
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid, iterative syntheses of unsymmetrical di- and triarylboranes from crystalline aryldifluoroboranes†
Douglas
Turnbull
 and
Marc-André
Légaré
*
and
Marc-André
Légaré
*
Department of Chemistry, McGill University, Otto Maass Chemistry Building, 801 Rue Sherbrooke O, Montreal, Quebec, Canada H3A 0B8. E-mail: ma.legare@mcgill.ca
First published on 17th November 2023
Abstract
A one-pot procedure to synthesise aryldifluoroboranes, ArBF2, from bench-stable arylsilanes is presented. These ArBF2 react conveniently with aryllithium reagents to form unsymmetrical ArAr′BF and BArAr′Ar′′ in high yield. Examples of all three classes of borane have been characterised crystallographically, allowing for elucidation of geometric and crystal packing trends in crystalline ArBF2.
Introduction
Triarylboranes, bolstered by over a century of continuous research, find importance across chemical disciplines such as in metal-free catalysis,1,2 anion sensing,3 optoelectronic materials,4 and cell imaging.5 Their uniquely advantageous properties are governed, in part, by the electron deficiency of the central boron atom, whose unoccupied p orbital can both mediate reactivity and be part of an extended conjugated π-system as an electron acceptor.Equally important is the nature of the aryl groups, which must be tailored to fine-tune the desired steric and electronic properties of triarylboranes, such as their Lewis acidity, solubility in aqueous and/or organic media, and kinetic stability to hydrolysis. As was recently highlighted by Marder and co-workers in an extensive synthetic review,6 numerous methods have been reported to synthesise triarylboranes by substitution reactions of aryl nucleophiles with commercial BX3 (X = H, OR, F, Cl, Br) sources. However, such substitutions overwhelmingly produce symmetrically substituted BAr3 or BAr2Ar′ (Ar = aryl) due to the presence of several complex equilibria,7 the importance of each being governed by the natures of the BX3 source, nucleophile, and extrinsic conditions. This inherently limits reaction control, and consequently the potential diversity, and thus applications, of the resultant boranes.
Further diversification to fully unsymmetrical BArAr′Ar′′, meanwhile, has hitherto proven to be a significant contemporary synthetic challenge, thereby limiting modern advances in our understanding and application of boron-containing compounds. A small number of BArAr′Ar′′ have been prepared using M/B (M = Li, MgBr) exchange with ArBX2 (X = H,8 Br,9 OR10), or Pd-catalysed cross-coupling to symmetric boranes,11–14 usually requiring multi-step protocols. These studies have demonstrated the possible applications of BArAr′Ar′′ in fluorescence imaging and small-molecule sensing for biological systems, though their synthetic routes were target-specific, rather than general. Jäkle and co-workers first approached a more iterative BArAr′Ar′′ synthesis via Me3Sn/B exchange with ArBBr2, followed by transmetallation with aryl-magnesium, -copper, or -tin reagents (Scheme 1, top).15–19 However, the toxicity of Me3Sn and ArCu reagents is a drawback to such an approach. Only recently, Marder and co-workers reported the stepwise transmetallation of K[ArBF3] complexes with 2,6-disubstituted aryllithium or Grignard reagents to generate trixylylboranes (Scheme 1, middle).20 In both this and the Sn-mediated cases, reactions can take days with poor-to-moderate overall yields.
 | ||
| Scheme 1 Iterative approaches to triarylborane synthesis. | ||
In light of the promising properties of triarylboranes highlighted above, iterative syntheses of BArAr′Ar′′ through stepwise arylation of a reactive boron centre represents an attractive avenue for development of new boron-containing molecules and materials. An ideal synthetic approach for the preparation of diverse triarylboranes would be capable of selectively substituting accessible boron sources rapidly and in high yields, using convenient aryl nucleophiles. This would improve both the simplicity and scope of modern borane synthesis, and notably enable the synthesis of BArAr′Ar′′ in which each aryl substituent possesses uniquely tailored, application-driven steric and electronic properties.
Herein, we demonstrate that crystalline ArBF2 are easily accessed from air-stable silanes in a one-pot protocol, which has allowed for their multigram-scale isolation and their use as privileged reagents in organometallic boron chemistry. Specifically, they react conveniently with aryllithium reagents in rapid and selective transmetallations to form unsymmetrical ArAr′BF and BArAr′Ar′′ in one pot, in high yield and within minutes, from which LiF is the sole by-product (Scheme 1, bottom). These boranes have been comprehensively characterised in the solid state and in solution, including the first crystal structures of unsymmetrical ArAr′BF.
Results and discussion
Syntheses of aryldifluoroboranes (3)
Our synthetic route was developed from a previous reaction of PhBCl2 with main-group fluoroanions, which gave PhBF2 in >95% yield after vacuum distillation.21 We have instead proceeded via reaction of bench-stable ArSiMe2R (1, R = Me except for 1b, where R = H) with BBr3 in CH2Cl2 to form ArBBr2 (2) in situ followed by solvent removal and direct addition of Na[BF4] (Scheme 2). The desired ArBF2, 3, are formed in moderate-to-high isolated yields and high purity but can be further purified by vacuum sublimation and/or recrystallisation. These conditions tolerate arenes with steric hindrance (3a–b), polycyclic aromatic systems (3c–e), multiple boryl substituents (3f–g), and heteroatoms (3h).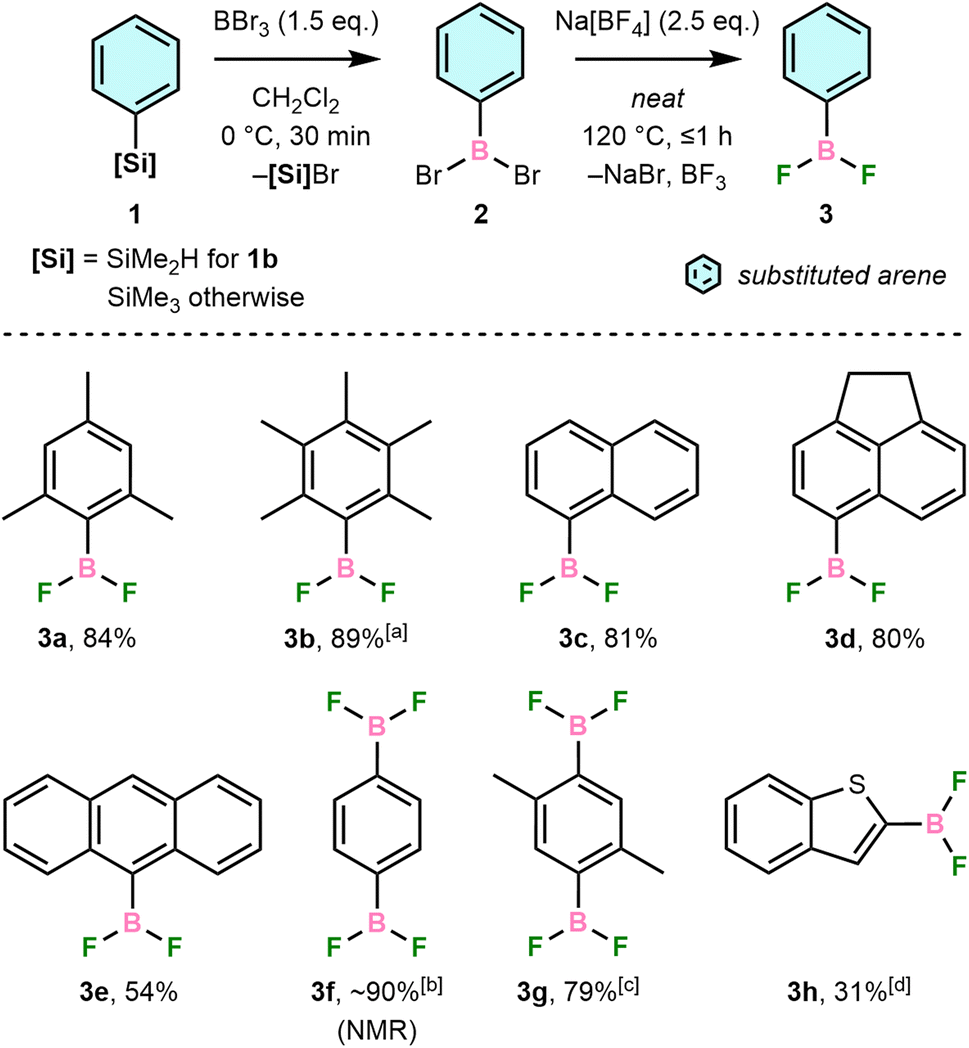 | ||
| Scheme 2 Isolated yields (unless noted otherwise) from one-pot syntheses of aryldifluoroboranes (3) via stepwise Si/B and Br/F exchange of arylsilanes (1). [a] Reaction of 1b with BBr3 (3.0 eq.) was run for 2 h. [b] Prepared directly from 2f. [c] Reaction of 1g with BBr3 was run for 3 h at 70 °C in 1,2-dichloroethane. [d] Prepared using 8.0 equiv. Na[BF4]. | ||
Isolated 3 are typically colourless, crystalline solids that sublime readily upon heating or in vacuo, including 3a, which was previously reported to be a colourless liquid.22 In all cases, they are highly soluble in common organic solvents and easily handled using standard glove box techniques. Exposure to moisture, however, results in fuming and formation of [ArBO]3, ArH, and BF3 as hydrolysis products, determined by NMR spectroscopy.
Boron tribromide was preferred for the Si/B exchange step due to greater reactivity towards ArSiMe3 than BCl3 and demonstrated reactivity with sterically demanding silanes, e.g., 1a.23 Reactions were performed with dimethylsilane 1b due to the steric hindrance of the C6Me5 group preventing preparation of the corresponding trimethylsilane. This increased bulk slowed the Si/B exchange step, with incomplete conversion revealing intermediate Si–H bromination. Electron-deficient arenes were previously found not to undergo the necessary Si/B exchange,24 and we have found that ArNMe2 derivatives are also incompatible due to the formation of inert BBr3 adducts (Fig. S1†).
No [ArBF3]− was observed even when using excess Na[BF4], indicating the weaker Lewis acidity of 3 than BF3, which is supported by F− exchange energy calculations (Table S1†) and a previous NMR study of related ArBF2.25 Our route thus complements a recent synthesis of [ArBF3]− reported by Perrin and co-workers using an ostensibly similar metathesis of ArB(OH)2 with [BF4]− in alcoholic solvents.26
Spectroscopic and crystallographic studies of 3
Consistent with previously reported NMR spectra,27 the 11B NMR spectra of 3 (C6D6, 298 K) give rise to broad triplets at approximately 25 ppm (1JBFca. 75 Hz), whereas the 19F NMR spectra give rise to pseudo-doublets (quadrupole-collapsed equal-intensity quartets) between ca. −57 and −90 ppm. The 11B and 19F chemical shifts calculated for 3 in C6H6 using DFT agree well with the experimental values (Table S2†).The crystal structures of 3 at 150 K (Fig. 1, top), refined to high accuracy using non-spherical atomic scattering factors (see the ESI†), reveal B–F bonds shorter than in BF3 (in Å, 1.26–1.31),28 similar to previously reported ArBF2 (1.307(2)–1.3146(15)),29,30 B–BF2 (1.24(3)–1.359(5)),31–34 and Pt–BF2 (1.324(7)–1.344(4))32,35–37 systems, but contracted in comparison to [ArBF3]−, e.g., [N(nBu)4][PhBF3] (1.409(3)–1.426(2)).38 When ordered in terms of steric bulk, the B–F bonds elongate only subtly from 3h (least hindered) to 3a (most), but are significantly shorter in 3b, comparably to EindBF2 and TerBF2.29,30 In contrast, a more conspicuous contraction of the B–C bond occurs with increasing bulk in 3. These trends are attributed to, at least in part, steric pressure caused by crystal packing (vide infra), as no obvious trends in geometric parameters were observed in solution-phase optimised geometries (Table S3†), which otherwise agree well with the experimental data.
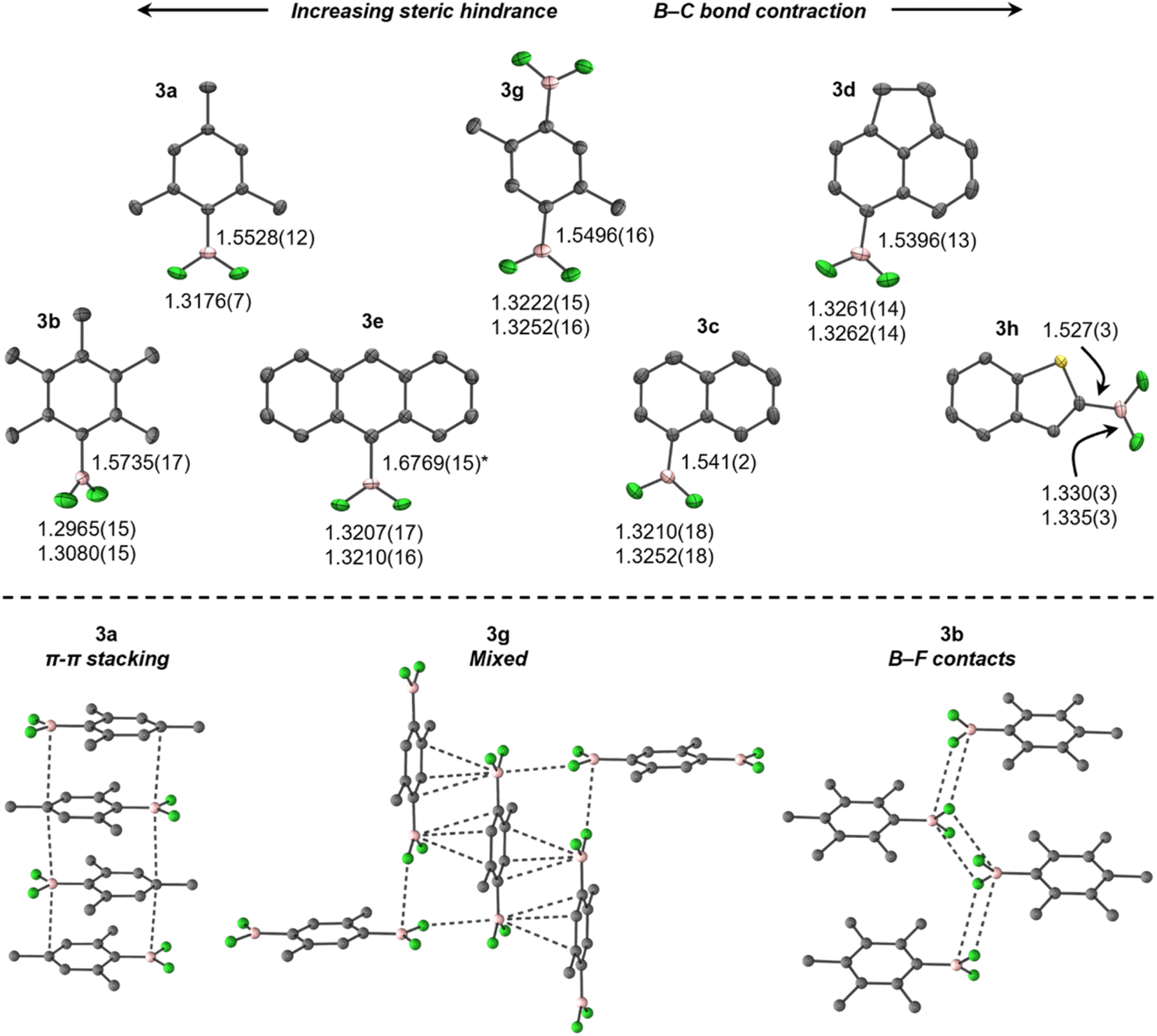 | ||
| Fig. 1 Top: Thermal ellipsoid plots (50% probability level) of aryldifluoroboranes (3), with B–C and B–F bond lengths (in Å). Colours denote F (green), B (pink), and C (grey), with H atoms omitted for clarity. Asterisk (*) denotes an artificially elongated B–C bond due to 50/50 H/BF2 disorder. Bottom: Selected crystal packing diagrams for 3. | ||
The crystal structures of 3 exhibit diverse solid-state interactions (Fig. 1, bottom). Their planarity facilitates π–π stacking wherein the BF2 moieties are situated over comparatively electron-rich aryl rings in adjacent molecules, except for 3b, which is non-planar (τ(F1–B–C1–C2) = 60.58(15)°) and prefers fluorous layers containing zig-zag chains of B–F contacts. Interestingly, 3g and 3h adopt B–F contacts at only one face of the boron atom, which is flanked by a π–π stacking/B–S interaction (Fig. 2). The contacts are weaker in 3b (in Å, 3.326), 3g (3.070) and 3h (3.210) than BF3 (2.68–2.71),28 but still within the sum of van der Waals radii (3.39).39 In contrast, steric crowding in EindBF2 and TerBF2 prevents any notable contacts to boron.29,30
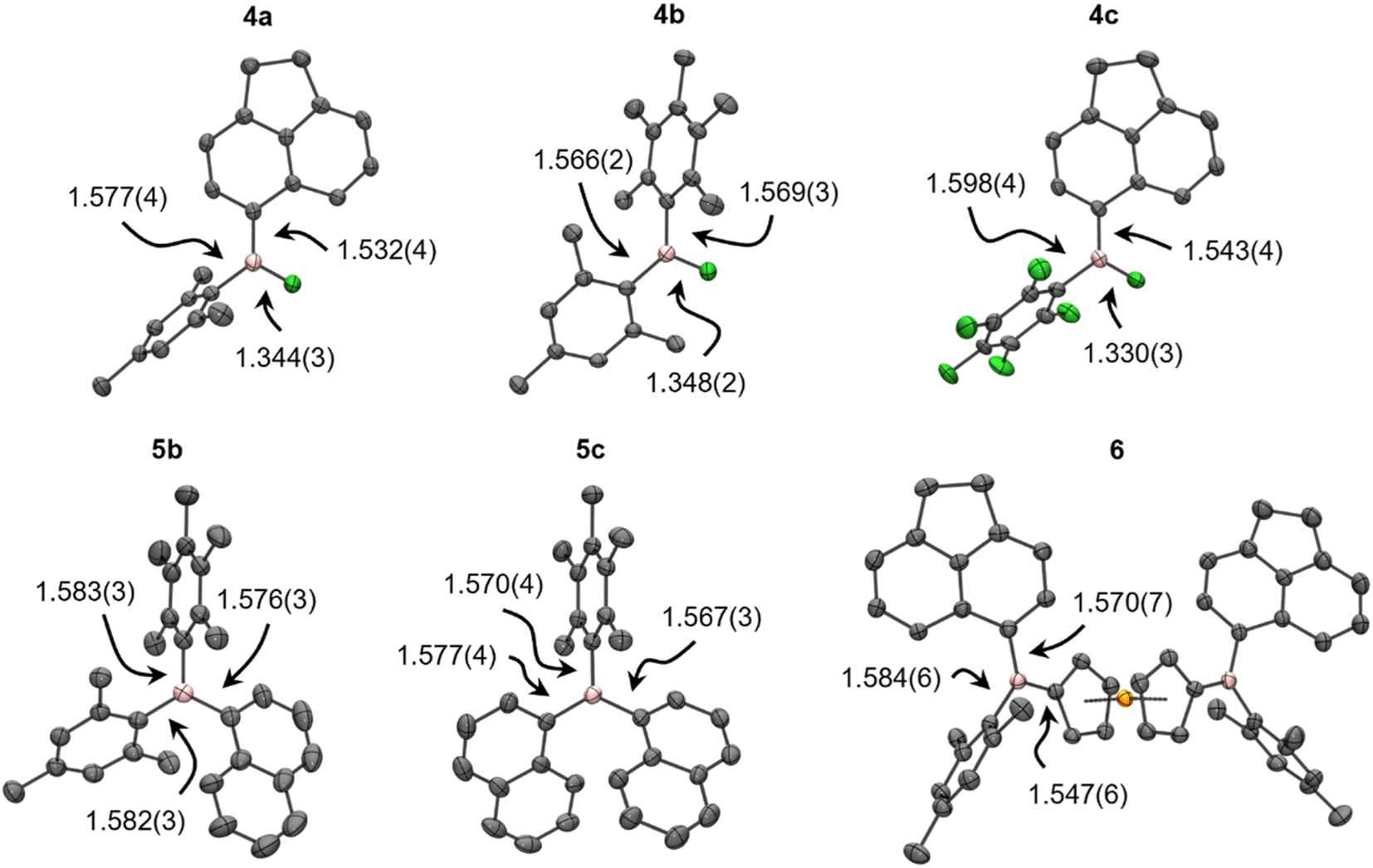 | ||
| Fig. 2 Thermal ellipsoid plots of diaryl- (4, 150 K, 50% probability level) and triarylboranes (5b, 5c and 6, 298 K, 20% probability level), with B–C and B–F bond lengths (in Å). Colours denote Fe (orange), F (green), B (pink), and C (grey), with H atoms omitted for clarity. | ||
The large difference in torsion angle and packing motif between 3a and 3b is notable, given that they differ only by the presence of meta-methyl groups in 3b. The Ci–Co–CMe angle in 3a (123.02(6)°) suggests a slight repulsion of the ortho-methyl groups from the BF2 moiety to facilitate planarity, but this is seemingly blocked in 3b (120.36(11), 120.60(11)°) and thus torsion occurs. This is attributed to a resonance effect from boron in 3a, rather than crystal packing, as it is reflected in the optimised geometries (123.04, 123.14° in 3avs. 120.67, 120.76° in 3b). The planarity and presence of π–π stacking in 3a may also rationalise its crystallinity versus heavier MesBX2, which are minimally volatile liquids at ambient temperature.40,41 This is supported by calculated solution-phase 3a/MesBX2 head-to-tail dimers, which show considerable disruption of stacking by twisting of the larger BX2 moieties out of plane (Fig. S3†). The enthalpies and Gibbs energies reflect this, being most exothermic and least endergonic for 3a (Fig. S3†).
Syntheses of di- (4) and triarylboranes (5/6)
To establish the utility of 3 in organoboron chemistry, derivatisation with aryl nucleophiles was performed (Scheme 3). Stoichiometric reactions with simple, moderately sterically shielded aryllithium or Grignard reagents, i.e., MesLi and C6F5MgBr, were found to selectively produce unsymmetrical ArAr′BF (4a–d) in quantitative yield after ca. 2 min at ambient temperature. These could be isolated in high yield after recrystallisation or derivatised in one pot to unsymmetrical naphthyl- (5a, 5b) and ferrocenyl-substituted (6) boranes with the respective aryllithium reagent in similarly rapid conversion and high yield. The generation of less hindered ArAr′BF using 3d shows that steric freedom in 3 does not negatively impact selectivity upon transmetallation under these circumstances. Furthermore, the convenient generation of unsymmetrical C6F5-substituted boron using C6F5MgBr in Et2O is especially notable considering the importance of electron-withdrawing groups in boron chemistry. By contrast, typical approaches have used C6F5Cu and introduced C6F5 as a final step.27 Overall, these reactions demonstrate that 4 are versatile precursors that, for the first time, allow for direct access to unsymmetrical boranes within minutes. | ||
| Scheme 3 Isolated yields of transmetallations with aryldifluoroboranes (3) to form di- (4) and triarylboranes (5–7). [a] Reaction was run in pentane at −35 °C. | ||
Reactions of 3 with less hindered NaphLi and ArFMgBr (ArF = 3,5-(CF3)2C6H3) were found to result in distributions of the desired ArAr′BF, symmetrical BAr(Ar′)2, and unreacted 3, and were investigated by NMR spectroscopy (Table S1†). Under typical conditions, i.e., in Et2O at ambient temperature, 3b and NaphLi afforded the desired ArAr′BF in only ca. 10% NMR yield, though symmetrical BAr(Ar′)25c was generated quantitatively with 2 equiv. of NaphLi. This yield increased to ca. 40 and 60% in benzene and pentane, respectively, though cooling the reaction in pentane to −35 °C had no effect on yield. The steric profile of 3 also began to influence product distribution, as 3a and 3d afforded the corresponding ArAr′BF in 40 and 10% yields upon reaction with NaphLi in pentane. Notably, the use of electron withdrawing ArFMgBr significantly improved selectively, as reactions of 3a with ArFMgBr in Et2O afforded ArAr′BF in 70% yield despite the more polar solvent and absence of ortho-substitution. In contrast, bulky dibromoborane 2b showed no selectivity during transmetallation with MesLi or NaphLi.
These findings highlight the specific utility of fluoroboranes in transmetallations with aryllithium and Grignard reagents. We attribute this to the high electrophilicity of boron improving the rate of B–C bond formation, while the strength of the B–F bond mitigates F− dissociation and subsequent overreaction.8 The observed effects of solvent, sterics of the borane and aryl nucleophile, and electronics of the nucleophile, on selectivity serve as complements to previous observations on solvent and electronic effects in reactions of B(OR)3 (R = alkyl) with Grignard reagents.8
Spectroscopic and crystallographic studies of 4–6
The asymmetry of 4–6 was established by complete assignment of the multi-nuclear NMR spectra (see the ESI†). The transformations of 3 to 4, and then 4 to 5/6, were easily monitored by 11B and 19F NMR spectroscopy, the former revealing high-frequency shifts to ca. 50, then 75 ppm, and the later showing shifts to >−40 ppm before disappearing entirely. The calculated 11B chemical shifts of 4–6 agree well with experimental data, though the 19F resonances of the B–F moiety in 4 are systematically overestimated to varying extents (≤20 ppm), which is attributed to dynamic aryl ring rotation not being captured in the static shielding tensor calculations.In addition to NMR spectroscopy, the asymmetry of 4–6 was confirmed, where possible, using X-ray crystallography (Fig. 2, Table S5†); to our knowledge, the crystal structures of 4a–c are the first to be reported for unsymmetrical ArAr′BF. The B–F bonds are significantly longer than in 3, more comparable to symmetric Ar2BF (1.312(3)–1.354(2) Å).15,29,42–44 The B–C bonds are, however, insignificantly different from their counterparts in 3. The trigonal plane at boron is coplanar with the less sterically hindered aryl group and the increased steric shielding prevents any notable solid-state contacts to boron. Meanwhile, increased strain in 5b, 5c results in B–C bonds elongated to a similar extent as in, e.g., BMes3 (1.58 Å).45 Likewise, the B–C bonds in 6 reflect those observed in unsymmetrical 1-borylferrocenes (1.546(5)–1.553(3) Å for B–C(Fc), 1.567(6)–1.585(5) Å otherwise).16
Conclusions
In conclusion, we have developed a facile and general synthetic route to solid aryldifluoroboranes, allowing for their crystallographic study. This has revealed subtle trends in solid-state geometric parameters as a function of the steric demands of the aryl group, as well as the diversity of their crystal packing motifs. Furthermore, they are shown to react selectively with aryllithium reagents to form unsymmetrical di- and triarylboranes within minutes, with a selectivity that arises from steric and electronic factors. Thus, aryldifluoroboranes may provide an accessible avenue for modular borane synthesis with broad potential application in organoboron chemistry; investigations into further preparation and functionalisation of such unsymmetrical species are currently underway.Data availability
The data that supports the findings of this article are included in the ESI.†Author contributions
Experiments and computations were designed and performed by D. T. Data analysis and manuscript preparation were performed by D. T. The project was overseen by M.-A. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Alexander S. Wahba for high-resolution mass-spectrometric analysis, Prof. Scott Bohle for access to his Bruker Apex II diffractometer, along with Thierry Maris and Daniel Chartrand at l'g Université de Montréal for access to their Bruker Metaljet X-ray diffractometer. We also thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for a Discovery Grant (M.-A. L.; RGPIN-2021-03814) and Postdoctoral Fellowship (D. T.), as well as McGill University, the Canada Foundation for Innovation (CFI), and the Digital Research Alliance of Canada for funding and resources.Notes and references
- J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC.
- V. Nori, F. Pesciaioli, A. Sinibaldi, G. Giorgianni and A. Carlone, Catalysts, 2022, 12, 5 CrossRef CAS.
- H. Zhao, L. A. Leamer and F. P. Gabbaï, Dalton Trans., 2013, 42, 8164–8178 RSC.
- S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS.
- S. M. Berger and T. B. Marder, Mater. Horiz., 2022, 9, 112–120 RSC.
- S. M. Berger, M. Ferger and T. B. Marder, Chem.– Eur. J., 2021, 27, 7043–7058 CrossRef CAS PubMed.
- T. Klis, A. Libura and J. Serwatowski, Main Group Met. Chem., 2002, 25, 479–484 CAS.
- R. J. Blagg and G. G. Wildgoose, RSC Adv., 2016, 6, 42421–42427 RSC.
- M. J. Kelly, R. Tirfoin, J. Gilbert and S. Aldridge, J. Organomet. Chem., 2014, 769, 11–16 CrossRef CAS.
- M. Ito, E. Ito, M. Hirai and S. Yamaguchi, J. Org. Chem., 2018, 83, 8449–8456 CrossRef CAS PubMed.
- J. Liu, S. Zhang, C. Zhang, J. Dong, C. Shen, J. Zhu, H. Xu, M. Fu, G. Yang and X. Zhang, Chem. Commun., 2017, 53, 11476–11479 RSC.
- J. Liu, C. Zhang, J. Dong, J. Zhu, C. Shen, G. Yang and X. Zhang, New J. Chem., 2017, 41, 4733–4737 RSC.
- J. Liu, C. Zhang, J. Dong, J. Zhu, C. Shen, G. Yang and X. Zhang, RSC Adv., 2017, 7, 14511–14515 RSC.
- X. Yin, K. Liu, Y. Ren, R. A. Lalancette, Y.-L. Loo and F. Jäkle, Chem. Sci., 2017, 8, 5497–5505 RSC.
- S. M. Cornet, K. B. Dillon, C. D. Entwistle, M. A. Fox, A. E. Goeta, H. P. Goodwin, T. B. Marder and A. L. Thompson, Dalton Trans., 2003, 4395–4405 RSC.
- H. Li, A. Sundararaman, T. Pakkirisamy, K. Venkatasubbaiah, F. Schödel and F. Jäkle, Macromolecules, 2011, 44, 95–103 CrossRef CAS.
- A. Sundararaman, M. Victor, R. Varughese and F. Jäkle, J. Am. Chem. Soc., 2005, 127(40), 13748–13749 CrossRef CAS PubMed.
- K. Parab, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2006, 128(39), 12879–12885 CrossRef CAS PubMed.
- K. Parab, A. Doshi, F. Cheng and F. Jäkle, Macromol., 2011, 44(15), 5961–5967 CrossRef CAS.
- M. Ferger, S. M. Berger, F. Rauch, M. Schönitz, J. Rühe, J. Krebs, A. Friedrich and T. B. Marder, Chem. Eur.– J., 2021, 27(35), 9094–9101 CrossRef CAS PubMed.
- O. Farooq, J. Fluorine Chem., 1995, 70, 225–227 CrossRef CAS.
- G. Bir, W. Schacht and D. Kaufmann, J. Organomet. Chem., 1988, 340, 267–271 CrossRef CAS.
- D. Kaufmann, Chem. Ber., 1987, 120, 853–854 CrossRef CAS.
- H.-J. Frohn, H. Franke, P. Fritzen and V. V. Bardin, J. Organomet. Chem., 2000, 598, 127–135 CrossRef CAS.
- M. M. Shmakov, S. A. Prikhod'ko, R. Y. Peshkov, V. V. Bardin and N. Y. Adonin, Mol. Catal., 2022, 521, 112202 CrossRef CAS.
- J. Lozada, W. Xuan Lin, R. M. Cao-Shen, R. Astoria Tai and D. M. Perrin, Angew. Chem., Int. Ed., 2023, 62, e202215371 CrossRef CAS PubMed.
- A. Sundararaman, K. Venkatasubbaiah, M. Victor, L. N. Zakharov, A. L. Rheingold and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 16554–16565 CrossRef CAS PubMed.
- D. Mootz and M. Steffen, Angew. Chem., Int. Ed. Engl., 1980, 19, 483–484 CrossRef.
- D. Duvinage, L. A. Malaspina, S. Grabowsky, S. Mebs and J. Beckmann, Eur. J. Inorg. Chem., 2023, 26, e202200482 CrossRef CAS.
- T. Murosaki, S. Kaneda, R. Maruhashi, K. Sadamori, Y. Shoji, K. Tamao, D. Hashizume, N. Hayakawa and T. Matsuo, Organometallics, 2016, 35, 3397–3405 CrossRef CAS.
- J. C. Jeffery, N. C. Norman, J. A. J. Pardoe and P. L. Timms, Chem. Commun., 2000, 2367–2368 RSC.
- J. H. Muessig, D. Prieschl, A. Deißenberger, R. D. Dewhurst, M. Dietz, J. O. C. Jiménez-Halla, A. Trumpp, S. R. Wang, C. Brunecker, A. Haefner, A. Gärtner, T. Thiess, J. Böhnke, K. Radacki, R. Bertermann, T. B. Marder and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 13056–13063 CrossRef CAS PubMed.
- N. Arnold, H. Braunschweig, A. Damme, R. D. Dewhurst, L. Pentecost, K. Radacki, S. Stellwag-Konertz, T. Thiess, A. Trumpp and A. Vargas, Chem. Commun., 2016, 52, 4898–4901 RSC.
- M. Arrowsmith, S. Endres, M. Heinz, V. Nestler, M. C. Holthausen and H. Braunschweig, Chem.– Eur. J., 2021, 27, 17660–17668 CrossRef CAS PubMed.
- A. Kerr, N. C. Norman, A. G. Orpen, M. J. Quayle, C. R. Rice, P. L. Timms, G. R. Whittell, A. Kerr and T. B. Marder, Chem. Commun., 1998, 319–320 RSC.
- N. Lu, N. C. Norman, A. Guy Orpen, M. J. Quayle, P. L. Timms and G. R. Whittell, J. Chem. Soc., Dalton Trans., 2000, 4032–4037 RSC.
- J. Bauer, H. Braunschweig, K. Kraft and K. Radacki, Angew. Chem., Int. Ed., 2011, 50, 10457–10460 CrossRef CAS PubMed.
- T. D. Quach, R. A. Batey and A. J. Lough, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o688–o689 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- W. Haubold, J. Herdtle, W. Gollinger and W. Einholz, J. Organomet. Chem., 1986, 315, 1–8 CrossRef CAS.
- W. Schacht and D. Kaufmann, Chem. Ber., 1987, 120, 1331–1338 CrossRef CAS.
- K. Samigullin, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 3564–3569 CrossRef CAS.
- S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. Blanchard-Desce, L. Meinel and T. B. Marder, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS PubMed.
- K. Samigullin, Y. Soltani, H.-W. Lerner, M. Wagner and M. Bolte, Acta Crystallogr., Sect. C: Struct. Chem., 2016, 72, 189–197 CrossRef CAS PubMed.
- J. F. Blount, P. Finocchiaro, D. Gust and K. Mislow, J. Am. Chem. Soc., 1973, 95, 7019–7029 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2292569–2292582 and 2305672. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05071h |
| This journal is © The Royal Society of Chemistry 2023 |