DOI:
10.1039/D3RA04721K
(Paper)
RSC Adv., 2023,
13, 26160-26168
Novel practical stereoselective synthesis of a bicyclic hydantoino-thiolactone as the key intermediate for production of (+)-biotin†‡
Received
14th July 2023
, Accepted 9th August 2023
First published on 4th September 2023
Abstract
Bicyclic hydantoinothiolactone (1), as the key intermediate for production of (+)-biotin, has been efficiently and high-stereoselectively synthesized from the cheap starting material L-cystine via nine steps in 44% overall yield. In this new practical synthesis, there are two characteristic steps worthy of note. One step is TMSOTf-catalyzed efficient cyanation of (3S,7aR)-6-benzyl-5-oxo-3-phenyltetrahydro-1H,3H-imidazo[1,5-c]thiazol-7-yl acetate, the other step is DBU-catalyzed rapid isomerization of trans-isomer to cis-isomer of the bicyclic hydantoinothiolactone.
Introduction
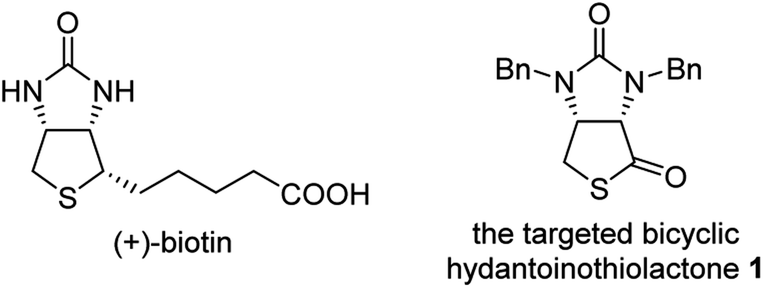
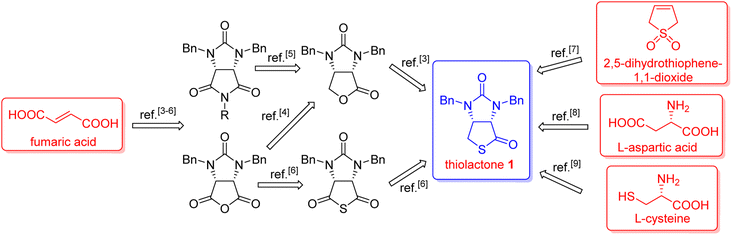
(+)-Biotin (see Fig. 1), an important member of the vitamin B family, has been widely used in the fields of medicines, health products, cosmetics and so on.1 It has persistently attracted widespread attention from the synthetic community because of unique physicochemical and pharmacological properties. It is noted that bicyclic hydantoinothiolactone 1 (Fig. 1) has emerged as the key common intermediate in many synthetic routes towards (+)-biotin.2 Due to the importance of this key intermediate 1 in production of (+)-biotin, considerable efforts have been devoted to developing an industrialized method for its preparation. Many syntheses of the key intermediate 1 have been reported to date, and they can be categorized into four classes according to the different starting materials (see Scheme 1): (a) syntheses of compound 1 starting from fumaric acid3–6 via meso-cyclic anhydride,4,6 meso-cyclic imide5 and meso-cyclic thioanhydride6 by desymmetrization and sulfurization; (b) syntheses of compound 1 starting from 2,5-dihydrothiophene-1,1-dioxide by desymmetrization and resolution;7 (c) diastereoselective syntheses of compound 1 starting from L-aspartic acid;8 (d) diastereoselective syntheses of compound 1 starting from L-cysteine.9 L-Cysteine might be the best starting material for synthesis of compound 1 in view of the following reasons: (i) L-cysteine is commercially cheap and readily available, (ii) the chirality of L-cysteine can be used to induce highly stereoselective construction of the neighbouring chiral centre, (iii) the sulfur atom of compound 1 could originate from L-cysteine with no need for sulfurization under drastic conditions. Although several syntheses9 of compound 1 starting from L-cysteine have been reported, there are some drawbacks such as moderate yields, severe reaction conditions, use of poisonous or toxic reagents, and complicated purification procedures. Herein, we would like to disclose an efficient, highly stereoselective and practical synthesis of the targeted bicyclic hydantoinothiolactone 1.
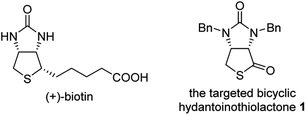 |
| | Fig. 1 The structure of related compounds. | |
 |
| | Scheme 1 The reported syntheses of bicyclic hydantoinothiolactone 1 from various starting material. | |
Results and discussion
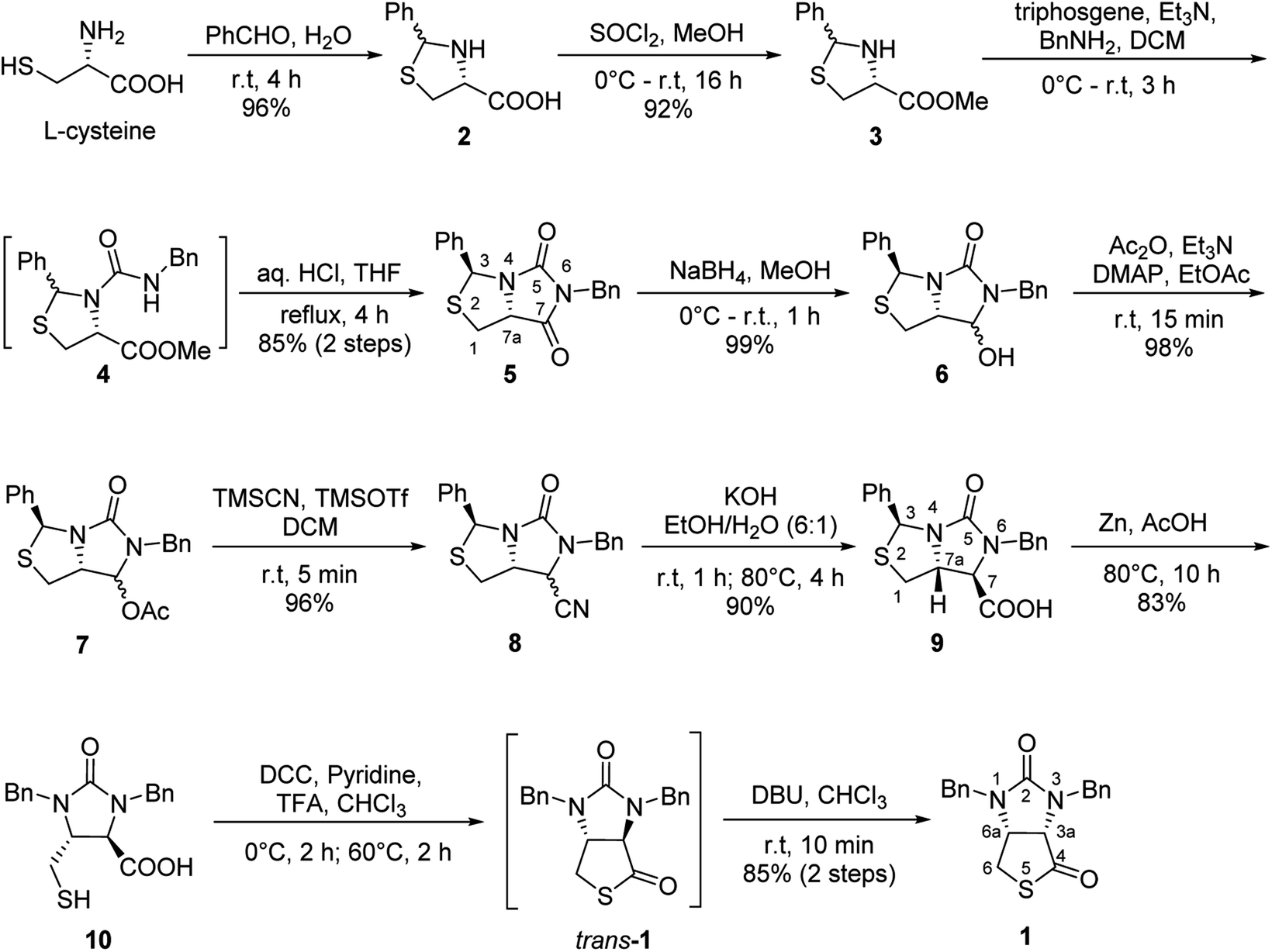
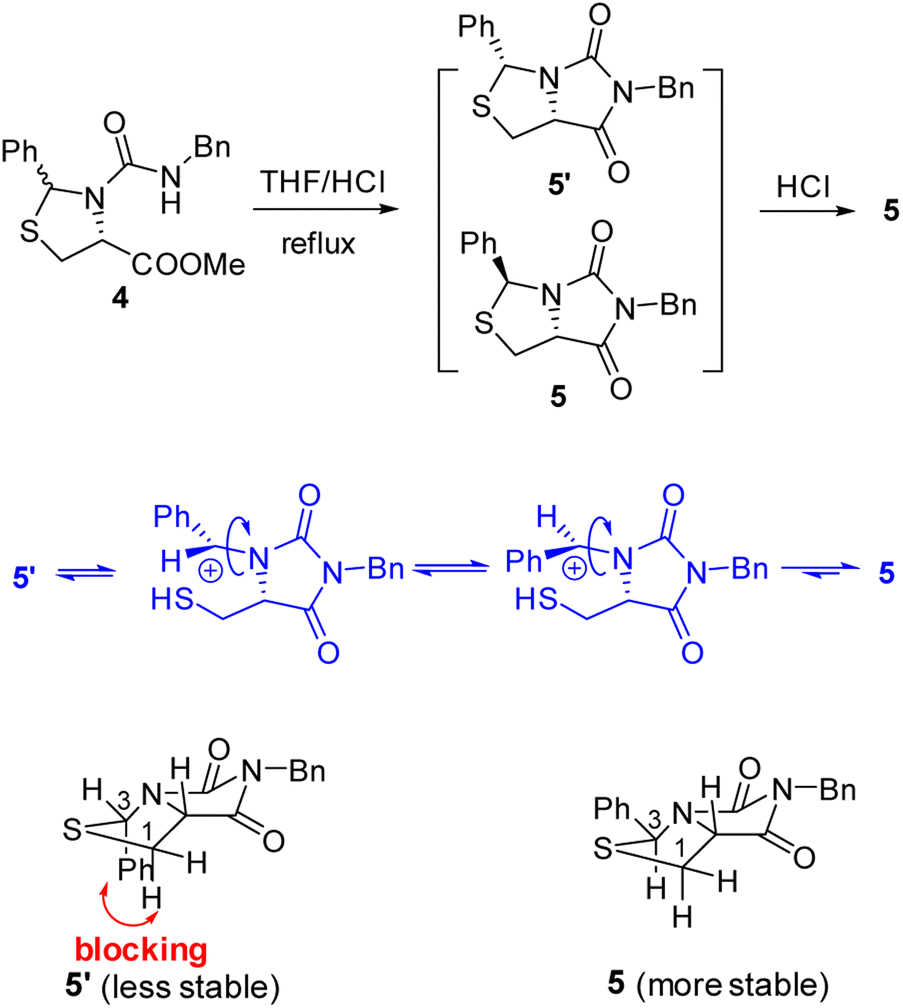
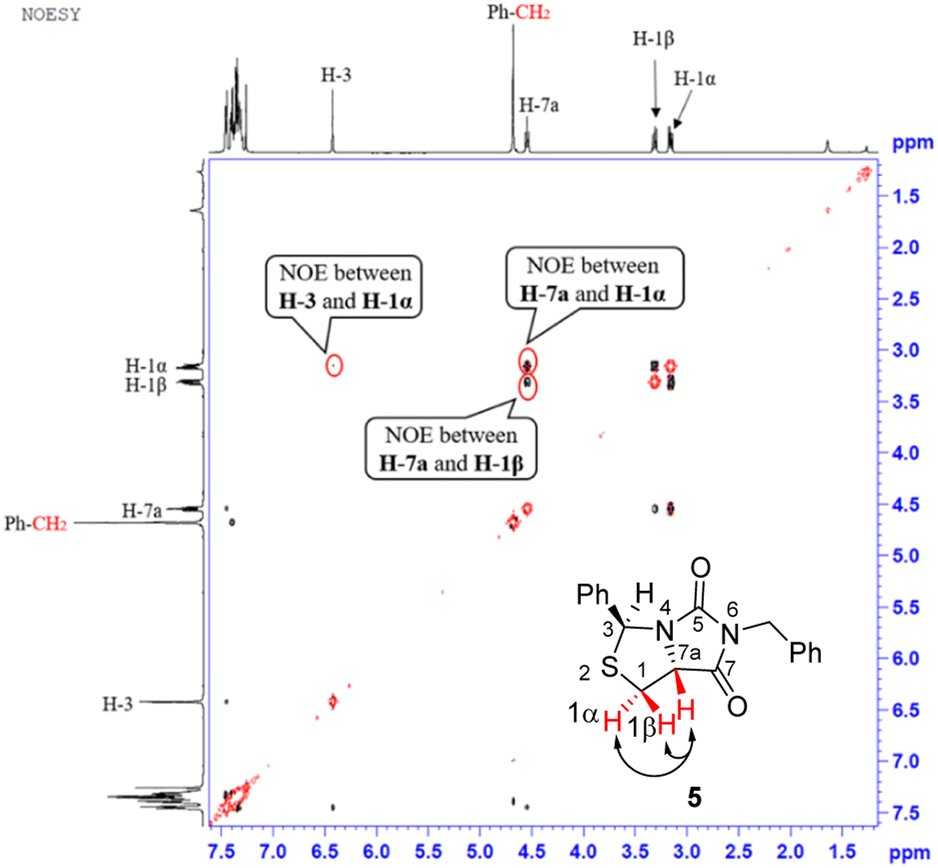
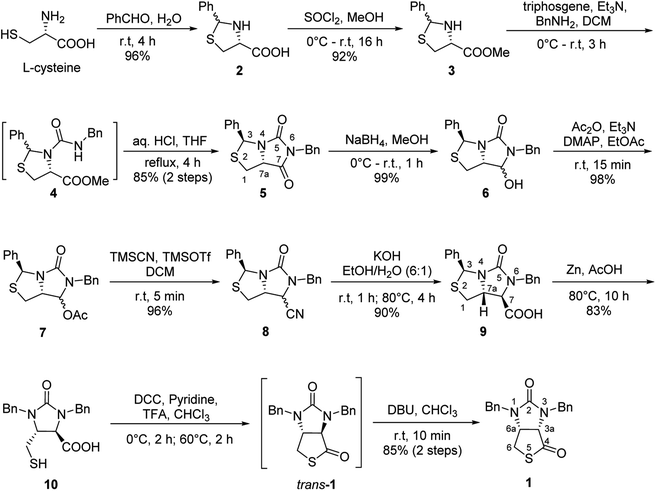
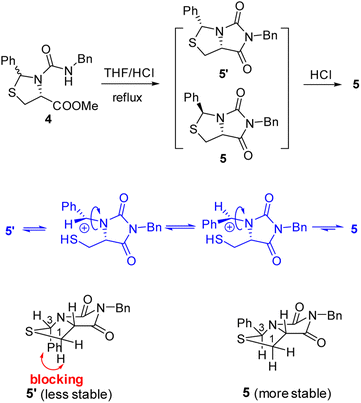
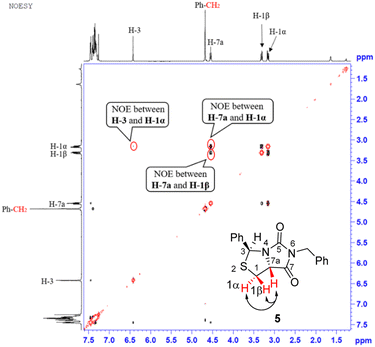
Our novel synthesis of bicyclic hydantoinothiolactone 1 is depicted in Scheme 2. As can be seen from the Scheme 2, the condensation of L-cysteine with benzaldehyde in water at room temperature (r.t.) first gave (R)-2-phenyl-thiazolidine-4-carboxylic acid 2 in 96% yield. Compound 2 was then exposed to thionyl chloride (SOCl2) in anhydrous methanol at 0 °C to r.t. to furnish methyl (R)-2-phenyl-thiazolidine-4-carboxylate 3 in 92% yield. After that, compound 3 was treated with triphosgene, triethylamine and benzylamine (BnNH2) in dichloromethane (DCM) at 0 °C to r.t. to produce a N,N-disubstituted urea (i.e. compound 4 in the parenthesis), which then underwent an acid-catalyzed cyclization under reflux for 4 h in a mixed solvent of aqueous HCl and tetrahydrofuran (THF) to afford (3S,7aR)-6-benzyl-3-phenyldihydro-3H,5H-imidazo[1,5-c]thiazole-5,7(6H)-dione 5 in 85% yield (over 2 steps from 3 to 5). Cyclization of compound 4 should produce an epimeric mixture of compounds 5 and 5′ (see Fig. 2), but herein it was observed that stereoisomer 5 was formed as the only product without contamination of its 3-epimer 5′ during the above HCl-catalyzed cyclization of compound 4. We deduced that HCl not only accelerated cyclization of compound 4, but also catalyzed epimerization between compounds 5 and 5′ via cleavage of the C–S bond, rotation of the C–N bond and regeneration of the C–S bond (see Fig. 2).10 Conformational analysis of compounds 5 and 5′ was also shown in Fig. 2, phenyl group (Ph) would occupy an equatorial position in the conformer of compound 5, whereas the phenyl group would occupy an axial position in the conformer of compound 5′. Conformer 5′ would be much less stable than conformer 5 due to blocking between axial Ph and H (enthalpy difference ΔH between conformers 5 and 5′ was estimated to be 125 kJ mol−1 by Cook's method11) stereochemistry of compound 5 has been further confirmed by 2D NMR technique. As can be seen from the 1H–1H NOESY spectrum (Fig. 3) of compound 5, the correlation spot between H-7a and vicinal H-1β is greater than the correlation spot between H-7a and vicinal H-1α, meaning that H-7a and H-1β have a cis relationship while H-7a and H-1α have a trans relationship; in addition, H-3 correlates with H-1α rather than H-1β, meaning that H-3 and H-1α have a cis relationship while H-3 and H-1β have a trans relationship.
 |
| | Scheme 2 The novel highly stereoselective synthesis of bicyclic hydantoinothiolactone 1 starting from L-cysteine. | |
 |
| | Fig. 2 The mechanism of configuration transformation and the conformational analysis of the compounds 5 and 5′. | |
 |
| | Fig. 3 1H–1H NOESY spectra of compound 5. | |
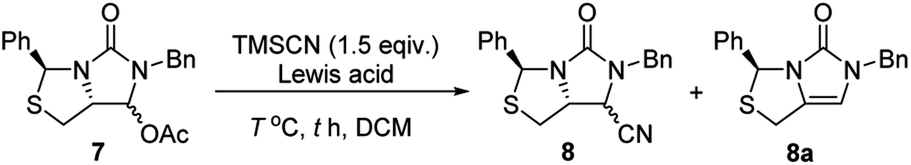
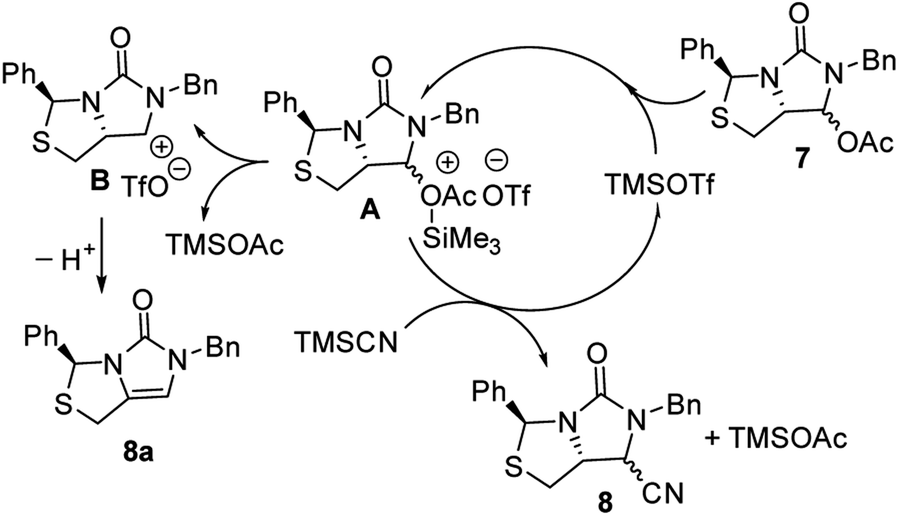
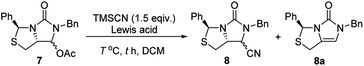
Subsequently, reduction of compound 5 with NaBH4 at 0 °C in MeOH produced (3S,7aR)-6-benzyl-7-hydroxy-3-phenyl-tetrahydro-3H,5H-imidazo[1,5-c]thiazol-5-one 6 in almost quantitative yield. Compound 6 was then treated with acetic anhydride (Ac2O), triethylamine and catalytic N,N-dimethyl-aminopyridine (DMAP) at room temperature in ethyl acetate to furnish (3S,7aR)-6-benzyl-5-oxo-3-phenyltetrahydro-1H,3H-imidazo[1,5-c]thiazol-7-yl acetate 7 in 98% yield. We then attempted Lewis acid-catalyzed cyanation of compound 7 with trimethylsilyl cyanide (TMSCN) under various conditions, the results were summarized in Table 1. As can be seen from the Table 1, cyanation of compound 7 with TMSCN did not take place in the absence of a Lewis acid (entry 1), while Lewis acids catalyzed the reaction to produce (3S,7aR)-6-benzyl-5-oxo-3-phenyltetrahydro-1H,3H-imidazo[1,5-c]thiazole-7-carbonitrile 8 and undesired (3S)-6-benzyl-3-phenyl-1,6-dihydro-3H,5H-imidazo[1,5-c]thiazol-5-one 8a (entries 2–12). Several Lewis acids such as aluminium chloride, zinc bromide, boron trifluoride etherate (BF3·Et2O), tributyltin chloride and trimethylsilyl trifluoromethanesulfonate (TMSOTf)12 have been tested as the catalyst for the reaction, and it was found that TMSOTf could dramatically catalyze the cyanation to furnish the compound 8 as the major product (entries 8–12), when 1 mol% (0.01 equiv.) of TMSOTf was used the catalyst, the desired product 8 was obtained in the best yield (Table 1, entry 12). A possible mechanism for the TMSOTf-catalyzed cyanation of compound 7 with TMSCN was proposed in Fig. 4, compound 7 would first react with TMSOTf to form an active intermediate A,13 which would then react with TMSCN to afford product 8, or might decompose to form by-product 8a via intermediate B. When TMSOTf was used as the catalyst, reaction of TMSCN with the active intermediate A to form desired product 8 would be much faster than the decomposition via intermediate B to form undesired by-product 8a. In contrast, when other lewis acids (see Table 1, entries 2–7) were used as the catalyst, reaction of TMSCN with the similar active intermediate A would be significantly slowed down, thus amount of the undesired by-product 8a would be significantly increased.
Table 1 Optimization of reaction conditions for the Lewis acid-catalyzed cyanation of compound 7
|
| Entry |
Lewis acid (equiv.) |
T (°C) |
t |
Yield% (8/8a) |
| 1 |
None |
25 |
10 h |
0/0 |
| 2 |
AlCl3 (0.5) |
25 |
10 h |
32/31 |
| 3 |
ZnBr2 (0.5) |
25 |
10 h |
35/32 |
| 4 |
Bu3SnCl (0.5) |
25 |
8 h |
46/35 |
| 5 |
BF3·Et2O (0.5) |
0 |
8 h |
45/50 |
| 6 |
BF3·Et2O (0.1) |
0 |
8 h |
60/38 |
| 7 |
BF3·Et2O (0.05) |
25 |
8 h |
65/30 |
| 8 |
TMSOTf (0.5) |
0 |
2 min |
88/9 |
| 9 |
TMSOTf (0.1) |
0 |
5 min |
90/8 |
| 10 |
TMSOTf (0.05) |
25 |
5 min |
92/6 |
| 11 |
TMSOTf (0.02) |
25 |
5 min |
94/3 |
| 12 |
TMSOTf (0.01) |
25 |
5 min |
96/1 |
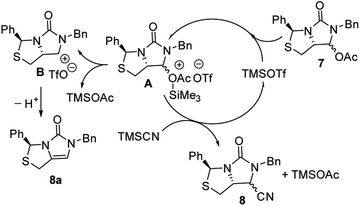 |
| | Fig. 4 Possible mechanism for TMSOTf-catalyzed cyanation of 7 with TMSCN. | |
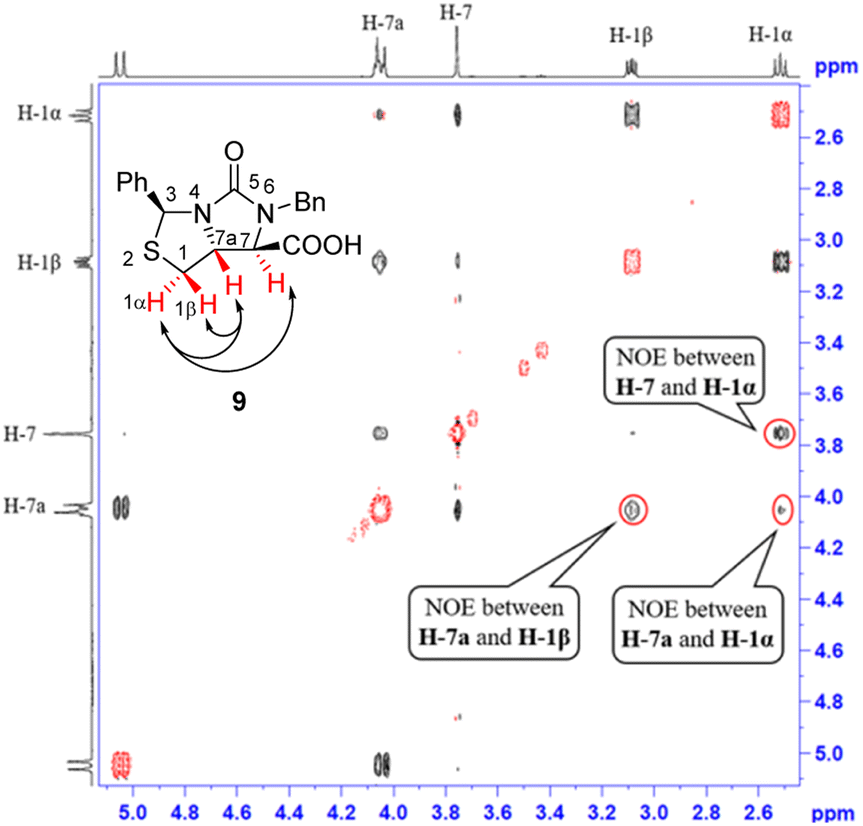
Next, carbonitrile 8 was treated with excess of KOH under refluxing (80 °C) in a mixed solvent of ethanol and water (EtOH/H2O = 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), hydrolysis of the cyano group at C-7 position occurred smoothly to afford a carboxylic acid 9 in 90% yield. The compound 8 was actually a mixture of two epimers of the chiral center at C-7 position, but herein both hydrolysis and reversible enolization happened to form the compound 9 as a single stereoisomer, in which COOH group has an upward orientation. The stereochemistry was also confirmed by 1H–1H NOESY spectrum. As can be seen from the 1H–1H NOESY spectrum of the compound 9 (see Fig. 5), the correlation spot between H-7a and vicinal H-1β is greater than the correlation spot between H-7a and vicinal H-1α, meaning that H-7a and H-1β have a cis relationship while H-7a and H-1α have a trans relationship; in addition, H-7 correlates with H-1α rather than H-1β, meaning that H-7 and H-1α have a cis relationship while H-7 and H-1β have a trans relationship, and thus COOH group should be cis with both of H-7a and H-1β. Afterwards, we performed reductive cleavage of C–S bond of the compound 9. When compound 9 was exposed to a large excess of zinc powder in acetic acid at 80 °C, cleavage of C–S bond occurred to afford (4R,5R)-1,3-dibenzyl-5-(mercaptomethyl)-2-oxoimidazo-lidine-4-carboxylic acid 10 in 83% yield.9e
:1), hydrolysis of the cyano group at C-7 position occurred smoothly to afford a carboxylic acid 9 in 90% yield. The compound 8 was actually a mixture of two epimers of the chiral center at C-7 position, but herein both hydrolysis and reversible enolization happened to form the compound 9 as a single stereoisomer, in which COOH group has an upward orientation. The stereochemistry was also confirmed by 1H–1H NOESY spectrum. As can be seen from the 1H–1H NOESY spectrum of the compound 9 (see Fig. 5), the correlation spot between H-7a and vicinal H-1β is greater than the correlation spot between H-7a and vicinal H-1α, meaning that H-7a and H-1β have a cis relationship while H-7a and H-1α have a trans relationship; in addition, H-7 correlates with H-1α rather than H-1β, meaning that H-7 and H-1α have a cis relationship while H-7 and H-1β have a trans relationship, and thus COOH group should be cis with both of H-7a and H-1β. Afterwards, we performed reductive cleavage of C–S bond of the compound 9. When compound 9 was exposed to a large excess of zinc powder in acetic acid at 80 °C, cleavage of C–S bond occurred to afford (4R,5R)-1,3-dibenzyl-5-(mercaptomethyl)-2-oxoimidazo-lidine-4-carboxylic acid 10 in 83% yield.9e
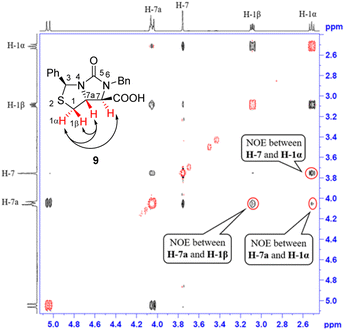 |
| | Fig. 5 1H–1H NOESY spectra of compound 9. | |
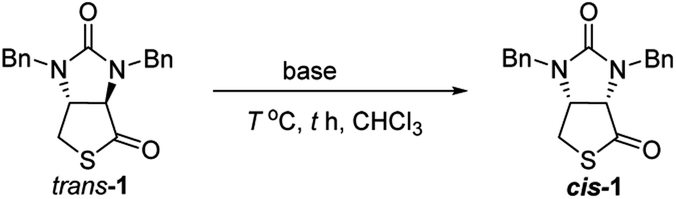
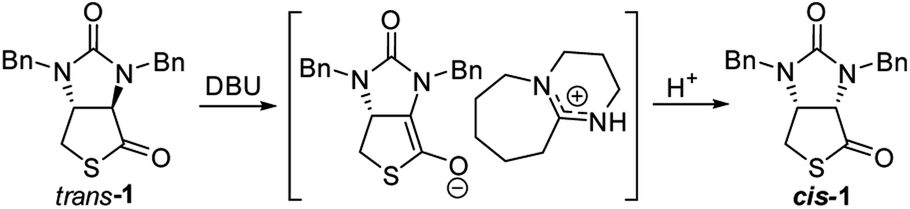
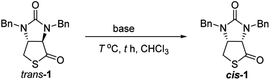
Finally, we tried to perform the cyclization of thiol carboxylic acid 10, when compound 10 was treated with dicyclohexylcarbodiimide (DCC), pyridine and trifluoracetic acid9b,e in chloroform, thiolactonization occurred to furnish intermediate compound (3aR,6aR)-1,3-dibenzyltetrahydro-1H-thieno[3,4-d]imidazole-2,4-dione trans-1, which was then treated in situ with 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) to afford thermodynamically much more stable (3aS,6aR)-1,3-dibenzyl-tetrahydro-1H-thieno[3,4-d]imidazole-2,4-di-one cis-1 in 85% yield. We have also tried amine-catalyzed isomerization of trans-1 to cis-1 under various conditions, the results are summarized in Table 2. As can be seen from the Table 2, isomerization from trans-1 to cis-1 did not take place at all in the absence of an amine (entry 1), while amines could catalyze the isomerization (entries 2–10). Several amines such as pyridine, triethylamine, 4-N,N-dimethyl-aminopyridine (DMAP) and DBU have been tested as the catalyst for isomerization, only DBU could rapidly and efficiently catalyze isomerization (Table 2, entries 8–10).
Table 2 Amine-catalyzed isomerization of trans-1 to cis-1 under various conditions
|
| Entry |
Base (equiv.) |
T (°C) |
t |
Yield% (trans/cis) |
| 1 |
None |
25 |
24 h |
100/0 |
| 2 |
Pyridine (5.0) |
25 |
72 h |
2/96 |
| 3 |
Pyridine (1.0) |
60 |
24 h |
1/96 |
| 4 |
DMAP (2.0) |
25 |
48 h |
2/96 |
| 5 |
DMAP (1.0) |
60 |
8 h |
1/95 |
| 6 |
Et3N (10.0) |
25 |
72 h |
5/90 |
| 7 |
Et3N (1.0) |
60 |
32 h |
5/91 |
| 8 |
DBU (0.5) |
25 |
5 min |
0/99 |
| 9 |
DBU (0.1) |
25 |
10 min |
0/99 |
| 10 |
DBU (0.05) |
25 |
10 min |
0/99 |
A possible mechanism for DBU-catalyzed isomerization from trans-1 to cis-1 was proposed in Fig. 6, isomerization would take place via ion pairs, an enolate anion and a delocalized DBU-derived cation.14
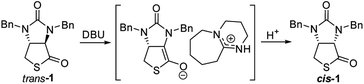 |
| | Fig. 6 Possible mechanism of DBU-catalyzed isomerization of trans-1 to cis-1. | |
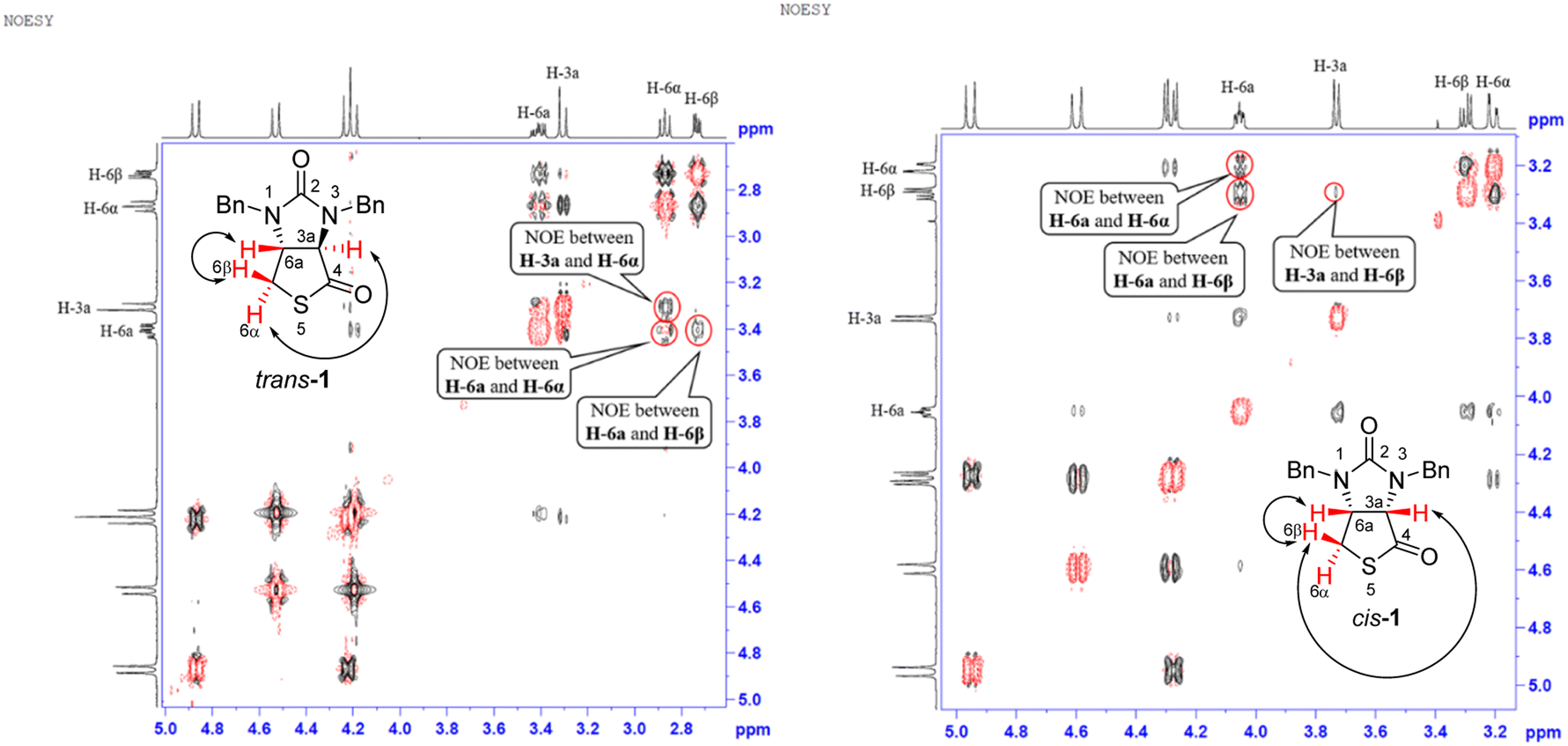
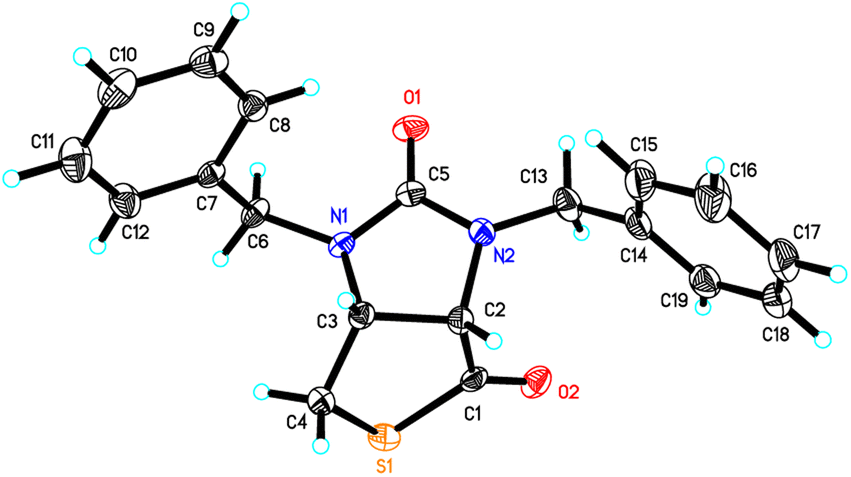
Stereochemistry of compounds trans-1 and cis-1 were confirmed by 2D NMR technique. As can be seen from the 1H–1H NOESY spectrum (see Fig. 7) of trans-1, the correlation spot between H-6a and vicinal H-6β is greater than the correlation spot between H-6a and vicinal H-6α, meaning that H-6a and H-6β have a cis relationship while H-6a and H-6α have a trans relationship; H-3a obviously correlates with H-6α rather than H-6β, meaning that protons H-3a and H-6α have a cis relationship. As can be seen from the 1H–1H NOESY spectrum (see also Fig. 7) of cis-1, the correlation spot between H-6a and vicinal H-6β is greater than the correlation spot between H-6a and vicinal H-6α, meaning that H-6a and H-6β have a cis relationship while H-6a and H-6α have a trans relationship; H-3a obviously correlates with H-6β rather than H-6α, meaning that protons H-3a and H-6β have a cis relationship. In addition, the stereochemistry of cis-1 was further unequivocally confirmed by X-ray diffraction analysis of its single crystal as shown in Fig. 8.
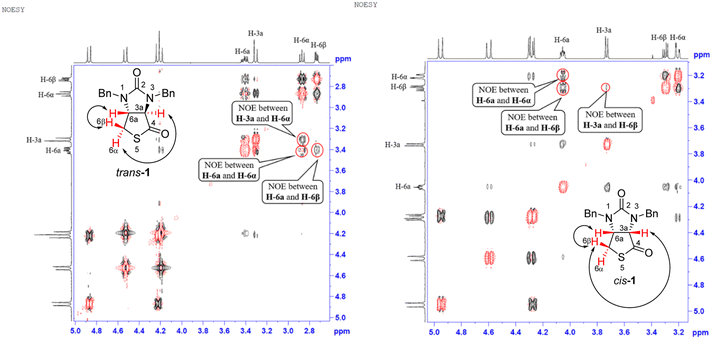 |
| | Fig. 7 1H–1H NOESY spectra of compounds trans-1 and cis-1. | |
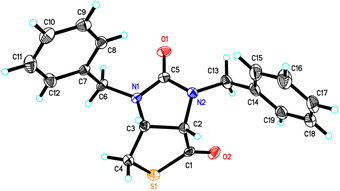 |
| | Fig. 8 ORTEP drawing of the compound cis-1. | |
Conclusions
A novel efficient and highly stereoselective synthesis of bicyclic hydantoinothiolactone cis-1 has been developed. It was synthesized starting from L-cysteine via nine steps in 44% overall yield. Two characteristic steps of this total synthesis have been extensively studied, and TMSOTf was found as the best catalyst for cyanation of compound 7 with trimethylsilyl cyanide, and DBU was found as the most appropriate catalyst for rapid isomerization of trans-1 to cis-1. Stereochemical structures of compounds 5, 9, trans-1 and cis-1 have been confirmed by 2D NMR technique; and the stereochemistry of cis-1 was further unequivocally confirmed by X-ray crystallographic analysis. The present synthesis has some advantages such as use of cheap starting material, good yields, mild reaction conditions, freeness of poisonous or toxic reagents, and easy purification procedures.
In addition, the stereochemical configuration of C-3 in compound 5 was lost during the process of reductive cleavage from compound 9 to 10 in present synthesis, meaning stereoseletive construction of the chiral centra at C-3 of compound 5 seemed unnecessary. However, 1,3-dihidro-imidazo[1,5-c]thiazole-5,7-dione derivatives like compound 5 are interesting in medicinal chemistry,15 so the above protocol for highly stereoselective preparation of compound 5 and its derivatives might be important.
Experimental
General method
NMR spectra were acquired on a Bruker AM-400 instrument. Chemical shifts are given on the δ scale as parts per million (ppm) with tetramethylsilane (TMS) as the internal standard. Mass spectra were performed with a HP1100 LC-MS spectrometer. Optical rotations of chiral compounds were measured on a PerkinElmer polarimeter at room temperature. Melting points were determined on a Mel-TEMP II apparatus. Column chromatography was performed on silica gel (Qingdao Ocean Chemical Corp.).
(R)-2-Phenylthiazolidine-4-carboxylic acid (2)
A round-bottom flask (500 mL) was charged with L-cysteine (20.00 g, 165.0 mmol) and H2O (200 mL). The mixture was stirred at room temperature for 20 min, and then, PhCHO (17.52 g, 165.1 mmol) was dropwise added. The reaction mixture was vigorously stirred for 4 h, during which white amorphous solids precipitated out from reaction solution. The reaction mixture was then filtered by suction, white amorphous crystals were collected on a buchner funnel, and washed successively with cold water (50 mL) and a mixed solvent of hexane and ethyl acetate (30 mL, hexane/EtOAc = 1:8). White solid compound 2 (33.15 g, 158.4 mmol) was thus obtained as an epimeric mixture in 96% yield after drying overnight in an infrared oven. 1H NMR (400 MHz, DMSO-d6) δ 3.09 (dd, J = 8.9, 10.0 Hz, 0.4H, H-5), 3.15 (dd, J = 4.5, 10.3 Hz, 0.6H, H-5), 3.30 (dd, J = 7.1, 10.2 Hz, 0.6H, H-5), 3.38 (dd, J = 7.2, 10.1 Hz, 0.4H, H-5), 3.91 (dd, J = 7.2, 8.8 Hz, 0.4H, H-4), 4.24 (dd, J = 4.5, 7.0 Hz, 0.6H, H-4), 5.67 (s, 0.6H, H-2), 5.51 (s, 0.4H, H-2), 7.25–7.57 (m, 5H, Ph–H). 13C NMR (101 MHz, DMSO) δ 173.47, 172.72, 141.69, 139.39, 128.96, 128.78, 128.71, 128.08, 127.76, 127.42, 72.24, 71.56, 65.91, 65.36, 38.92, 38.46.
Methyl (R)-2-phenylthiazolidine-4-carboxylate (3)
Thionyl chloride (18.19 g, 152.9 mmol) was dropwise added into anhydrous MeOH (120 mL) at 0 °C. After the addition was finished, the resulting solution was continuously stirred for 10 min in an ice bath. Compound 2 (16.00 g, 76.46 mmol) was added into the solution in portions. After the addition was finished, the ice bath was removed, and the reaction mixture was further stirred at room temperature for 16 h. Removal of the solvent by vacuum distillation gave a yellow oil. Dichloromethane (100 mL) and water (100 mL) were added, and the biphasic mixture was vigorously stirred, then NaHCO3 was slowly added in portions until the pH was adjusted to 7–8. Two phases were separated. The aqueous layer was reextracted twice with dichloromethane (2 × 30 mL). The organic extracts were combined, washed with H2O (20 mL) and dried over anhydrous MgSO4. Evaporation of solvent gave a residue, which was purified by flash chromatography (eluent: EtOAc/hexane = 1:4) to give colorless oily compound 3 (15.71 g, 70.36 mmol) as an epimeric mixture in 92% yield. 1H NMR (400 MHz, CDCl3) δ 3.14 (dd, J = 9.0, 10.2 Hz, 0.6H, H-5), 3.23 (dd, J = 5.8, 10.6 Hz, 0.4H, H-5), 3.42 (dd, J = 7.1, 10.6 Hz, 0.4H, H-5), 3.49 (dd, J = 7.1, 10.3 Hz, 0.6H, H-5), 3.82 (s, 1.2H, OCH3), 3.83 (s, 1.8H, OCH3), 4.02 (dd, J = 7.1, 8.9 Hz, 0.6H, H-4), 4.24 (dd, J = 7.5, 8.6 Hz, 0.4H, H-4), 5.59 (s, 0.6H, H-2), 5.84 (s, 0.4H, H-2), 7.28–7.58 (m, 5H, Ph–H). 13C NMR (101 MHz, CDCl3) δ 171.20, 170.58, 140.12, 137.13, 127.71, 127.67, 127.42, 126.90, 126.42, 125.89, 71.59, 69.78, 64.54, 63.29, 51.59, 51.55, 38.22, 37.13.
(3S,7aR)-6-Benzyl-3-phenyldihydro-3H,5H-imidazo[1,5-c]thiazole-5,7(6H)-dione (5)
Compound 3 (12.00 g, 53.73 mmol) and Et3N (11.70 g, 115.6 mmol) were dissolved in dichloromethane (60 mL), and the solution was cooled to 0 °C by an ice-bath. A freshly prepared solution of triphosgene (6.380 g, 21.50 mmol) in dichloro-methane (40 mL) was then dropwise added over 10 min at 0 °C. After addition was finished, the reaction mixture was further stirred for 0.5 h until compound 3 was completely consumed as determined by thin-layer chromatography (TLC) analysis (eluent: EtOAc/hexane = 1:3). BnNH2 (6.330 g, 59.07 mmol) was added, and then the ice-bath was removed. The reaction mixture was further stirred at room temperature for 2.5 h. Removal of the dichloromethane by vacuum distillation gave an oily residue, which was dissolved in THF (120 mL). Concentrated HCl aqueous solution (12 mL) was added, and the reaction mixture was heated to reflux (70 °C), and was further stirred under refluxing for 4 h. After the reaction was complete (checked by TLC; eluent, EtOAc/hexane = 1:3), THF was removed by vacuum distillation. EtOAc (100 mL) and H2O (100 mL) were added, and the biphasic mixture was vigorously stirred for 5 min. Two layers were separated, and the aqueous layer was twice extracted with EtOAc (2 × 50 mL). The organic extracts were combined and washed with brine (50 mL), and then dried over anhydrous MgSO4. Evaporation of the solvent under reduced pressure gave crude product which was purified by flash chromatography (eluent: EtOAc/hexane = 1:20–1:3) to furnish pure compound 5 (14.82 g, 45.68 mmol) as white crystals in 85% yield. M.p. 77–79 °C. [α]20D = −251 (c 1.0, CHCl3) {lit.16 [α]20D = −250 (c 1.1, CHCl3)}. 1H NMR (500 MHz, CDCl3) δ 7.48–7.27 (m, 10H, Ph–H), 6.42 (s, 1H, H-3), 4.68 (s, 2H, Ph–CH2–N), 4.55 (t, J = 7.4 Hz, 1H, H-7a), 3.31 (dd, J = 11.6, 7.8 Hz, 1H, H-1), 3.16 (dd, J = 11.5, 6.9 Hz, 1H, H-1). 13C NMR (101 MHz, CDCl3) δ 170.06, 157.64, 137.90, 134.33, 127.76 (2C), 127.65 (2C), 127.54 (2C), 127.38, 127.12, 125.37 (2C), 64.93, 64.22, 41.93, 32.36. HRMS (ESI) m/z calcd for C18H17N2O2S [M + H]+: 325.1011, found: 325.1009.
(3S,7aR)-6-Benzyl-7-hydroxy-3-phenyltetrahydro-3H,5H-imidazo[1,5-c]thiazol-5-one (6)
Compound 5 (11.00 g, 33.91 mmol) was dissolved in methanol (35 mL), and the solution was cooled to 0 °C by an ice-bath. Sodium borohydride (1.926 g, 50.91 mmol) was gradually added in portions. After the addition was finished, the reaction mixture was further stirred at 0 °C for 30 min. The ice-bath was removed, and stirring was continued at room temperature for additional 30 min. Once the starting material was completely consumed as monitored by TLC (eluent: EtOAc/hexane = 1:3), the reaction was quenched by addition of H2O (5 mL). Methanol was removed by evaporation under reduced pressure. EtOAc (80 mL) and H2O (40 mL) were added, and the mixture was vigorously stirred for 10 min. Two layers were separated, and the aqueous layer was reextracted twice with EtOAc (2 × 40 mL), The organic extracts were combined, washed with brine (40 mL) and dried over anhydrous MgSO4. Evaporation of EtOAc under vacuum gave white solid product, which was washed with a mixed solvent EtOAc and hexane (EtOAc/hexane = 1:8) to give pure compound 6 (10.95 g, 33.55 mmol) as white crystals in 99% yield. M.p. 134–135 °C. 1H NMR (400 MHz, acetone-d6) δ 7.50–7.19 (m, 10H, Ph–H), 6.38 (s, 1H, H-3), 5.46–5.36 (m, 1H, H-7), 4.69 (d, J = 15.4 Hz, 1H, Ph–CH2–N), 4.37 (dt, J = 8.6, 6.7 Hz, 1H, H-7a), 4.21 (d, J = 15.4 Hz, 1H, another Ph–CH2–N), 3.36 (dd, J = 11.1, 8.5 Hz, 1H, H-1), 3.03 (dd, J = 11.1, 6.5 Hz, 1H, H-1). 13C NMR (101 MHz, acetone-d6) δ 158.94, 142.51, 137.55, 128.46 (2C), 128.33 (2C), 127.99 (2C), 127.48, 127.23, 126.23 (2C), 77.36, 77.26, 65.82, 64.96, 43.48, 31.92. HRMS (ESI) m/z calcd for [C18H18N2O2NaS]: 349.0987, found: 349.0985.
(3S,7aR)-6-Benzyl-5-oxo-3-phenyltetrahydro-1H,3H-imidazo[1,5-c]thiazol-7-yl acetate (7)
To a solution of compound 6 (10.50 g, 32.17 mmol), Et3N (4.897 g, 48.39 mmol) and DMAP (10.0 mg, 0.082 mmol) in dried EtOAc (60 mL), Ac2O (3.952 g, 38.71 mmol) was dropwise added at room temperature, the reaction was complete in 15 min as monitored by TLC (eluent: EtOAc/hexane = 1:3). The reaction was then quenched by addition of H2O (30 mL). After the biphasic mixture was vigorously stirred for 10 min, two layers were separated, and the aqueous layer was reextracted twice with EtOAc (2 × 30 mL). Organic extracts were combined, washed successively with saturated aqueous solution of NaHCO3 (25 mL) and brine (30 mL), and then dried over anhydrous MgSO4. Evaporation of EtOAc under vacuum gave white solid product, which was washed with a mixed solvent of ethyl ether and hexane (Et2O/hexane = 1:2) to give pure compound 7 (11.62 g, 31.54 mmol) in 98% yield. M.p. 115–116 °C. 1H NMR (400 MHz, acetone-d6) δ 7.52–7.16 (m, 10H, Ph–H), 6.34 (s, 1H, H-3), 6.29 (d, J = 7.0 Hz, 1H, H-7), 4.65 (dd, J = 7.6, 7.2 Hz, 1H, H-7a), 4.59 (d, J = 15.4 Hz, 1H, Ph–CH2–N), 4.30 (d, J = 15.4 Hz, 1H, another Ph–CH2–N), 3.21 (dd, J = 11.4, 7.7 Hz, 1H, H-1), 3.04 (dd, J = 11.4, 6.5 Hz, 1H, H-1), 1.97 (s, 3H, CH3 in OAc). 13C NMR (101 MHz, acetone-d6) δ 169.89, 158.48, 141.84, 137.26, 128.50 (2C), 128.39 (2C), 128.05 (2C), 127.66, 127.43, 126.22 (2C), 79.56, 65.43, 63.17, 45.10, 31.86, 19.67. HRMS (ESI) m/z calcd for [C20H20N2O3NaS]: 391.1092, found: 391.1091.
(3S,7aR)-6-Benzyl-5-oxo-3-phenyltetrahydro-1H,3H-imidazo[1,5-c]thiazole-7-carbonitrile (8)
Compound 7 (11.50 g, 31.21 mmol) and TMSCN (4.640 g, 46.77 mmol) were dissolved in dichloromethane (50 mL). TMSOTf (70.0 mg, 0.31 mmol) was added by a micro injector at room temperature. The reaction was complete in 5 min as monitored by TLC (eluent: Et2O/hexane = 2:3). Dichloromethane was removed by evaporation under vacuum. EtOAc (60 mL), H2O (40 mL) and a saturated aqueous solution of NaHCO3 (15 mL) were added. The biphasic mixture was vigorously stirred for 10 min, two layers were separated, and the aqueous layer was twice extracted with EtOAc (2 × 30 mL). The organic extracts were combined, washed with brine (30 mL), and then dried over anhydrous MgSO4. Evaporation of EtOAc under vacuum gave a residue, which was purified by flash column chromatography on silica gel (eluent: EtOAc/hexane = 1:7) to furnish compound 8 (10.05 g, 29.96 mmol) as a colorless oil in 96% yield. Two diastereomers of compound 8 could be separated by a very careful chromatography. Minor diastereomer: 1H NMR (400 MHz, CDCl3) δ 7.50–7.04 (m, 10H, Ph–H), 6.32 (s, 1H, H-3), 5.02 (d, J = 15.1 Hz, 1H, Ph–CH2–N), 4.29 (d, J = 7.6 Hz, 1H, H-7), 4.12 (ddd, J = 9.5, 7.6, 5.9 Hz, 1H, H-7a), 3.99 (d, J = 15.1 Hz, 1H, another Ph–CH2–N), 3.21 (dd, J = 10.9, 5.9 Hz, 1H, H-1), 3.10 (dd, J = 10.9, 9.5 Hz, 1H, H-1). 13C NMR (101 MHz, CDCl3) δ 158.55, 140.42, 134.24, 129.27 (2C), 128.76 (2C), 128.69 (2C), 128.64, 128.30, 126.47 (2C), 65.85, 59.56, 46.73, 46.70, 35.39. Major diastereomer: 1H NMR (400 MHz, CDCl3) δ 7.58–6.99 (m, 10H, Ph–H), 6.37 (s, 1H, H-3), 5.02 (d, J = 14.9 Hz, 1H, Ph–CH2–N), 4.15 (dd, J = 9.3, 6.2 Hz, 1H, H-7a), 4.04–3.97 (m, 2H, H-7 and anther Ph–CH2–N), 3.10 (dd, J = 10.6, 6.2 Hz, 1H, H-1), 2.53 (dd, J = 10.5, 9.4 Hz, 1H, H-1). 13C NMR (101 MHz, CDCl3) δ 158.87, 140.32, 134.38, 129.29 (2C), 128.74 (2C), 128.66, 128.46 (2C), 128.21, 126.17 (2C), 115.51, 65.32, 62.34, 47.44, 46.47, 36.17. HRMS (ESI) m/z calcd for [C19H17N3ONaS]: 358.0990, found: 358.0992.
(3S,7R,7aR)-6-Benzyl-5-oxo-3-phenyltetrahydro-1H,3H-imidazo[1,5-c]thiazole-7-carboxylic acid (9)
A round-bottom flask (100 mL) was charged with compound 8 (10.00 g, 29.81 mmol), EtOH (60 mL) and H2O (10 mL). KOH (8.360 g, 149.0 mmol) was added, and the mixture was stirred at room temperature for 1 h. The reaction mixture was then heated to reflux (80 °C), and was further stirred under refluxing for 4 h. After the reaction was complete as determined by TLC (eluent: DCM/MeOH = 10:1), EtOH was removed by vacuum distillation. EtOAc (40 mL) and H2O (40 mL) were added. The biphasic mixture was vigorously stirred for 5 min, and then the organic layer was extracted twice with H2O (2 × 20 mL). The aqueous extracts were combined, and then acidified with 4N aqueous solution of HCl until the pH value was adjusted to 1–2. EtOAc (100 mL) was added, and the biphasic mixture was vigorously stirred for 10 min. Two layers were separated, and the aqueous layer was twice extracted with EtOAc (2 × 50 mL). The organic extracts were combined, washed with brine (30 mL), and then dried over anhydrous MgSO4. Solvent was removed under vacuum to give light yellow solid product, which was washed with a mixed solution of ethyl acetate and hexane (EtOAc/hexane = 1:6) to furnish pure compound 9 (9.509 g, 26.83 mmol) in 90% yield. M.p. 182–183 °C. [α]20D = −210.38 (c 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.04 (s, 1H, COOH), 7.42–6.96 (m, 10H, Ph–H), 6.39 (s, 1H, H-3), 5.05 (d, J = 14.9 Hz, 1H, Ph–CH2–N), 4.10–4.00 (m, 2H, H-7a and another Ph–CH2–N), 3.76 (s, 1H, H-7), 3.08 (dd, J = 10.5, 6.1 Hz, 1H, H-1), 2.51 (t, J = 9.9 Hz, 1H, H-1). 13C NMR (101 MHz, CDCl3) δ 172.64, 160.58, 140.99, 135.33, 129.04 (2C), 128.59 (2C), 128.43 (2C), 128.21, 127.93, 126.17 (2C), 65.32, 62.67, 58.27, 46.47, 37.20. HRMS (ESI) m/z calcd for [C19H17N2O3S]: 353.0960, found: 353.0959.
(4R,5R)-1,3-Dibenzyl-5-(mercaptomethyl)-2-oxoimidazolidine-4-carboxylic acid (10)
To a solution of compound 9 (9.000 g, 25.39 mmol) in acetic acid (30 mL) was added zinc powder (8.300 g, 126.9 mmol). The reaction mixture was heated to 80 °C, and then stirring was continued at this temperature for 10 h under a nitrogen atmosphere. After the reaction was complete (checked by TLC, eluent: DCM/MeOH = 1:20), the mixture was cooled to room temperature and filtered to remove insoluble solid, and the filter cake was washed three times with EtOH (3 × 30 mL). The filtrates were combined and concentrated under vacuum to give a viscous residue. EtOAc (60 mL) and 2N aqueous solution of HCl (40 mL) were added, the biphasic mixture was vigorously stirred for 10 min. Two layers were separated, the aqueous layer was twice extracted with EtOAc (2 × 30 mL). Organic extracts were combined, washed with brine (30 mL), and then dried over anhydrous MgSO4. The solution was concentrated under a reduced pressure to give solid residue which was washed with ethyl ether to furnish pure compound 10 (7.514 g, 21.08 mmol) as white crystals in 83% yield. M.p. 160–161 °C {lit.9c mp. 158–160 °C}. [α]20D = +49.5 (c 1.0, DMF) {lit.9b [α]20D = +48.8 (c 0.62, DMF)}. 1H NMR (400 MHz, DMSO-d6) δ 13.58–12.79 (brs., 1H, COOH), 7.43–7.15 (m, 10H, Ph–H), 4.83 (d, J = 15.4 Hz, 1H, Ph–CH2–N), 4.65 (d, J = 15.6 Hz, 1H, Ph–CH2–N), 4.13 (d, J = 15.6 Hz, 1H, Ph–CH2–N), 4.06 (d, J = 15.4 Hz, 1H, Ph–CH2–N), 3.82 (d, J = 5.2 Hz, 1H, H-4), 3.60 (td, J = 5.4, 3.1 Hz, 1H, H-5), 2.84–2.61 (m, 2H, CH2S), 2.11 (t, J = 8.3 Hz, 1H, SH).·13C NMR (101 MHz, DMSO-d6) δ 172.27, 159.36, 137.88, 137.22, 129.01 (2C), 128.93 (2C), 128.36 (2C), 128.11 (2C), 127.77 (2C), 57.97, 57.39, 46.32, 44.89, 26.23. HRMS (ESI) m/z calcd for [C19H21N2O3S]: 357.1273, found: 357.1275.
(3aR,6aR)-1,3-Dibenzyltetrahydro-1H-thieno[3,4-d]imidazole-2,4-dione (trans-1)
To a solution of compound 10 (5.100 g, 14.31 mmol) in CHCl3 (15 mL) was added pyridine (1.582 g, 20.00 mmol) and TFA (680.0 mg, 5.964 mmol), and the reaction mixture was stirred at 0 °C for 10 min. A freshly prepared solution of DCC (4.450 g, 21.56 mmol) in CHCl3 (10 mL) was added, and then the reaction mixture was further stirred at 0 °C for 2 h. After that, the reaction mixture was warmed to 20 °C, and then stirred for 2 h. After the reaction was complete (checked by TLC, eluent: EtOAc/hexane = 1:3), the reaction mixture was filtered to remove insoluble substance, which was rinsed twice with CHCl3 (2 × 20 mL). The filtrates were combined, and then evaporated under vacuum. The residue was diluted with EtOAc (100 mL) and H2O (50 mL), 2N aqueous solution of HCl was added to adjust pH to 1–2. After the biphasic mixture was vigorously stirred for 10 min, two layers were separated, organic layer was successively washed with saturated aqueous NaHCO3 solution (20 mL) and brine (10 mL). After being dried over anhydrous MgSO4, the solution was concentrated under reduced pressure to give a residue, which was purified by flash column chromatography on silica gel (eluent: EtOAc/hexane = 1:10) to afford compound trans-1 (3.440 g, 10.16 mmol) as white crystals in 71% yield. M.p. 116–117 °C. [α]20D = + 103.2 (c 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.31–7.19 (m, 10H, Ph–H), 4.87 (d, J = 14.4 Hz, 1H, Ph–CH2–N), 4.53 (d, J = 14.5 Hz, 1H, Ph–CH2–N), 4.23 (d, J = 11.4 Hz, 1H, Ph–CH2–N), 4.19 (d, J = 11.5 Hz, 1H, Ph–CH2–N), 3.41 (ddd, J = 14.0, 10.5, 4.6 Hz, 1H, H-6a), 3.30 (d, J = 14.0 Hz, 1H, H-3a), 2.87 (dd, J = 10.5, 9.5 Hz, 1H, H-6), 2.73 (dd, J = 9.5, 4.5 Hz, 1H, H-6). 13C NMR (101 MHz, CDCl3) δ 194.18, 165.25, 136.03, 135.28, 129.35 (2C), 128.90 (2C), 128.70 (2C), 128.21 (2C), 128.10 (2C), 65.19, 59.57, 48.99, 46.97, 34.66.
(3aS,6aR)-1,3-Dibenzyltetrahydro-1H-thieno[3,4-d]imidazole-2,4-dione (cis-1)
To a mixture of compound 10 (2.400 g, 6.733 mmol) in CHCl3 (10 mL) was added pyridine (746.0 mg, 9.431 mmol) and TFA (307.0 mg, 2.692 mmol), and the reaction mixture was stirred at 0 °C for 10 min. A freshly prepared solution of DCC (2.070 g, 10.03 mmol) in CHCl3 (6 mL) was added, and reaction mixture was stirred at 0 °C for 2 h. After that, the reaction mixture was warmed to 60 °C, and then stirred for 2 h. The mixture was cooled down to room temperature, DBU (52.0 mg, 0.341 mmol) was added, and the reaction was further stirred for 10 min. After the reaction was complete (checked by TLC, EtOAc/hexane = 1:3), the reaction mixture was filtered to remove insoluble substance, which was rinsed twice with CHCl3 (2 × 15 mL). The filtrates were combined, and then evaporated under vacuum. The residue was diluted with EtOAc (50 mL) and H2O (20 mL), 2N aqueous solution of HCl was added to adjust pH to 1–2. After the biphasic mixture was vigorously stirred for 10 min, two layers were separated, the aqueous layer was extracted with EtOAc (2 × 20 mL). Organic extracts were combined, and washed successively with saturated aqueous NaHCO3 solution (20 mL) and brine (10 mL). After being dried over anhydrous MgSO4, the solution was concentrated under reduced pressure to give a solid residue, which was washed with aqueous methanol (MeOH/H2O = 4:1) to afford pure compound 1 (1.937 g, 5.724 mmol) as white crystals in 85% yield. M.p. 122–123 °C {lit.9b m.p. 122–123 °C }. [α]20D = + 90.2 (c 1.0, CHCl3) {lit.9b [α]25D = + 90.5 (c 1.0, CHCl3)}. 1H NMR (400 MHz, CDCl3) δ 7.37–7.07 (m, 10H, Ph–H), 4.95 (d, J = 14.8 Hz, 1H, Ph–CH2–N), 4.60 (d, J = 15.4 Hz, 1H, Ph–CH2–N), 4.30 (d, J = 4.5 Hz, 1H, Ph–CH2–N), 4.27 (d, J = 3.9 Hz, 1H, Ph–CH2–N), 4.06 (ddd, J = 7.7, 5.5, 2.1 Hz, 1H, H-6a), 3.73 (d, J = 7.8 Hz, 1H, H-3a), 3.30 (dd, J = 12.4, 5.5 Hz, 1H, H-6), 3.21 (dd, J = 12.5, 2.1 Hz, 1H, H-6). 13C NMR (101 MHz, CDCl3) δ 203.61, 158.35, 136.42, 136.23, 128.93 (2C), 128.83 (2C), 128.71 (2C), 128.04 (2C), 127.97, 127.80, 62.16, 55.89, 46.53, 45.28, 33.05. HRMS (ESI) m/z calcd for [C19H19N2O2S]: 339.1167, found: 339.1168.
Synthesis of cis-1 from trans-1
Compound trans-1 (1.250 g, 3.694 mmol) was dissolved in CHCl3 (5 mL), and DBU (29.2 mg, 0.192 mmol) was added. The reaction solution was then stirred at room temperature for 10 min. The reaction was complete (checked by TLC, EtOAc/hexane = 1:3), and then chloroform was removed by evaporation under vacuum. The residue was diluted with EtOAc (40 mL) and H2O (6 mL), 2N aqueous solution of HCl (2 mL) was added. After the biphasic mixture was vigorously stirred for 5 min, two layers were separated, the aqueous layer was extracted again with EtOAc (15 mL). Organic extracts were combined, and washed successively with saturated aqueous NaHCO3 solution (10 mL) and brine (10 mL). After being dried over anhydrous MgSO4, the solution was concentrated under reduced pressure to give a solid residue, which was washed with a small amount of the mixed solvent of ethyl ether and hexane (Et2O/hexane = 1:2) to afford pure compound cis-1 (1.237 g, 3.655 mmol) as white crystals in 99% yield. The characterization data of the present sample was identical with the data of above-obtained sample from compound 10.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 20972048, 20172015) for the financial support of this work.
Notes and references
-
(a) D. Pacheco-Alvarez, R. S. Solorzano-Vargas and A. L. Del Rio, Arch. Med. Res., 2002, 33, 439 CrossRef CAS PubMed;
(b) J.-D. Zhang, A.-C. Liu, J. Chen, G.-H. Yuan and H.-F. Jin, Prog. Chem., 2020, 32, 594 CAS.
-
(a) P. J. De Clercq, Chem. Rev., 1997, 97, 1755 CrossRef CAS PubMed;
(b) Z. Zhong, X.-F. Wu and F.-E. Chen, Chin. J. Org. Chem., 2012, 32, 1792 CrossRef CAS.
-
(a) M. Gerecke, J. P. Zimmermann and W. Aschwanden, Helv. Chim. Acta, 1970, 53, 991 CrossRef CAS;
(b) Y. Aoki, H. Suzuki, H. Akiyama and S. Okano, US Pat, 3876656, Apr. 8, 1975 Search PubMed;
(c) F.-E. Chen, X.-X. Chen, H.-F. Dai, Y.-Y. Kuang, B. Xie and J.-F. Zhao, Adv. Synth. Catal., 2005, 347, 549 CrossRef CAS.
-
(a) K. Matsuki, H. Inoue and M. Takeda, Tetrahedron Lett., 1993, 34, 1167 CrossRef CAS;
(b) J. Huang, F. Xiong and F.-E. Chen, Tetrahedron: Asymmetry, 2008, 19, 1436 CrossRef CAS;
(c) F. Xiong, X.-X. Chen and F.-E. Chen, Tetrahedron: Asymmetry, 2010, 21, 665 CrossRef CAS;
(d) H.-F. Dai, W.-X. Chen, L. Zhao, F. Xiong, H. Sheng and F.-E. Chen, Adv. Synth. Catal., 2008, 350, 1635 CrossRef CAS;
(e) F. Xiong, J. Li, G. Li, B. Song, Y.-X. Zhou, F.-E. Chen and D. Li, Heterocycles, 2016, 92, 544 CrossRef CAS;
(f) X.-X. Chen, F. Xiong, H. Fu, Z.-Q. Liu and F.-E. Chen, Chem. Pharm. Bull., 2011, 59, 488 CrossRef CAS PubMed;
(g) F. Xiong, F.-J. Xiong, W.-X. Chen, H.-Q. Jia and F.-E. Chen, J. Heterocycl. Chem., 2013, 50, 1078 CAS;
(h) F.-E. Chen, X.-H. Ling, Y.-X. Lu, X.-Y. Zhang and X.-H. Peng, Chem. J. Chin. Univ., 2001, 22, 1141 CAS;
(i) C. Wehrli, WO Pat. 2004094367, Apr. 20, 2004;
(j) M. Schwarz, and J. Eckstein, WO Pat. 01/25215, Oct. 02, 2000;
(k) C. Choi, S.-K. Tian and L. Deng, Synthesis, 2001, 1737 CrossRef CAS;
(l) S.-X. Wang and F.-E. Chen, Adv. Synth. Catal., 2009, 351, 547 CrossRef CAS.
-
(a) F.-E. Chen, H.-F. Dai, Y.-Y. Kuang and H.-Q. Jia, Tetrahedron: Asymmetry, 2003, 14, 3667 CrossRef CAS;
(b) M. Seki and Y. Takahashi, Org. Process Res. Dev., 2021, 25, 1950 CrossRef CAS;
(c) F.-E. Chen, H.-Q. Jia, X.-X. Chen, H.-F. Dai, B. Xie, Y.-Y. Huang and J.-F. Zhao, Chem. Pharm. Bull., 2005, 53, 743 CrossRef CAS;
(d) F.-E. Chen, J.-L. Yuan, H.-F. Dai, Y.-Y. Kuang and Y. Chu, Synthesis, 2003, 2155 CrossRef CAS;
(e) M. Shimizu, Y. Nishigaki and A. Wakabayashi, Tetrahedron Lett., 1999, 40, 8873 CrossRef CAS;
(f) F.-E. Chen, Z.-Z. Peng, L.-Y. Shao and Y. Cheng, Acta Pharm. Sin., 1999, 34, 822 CAS.
-
(a) F.-E. Chen, Y.-D. Huang, H. Fu, Y. Cheng, D.-M. Zhang, Y.-Y. Li and Z.-Z. Peng, Synthesis, 2000, 2004 CrossRef CAS;
(b) F. Xiong, X.-X. Chen, Z.-Q. Liu and F.-E. Chen, Tetrahedron Lett., 2010, 51, 3670 CrossRef CAS.
-
(a) H. A. Bates and S. B. Rosenblum, J. Org. Chem., 1986, 51, 3447 CrossRef CAS;
(b) H. A. Bates, L. Smilowitz and J. Lin, J. Org. Chem., 1985, 50, 899 CrossRef CAS;
(c) H. A. Bates, L. Smilowitz and S. B. Rosenblum, J. Chem. Soc., Chem. Commun., 1985, 353 RSC;
(d) S. Tokuyama, T. Yamano, I. Aoki, K. Takanohashi and K. Nakahama, Chem. Lett., 1993, 741 CrossRef CAS.
- M. Seki, T. Shimizu and K. Inubushi, Synthesis, 2002, 361 CrossRef CAS.
-
(a) M. Seki, M. Kimura, M. Hatsuda, S. Yoshida and T. Shimizu, Tetrahedron Lett., 2003, 44, 8905 CrossRef CAS;
(b) M. Seki, M. Hatsuda, Y. Mori, S. Yoshida, S. Yamada and T. Shimizu, Chem. - Eur. J., 2004, 10, 6102 CrossRef CAS;
(c) Y. Mori, M. Kimura and M. Seki, Synthesis, 2003, 2311 CAS;
(d) M. Seki, M. Hatsuda and S. Yoshida, Tetrahedron Lett., 2004, 45, 6579 CrossRef CAS;
(e) E. Poetsch and M. Casutt, Chimia, 1987, 41, 148 CAS.
-
(a) J. J. Pesek and J. H. Frost, Tetrahedron, 1975, 31, 907 CrossRef CAS;
(b) C. Jin, J. P. Burgess, M. B. Gopinathan and G. A. Brine, Tetrahedron Lett., 2006, 47, 943 CrossRef CAS;
(c) L. Szilagyi and Z. Gyorgydeak, J. Am. Chem. Soc., 1979, 101, 427 CrossRef CAS;
(d) R. G. Kallen, J. Am. Chem. Soc., 1971, 93, 6227 CrossRef CAS;
(e) R. Parthasarathy, B. Paul and W. Korytnyk, J. Am. Chem. Soc., 1976, 98, 6634 CrossRef CAS.
- M. L. Van Linn and J. M. Cook, J. Org. Chem., 2010, 75, 3587 CrossRef CAS PubMed.
-
(a) A. D. Dilman and S. L. Ioffe, Chem. Rev., 2003, 103, 733 CrossRef CAS;
(b) P.-Z. Wang, Y. Gao, J. Chen, X.-D. Huan, W.-J. Xiao and J.-R. Chen, Nat. Commun., 2021, 12, 1815 CrossRef CAS;
(c) J. Chen, P.-Z. Wang, B. Lu, D. Liang, X.-Y. Yu, W.-J. Xiao and J.-R. Chen, Org. Lett., 2019, 21, 9763 CrossRef CAS.
-
(a) K. Toshima, G. Matsuo, T. Ishizuka, Y. Ushiki, M. Nakata and S. Matsumura, J. Org. Chem., 1998, 63, 2307 CrossRef CAS;
(b) R. Liu, Q. Hua, Q. Lou, J. Wang, X. Li, Z. Ma and Y. Yang, J. Org. Chem., 2021, 86, 4763 CrossRef CAS;
(c) K. Crossey, R. N. Cunningham, P. Redpath and M. E. Migaud, RSC Adv., 2015, 5, 58116 RSC;
(d) C.-K. Chan, Y.-H. Chung and C.-C. Wang, RSC Adv., 2022, 12, 8263 RSC.
-
(a) J. Dong, T.-Z. Meng, X.-X. Shi, W.-H. Zou and X. Lu, Tetrahedron: Asymmetry, 2013, 24, 883 CrossRef CAS;
(b) S. Xiao, X.-X. Shi, F. Ni, J. Xing, J.-J. Yan and S.-L. Liu, Eur. J. Org Chem., 2010, 1711 CrossRef CAS;
(c) L. Wang, D. Zhang, J. Li, G. Xu and J. Sun, RSC Adv., 2014, 4, 44193 RSC;
(d) X. Qin, J. Zhang, Z.-Y. Wang, Y. Song, Y. Yang, W. Zhang and H. Liu, RSC Adv., 2023, 13, 4782 RSC.
-
(a) P. Campiglia, M. Scrima, M. Grimaldi, G. Cioffi, A. Bertamino, M. Sala, C. Aquino, I. Gomez-Monterrey, P. Grieco, E. Novellino and A. M. D'Urisi, Chem. Biol. Drug Des., 2009, 74, 224 CrossRef CAS PubMed;
(b) B. Refouvelet, S. Harraga, L. Nicod, J.-F. Robert, E. Seilles, J. Couquelet and P. Tronche, Chem. Pharm. Bull., 1994, 42, 1076 CrossRef CAS.
- S. P. Chavan, A. G. Chittiboyina, G. Ramakrishna, R. B. Tejwani, T. Ravindranathan, S.-K. Kamat, B. Rai, L. Sivadasan, K. Balakrishnan, S. Ramalingam and V. H. Deshpande, Tetrahedron, 2005, 61, 9273 CrossRef CAS.
Footnotes |
| † Dedicated to esteemed Professor Li-Xin Dai on the occasion of his 100th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04721k |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a