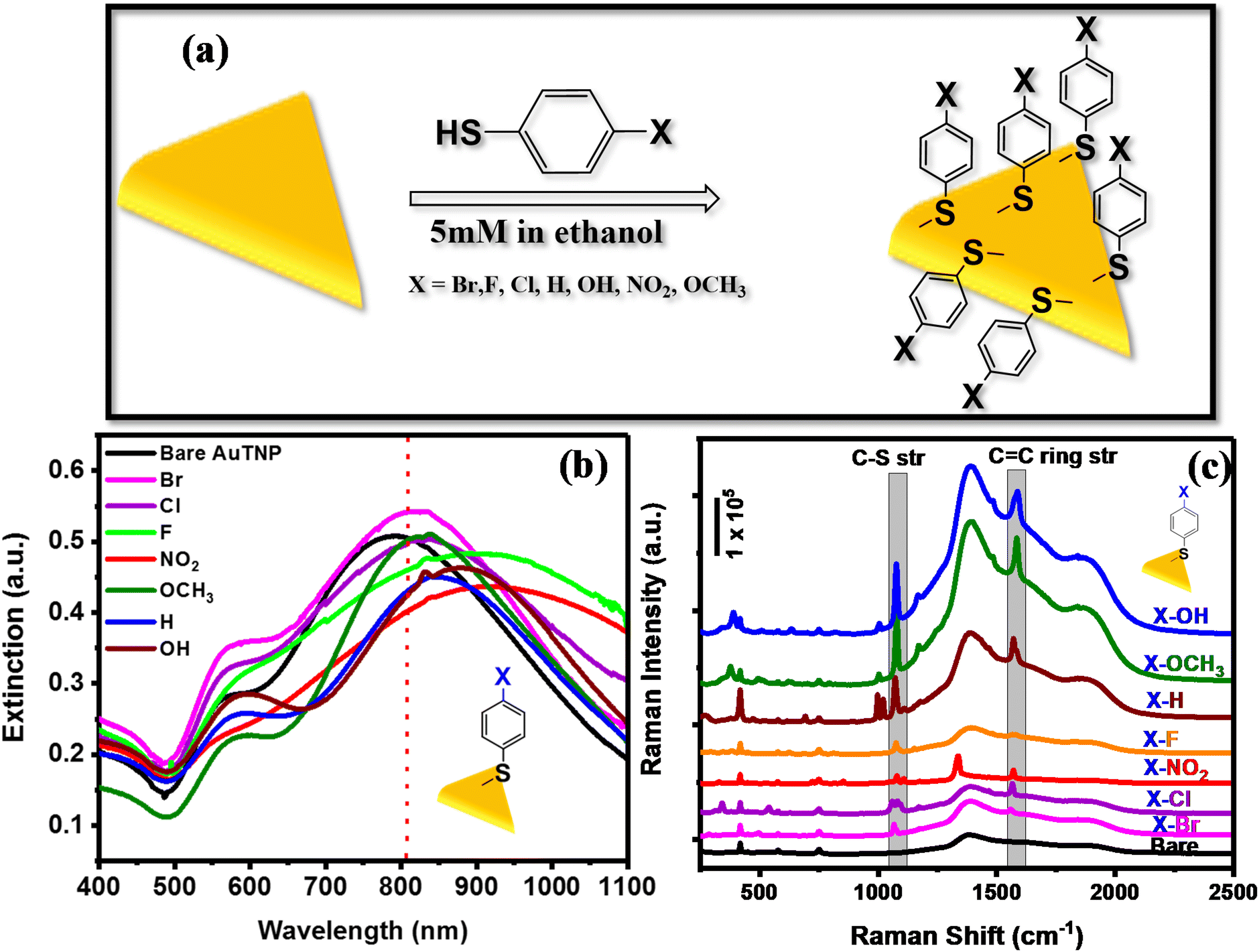 | ||
| Fig. 2 (a) Schematic representation of the AuTP substrate functionalization with the various p-substituted benzene thiol ligands (X-Ph-SH). The figure is not to scale. (b) LSPR spectra of Bare AuTP (black) and after functionalization with different ligands. LSPR peak red-shift observed after each thiol functionalization. (c) SERS analysis of bare AuTP (black) and after functionalization with X-Ph-SH. | ||
To understand the effect of the ligands on the photocatalytic activity of AuTP, the AuTP substrates modified with X-Ph-SH were used for photocatalytic HER from water at neutral pH under 808 nm light illumination. The same excitation power density of 100 mW cm−2 was applied to all samples. The photocatalytic HER performance of all samples was monitored for 6 h under continuous illumination (Fig. 3a). We noticed that the HER performance strongly depends on the surface ligands. Bare AuTP (without any surface coating ligand) and AuTPs coated with F/Cl/NO2 substituted benzene thiols produced only a negligible amount of hydrogen. HER rate increases significantly for AuTPs with H/OH/OCH3 substituted benzene thiols (Fig. 3b). The AuTPs with Br-substituted benzene thiols showed the best HER performance, producing hydrogen at a rate of 4.1 mL g−1 h−1, which is approximately ∼7 times higher than AuTPs with H-Ph-SH ligand and ∼4500 times higher than the bare AuTPs. Overall, the HER rate among different substituted benzene thiols coated AuTPs were found in the following order: bare ≈ Cl∼F < NO2 < H < OH < OCH3 < Br. The incident photon to hydrogen conversion efficiency was calculated for each substrate in the range of 1 × 10–4 to 7.5 × 10–3%, from NO2 to Br-substituted substrates (Fig. 3c). The UV-vis spectral (Fig. S3†) and SERS (Fig. S4†) analysis of ligand-bound AuTP substrate before and after 6 h of photocatalysis confirms that the AuTP are stable with ligand intake. The SEM analysis of AuTP after photocatalysis also supports the stability of AuTP substrates (Fig. S5†). To confirm that the HER is a hot-electron-driven process, the photocatalytic HER production for AuTP surface modified with Br-substituted benzene thiol was monitored in the presence of a hole scavenger (10% methanol) at 808 nm light illumination (Fig. S6a†). The HER rate increased by ∼1.4 folds in the presence of a hole scavenger, confirming the hot-electron-driven process (Fig. S6b†).
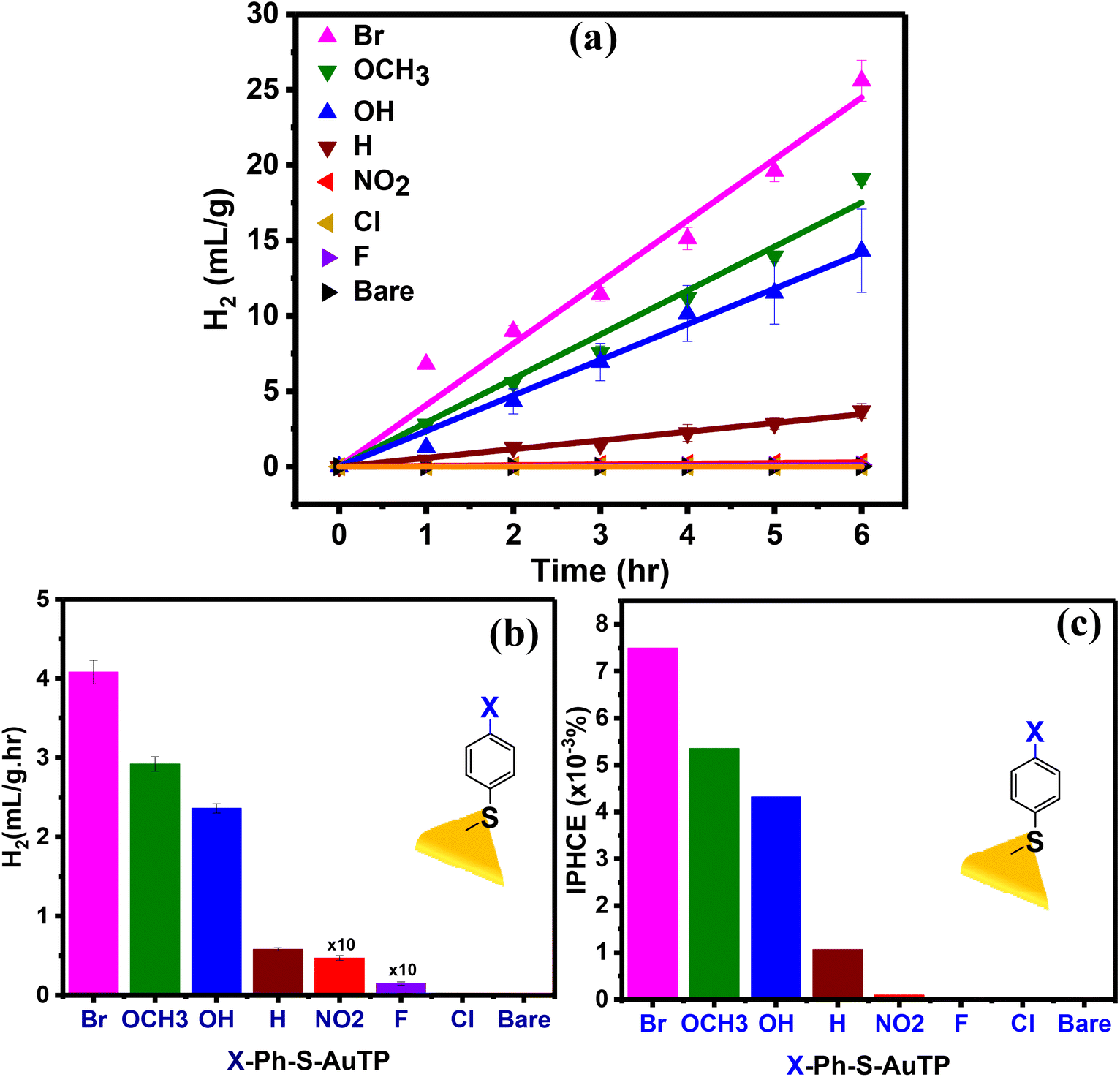 | ||
| Fig. 3 (a) Photocatalytic H2 production observed over time for various X-Ph-S-AuTP substrates, (b) the H2 production rate, and (c) IPHCE calculated for each substrate. The experiment was performed in neutral water under 808 nm laser illumination with a power density of 100 mW cm−2. | ||
The influence of the Br-substituted ligands on the photocatalytic HER was further investigated using the ortho- and meta-substituted bromobenzene thiols as well as 4-Br-Ph-CH2-SH. The AuTP substrates were functionalized with these ligands following the same procedure (Experimental section and Fig. S7†). The ligand attachments were also confirmed through LSPR peak red-shift (Fig. S8†) and SERS analysis (Fig. S9†). The ortho and meta-substituted ligand functionalized AuTP substrates were further utilized for photocatalytic HER under 808 nm illumination and compared the results with 4-Br-Ph-S-AuTP (Fig. 4a). We noticed that HER rate decreased by approximately ∼3 folds when 4-Br-Ph-SH ligand was replaced with 4-Br-Ph-CH2-SH (Fig. 4b). HER rate decreased drastically for ortho- (∼90 folds), and meta-substituted (∼200 folds) bromobenzene thiols and negligible hydrogen was obtained. These results demonstrate that Br position in ligands plays a key role in determining HER performance of the functionalized nanostructures.
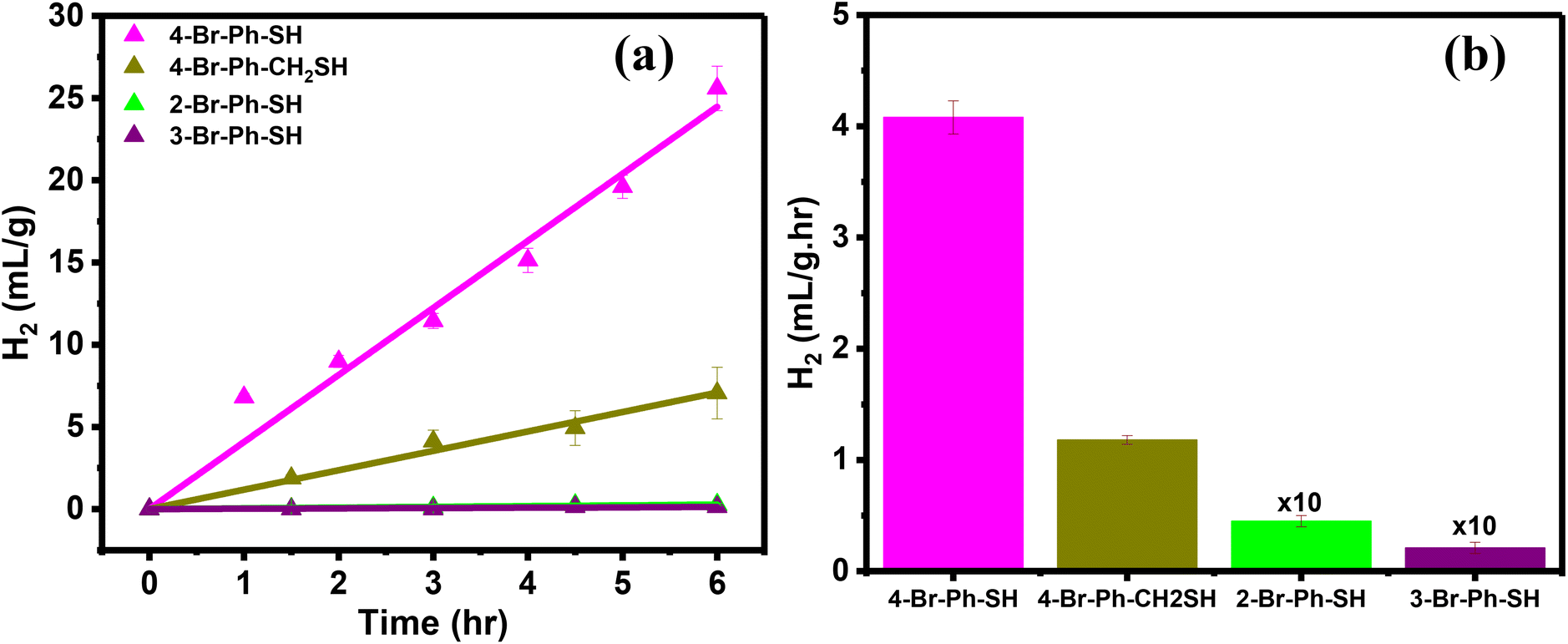 | ||
| Fig. 4 (a) Photocatalytic H2 production was observed over time for ortho-, meta-, and para-substituted benzene-thiols and with 4-Br-Ph-CH2-SH functionalized AuTP substrates. (b) Comparison of the H2 production rate observed with different Br substituted substrates. The experiment was performed in neutral water under 808 nm laser illumination with a 100 mW cm−2 power density. | ||
Now we discuss on how ligands can facilitate the hot electron-driven HER. Firstly, the surface charge of the ligand may facilitate HER reaction by allowing the protons to be near the nanoparticles.62–65 We measured the surface zeta potentials of all AuTP samples with different p-substituted benzene thiols and found them within a narrow range of 12 mV to 30 mV. Thus, it is unlikely that the electrostatic interaction between the ligands and reactants will significantly contribute to the observed HER rate difference among different ligands. Secondly, the ligands can enhance HER rate by facilitating the charge separation in plasmonic nanoparticles. A ligand LUMO may accept an electron from the AuTP, creating an n-type channel for the hot carriers. This may prolong the carriers’ lifetime, providing them a better chance to participate in the HER process. To further support the charge transfer process, we performed electrochemical impedance spectroscopy on the p-substituted benzene thiol-coated AuTP substrates. We find that the AuTP coated with 4-Br-Ph-SH showed the lowest charge transfer resistance among all the thiol functionalized samples, indicating favorable charge transfer at the AuTP-ligand interface (Fig. S10†).
Density functional theory-based calculations were performed on all nanoparticle-ligand systems to understand the microscopic factors determining the variation in HER rates and the high hydrogen generation with a Br-substituted catalyst. The ligand's dipole moment has been considered a significant parameter governing electron transfer to the reactants by altering the work function of the Au surface.66Fig. 5 shows the correlation between the dipole moment of the Au-ligand system (DT) for various substituents and hydrogen production. All values of DT are summarized in Table S1.† It is understood that a low DT shifts the work function to lower values, favoring electron transfer.67 However, DT for catalysts with F-, Cl-, and Br-substituted ligands is 1.768 Dy, 1.975 Dy, and 2.085 Dy, respectively, which does not correspond to the trends observed in HER rates. Thus, it is apparent that the ligand's polarity alone is insufficient to determine the catalyst's reactivity.
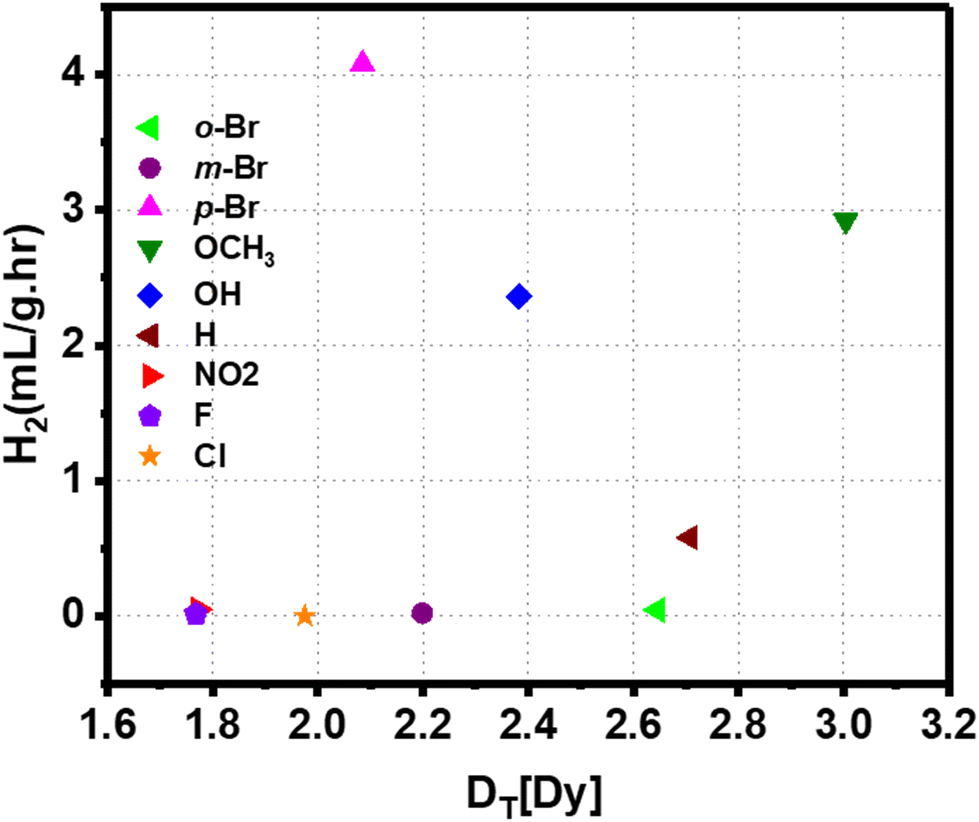 | ||
| Fig. 5 Correlation between the dipole moment of the Au-ligand system for various substituents and hydrogen production rate. | ||
As per the Marcus equation,68 the electron transfer rate between an initial and final state is governed by the interactions between the orbitals representing the two states. We examined the spatial distribution of relevant orbitals and their interactions in nanoparticle-ligand systems to discern the orbital contributions in electron transfer from the nanoparticle to the ligand. Fig. 6 shows the position of the lowest unoccupied molecular orbitals (LUMO) obtained by density functional theory (DFT) based computations. LUMOs are the pertinent orbitals for the transfer of electrons in a photo-excited catalyst. As can be seen, the LUMO of the Au-ligand system with OH-substitution is more delocalized than NO2-substitution. This corresponds to the high difference in reactivity observed for respective catalysts. To explore this further, the charge transfer integral (J) was calculated for the LUMO of nanoparticle and ligand as per the equation:
| J = ∫ψiHψjdr3 |
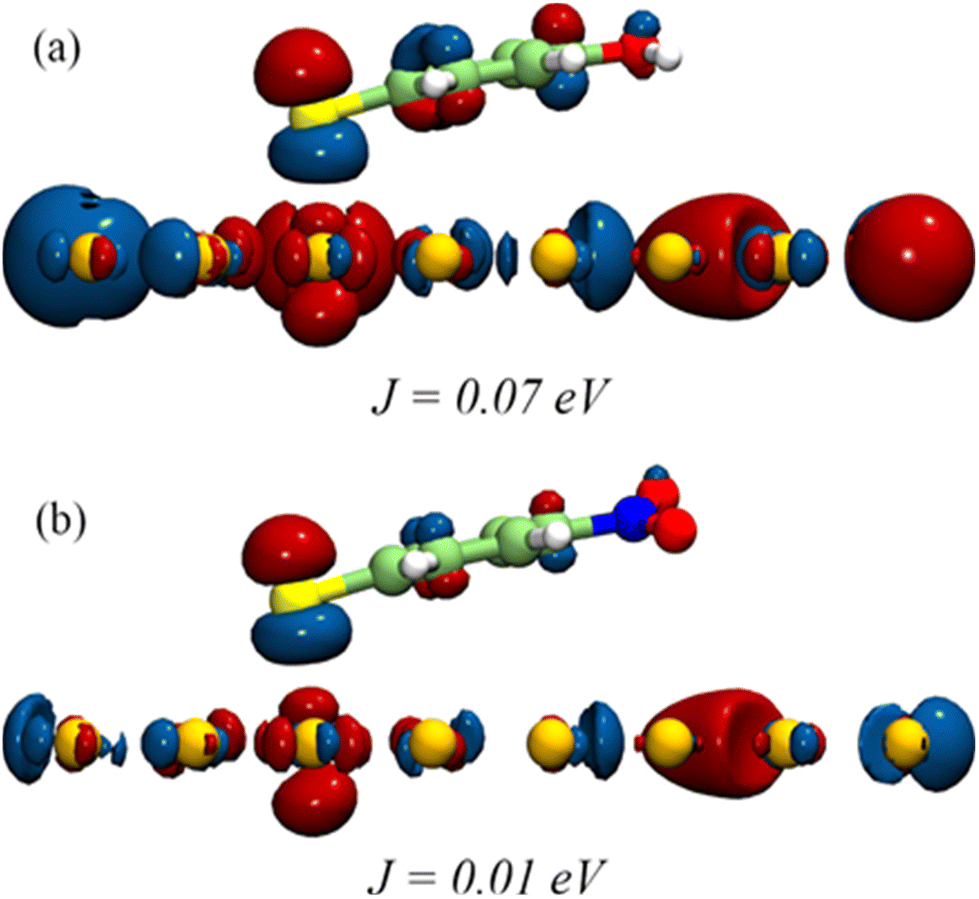 | ||
| Fig. 6 Frontier orbital distribution into (a) OH and (b) NO2 substituted Au-ligand systems showing the difference in their computed J values. | ||
J is one of the factors that determines the charge transfer rate between orbitals given by ψiand ψj.68 Here J is calculated based on DFT computations for all catalysts, with ψi and ψj representing the LUMO of the nanoparticle surface and ligand, respectively. A high value of J corresponds to the higher feasibility of electron transfer. J for catalysts with F-, Cl-, and Br- substituted ligands is 2.04 × 10–2 eV, 1.53 × 10–2 eV, and 3.31 × 10–2 eV, respectively, corresponding to the HER rates for ligands with halogens. Values of J for all systems are summarized in Table S1.†
We observed that both polarities of the ligand and interfacial orbitals interactions affect the electron transfer in the Au-ligand system and, consequently, the catalyst's efficiency. Therefore, the reactivity of the ligands can be described by a comprehensive model that combines these parameters. This is given by the function DT2/J, such that a high function value corresponds to high H2 generation. Fig. 7 shows the H2 generation measured for the catalysts plotted against the value of DT2/J. An increase in the value of the function corresponds to a steep decrease in reactivity. The effect of DT and J is to compensate for each other. A low value of the function corresponds to a system with low DT and (or) high J, both of which are favorable to electron transfer. The correlation between computed DT2/J and measured reactivity shows that polarity and orbital effects significantly determine the catalyst's efficiency. The high reactivity of the Br-substituted ligand can be explained based on the function. HER for catalyst with p-Br substituted ligand is higher than that of Cl- and F-substituted catalyst despite its higher D. However, the high value of J for Br-substituted ligand facilitates the electron transfer and enhances its activity. The combined effect of dipole and orbital interaction enhances reactivity for Br-substituted catalysts more extensively than others. Thus, we show that HER activity for ligand-nanoparticle can be predicted based on the combination of dipole and orbital couplings in the catalyst, as captured in the function DT2/J.
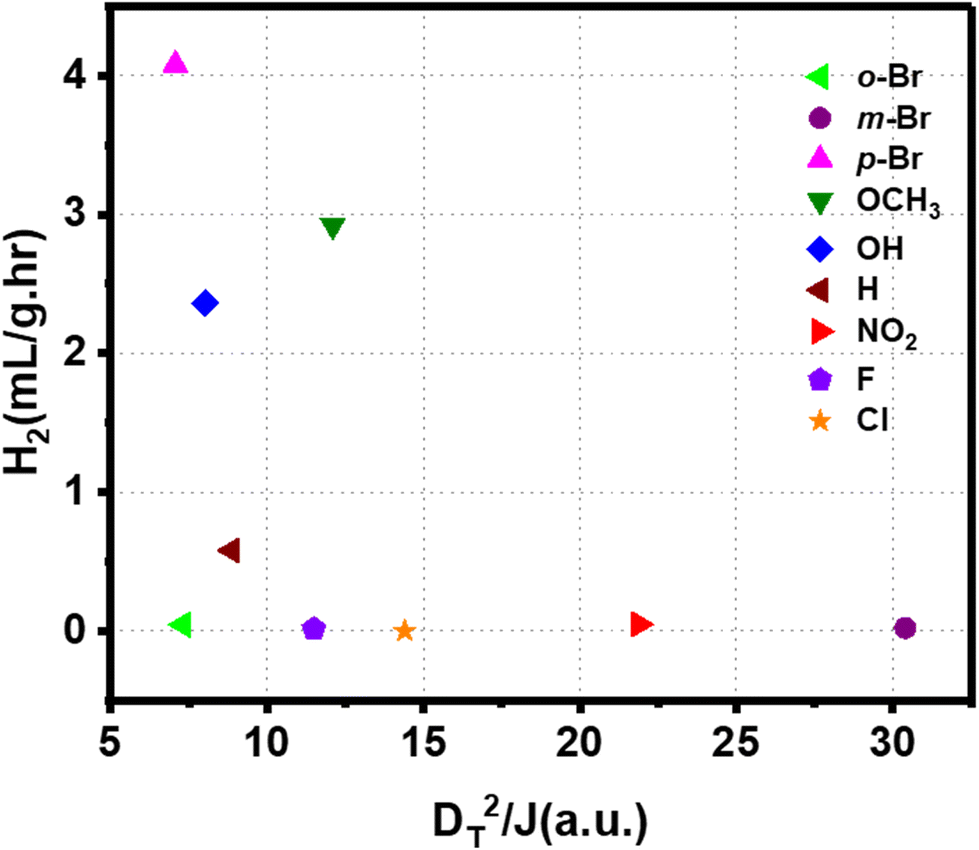 | ||
| Fig. 7 Correlation between the combined function comprised of dipole moment and J with net hydrogen production rate by different Au-ligand system. | ||
Now, we discuss the possible origin of the hydrogen gas observed in our experiments. Hydrogen formation may happen from the decomposition of impurities such as unreacted thiols and silanes or from reduction of water. To check this aspect, we first repeated our photocatalytic experiment under 808 nm excitation with 4-Br-Ph-S-AuTP substrate but replaced water with acetonitrile. We note that we have not seen any hydrogen production in this experiment (Fig. S13†). This result clearly indicates that the hydrogen originated from water and not from the decomposition of any thiol impurities [SERS measurement on our sample also did not show any S–H stretching peak (Fig. S11†)]. Now, water may produce hydrogen via water reduction or via hydrolytic activation of Si–H impurities that may be remaining from the synthesis procedure.3 We cleaned our samples thoroughly and removed any Si–H impurities. This was consistent with the absence of Si–H stretching in our SERS measurement (Fig. 2c). Nonetheless, we performed a control photocatalytic experiment using 4-Br-Ph-S-AuTP substrate and a broadband light with wavelengths between 1000–1600 nm as an excitation source (Fig. S18†). This wavelength range was selected as the associated photons would not have energy for water splitting but could drive a photothermal Si–H activation, as demonstrated previously.69 We also did not see any hydrogen formation in this study, confirming that Si–H activation is not the source of hydrogen. Furthermore, simultaneous measurement of oxygen reveals that hydrogen and oxygen are being produced at a ratio of 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is very close to what one would expect from a water splitting reaction (Fig. S12†). These observations confirm that hydrogen is being produced from water. The excitation photon energy is just above the thermodynamic potential requirement for water splitting(Fig. S16†), but the fast charge carrier recombination and the inert nature of gold (high overpotential) make water splitting highly unlikely on a pristine nanoparticle surface, which is also consistent with our observation that no hydrogen formation is seen with pristine AuTP substrates (Fig. 3). In the presence of an appropriate ligand, the charge separation process is greatly enhanced, making it favorable for the energetic carriers to participate in the reaction. Also, it may be noted that the reaction is likely to happen at the ligand-water interface, where the overpotential requirement may be different than the pristine Au surface. There is a possibility that the ligands can interact with water through dipole–dipole interaction or hydrogen bonding and may have an influence on photocatalytic activity in terms of the proximity of the reactant to the surface of the photocatalyst, although this aspect needs further theoretical and experimental studies.
:1, which is very close to what one would expect from a water splitting reaction (Fig. S12†). These observations confirm that hydrogen is being produced from water. The excitation photon energy is just above the thermodynamic potential requirement for water splitting(Fig. S16†), but the fast charge carrier recombination and the inert nature of gold (high overpotential) make water splitting highly unlikely on a pristine nanoparticle surface, which is also consistent with our observation that no hydrogen formation is seen with pristine AuTP substrates (Fig. 3). In the presence of an appropriate ligand, the charge separation process is greatly enhanced, making it favorable for the energetic carriers to participate in the reaction. Also, it may be noted that the reaction is likely to happen at the ligand-water interface, where the overpotential requirement may be different than the pristine Au surface. There is a possibility that the ligands can interact with water through dipole–dipole interaction or hydrogen bonding and may have an influence on photocatalytic activity in terms of the proximity of the reactant to the surface of the photocatalyst, although this aspect needs further theoretical and experimental studies.
To check for the contribution from a possible photothermal effect, we repeated hydrogen production studies with 4-Br-Ph-S-AuTP substrates under dark conditions but at a higher temperature. First, we measured the temperature on the substrate after 6 hours of laser excitation to be 31.4 °C. We used three 4-Br-Ph-S-AuTP substrates and performed the experiment at 32 °C. We did not see any hydrogen formation at 32 °C for two samples. No hydrogen was also observed when the temperature was raised to even 40 °C (Fig. S15†). However, in one substrate, we noticed a small but detectable amount of H2 production (∼2% of the hydrogen produced under 32 °C conditions, Fig. S13†). However, it is important to note that after further washing with ethanol, this substrate did not show any hydrogen production at 32 °C while maintaining its activity under photocatalytic conditions (Fig. S14†). The small amount of hydrogen formation under thermal condition seen in that specific sample could originate from degradation of some unreacted thiol or silane impurities remaining from improper sample cleaning. Finally, to further demonstrate the important role of the ligand's electronic structure on the photocatalysis, we repeated the photocatalytic hydrogen production reaction using benzenethiol-modified AuTP and cyclohexanethiol-modified AuTP photocatalytic substrates (Fig. S17†) under same experimental condition (808 nm excitation at 100 mW cm−2 power density). We found that the hydrogen production rates drastically differed between these substrates, and the cyclohexanethiol-modified AuTP substrate did not produce any hydrogen. In contrast, the benzenethiol-modified AuTP produced hydrogen at the rate of 0.58 mL g−1 h−1.
Conclusion
In summary, we show that organic ligands can drastically enhance the photocatalytic activity of plasmonic nanoparticles by providing electron transport channels that facilitate hot electron extraction from the nanoparticles. The experimental evidence and the theoretical calculations suggest that the ligand's polarity and the orbital coupling between the nanoparticle and ligands influence the overall photocatalytic efficiency. Easy tunability of the ligand orbital energy, favorable economic implications, and ease of fabrication will likely establish various metal nanoparticle-ligand constructs as preferred next-generation photocatalysts for a wide range of applications.Author contributions
G. J. and S. K. conceptualized the work. R. K. and G. J. performed the preparation and characterization of AuTP substrates, photocatalytic study, and analysis of data. K. P. and A. M. performed the theoretical DFT calculations. G. J., A. M., and S. K. wrote the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge financial support from Science and Engineering research board, India (CRG/2020/003471) and Gujarat Council of Science and Technology (GUJCOST/2020-21/872), India. We thank the central instrumentation facility (CIF) at IIT Gandhinagar for providing access to SEM, ICP-OES, Prof. Sameer Dalvi for the Raman Instrument, and Prashant Sharma for his assistance in SEM analysis. AM and KP acknowledge PARAM Ananta for computational resources.References
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS PubMed.
- Y.-C. Chen, Y.-K. Hsu, R. Popescu, D. Gerthsen, Y.-G. Lin and C. Feldmann, Nat. Commun., 2018, 9, 232 CrossRef PubMed.
- G. Joshi, A. Saha, A. Dutta and S. Khatua, ACS Appl. Mater. Interfaces, 2022, 14, 38815–38823 CrossRef CAS PubMed.
- H. Kim, H. Park, M. Kang and J. Y. Park, J. Chem. Phys., 2022, 157, 084701 CrossRef CAS.
- V. Jain, R. K. Kashyap and P. P. Pillai, Adv. Opt. Mater., 2022, 10, 2200463 CrossRef CAS.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- C. Ng, J. J. Cadusch, S. Dligatch, A. Roberts, T. J. Davis, P. Mulvaney and D. E. Gómez, ACS Nano, 2016, 10, 4704–4711 CrossRef CAS PubMed.
- P. Christopher and M. Moskovits, Annu. Rev. Phys. Chem., 2017, 68, 379–398 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- E. Cortés, W. Xie, J. Cambiasso, A. S. Jermyn, R. Sundararaman, P. Narang, S. Schlücker and S. A. Maier, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- P. Ghosh, A. Kar, S. Khandelwal, D. Vyas, A. Q. Mir, A. L. Chakraborty, R. S. Hegde, S. Sharma, A. Dutta and S. Khatua, ACS Appl. Nano Mater., 2019, 2, 5795–5803 CrossRef CAS.
- S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef CAS.
- T. Takata and K. Domen, ACS Energy Lett., 2019, 4, 542–549 CrossRef CAS.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- G. Joshi, A. Q. Mir, A. Layek, A. Ali, Sk. T. Aziz, S. Khatua and A. Dutta, ACS Catal., 2022, 1052–1067 CrossRef CAS.
- A. Q. Mir, G. Joshi, P. Ghosh, S. Khandelwal, A. Kar, R. Hegde, S. Khatua and A. Dutta, ACS Energy Lett., 2019, 4, 2428–2435 CrossRef CAS.
- U. Aslam, S. Chavez and S. Linic, Nat. Nanotechnol., 2017, 12, 1000–1005 CrossRef CAS.
- E. Cortés, L. V. Besteiro, A. Alabastri, A. Baldi, G. Tagliabue, A. Demetriadou and P. Narang, ACS Nano, 2020, 14, 16202–16219 CrossRef PubMed.
- L. Liu, S. Ouyang and J. Ye, Angew. Chem., Int. Ed., 2013, 52, 6689–6693 CrossRef CAS PubMed.
- X. Yu, F. Liu, J. Bi, B. Wang and S. Yang, Nano Energy, 2017, 33, 469–475 CrossRef CAS.
- X. Yang, L. Wu, L. Du and X. Li, Catal. Lett., 2015, 145, 1771–1777 CrossRef CAS.
- K. Song, H. Lee, M. Lee and J. Y. Park, ACS Energy Lett., 2021, 6, 1333–1339 CrossRef CAS.
- Q. Zhang, D. Thrithamarassery Gangadharan, Y. Liu, Z. Xu, M. Chaker and D. Ma, J. Materiomics, 2017, 3, 33–50 CrossRef.
- C.-H. Hao, X.-N. Guo, Y.-T. Pan, S. Chen, Z.-F. Jiao, H. Yang and X.-Y. Guo, J. Am. Chem. Soc., 2016, 138, 9361–9364 CrossRef CAS PubMed.
- D. B. Ingram and S. Linic, J. Am. Chem. Soc., 2011, 133, 5202–5205 CrossRef CAS PubMed.
- A. Manjavacas, J. G. Liu, V. Kulkarni and P. Nordlander, ACS Nano, 2014, 8, 7630–7638 CrossRef CAS PubMed.
- J.-J. Chen, J. C. S. Wu, P. C. Wu and D. P. Tsai, J. Phys. Chem. C, 2011, 115, 210–216 CrossRef CAS.
- P. Christopher, D. B. Ingram and S. Linic, J. Phys. Chem. C, 2010, 114, 9173–9177 CrossRef CAS.
- Y. Guo, Q. Zhou, X. Chen, Y. Fu, S. Lan, M. Zhu and Y. Du, J. Mater. Sci. Technol., 2022, 119, 53–60 CrossRef CAS.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef CAS PubMed.
- S. Lee, H. Hwang, W. Lee, D. Schebarchov, Y. Wy, J. Grand, B. Auguié, D. H. Wi, E. Cortés and S. W. Han, ACS Energy Lett., 2020, 5, 3881–3890 CrossRef CAS.
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2017, 8, 27 CrossRef PubMed.
- K. H. Leong, A. A. Aziz, L. C. Sim, P. Saravanan, M. Jang and D. Bahnemann, Beilstein J. Nanotechnol., 2018, 9, 628–648 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- H. H. Arefi, M. Nolan and G. Fagas, J. Phys. Chem. C, 2015, 119, 11588–11597 CrossRef CAS.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulović, ACS Nano, 2014, 8, 5863–5872 CrossRef CAS PubMed.
- A. Cirri, A. Silakov, L. Jensen and B. J. Lear, Phys. Chem. Chem. Phys., 2016, 18, 25443–25451 RSC.
- M. Hollerer, D. Lüftner, P. Hurdax, T. Ules, S. Soubatch, F. S. Tautz, G. Koller, P. Puschnig, M. Sterrer and M. G. Ramsey, ACS Nano, 2017, 11, 6252–6260 CrossRef CAS PubMed.
- K. Homma, S. Kaneko, K. Tsukagoshi and T. Nishino, ACS Appl. Nano Mater., 2023, 6, 8135–8140 CrossRef CAS.
- V. Jain, R. K. Kashyap and P. P. Pillai, Adv. Opt. Mater., 2022, 10, 2200463 CrossRef CAS.
- V. Jain, S. Roy, P. Roy and P. P. Pillai, Chem. Mater., 2022, 34, 7579–7597 CrossRef CAS.
- B. Kafle, M. Poveda and T. G. Habteyes, J. Phys. Chem. Lett., 2017, 8, 890–894 CrossRef CAS PubMed.
- N. Langer and O. Kedem, J. Phys. Chem. C, 2022, 126, 13705–13713 CrossRef CAS.
- T. Liyanage, M. Nagaraju, M. Johnson, B. B. Muhoberac and R. Sardar, Nano Lett., 2020, 20, 192–200 CrossRef CAS PubMed.
- M. Malicki, G. Heimel, Z.-L. Guan, S. D. Ha, S. Barlow, A. Kahn and S. R. Marder, J. Phys. Chem. C, 2011, 115, 7487–7495 CrossRef CAS.
- S. Volk, N. Yazdani, O. Yarema, M. Yarema, D. Bozyigit and V. Wood, J. Phys. Chem. Lett., 2018, 9, 7165–7172 CrossRef CAS PubMed.
- Y. Kim, A. J. Wilson and P. K. Jain, ACS Catal., 2017, 7, 4360–4365 CrossRef CAS.
- R. Schürmann, K. Ebel, C. Nicolas, A. R. Milosavljević and I. Bald, J. Phys. Chem. Lett., 2019, 10, 3153–3158 CrossRef PubMed.
- R. Schürmann, E. Titov, K. Ebel, S. Kogikoski, A. Mostafa, P. Saalfrank, A. R. Milosavljević and I. Bald, Nanoscale Adv., 2022, 4, 1599–1607 RSC.
- R. Schürmann and I. Bald, Nanoscale, 2017, 9, 1951–1955 RSC.
- S. A. Lee and S. Link, Acc. Chem. Res., 2021, 54, 1950–1960 CrossRef CAS PubMed.
- B. Foerster, V. A. Spata, E. A. Carter, C. Sönnichsen and S. Link, Sci. Adv., 2019, 5, eaav0704 CrossRef CAS PubMed.
- Z. Li, C. Zhang, H. Sheng, J. Wang, Y. Zhu, L. Yu, J. Wang, Q. Peng and G. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 38302–38310 CrossRef CAS PubMed.
- S. Roy, V. Jain, R. K. Kashyap, A. Rao and P. P. Pillai, ACS Catal., 2020, 10, 5522–5528 CrossRef CAS.
- S. Roy, S. Roy, A. Rao, G. Devatha and P. P. Pillai, Chem. Mater., 2018, 30, 8415–8419 CrossRef CAS.
- Z. Zhang and J. Kneipp, J. Phys. Chem. Lett., 2021, 12, 1542–1547 CrossRef CAS PubMed.
- R. Schürmann, A. Dutta, K. Ebel, K. Tapio, A. R. Milosavljević and I. Bald, J. Chem. Phys., 2022, 157, 084708 CrossRef PubMed.
- Y. Chen, Y. Zhu, H. Sheng, J. Wang, C. Zhang, Y. Chen, W. Huang and G. Lu, ACS Catal., 2022, 12, 2938–2946 CrossRef CAS.
- T. Liyanage, M. Nagaraju, M. Johnson, B. B. Muhoberac and R. Sardar, Nano Lett., 2020, 20, 192–200 CrossRef CAS PubMed.
- R. Schürmann, A. Dutta, K. Ebel, K. Tapio, A. R. Milosavljević and I. Bald, J. Chem. Phys., 2022, 157, 084708 CrossRef PubMed.
- Y. Kim, A. J. Wilson and P. K. Jain, ACS Catal., 2017, 7, 4360–4365 CrossRef CAS.
- V. Jain, S. Roy, P. Roy and P. P. Pillai, Chem. Mater., 2022, 34, 7579–7597 CrossRef CAS.
- S. Roy, V. Jain, R. K. Kashyap, A. Rao and P. P. Pillai, ACS Catal., 2020, 10, 5522–5528 CrossRef CAS.
- S. Roy, S. Roy, A. Rao, G. Devatha and P. P. Pillai, Chem. Mater., 2018, 30, 8415–8419 CrossRef CAS.
- B. de Boer, A. Hadipour, M. M. Mandoc, T. van Woudenbergh and P. W. M. Blom, Adv. Mater., 2005, 17, 621–625 CrossRef CAS.
- Y. Xu, H. Sun and Y.-Y. Noh, IEEE Trans. Electron Devices, 2017, 64, 1932–1943 CAS.
- K. Patrikar, N. Jain, D. Chakraborty, P. Johari, V. R. Rao and D. Kabra, Adv. Funct. Mater., 2019, 29, 1805878 CrossRef.
- N. Kumar, A. Sharma, A. Bahirat, G. Joshi and S. Khatua, J. Phys. Chem. C, 2023, 127, 13484–13492 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and theoretical calculation details, representative LSPR spectra, SERS, GC analysis, SEM images, and Table containing calculated D and J values. See DOI: https://doi.org/10.1039/d3nr02829a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |