
Compositing redox-rich Co–Co@Ni–Fe PBA nanocubes into cauliflower-like conducting polypyrrole as an electrode material in supercapacitors†
Poulami
Mukherjee‡
 ,
Vishwanath
R. S.‡
*,
Arie
Borenstein
and
Tomer
Zidki
*
,
Vishwanath
R. S.‡
*,
Arie
Borenstein
and
Tomer
Zidki
*
Department of Chemical Sciences and the Center for Radical Reactions and Materials Research, Ariel University, Ariel, 4077625, Israel. E-mail: rs.vishwanath234@gmail.com; tomerzi@ariel.ac.il
First published on 13th January 2023
Abstract
Supercapacitors (SCs) coupled with redox materials have been extensively investigated to maximize the energy density of SCs. Common metal–organic frameworks explicitly used for this purpose are Prussian blue analogs (PBAs). However, their low conductivity and number of external electroactive sites hinder their capacitance and reaction rate. Our work focuses on a material-level optimization strategy, including morphology modification and superstructure fabrication of PBA. We report the synthesis of Co–Co@Ni–Fe trimetallic core–shell PBA nanocubes containing an FeII/III redox couple in the Ni–Fe PBA shell over the conductive Co–Co PBA core. This complex underwent a reversible redox reaction and was further encapsulated in a polypyrrole (PPy) network as a redox additive to prepare a novel polymer-encapsulated double-PBA nanocube composite (Co–Co@Ni–Fe PBA–PPy). The composite exhibited a faradaic non-capacitive diffusion-dominated charge storage ability. It yielded an improved specific capacity of 318.1 C g−1 at 1 A g−1 with a capacity retention of 90% over 2000 cycles @15 A g−1. Furthermore, an asymmetric supercapacitor based on Co–Co@Ni–Fe PBA–PPy and activated carbon electrodes delivered a maximum energy density of 20 W h kg−1 at a power density of 808.9 W kg−1 within a 1.6 V voltage window. The electrochemical analysis demonstrated a considerable improvement in the charge-storage performance due to an increase in electron transfer, electrolyte diffusion, and electroactive area via strong electronic coupling between the Co–Co@Ni–Fe core–shell PBA and the PPy. Furthermore, this work helps to differentiate the composite's current contributions from the PBA and PPy. The detailed electrochemical characterization steps of these methods concerning redox additives integrated into conducting polymers are provided in this work.
1. Introduction
Electrochemical energy storage systems such as batteries and supercapacitors (SCs) are primary devices that efficiently store and deliver energy produced from sustainable sources.1 Compared to SCs, batteries can provide high energy density; however, their power density is relatively low due to the slow ion transport through the electrode. SCs, on the other hand, having higher power density, can quickly store and deliver significant energy and demonstrate a higher cycle life than batteries.2 Hence, SCs have been widely investigated, aiming to achieve high energy and power densities in one system (electrode) for better performance in practical applications.Integrating active redox species and capacitive materials in one electrode is one of the most straightforward methods to maximize the energy density of SCs by preserving their power density and cyclic stability.3–5 However, such integration results in a hybrid energy storage system with the co-existence of faradaic, capacitive, or pseudocapacitive energy storage mechanisms.6,7 The possible co-existing mechanisms are mainly of three kinds: (1) the electric-double-layer (EDL) capacitance that arises from the electrostatic charge storage via a thin electron-ion partition layer at the electrode–electrolyte interface; (2) the faradaic capacitive (pseudocapacitive) charge storage by the non-diffusion-controlled reversible redox reactions at the electrode–electrolyte interface; and (3) the faradaic non-capacitive process by diffusion-controlled reversible redox reactions at the electrode–electrolyte interface or within the solid electrode (solid-state diffusion in battery electrodes).2,6,7 In this mixed regime, the kinetics study can only provide a rough differentiation between capacitive and diffusive current contributions to the energy storage mechanism.6
SCs are categorized as EDL capacitors and pseudo capacitors based on the energy storage mechanism. In EDL SCs, many carbon-based electrode materials, such as activated carbon (AC), carbon nanotubes, graphene, and carbon nanofibers, have been used.8–11,12 The capacitance is limited to EDL formation; hence the specific capacitance (Cs) is lower than that of the pseudocapacitive electrode materials.8 As a result, numerous pseudocapacitive compounds, including transition metal oxides, sulfides, phosphides, selenides, nitrides, and conducting polymers, are employed.8,13–15 Among them, porous crystalline metal–organic frameworks (MOFs) are mainly attractive due to their high surface area with open frameworks for insertion/desertion of ions to associate the faradaic redox reactions.
Accordingly, Prussian blue analogs (PBAs) are valuable materials due to their high surface area, 3D open framework, high charge-transfer kinetics, and high ionic conductivity required for hybrid SCs.4,16–20 On the other hand, most PBAs have low electrical conductivity, a short cycle life, and poor stability in alkaline and neutral solutions, preventing them from achieving the desired energy/power densities.21,22 Mainly, the electrochemical stability deteriorates with increasing cycles due to the rapid redox reactions at the electrode–electrolyte interface. Integrating PBAs with conducting polymers has been proposed as an effective way to overcome these challenges.4,16,20 The coated conducting polymers anchored on the PBA nanocubes serve as a protective layer to avoid electrode materials dissolution during cycling. Also, conducting polymers act as electronic conductors to improve the electrical conductivity of PBAs by forming a linkage between separated nanocubes. Furthermore, the synergistic effect of conducting polymers and multimetallic structures with redox reactions (pseudocapacitance) further improves the specific capacitance.4,23 Accordingly, electrode materials composited of PBAs and conducting polymers resulted in SCs with enhanced electrochemical performance corresponding to their constituents.4,23
Among various conducting polymers, polypyrrole (PPy) is commonly used for compositing due to its feasible preparation, high conductivity, and superior pseudocapacitance.24–26 Nevertheless, these good intrinsic properties depend on polymerization (chemical or electrochemical); compared to chemical oxidation, the electrochemical methods allow more control over coating thickness and morphology.24,27 In the electrochemical process, a solvent containing the pyrrole monomer is oxidized by the applied anodic potential on the electrode surface. Thus, the PPy properties rely on the solvent, supporting electrolyte, electropolymerization techniques, and substrate.24,27–29 The two main drawbacks that hinder the use of PPy in high-performance SCs are the lower practical capacitance (than theoretical capacitance) and the lack of cycling stability.15,30 Frequently, PPy-based electrodes retain <50% of their initial capacitance after 1000 cycles.31,32 Compositing using various materials, including carbon-based materials, metal oxides, metal–organic frameworks, and redox probes, can address these issues.15,30,31 In this work, we increased the cyclic stability of both the constituents (PBA and PPy) by PBA nanocubes deposition on carbon paper (CP) and completely enfolded them in the PPy network. Moreover, inert substrates, such as CP, are favorable over metals for electropolymerization since the oxidation potential of the pyrrole monomers is higher than that of oxidizable metals.29,33
Herein, we employed Co–Co PBA as a template for synthesizing well-defined Co–Co@Ni–Fe core–shell PBA nanocubes (Co–Co@Ni–Fe PBA). The rational integration of Fe, Co, and Ni in the hierarchical core–shell nanocubes played an essential role in manipulating the charge transfer capability and conductivity of the catalyst to achieve its activity. Later we electro-deposited PPy to enwrap the core–shell PBA nanocubes to increase the energy density, conductivity, and cyclic stability of the Co–Co@Ni–Fe PBA–PPy composite. This refinement in the electrochemical performance in the composite was assigned to an upsurge in electron transfer, electrolyte diffusion, and electroactive area through strong electronic coupling and interfacial phenomena between Co–Co@Ni–Fe core–shell PBA and PPy. However, including the redox behavior of the Co–Co@Ni–Fe core–shell PBA into PPy increased the complexity of determining the charge storage mechanisms. Our analysis roughly differentiated that around ∼93% of the Cs arrived from the diffusion-controlled inner capacitance and ∼7% from the non-diffusion-controlled outer-surface capacitance.
2. Experimental procedure
2.1 Materials
Potassium hexacyanocobaltate (K3[Co(CN)6]), cobalt nitrate hexahydrate (Co(NO3)2·6H2O), trisodium citrate dihydrate (C6H5Na3O7·2H2O), potassium ferrocyanide (K3[Fe(CN)6]), nickel nitrate hexahydrate (Ni(NO3)2·6H2O), 5 wt% Nafion, and tetrabutylammonium hexafluorophosphate (TBAClO4) were purchased from Alfa Aesar. Polyvinylpyrrolidone (PVP-M.W. 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Holland Moran. Acetonitrile, pyrrole, and AC were purchased from Sigma Aldrich. All chemical reagents were of analytical grade and were used as received without further purification. Milli-pore water (deionized water, DI) with a resistivity of >15 MΩ cm was used throughout the experiments.
000) was purchased from Holland Moran. Acetonitrile, pyrrole, and AC were purchased from Sigma Aldrich. All chemical reagents were of analytical grade and were used as received without further purification. Milli-pore water (deionized water, DI) with a resistivity of >15 MΩ cm was used throughout the experiments.
2.2 Synthesis of Co–Co PBA nanocubes
The Co–Co PBA nanocubes were synthesized by the co-precipitation method following a reported protocol.34 Briefly, solution A was prepared by combining 0.6 mmol Co(NO3)2 and 0.9 mmol sodium citrate in 20 mL DI water. Solution B contained 0.4 mmol of K3[Co(CN)6] dissolved in 20 mL DI water. Then, solution B was added dropwise to solution A under magnetic stirring for 15 min to distribute the particles uniformly. The resulting mixed solution was aged for 24 h at room temperature. The pink-colored product was centrifuged, rinsed thrice with DI water and absolute ethanol, and dried overnight at 70 °C.2.3 Synthesis of Co–Co@Ni–Fe core–shell PBA nanocubes
20 mg of Co–Co nanocubes, 142 mg of Ni(NO3)2·6H20, 0.3 mg of sodium citrate, and 0.3 g of PVP (K30) were dissolved in 30 mL DI water to obtain a transparent green solution. Meanwhile, solution B was prepared by dissolving 66 mg of K3[Fe(CN)6] in 20 mL of DI water. Then, solution B was added to solution A under constant magnetic stirring for 15 min. The resulting solution was aged for 24 h at room temperature without interruption. The obtained, yellow-colored precipitate was centrifuged, washed thrice with DI water and absolute ethanol, and dried overnight at 70 °C.2.4 Preparation of electrodes
3. Results and discussion
Since regulating the morphologies and sizes of PBA nanocubes in an aqueous medium is complex, the straightforward co-precipitation approach was used to generate regular Co–Co PBA nanocubes.35 Because of its similar crystal structure and lattice constant,36 a Ni–Fe PBA shell could be easily formed atop the Co–Co PBA nanocubes through epitaxial deposition at ambient temperature. The resulting nanocubes are marked as Co–Co@Ni–Fe core–shell PBA. Later, the core–shell structure was enwrapped by PPy, denoted as Co–Co@Ni–Fe PBA–PPy. The synthesis procedure of the composite is illustrated in Scheme 1.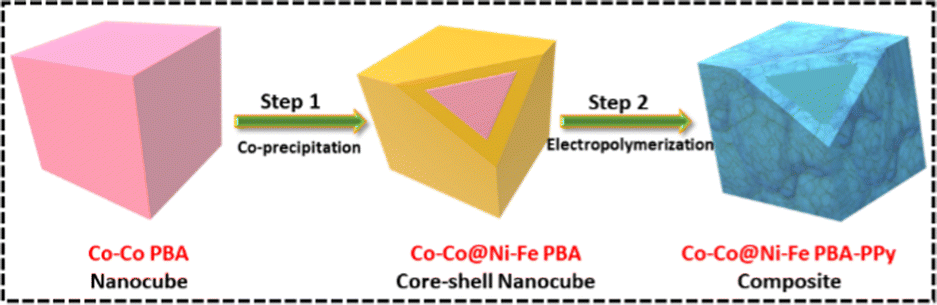 | ||
| Scheme 1 Schematic illustration of the synthesis process of the Co–Co@Ni–Fe PBA–PPy composite. | ||
3.1 Structural characterization
The structural morphology of the nanocubes was analyzed by field-emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM). Fig. 1(a) shows the HR-SEM image of the Co–Co@Ni–Fe core–shell PBA nanocubes with an average particle size of 400 nm. The core–shell nanocubes retain their cubic structure due to the low-speed co-precipitation synthesis method. Adding citrate as a chelating agent slowed the crystallization process and improved the catalyst crystallinity.37 The Ni–Fe PBA's homogeneous coating over the Co–Co PBA template was demonstrated by the contrast in the TEM images (Fig. 1(b)). Fig. 1(c) depicts a shell thickness of ∼30 nm over the Co core. The elemental mapping images of a single core–shell nanocube obtained using energy dispersive X-ray (EDX) reveal a Co-riched core and a shell rich in Ni and Fe, Fig. 1(d), illustrating the compositional variability of the Co–Co@Ni–Fe core–shell PBA nanocubes. In supercapacitors, the synergistic combination of Ni and Co provides excellent electrical conductivity and electrochemical performance.38,39 The Fe species improve the conductivity of the electrode40,41 or act as an electron promoter36 to enable high activities, and Ni stabilizes the crystal structure.42 | ||
| Fig. 1 (a) HR-SEM image; (b), (c) TEM images; (d) STEM image of core–shell PBA and the corresponding elemental mapping of the same frame showing Co, Fe, and Ni with their overlay; (e) XRD patterns of Co–Co@Ni–Fe core–shell PBA. | ||
Fig. S3(a) and (b) (ESI†) show the HR-SEM and TEM images of the initial Co–Co PBA with a uniform particle size of around 300 nm. The HR-SEM image of PPy shows a net-like structure (Fig. 2(a)), but the magnified SEM image reveals a homogenous granular structure reminding one of cauliflower (Fig. 2(b)). The larger and smaller granules appear to form a dense, non-porous layer. Fig. 2(c) depicts the PPy enveloped core–shell PBA nanocubes. The cauliflower-like arrangement on the nanocubes’ surface in the composite is highlighted in Fig. 2(d). Roughness, a critical physical-chemical surface property, increased implicitly with polymerization, which benefits the electrode/electrolyte contact (Fig. 2(d)).
 | ||
| Fig. 2 HR-SEM images of (a), (b) PPy; and (c), (d) Co–Co@Ni–Fe PBA–PPy composite. | ||
Powder X-ray diffraction (XRD) analysis validated the effective production of Co–Co PBA nano-cubes (Fig. S3c, ESI†). The prominent peaks suggest that the nanocubes created exhibited excellent crystallinity and could be indexed to Co3[Co(CN)6]2 (JCPDS no. 77-1161).43 The diffraction peaks of the core–shell PBA relocated at slightly low angles due to the Ni–Fe PBA shell formation compared to the Co–Co PBA core (Fig. 1(e)). The crystalline nature of the nanocubes proved advantageous because faster ion diffusion and a greater depth could be achieved in well-crystalline PBAs with smaller dimensions and a higher specific surface area.44 With a higher Ni content, the atomic ratio of Ni/Fe was proven to be 1.2 in the EDX spectrum (Fig. S4, ESI†). Co, as the core element, accounted for 80% of the total composition of the core–shell nanocube. The ICP results were consistent with the EDX study, which showed that the Ni/Fe ratio for Co–Co@Ni–Fe PBA was 1.5 (Table S1, ESI†), i.e., the theoretical ratio for Ni–Fe PBA. We performed Fourier transform infrared (FTIR) spectroscopy to characterize the chemical composition of PPy, Co–Co@Ni–Fe PBA, and the Co–Co@Ni–Fe PBA–PPy composite (Fig. S5, ESI†). The detailed study is provided in the ESI.† Fig. S6 (ESI†) shows the thermal gravimetric analysis (TGA) plot for the Co–Co@Ni–Fe PBA–PPy composite deposited on the CP substrate.
X-ray photoelectron spectroscopy (XPS) analysis of core–shell PBA, PPy, and Co–Co@Ni–Fe PBA–PPy composite was performed to characterize the chemical composition and element valence states. In the high-resolution spectra, Fig. 3(b)–(d) and Fig. S7, S8b–d, and S9b, c (ESI†), the black and red colors stand for experimental and fitting data, respectively, and the other colors for deconvoluted data. Fig. 3(a) and Fig. S8a (ESI†) confirm the presence of Ni, Co, Fe, O, N, and C in the Co–Co@Ni–Fe PBA–PPy composite and core–shell PBA, respectively. It is worth noting that the peak intensities of O, C, and N increased in the PBA–PPy composite compared with core–shell PBA, indicating the successful polymerization of PPy over core–shell PBA (compare Fig. 3(a) and Fig. S8a, ESI†). In the high-resolution Co 2p spectrum, the binding energies (BE) at 785.3 eV and 798.6 eV corresponded to the Co 2p3/2 and Co 2p1/2 of Co2+ (Fig. 3(b)).45,46 These peaks were upshifted compared to core–shell PBA nanocubes (Fig. S8b, ESI†). The peak at 789.3 eV was attributed to a satellite of Co2+.47 The prominent Ni 2p3/2 peak at 856.5 eV belonged to Ni2+, which was downshifted with respect to core–shell PBA. The other peaks at 858.4 eV and 863.4 eV were satellite peaks (Fig. 3(c)).48 The peak at 874.4 eV of Ni 2p1/2 was attributed to NiOOH (Ni 2p1/2 at 875.6 eV for core–shell PBA, Fig. S8c, ESI†).49 The presence of oxygen causes the formation of Ni(OH)2, which may be electrochemically oxidized to NiOOH by the presence of Co.50 It is known that NiOOH is much more conductive than Ni(OH)2, and its conductivity can be significantly increased by the presence of Fe.50 Therefore, the formation of NiOOH in the Co–Co@Ni–Fe PBA–PPy composite can further increase the conductivity of the catalyst. In the high-resolution Fe 2p spectrum (Fig. 3(d)), the BE peaks at 710.2 eV and 723.6 eV corresponded to Fe 2p3/2 and Fe 2p1/2 of Fe2+, respectively.51 The peak at 712.1 eV was ascribed to Fe 2p3/2, representing Fe3+.52 All the Fe peaks also shifted towards higher BEs with respect to Co–Co@Ni–Fe core–shell PBA. Hence, the iron in the Ni–Fe PBA was present in mixed oxidation states of Fe2+ and Fe3+ as both Ni2+–CN–Fe3+ and Ni2+–CN–Fe2+. The Ni2+–CN–Fe2+ sites were mainly located at the surfaces of the nanocubes, while the Ni2+–CN–Fe3+ were at the body center.17 Since XPS is a surface analysis, we mainly saw the Fe2+ peaks, not Fe3+. The BE shifts of Co, Fe, and Ni in the Co–Co@Ni–Fe PBA–PPy composite relative to core–shell PBA implied synergistic electronic interactions between Fe, Co, and Ni atoms due to the Ppy deposition in the Co–Co@Ni–Fe PBA–PPy composite. The deconvoluted N 1s spectrum showed three peaks at 399.9, 401.4, and 402.4 eV, belonging to C–N or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, C
N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, and N–O bonds, respectively (Fig. S7, ESI†).53 The oxidation states and electronic environment of all the elements of core–shell PBA and PPy are shown in Fig. S8 and S9 (ESI†).
N, and N–O bonds, respectively (Fig. S7, ESI†).53 The oxidation states and electronic environment of all the elements of core–shell PBA and PPy are shown in Fig. S8 and S9 (ESI†).
 | ||
| Fig. 3 (a) XPS survey spectrum of the Co–Co@Ni–Fe PBA–PPy composite; high-resolution XPS spectra of (b) Co 2p; (c) Ni 2p; (d) Fe 2p. | ||
3.2 Electrochemical studies
Initially, cyclic voltammetry (CV) measurements of the as-prepared Co–Co@Ni–Fe core–shell PBA (Fig. 4(a)) and other bimetallic PBA complexes with different metal combinations (Fig. S10, ESI†) were executed to observe the metal-centered electrochemical behavior. As shown in Fig. 4(a), the CVs of Co–Co@Ni–Fe core–shell PBA showed well-defined, metal-based reversible FeII/III redox couples, with the ratio of anodic to the cathodic current being close to unity. The FeII/III sites in the Ni–Fe PBA outer shell (over the Co–Co PBA core) were the source of this redox couple wave, and the Co–Co PBA showed no prominent peaks (a detailed explanation is provided with Fig. S10, ESI†). The CV integral area increased gradually with the scan rate. Fig. 4(b) exhibits the linear dependence of both anodic (Ipa) and cathodic peak currents (Ipc) against the square root of the scan rate (ν1/2), indicating that the FeII/III faradaic redox reaction was diffusion-controlled and obeyed the Randles–Sevcik equation. | ||
| Fig. 4 (a) CV curves of Co–Co@Ni–Fe core–shell PBA at different scan rates in 1.0 M Na2SO4; (b) the dependency of the peak currents on the square root of scan rates; (c) CV profiles of the Co–Co@Ni–Fe PBA–PPy composite at various scan rates in 1.0 M Na2SO4; (d) the plot of peak currents against the square root of scan rates. | ||
After the electropolymerization of PPy on Co–Co@Ni–Fe core–shell PBA, the CV curves of the Co–Co@Ni–Fe PBA–PPy composite depicted redox peaks between 0 to 0.7 V due to faradaic FeII/III redox reactions (Fig. 4(c)). The CV curve area increased with the scan rates, and the ratio of the anodic peak current to the cathodic peak current was almost identical. Also, the CV shapes were retained at the higher scan rates showing the faster charge/discharge rates supported by PPy conducting polymer. The order-of-magnitude increase in CV current for the Co–Co@Ni–Fe PBA–PPy composite compared to Co–Co@Ni–Fe core–shell PBA revealed the high conductivity, enhanced electron transport, and charge–discharge rates due to the synergistic effect of PBA and PPy constituents. The polymerization of PPy in the spaces between the PBA nanocube layers provided improved conductivity, interfacial effects, and electrolyte diffusion. The linear dependence of the peak currents as a function of the scan rate's square root was retained even after the PPy deposition. The linear curve intercepts close to the origin ensured the FeII/III reversible redox couple was diffusion-controlled and followed the Randles–Sevcik equation (Fig. 4(d)).
According to Lindström et al.,54 the power-law (i = aνb) relates to the non-diffusion-limited capacitive/pseudocapacitive and diffusion-limited faradaic current contribution of the redox peaks by the relationship between the peak current response (i) with its corresponding scan rate (ν). The b is the exponential component (slope) obtained from the plot of log (peak current) vs. log (scan rate); b = 0.5 for the diffusion-dominated charge storage phenomenon, and b = 1 for the capacitive type charge-storage process. The transition range from 0.5 to 1 indicates the change from the diffusion-controlled reaction to the capacitive type (surface-controlled reaction).5,16,55,56
From Fig. 5(a), the derived b values of the Co–Co@Ni–Fe PBA–PPy composite were 0.6443 and 0.6440, corresponding to the anodic and cathodic peaks, respectively, showing the co-existence of capacitive and diffusion-controlled contributions to the energy storage mechanism. In this mixed regime, the current was regulated by diffusion and electrochemical redox reaction rate. However, the b values are close to 0.5, indicating that the diffusion-controlled charge-storage process was dominant. The concentration gradient could arise within the aqueous electrolyte at the electrode–electrolyte interface and inside the solid composite electrode. Additionally, Trasatti's analysis was used to quantify the contribution of capacitive and diffusive currents to the total capacitance value.54,57–61 According to this approach, the total charge (qT) is the sum of the diffusion-controlled inner (qi) and non-diffusion-controlled outer surface charge (qo); by dividing the charge with the CV potential window, total capacitance (CT), diffusion-controlled inner capacitance (Ci), and non-diffusion controlled outer-surface capacitance (Co) can be determined.54,62,63 The CT equals the reciprocal y-intercept of the Cs−1vs. ν1/2 plot (Fig. S11a, ESI†), and Co is equal to the y-intercept of the Csvs. ν−1/2 plot (Fig. S11b, ESI†). The subtraction of Co from CT gives the maximum diffusion-controlled inner capacitance (Ci). Over ∼92.9% of the Co–Co@Ni–Fe PBA–PPy composite's Cs came from the diffusion-controlled inner capacitance contribution, and the remaining ∼7.1% from the non-diffusion controlled outer-surface capacitance. All the electrode–electrolyte interfaces formed the EDL. In faradaic energy storage systems, EDL capacitance does not contribute significantly; hence it can be neglected.7
 | ||
| Fig. 5 (a) The plot of the logarithm of peak current vs. the logarithm of scan rate; (b) galvanostatic charge–discharge curves of Co–Co@Ni–Fe PBA–PPy composite at different current densities; (c) comparing the specific capacitance of all materials at increasing current densities; and (d) Nyquist plots of Co–Co@Ni–Fe PBA–PPy and Co–Co@Ni–Fe core–shell PBA composites at a fully charged state. | ||
In the case of bare PPy, the CVs seemed nearly quasi-rectangular when scanned up to 0.8 V at different scan rates (Fig. S12a, ESI†). The CV integral area increased with the scan rate; however, this behavior diminished when scanned up to 1.0 V (Fig. S12b, ESI†). PPy underwent irreversible oxidative degradation in aqueous solutions at higher oxidation potentials, resulting in a loss of electrochemical activity, conjugation, and conductivity.64–66 Hence, the gradual decrease in current with subsequent scan rate increase (5 to 120 mV s−1) was observed in a wider potential window (−0.4 to 1 V). In contrast, the Co–Co@Ni–Fe PBA–PPy composite did not show a decrease in the peak current with the scan rate in the wider potential window (up to 1.0 V in Fig. 4(c)). Still, for further galvanostatic charge–discharge (GCD) measurements, the potential range was limited between −0.4 to 0.8 V to avoid partial/overoxidation of PPy. GCD measurements were carried out at different current densities to estimate the Cs of Co–Co@Ni–Fe PBA–PPy composite (Fig. 5(b)), Co–Co@Ni–Fe core–shell PBA (Fig. S13a, ESI†), and PPy electrodes (Fig. S13b, ESI†), using eqn (S2) (ESI†). As shown in Fig. 5(b), the discharge curve of the composite had two regions; the first region (0.8 to 0.4 V) was designated for the linear discharge, including the iR-drop that discharged the charge near the surface. Similar behavior was seen with the PBA core–shell nanocubes (Fig. S13a, ESI†). Conversely, the second region, from 0.4 to −0.4 V, was assigned to the extended discharge curve, where the diffusion-limited de-intercalation of the ions from the Co–Co@Ni–Fe PBA–PPy composite dominates. Fig. 5(c) shows that the Co–Co@Ni–Fe PBA–PPy composite showed the best performance, and the calculated Cs values were 265, 258, 251, 245, 239, 236, 233, and 225 F g−1 at the current densities 1, 2, 3, 4, 5, 6, 7, and 8 A g−1, respectively. For PPy, the values are 183, 168, 159, 148, 143, 141, 135, and 133 F g−1, and Co–Co@Ni–Fe core–shell PBA were 38, 20, 14, 12, 10, 9, 8.4, and 8 F g−1 (Fig. 5(c)), at identical current densities. The Cs of all materials decreased with an increase in the current density because the electrolyte ions did not get adequate time to diffuse inside the material. Note that the Co–Co@Ni–Fe PBA–PPy and Co–Co@Ni–Fe PBA exhibit battery-type behavior, so their electrochemical performance can be compared and presented in terms of specific capacity (Qs, C g−1). The Qs values of Co–Co@Ni–Fe PBA–PPy were 318.1, 308.3, 301.7, 295.1, 287.6, 285.7, 284.1, and 271 C g−1 at current densities of 1, 2, 3, 4, 5, 6, 7, and 8 A g−1, respectively. In contrast, the Qs of Co–Co@Ni–Fe PBA were 46.5, 24.9, 17.5, 14.5, 12.6, 11.3, 10, and 9.6 C g−1 at the same current densities as Co–Co@Ni–Fe PBA–PPy. Furthermore, the estimated Cs value of Co–Co@Ni–Fe PBA–PPy was compared with other reported PBA-based electrodes in Table S2 (ESI†). Fig. 5(d) exhibits the Nyquist plots with the inset of the fitted equivalent circuit model. Rs is the equivalent series resistance arising from the combined resistance of the electrolyte, active material, current collector, and the contact between the active material and current collector. In the high-frequency region, the Co–Co@Ni–Fe PBA–PPy composite showed a negligible increase in the Rs value compared to the Co@Ni–Fe core–shell PBA (Table S3, ESI†). The semicircle diameter near the high-to-medium frequency area presented the charge-transfer resistance (Rct) across the electrode/electrolyte interface, and the inclined slope at the low-frequency part confines the Warburg diffusion resistance (W), which emerged due to the frequency-dependent ion diffusion inside the electrolyte and the electrode composite material. The lower Rct value and the high slope (plot shift towards the −Z′′) for Co–Co@Ni–Fe PBA–PPy composite indicated the fast charge transfer rate at the electrode/electrolyte interface and effective ions diffusion (Table S3, ESI†). The EIS study revealed that PPy deposition on core–shell PBA significantly enhanced the conductivity and ion diffusion, which set the superior capacitive performance of the Co–Co@Ni–Fe PBA–PPy composite. Fig. S14 (ESI†) depicts the cyclic stability test of the PBA–PPy composite performed over 3000 charge–discharge cycles at a current density of 15 A g−1. The Co–Co@Ni–Fe PBA–PPy composite demonstrated excellent cycling stability over 2000 cycles with a 90% capacity retention; the capacitance dropped to 71.4% after 3000 cycles. This rapid decline was primarily due to the loss of active capacitive sites of the composite, either redox sites of Co–Co@Ni–Fe core–shell PBA or degradation of the PPy network by swelling and shrinking upon cycling. The Co–Co@Ni–Fe PBA–PPy composite's HR-SEM images are depicted in Fig. S15a and b (ESI†), and they indicate some structural alteration that may be responsible for the modest reduction in activity after 3000 cycles.
To assess the practical utility of the Co–Co@Ni–Fe PBA–PPy composite, an asymmetric supercapacitor (ASC) device (denoted Co–Co@Ni–Fe PBA–PPy//AC) was assembled using Co–Co@Ni–Fe PBA–PPy as a positive electrode and AC as a negative electrode. The fabrication procedure is given in the electrochemical measurements section (in the the ESI†). Before the device assembly evaluation, electrochemical measurements of AC were conducted in the similar three-electrode configuration used for Co–Co@Ni–Fe PBA–PPy (Fig. S16, ESI†), and the comparison CVs are depicted in Fig. 6(a). The charge/mass balance between positive and negative electrodes was assessed following eqn (S4) (ESI†) to be 0.75. The predicted operating potential window of the Co–Co@Ni–Fe PBA–PPy//AC device was 2.0 V, ascribed to the combined potential window of the positive and negative electrodes. However, the CVs were scanned up to 1.6 V (Fig. 6(b)) to avoid electrolyte decomposition and oxidative degradation of PPy at 1.7 V, suggesting that the stable potential window of the device is up to 1.6 V. Fig. 6(b) illustrates the CV curves of the ASC device at different scan rates; the anodic peaks were visible with less distinguishable cathodic peaks. This trend was almost retained with increasing scan rates, indicating the good electrochemical reversibility associated with the fast charge/discharge properties.
 | ||
| Fig. 6 (a) CV curves of AC and Co–Co@Ni–Fe PBA–PPy composite at 40 mV s−1 in 1.0 M Na2SO4. (b) CV curves, (c) GCD curves, and (d) Ragone plot of the Co–Co@Ni–Fe PBA–PPy//AC device. | ||
Nevertheless, both anodic and cathodic peaks were visible only when a symmetric SC cell configuration was formed by taking positive and negative electrodes containing an equal amount of Co–Co@Ni–Fe PBA–PPy (Fig. S17, ESI†). Based on the GCD curves of the ASC device at various current densities (Fig. 6(c)), the Qs values were 90, 67, 61.9, 45.6, 44.5, 40.2, and 31.5 C g−1 at current densities of 1, 2, 3, 4, 5, 6, and 7 A g−1, respectively. The long-term stability of the device showed 79% capacity retention at 10 A g−1 over 2000 cycles (Fig. S18, ESI†), indicating good cyclic stability. Moreover, the energy density and power density of the ASC device are estimated using eqn (S5) and (S6) (ESI†), and the relationship between them is shown in the Ragone plot (Fig. 6(d)). The device delivered a high energy density of 20 W h kg−1 at a power density of 808.9 W kg−1 and maintained 7.0 W h kg −1 at a higher power density of 1996.1 W kg−1. The estimated energy and power density were compared with other PBA composite-based ASC devices in the reported literature (Table S4, ESI†).
4. Conclusions
In summary, Co–Co@Ni–Fe trimetallic core–shell PBA nanocubes were synthesized using the co-precipitation method, where Ni–Fe PBA uniformly coated the Co–Co PBA core. The Co-rich core facilitated the conduction of electrons, whereas the FeII/III sites in the Ni–Fe PBA shell acted as a redox couple. The Co–Co@Ni–Fe PBA redox nanocube was encapsulated in a PPy network to boost core–shell PBA stability, redox response and electrolyte diffusion to increase the Qs of the Co–Co@Ni–Fe PBA–PPy composite. Profiting from the abovementioned properties, the Co–Co@Ni–Fe PBA–PPy composite showed a Qs of 318.1 C g−1 at 1 A g−1, higher than the individual constituents. Also, the composite exhibited 90% of the initial capacity over 2000 cycles at 15 A g−1. Furthermore, the ASC device, Co–Co@Ni–Fe PBA–PPy//AC, held the maximal Qs of 90 C g−1 at 1 A g−1 and provided a high energy density of 20 W h kg−1 at a power density of 808.9 W kg−1 with 79% initial capacity retention at 10 A g−1 over 2000 cycles. The Co–Co@Ni–Fe PBA–PPy composite showed the hybrid energy storage mechanism involving capacitive and diffusion-controlled current contributions. However, the significant capacitance came from the diffusion-controlled inner capacitance of the composite. We believe this straightforward approach can be used for most redox-active metal–organic frameworks suffering poor conductivity, limited cyclic stability, and low energy density for supercapacitor applications. Furthermore, this work assists in differentiating co-existing energy storage mechanisms based on the current contributions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PM thanks Ariel University for a PhD fellowship. VRS acknowledges Ariel University for a post-doctoral fellowship. This work was partially supported by the Israeli Atomic Energy Commission – Prof. A. Pazy Joint Foundation under Grant number ID126-2020. The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Notes and references
- V. Strauss, M. Muni, A. Borenstein, B. Badamdorj, T. Heil, M. D. Kowal and R. Kaner, Patching laser-reduced graphene oxide with carbon nanodots, Nanoscale, 2019, 11, 12712–12719 RSC.
- D. P. Chatterjee and A. K. Nandi, A review on the recent advances in hybrid supercapacitors, J. Mater. Chem. A, 2021, 9, 15880–15918 RSC.
- A. M. P. Sakita, P. F. R. Ortega, G. G. Silva, R. Della Noce and R. L. Lavall, Unveiling the performance metrics for supercapacitor electrodes with adsorbed redox additives, Electrochim. Acta, 2021, 390, 138803 CrossRef CAS.
- E. S. Goda, S. Lee, M. Sohail and K. R. Yoon, Prussian blue and its analogues as advanced supercapacitor electrodes, J. Energy Chem., 2020, 50, 206–229 CrossRef.
- S. Ryu, E. Kim and J. Yoo, Optimization of redox-active anthraquinone as electrode and electrolyte materials in supercapacitors, Electrochim. Acta, 2021, 370, 137809 CrossRef CAS.
- F. Yu, T. Huang, P. Zhang, Y. Tao, F.-Z. Cui, Q. Xie, S. Yao and F. Wang, Design and synthesis of electrode materials with both battery-type and capacitive charge storage, Energy Storage Mater., 2019, 22, 235–255 CrossRef.
- T. Schoetz, L. W. Gordon, S. Ivanov, A. Bund, D. Mandler and R. J. Messinger, Disentangling faradaic, pseudocapacitive, and capacitive charge storage: A tutorial for the characterization of batteries, supercapacitors, and hybrid systems, Electrochim. Acta, 2022, 412, 140072 CrossRef CAS.
- P. E. Lokhande, U. S. Chavan and A. Pandey, Materials and Fabrication Methods for Electrochemical Supercapacitors: Overview, Electrochem. Energy Rev., 2020, 3, 155–186 CrossRef CAS.
- Z. Yang, J. Tian, Z. Yin, C. Cui, W. Qian and F. Wei, Carbon nanotube- and graphene-based nanomaterials and applications in high-voltage supercapacitor: A review, Carbon N. Y., 2019, 141, 467–480 CrossRef CAS.
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, Carbon-based composite materials for supercapacitor electrodes: a review, J. Mater. Chem. A, 2017, 5, 12653–12672 RSC.
- C. Wang, M. Muni, V. Strauss, A. Borenstein, X. Chang, A. Huang, S. Qu, K. Sung, T. Gilham and R. B. Kaner, Graphene's Role in Emerging Trends of Capacitive Energy Storage, Small, 2021, 17, 2006875 CrossRef CAS PubMed.
- A. Borenstein, V. Strauss, M. D. Kowal and M. Anderson, Laser-Assisted Lattice Recovery of Graphene by Carbon Nanodot Incorporation, Small, 2019, 15(52), 190498 Search PubMed.
- A. Pramitha and Y. Raviprakash, Recent developments and viable approaches for high-performance supercapacitors using transition metal-based electrode materials, J. Energy Storage, 2022, 49, 104120 CrossRef.
- R. B. Choudhary, S. Ansari and M. Majumder, Recent advances on redox active composites of metal-organic framework and conducting polymers as pseudocapacitor electrode material, Renewable Sustainable Energy Rev., 2021, 145, 110854 CrossRef CAS.
- Y. Huang, H. Li, Z. Wang, M. Zhu, Z. Pei, Q. Xue, Y. Huang and C. Zhi, Nanostructured Polypyrrole as a flexible electrode material of supercapacitor, Nano Energy, 2016, 22, 422–438 CrossRef CAS.
- P. reddy Bommireddy, M. Kumar, Y. W. Lee, R. Manne, Y. Suh and S. H. Park, Prussian blue analogue Co3(Co(CN)6)2 cuboids as an electrode material for high-performance supercapacitor, J. Power Sources, 2021, 513, 230521 CrossRef.
- J. Nai and X. W. (David) Lou, Hollow Structures Based on Prussian Blue and Its Analogs for Electrochemical Energy Storage and Conversion, Adv. Mater., 2019, 31, 1706825 CrossRef PubMed.
- J. Chen, L. Wei, A. Mahmood, Z. Pei, Z. Zhou, X. Chen and Y. Chen, Prussian blue, its analogues and their derived materials for electrochemical energy storage and conversion, Energy Storage Mater., 2020, 25, 585–612 CrossRef.
- Y. Xu, S. Zheng, H. Tang, X. Guo, H. Xue and H. Pang, Prussian blue and its derivatives as electrode materials for electrochemical energy storage, Energy Storage Mater., 2017, 9, 11–30 CrossRef.
- Y. Lin, L. Zhang, Y. Xiong, T. Wei and Z. Fan, Toward the Design of High-performance Supercapacitors by Prussian Blue, its Analogues and their Derivatives, Energy Environ. Mater., 2020, 3, 323–345 CrossRef CAS.
- P. Mukherjee, K. Sathiyan, R. S. Vishwanath and T. Zidki, Anchoring MoS2 on an ethanol-etched Prussian blue analog for enhanced electrocatalytic efficiency for the oxygen evolution reaction, Mater. Chem. Front., 2022, 6, 1770–1778 RSC.
- P. Mukherjee, K. Sathiyan, A. V. Kottapurath, R. Bar-Ziv and T. Zidki, Hybrid Nanostructure of Mixed Transition Metal Oxysulfides Supported by Porous PBA as Efficient Electrocatalysts for the Oxygen Evolution Reaction, Isr. J. Chem., 2021, 62(3–4), e202100110 Search PubMed.
- Y. Zou, Q. Wang, C. Xiang, Z. She, H. Chu, S. Qiu, F. Xu, S. Liu, C. Tang and L. Sun, One-pot synthesis of ternary polypyrrole-Prussian-blue-graphene-oxide hybrid composite as electrode material for high-performance supercapacitors, Electrochim. Acta, 2016, 188, 126–134 CrossRef CAS.
- P. Bocchetta, D. Frattini, M. Tagliente and F. Selleri, Electrochemical Deposition of Polypyrrole Nanostructures for Energy Applications: A Review, Curr. Nanosci., 2020, 16, 462–477 CrossRef CAS.
- G. Shimoga, R. R. Palem, D.-S. Choi, E.-J. Shin, P.-S. Ganesh, G. D. Saratale, R. G. Saratale, S.-H. Lee and S.-Y. Kim, Polypyrrole-Based Metal Nanocomposite Electrode Materials for High-Performance Supercapacitors, Metals, 2021, 11, 905 CrossRef CAS.
- A. Afzal, F. A. Abuilaiwi, A. Habib, M. Awais, S. B. Waje and M. A. Atieh, Polypyrrole/carbon nanotube supercapacitorsTechnological advances and challenges, J. Power Sources, 2017, 352, 174–186 CrossRef CAS.
- G. Sabouraud, S. Sadki and N. Brodie, The mechanisms of pyrrole electropolymerization, Chem. Soc. Rev., 2000, 29, 283–293 RSC.
- C. Weidlich, K.-M. Mangold and K. Jüttner, EQCM study of the ion exchange behaviour of polypyrrole with different counterions in different electrolytes, Electrochim. Acta, 2005, 50, 1547–1552 CrossRef CAS.
- M. Bazzaoui, L. Martins, E. A. Bazzaoui and J. I. Martins, New single-step electrosynthesis process of homogeneous and strongly adherent polypyrrole films on iron electrodes in aqueous medium, Electrochim. Acta, 2002, 47, 2953–2962 CrossRef CAS.
- J. Li, X. Li, W. Wei, D. Wang and P. Liu, Hollow core–shell polypyrrole@poly(1,5-diaminoanthraquinone) composites with superior electrochemical performance for supercapacitors, Electrochim. Acta, 2021, 395, 139193 CrossRef CAS.
- T. Liu, L. Finn, M. Yu, H. Wang, T. Zhai, X. Lu, Y. Tong and Y. Li, Polyaniline and Polypyrrole Pseudocapacitor Electrodes with Excellent Cycling Stability, Nano Lett., 2014, 14, 2522–2527 CrossRef CAS PubMed.
- G.-F. Chen, Y.-Z. Su, P.-Y. Kuang, Z.-Q. Liu, D.-Y. Chen, X. Wu, N. Li and S.-Z. Qiao, Polypyrrole Shell@3D-Ni Metal Core Structured Electrodes for High-Performance Supercapacitors, Chem. – Eur. J., 2015, 21, 4614–4621 CrossRef CAS PubMed.
- T. Zalewska, A. Lisowska-Oleksiak, S. Biallozor and V. Jasulaitiene, Polypyrrole films polymerized on a nickel substrate, Electrochim. Acta, 2000, 45, 4031–4040 CrossRef CAS.
- L. Ji, J. Wang, X. Teng, T. J. Meyer and Z. Chen, CoP Nanoframes as Bifunctional Electrocatalysts for Efficient Overall Water Splitting, ACS Catal., 2020, 10, 412–419 CrossRef CAS.
- C. Li, W. Yan, S. Liang, P. Wang, J. Wang, L. Fu, Y. Zhu, Y. Chen, Y. Wu and W. Huang, Achieving a high-performance Prussian blue analogue cathode with an ultra-stable redox reaction for ammonium ion storage, Nanoscale Horiz., 2019, 4, 991–998 RSC.
- D. Cai, B. Liu, D. Wang, L. Wang, Y. Liu, B. Qu, X. Duan, Q. Li and T. Wang, Rational synthesis of metal–organic framework composites, hollow structures and their derived porous mixed metal oxide hollow structures, J. Mater. Chem. A, 2016, 4, 183–192 RSC.
- X. Wu, C. Wu, C. Wei, L. Hu, J. Qian, Y. Cao, X. Ai, J. Wang and H. Yang, Highly Crystallized Na2CoFe(CN)6 with Suppressed Lattice Defects as Superior Cathode Material for Sodium-Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 5393–5399 CrossRef CAS PubMed.
- S. P. Mardikar, S. D. Balgude and S. J. Uke, Supercapacitors for the Next Generation, IntechOpen, 2022.
- S. Cheng, Y. Zhang, Y. Liu, Z. Sun, P. Cui, J. Zhang, X. Hua, Q. Su, J. Fu and E. Xie, Energizing Fe2O3-based supercapacitors with tunable surface pseudocapacitance via physical spatial-confining strategy, Chem. Eng. J., 2021, 406, 126875 CrossRef CAS.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Spinels: Controlled Preparation, Oxygen Reduction/Evolution Reaction Application, and Beyond, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- L.-M. Luo, R.-H. Zhang, D. Chen, Q.-Y. Hu and X.-W. Zhou, Synthesis of 3D Thornbush-like Trimetallic CoAuPd Nanocatalysts and Electrochemical Dealloying for Methanol Oxidation and Oxygen Reduction Reaction, ACS Appl. Energy Mater., 2018, 1, 2619–2629 CrossRef CAS.
- T. Huang, G. Du, Y. Qi, J. Li, W. Zhong, Q. Yang, X. Zhang and M. Xu, A Prussian blue analogue as a long-life cathode for liquid-state and solid-state sodium-ion batteries, Inorg. Chem. Front., 2020, 7, 3938–3944 RSC.
- T. Zhu, Z. Li, Z. Chen, E. Hu, L. Wang, Y. Huang, S. Liu, Y. Liu, X. Gao and Z. Lin, Lithiophilic hollow Co3[Co(CN)6]2 embedded carbon nanotube film for dendrite-free lithium metal anodes, J. Colloid Interface Sci., 2022, 623, 532–540 CrossRef CAS PubMed.
- J. Peng, W. Zhang, Q. Liu, J. Wang, S. Chou, H. Liu and S. Dou, Prussian Blue Analogues for Sodium-Ion Batteries: Past, Present, and Future, Adv. Mater., 2022, 34, 2108384 CrossRef CAS PubMed.
- H.-M. Zhang and C. Zhu, Co nanoparticles-embedded N, S-codoped hierarchically porous graphene sheets as efficient bifunctional electrocatalysts for oxygen reduction reaction and hydrogen evolution reaction, J. Mater. Res. Technol., 2020, 9, 16270–16279 CrossRef CAS.
- J. Han, J. Zhang, T. Wang, Q. Xiong, W. Wang, L. Cao and B. Dong, Zn Doped FeCo Layered Double Hydroxide Nanoneedle Arrays with Partial Amorphous Phase for Efficient Oxygen Evolution Reaction, ACS Sustainable Chem. Eng., 2019, 7, 13105–13114 CrossRef CAS.
- R. Yan, X. Gao, W. He, R. Guo, R. Wu, Z. Zhao and H. Ma, A simple and convenient method to fabricate new types of phytic acid–metal conversion coatings with excellent anti-corrosion performance on the iron substrate, RSC Adv., 2017, 7, 41152–41162 RSC.
- B. Malik, S. Majumder, R. Lorenzi, I. Perelshtein, M. Ejgenberg, A. Paleari and G. D. Nessim, Promising Electrocatalytic Water and Methanol Oxidation Reaction Activity by Nickel Doped Hematite/Surface Oxidized Carbon Nanotubes Composite Structures, ChemPlusChem, 2022, 87, e2022000 CrossRef PubMed.
- L. Zhu, S. Shan, V. Petkov, W. Hu, A. Kroner, J. Zheng, C. Yu, N. Zhang, Y. Li, R. Luque, C.-J. Zhong, H. Ye, Z. Yang and B. H. Chen, Ruthenium–nickel–nickel hydroxide nanoparticles for room temperature catalytic hydrogenation, J. Mater. Chem. A, 2017, 5, 7869–7875 RSC.
- X. Shi, R. Hua, Y. Xu, T. Liu and G. Lu, Trimetallic conductive metal–organic frameworks as precatalysts for the oxygen evolution reaction with enhanced activity, Sustain. Energy Fuels, 2020, 4, 4589–4597 RSC.
- S. S K, S. Kalal, H. P. Perumal, D. Kumar, M. Gupta and J. Sinha, X-ray photoelectron spectroscopy investigation of Ta/CoFeB/TaOx heterostructures, Mater. Sci. Eng., B, 2021, 272, 115367 CrossRef CAS.
- L. Wang, Y. He, Y. Mu, M. Liu, Y. Chen, Y. Zhao, X. Lai, J. Bi and D. Gao, Sintering Temperature Induced Evolution of Microstructures and Enhanced Electrochemical Performances: Sol-Gel Derived LiFe(MoO4)2 Microcrystals as a Promising Anode Material for Lithium-Ion Batteries, Front. Chem., 2018, 6, 492 CrossRef CAS PubMed.
- X. Yan, T. Xu, G. Chen, S. Yang, H. Liu and Q. Xue, Preparation and characterization of electrochemically deposited carbon nitride films on silicon substrate, J. Phys. D: Appl. Phys., 2004, 37, 907–913 CrossRef CAS.
- A. Cymann-Sachajdak, M. Graczyk-Zajac, G. Trykowski and M. Wilamowska-Zawłocka, Understanding the capacitance of thin composite films based on conducting polymer and carbon nanostructures in aqueous electrolytes, Electrochim. Acta, 2021, 383, 138356 CrossRef CAS.
- Y. Zhang, J. Fu, P. Cui, S. Cheng, X. Cui, T. Qin, J. Zhou, Z. Zhang, Q. Su and E. Xie, Low-crystalline birnessite-MnO2 nanograins for high-performance supercapacitors, Electrochim. Acta, 2021, 389, 138761 CrossRef CAS.
- V. Kumar, R. Aepuru, A. Faizal and H. S. Panda, Elucidating the pseudocapacitive mechanism of ternary Co-Ni-B electrodes–Towards miniaturization and superior electrochemical performance for building outmatched supercapacitors, Electrochim. Acta, 2022, 409, 140003 CrossRef CAS.
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. Jiang, V. Presser and V. Augustyn, Pseudocapacitance: From Fundamental Understanding to High Power Energy Storage Materials, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS PubMed.
- B. K. Satpathy, S. Patnaik and D. Pradhan, Room-Temperature Growth of Co(OH)2 Nanosheets on Nanobelt-like Cu(OH)2 Arrays for a Binder-Free High-Performance All-Solid-State Supercapacitor, ACS Appl. Energy Mater., 2022, 5, 77–87 CrossRef CAS.
- M. Girirajan, N. B. Alagarsamy, K. Ramachandran, R. P. Manimuthu, D. Pazhanivel, K. K. Muthusamy and S. Sakkarapani, Two dimensional layered bismuthene nanosheets with ultra-fast charge transfer kinetics as a superior electrode material for high performance asymmetric supercapacitor, Electrochim. Acta, 2022, 426, 140838 CrossRef CAS.
- M. Girirajan, V. Arumugam, S. Subramaniyan, R. P. Manimuthu and S. Sakkarapani, Two-Dimensional Layered Bismuthene/Antimonene Nanocomposite as a Potential Electrode Material for the Fabrication of High-Energy Density Hybrid Supercapacitors, Energy Fuels, 2022, 36, 12299–12309 CrossRef CAS.
- L. Wang, Z. Wang, L. Xie, L. Zhu and X. Cao, An enabling strategy for ultra-fast lithium storage derived from micro-flower-structured NiX (X = O, S, Se), Electrochim. Acta, 2020, 343, 136138 CrossRef CAS.
- S. Ardizzone, G. Fregonara and S. Trasatti, “Inner” and “outer” active surface of RuO2 electrodes, Electrochim. Acta, 1990, 35, 263–267 CrossRef CAS.
- H. Vogt, Note on a method to interrelate inner and outer electrode areas, Electrochim. Acta, 1994, 39, 1981–1983 CrossRef CAS.
- C. Debiemme-Chouvy and T. T. M. Tran, An insight into the overoxidation of polypyrrole materials, Electrochem. Commun., 2008, 10, 947–950 CrossRef CAS.
- I. Rodríguez, B. R. Scharifker and J. Mostany, In situ FTIR study of redox and overoxidation processes in polypyrrole films, J. Electroanal. Chem., 2000, 491, 117–125 CrossRef.
- K. Cysewska, S. Virtanen and P. Jasiński, Study of the electrochemical stability of polypyrrole coating on iron in sodium salicylate aqueous solution, Synth. Met., 2016, 221, 1–7 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qm01162j |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2023 |